

Abstract
Both GnRH and activin are crucial for the correct function of pituitary gonadotrope cells. GnRH regulates LH and FSH synthesis and secretion and gonadotrope proliferation, whereas activin is essential for expression of FSH. Little is known, however, about the interplay of signaling downstream of these two hormones. In this study, we undertook expression profiling to determine how activin pre-treatment alters the transcriptional response of LβT2 gonadotrope cells to GnRH stimulation. Activin treatment alone altered the transcriptional profile of 303 genes including inducing that of the 17β-hydroxysteroid dehydrogenase B1 gene that converts estrone to 17β-estradiol, altering the sensitivity of the cells to estrone. Furthermore, activin had a dramatic effect on the response of LβT2 cells to GnRH. Hierarchical clustering of 2453 GnRH-responsive genes identified groups of genes the response of which to GnRH was either enhanced or blunted after activin treatment. Mapping of these genes to gene ontology classifications or signaling pathways highlighted significant differences in the classes of altered genes. In the presence of activin, GnRH regulates genes in pathways controlling cell energetics, cytoskeletal rearrangements, organelle organization, and mitosis in the absence of activin, but genes controlling protein processing, cell differentiation, and secretion. Therefore, we demonstrated that activin enhanced GnRH induction of p38MAPK activity, caused GnRH-dependent phosphorylation of p53, and reduced the ability of GnRH to cause G1 arrest. Thus, although activin alone changes a modest number of transcripts, activin pretreatment dramatically alters the response to GnRH from an antiproliferative response to a more differentiated, synthetic response appropriate for a secretory cell.
GnRH is a hypothalamic peptide central to normal mammalian reproductive function. It acts on anterior pituitary gonadotrope cells to trigger the synthesis and secretion of the gonadotropins LH and FSH, which together stimulate the production of gonadal steroids essential for reproductive development and function (1, 2). The gonadotropins are members of the glycoprotein hormone family that is characterized by a shared α-subunit and a unique, hormone-defining, β-subunit (LHβ and FSHβ). GnRH is also essential for gonadotrope proliferation and development because the hpg mouse, which harbors a deletion in the mouse GnRH gene, has fewer gonadotropes in the anterior pituitary, as well as decreased circulating levels of LH and FSH, and is infertile (3). The hypothalamic-pituitary-gonadal axis is also critical to human fertility. Cases of hypogonadotropic hypogonadism in humans can be traced to mutations in the GnRH receptor gene on chromosome 4q21 that cause GnRH resistance (4–6).
In vitro promoter studies have suggested that the effects of GnRH on LHβ and FSHβ are distinct. The induction of the LHβ promoter by GnRH is mediated by synergistic interactions between steroidogenic factor 1 (SF-1) and Egr-1 (7–9). SF-1 null mice are infertile because SF-1 is essential for adrenal and gonadal development (10–12); however, a pituitary-specific deletion of SF-1 in mice causes hypogonadotropic hypogonadism with decreased LH and FSH in both males and females (13). Egr-1 knockout mice are sub-fertile due to LH deficiency but have normal FSH levels (14). Male Egr-1-deficient mice, however, are fertile due to partial compensation by Egr-4 in the pituitary (15). This Egr isoform appears to be more important in males, because female Egr-4 knockout mice are fertile but males are infertile due to testicular abnormalities (16). A second Egr-1 knockout demonstrates a more severe phenotype with both male and female infertility (17). The phenotype of the Egr-1 null mouse is less severe than LH receptor null mice, which demonstrate underdeveloped gonads and external genitalia (18, 19), as basal expression of LHβ by the gonadotrope is sufficient for normal gonadal development.
In contrast, induction of the FSHβ promoter appears to be mediated by the activator protein 1 (AP-1) family of factors (20, 21). Multiple complexes of c-fos, FosB, c-jun, and JunB have been detected on a novel AP-1 site in the mouse FSHβ promoter (22). Mice null for various AP-1 subunits have been reported, but very few have reproductive phenotypes due to redundant functions (23). More importantly, activin signaling is essential for FSHβ expression (21, 24, 25). Activin is a member of the TGFβ family and is composed of dimers of inhibin β-subunits (Inhb) (26). The action of activin is counteracted by inhibin produced by the gonads. Inhibin is a heterodimer of an inhibin α-subunit (Inha) with an inhibin β-subunit (Inhb). The activin receptor signals via both Sma- and Mad-related proteins (Smad)2/4 and Smad3/4 heterodimers to regulate the FSHβ promoter through multiple sites (27–29). Smad null mice are embryonic lethal with multiple developmental defects (30–33), but deficiency of the activin type IIA receptor (Acvr2) results in female infertility and delayed fertility in males with decreased FSH but normal LH (34). The phenotype is very similar to the FSHβ knockout mouse (35) or to adenoviral over-expression of the extracellular domain of Acvr2 (36). Deletion of the activin/inhibin subunit βB (Inhbb) or overexpression of follistatin (Fst) also causes female subfertility (37–39), but inhibin α (Inha) null mice have elevated levels of FSH and infertility due to hyper-stimulation, as well as granulosa and Sertoli cell tumors (40). All of these genetic models point to an essential role for activin in selective stimulation of FSHβ expression.
Although the transcriptional response of LβT2 cells to GnRH has been published previously (41, 42), there have been few reports of the modulating effect of activin on this response. Because these hormones are the critical regulators of gonadotrope function, we initiated studies to investigate how activin exposure modulates the transcriptional response of LβT2 pituitary gonadotropes to GnRH. We demonstrate that whereas activin alone has little effect on gene expression in the gonadotrope, it dramatically alters the cellular response to subsequent GnRH stimulation. The data also indicate a role for activin in modulating the cell-cycle effects of GnRH, which may be important for gonadotrope proliferation during pituitary development.
RESULTS
Effect of Activin Treatment on Gene Expression in LβT2 Gonadotrope Cells
Initially, the effect of 24 h of activin treatment on basal gene expression was determined. RNA from duplicate experiments was hybridized to Affymetrix MU74Av2 oligonucleotide chips, which sample 12,000 genes and expressed sequence tags. Data were analyzed by four independent statistical approaches, as described in Materials and Methods. Of the 12,000 genes and expressed sequence tags, 6,291 are expressed in basal LβT2 cells. The normalized log mean expression in activin-treated cells is plotted against the log mean expression in untreated cells (Fig. 1A). Genes showing greater than 2-fold up- or down-regulation by activin are indicated. Statistical analysis of activin-regulated gene expression using either standard or permuted t test approaches did not reveal any significant changes after multiple testing correction (MTC). Normalization of the data to reduce noise at low expression levels did not improve our ability to detect changes. However, analysis of differential expression using the VAMPIRE Bayesian variance modeling approach (43) identified 94 genes that altered with activin treatment using Bonferroni multiple testing correction (αBonf < 0.05), or 303 genes using the less stringent false discovery rate (FDR, P < 0.05) (supplemental Tables 1 and 2, published as supplemental data on The Endocrine Society’s Journals Online web site at http://mend.endojournals.org). Eight genes identified on the arrays (Inhbb, RAD50, PN-1, Eph2, Crp2, Inha, GDF-9, and Tn1) were verified by quantitative PCR (Q-PCR) (Fig. 1B). All genes showed similar changes by Q-PCR and microarray. We also inspected the gonadotropin subunit genes and the GnRH receptor gene. Both LHβ and GnRH receptors were up-regulated 2-fold, but the α-subunit gene (Cga) was unchanged (Fig. 1C). The gene encoding the FSHβ subunit was not detectable by microarray, but was strongly induced by activin (20-fold) by Q-PCR. A similar magnitude 20-fold induction was observed for only one other gene, the 17 β-hydroxysteroid dehydrogenase (HSD) B1 gene (supplemental Table 1). This gene product converts estrone to 17 β-estradiol, a more potent estrogen (44). This observation suggested that activin may enhance estrone action in the pituitary. To test this hypothesis, LβT2 cells were transfected with an estrogen-responsive reporter gene (ERE-tk-luc) plus an estrogen receptor-α expression vector, and dose-response curves were determined for activation of the reporter by estradiol or estrone in the absence or presence of activin. In the absence of activin, estrone is a weaker estrogen and the dose-response curve is right-shifted as expected (Fig. 2A). In contrast, in the presence of activin, the dose-response curves for estradiol and estrone were superimposable (Fig. 2B).
Fig. 1. Gene Expression Profiling in LβT2 Cells in Response to Activin.

Cells were treated with 25 ng/ml activin or vehicle for 24 h. Total RNA was profiled on Affymetrix U74Av2 chips. A, Scatterplot of expression levels from activin-treated (y-axis) or basal cells (x-axis). Data are normalized using CORGON and log transformed. Parallel lines indicate 2-fold changes. Units are relative fluorescence. B, Validation of selected genes by Q-PCR. Graph shows fold change with activin treatment as measured by microarray (open bars) or Q-PCR (black bars). RNAs are from four independent experiments. C, Induction of gonadotropin and GnRH-receptor genes by activin by microarray (open bars) or Q-PCR (black bars). Gene symbols are consistent with National Center for Biotechnology Information Entrez Gene nomenclature. ND, Not detectable.
Fig. 2. Activin Treatment Enhances the Response of LβT2 Cells to Estrone.

Cells were transfected with an estrogen response element (ERE)-tk-luc reporter gene and an estrogen receptor-α expression vector along with a β-galactosidase control plasmid. After transfection, cells were treated with vehicle (panel A) or 25 ng/ml activin (panel B) for 24 h, and then with increasing doses of estradiol or estrone for a further 24 h. Luciferase activity is normalized for the cotransfected β-galactosidase activity. The experiment was repeated four times in duplicate, and the results show the mean ± SEM. Asterisks indicate P < 0.05 for estrone vs. estradiol.
Mapping of the differentially expressed genes to gene ontology classifications or metabolic and signaling pathways was uninformative, and no statistical overrepresentation was found. Gene Set Enrichment Analysis (GSEA) on all gene data (45), however, highlighted six pathways that are overrepresented in the activin-treated data set using a FDR of 0.05 (Table 1). Two of the six pathways involve mRNA splicing and processing (48% and 44% genes enriched, respectively). A third involves regulation of protein translation by the mTOR (mammalian target of rapamycin) pathway (32% enriched). The overrepresentation of these three pathways suggests that activin treatment increases the capacity of the cells for protein synthesis. The other pathways contain cell cycle and DNA damage repair proteins (38% and 40% enriched, respectively). A large number of stem cell genes (Fortunel_Stemness) are also enriched (138 of 380, or 36%). This list of genes was compiled by Fortunel et al. (46) from a study of embryonic stem cells, neural stem cells, and retinal progenitor/stem cells compared with adult brain and retina. The genes are common to all three stem cell lineages and have been termed putative “stemness” genes. We conclude that activin treatment per se does not greatly alter the transcriptional network of LβT2 gonadotrope cells because only two genes show large changes in expression; however, many genes show small but coordinate changes leading to significant alterations in cellular pathways.
Table 1.
Gene Set Enrichment Analysis of Activin-Treated Lβ T2 Cells
| Name | Size | ES | NES | NOM P Value | FDR q Value | FWER P Value |
|---|---|---|---|---|---|---|
| mRNA_splicing | 50 | −0.597 | −2.067 | 0 | 0 | 0 |
| CR_REPAIR | 45 | −0.624 | −2.062 | 0 | 0 | 0 |
| FORTUNEL_STEMNESS | 380 | −0.450 | −2.034 | 0 | 0 | 0 |
| MRNA_processing | 48 | −0.592 | −1.922 | 0 | 0.014 | 0.06 |
| igf1mtorPathway | 27 | −0.598 | −1.858 | 0 | 0.028 | 0.14 |
| Cell_cycle_checkpoint | 32 | −0.606 | −1.802 | 0 | 0.046 | 0.24 |
Size, Number of genes in set; ES, enrichment score; NES, normalized enrichment score; NOM, nominal; FDR, false discovery rate; FWER, family wise error rate.
Effect of Activin Pretreatment on the Subsequent Transcriptional Response of LβT2 Cells to GnRH
The following experiments were designed to test whether activin pretreatment would alter the response of the cells to a subsequent challenge with GnRH. Both acute (4-to 6-h) and chronic (24-h) effects on gonadotropin subunit gene transcription have been described. Therefore, cells were pretreated with serum-free medium with or without 25 ng/ml activin for 24 h and then stimulated with 100 nM GnRH for 4, 8, or 24 h. RNA from duplicate experiments was hybridized to independent MU74Av2 arrays. When all conditions are combined, 8395 genes have measurable expression in two or more samples. The quality of the sample chip data was inspected using principal component analysis. The chips clustered according to both activin pretreatment and time of GnRH treatment (Fig. 3A). None of the chips are outliers by this metric.
Fig. 3. Gene Expression Profiling in LβT2 Cells in Response to GnRH.

Cells were treated with 25 ng/ml activin or vehicle for 24 h and then with GnRH for 0, 4, 8, and 24 h. Total RNA was profiled on Affymetrix U74Av2 chips. A, Principal Component (PC) Analysis on all chip data. Left panel shows chips clustered by activin pretreatment. Right panel shows chips clustered by time of GnRH treatment. Chips are shown by red spots. Ellipsoids indicate 2 SDS from the mean. B, Data analysis scheme. Four independent statistical approaches were taken. The resulting gene lists were compared to generate the final list of all GnRH-regulated genes. C, Hierarchical clustering of the 2453 GnRH-regulated genes. Data are log transformed and median normalized. Gene tree in blue to left indicates relationship of clusters. Green indicates decreased expression; red indicates increased. Clusters are indicated by white boxes to the right of the heat map. D, Expression profiles of the indicated clusters. Data are log-transformed, median-normalized MAS data. Horizontal axis shows time of GnRH treatment; vertical axis shows relative fluorescence signal. Lists of genes in the clusters are given in supplemental Table 4. Act., Activin; PC, Principal component; reg., regulation.
Data were analyzed by multiple independent approaches (Fig. 3B). First, individual times were compared with their respective zero times using the variance modeling approach in VAMPIRE and combined to generate the sets of GnRH-regulated genes in the absence or presence of activin pretreatment. Using Bonferroni MTC, 2453 genes were regulated by GnRH treatment (αBonf < 0.05). Of these, 1991 genes were altered by GnRH in the absence of activin, 1265 genes were altered in the presence of activin, and 803 genes were regulated by GnRH under both conditions. Second, the data were normalized by the Robust Multichip Average method (47) consisting of background adjustment and quantile normalization and summarization, and then analyzed by mixed-model three-way analysis of covariance (ANCOVA) with activin and GnRH treatments as categorical variables, and time of treatment as a quantitative covariant. Inspection of the source of variation showed that GnRH and time were the major contributors to variance with activin barely exceeding the background error, in agreement with our previous conclusion that activin treatment per se does not greatly alter the transcriptional profile. Using a Bonferroni MTC (αBonf < 0.05), 454 genes were altered; the majority were GnRH and time, but not activin, dependent. This number increased to 3952 using a less stringent FDR correction.
Third, data were normalized using the CORGON multiplicative noise model (48) and then analyzed by two-way ANOVA to identify genes regulated by GnRH or activin. Only 254 genes are GnRH or activin dependent using Bonferroni MTC. Lastly, individual time courses of GnRH treatment, in the presence or absence of activin, were analyzed by a permuted multi-class t test implemented in Significance Analysis of Microarrays (49). After the FDR was minimized, 559 genes were regulated by GnRH.
By inspecting the overlap of the statistically significant changes derived from the four different approaches, we determined that the variance modeling approach using Bonferroni multiple testing correction on the raw expression data gave the greatest number of significant genes and contained all of genes found by ANCOVA, ANOVA or permuted t test. Surprisingly, the variance modeling method with αBonf < 0.05 also found 47%, 52%, and 48% of the genes found by the other methods using less stringent FDR correction. Therefore, the list of 2453 genes generated by the variance modeling approach was used for further analysis (supplemental Table 3).
The 2453 regulated genes were subjected to hierarchical clustering (Fig. 3C). Many genes were coordinately regulated by GnRH in both the presence and absence of activin, but a number of gene clusters were evident that showed differential expression in the presence of activin. The gene tree showing the relationship of the clusters is depicted to the left. Expression profiles of four clusters are presented graphically. The top cluster contains genes induced by GnRH only in the absence of activin. The second cluster contains genes that are induced by GnRH in the absence or presence of activin. The third cluster contains genes that are repressed by GnRH in the absence of activin but that are independent of GnRH in the presence of activin. This cluster has a statistical overrepresentation of mitochondrial genes. The bottom cluster contains genes induced by GnRH only in the presence of activin. The genes present in these clusters are listed in the supplemental Table 4. Ten representative genes (Actg, Phlda1, Spp1, IL-1rn, Wdr1, IGFBP5, PAI-1, RhoB, Tubb5, and Car2) were verified by Q-PCR. All genes showed similar changes by Q-PCR and GeneChip (Fig. 4).
Fig. 4. Validation of Microarray Data by Q-PCR.

Ten genes were chosen from the list of differentially regulated genes for validation on RNA from four independent experiments. Each Q-PCR measurement was made in triplicate. Data are standardized to the ATPase6d1 gene (vacuolar ATP synthase subunit d1 or physophilin). Left-hand panel for each gene shows the microarray results; right-hand panel shows the Q-PCR data. Black bars (■) denote activin-treated cells; open bars (□) denote untreated cells. Vertical axis shows relative fluorescence units; horizontal axis shows time of GnRH treatment.
To identify genes that show a differential response to GnRH when cells are exposed to activin, we analyzed the 2453 GnRH-regulated genes using EDGE (50). This program fits a natural cubic spline curve to the time course data and then tests whether the actual curve is a significantly better fit than a null curve generated by averaging the data between treatments. The program also implements multiple testing correction via the FDR method to correct for the large number of genes analyzed. Using this approach, 406 genes show differential time courses in the presence or absence of activin (supplemental Table 5). The overlap of these genes with the GnRH-regulated data sets is shown (Fig. 5A). The genes were subjected to hierarchical clustering, and a number of clusters are apparent (Fig. 5B). Expression profiles of these clusters are shown (Fig. 5C). Genes present in these clusters are listed in supplemental Table 6. Many clusters resemble those derived earlier from the complete set of 2453 genes (Fig. 3C).
Fig. 5. Clustering of Differential Time Course Genes.

Genes that demonstrated differential GnRH time courses in the presence or absence of activin were determined using a natural cubic spline curve fit method in EDGE. A FDR of 0.05 was applied. A, Venn diagram showing overlap of the differentially expressed genes with the GnRH-regulated genes in the absence or presence of activin. B, Hierarchical clustering of the 406 differentially expressed genes. Data are log transformed and median normalized. Gene tree in blue to left indicates relationship of clusters. Green indicates decreased expression; red indicates increased expression. Clusters are indicated by white boxes to the right of the heat map. C, Expression profiles of the highlighted clusters. Data are log-transformed, median-normalized MAS data. Horizontal axis shows time of GnRH treatment; vertical axis shows relative fluorescence signal. Lists of genes in the clusters are given in supplemental Table 6.
Mapping Altered Genes to Gene Ontology Terms, Gene Set Enrichment Analysis, and Functional Pathways
The 2453 genes altered by GnRH were mapped to Gene Ontology classifications to determine whether particular groups were overrepresented (Table 2). At 4 h of GnRH, the overrepresented classifications contain immediate early genes that are implicated in cell cycle and metabolic control. At 8 h, the classifications began to diverge. The GnRH-regulated genes in the absence of activin are predominantly involved in cytoskeletal and organelle reorganization, whereas those regulated in the presence of activin are involved in protein processing and differentiation. After 24 h of GnRH treatment, the GnRH-regulated genes are involved in mitosis and cell division without activin, but in the presence of activin, they are involved in secretion and processing of extracellular proteins. This divergence suggested that activin might be conferring a more differentiated, less proliferative, secretory phenotype on the LβT2 cells. The 406 differential time course genes showed overrepresentation of genes involved in lipid metabolism and protein synthesis in the endoplasmic reticulum.
Table 2.
Assignment of GnRH-Regulated Genes to Gene Ontology Terms
| GO Group ID | Group Name | Gene Count | NOM P Value | FDR q Value |
|---|---|---|---|---|
| 4 h GnRH, no activin | ||||
| GO:0016043 | Cell organization and biogenesis | 102 | 1.75E −05 | 2.10E −04 |
| GO:0005856 | Cytoskeleton | 77 | 1.69E −05 | 1.16E −03 |
| GO:0000279 | M phase | 28 | 1.18E −05 | 7.12E −03 |
| 4 h GnRH, activin-treated | ||||
| GO:0019222 | Regulation of metabolism | 84 | 5.19E −05 | 5.97E −06 |
| GO:0000074 | Regulation of cell cycle | 29 | 4.09E −06 | 4.37E −03 |
| 8 h GnRH, no activin | ||||
| GO:0008092 | Cytoskeletal protein binding | 50 | 9.77E −08 | 9.04E −07 |
| GO:0005489 | Electron transporter activity | 27 | 1.23E −05 | 2.27E −06 |
| GO:0005856 | Cytoskeleton | 99 | 7.54E −08 | 4.70E −06 |
| GO:0003924 | GTPase activity | 29 | 2.83E −05 | 1.22E −05 |
| GO:0016043 | Cell organization and biogenesis | 123 | 4.91E −06 | 1.46E −05 |
| GO:0043228 | Non-membrane-bound organelle | 151 | 2.75E −06 | 9.45E −04 |
| GO:0003954 | NADH dehydrogenase activity | 11 | 9.42E −06 | 7.21E −03 |
| 8 h GnRH, activin-treated | ||||
| GO:0030333 | Antigen processing | 14 | 3.40E −06 | 1.65E −06 |
| GO:0019882 | Antigen presentation | 14 | 2.70E −05 | 1.51E −05 |
| GO:0030154 | Cell differentiation | 56 | 2.61E −05 | 2.34E −05 |
| GO:0008092 | Cytoskeletal protein binding | 34 | 9.28E −06 | 6.32E −05 |
| 24 h GnRH, no activin | ||||
| GO:0008283 | Cell proliferation | 94 | 9.04E −08 | 9.41E −07 |
| GO:0000279 | M phase | 32 | 4.00E −10 | 2.23E −05 |
| GO:0000910 | Cytokinesis | 27 | 2.25E −08 | 1.76E −04 |
| GO:0000278 | Mitotic cell cycle | 31 | 7.90E −09 | 2.71E −04 |
| GO:0007051 | Spindle organization and biogenesis | 6 | 5.21E −06 | 3.03E −04 |
| 24 h GnRH, activin-treated | ||||
| GO:0005576 | Extracellular region | 118 | 0.00E+00 | 0.00E+00 |
| GO:0005783 | Endoplasmic reticulum | 28 | 2.63E −05 | 2.26E −07 |
| GO:0016020 | Membrane | 120 | 3.03E −03 | 3.73E −05 |
| Differential time course genes | ||||
| GO:0005737 | Cytoplasm | 110 | 4.54E −04 | 2.10E −07 |
| GO:0006629 | Lipid metabolism | 28 | 1.67E −04 | 1.51E −05 |
| GO:0005783 | Endoplasmic reticulum | 30 | 1.09E −04 | 2.18E −05 |
NOM, Nominal; FDR, false discovery rate.
GSEA was performed by comparing each basal data set, with or without activin pretreatment, with its corresponding GnRH-stimulated data sets (Table 3). This analysis used all 12,000 probes rather than the list of GnRH-dependent genes. In the absence of activin, eight of 10 gene sets that are enriched in the basal data set encode metabolic or mitochondrial genes. The remaining two gene sets are the Fortunel_Stemness set of stem cell genes and a set of DNA repair genes. The finding of stem cell genes in LβT2 cells is consistent with immortalization of the gonadotropes at embryonic d 17 when LHβ is first expressed (51) and the decrease with GnRH stimulation is consistent with a role for GnRH to promote gonadotrope differentiation. The hpg mouse carrying a mutation in the GnRH gene is infertile and has few gonadotropes in the anterior pituitary, underscoring the role of GnRH in gonadotrope differentiation (3). Under GnRH stimulation, many signaling pathways are up-regulated including the mTOR, vascular endothelial growth factor, integrin, rho, calpain, cell motility, MAPK, and TGFβ Downstream of these pathways serum-, K-ras-, CCAAT enhancer binding protein α- and NF-κB dependent genes are induced. We also observe up-regulation of p53-dependent genes and the p53-hypoxia pathway. Cells that have been exposed to activin before GnRH have slightly different significant gene sets. Basally enriched gene sets include four mitochondrial or metabolic pathways and the DNA repair gene set, which are observed in the absence of activin, and, interestingly, a set of genes down-regulated by CCAAT enhancer binding protein α that are not seen in the absence of activin. When cells are stimulated with GnRH in the presence of activin, many of the same signaling pathways and gene sets are enriched as seen in the absence of activin, but there are some notable additions and omissions. Two immune related gene sets are enriched as is the p38MAPK pathway. The stress, vascular endothelial growth factor, and mTOR pathways are not enriched; neither are the hTERT-dependent genes. Thus, although there are many similarities in GnRH-regulated gene expression in the presence and absence of activin, there are distinct differences that may be important for gonadotrope biology.
Table 3.
Gene Set Enrichment Analysis of GnRH and Activin-Treated LβT2 Cells
| Name | Size | ES | NES | NOM P Value | FDR q Value | FWER P Value |
|---|---|---|---|---|---|---|
| Basal enriched, no activin | ||||||
| FORTUNEL_STEMNESS | 380 | 0.451 | 2.069 | 0.00E+00 | 1.42E−03 | 2.00E−03 |
| VOXPHOS | 88 | 0.534 | 2.057 | 0.00E+00 | 7.12E−04 | 2.00E−03 |
| MAP00280_Val/Leu/Iso_degradation | 28 | 0.667 | 2.044 | 0.00E+00 | 7.19E−04 | 3.00E−03 |
| Electron_Transport_Chain | 94 | 0.519 | 2.012 | 0.00E+00 | 1.10E−03 | 6.00E−03 |
| CR_REPAIR | 45 | 0.542 | 1.842 | 0.00E+00 | 1.10E−02 | 7.30E−02 |
| mitochondr | 418 | 0.391 | 1.826 | 0.00E+00 | 1.07E−02 | 8.30E−02 |
| GnRH enriched, no activin | ||||||
| SERUM_INDUCED_MKL_INDEP_SRF | 101 | −0.711 | −2.574 | 0.00E+00 | 0.00E+00 | 0.007E+00 |
| LEM_HSC | 182 | −0.577 | −2.265 | 0.00E+00 | 0.00E+00 | 0.00E+00 |
| NFKB_INDUCED | 102 | −0.614 | −2.226 | 0.00E+00 | 0.00E+00 | 0.00E+00 |
| DOWN_MEGS | 52 | −0.669 | −2.169 | 0.00E+00 | 0.00E+00 | 0.00E+00 |
| integrinPathway | 46 | −0.624 | −2.000 | 0.00E+00 | 1.25E−03 | 5.00E−03 |
| P53_UP | 33 | −0.659 | −1.943 | 0.00E+00 | 3.82E−03 | 1.90E−02 |
| mcalpainPathway | 28 | −0.674 | −1.906 | 0.00E+00 | 6.82E−03 | 3.90E−02 |
| KRAS_TOP100_KNOCKDOWN | 74 | −0.549 | −1.872 | 0.00E+00 | 8.59E−03 | 5.60E−02 |
| cell_motility | 105 | −0.509 | −1.860 | 0.00E+00 | 9.21E−03 | 6.80E−02 |
| TGF_Beta_Signaling_Pathway | 40 | −0.600 | −1.856 | 0.00E+00 | 8.52E−03 | 6.90E−02 |
| MELTON_200_HEMO | 174 | −0.475 | −1.844 | 0.00E+00 | 9.15E−03 | 8.20E−02 |
| ST_Integrin_Signaling_Pathway | 82 | −0.523 | −1.837 | 1.63E+03 | 9.47E−03 | 9.20E−02 |
| HTERT_UP | 115 | −0.495 | −1.831 | 0.00E+00 | 9.29E−03 | 9.80E−02 |
| Basal enriched, activin-treated | 94 | 0.685 | 2.126 | 0.00E+00 | 0.00E+00 | 0.00E+00 |
| Electron_Transport_Chain | ||||||
| VOXPHOS | 88 | 0.670 | 2.071 | 0.00E+00 | 5.42E−04 | 1.00E−03 |
| GnRH enriched, activin-treated | 101 | −0.764 | −2.032 | 0.00E+00 | 0.00E+00 | 0.00E+00 |
| SERUM_INDUCED_MKL_INDEP_SRF | ||||||
| P53_UP | 33 | −0.832 | −1.935 | 0.00E+00 | 0.00E+00 | 0.00E+00 |
| NFKB_INDUCED | 102 | −0.703 | −1.894 | 0.00E+00 | 0.00E+00 | 0.00E+00 |
| DOWN_MEGS | 52 | −0.742 | −1.820 | 0.00E+00 | 1.85E−03 | 7.00E−03 |
| cell_motility | 105 | −0.664 | −1.780 | 0.00E+00 | 4.11E−03 | 1.90E−02 |
| il1rPathway | 36 | −0.758 | −1.765 | 1.45E−03 | 4.88E−03 | 2.70E−02 |
| LEM_HSC | 182 | −0.618 | −1.749 | 0.00E+00 | 5.23E−03 | 3.40E−02 |
| CEBPA_UPREG | 46 | −0.718 | −1.717 | 0.00E+00 | 8.91E−03 | 6.30E−02 |
| EMT_DOWN | 48 | −0.711 | −1.710 | 0.00E+00 | 9.47E−03 | 7.60E−02 |
| EMT_UP | 83 | −0.650 | −1.705 | 0.00E+00 | 9.27E−03 | 8.20E−02 |
| tollPathway | 35 | −0.723 | −1.695 | 0.00E+00 | 1.00E−02 | 9.80E−02 |
Size, Number of genes in set; ES, enrichment score; NES, normalized enrichment score; NOM, nominal; FDR, false discovery rate; FWER, family wise error rate.
The 2453 GnRH-dependent genes were also mapped to known pathways using MetaCore (Table 4). In the absence of activin, the most significant GnRH-regulated pathway contains the anaphase-promoting complex (APC), which is a multisubunit E3 ubiquitin ligase that targets cell cycle proteins for destruction by the 26S proteasome (52) (Fig. 6A). Of the 52 genes in this pathway, 20 (or 38%) were altered by GnRH (P = 9.9 × 10−6). The background occurrence of genes in the nonsignificant curated pathways was 16.3 ± 0.1% (mean and SD). Of the 20 genes that are regulated, 18 decreased and only two are increased by GnRH stimulation. The repressed genes include cyclins B1, B2, and A2, four subunits of the APC, five cell cycle-associated kinases Cdk1, Bub1, Nek2a, STK6, and PLK1, the kinetochore-associated proteins KNSL4 and MAD2a, and the pituitary tumor transforming protein Pttg1. One of the two induced proteins, TCP1, is a molecular chaperone, whereas the other, Geminin, inhibits DNA replication by blocking formation of the prereplication complex. Other cell cycle pathways were similarly altered including ATM/ATR regulation, p53 signaling, and the DNA damage response. The genes in these cell cycle-related groups were combined and used to initiate nuclear interaction network generation. Molecular interactions were restricted to the nucleus, and path lengths were limited to one step to include only direct interactions. Of the 51 genes in the most significant network, 31 (P = 6.4 × 10−41; G score, −65.5) are root nodes derived from our data set of cell cycle-related genes (Fig. 6B). The network centers on the tumor suppressor p53 and the transcription factor c-Fos, which is induced by GnRH. Higher resolution images of the networks are published as supplemental data (supplemental networks).
Table 4.
Pathway Assignment of GnRH-Regulated Genes by MetaCore
| Pathway | NOM P Value | Gene Count | Total | % |
|---|---|---|---|---|
| GnRH-regulated, no activin | ||||
| Role APC in cell cycle regulation | 9.94E−06 | 20 | 52 | 38 |
| Putative integrins pathway | 2.27E−05 | 24 | 72 | 33 |
| A2BR signaling via G-α-q: PRKCB/G/Q | 1.15E−04 | 17 | 47 | 36 |
| Nuclear function of p38-MAPK | 2.08E−04 | 17 | 49 | 35 |
| DNA damage response: inhibition of CDK1 in G2 | 2.69E−04 | 14 | 37 | 38 |
| ATM/ATR regulation of G2/M checkpoint | 3.42E−04 | 15 | 42 | 36 |
| Mitochondrial unsaturated fatty acid β-oxidation | 3.75E−04 | 8 | 15 | 53 |
| P53 signaling pathway | 6.07E−04 | 15 | 44 | 34 |
| RhoA regulation pathway | 6.81E−04 | 14 | 40 | 35 |
| RalA regulation pathway | 7.51E−04 | 13 | 36 | 36 |
| c-KIT-pleckstrin homology (PH) proteins interactions | 1.51E−03 | 12 | 34 | 35 |
| Notch signaling pathway | 1.53E−03 | 14 | 43 | 33 |
| Regulation of actin cytoskeleton by ρ GTPases | 1.55E−03 | 18 | 62 | 29 |
| Mitochondrial long chain fatty acid β-oxidation | 1.73E−03 | 8 | 18 | 44 |
| G-Protein α-q signaling cascades | 2.28E−03 | 17 | 59 | 29 |
| Angiotensin signaling via β-Arrestin | 3.14E−03 | 14 | 46 | 30 |
| Cytoplasm/mitochondrial transport of Bad and Bax | 4.90E−03 | 15 | 53 | 28 |
| A2BR signaling via G-α-q: PRKCE-RASGRP | 5.78E−03 | 13 | 44 | 30 |
| ATM/ATR regulation of G1/S checkpoint | 7.18E−03 | 14 | 50 | 28 |
| Start of the mitosis | 8.00E−03 | 17 | 66 | 26 |
| Role PKA in cytoskeleton reorganization | 9.26E−03 | 20 | 83 | 24 |
| Ubiquinone metabolism | 9.76E−03 | 16 | 62 | 26 |
| GnRH-regulated, activin-treated | ||||
| Nuclear function of p38-MAPK | 1.52E−06 | 16 | 49 | 33 |
| Cholesterol biosynthesis | 3.03E−04 | 8 | 22 | 36 |
| Brca1 as transcription regulator | 6.55E−04 | 9 | 30 | 30 |
| DNA damage response: inhibition of CDK1 in G2 | 8.25E−04 | 10 | 37 | 27 |
| PLAU signaling | 1.68E−03 | 11 | 47 | 23 |
| ATM/ATR regulation of G2/M checkpoint | 2.36E−03 | 10 | 42 | 24 |
| Notch signaling pathway | 2.85E−03 | 10 | 43 | 23 |
| VEGF signaling via VEGFR2 | 3.42E−03 | 10 | 44 | 23 |
| P53 signaling pathway | 3.42E−03 | 10 | 44 | 23 |
| Cytoplasm/mitochondrial transport of Bad and Bax | 4.60E−03 | 11 | 53 | 21 |
| Integrin pathway | 7.49E−03 | 13 | 72 | 18 |
| Erk interactions: activation and selected Erk targets | 8.17E−03 | 11 | 57 | 19 |
| HLA-DR apoptosis regulation | 9.88E−03 | 8 | 36 | 22 |
| Differential time course genes | ||||
| Cholesterol biosynthesis | 1.08E−03 | 5 | 22 | 22 |
| Mitochondrial unsaturated fatty acid β-oxidation | 1.86E−03 | 4 | 15 | 27 |
| Macrophage migration inhibitory factor | 3.00E−03 | 10 | 97 | 10 |
| Role of APC in cell cycle regulation | 3.58E−03 | 7 | 54 | 13 |
| Prostaglandin 2 biosynthesis and metabolism | 4.71E−03 | 4 | 19 | 21 |
NOM, Nominal.
Fig. 6. Regulation of the APC by GnRH.

A, Highest scoring curated pathway regulated by GnRH in the absence of activin (P = 9.9 × 10−6). Black arrows indicate stimulatory interactions; black bars indicate inhibitory interactions. Green arrows indicate decreased expression; red arrows increased expression over the GnRH time course. B, Transcriptional interaction network generated from the combined list of cell cycle-related genes (P = 6.4 × 10−41, G score, −65.5). Yellow and white stars on the icons indicated multicomponent complexes or families. Changes in gene expression are indicated by the red arrows (up) or green arrows (down) next to the icon. Known interactions are shown by arrows. Red circles indicate the transcription factors that drive the network. Red spots indicate termination of the network due to the applied one-step filter. C, Legend of icons used in transcriptional network map. Original network is provided in the supplemental data.
A number of signaling pathways involving Gαq were also significantly repressed by GnRH, consistent with our previous data showing that chronic GnRH desensitizes LβT2 cells at the level of downstream signaling (53). Mitochondrial pathways are repressed. Expression of components of fatty acid oxidation is decreased. Carnitine-phosphoryl transferase 1a, which mediates fatty-acid uptake into mitochondria, is decreased, as is acyl-coenzyme A (CoA) acetyl-transferase, double-bond isomerase, enoyl-CoA hydratase, and acyl-CoA dehydrogenase. Of the 44 subunits of the reduced nicotinamide adenine dinucleotide-ubiquinone oxidoreductase complex (electron transport chain, complex I), 15 are also decreased (Fig. 7A). These data and the earlier GSEA results suggested that mitochondrial β-oxidation and electron transport were reduced by GnRH. Indeed measurement of mitochondrial activity using the redox-sensitive dye, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS), confirmed a 30% decrease after 24 h of GnRH in the absence but not presence of activin (Fig. 7B). Because many mitochondrial genes are regulated by the transcription factor Nrf-2, we tested whether GnRH regulates Nrf-2-dependent transcription (54). Two Nrf-2 dependent reporter genes containing one or four copies of an antioxidant response element that binds Nrf-2 (1xARE-tk-luc and 4xARE-tk-luc) were transfected into LβT2 cells and stimulated with GnRH for increasing times. GnRH caused a time-dependent decrease in reporter activity, which paralleled the decrease in mitochondrial gene expression (Fig. 7C).
Fig. 7. GnRH Represses Mitochondrial Function.
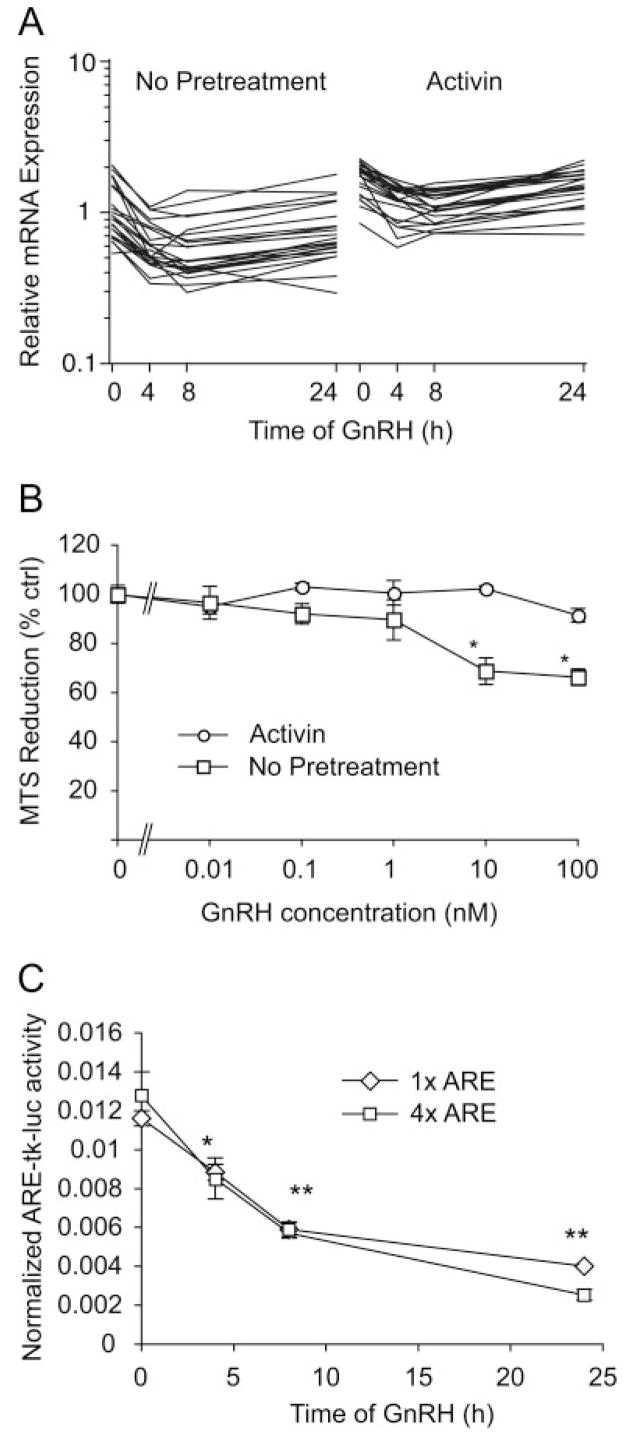
A, Expression profiles of mitochondrial genes that were found to be repressed by GnRH. Left panel shows expression data in cell not treated with activin; right panel shows data in activin-treated cells. Data are log-transformed, median-normalized MAS data. Horizontal axis shows time of GnRH treatment; vertical axis shows relative fluorescence signal. B, Mitochondrial activity is repressed by GnRH. Cells were treated with activin for vehicle for 24 h and then with increasing doses of GnRH for a further 24 h. Mitochondrial activity was measured with the redox-sensitive dye MTS by the increase in absorbance at 490 nm. Experiment was performed in quadruplicate on three independent occasions. C, Cells were transfected with 1xARE-tk-luc or 4xARE-tk-luc reporter genes and a β-galactosidase control plasmid. After transfection, cells were treated with 100 nM GnRH for increasing times. Luciferase activity is normalized for the cotransfected β-galactosidase activity. The experiment was repeated four times in duplicate. The results show the mean ± SEM; *, P < 0.05; **, P < 0.01 vs. zero time control. ARE, Antioxidant response element.
When the genes regulated by GnRH in the presence of activin were mapped to these pathways, a different set was obtained (Table 3). Notably, the APC, Gαq signaling, and mitochondrial pathways were not significantly altered by GnRH in the presence of activin. In contrast, new pathways such as cholesterol biosynthesis, the transcriptional regulator Brca1, urokinase signaling, and apoptosis regulation were significant using this gene set. The most significant pathway contains the nuclear functions of p38MAPK (P = 1.5 × 10−6). Thirty-three percent (16/49) of the genes are changed; 15 are increased (GADD45 α/β/γ, c-jun, NF-κB1/2, Sp1, Fra-1, jun-B, CEBP α/β/γ, CHOP, 4E-BP1, and Egr1) and only one (Cdc25C) is decreased (Fig. 8A). Fifteen of these gene products are known targets for p38MAPK phosphorylation. This agrees with the GSEA results that identified the p38MAPK pathway as enriched in GnRH and activin-cotreated cells. These genes were used to initiate nuclear interaction networks. The most significant network (P = 4.1 × 10−26; G score, −41.6) contains 18 root nodes from the initial data set and centers on Smad3, NF-κB1/2, Egr-1, c-Myc, AP-1, Sp1/3, and p53 [Fig. 8B and supplemental data (supplemental networks)]. Smad3 is a known target for activin signaling in these cells so its presence in the predicted network might be expected.
Fig. 8. Regulation of Nuclear p38MAPK Function by GnRH.

A, Highest scoring curated pathway regulated by GnRH in the presence of activin (P = 1.5 × 10−6). Black arrows indicate stimulatory interactions; black bars indicate inhibitory interactions. Green arrows indicate decreased expression; red arrows indicate increased expression over the GnRH time course. B, Transcriptional interaction network generated from the combined list of p38MAPK interacting genes (P = 4.1 × 10−26, G score, −41.6). Yellow and white stars on the icons indicated multicomponent complexes or families. Changes in gene expression are indicated by the red arrows (up) or green arrows (down) next to the icon. Known interactions are shown by arrows. Red circles indicate the transcription factors that drive the network. Red spots indicate termination of the network due to the applied one-step filter. Icons used in the network are the same as shown in the legend to Fig. 6. Original network is provided in the supplemental data.
We also generated a nuclear interaction network using the 406 differential time-course genes. This created a large network of transcription factors that are known to interact with these genes (Fig. 9). This network contained 278 nodes and 783 edges. The original network is provided (Supplemental Material Networks) along with the top 25 transcription factors and their connecting edges. The transcription factors with the greatest number of interactions are Sp1/Sp3, AP-1, p53, c-Myc, and Stat3 (63, 52, 41, 35, and 24 edges, respectively). Not surprisingly, many transcription factors are documented targets for GnRH and activin signaling including Smad3, Smad4, Egr-1, AP-1, cAMP response element-binding protein, and Sp1/Sp3. Others, such as the estrogen, glucocorticoid, and progesterone receptors, have known modulatory effects on gonadotrope gene expression or are involved in pituitary gonadotrope lineage specification, e.g. GATA-2, Pax-6, NeuroD2, and SF-1. The network also contains transcription factors involved in lipid metabolism including sterol regulatory element-binding protein 1, retinoic acid receptor, and liver X receptor/retinoic X receptor, and the nuclear respiratory factors Nrf1/2, which regulate mitochondrial gene expression and biogenesis. These transcription factors likely underlie the repression of mitochondrial genes and function by GnRH that we observed (Fig. 7). The presence of transcription factors mediating classical inflammatory responses was unexpected, however, as the network includes Stat1/3/5, Oct1/2, NF-κB, and IRF1/2. The appearance of these factors correlates with the enrichment of immune-related gene sets found by GSEA. Although GnRH is not known to signal via these proteins, their presence is reasonable given that GnRH has been demonstrated to modulate immune function in peripheral blood mononuclear cells (55). Components of the circadian clock, Clock, Bmal1/2, Dec1, and DBP, are also present. The circadian clock is essential for pulsatile GnRH release from GT1–7 hypothalamic neurons, so may also be required in the pituitary gonadotrope (56).
Fig. 9. Transcriptional Interaction Network for Differential Time Course Genes.

Transcriptional interaction network generated from the list of 406 differential time course genes. Root nodes are indicated by pale blue circles; multicomponent complexes or families are indicated by yellow and white stars on the icons. Changes in gene expression with GnRH are indicated by the red arrows (up) or green arrows (down) next to the icon. The GnRH regulation shown is the predominant effect, either in the presence or absence of activin. Known interactions are shown by connecting arrows. Icons used in the network are the same as shown in the legend to Fig. 6. Original network is provided in the supplemental data.
GnRH has Antiproliferative Effects on LβT2 Cells
The GO and pathway mapping results suggested that pathways and ontologies involved in cell proliferation were altered by GnRH depending on prior exposure to activin. Inspection of the microarray results points to the strong induction of Cdkn1a (p21Cip1/Waf1) and Cdkn2b (p15INK) by GnRH (P = 3 × 10−13 and 4 × 10−14, respectively) but a repression of Cdkn2c (p18INK) and Cdkn2d (p19INK) by GnRH (P < 3 × 10−7 and 3 × 10−11, respectively). The induction of p21 was confirmed by Q-PCR and at the protein level (Fig. 10, A and B). This gene and many of the others identified by pathway mapping are known targets for the tumor suppressor p53. Therefore, we tested whether p53 was activated and found that GnRH caused an increase in phosphorylation of p53 on serines 15 and 46 over 60 min only when the cells had been pretreated with activin (Fig. 10C). A number of kinases phosphorylate p53 including p38MAPK. We have previously shown that GnRH activates ERK and p38MAPK within 5 min; therefore we tested whether activin treatment altered the phosphorylation of these kinases. GnRH activates ERK in the absence and presence of activin; however, phosphorylation of ERK is decreased with activin treatment. In contrast, the activation of p38MAPK was stronger with activin (Fig. 10D). Phosphorylation of JNK is also enhanced by activin exposure, but the kinetics are slower with maximal activation at 30 min (Fig. 10E). A pharmacological inhibitor of p38MAPK was used to test whether phosphorylation of p53 depended on activation of p38MAPK. Pretreatment of cells with PD169316 blocked the GnRH-stimulated increase in p53 serine15 phosphorylation (Fig. 10F), indicating that the enhanced activation of p38MAPK is likely responsible for the observed phosphorylation of p53.
Fig. 10. Activin Enhances the GnRH-Induced Activation of p38MAPK, JNK, and p53.

Cells were stimulated with GnRH for the indicated times and doses. A, Induction of p21Cip1/Waf1 mRNA by 100 nM GnRH by microarray and Q-PCR. Black bars (■) indicate cells pretreated with 25 ng/ml activin for 24 h; open bars (□) are control untreated cells. Q-PCR results are mean and SEM of four independent experiments. Horizontal axis shows time of GnRH treatment; vertical axis shows p21 mRNA levels (arbitrary units). B, Induction of p21Cip1/Waf1 protein by Western blot. Experiment was repeated three times with similar results. C, Phosphorylation of p53 on Ser15 and Ser46 after GnRH stimulation in the presence of activin. Experiment was repeated twice with similar results. D, Phosphorylation of ERK and p38MAPK by GnRH in the presence or absence of activin. Cells were stimulated for 10 min with 0, 10, and 100 nM GnRH. Whole-cell extracts were blotted for phospho-ERK. Blot was stripped and reblotted for phospho-p38MAPK and then for p38MAPK protein as a loading control. Experiment was repeated four times with similar results. E, Stimulation of JNK by GnRH in the presence or absence of activin. Cells were stimulated for increasing times with 100 nM GnRH. Whole-cell extracts were blotted for phospho-JNK and then stripped and blotted for JNK. Experiment was repeated twice with similar results. F, Pharmacological blockade of p38MAPK prevents the phosphorylation of p53. Cell were treated with activin for 24 h and then with 500 nM PD169316 or vehicle for 30 min before being stimulated with GnRH for 30 or 60 min. Whole-cell extracts were blotted for phospho-p53(Ser15), and then stripped and blotted for p53. Experiment was repeated twice with similar results. AU, Arbitrary units.
As p21 is critical to many cell cycle checkpoints, we investigated the effects of GnRH on cell cycle progression. GnRH caused a dose-dependent increase in bromodeoxyuridine labeling of αT3–1 cells but had no effect in LβT2 cells (data not shown). The lack of S-phase progression was confirmed by proliferation assays using the mitochondrial dye MTS (data not shown). Flow cytometry was used to determine whether GnRH caused growth arrest. Treatment with GnRH over 4 d caused an increase in the number of cells in G1 (Fig. 11A), as has been published previously (57), whereas treatment with activin reduced the number in G1 compared with G2/M (Fig. 11B). Addition of GnRH after activin treatment reverses the effect of the activin and restores the number of cells in G1 to the level seen in normal cycling cells but does not cause the G1 arrest seen with GnRH alone (Fig. 11C). Therefore, activin exposure alters the cell cycle response of the cells to GnRH.
Fig. 11. Effect of GnRH on Cell Cycle by Flow Cytometry.

A, Cells were cultured in complete medium containing 10% fetal calf serum and 100 nM GnRH for increasing times. Cells were fixed in ethanol and stained with propidium iodide. DNA content was measured by flow cytometry. B, Cells were treated with 25 ng/ml activin for increasing times, and then fixed and stained with propidium iodide. C, Effect of activin plus GnRH on cell cycle. Cells were treated with 25 ng/ml activin for 24 h before 100 nM GnRH for increasing times. Cells were fixed and stained. Experiment was repeated three times with similar results. G1 and G2/M peaks are indicated. Horizontal axis shows DNA content; vertical axis shows number of events. Peak at higher DNA content is due to tetraploidy.
DISCUSSION
We have presented evidence that activin treatment of LβT2 gonadotropes does not greatly alter their expression profile but, more importantly, it modulates their transcriptional response to GnRH. Notwithstanding, a number of the genes that are altered by activin do have well defined roles in the regulation of reproduction. Both the inhibin α and β-B genes are induced, suggesting that activin treatment auto-regulates both activin B and inhibin B production in the pituitary. The LHβ subunit and GnRH-receptor genes are induced modestly (2-fold), but the FSHβ subunit gene is strongly induced (20-fold) as is the17β-hydroxysteroid dehydrogenase B1 gene. The induction of this gene correlates with an increase in the sensitivity of the LβT2 cells to estrone. We do not know whether induction of hydroxysteroid dehydrogenase (HSD)17β1 causes the increased sensitivity as the cells express a number of other HSD genes including HSD17β4, HSD17β12, HSD3β7, HSD11β1, and HSD11β2, but not the β2, β3, or β7 isoforms of HSD17 nor the β1, β2, β3, β4, β5, or β6 isoforms of HSD3. The HSD17β1, β7, and β12 isoforms promote the conversion of estrone to estradiol, whereas the β2 and β4 isoforms catalyze the reverse reaction (44). Interestingly, HSD17β1 is the only isoform to increase with activin. There is evidence for activin regulation of HSD17β1 in other cells. Treatment of rat granulosa cells with activin A increases both 17HSD activity and HSD17β1 mRNA levels (58). Induction of the gene was maximal at 100 ng/ml activin A, but significant induction was seen at concentrations of 20 ng/ml, similar to those used in our study. This gene is also induced by FSH or cAMP in parallel with the p450aromatase gene in granulosa cells, but is repressed by protein kinase C (PKC) signaling. In contrast, the HSD17β1 gene is induced by activin but significantly repressed (90%) by GnRH in LβT2 pituitary cells. We have previously shown that the GnRH receptor couples to both Gs and Gq/11 pathways in LβT2 cells (59). Even though GnRH elevates cAMP, the strong activation of PKC downstream of Gq/11 likely leads to overall repression of 17HSD1 expression. Furthermore, HSD17β1 expression has been demonstrated in mouse pituitaries by in situ hybridization, although the majority of the staining was in the intermediate lobe rather than the anterior lobe. Interestingly, the HSD17β4 isoform is repressed by GnRH but only in the absence of activin, which is the opposite of the regulation seen for HSD17β1. This suggests that in the absence of activin, GnRH favors the production of estradiol from estrone, but in the presence of activin GnRH favors the opposite reaction. The biological relevance of this regulation is not known. Inhibin levels are known to vary throughout the estrous cycle, however, even though serum activin does not (60, 61). Indeed, serum inhibin B is elevated during the follicular phase of the cycle and inhibin A is elevated during the luteal phase. There is a nadir in serum inhibin during the LH surge and ovulation. It is interesting to speculate that increased activin tone during this period might allow the pituitary to respond to the elevated estrone modulating the LH surge or facilitating the coincident FSH increase.
A number of the GnRH-regulated genes described here have been previously reported by Wurmbach et al. (41) and Kakar et al. (42). Wurmbach et al. (41) used a focused microarray (FMA) containing 956 immediate early genes to identify 31 genes that were regulated by GnRH. The majority of these genes were transcription factors (c-fos, Klf4, Egr1, Egr2, c-jun, Nrf2, LRG21, TSC22, Nr4a1) that are acutely induced by many hormones, but only a few signaling and structural proteins were altered (Rgs2, IκB, gem, γ-actin, MKP1). Kakar et al. (42) used Affymetrix MU74Av2 chips but were limited to using a comparison call in the Affymetrix software [Microarray Suite (MAS) 4] to assess differential expression due to the lack of replicates. However, given that limitation, they were able to identify 453 genes altered at 1 h of GnRH treatment and 468 genes altered with 24 h of GnRH. Many of the genes altered at 1 h are those found by Wurmbach et al. (41). The study reported here has the advantage over the previous studies in that multiple, independent statistical methods were applied to the data set to identify the sets of genes with the most robust and reliable changes. In particular, we used Bonferroni multiple-testing correction to correct for false positives that might arise by chance in such a large data set. Although we are undoubtedly missing many regulated genes by focusing on the false positive rather than the false negative rate, we obtained sufficient genes for further study. We found that the most informative approach was the Bayesian variance modeling implemented in VAMPIRE, which has the advantage that it works well with few replicates, unlike most t test-based approaches. Comparing the 1 h data from Kakar et al. (42) with our 4 h data, only 123 of their 453 genes are altered at 4 h in our data set. These common 123 genes include many of the immediate early genes (crem, Atf3, Egr2, c-jun, Gadd45b, Zfp36, Rrad). After 24 h of GnRH treatment, only 118 genes of the 468 genes found by Kakar et al. are present in our 24 h data set. There may be many explanations for this discrepancy. First, Kakar et al. (42) used an earlier time point, and many of the genes altered at 1 h may have returned to basal by 4 h as observed by Wurmbach et al. (41). Second, we have used strict multiple-testing correction, and many genes may be false negatives in our analysis. Indeed, using a less stringent 5% FDR cut-off, 225 of their 453 genes are found in our 4 h data set, and 215 genes of 468 are in our 24 h data set. Lastly, many of the remaining genes reported by Kakar et al. (42) may be false positives due to the lack of replicates. A recent publication by Mazhawidza et al. (62) presented microarray data from LβT2 cells treated with activin for 48 h and then with GnRH for 1 h. They observed 70 genes the expression of which changed more than 2-fold by activin treatment alone but without multiple testing correction. If this list is pruned to eliminate untrustworthy genes (errors > 50% of the mean fold change), 25 genes show robust greater than 2-fold changes. This is similar to our findings. Manual inspection of this list showed a number of agreements with our data, including Ephx2, Csrp2, Hsd17b1, Cdkn2b, and Tnni3. Treatment with the GnRH agonist [D-Ala6]GnRH for 1 h altered the expression of 213 genes. Cotreatment with activin and the GnRH agonist altered 169 genes. After comparison with our data set, 144 (54%) of the genes identified by Mazhawidza et al. are present in our set of 2453 GnRH-regulated genes. The remaining genes not present in our data set may reflect the shorter time of stimulation, the use of a GnRH agonist rather than GnRH itself, or genes with high variability that are not statistically significant.
The ability of the activin system to modulate the expression and secretion of the gonadotropins by the pituitary has been well established both in vitro and in vivo. Hierarchical clustering of our gene expression data highlighted groups of genes the response to GnRH of which is dependent on prior exposure to activin. Some genes are induced more strongly in the absence of activin, whereas others are induced more strongly after activin treatment. A large group of genes is repressed by GnRH, but only in the absence of activin. These clusters are evidence that activin is modulating the transcriptional response of the gonadotrope to GnRH and priming the gonadotrope to respond to GnRH. This coregulation has been investigated extensively on the FSHβ promoter where GnRH and activin act synergistically (25, 63). As these two hormones are critical for controlling the reproductive cycle, the genes in these clusters are likely important for normal pituitary function. The transcriptional network mapping indicated a potential role for both activin-dependent (Smad3 and Smad4) as well as GnRH-dependent (c-fos, Egr-1, cAMP-response element binding protein, serum response factor) transcription factors on a number of these promoters. Not surprisingly, a number of these genes are known targets for TGFβ and bone-morphogenic protein signaling. For example, the osteopontin gene (Spp1) is activated by phorbol esters, UTP, cytokines, and growth factors via upstream stimulatory factor and AP-1 (64–66). It is also subject to up-regulation in response to TGFβ and bone-morphogenic protein-2 via a Smad binding element (AGACTGTCTGGAC) (67). Unlike TGFβ, however, activin reduces the induction of Spp1. Similar effects are seen on the plasminogen-activator inhibitor-1 (PAI-1) gene, which is another target for TGFβ (68). This may represent the differential use of Smads downstream of the activin or TGFβ receptors.
Mapping the altered genes to Gene Ontology classifications suggested that different classes of genes were being regulated by GnRH with and without activin treatment. This was particularly apparent at 8 and 24 h when GnRH alters genes in pathways controlling cell energetics, cytoskeletal rearrangements, organelle organization, and mitosis in the absence of activin, but genes controlling protein processing, cell differentiation, and secretion in the presence of activin. Similar effects were observed when we mapped the altered genes to known signaling and metabolic pathways or analyzed the data with GSEA. These differences correlated with altered proliferation and cell cycle regulation and with differential activation of ERK, p38MAPK, and p53. Many of these findings are consistent with the G0/G1 arrest that we observe with GnRH treatment in the absence of activin. The reduction in mitochondrial and metabolic pathways is due to the reduced metabolic and energy requirements of quiescent cells and the reduction in the anaphase-promoting complex components and cell cycle genes is consistent with cells arresting in G0/G1. Activin treatment causes a G2/M arrest that is consistent with the enrichment of DNA repair genes and cell-cycle checkpoint genes by activin. Genes involved in mRNA and protein synthesis are also induced by activin, and this may reflect the gene expression required during DNA replication and the G2 phase of the cell cycle. A surprising finding is the induction of immune-related genes by GnRH in the presence of activin. Some of these are secreted proteins, such as lipopolysaccharide-induced TNF, TGFβ3, TGFβ1, IL-1 receptor antagonist, endothelin 1, and Ccl5, that potentially signal to other cell types in the anterior pituitary.
GnRH is known to inhibit the proliferation of gynecological tumors although the mechanisms involved are not clear (69, 70). Repression of gonadal steroids is certainly a contributing factor, but direct effects on target cells have also been demonstrated. Our results are consistent with a direct effect of GnRH on cell proliferation and survival. We observe the phosphorylation of p53 in response to GnRH, especially in the presence of activin, and the regulation of many p53 target genes in these cells. Previously our laboratory and others have shown that GnRH is a strong activator of p38MAPK. In this study, we found that activation of the p38MAPK stress response pathway is enhanced in activin-treated cells but ERK activation is impaired. Pharmacological inhibition of p38MAPK reduced the phosphorylation of p53; hence, this may be the link to p53 activation because p38MAPK can phosphorylate p53 in human fibroblasts (71). The signal leading to p38MAPK activation in LβT2 cells is not known, but PKC signaling is required in the less-differentiated αT3–1 cells (72). Studies in ovarian tumor cells using pertussis toxin have also implicated the Gi pathway in growth suppression, but it is not known whether this leads to p38MAPK activation (73).
Analysis of cell cycle regulation in LβT2 cells is complicated by the presence of the transforming SV40 large T antigen that was used to immortalize the cells (51). Large T antigen is known to bind and sequester p53 and therefore could be a confounding factor in our analysis, especially of genes involved in proliferation, if this interaction is altered by phosphorylation of p53 (74). We do not believe this is the case, however, for two reasons. First, we have coprecipitated SV40 large T antigen with p53 and do not see a difference in the interaction in the presence or absence of activin (data not shown). Second, we have compared our data set not only with data generated by studies using temperature-sensitive large T antigen alleles (75) or transgenic expression of T antigen in mammary epithelium (76), but also with data on p53 target genes (77) and p53 genomic binding sites (78). Of the 2453 GnRH-regulated genes identified in this study, 375 (15%) are known to be targets for SV40 Large T antigen (of 1841 total). In contrast, 1466 known SV40 large T responsive genes are not altered in our data set. Furthermore, only 66 large T responsive genes (0.3%) are found in our set of differentially expressed genes. Therefore, more than 99% of the large T responsive genes are not found in our analysis of differential regulation. If activin were altering the ability of SV40 large T to sequester p53, we would expect to see a greater proportion of large T responsive genes in our data set. Of the 515 known p53 targets, only 210 genes are represented on the arrays used in this study. In our set of 2453 GnRH-regulated genes, 39 are p53 targets, and only 40 genes of the 1841 large T responsive genes bind p53. Comparing the overlap of these gene sets, only seven genes are p53, SV40 large T antigen, and GnRH responsive. Therefore, we do not believe the presence of SV40 large T antigen greatly influences the results.
The modulation of the GnRH response by activin costimulation is intriguing. The TGFβ family also has well-documented antiproliferative effects (79). The common signaling partner Smad4 is known to be a tumor suppressor and is found mutated or deleted in pancreatic, colorectal, or cervical cancers. Loss of function mutations in ActR-1B (ALK-4), TGFβ-RI, TGFβ-RII, or Smad2 also occur in various human tumors. A mechanistic understanding of the antiproliferative effects of activin has been derived from in vitro studies in various cell lines. Activin inhibits the growth of prostate and breast cancer, B-cell leukemia, vascular endothelial cells, vascular smooth muscle, hematopoietic progenitor cells, fetal adrenal cells, rat liver, mouse plasmacytoma, and hepatocytes. Many of these effects are mediated by induction of p21Cip1/Waf1 and suppression of cyclins D2 or A leading to hypophosphorylation of Rb and a G0/G1 arrest (80–82). The LβT2 cells were previously shown to express inhibin βB, inhibin α, follistatin, and the type IA, IB, IIA, and IIB activin receptors. Our microarray data presented here confirm this expression pattern, but we also found evidence for expression of inhibins βA, βC, and βE. Not only do the cells have a functional activin receptor system, but they also make endogenous activin A, activin B, and inhibin. Inhibin α and βB are induced 2-fold by activin treatment suggesting negative feedback. The growth-inhibitory effects of activin in LβT2 cells are somewhat different from other cells as activin has little effect on p21Cip1/Waf1 expression and does not cause G1 arrest. In contrast, activin induces the DNA damage response genes RAD50, Xpc, and GADD45β that are involved in DNA repair, telomere maintenance, nucleotide excision, and G2 arrest. Further studies will be needed to unravel the mechanism of this G2/M arrest in LβT2 cells.
In conclusion, we have shown that activin treatment modulates the transcriptional response of pituitary LβT2 gonadotrope cells to GnRH. This modulation has important consequences for the antiproliferative action of GnRH and suggests that activin tone might be an important regulator of pituitary estrogen sensitivity and the ability of GnRH to inhibit tumor growth. Further studies will be needed to delineate the exact pathways used by GnRH and activin to regulate proliferation.
MATERIALS AND METHODS
Materials
GnRH was purchased from Sigma Chemical Co. (St. Louis, MO). Activin A was purchased from R&D Systems (Minneapolis, MN). Rabbit polyclonal anti-p21, anti-p53, and horse-radish peroxidase-linked antirabbit antibodies were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Rabbit polyclonal anti-ACTIVE MAPK antibody and rabbit polyclonal anti-ACTIVE JNK antibody were from Promega Corp. (Madison, WI). Rabbit polyclonal antibodies to phospho-p38 MAPK (Thr180/Tyr182), p38MAPK, phospho-p53(Ser15) phospho-p53(Ser46), acetylated-p53(Lys382) were from Cell Signaling Technology (Beverly, MA). DMEM and fetal bovine serum (FBS) were purchased from Life Technologies (Gaithersburg, MD). All other reagents were purchased from either Sigma or Fisher Scientific (Pittsburgh, PA). LβT2 cells were maintained in monolayer cultures in DMEM supplemented with 10% fetal bovine serum and antibiotics in a humidified 10% CO2 atmosphere at 37 C.
Gene Expression Profiling in LβT2 Gonadotrope Cells
LβT2 cells were plated in 10-cm dishes and starved of serum for 24 h in the absence or presence of activin (25 ng/ml) before stimulation with 100 nM GnRH for 4, 8, or 24 h. Total RNA was harvested using RNAzolB (Tel-Test, TX) and purified on RNeasy spin columns (QIAGEN, Valencia, CA). The mRNA was quantified and its integrity checked by agarose gel electrophoresis. Ten micrograms of mRNA from basal cells, activin-treated cells, or cells treated with GnRH were analyzed on Affymetrix mouse MU74Av2 gene chips. Duplicates were run for each condition with independently isolated RNA from independent experiments.
Data were analyzed using multiple independent strategies. First, the raw expression values derived from Affymetrix Microarray Suite software (MAS 5) were imported into VAMPIRE without prior normalization. This program uses a Bayesian approach to identify altered genes (43). Statistical analysis by VAMPIRE requires two distinct steps: 1) modeling of the error structure of sample groups; and 2) significance testing with a priori-defined significance thresholds. VAMPIRE models the existing error structure to distinguish signal from noise and identify the coefficients of expression-dependent and expression-independent variance. These models are then used to identify microarray features that are differentially expressed between treatment groups. Initially we compared cells treated with or without activin in the absence of GnRH. A Bonferroni multiple testing correction (αBonf < 0.05) was applied to identify only those genes with the most robust changes. This approach was then used for pair-wise comparisons for each time of GnRH treatment against its own zero time control either pretreated with activin or not. The genes altered at each of the three times (4, 8, 24 h) were combined to give lists of GnRH-regulated genes in the absence of activin or with activin pretreatment.
Second, the data were normalized using the Robust Multichip Average method consisting of background adjustment, quantile normalization, and summarization (47). Normalized data were imported into Partek Pro Genomic Solutions to perform multivariate, mixed-model ANOVA. This software allows multivariate ANOVA using unbalanced data. We used two categorical variables (activin treatment, GnRH treatment) and one quantitative variable (time). The software also allowed us to factor out random batch effects such as the date the chips were scanned. Analysis of the data by three-way ANCOVA identified genes dependent on activin pretreatment, GnRH treatment, or time. Multiple testing correction using either Bonferroni or the less stringent Benjamini-Hochberg FDR were applied.
Third, data were normalized using the CORGON multiplicative noise model (48). This is a model-based method assuming multiplicative rather than additive noise and eliminating statistically significant outliers. Normalized data were imported into GeneSpring version 6 (Silicon Genetics, Redwood City, CA) and Statistical Analysis of Microarrays (SAM, version 1.21) in Microsoft Excel (Redmond, WA) for statistical analysis (49). Genes were filtered to exclude genes that did not have a P value of 0.1 or less (equivalent to a present call by MAS 5) on at least two chips, and the expression data were averaged between the duplicate chips. Standard two-sample t test (GeneSpring) or permuted t test (SAM) approaches were used to identify genes altered by activin treatment alone. The time courses of GnRH treatment, with and without activin, were analyzed in GeneSpring by two-way ANOVA, or individually in Significance Analysis of Microarrays using multiclass analysis with 500 permutations. The overlap of these gene lists was examined in GeneSpring to give the complete repertoire of genes that are regulated by GnRH and their dependence on activin pretreatment. Multiple-testing correction using the Bonferroni or FDR method was applied in all cases. The final gene list was subjected to hierarchical clustering using Pearson correlation, Spearman correlation, or Euclidean distance measurements. The expression profiles of significant clusters were generated based on the median-normalized MAS 5 data.
Lists of genes altered at each time point and treatment generated by VAMPIRE were mapped to gene ontology terms using the program GOby to determine whether any GO classifications were overrepresented (43). MTC (αBonf < 0.05) was applied to minimize GO terms arising as false positives. Significant genes were also mapped to known manually curated metabolic and signaling pathways using MetaCore (GeneGo, St. Joseph, MI). Interaction networks were generated de novo using lists of target genes as starting root nodes in MetaCore. Only nuclear interactions were considered, and path lengths were limited to one step to avoid secondary effects. The networks were ranked based on P values and G scores (a modified Z score). Gene Set Enrichment Analysis was performed using the GSEA software on the complete data set as described elsewhere (45).
Verification of RNA Expression by Quantitative RT-PCR
The experimental treatments were repeated in four independent experiments. RNA was obtained with RNAzol-B. Contaminating DNA was removed with DNA-free reagent (Ambion, Inc., Austin, TX) and 2 μg RNA was reverse transcribed using Superscript III First-Strand Synthesis System (Invitrogen, Carlsbad, CA). Quantitative real-time PCR was performed on a MJ Research PTC 200 Peltier Thermal Cycle with a Chromo4 fluorescence detector (Bio-Rad Laboratories, Inc., Hercules, CA) under the following conditions: 95 C for 7 min, followed by 40 cycles at 94 C for 20 sec, 60 C for 60 sec, and 72 C for 60 sec. Amplification was detected using either the QuantiTect SYBR Green PCR Kit (QIAGEN), or using FAM-labeled fluorescent-locked nucleic acid probes (Exiqon, Vedbaek, Denmark). Ct values were extracted by manually setting the threshold midway between basal and maximal fluorescence on a log10 scale. Each sample was assayed in triplicate, and the experiment was repeated four times. Serial dilutions of plasmid or positive control RNA were run in parallel to determine amplification efficiency for each gene, and statistically significant changes were determined using the software REST using the following formula: r = (Etarget)ΔCPtarget(Sample−Control)/(Eref)ΔCPref(Sample−Control) (83). In this formula, R represents a relative expression ratio of target gene, E is PCR efficiency, ΔCP is a crossing point difference of an unknown sample vs. a control, and ref represents a reference gene. We used three housekeeping genes (Atp6v0d1, cyclophilin, and β-actin) for normalization.
Luciferase Activity Assay
Cells in six-well plates were transfected with 0.5 μg of reporter gene, 50 ng of tk-LacZ control, and 100 ng of expression vector using Fugene6 (Roche, Indianapolis, IN) following the manufacturer’s instructions. Cells were stimulated with increasing doses of estrogens for 24 h or with 100 nM GnRH for increasing times. Luciferase and β-galactosidase activities were measured using the Promega Firefly Luciferase Assay or Tropix Galactolight Assay (Tropix, Bedford, MA). Luciferase activity was normalized to the β-galactosidase activity.
Mitochondrial Activity Assay
To assess mitochondrial activity, LβT2 cells were plated at confluence in 96-well plates. Cells were pretreated with follistatin (250 ng/ml), to block endogenous activin, or activin (25 ng/ml) for 24 h in the presence or absence of 10% FBS, and then increasing doses of GnRH (0, 0.1, 1, 10, 100 nM) for 24 h. Mitochondrial activity was assessed by adding the redox sensitive dye 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxyme-thoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) and the electron-coupling reagent phenazine methosulfate and measuring the increase in absorbance at 490 nm over time (Cell-Titer, Promega Corp.).
Western Blotting
LβT2 cells were grown to confluence in six-well plates, washed once with PBS, and incubated in serum-free medium overnight. Cells were stimulated with agonists for various times at 37 C. Thereafter, cells were washed with ice-cold PBS, and then lysed on ice in sodium dodecyl sulfate sample buffer (50 mM Tris; 5% glycerol; 2% sodium dodecyl sulfate; 0.005% bromophenol blue; 84 mM dithiothreitol; 100 mM sodium fluoride; 10 mM sodium pyrophosphate; and 2 mM sodium orthovanadate, pH 6.8), boiled for 5 min to denature proteins, and sonicated for 5 min to shear the chromosomal DNA. Equal volumes (30–40 μl) of these lysates were separated by SDS-PAGE on 7.5 or 10% gels, electrotransferred to polyvinylidene difluoride membranes (Immobilon-P, Millipore Corp., Bedford, MA). The membranes were blocked with 5% nonfat dried milk in TBS-Tween (50 mM Tris-HCl, pH 7.4; 150 mM NaCl; 0.1% Tween 20). Blots were incubated with primary antibodies in blocking buffer for 60 min at room temperature, and then incubated with horseradish peroxidase-linked secondary antibodies followed by chemiluminescent detection. For the phospho-specific antibodies, the polyvinylidene difluoride membranes were immediately stripped by placing the membrane in stripping buffer (0.5 M NaCl and 0.5 M acetic acid) for 10 min at room temperature. The membrane was then washed once for 10 min in TBS-Tween, reblocked, and blotted with antibodies to the unphosphorylated form of the protein to control for equal protein loading.
Flow Cytometry
Cells were plated into 6-cm dishes in complete DMEM supplemented with 10% FBS for 24 h, and then treatments were performed in complete DMEM supplemented with 10% FBS for 96 h. Culture medium, GnRH, and activin were changed every 24 h. Cells were harvested, washed with PBS, and fixed in ice-cold 70% ethanol for 1 h and then kept at 4 C for 24 h. Cells were washed and incubated in PBS containing ribonuclease A (0.2 mg/ml) and Triton X-100 (0.2%) at 37 C for 30 min. After a final PBS wash, cells were incubated with propidium iodide (50 mg/ml) for a minimum of 30 min at 4 C. Cells were filtered through a 0.3-μm mesh. Samples were then processed using a FACSCalibur flow cytometer (BD Biosciences, Palo Alto, CA) and analyzed using ModFit LT (version 3.1). G0/G1 and G2/M peaks were identified by arresting the cells with aphidicolin (10 μg/ml) or nocodazole (1 μg/ml), respectively.
Statistical Analysis
Unless otherwise noted, data were analyzed by ANOVA followed by Tukey post hoc tests. Individual pairwise comparisons were performed using two-tailed t tests.
Supplementary Material
Acknowledgments
We thank Dr. David Rose (University of California, San Diego) and Dr. Mark Hannink (University of Missouri,) for the ERE-tk-luc and ARE-tk-luc reporter plasmids.
This work was supported by National Institute of Child Health and Human Development/National Institutes of Health (NIH) through a cooperative agreement (U54 HD012303) as part of the Specialized Cooperative Centers Program in Reproduction Research (to N.J.G.W., M.A.L., P.L.M.). This work was also supported by NIH Grant R37 HD020377 (to P.L.M.), R01 HD 43758 and K02 40803 (to M.A.L.). H.Z. was supported by NIH Grant T32 HD007203. J.S.B. was supported in part by NIH Grant T32 GM08666. D.C. was partially supported by NIH Grant NRSA F32 HD41301, NIH T32 DK07044, and the Lalor Foundation. N.J.G.W., M.A.L, and P.L.M. are faculty members of the UCSD Biomedical Sciences Graduate Program.
Abbreviations
- ANCOVA
Analysis of covariance
- AP-1
activator protein 1
- APC
anaphase-promoting complex
- CoA
coenzyme A
- FBS
fetal bovine serum
- FDR
false discovery rate
- GSEA
Gene Set Enrichment Analysis
- MAS
Microarray Suite
- MTC
multiple testing correction
- mTOR
mammalian target of rapamycin
- MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- NF-κB
nuclear factor-κB
- PKC
protein kinase C
- Q-PCR
quantitative PCR
- SF-1
steroidogenic factor 1
- Smad
Sma-and Mad-related protein
Footnotes
Disclosure Summary: The authors have nothing to disclose.
Molecular Endocrinology is published monthly by The Endocrine Society (http://www.endo-society.org), the foremost professional society serving the endocrine community.
References
- 1.Conn PM, Crowley WFJ. Gonadotropin-releasing hormone and its analogs. Annu Rev Med. 1994;45:391–405. doi: 10.1146/annurev.med.45.1.391. [DOI] [PubMed] [Google Scholar]
- 2.Kaiser UB, Conn PM, Chin WW. Studies of gonadotropin-releasing hormone (GnRH) action using GnRH receptor-expressing pituitary cell lines. Endocr Rev. 1997;18:46–70. doi: 10.1210/edrv.18.1.0289. [DOI] [PubMed] [Google Scholar]
- 3.McDowell IF, Morris JF, Charlton HM. Characterization of the pituitary gonadotroph cells of hypogonadal (hpg) male mice: comparison with normal mice. J Endocrinol. 1982;95:321–330. doi: 10.1677/joe.0.0950321. [DOI] [PubMed] [Google Scholar]
- 4.Bedecarrats GY, Linher KD, Janovick JA, Beranova M, Kada F, Seminara SB, Michael Conn P, Kaiser UB. Four naturally occurring mutations in the human GnRH receptor affect ligand binding and receptor function. Mol Cell Endocrinol. 2003;205:51–64. doi: 10.1016/s0303-7207(03)00201-6. [DOI] [PubMed] [Google Scholar]
- 5.Achermann JC, Jameson JL. Advances in the molecular genetics of hypogonadotropic hypogonadism. J Pediatr Endocrinol Metab. 2001;14:3–15. doi: 10.1515/jpem.2001.14.1.3. [DOI] [PubMed] [Google Scholar]
- 6.Beranova M, Oliveira LM, Bedecarrats GY, Schipani E, Vallejo M, Ammini AC, Quintos JB, Hall JE, Martin KA, Hayes FJ, Pitteloud N, Kaiser UB, Crowley WFJ, Seminara SB. Prevalence, phenotypic spectrum, and modes of inheritance of gonadotropin-releasing hormone receptor mutations in idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2001;86:1580–1588. doi: 10.1210/jcem.86.4.7395. [DOI] [PubMed] [Google Scholar]
- 7.Kaiser UB, Halvorson LM, Chen MT. Sp1, steroidogenic factor 1 (SF-1), and early growth response protein 1 (egr-1) binding sites form a tripartite gonadotropin-releasing hormone response element in the rat luteinizing hormone-β gene promoter: an integral role for SF-1. Mol Endocrinol. 2000;14:1235–1245. doi: 10.1210/mend.14.8.0507. [DOI] [PubMed] [Google Scholar]
- 8.Dorn C, Ou Q, Svaren J, Crawford PA, Sadovsky Y. Activation of luteinizing hormone β gene by gonadotropin-releasing hormone requires the synergy of early growth response-1 and steroidogenic factor-1. J Biol Chem. 1999;274:13870–13876. doi: 10.1074/jbc.274.20.13870. [DOI] [PubMed] [Google Scholar]
- 9.Mouillet JF, Sonnenberg-Hirche C, Yan X, Sadovsky Y. p300 regulates the synergy of steroidogenic factor-1 and early growth response-1 in activating luteinizing hormone-β subunit gene. J Biol Chem. 2004;279:7832–7839. doi: 10.1074/jbc.M312574200. [DOI] [PubMed] [Google Scholar]
- 10.Sadovsky Y, Crawford PA, Woodson KG, Polish JA, Clements MA, Tourtellotte LM, Simburger K, Milbrandt J. Mice deficient in the orphan receptor steroidogenic factor 1 lack adrenal glands and gonads but express P450 side-chain-cleavage enzyme in the placenta and have normal embryonic serum levels of corticosteroids. Proc Natl Acad Sci USA. 1995;92:10939–10943. doi: 10.1073/pnas.92.24.10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parker KL, Rice DA, Lala DS, Ikeda Y, Luo X, Wong M, Bakke M, Zhao L, Frigeri C, Hanley NA, Stallings N, Schimmer BP. Steroidogenic factor 1: an essential mediator of endocrine development. Recent Prog Horm Res. 2002;57:19–36. doi: 10.1210/rp.57.1.19. [DOI] [PubMed] [Google Scholar]
- 12.Morohashi KI, Omura T. Ad4BP/SF-1, a transcription factor essential for the transcription of steroidogenic cytochrome P450 genes and for the establishment of the reproductive function. FASEB J. 1996;10:1569–1577. doi: 10.1096/fasebj.10.14.9002548. [DOI] [PubMed] [Google Scholar]
- 13.Zhao L, Bakke M, Hanley NA, Majdic G, Stallings NR, Jeyasuria P, Parker KL. Tissue-specific knockouts of steroidogenic factor 1. Mol Cell Endocrinol. 2004;215:89–94. doi: 10.1016/j.mce.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 14.Lee SL, Sadovsky Y, Swirnoff AH, Polish JA, Goda P, Gavrilina G, Milbrandt J. Luteinizing hormone deficiency and female infertility in mice lacking the transcription factor NGFI-A (Egr-1) Science. 1996;273:1219–1221. doi: 10.1126/science.273.5279.1219. [DOI] [PubMed] [Google Scholar]
- 15.Tourtellotte WG, Nagarajan R, Bartke A, Milbrandt J. Functional compensation by Egr4 in Egr1-dependent luteinizing hormone regulation and Leydig cell steroidogenesis. Mol Cell Biol. 2000;20:5261–5268. doi: 10.1128/mcb.20.14.5261-5268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tourtellotte W, Nagarajan R, Auyeung A, Mueller C, Milbrandt J. Infertility associated with incomplete spermatogenic arrest and oligozoospermia in Egr4-deficient mice. Development. 1999;126:5061–5071. doi: 10.1242/dev.126.22.5061. [DOI] [PubMed] [Google Scholar]
- 17.Topilko P, Schneider-Maunoury S, Levi G, Trembleau A, Gourdji D, Driancourt MA, Rao CV, Charnay P. Multiple pituitary and ovarian defects in Krox-24 (NGFI-A, Egr-1)-targeted mice. Mol Endocrinol. 1998;12:107–122. doi: 10.1210/mend.12.1.0049. [DOI] [PubMed] [Google Scholar]
- 18.Zhang FP, Poutanen M, Wilbertz J, Huhtaniemi I. Normal prenatal but arrested postnatal sexual development of luteinizing hormone receptor knockout (LuRKO) mice. Mol Endocrinol. 2001;15:172–183. doi: 10.1210/mend.15.1.0582. [DOI] [PubMed] [Google Scholar]
- 19.Lei ZM, Mishra S, Zou W, Xu B, Foltz M, Li X, Rao CV. Targeted disruption of luteinizing hormone/human chorionic gonadotropin receptor gene. Mol Endocrinol. 2001;15:184–200. doi: 10.1210/mend.15.1.0586. [DOI] [PubMed] [Google Scholar]
- 20.Huang HJ, Sebastian J, Strahl BD, Wu JC, Miller WL. Transcriptional regulation of the ovine follicle-stimulating hormone-β gene by activin and gonadotropin-releasing hormone (GnRH): involvement of two proximal activator protein-1 sites for GnRH stimulation. Endocrinology. 2001;142:2267–2274. doi: 10.1210/endo.142.6.8203. [DOI] [PubMed] [Google Scholar]
- 21.Miller WL, Shafiee-Kermani F, Strahl BD, Huang HJ. The nature of FSH induction by GnRH. Trends Endocrinol Metab. 2002;13:257–263. doi: 10.1016/s1043-2760(02)00614-8. [DOI] [PubMed] [Google Scholar]
- 22.Coss D, Jacobs SBR, Bender CE, Mellon PL. A novel AP-1 site is critical for maximal induction of the follicle-stimulating hormone β gene by gonadotropin-releasing hormone. J Biol Chem. 2004;279:152–162. doi: 10.1074/jbc.M304697200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jochum W, Passegue E, Wagner EF. AP-1 in mouse development and tumorigenesis. Oncogene. 2001;20:2401–2412. doi: 10.1038/sj.onc.1204389. [DOI] [PubMed] [Google Scholar]
- 24.Gregory SJ, Kaiser UB. Regulation of gonadotropins by inhibin and activin. Semin Reprod Med. 2004;22:253–267. doi: 10.1055/s-2004-831901. [DOI] [PubMed] [Google Scholar]
- 25.Pernasetti F, Vasilyev VV, Rosenberg SB, Bailey JS, Huang HJ, Miller WL, Mellon PL. Cell-specific transcriptional regulation of follicle-stimulating hormone-β by activin and gonadotropin-releasing hormone in the LβT2 pituitary gonadotrope cell model. Endocrinology. 2001;142:2284–2295. doi: 10.1210/endo.142.6.8185. [DOI] [PubMed] [Google Scholar]
- 26.Bilezikjian LM, Blount AL, Corrigan AZ, Leal A, Chen Y, Vale WW. Actions of activins, inhibins and follistatins: implications in anterior pituitary function. Clin Exp Pharm Physiol. 2001;28:244–248. doi: 10.1046/j.1440-1681.2001.03422.x. [DOI] [PubMed] [Google Scholar]
- 27.Suszko MI, Lo DJ, Suh H, Camper SA, Woodruff TK. Regulation of the rat follicle-stimulating hormone β-subunit promoter by activin. Mol Endocrinol. 2003;17:318–332. doi: 10.1210/me.2002-0081. [DOI] [PubMed] [Google Scholar]
- 28.Bernard DJ. Both SMAD2 and SMAD3 mediate activin-stimulated expression of the follicle-stimulating hormone β subunit in mouse gonadotrope cells. Mol Endocrinol. 2004;18:606–623. doi: 10.1210/me.2003-0264. [DOI] [PubMed] [Google Scholar]
- 29.Dupont J, McNeilly J, Vaiman A, Canepa S, Combarnous Y, Taragnat C. Activin signaling pathways in ovine pituitary and LβT2 gonadotrope cells. Biol Reprod. 2003;68:1877–1887. doi: 10.1095/biolreprod.102.012005. [DOI] [PubMed] [Google Scholar]
- 30.Chang H, Brown CW, Matzuk MM. Genetic analysis of the mammalian transforming growth factor-β superfamily. Endocr Rev. 2002;23:787–823. doi: 10.1210/er.2002-0003. [DOI] [PubMed] [Google Scholar]
- 31.Moustakas A, Souchelnytskyi S, Heldin CH. Smad regulation in TGF-β signal transduction. J Cell Sci. 2001;114:4359–4369. doi: 10.1242/jcs.114.24.4359. [DOI] [PubMed] [Google Scholar]
- 32.Mehra A, Wrana JL. TGF-β and the Smad signal transduction pathway. Biochem Cell Biol. 2002;80:605–622. doi: 10.1139/o02-161. [DOI] [PubMed] [Google Scholar]
- 33.Chang H, Lau AL, Matzuk MM. Studying TGF-β superfamily signaling by knockouts and knockins. Mol Cell Endocrinol. 2001;180:39–46. doi: 10.1016/s0303-7207(01)00513-5. [DOI] [PubMed] [Google Scholar]
- 34.Kumar TR, Agno J, Janovick JA, Conn PM, Matzuk MM. Regulation of FSHβ and GnRH receptor gene expression in activin receptor II knockout male mice. Mol Cell Endocrinol. 2003;212:19–27. doi: 10.1016/j.mce.2003.09.019. [DOI] [PubMed] [Google Scholar]
- 35.Kumar TR, Wang Y, Lu N, Matzuk MM. Follicle stimulating hormone is required for ovarian follicle maturation but not male fertility. Nat Genet. 1997;15:201–204. doi: 10.1038/ng0297-201. [DOI] [PubMed] [Google Scholar]
- 36.Leal AM, Takabe K, Wang L, Donaldson CJ, MacConell LA, Bilezikjian LM, Verma IM, Vale W. Effect of adenovirus-mediated overexpression of follistatin and extracellular domain of activin receptor type II on gonadotropin secretion in vitro and in vivo. Endocrinology. 2002;143:964–969. doi: 10.1210/endo.143.3.8667. [DOI] [PubMed] [Google Scholar]
- 37.Schrewe H, Gendron-Maguire M, Harbison ML, Gridley T. Mice homozygous for a null mutation of activin β B are viable and fertile. Mech Dev. 1994;47:43–51. doi: 10.1016/0925-4773(94)90094-9. [DOI] [PubMed] [Google Scholar]
- 38.Vassalli A, Matzuk MM, Gardner HA, Lee KF, Jaenisch R. Activin/inhibin β B subunit gene disruption leads to defects in eyelid development and female reproduction. Genes Dev. 1994;8:414–427. doi: 10.1101/gad.8.4.414. [DOI] [PubMed] [Google Scholar]
- 39.Guo Q, Kumar TR, Woodruff T, Hadsell LA, DeMayo FJ, Matzuk MM. Overexpression of mouse follistatin causes reproductive defects in transgenic mice. Mol Endocrinol. 1998;12:96–106. doi: 10.1210/mend.12.1.0053. [DOI] [PubMed] [Google Scholar]
- 40.Matzuk MM, Finegold MJ, Mather JP, Krummen L, Lu H, Bradley A. Development of cancer cachexia-like syndrome and adrenal tumors in inhibin-deficient mice. Proc Natl Acad Sci USA. 1994;91:8817–8821. doi: 10.1073/pnas.91.19.8817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wurmbach E, Yuen T, Ebersole BJ, Sealfon SC. Gonadotropin-releasing hormone receptor-coupled gene network organization. J Biol Chem. 2001;276:47195–47201. doi: 10.1074/jbc.M108716200. [DOI] [PubMed] [Google Scholar]
- 42.Kakar SS, Winters SJ, Zacharias W, Miller DM, Flynn S. Identification of distinct gene expression profiles associated with treatment of LβT2 cells with gonadotropin-releasing hormone agonist using microarray analysis. Gene. 2003;308:67–77. doi: 10.1016/s0378-1119(03)00446-3. [DOI] [PubMed] [Google Scholar]
- 43.Hsiao A, Ideker T, Olefsky JM, Subramaniam S. VAMPIRE microarray suite: a web-based platform for the interpretation of gene expression data. Nucleic Acids Res. 2005;33:W627–W632. doi: 10.1093/nar/gki443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Labrie F, Luu-The V, Lin SX, Simard J, Labrie C, El-Alfy M, Pelletier G, Belanger A. Intracrinology: role of the family of 17 β-hydroxysteroid dehydrogenases in human physiology and disease. J Mol Endocrinol. 2000;25:1–16. doi: 10.1677/jme.0.0250001. [DOI] [PubMed] [Google Scholar]
- 45.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 46.Fortunel NO, Otu HH, Ng HH, Chen J, Mu X, Chevassut T, Li X, Joseph M, Bailey C, Hatzfeld JA, Hatzfeld A, Usta F, Vega VB, Long PM, Libermann TA, Lim B. Comment on “‘Stemness’: transcriptional profiling of embryonic and adult stem cells” and “a stem cell molecular signature”. Science. 2003;302:393. doi: 10.1126/science.1086384. [DOI] [PubMed] [Google Scholar]
- 47.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 48.Sasik R, Calvo E, Corbeil J. Statistical analysis of high-density oligonucleotide arrays: a multiplicative noise model. Bioinformatics. 2002;18:1633–1640. doi: 10.1093/bioinformatics/18.12.1633. [DOI] [PubMed] [Google Scholar]
- 49.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Storey JD, Xiao W, Leek JT, Tompkins RG, Davis RW. Significance analysis of time course microarray experiments. Proc Natl Acad Sci USA. 2005;102:12837–12842. doi: 10.1073/pnas.0504609102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alarid ET, Windle JJ, Whyte DB, Mellon PL. Immortalization of pituitary cells at discrete stages of development by directed oncogenesis in transgenic mice. Development. 1996;122:3319–3329. doi: 10.1242/dev.122.10.3319. [DOI] [PubMed] [Google Scholar]
- 52.Castro A, Bernis C, Vigneron S, Labbe JC, Lorca T. The anaphase-promoting complex: a key factor in the regulation of cell cycle. Oncogene. 2005;24:314–325. doi: 10.1038/sj.onc.1207973. [DOI] [PubMed] [Google Scholar]
- 53.Liu F, Austin DA, Webster NJ. Gonadotropin-releasing hormone-desensitized LβT2 gonadotrope cells are refractory to acute protein kinase C, cyclic AMP, and calcium-dependent signaling. Endocrinology. 2003;144:4354–4365. doi: 10.1210/en.2003-0204. [DOI] [PubMed] [Google Scholar]
- 54.Lo S-C, Hannink M. CAND1-mediated substrate adaptor recycling is required for efficient repression of Nrf2 by Keap1. Mol Cell Biol. 2006;26:1235–1244. doi: 10.1128/MCB.26.4.1235-1244.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tanriverdi F, Silveira LF, MacColl GS, Bouloux PM. The hypothalamic-pituitary-gonadal axis: immune function and autoimmunity. J Endocrinol. 2003;176:293–304. doi: 10.1677/joe.0.1760293. [DOI] [PubMed] [Google Scholar]
- 56.Chappell PE, White RS, Mellon PL. Circadian gene expression regulates pulsatile gonadotropin-releasing hormone (GnRH) secretory patterns in the hypothalamic GnRH-secreting GT1–7 cell line. J Neurosci. 2003;23:11202–11213. doi: 10.1523/JNEUROSCI.23-35-11202.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miles LEC, Hanyaloglu AC, Dromey JR, Pfleger KDG, Eidne KA. Gonadotropin-releasing hormone receptor-mediated growth suppression of immortalized LβT2 gonadotrope and stable HEK293 cell lines. Endocrinology. 2004;145:194–204. doi: 10.1210/en.2003-0551. [DOI] [PubMed] [Google Scholar]
- 58.Ghersevich S, Akinola L, Kaminski T, Poutanen M, Isomaa V, Vihko R, Vihko P. Activin-A, but not inhibin, regulates 17β-hydroxysteroid dehydrogenase type 1 activity and expression in cultured rat granulosa cells. J Steroid Biochem Mol Biol. 2000;73:203–210. doi: 10.1016/s0960-0760(00)00079-0. [DOI] [PubMed] [Google Scholar]
- 59.Liu F, Usui I, Evans LG, Austin DA, Mellon PL, Olefsky JM, Webster NJ. Involvement of both G(q/11) and G(s) proteins in gonadotropin-releasing hormone receptor-mediated signaling in L β T2 cells. J Biol Chem. 2002;277:32099–32108. doi: 10.1074/jbc.M203639200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lockwood GM, Muttukrishna S, Ledger WL. Inhibins and activins in human ovulation, conception and pregnancy. Hum Reprod Update. 1998;4:284–295. doi: 10.1093/humupd/4.3.284. [DOI] [PubMed] [Google Scholar]
- 61.Fraser HM, Groome NP, McNeilly AS. Follicle-stimulating hormone-inhibin B interactions during the follicular phase of the primate menstrual cycle revealed by gonadotropin-releasing hormone antagonist and antiestrogen treatment. J Clin Endocrinol Metab. 1999;84:1365–1369. doi: 10.1210/jcem.84.4.5586. [DOI] [PubMed] [Google Scholar]
- 62.Mazhawidza W, Winters SJ, Kaiser UB, Kakar SS. Identification of gene networks modulated by activin in LβT2 cells using DNA microarray analysis. Histol Histopathol. 2006;21:167–178. doi: 10.14670/HH-21.167. [DOI] [PubMed] [Google Scholar]
- 63.Weiss J, Guendner MJ, Halvorson LM, Jameson JL. Transcriptional activation of the follicle-stimulating hormone β-subunit gene by activin. Endocrinology. 1995;136:1885–1891. doi: 10.1210/endo.136.5.7720634. [DOI] [PubMed] [Google Scholar]
- 64.Renault MA, Jalvy S, Belloc I, Pasquet S, Sena S, Olive M, Desgranges C, Gadeau AP. AP-1 is involved in UTP-induced osteopontin expression in arterial smooth muscle cells. Circ Res. 2003;93:674–681. doi: 10.1161/01.RES.0000094747.05021.62. [DOI] [PubMed] [Google Scholar]
- 65.Bidder M, Shao JS, Charlton-Kachigian N, Loewy AP, Semenkovich CF, Towler DA. Osteopontin transcription in aortic vascular smooth muscle cells is controlled by glucose-regulated upstream stimulatory factor and activator protein-1 activities. J Biol Chem. 2002;277:44485–44496. doi: 10.1074/jbc.M206235200. [DOI] [PubMed] [Google Scholar]
- 66.Chang PL, Tucker MA, Hicks PH, Prince CW. Novel protein kinase C isoforms and mitogen-activated kinase kinase mediate phorbol ester-induced osteopontin expression. Int J Biochem Cell Biol. 2002;34:1142–1151. doi: 10.1016/s1357-2725(02)00035-3. [DOI] [PubMed] [Google Scholar]
- 67.Hullinger TG, Pan Q, Viswanathan HL, Somerman MJ. TGFβ and BMP-2 activation of the OPN promoter: roles of smad- and hox-binding elements. Exp Cell Res. 2001;262:69–74. doi: 10.1006/excr.2000.5074. [DOI] [PubMed] [Google Scholar]
- 68.Dong C, Zhu S, Wang T, Yoon W, Goldschmidt-Clermont PJ. Upregulation of PAI-1 is mediated through TGF-β/Smad pathway in transplant arteriopathy. J Heart Lung Transplant. 2002;21:999–1008. doi: 10.1016/s1053-2498(02)00403-5. [DOI] [PubMed] [Google Scholar]
- 69.Emons G, Grundker C, Gunthert AR, Westphalen S, Kavanagh J, Verschraegen C. GnRH antagonists in the treatment of gynecological and breast cancers. Endocr Relat Cancer. 2003;10:291–299. doi: 10.1677/erc.0.0100291. [DOI] [PubMed] [Google Scholar]
- 70.Limonta P, Moretti RM, Marelli MM, Motta M. The biology of gonadotropin hormone-releasing hormone: role in the control of tumor growth and progression in humans. Front Neuroendocrinol. 2003;24:279–295. doi: 10.1016/j.yfrne.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 71.Kishi H, Nakagawa K, Matsumoto M, Suga M, Ando M, Taya Y, Yamaizumi M. Osmotic shock induces G1 arrest through p53 phosphorylation at Ser33 by activated p38MAPK without phosphorylation at Ser15 and Ser20. J Biol Chem. 2001;276:39115–39122. doi: 10.1074/jbc.M105134200. [DOI] [PubMed] [Google Scholar]
- 72.Roberson MS, Zhang T, Li HL, Mulvaney JM. Activation of the p38 mitogen-activated protein kinase pathway by gonadotropin-releasing hormone. Endocrinology. 1999;140:1310–1318. doi: 10.1210/endo.140.3.6579. [DOI] [PubMed] [Google Scholar]
- 73.Grundker C, Emons G. Role of gonadotropin-releasing hormone (GnRH) in ovarian cancer. Reprod Biol Endocrinol. 2003;1:65. doi: 10.1186/1477-7827-1-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ali SH, DeCaprio JA. Cellular transformation by SV40 large T antigen: interaction with host proteins. Semin Cancer Biol. 2001;11:15–23. doi: 10.1006/scbi.2000.0342. [DOI] [PubMed] [Google Scholar]
- 75.Hardy K, Mansfield L, Mackay A, Benvenuti S, Ismail S, Arora P, O’Hare MJ, Jat PS. Transcriptional networks and cellular senescence in human mammary fibroblasts. Mol Biol Cell. 2005;16:943–953. doi: 10.1091/mbc.E04-05-0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Klein A, Guhl E, Zollinger R, Tzeng YJ, Wessel R, Hummel M, Graessmann M, Graessmann A. Gene expression profiling: cell cycle deregulation and aneuploidy do not cause breast cancer formation in WAP-SVT/t transgenic animals. J Mol Med. 2005;83:362–376. doi: 10.1007/s00109-004-0625-1. [DOI] [PubMed] [Google Scholar]
- 77.Sax JK, Stoddard A, Murphy ME, Chodosh L, El-Deiry WS. Microarray expression profiling of p53-dependent transcriptional changes in an immortalized mouse embryo fibroblast cell line. Cancer Biol Ther. 2003;2:416–430. doi: 10.4161/cbt.2.4.477. [DOI] [PubMed] [Google Scholar]
- 78.Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, Shahab A, Yong HC, Fu Y, Weng Z, Liu J, Zhao XD, Chew JL, Lee YL, Kuznetsov VA, Sung WK, Miller LD, Lim B, Liu ET, Yu Q, Ng HH, Ruan Y. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124:207–219. doi: 10.1016/j.cell.2005.10.043. [DOI] [PubMed] [Google Scholar]
- 79.Miyazono K, Suzuki H, Imamura T. Regulation of TGF-β signaling and its roles in progression of tumors. Cancer Sci. 2003;94:230–234. doi: 10.1111/j.1349-7006.2003.tb01425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen YG, Lui HM, Lin SL, Lee JM, Ying SY. Regulation of cell proliferation, apoptosis, and carcinogenesis by activin. Exp Biol Med (Maywood) 2002;227:75–87. doi: 10.1177/153537020222700201. [DOI] [PubMed] [Google Scholar]
- 81.Burdette JE, Jeruss JS, Kurley SJ, Lee EJ, Woodruff TK. Activin A mediates growth inhibition and cell cycle arrest through Smads in human breast cancer cells. Cancer Res. 2005;65:7968–7975. doi: 10.1158/0008-5472.CAN-04-3553. [DOI] [PubMed] [Google Scholar]
- 82.Dalkin AC, Gilrain JT, Bradshaw D, Myers CE. Activin inhibition of prostate cancer cell growth: selective actions on androgen-responsive LNCaP cells. Endocrinology. 1996;137:5230–5235. doi: 10.1210/endo.137.12.8940339. [DOI] [PubMed] [Google Scholar]
- 83.Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.