

Abstract
The proteomic profiles from two distinct ovarian endometrioid tumor derived cell lines, (MDAH-2774 and TOV-112D) each with different morphological characteristics and genetic mutations, have been studied. Characterization of the differential global protein expression between these two cell lines has important implications for the understanding of the pathogenesis of ovarian endometrioid carcinoma. In this comparative proteomic study, extensive fractionation of peptides generated from whole cell trypsin digestion was achieved by coupling capillary isoelectric focusing (cIEF) in the first dimensional separation with capillary liquid chromatography (RP-HPLC) in the second dimensional separation. On-line analysis was performed using tandem mass spectra acquired by a linear ion trap mass spectrometer from triplicate runs. A total of 1749 and 1955 proteins with protein probability above 0.95 were identified from MDAH-2774 and TOV-112D after filtering through Peptide Prophet/ Protein Prophet software. Differentially expressed proteins were further investigated by Ingenuity Pathway Analysis (IPA) to reveal the association with important biological functions. Canonical pathway analysis using IPA demonstrates that important signaling pathways are highly associated with one of these two cell lines versus the other, such as the PI3K/AKT pathway which is found to be significantly predominant in MDAH-2774 but not in TOV-112D. Also, protein network analysis using IPA highlights p53 as a central hub relating to other proteins from the connectivity map. These results illustrate the utility of high throughput proteomics methods using large scale proteome profiling combined with bioinformatics tools to identify differential signaling pathways, thus contributing to the understanding of mechanisms of deregulation in neoplastic cells.
Keywords: capillary isoelectric focusing, proteins, ovarian cancer, pathway analysis, quantitation, spectral count
1. Introduction
Ovarian cancer is the fifth leading cause of cancer-related death in the Western world and causes more deaths of women in the United States than any other gynecological malignancy [1]. The five-year survival rate can be as high as 90% with early detection; however, early detection of ovarian cancer is rare and known markers have limited utility for general population screening. The most common form of ovarian cancer is epithelial ovarian cancer which can be further divided into four major histological subtypes: serous, clear cell, mucinous and endometrioid[2]. Ovarian endometrioid adenocarcinoma (OEA) represents approximately 20% of common epithelial tumors.
In the present comparative study, we have employed two closely related OEA cell lines, MDAH-2774 and TOV-112D[3,4]. Both of these two cell lines were derived from female Caucasian patients with OEA. In particular, the TOV-112D cell line originates from an aggressive ovarian endometrioid tumor (stage 3, grade 3). The growth characteristics and tumorigenic potential of this cell line parallels the clinical behavior of aggressive OEAs. Categorization of the tumor grade/stage of MDAH-2774 is not available. Differential global gene expression analyses have been performed, and different genetic defects have been previously detected between these two cell lines, possibly leading to different levels of deregulation of important signaling pathways[5]. It has been shown that both the MDAH-2774 and TOV-112D cell lines have elevated constitutive Wnt signaling deregulation. A missense AXIN1 sequence alteration was identified in MDAH-2774 and mutant beta-catenin was identified in TOV-112D. A mutated K-ras gene, involved in the PI3K/AKT signaling pathway, was detected in MDAH-2774 but not in TOV-112D. Both the MDAH-2774 and the TOV-112D OEA cell lines have a mutant p53 gene[6].
Protein expression and gene expression data, while being mutually exclusive, are complimentary to each other. A lack of direct correlation between protein expression and gene expression has been reported [7]. Protein over/under expression is expected to relate to deregulated tumor cell behavior more directly than would gene expression. The proteomic profiles of these two cell lines have been generated in previous work using different methods. In one study, Rotofor IEF and nonporous (NPS) reversed phase separation was coupled with ESI-TOF-MS and MALDI-TOF-MS to analyze the proteome of MDAH-2774 via intact protein fractionation [8]. In a second study, 2D-PAGE coupled to MALDI-TOF-MS and SDS-PAGE coupled to LC-MS/MS were both used to obtain protein profiles from TOV-112D [9]. Alternatively, a shotgun [10] proteomics strategy of a whole cell digest can be used to compare the global proteome profile of MDAH-2774 and TOV-112D (both qualitatively and quantitatively) in order to analyze protein expression differences in neoplastic dedifferentiation. Within, we have utilized capillary isoelectric focusing (cIEF) to separate peptides based on pH[11-12], followed by capillary reversed phase separation with on-line nanoESI-ion trap mass spectrometer analysis. This method is capable of identifying large numbers of proteins over an extended pH range where 1749 and 1955 proteins from triplicate runs of MDAH-2774 and TOV-112D, respectively, have been identified in this work.
Quantitation is always an important issue in pathway analysis using either isotopic labeling or label free methods. Label-free quantitation has gained increasing popularity in recent years and has been successfully applied in large quantitative studies [13-14] due to the development of computational and statistical methods and advances in LC-MS/MS systems. Extraction of peptide ion intensities and spectral counting (defined as the number of MS/MS spectra identified per protein) are two widely adopted methods for performing comparative quantitative analysis of LC-MS proteomics experiments. It has been shown that spectral counting is highly reproducible and is sensitive to protein abundance changes[15]. Further, in controlled experiments it was found that the correlation of protein abundance with spectral count is superior to that of protein sequence coverage or peptide count[14]. Thus, we have utilized spectral counting to measure protein abundance. The ratio of the spectral count of the same protein represents the relative expression level between two samples. Spectral sampling can enable protein ratios larger than ∼2-fold to be determined with high confidence.
The large number of identified proteins between these two cell lines provides a means for qualitative and quantitative bioinformatics pathway analysis. Differentially expressed proteins can be further investigated to reveal the associated biological pathways using bioinformatic tools such as Ingenuity Pathway Analysis (IPA). The IPA program uses a knowledge base derived from the literature containing information on interactions between genes, proteins and other biological molecules. After uploading differentially expressed protein lists to the IPA server, IPA uses these focused proteins to extract connectivity networks which relate candidate proteins to each other based on their interactions and generates global canonical pathways which are shown to be significantly associated with these candidates[16]. As illustrated in Figure 1, we have used a strategy of shotgun proteomics with subsequent bioinformatics analysis to study pathways in the TOV-112D and MDAH-2774 cell lines in order to understand the different interactions in these two OEA cell lines.
Figure1.
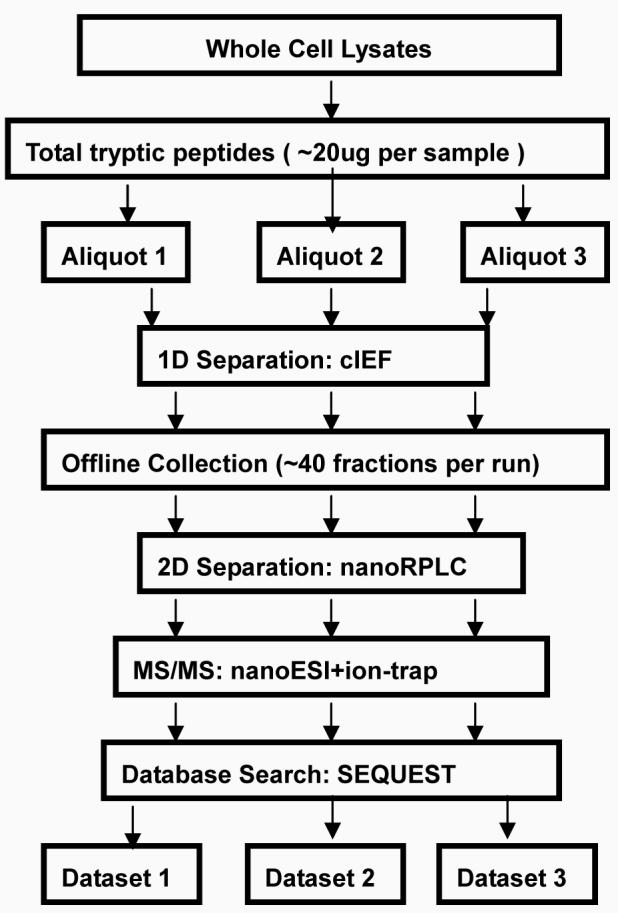

Figure1a: Experimental flowchart
Fig1bDataProcessingStrategy:
2. Materials and Methods
1. Sample preparation
MDAH-2774 and TOV-112D cells were gently washed 3 times with PBS (pH 7.4) by repetitive pipetting, followed each time by centrifugation at 1,500×g for 5min at 4 C. The cell pellets were resuspended with 1 ml lysis-denaturing buffer (7.5M urea, 2.5M thiourea, 12.5 v/v glycerol, 62.5M Tris-HCl, 2.5%(w/v) n-octylglucoside (n-OG) and 1% v/v protease inhibitor cocktail). All chemicals were purchased from Sigma (St. Louis, MO) unless otherwise noted. The lysates were vortexed and then centrifuged at 35,000×g for 1hr at 4 C. The supernatant was collected and dialyzed against 50 mM ammonia bicarbonate overnight using Slide-A-Lyzer dialysis cassettes with a 3,500 Da molecular cutoff from Pierce (Rockford, IL). The proteins were quantified with the micro-BCA assay kit from Pierce (Rockford, IL), and then lyophilized to 100 μl using a SpeedVac concentrator (Labconco, Kansas City, MO) operating at 45 C.
2. Trypsin Digestion
5mM dithiothreitol (DTT) was added and the mixture was incubated at 60C for 30min. After cooling, 5mM iodoacetamide (IAA) was added and the mixture was placed in the dark at room temperature for 30 min in order to carboxamidomethylate the Cysteine residues. Then, 1:50 w/v L-1-tosylamido-2-phenylethyl chloromethylketone (TPCK) modified sequencing-grade porcine trypsin from Promega (Madison, WI) was added. Following vortexing, the mixture was incubated overnight at 37°C in a water bath with agitation, followed by addition of 2% formic acid (FA) to terminate the reaction.
3. First dimension separation: cIEF
Peptides were sequentially resolved based on their different isoelectric points (pI) and hydrophobicity. cIEF was performed on a Beckman CE instrument with sample collection as shown in figure 2. A 70cm cIEF (100um i.d. 365um i.d.) capillary was coated with hydropropyl cellulose for eliminating electroosmotic flow and absorption of peptides onto the capillary wall. The capillary was initially filled with sample gel buffer containing 2% ampholyte 3-10 and 1μg tryptic peptides. Sodium hydroxide solution at pH 10.8 and 0.1M phosphate acid solution were employed as catholyte and anolyte, respectively. One end of the capillary was emerged in the anolyte, while the other end was kept in coaxial metal tubing with a sheath flow composed of catholyte eluting flush with the exit of the capillary. The flow rate was controlled by a syringe pump at 2 μl/min, and was adjusted to ensure that a proper droplet formed at the exit to carry the peptides fractionated into individual wells in the sample plate. Isoelectric focusing was performed at 21kV (300V/cm) over the entire capillary. The current decreased continuously as the peptides were focused and the process was considered complete after the current no longer changed. The focused bands of peptides were sequentially mobilized slowly under pressure towards the cathode and delivered as droplets with catholyte sheath flow into individual wells on a sample plate, where the fractions were collected with a modified Beckman HPLC sample collector. Each cIEF separation runs approximately 90 min. One-third of the run time is spent focusing the peptides in the capillary while the remaining time is used to deposit the off-line fractions.
Figure2.

CIEF-autocollection Instrument Layout
4. Second dimensional separation: nanoRPLC+nanoESI-MS/MS
When cIEF separation was completed, each pI fraction of tryptic-digested sample was injected via Paradigm autosampler (Michrom Biosciences, Auburn, CA) and loaded onto a desalting Nano trap (150μm×50mm) (Michrom) connected to a nano RP column(C18AQ, 5μm 200A 0.1×150mm) (Michrom) by a Paradigm AS1 micropump (Michrom). The mobile phase A and B were composed of 0.3% FA in water and 0.3% FA in acetonitrile (ACN), respectively. Peptides were first desalted and enriched starting at 100%A with a flow rate of 50 μl/min for 5 min. Sample was subsequently separated by a Nano RP column with a flow rate of 0.3 μl/min after splitting. The linear gradient for separation was as follows: from 3% ACN to 12% ACN in 5 min, from 12% ACN to 40% ACN in 30 min, from 40% ACN to 80% ACN in 15 min and finally decreased from 80% ACN to 3% ACN in 10 min. The resolved peptides were then introduced into a ThermoFinnigan LTQ mass spectrometer (Thermo Electron Corp., San Jose, CA) equipped with a nanospray ion source (Thermo). The LTQ was operated in data-dependent mode in which one cycle of experiments consisting of one full MS scan was followed by 5 pairs of zoom scans and MS/MS scans with dynamic exclusion set to 30 sec. The capillary temperature was set at 175 C, spray voltage was 2.8kV, capillary voltage was 30V and the normalized collision energy was 35% for the fragmentation.
5. Database Search and Protein Identification
MS/MS spectra were then searched against the human UniProt FASTA database (updated in Dec.2007) by TurboSEQUEST provided by Bioworks ver3.1 SR1 (ThermoFinnigan). The search was performed using the following parameters: (1) Enzyme: trypsin; (2) one missed cleavage allowed; (3) peptide ion mass tolerance: 1.5Da; (3) fragment ion mass tolerance 0.0 Da; (4)mass tolerance for precursor ions 1.4Da; (5) peptide charges +1, +2, +3; (6) possible modifications: 15.99 Da shift for oxidized Met residues; 79.97 Da for phosphorylated Ser, Thr, Tyr residues respectively; 58.1 Da shift for carboxymethylated Cys residues. The identified peptides were subsequently processed through Peptide Prophet and Protein Prophet incorporated in the Trans Proteomic Pipeline (TPP)[17]. In TPP, the search results were first validated by Peptide Prophet, which converts various SEQUEST parameters to a discriminant score and uses Bayesian statistics to compute the probability that each identified peptide is correct. Protein Prophet reads in peptides and assigned probabilities to compute the probabilities of proteins that are present in the original sample(http://proteinprophet.sourceforge.net/prot-software.html). In this study, we use a protein probability score of ≥0.95 as the threshold for protein identification, to ensure that the minimized overall error rate is below 0.05.
6. Label-free Quantitation and Normalization
After processing the Sequest data through TPP, the spectral counts were parsed out of TPP protXML files using a perl script(see figure 1b). Three datasets of identified proteins with 0.95 protein probability and their associated spectral count have been generated for each sample. We divided the data into two groups. Qualitative data consists of proteins that are only identified in one of these two cell lines, whereas quantitative data consists of proteins that are identified in both of these two cell lines with their expression values. In the first group one cannot compare the same protein expression level between two cell lines. In the second group the relative protein abundance fold change of the same protein can be calculated by the ratio of their spectral count in two samples as explained later in this work. The data was processed in two different ways. For the qualitative analysis, only the qualitative data was used (i.e. only different protein names from two samples were included). We also performed a quantitative analysis in which we combined the qualitative data and quantitative data by replacing any missing value with zero. For example, protein “A” is only detected in MDAH-2774 with its assigned spectral count. In order to make the comparison of the differential expression of this protein plausible, we assume protein “A” is also present in TOV-112D but at a very low level which is not detectable and assign a spectral count of zero to protein “A” in TOV-112D.
Subsequent normalization was used to reduce technical bias when acquiring spectral count data from different runs across the two different cell lines. The bias may come from instrument error or the inherent random sampling nature of the LTQ. In order to normalize the data, we first calculated the ratio of the total spectral count of 3 runs between MDAH-2774 and TOV-112D and then multiplied the spectral count of each protein in the numerator by this ratio. Statistical significance levels of the pair-wise comparison were then adjusted for multiple testing using the false discovery rate (FDR), q-value method. Differentially expressed genes used to learn network structure were declared at a FDR q-value threshold of 0.3. The FDR q-values were calculated using the R package qvalue [18].
7. Ingenuity Pathway Analysis
To infer global network functions between all differentially expressed proteins from MDAH-2774 and TOV-112D, we conducted two types of analysis using Ingenuity Pathway Analysis software (IPA). For the qualitative analysis, we uploaded into the IPA database two sets of proteins with corresponding primary accession number which were only identified from one of these two cell lines. Out of 652 and 838 proteins uploaded from MDAH-2774 and TOV-112D, the IPA software identified 515 and 665 “focus genes” that were eligible for generating connectivity networks and 443 and 582 “focus genes” that were eligible for generating biological functions/disease and associated pathways.
In order to gain further insight into the dynamic changes of the cell states between these two cell lines at the molecular level, we performed a quantitative analysis by incorporating quantitative data in addition to qualitative data. In this analysis, we uploaded a list of 828 differentially expressed proteins with fold change larger than 2 based on normalized spectral count data. The relevant proteins with their fold change, qv-value and corresponding primary accession number were uploaded as an Excel spreadsheet file. 609 proteins were eligible for generating networks and 532 proteins were used to retrieve functions/pathways after applying a threshold of qv-value of <0.3.
The significance values for analyses of network and pathway generation were calculated using the right-tailed Fisher's Exact Test by comparing the number of proteins that participate in a given function or pathway relative to the total number of occurrences of these proteins in all functional/pathway annotations stored in the Ingenuity Pathway Knowledge Base (IPKB).
8. Western Blot Analysis
MDAH-2774 and TOV-112D cell lines were lysed in lysis buffer as described above. 100 μg of total protein from each of the cell lysates was separated by 10% SDS-PAGE in parallel. The resolved proteins were transferred to PVDF membranes (Immobilon-P, Millipore) by conventional procedures using a TE70 semi-dry transfer unit (Amersham Biosciences, Princeton, NJ). Beta-actin protein expression levels were used as an internal control to ensure equal loading between lanes. After transfer, membranes were incubated with a blocking buffer consisting of PBS and 0.1% Tween 20 containing 5% nonfat dry milk overnight. The membranes were incubated for 1 h at room temperature with primary antibodies against UCHL1 (rabbit polyclonal antibody, Biogenesis, NH), SFN (mouse monoclonal antibody; Abcam, Cambridge, MA), MARKS (mouse monoclonal antibody, (Abcam) and beta-actin (mouse monoclonal antibody, (Sigma, St. Louis, MO)) for 1hr at 1:5000 dilution in Tris-buffered saline. Membranes were simultaneously incubated with the mouse anti-beta actin antibody and either the rabbit anti-UCHL1 antibody, the mouse anti-SFN antibody or the mouse anti-MARKS antibody. After three washes with washing buffer (PBS containing 0.1% Tween 20), the membranes were incubated with the appropriate secondary antibody (highly cross absorbed HRP-conjugated goat anti mouse and/or highly cross absorbed HRP-conjugated goat anti rabbit; Abcam) for 1hr at 1:2000 dilution. Immunodetection was accomplished by enhanced chemiluminescence (Amersham Biosciences) followed by autoradiography on Hyperfilm MP (Amersham Biosciences).
3. Results and Discussion
1. cIEF performance
20ug of whole cell lysate was extracted from MDAH-2774 and TOV-112D followed by trypsin digestion. Each aliquot of tryptic peptides (∼5ug) was then loaded to cIEF separation. About 40 fractions were collected per run and each fraction was further subjected to the second dimensional separation coupled with nanoESI-ion trap.
The theoretical pI value for each identified peptide within each fraction was calculated after database searching. The pI distribution plot from the second run of MDAH-2774 is shown in Figure 3. As expected, the pI trend follows a non-perfect linear velocity. Peptides with pI in the region 3.5 to 8 tend to show improved separation performance compared to basic peptides with pI from 8 to 10. Peptides with pI above 10 or below 3 are not expected to be resolved as they fall outside the pH range of the ampholytes (pH 3-10) used in these experiments. Overall, cIEF exhibits high separation resolution with little overlap of the same peptides identified between adjacent fractions.
Figure 3.

Theoretical pI distribution plot of the second run of MDAH-2774. Fraction number shown in the X-axis is plotted against the average of peptides pI value within each fraction shown in the Y-axis.
An important issue here is the use of offline collection of cIEF fractions coupled to nano-RPLC. With the use of on-line cIEF coupled to nano-RPLC one can directly load each fraction to a nano RP-column by sacrificing the separation resolution due to the transfer of cIEF fractions to the 2nd dimension from the increased back pressure and dead volume. In the offline collection method, the sheath-flow eluting from the coaxial tubing was adjusted flush with the exit of the capillary in order to eliminate back pressure and dead volume as shown in figure 2b. Compared to the online integration of cIEF/nano-RPLC, the offline collection mode does not degrade the cIEF separation, significantly reducing the mixing of separated peptides during the transfer process. This is also central for precise quantification by spectral counting in this work.
2. Proteomic Profiling
a). Number of proteins identified
MS/MS spectra were searched against the UniProt database using SEQUEST software and search results were then validated using the Peptide Prophet program. Peptide Prophet provides an empirical statistical model that estimates the accuracy of peptide identifications made by SEQUEST. For each tandem mass spectrum, Peptide Prophet determines the probability that the spectrum is correctly assigned to a peptide based upon its SEQUEST scores. A second program, Protein Prophet was subsequently used to group peptides by their corresponding proteins to compute probabilities that those proteins were present in the original sample. A stringent cutoff of 0.95 was used to filter all the SEQUEST results based on Protein Prophet's estimate of error rate. For each cell line, we have repeated the same experimental procedure and combined the results from all three runs instead of selecting only the overlapping proteins. This is done since some proteins can only be identified in a single run of a sample due to the random sampling nature of tandem MS. The Venn diagram in Figure 4a summarizes the intersection of proteins identified from all three runs of MDAH-2774. In the first run of MDAH-2774, 656 distinct proteins were identified from 25 fractions. 1181 and 1095 distinct proteins were identified in the second run and the third run of MDAH-2774, respectively, when we increased the fractionation number to approximately 40. The total number of proteins from the combined list is 1749 for MDAH-2774 and 1955 for TOV-112D with an overlap of 1092 as shown in Figure 4b.
Figure 4.

Venn diagram of the number of proteins identified from: all three runs of MDAH-2774-2774(3a); MDAH-2774-2774 and TOV-112D-112D (3b) with a minimum protein probability of 0.95 as given by ProteinProphet™.
b) Cellular Localization
Each identified protein was assigned a cellular localization based on information from the Swiss-Prot, Entrez Gene, and Genome Ontology (GO) databases. Figure 5 shows the cellular distribution of 1749 identified proteins from MDAH-2774 and 1092 identified proteins from TOV-112D. The majority are cytoplasmic and nuclear proteins for both of these two cell lines. Membrane proteins only occupy 6% of each total proteome, which is not surprising since the protein extraction method used in this study is not optimized for hydrophobic proteins.
Figure 5.

Cellular Distribution of identified proteins from MDAH-2774 and TOV-112D
3. Label-free Quantitation
Detecting protein quantity and the changes in this quantity between various stages or different samples is central to understanding the molecular process of the cell. We used the spectral count as the measurement of relative protein abundance because it has been shown to accurately reflect relative protein abundance with a linear correlation of over two orders of magnitude of dynamic range [15]. Spectral count was assigned to each identified protein followed by normalization and log transformation. The signal distribution in Figure 6 shows that the ratio of protein expression level between MDAH-2774 and TOV-112D follows a symmetric distribution. These two cell lines have approximately an equal number of proteins that are up-regulated or down-regulated when compared to each other. Only proteins with fold change larger than 2 between MDAH-2774 and TOV-112D, which is equal to 1 in log2 scale, are shown in Figure 6 and were used for further comparative analysis. About two thirds of the identified proteins fall into the column with fold change range between 2 to 4. The rest of proteins fall into a fold change from 4 to 32, with a few exceptions over 2 orders of magnitude.
Figure 6.

Distribution of the protein abundance ratio between MDAH-2774 and TOV-112D on log2 scale. 828 differentially expressed proteins with fold changes larger than 2 based on normalized spectral count data were used to generate this histogram. Horizontal axis shows the ratio of the relative abundance between filtered proteins from MDAH and TOV on log2 scale. Vertical axis shows the number of proteins within each column.
Table 1 lists the 10 most abundant proteins in MDAH-2774 and in TOV-112D and the 10 most differentially expressed proteins based on the ratio of their spectral count from MDAH-2774 over TOV-112D. From the quantitation list, we observed that the most abundant proteins from both of these two cell lines are proteins related to structural elements like vimentin, actin and tubulin, as well as chaperone proteins and members of the heat shock protein family. Proteins that are most differentially expressed between these two cell lines cover a wide range of molecular functions and associations with different diseases. For example, collagen3 alpha1 is a structural constituent of intracellular matrix. Tubulin beta4 is the major constituent of the microtubules. They have both been shown to be associated with epithelial ovarian cancer. Stratifin, a protein kinase C inhibitor, is involved in regulation of progression through the cell cycle and has been shown to be associated with breast cancer and prostate cancer. Eukaryotic translation elongation factor 1 alpha 2 has been shown to be associated with breast cancer. Myristoylated alanine-rich protein kinase C substrate has been shown to be associated with endometriosis.
Table1.
Top10 Expression molecules and top10 differentially expression molecules. The expression values for top10 expression proteins in MDAH-2774 and TOV-112D are normalized mean spectral counts across different runs and are shown in log2 scale. The expression values for top10 differentially expressed proteins are normalized spectral count ratio in log2 scale. Qv-value indicates the significance of difference from multiple test correction.
| Protein Accession Number |
Gene Name |
Exp.Value (log) |
qv-value | Description | |
|---|---|---|---|---|---|
| Top10 Expression Molecules in MDAH-2774 |
P60709 | ACTB | 8.1 | actin, beta | |
| P08670 | VIM | 8.04 | vimentin | ||
| P38646 | HSPA9 | 7.86 | heat shock 70kDaprotein9 (mortalin) | ||
| P68032 | ACTC1 | 7.84 | actin,alpha, cardiac muscle1 | ||
| P11142 | HSPA8 | 7.75 | heat shock 70kDa protein8 | ||
| P62736 | ACTA2 | 7.74 | actin, alpha2, smooth muscle, | ||
| P10809 | HSPD1 | 7.67 | heat shock 60KDa protein1 (chaperonin) | ||
| P043350 | TUBB4 | 7.44 | tublin, beta4 | ||
| P11021 | HSPA5 | 7.43 | heat shock 70KDa protein5 (glucose-regulated protein,78kD) |
||
| P68104 | EEF1A1 | 7.39 | eukaryotic translation elongation factor 1 alpha 1) |
||
| Top10 Expression Molecules in TOV-112D |
P08670 | VIM | 10.67 | vimentin | |
| P07737 | PFN1 | 8.52 | profilin1 | ||
| P15531 | NME1 | 8.3 | non-metastatic cells 1 | ||
| P60709 | ACTB | 8.2 | actin, beta | ||
| P22392 | NME2 | 8.15 | non-metastatic cells 2 | ||
| P11142 | HSPA8 | 8.03 | heat shock 70kDa protein8 | ||
| P68104 | EEF1A1 | 7.9 | eukaryotic translation elongation factor 1 alpha 1) |
||
| P04083 | ANXA1 | 7.81 | annexin A1 | ||
| P02461 | COL3A1 | 7.78 | collagen, type3, alpha1 | ||
| P61978 | HNRPK | 7.65 | heterogeneous nulear ribonucleoproteinK | ||
| Top10 Differentially Expressed Molecules between MDAH-2774 and TOV-112D |
P02461 | COL3A1 | −7.78 | 0 | collagen, type3, alpha1 |
| P04350 | TUBB4 | 7.44 | 0 | tublin, beta4 | |
| P09936 | UCHL1 | −6.77 | 0 | ubiquitin carboxyl-terminal esterase L1 |
|
| P31947 | SFN | 6.56 | 0 | stratifin | |
| P20671 | HIST1H2 AD |
6.15 | 0□26 | histone cluster1, H2ad | |
| Q05639 | EEF1A2 | −5.38 | 0□26 | Eukaryotic translation elongation factor 1 alpha 2) |
|
| P29966 | MARCKS | −5.24 | 0□01 | myristoylated alanine-rich protein kinase C substrate |
|
| P31949 | S100A11 | 5.19 | 0 | S100 calcium binding protein A11 | |
| P37840 | SNCA | −4.94 | 0□26 | synuclein, alpha | |
| Q9BQE3 | TUBA1C | 4.55 | 0 | tublin,alpha 1c | |
Other differentially expressed proteins that are not shown in this table also have important implications on the mechanisms of ovarian endometrioid adenocarcinoma (OEA). For example, beta-catenin (CTNNB1), a critical component of the Wnt signaling pathway, was found to be over-expressed 4.2-fold in TOV-112D as compared to MDAH-2774 based on our spectral count data. This compares favorably with previously reported data by Wu et al. in which CTNNB1 was expressed 4.4-fold in TOV-112D over MDAH-2774 from the CTNNB1/TCF transcription reporter assay [5]. Although CTNNB1/TCF transcriptional activity in MDAH-2774 is modest compared with TOV-112D, it is known to be present at elevated levels in both these two cell lines compared to other ovarian cell lines leading to constitutive activation of the Wnt signaling pathway. Notably, the CTNNB1 missense mutation was detected in TOV-112D by PCR sequencing [5]. It has a mutation in its NH2-terminal regulatory domain, thereby rendering the mutant protein resistant to degradation thus resulting in a higher CTNNB1 level in TOV-112D than in MDAH-2774.
The most significant drawback of spectral counting is that it is more likely to be influenced by the acquisition program of the mass spectrometer compared to other label free comparative quantitation methods such as peptide ion intensity-based quantification. High abundance peptides can mask low abundant peptides if the data dependent MS/MS acquisition exclusion list is too small. If the exclusion list is too large, the spectral count can become rapidly saturated, resulting in reduced sensitivity. We have optimized the conditions in this case by extensive fractionation and setting the exclusion list time to 30 sec.
4. Comparison with Previously Reported Proteins
The proteome of both MDAH-2774 and TOV-112D cell lines have been previously analyzed by different methods. In the first study, a 2D all liquid phase (Rotofor IEF nonporous silica (NPS) RP-HPLC) separation method was used combined with ESI-TOF-MS and MALDI-MS/MS to compare the proteome profile of cultured ovarian cancer cell lines[8]. In this study, 161 unique proteins from MDAH-2774 were identified from five fractions with pH range from 5.8 to 8.3 by using PMF and peptide sequencing analysis after applying a 0.95 probability from the Mascot Search Engine. Around 70% of the proteins identified in the first study were also observed in the current study, including some important cancer-associated proteins such as the Oncoprotein 18/stathmin, ezrin and p53 protein. Oncoprotein 18/stathmin, a conserved cytosolic phosphoprotein that regulates microtubule dynamics, was identified in two of the three runs of the MDAH-2774 cell line. It was previously reported that over-expression of OP18 is associated with a variety of human cancers, including breast cancer and lung cancer[19,20]. Ezrin is a member of the ezrin/radixin/moesin family of membrane-axin cross-linking proteins that also transduces signals from growth factors. Previous studies have shown frequent ezrin over-expression in ovarian carcinomas, particularly in metastatic lesions[21]. Mutant P53 is also known to be over-expressed in MDAH-2774[22].
In another publication [9], the proteome profiling of TOV-112D has been examined by two complementary proteomic approaches, two-dimensional gel electrophoresis (2D PAGE) protein separation coupled to MALDI-TOF/MS and SDS-PAGE coupled to LC-MS/MS. 172 proteins were identified from 2D PAGE and a total of 589 proteins were identified from SDS-PAGE LC-MS/MS after applying a 0.9 probability cutoff by Protein Prophet, of which 436 proteins are also found in the current study. Relatively high expression of stress proteins like HSP90 and HSP71 were observed when compared to other proteins in both studies, as well as in numerous malignant tumors [23]. Two forms of aldehyde dehydrogenase1 which have previously been shown to be over-expressed in aggressive EOC versus non-aggressive EOC or normal ovarian surface epithelia cells at the RNA level were also observed in both studies[24]. Proteins that have been previously proposed as biomarkers or targets for diagnostic studies of invasive ovarian cancer because of their over-expression in invasive carcinomas as compared with benign tumors have been identified in previous studies[25] including FK506-binding protein 4 and several reported differentially expressed proteins such as proliferating cell nuclear antigen (PCNA); leukemia-associated phosphoprotein (stathmin); glutathione S-transferase π (GST π); triose-phosphate isomerase (TPI) and tumor metastatic process-associated protein (Nm23), which have been the subject of extensive investigation in ovarian cancer. In addition, Cytokeratin 18 and Cytokeratin 8 reported as biomarkers by Alaiya et al. [26]were also identified in our study, but not in the work of reference [9].
Overall, more than 70% of the proteins identified in previous work [8,9] were also found in our study when comparing our proteomic profiling results to previously reported data. Coupling of off-line cIEF with online nano-RPLC and nano-ESI-LTQ in our study has enabled a more comprehensive proteomic profiling of differentially expressed proteins between MDAH-2774 and TOV-112D.
5. Ingenuity Pathway Analysis
a) Qualitative Analysis
The 15 most variant canonical signaling pathways between these two cell lines were generated by IPA and are shown in Figure 7 with a threshold p-value<0.1 indicated. The length of the bar only indicates that the differentially expressed proteins are related to this pathway, but is by no means indicative of the pathway being either up-regulated or down-regulated. It is possible that the overall activity of a pathway is up-regulated or down-regulated, but it is not sufficient to draw a conclusion of the direction of change based on the data forming network alone. It is shown that MDAH-2774 and TOV-112D have different levels of association with different signaling pathways. For example, PI3K/AKT signaling was found to be more significant in MDAH-2774 than in TOV-112D from this figure. Previous studies have shown that frequent activation and over-expression of PI3K are associated with ovarian carcinoma[27]. Specifically, amplification of the catalytic subunit alpha of PI3K (PIK3CA) is detected in most ovarian cancer cell lines and primary tumors, as well as the somatic mutations in the gene encoding the p85α regulatory subunit of PI3K (PIK3R1) which leads to constitutive activation of PI3K. PIK3R1 was identified from MDAH-2774 but not in TOV-112D in this study, implying that PI3K/Akt signaling up-regulation is potentially more activated in MDAH-2774 than TOV-112D.
Figure 7.

Comparison of canonical signaling pathways between MDAH-2774 and TOV-112D. Only the 14 most different pathways are shown in the Figure, as ranked by the significance in MDAH-2774. The vertical line indicates a threshold of p<0.1.
It has also been shown that estrogen signaling was found to have a stronger connection with TOV-112D than MDAH-2774 from our IPA analysis. The estrogen receptor (ER) was found to be over-expressed in most ovarian cancers and anti-estrogen drugs have been used to inhibit the growth of ER positive epithelia ovarian cancer cells, implying a strong connection between ER signaling and the tumor, but little is known about the detailed mechanism[28]. The stronger connection with ER signaling in TOV-112D is probably due to the activation of K-ras which has been detected in TOV-112D but not in MDAH-2774 according to our analysis. K-ras is known to be present as the wild type in TOV-112D and mutated in MDAH-2774. Active K-ras can activate the ER through Erk-mediated ER phosphorylation and enhance the steady level of ER. Therefore, ER signaling may turn out to be more pronounced in TOV-112D than MDAH-2774. Also, VEGF and chemokine signaling, which are both related to metastasis, were both shown to be more significant in TOV-112D than MDAH-2774. Other pathways such as insulin-like growth factor-1 (IGF-1) signaling, ERK/MAPK signaling, integrin signaling, cAMP-mediated signaling have all been previously reported to be involved in ovarian cancer[28].
Signature proteins were further characterized based on protein-protein interaction. The de novo network constructed by IPA shown in Figure 8 produced a network comprised of network eligible molecules which have been combined to maximize their specific connectivity. Additional molecules are imported from the ingenuity knowledge base (IPKB) to connect two or more smaller sub-networks by merging them into a larger one. The proteins in the network of MDAH-2774 and TOV-112D and their major functions are listed in Table 2. The network for MDAH-2774 is a compact one, centering on P53□hile the network for TOV-112D is more scattered, composed of small sub-networks with ATM, Jnk and GLI1 in the center.
Figure 8.

Top connectivity map for MDAH-2774 with p values of <10−49(left) and TOV-112D (right) with p values of <10−45. Nodes with gray background are network eligible molecules and nodes with plain background (TOP2, DAN-directed RNA polymerase for MDAH-2774 and 14-3-3, Jnk for TOV-112D) are imported from IPKB. A line indicates interactions, with the arrow-head indicating directionality. The absence of arrowheads refers to a binding interaction. A dotted line indicates an inferred or indirect interaction. The score is based on a p-value calculation, which calculates the likelihood that the Network Eligible Molecules that are part of a network are found therein by random chance alone.
Table2.
Molecules in the top1 network of MDAH-2774 and TOV-112D
| Analysis | Molecules in the network | Score | Top Functions |
|---|---|---|---|
| MDAH-2774 | BANF1,CIRH1A,Ck2,CKAP2 (includes EG:26586),CSNK2A2, CXXC1,DNA-directedRNApolymerase,F11R,FAM3C,GNL3, HIST1H1C,HIST1H1D,MTDH,PDRG1,PLXNB2,POLR1C,POLR2B, POLR3F,POLR3G,PRIM2,RBBP5,S100A16,SAE1,SARS,SUB1, TCOF1 (includesEG:6949),TEP1,TMED7,Top2,TP53, TUBB4,UBE1L2,UBE2I,UBTF,UPP1 |
−49 | Cell Cycle, Cellular Assembly And organization, DNA Replication, Recombinatio n and Repair |
| TOV-112D | 14-3-3,AOF2,ATM,BNC1,Calcineurin protein(s), CD72,CDC25A,CTBP1,DCTN1,DCX,GATA5(includesEG:140628), GLI1,GLI2,GMFG,GSTM2,H2AFX,HDAC4,HMGA2,Jnk,KRT15, MAP3K3,MAP3K5,MRE11A,NEK2,PKD1,PTHR1,REM1,RFC2, RFC4,RFC5,RFXANK,RPA1,SKI,TP53BP1,ZEB2 |
−45 | Cancer, Cell Cycle, DNA Replication, Recombinatio n and Repair |
b) Quantitative Analysis
In order to gain further insight into the dynamic changes of the cell state between these two cell lines at the molecular level, we performed another analysis by incorporating quantitative data along with qualitative data. In this analysis, we uploaded a list of 828 differentially expressed proteins with fold changes larger than 2 based on normalized spectral count data. 609 proteins were eligible for generating networks and 532 proteins were used to retrieve functions/pathways after applying a threshold qv-value of <0.3. The canonical signaling pathways enriched with differentially expressed proteins were constructed and ranked by significance. 14 pathways have been calculated to be significant (Figure 9), where 8 of these pathways are also shown in Figure 7. In addition, some new pathways were also uncovered, such as protein ubiquitination signaling and the hypoxia signaling pathway. We have also sought to examine the differentially expressed components of these pathways in depth. For example, the detailed signaling cascade of PI3K/AKT is depicted in Figure 10. By incorporating the normalized spectral count results, we have been able to calculate the relative expression level of identified proteins under this pathway in addition to detecting their presence or absence.
Figure 9.

Canonical signaling pathways enriched with differential expressed proteins from quantitative analysis. A threshold of P-value <0.1 is applied.
Figure 10.
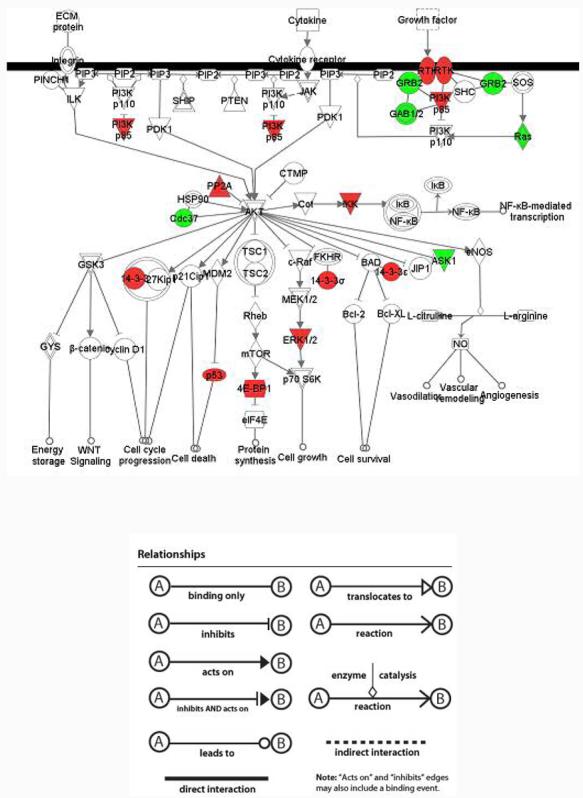
Signaling cascade of PI3K/AKT pathway. Green nodes represent over-expression in MDAH-2774 and red nodes represent over-expression in TOV-112D. Plain nodes are imported from IPKB. This figure has been manually modified from integrative analysis by adding some proteins which were identified from only one cell line but did not meet the threshold of integrative analysis.
The top connectivity network from integrative analysis for MDAH and TOV depicting protein-protein interactions is shown in figure 11. The major network, which is comprised of 34 identified differentially expressed proteins and two imported from IPKB, is displayed in Figure10 with a p-value of <10−49. The major functions extracted from this network are related to cancer, reproductive system disease, and skeletal and muscular disease. P53 is the hub of this network, implying that the differential expression level of P53 in these two cell lines is one of the major driving forces for their differentiation in tumor growth.
Figure 11.

Top connectivity network from integrative analysis. Red and green nodes represent proteins that are identified to be over-expressed in MDAH-2774 and TOV-112D respectively. Darker color indicates larger fold-change. The detailed description of these molecules and their relative expression values can be found from supplemental material.
Pathway analyses of the qualitative data and the quantitative data partially coincide with each other by using IPA. Qualitative data represents a group of proteins with enriched difference between MDAH-2774 and TOV-112D, as they are only detectable in either of these two cell lines. Quantitative data consists of proteins that are detected in both of these two cell lines with a fold change larger than two in addition to those detected in only one of these two cell lines. Analysis based on qualitative data alone is simple to handle, meanwhile it is biased as it excludes the information containing dynamic change in protein abundance whereas quantitative analysis is more comprehensive. The combination of qualitative data and quantitative data is based on the assumption that the spectral count of the protein detected in only one sample is assigned to 0 in the other one. However, we have observed a decrease of sensitivity induced by replacing any missing values with zero. After multiple testing corrections, fold-changes of some proteins between MDAH and TOV are decreased and the q-values are increased, which suggests global signal suppression by this replacement method. One of the explanations could be these missing values are not truly zero, simply because we can not detect them by the current technique. This is especially the case for low abundance peptides which could be masked by their co-eluting high abundance peptides. In the future, target proteomics method (e.g. multiple reaction monitoring methods) will be adopted to verify important proteins.
6. Western Blot Validation
It is becoming increasingly important to validate the proteome profiling results using alternative technologies. In this study, we used one-dimensional western blot analyses to confirm some of the differential expression results inferred by spectral counting. Three proteins were selected from Table 1: UCHL1, Stratifin and MARKS. As can be seen from Figure 12, the intensities from these three proteins correlate well with the spectral counting results shown in the left panel.
Figure12.
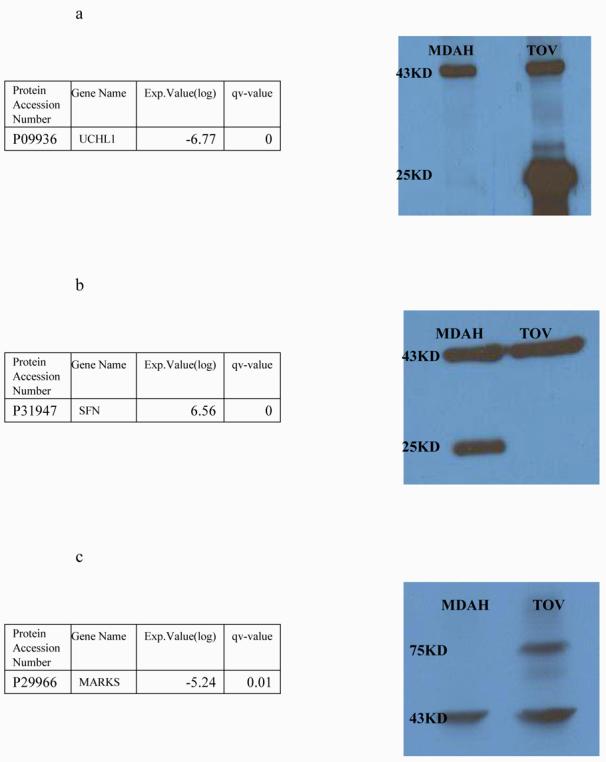
Western Results of UCHL1(a), Stratifin(1433-sigma)(b) and MARKS(c). Expression values are normalized spectral count ratio in log2 scale. Positive value indicates over-expression in MDAH-2774 and negative value indicates over-expression in TOV-112D. Band shown on 37KD is Beta-actin which was used as a control.
4. Concluding Remarks
Proteomic profiles from two ovarian endometrioid derived cell lines with different genetic mutations have been studied using a shotgun proteomic approach. This involved whole lysate digestion by trypsin with extensive fractionation in the first dimension using cIEF based upon a pH-based separation followed by capillary RP-HPLC. On-line analysis was performed using tandem mass spectrometry acquired by a linear ion trap mass spectrometer. A large number of proteins were identified after filtering through the Peptide Prophet/Protein Prophet Trans Proteomic pipeline. Differentially expressed proteins were quantitated using label free methods and studied by Ingenuity Pathway Analysis (IPA) to reveal the association with important biological functions. It was shown that some important signaling pathways may be highly associated with one of the two cell lines. The PI3K/AKT pathway was found to be significantly predominant in MDAH-2774 but not in TOV-112D. The p53 pathway is shown by network analysis to be important in both cell lines but the network in MDAH-2774 is a more compact one centered on p53 while the network for TOV-112D is more scattered and composed of small networks with ATM, Jnk and GLI1 in the center. The fact that p53 is an important hub of this network implies that this pathway is a major driving force for differentiation and growth. Other pathways such as estrogen signaling were found to have a stronger connection to TOV-112D than MDAH-2774 and activation of K-ras has been detected in TOV-112D but not in MDAH-2774. Thus, the method described can define the important pathways involved in cancer development and how it may differ between samples. This strategy may be important for biomarker discovery and may lead to development of candidates for drug treatment of disease.
Supplementary Material
Acknowledgements
This work was supported in part by the National Cancer Institute under grant R01CA100104 (D.M.L.) and the National Institutes of Health under grant R01GM49500 (D.M.L.). We would also like to thank Dr Rong Wu and Dr Kathleen Cho for helpful suggestions during this work. We also thank Dr Alexei Nesvizshkii and Damian Fermin for assistance in the spectral count work.
Abbreviations
- IPA
Ingenuity Pathway Analysis
- OEA
Ovarian Endometrioid Adenocarcinoma
- LTQ
linear ion trap mass spectrometer
- TPP
Transproteomic Pipeline
- NPS
nonporous silica
- FDR
false discovery rate
- IPA
Ingenuity Pathway Analysis
- IPKB
Ingenuity Pathway Knowledge Base
- GO
Genome Ontology
- ER
Estrogen Receptor
5. References
- 1.McCluskey LL, Dubeau L. Curr Opin Oncol. 1997;9(5):465–70. doi: 10.1097/00001622-199709050-00011. [DOI] [PubMed] [Google Scholar]
- 2.Scully RE, Young RH, Clement PB. Atlas of Tumor Pathology. Fascicle; 1998. p. 23. (Third Series). [Google Scholar]
- 3.Freedman RS, Pihl E, Kusyk C, Gallager HS, Rutledge F. Cancer. 1978;42:2352–2359. doi: 10.1002/1097-0142(197811)42:5<2352::aid-cncr2820420536>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 4.Provencher DM, Lounis H, Champoux L, Tetrault M, et al. In Vitro Cell Dev. Biol. Anim. 2000;36:357–361. doi: 10.1290/1071-2690(2000)036<0357:COFNEO>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 5.Wu R, Zhai Y, Fearon ER, Cho KR, et al. Cancer Research. 2001;61:8247–8255. [PubMed] [Google Scholar]
- 6.unpublished data from R.W and K.R.C
- 7.Wang JH, Hewick RM, et al. Drug Discovery Today. 1999;4:129–133. doi: 10.1016/s1359-6446(99)01306-9. [DOI] [PubMed] [Google Scholar]
- 8.Wang H, Kachman MT, Schwartz DR, Cho KR, Lubman DM. Proteomics. 2004;4(8):2476–2495. doi: 10.1002/pmic.200300763. [DOI] [PubMed] [Google Scholar]
- 9.Gagne JP, Gagne P, Hunter JM, Bonicalzi ME, et al. Mol. Cell. Biochemistry. 2005;275:25–55. doi: 10.1007/s11010-005-7556-1. [DOI] [PubMed] [Google Scholar]
- 10.Washburn MP, Wolters D, Yates JR. Nature Biotechnology. 2001;19(3):242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Balgley BM, Rudnick PA, Evans EL, DeVoe DL, Lee CS. J. Proteome Res. 2005;4:36–42. doi: 10.1021/pr049876l. [DOI] [PubMed] [Google Scholar]
- 12.Zhou F, Johnston MV. Electrophoresis. 2005;26:1383–1388. doi: 10.1002/elps.200410125. [DOI] [PubMed] [Google Scholar]
- 13.Andreev VP, Li L, Cao L, Gu Y, Rejtar T, Wu S, Karger BL. J. Proteome Research. 2007;6:2186–2194. doi: 10.1021/pr0606880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wong JWH, Sullivan MJ, Cagney G. Briefings in BioInformatics. 2008;9:156–165. doi: 10.1093/bib/bbm046. [DOI] [PubMed] [Google Scholar]
- 15.Liu H, Sadygov RG, Yates JR. Anal. Chem. 2004;76:4193–4201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 16.Uriarte Silvia M., Powell David W., Luerman Gregory C., Michael L, et al. J. Immunol. 2008;180:5575–5581. doi: 10.4049/jimmunol.180.8.5575. [DOI] [PubMed] [Google Scholar]
- 17.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 18. http://cran.r-project.org/web/packages/qvalue/
- 19.Curmi PA, Nogues C, Lachkar S, Carelle N, Gonthier MP, et al. Br. J. Cancer. 2000;82:142–150. doi: 10.1054/bjoc.1999.0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishio K, Nakamura T, Koh Y, Kanzawa F, Tamura T, et al. Cancer. 2001;91:1494–1499. doi: 10.1002/1097-0142(20010415)91:8<1494::aid-cncr1157>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 21.Chen Z. Cancer. 2001;92:3068–3075. doi: 10.1002/1097-0142(20011215)92:12<3068::aid-cncr10149>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 22.Jacobberger JW, Sramkoski RM, Zhang DS, Zumstein LA, Doerksen LD, et al. Cytometry. 1999;38(5):201–213. doi: 10.1002/(sici)1097-0320(19991015)38:5<201::aid-cyto2>3.3.co;2-6. [DOI] [PubMed] [Google Scholar]
- 23.Conroy SE, Latchman DS, et al. Br J Cancer. 1996;74:717–721. doi: 10.1038/bjc.1996.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tonin PN, Hudson TJ, Rodier F, Bossolasco M, Lee PD, Novak J, Manderson EN, Provencher D, Mes-Masson AM, et al. Oncogene. 2001;20:6617–6626. doi: 10.1038/sj.onc.1204804. [DOI] [PubMed] [Google Scholar]
- 25.Jones MB, Krutzsch H, Shu H, Zhao Y, Liotta LA, Kohn EC, Petricoin EF, et al. Proteomics. 2002;2:76–84. [PubMed] [Google Scholar]
- 26.Alaiya AA, Franzen B, Fujioka K, Moberger B, Schedvins K, et al. Int. J. Cancer. 1997;73:678–683. doi: 10.1002/(sici)1097-0215(19971127)73:5<678::aid-ijc11>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 27.Campbell IG, Russell SE, Choong DY, Montgomery KG, Ciavarella ML, Hooi CS, Cristiano BE, Pearson RB, Phillips WA. Cancer Res. 2004;64:7678–7681. doi: 10.1158/0008-5472.CAN-04-2933. [DOI] [PubMed] [Google Scholar]
- 28.Nicosia SV, Bai W, Cengu JQ, Coppola D, Kurk PA, et al. Hematol Oncol Clin N Am. 2003;17:927–943. doi: 10.1016/s0889-8588(03)00056-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.