

Abstract
Mutations of the ZIC2 transcription factor gene are among the most common heterozygous variations detected in holoprosencephaly (HPE) patients, a patient group who lack critical midline forebrain specification due to defective embryonic signaling during development. Recent studies indicate that complete deficiency of the related murine Zic2 transcription factor can also be a contributing factor to variable midline deficiencies, presenting during mid-gastrulation, that could explain similar forebrain anomalies in this model system. Here we collect and summarize all available mutations in the human ZIC2 gene detected in HPE patients (21 published and 62 novel). Our analysis corroborates this mechanism proposed in mice by predicting loss-of-function as the likely pathogenetic mechanism common to most, if not all, of these mutations in HPE.
Keywords: holoprosencephaly, mutation spectrum, ZIC2
Introduction
ZIC2 (MIM# 603073; HPE5 609637) accounts for at least 3% of detectable holoprosencephaly (HPE) associated genetic variation and is one of four genes, including SHH (Roessler et al., 1996; MIM# 600725; HPE3 142945), SIX3 (Wallis et al., 1999; MIM# 603714; HPE2 157170) and TGIF (Gripp et al., 2000; MIM# 602630; HPE4 142946), commonly screened in clinical laboratories as part of the molecular evaluation of new sporadic or familial cases (reviewed in Muenke and Beachy, 2001; Cohen, 2006; Dubourg et al., 2007). This transcription factor gene was originally implicated in causing this forebrain malformation disorder by mutational screening of candidate genes mapping within a minimal critical region on chromosome 13q32 determined to be causally responsible for HPE cases involving 13q- deletions (Brown et al, 1995; 1998). The original descriptions of the types of mutations observed noted a high prevalence of frameshifts or trunctions indicative of haploinsufficiency of this transcription factor as the consequence of either major genetic alterations of the coding region, or alternatively gross cytogenetic or recently described microdeletions of this gene eliminating normal gene function of one allele (Brown et al., 1998, 2001, 2002, 2005; Bendavid et al., 2005a, b; Paulussen et al., submitted). A second category of mutation prevalent among HPE cases involves an expansion of a poly-Alanine tract in the COOH terminus of the protein that can be shown to interfere with DNA binding and transcriptional activation and likely occurs by somatic recombination (Brown et al., 2001, 2005).
Recent studies of the developmental roles of the Zic2 ortholog in the mouse have elucidated several distinct requirements for this transcription factor during normal development: an early role in the development of the axial midline revealed by a Zic2 null allele (Warr et al., 2008) and a later role in dorsal telencephalic development manifesting as the anatomically distinct middle-hemispheric variant of HPE (Cheng et al., 2006; reviewed in Monuki 2007) provisionally considered characteristic of a hypomorphic allele. Here we tabulate the nature of the detected mutations seen in HPE patients in a classical structure/function analysis and relate these findings to the proposed mechanisms of forebrain pathogenesis suggested by these model systems.
Materials and Methods
Study population
At the NIH, we analyzed approximately 600 HPE patients (collectively comprising the entire spectrum of HPE brain malformations and prospectively collected over 17 years) for potential sequence variations in the ZIC2 gene under our NHGRI approved brain research protocol and newly established CLIA laboratory. In addition, we also studied 125 unrelated individual normal controls obtained as anonymous samples from the Coriell Institute for Medical Research that matched the predominant Northern European ethnicity of our HPE cases. In the cases extracted from literature reports the nature of the mutation was known to us only through these published sources (cited in Table 1). Similarly, in Rennes, 500 HPE patients were analyzed, prospectively collected over 12 years at the Laboratoire de Génétique Moléculaire (Rennes, France); we also include anonymous instances of mutations in the ZIC2 gene shared with us from prospective studies performed under CLIA standards by GeneDx (Gaithersburg, MD), or from investigators in Maastricht, the Netherlands as well as Göttingen, Germany.
Table 1. Summary of human ZIC2 mutations.
| Mutation number | Base-pair alteration | Coding region alteration | Functional activity | Comments | Reference for report |
|---|---|---|---|---|---|
| 1 | -24C>T | N/A | N/A | 5′ non-coding region (immediately preceding the ATG) of unknown importance | GeneDx, this report |
| 2 | c.21delG | p.Gln8SerfsX33 | Predicted null | lacks homeodomain | Brown 2001 (#1) |
| 3 |
c.81_86delGGCGGC insTCGGT |
p.Ala28ArgfsX13 | Predicted null | lacks homeodomain | NIH, this report CLIA; GeneDx confirmed |
| 4 | c.107A>C | p.Gln36Pro | 170% | NH2-terminus | Dubourg 2004; Brown 2005 |
| 5 | c.109G>A | p.Asp37Asn | unknown | NH2-terminus. Also c.1059C>T (p.H353H) | NIH, this report CLIA; GeneDx confirmed |
| 6 | c.129_184dup56 | p.Leu62Argfs175 | Predicted null | NH2-terminus truncation | NIH, this report |
| 7 | c.136C>T | p.Gln46X | Predicted null | NH2-terminus truncation | Dubourg 2004; and GeneDx confirmed |
| 8 | c.172G>T | p.Gly58X | Predicted null | NH2-terminus truncation | Dubourg 2004 |
| 9 | c.177ins56 | p.Phe60GlnfsX176 | Predicted null | lacks homeodomain | Brown 1998 (#a) |
| 10 | c.191dupC | p.Ala66ArgfsX301 | Predicted null | lacks homeodomain | U. Rennes, this report |
| 11 | c.217C>T | p.Gln73X | Predicted null | lacks homeodomain | U. Rennes, this report |
| 12 | c.217delC | p.Gln73Argfs145 | Predicted null | lacks homeodomain | NIH, this report |
| 13 | c.367delA | p.Ser123AlafsX95 | Predicted null | lacks homeodomain | U. Rennes, this report |
| 14 | c.382G>A | p.Asp128Asn | unknown | Mis-sense | NIH, this report |
| 15 | c.386_392delCGGCGCC | p.Ser129TrpfsX87 | Predicted null | lacks homeodomain | NIH, this report |
| 16 | c.392_398del7 | p.Gly133SerfsX83 | Predicted null | lacks homeodomain | GeneDx, this report |
| 17 | c.454_455delinsTT | p.Asp152Phe | 60% | Mis-sense NH2-terminus | Brown 2001 (#3); Dubourg 2004 |
| 18 | c.479delC | p.Pro160ArgfsX58 | Predicted null | lacks homeodomain | Maastricht |
| 19 | c.490G>T | p.Glu164X | Predicted null | lacks homeodomain | NIH, this report; confirmed GeneDx |
| 20 | c.557_572dup16 | p.Glu192GlyfsX180 | Predicted null | lacks homeodomain | GeneDx, this report |
| 21 | c.577delC | p.Gln193AsnfsX25 | Predicted null | lacks homeodomain | GeneDx, this report |
| 22 | c.582C>A | p.Tyr194X | Predicted null | lacks homeodomain | U. Rennes, this report |
| 23 | c.612delC | p.Tyr205ThrfsX13 | Predicted null | lacks homeodomain | NIH, this report CLIA |
| 24 | c.659delA | p.Asn220ThrfsX4 | Predicted null | lacks homeodomain | U. Göttingen, this report |
| 25 | c.665_676dup12 | p.Gly222_Met225dup | unknown | unknown | U. Rennes, this report |
| 26 | c.710_718dupCACCACCAC | H12 variant | unknown | Observed length variation of 8 up to 12 H's | Brown 2001; and NIH, this report CLIA |
| 27 | c.716_718del | H8 variant | unknown | Observed length variation in controls | Dubourg 2004 |
| 28 | c.716_718dupACC | H10 variant | unknown | Observed length variation in controls | Brown 2001; Orioli 2001; Dubourg 2004 GeneDx, and NIH this report CLIA |
| 29 | c.748C>T | p.Gln250X | Predicted null | Truncation in ZF-NC; lacks homeodomain | NIH, this report |
| 30 | c.779G>A | p.Trp260X | Predicted null | Truncation in ZF#1 | U. Göttingen, this report |
| 31 | c.793C>T | p.Gln265X | Predicted null | lacks homeodomain | GeneDx, this report |
| 32 | c.808_809ins17 | p.K270ThrfsX2 | Predicted null | Truncation in ZF#1 | Dubourg 2004 |
| 33 | c.815G>A | p.Ser272Asn | Predicted null | Mis-sense in ZF#1 | U. Rennes, this report |
| 34 | c.825_826delAA | p.Lys275AsnfsX91 | Predicted null | Fs in ZF#1; lacks homeodomain | GeneDx, this report |
| 35 | c.829_830dupTT | p.Thr279AlafsX7 | Predicted null | FS in ZF#1; lacks homeodomain | GeneDx, this report |
| 36 | c.862_863delTC | p.Ser288GlyfsX78 | Predicted null | Fs in ZF#1; lacks homeodomain | Orioli 2001 |
| 37 | c.857A>T | p.His286Leu | Predicted null | Invariant C2H2 structure ZF#1 | GeneDx, this report |
| 38 |
c.858C>G also c.1059C>T |
p.His286Gln also p.His353His | Predicted null also Common variant | Invariant C2H2 structure ZF#1; | NIH, this report; GeneDx confirmed |
| 39 | c.856C>T | p.His286Tyr | Predicted null | Invariant C2H2 structure ZF#1 | U. Rennes, this report |
| 40 | c.871C>T | p.His291Tyr | Predicted null | Invariant C2H2 structure ZF#1 | U. Rennes, this report |
| 41 | c.910T>A | p.Trp304Arg | Predicted null | ZF#2 | GeneDx, this report |
| 42 | c.912G>A | p.Trp304X | Predicted null | Truncation in ZF#2 | GeneDx and U. Rennes, this report |
| 43 | c.928G>T | p.Glu310X | Predicted null | ZF#2 truncation | NIH and U. Rennes, this report |
| 44 | c.932delG | p.Gly311AlafsX102 | Predicted null | ZF#2 frameshift | Brown 2001 (#4) |
| 45 | c.941T>G | p.Phe314Cys | Predicted null | Conserved ZF#2 | Rennes, this report |
| 46 | c.973C>A | p.Arg325Ser | Predicted null | Conserved ZF#2 | NIH, this report; GeneDx confirmed |
| 47 | c.974G>T | p.Arg325Leu | Predicted null | Conserved ZF#2 | U. Rennes, this report |
| 48 | c.979C>T | p.His327Tyr | Predicted null | ZF#2 invariant C2H2 motif | NIH, this report |
| 49 | c.1004G>T | p.Cys335Phe | Predicted null | ZF#3 invariant C2H2 motif | NIH, this report |
| 50 | c.1025_1026delAA | p.Lys342SerfsX24 | Predicted null | ZF#3 frameshift | Dubourg 2004 |
| 51 | c.1031_1032delTC | p.Phe344CysfsX22 | Predicted null | ZF#3 frameshift | Brown 2001 (#5) |
| 52 | c.1040_1046del | p.Glu348SerfsX63 | Predicted null | ZF#3 frameshift | Brown 1998 (#d) and NIH, this report |
| 53 | c.1051A>T | p.Lys351X | Predicted null | ZF#3 truncation | NIH, this report |
| 54 |
c.1052_1053insAAGGTT CACACAGAACCTCAA |
p.Lys351_Ile352ins7 | Predicted null | ZF#3 deletion/insertion | U. Rennes, this report |
| 55 |
c.1075+2T>A (IVS1+2T>A) |
Gly359fsX62 | Predicted null | Eliminates ZF#4 and 5 | U. Rennes, this report |
| 56 |
c.1076-1G>A (IVS1-1G>A) |
alternative splicing | Predicted null | ZF#1-3 intact; likely eliminates ZF#4-5 | NIH, this report; GeneDx confirmed |
|
c.1076-1G>A (IVS1-1G>A) |
alternative splicing | Predicted null | ZF#1-3 intact; likely eliminates ZF#4-5 | NIH, this report | |
|
c.1076-1G>A (IVS1-1G>A) |
alternative splicing | Predicted null | ZF#1-3 intact; likely eliminates ZF#4-5 | NIH, this report | |
| 57 |
c.1239+1G>C (IVS2+1G>C) |
Possible inclusion of intron 2 codons or alternative splicing | Predicted null | Disrupts ZF#5 | GeneDx, this report |
| 58 |
c.1240-2A>G (IVS2-2A>G) |
alternative splicing | Predicted null | ZF#1-3 intact; likely eliminates ZF#4-5 | NIH, this report |
| 59 | c.1090C>T | p.Gln364X | Predicted null | Eliminates all of ZF#4-5 | Maastricht |
| 60 | c.1091_1092delAG | p.Gln364LeufsX2 | Predicted null | Eliminates all of ZF#4-5 | Dubourg 2004 |
| 61 | c.1095_1096delTG | p.Cys365X | Predicted null | Eliminates all of ZF#4-5 | NIH, this report (case #1) |
| c.1095_1096delTG | p.Cys365X | Predicted null | Eliminates all of ZF#4-5 | NIH, this report (apparently unrelated case #2) | |
| 62 | c.1097_1098delAG | p.Glu366Valfs2 | Predicted null | ZF#4 truncation | NIH, this report; GeneDx confirmed; Brown 2001 (#7) |
| 63 | c.1119_1120delCT | p.Phe374ArgfsX17 | Predicted null | ZF#4 frameshift | NIH, this report |
| 64 | c.1118G>C | p.Arg373Pro | Predicted null | ZF#4 conserved | GeneDx, this report |
| 65 | c.1206C>G | p.Tyr402X | Predicated null | ZF #5 truncation | NIH, this report CLIA |
| 66 | c.1204T>A | p.Tyr402Asn | Predicted null | ZF#5 core sequence | NIH, this report; GeneDx confirmed |
| 67 | c. 1208C>A | p.Thr403Lys | Predicted null | ZF#5 core sequence | NIH, this report; GeneDx confirmed |
| 68 | c.1211A>G | p.His404Arg | Predicted null | ZF#5 core sequence | GeneDx, this report |
| 69 | c.1225C>T | p.Arg409Trp | Predicted null | ZF#5 core sequence | NIH, this report |
| 70 | c.1245T>G | p.His415Gln | Predicted null | Invariant C2H2 structure ZF#5 | U. Rennes, this report |
| 71 | c.1277delC | p.Pro426ArgfsX129 | Predicted null | Intact ZF#1-5 | NIH, this report; GeneDx confirmed |
| 72 | c.1313dupC | p.Leu440AlafsX90 | Predicted null | Intact ZF#1-5 | Brown 1998 (#b) |
| 73 | c.1323dupG | p.Ser442Valfs88 | 2% | Intact ZF#1-5 | NIH, this report; Brown 2001 (#8) and 2005 |
| 74 | c.1330_1365del | p.444_455del In frame deletion | 60% | Intact ZF#1-5 and intact polyalanine tract | Brown 2001 (#9) and 2005 |
| 75 | c.1377_1406dup30 | p.Ala461_470dup In frame duplication | 5% with reduced DNA binding | 25 alanine variant | Brown 1998 (#c) and 2001 (#10-16) and 2005; NIH, this report CLIA; Maastricht, this report (one case maternal and one de novo). |
| 76 | c.1392_1406del | In frame deletion | Predicted null | 10 alanine variant | NIH, this report; GeneDx confirmed |
| 77 | c.1377_1406del | In frame deletion | Predicted null | 5 alanine variant | U. Rennes, this report |
| 78 |
c.[1420-1427dup; 1428-1433delinsCG] |
p.Gly477CysfsX54 | Predicted null | Novel COOH terminus | U. Rennes, this report |
| 79 | c.1445_1461del17 | p.Ser482ArgfsX42 | Predicted null | Novel COOH-terminus | NIH, this report; GeneDx confirmed |
| 80 | c.1452_1456delCGCGG | p.Ala485ArgfsX43 | Predicted null | Novel COOH terminus | U. Rennes, this report |
| 81 | c.1455_1461delinsCG | p.Gly487LeufsX41 | Predicted null | Novel COOH terminus | U. Rennes, this report |
| 82 |
c.1508_1520delGCGGCGG GGGCGG |
p.Gly503AlafsX48 | Predicted null | Novel COOH terminus | Maastricht |
| 83 | c.1559delA | p.His520ProfsX35 | Predicted null | Novel COOH terminus | U. Rennes, this report |
Sequence variant numbering is based on ZIC2 cDNA sequence (GenBank NM_007129.2). Nucleotide numbering uses the A of the ATG translation initiation site as nucleotide +1 and the 5′ untranslated variant is numbered on this basis, rather than by its genomic position.
Mutation screening, PCR amplification and DNA sequencing
A strategy for screening the ZIC2 gene has previously been described (Brown et al., 1998; Dubourg et al., 2004). Descriptions of mutations (Table 1) are based on the NM_007129.2 reference sequence utilizing current nomenclature recommendations www.hgvs.org/mutnomen. Nucleotide numbering reflects cDNA numbering with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence. The initiation codon is codon 1. Our current approach for CLIA testing for ZIC2 differs slightly from previous reports and is available upon request. Amplification of human genomic DNA is performed in a 30 μl reaction volume, using 60-100 ng DNA template, 50 μM each of deoxynucleotide triphosphate, 0.25 μM of each primer, 3 μl of 10X PCR Amplification buffer (Invitrogen), 1.5 μl 10X Enhancer buffer (Invitrogen) and 0.3μl Taq polymerase. All reactions were performed using a PTC-255 thermocycler (MJ Research, MA). Typical PCR cycling parameters were 95°C for 4 minutes followed by 30 cycles at 95°C, annealing at 62°C, extension at 72°C for 1 minute, and a final extension step of 72°C for 5 minutes. One half of the PCR product was used for denaturing high-pressure liquid chromatography (dHPLC) analysis (Trangenomics WAVE™) and the remainder was retained for direct DNA sequencing. Amplicons displaying heterozygous profiles were purified using a high pure PCR purification kit (Roche, IN) and bi-directionally sequenced using the BigDye™ version 3.1 terminator cycle sequencing kit according to the manufacturer's protocol (Applied Biosystems, CA) on an ABI 3100 automated sequencer.
Results
In order to derive insight into the important functions and domains of the ZIC2 protein inferred from our mutational analysis, we have compiled all 83 of the known variants detected exclusively among HPE cases and not in controls into Table 1. Several important findings emerge from this compilation (broken down for convenience by mutation type in Figure 1A-D). Regardless of mutation type these categories of variation have either been directly shown to reduce biological activity (Brown et al., 2005) or would be predicted to lose activity by analysis of the effects on key structural motifs of virtually translated protein products.
Figure 1. A schematic representation of all of the mutations affecting the ZIC2 coding region sorted by their position and category.
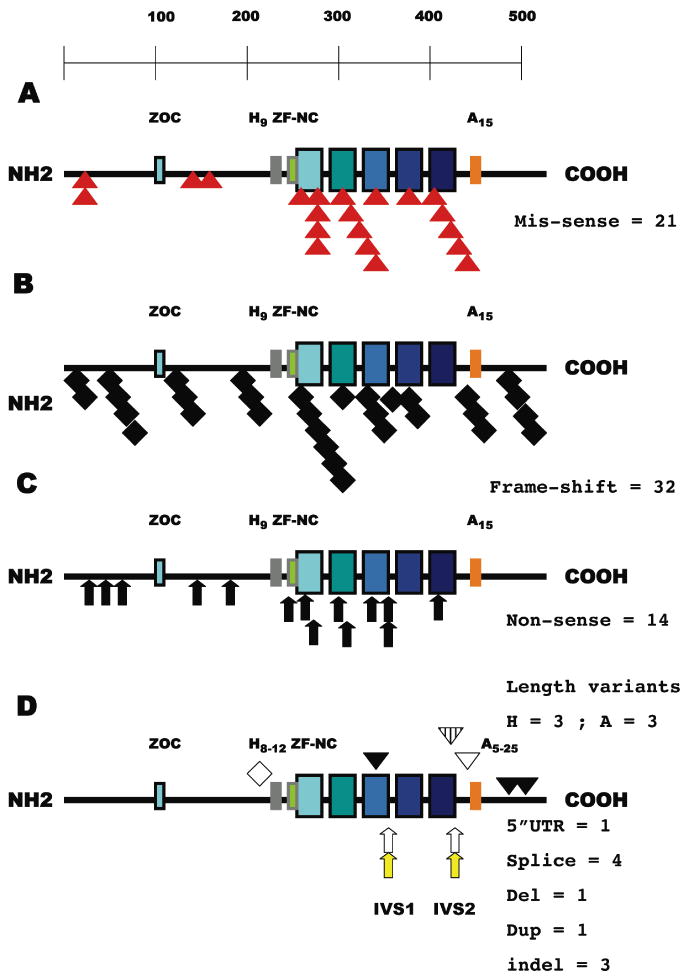
A: Mis-sense variations (red triangles, 25% overall) cluster within the zinc-finger motifs (17/21 = 81%). The ZIC2 transcription factor is illustrated as a line of aminoacids (from left to right, NH2 to COOH) with key motifs (Aruga et al., 2004, 2006) depicted by rectangular boxes: ZOC homology block (blue), His-9 variable region (grey, range 8-12), homology block ZF-NC (green) preceding the first of five zinc-fingers (increasing shades of blue to indicate that each finger has a unique structure) and Ala-15 variable region (orange, range 5-25). B: 32/83 = 38.5% of the mutations (black diamond) would predict a significant alteration of the reading frame and variable truncations primarily affecting DNA binding. C: Note that all non-sense truncations (black arrows, 17% overall) occur prior to zinc-fingers 4 and 5 primarily implicated in DNA binding by Krüppel/GLI-related transcription factors (Kinzler et al., 1990; Pavletich and Pabo, 1993). D: The typical observed range of poly-Alanine and poly-Histidine segments are shown (Brown et al., 1998, 2001, 2002; Dubourg et al., 2004). All four invariant splice sites including both donor (arrow, no fill) and both acceptor (yellow arrow) sites are subject to mutation. Note that the failure to initiate a splice from IVS2 donor site nevertheless maintains the reading frame predicting a protein with 71 additional aminoacids and a distortion of the zinc-finger 5 (inverted hatched triangle, see also Figure 2). Duplications that retain or resume frame are shown as diamonds with no fill while the inverted open triangle illustrates an in frame deletion. Complex insertion/deletion sequence variants causing a shift of the reading frame are the black inverted triangles. The non-coding 5′ UTR variation is not indicated by any symbol.
First, except for the poly-Alanine tract expansion/contraction cases, that continue to represent a prevalent mutation type (Fig. 1D, see below), we observe that most mutations are detected in only a single family (i.e. family-specific alterations) and at least 50% can be demonstrated to be de novo within the HPE proband's DNA (Dubourg, this study). There is a strikingly similar molecular pathology elucidated for the zinc-finger mutations in the X-linked ZIC3 gene and their proven effects on diminished DNA binding and reporter gene activation (Ware et al., 2004; Zhu et al., 2008). In this respect, heterozygous ZIC2 mutations differ from the hemizygocity of the X-linked ZIC3 gene or the homozygous hypomorph or null models of HPE in mice (Nagai et al., 2000; Warr et al., 2008).
Second, our findings clearly illustrate that there is a common feature of predicted loss-of-function attributable to virtually all of the HPE-specific variants, especially evident in our continued detection of a high frequency of frameshifts and truncations (>55%) also noted in the original studies (Brown et al. 1998, 2001; Dubourg 2004). None of the truncation mutations when hypothetically translated are consistent with a complete set of zinc-fingers; this is also true of the majority of the predicted frameshift variants (compare Fig. 1B to C). However, a significant number of frameshifts (Fig. 1B) or insertion/deletions (Fig. 1D) occur in the extreme COOH terminus of the protein indicating that simple variations affecting the zinc-finger domain are not the only mechanism affecting ZIC2 function and confirming previous interpretations (Brown et al. 2005).
Thirdly, almost all of the mis-sense mutations occur in residues known to be either extensively conserved among the zinc-finger regions of all Zic-related genes (Fig. 1A; see also Aruga et al., 2006) or intrinsic to their ability to fold and interact with DNA. Specifically, each of the five zinc fingers is defined by a Cystidine2-Histidine2 (C2H2 motif) comprising four bonds that coordinate with a zinc-atom into a unique three-dimensional configuration (Redemann et al., 1988). We observe that 5 of these 20 key residues are either directly mutated through codon changes or displaced by insertions (Fig. 2). In addition, we demonstrate that by compiling additional cases there are now numerous examples of codons or genetic elements important to the function of the protein that are targets of independent mutational damaging events in different families. For example, p.286His is not only an invariant residue within Zinc-finger #1 (ZF #1 in Table 1, Fig. 1A and Fig. 2) but it is also found mutated to different codons in three different HPE subjects, strengthening the interpretation that the zinc-fingers and their integrity are key to the normal function of the ZIC2 protein.
Figure 2. Detailed structure of the zinc-finger region.
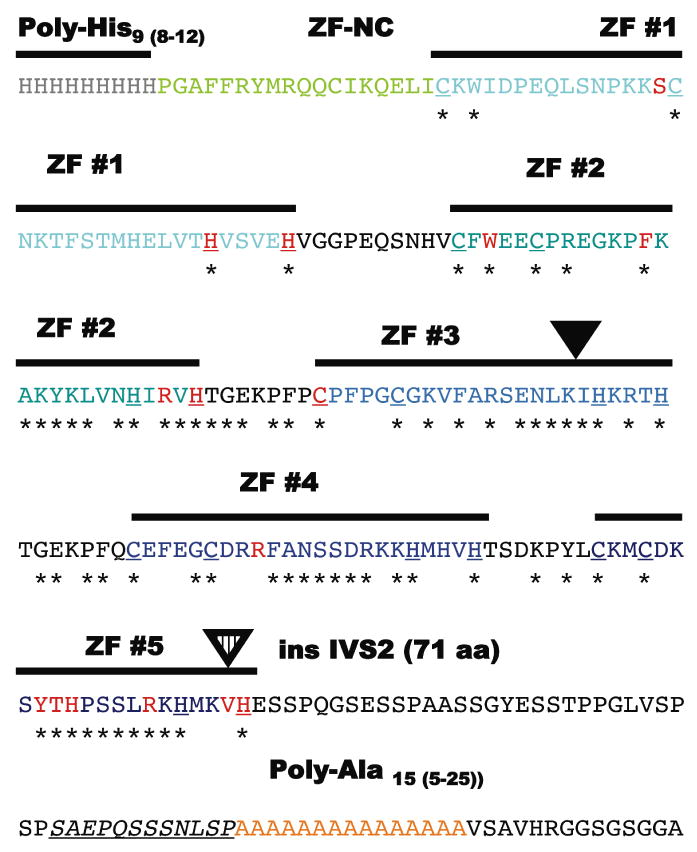
The aminoacid sequence (single aminoacid code) of the region from approximately His-9 to Ala-15 that contains the individually numbered Zinc-finger (ZF domains) and the most mutations with individual motifs colored as in Figure 1. The C2H2 components of each zinc-finger are bold and underlined and residues invariably present in the ZIC-family alignment are identified with an asterix. Note the non-random clustering of the mis-sense mutations (red) within these conserved aminoacid residues and particularly those that define the zinc-binding motif of the C2H2 zinc fingers (Redemann et al., 1988). Similarly, the addition of 7 novel aminoacids within the ZF #3 likely results in distorted alignment (inverted black triangle). The c.1239+1G>C (or IVS2+1G>C) allows for the incorporation in toto of the IVS2 into the reading frame of the transcript and adding an additional 71 aminoacids. This insertion also removes the ValHis sequence from its normal context within the C2H2 motif of ZF #5 and distorting its structure; this same key C2H2 Histidine is a target of mutation in another HPE individual.
Of the four mis-sense alterations detected outside of the zinc-finger domain there is evidence for apparent clustering. The c.107A>C Gln36Pro variant has previously been shown to retain 170% activity in cell-based luciferase assays (Brown et al., 2005). Interestingly, we now report that the adjacent residue c.109G>A Asp37Asn is altered in a novel HPE proband potentially supporting a role for this region of the NH2-terminus in ZIC2 function. Similarly, c.454_455delGAinsTT Asp152Phe has been shown to deleteriously affect function, while the nearby mutation c.382G>A Asp128Asn is presently untested. It is also worth noting that although the ZOC (Zic/Opa [Odd-paired] Conserved) domain is highly conserved among ZIC proteins (Aruga et al., 2006) it has not been implicated in our mutational analysis thus far.
Defects in splicing are predicted to follow from independent mutations in both donor and both acceptor sites (Fig. 1D); the common consequence of these changes is to deprive the transcript from the ability to encode an intact set of zinc-fingers. Note that three apparently unrelated families have the same alteration in the splice acceptor site of the first intervening sequence (c.1076-1G>A or IVS1-1G>A). Even in-frame insertions tend to occur in key structural elements, such as ZF #3 and ZF #5 predicting altered DNA binding or its absence as the most common component of the molecular pathology.
One of the principal conclusions of the most rigorous functional analysis to date (Brown et al., 2005) was that beyond the obvious importance of the zinc-finger domain (see above) there remained a significant fraction of all cases demonstrating length variations of either the poly-Histidine tract or especially the poly-Alanine tract (Brown et al. 2001, 2002, 2005). While the former His length variation is currently postulated to be a simple population variant (stretches of 8-12 residues have been observed in both patients and controls) there is overwhelming evidence that an expansion of this poly-Alanine tract to 25 affects both DNA binding and reporter gene activation. We now show that length reductions to 5 or 10 aminoacids are also detected. While these two length reduction variations will require additional study, it is intriguing that the spacing between the zinc-fingers and the poly-Alanine tract is proven to be important for ZIC2 function, since the in-frame deletion of 12 aminoacids, variant #74 c.1330_1365del36, between the end of ZF #5 and the beginning of the poly-Alanine tract clearly reduces biological activity (Brown et al., 1998, 2005).
Consistent with the fact that ZIC2 was initially described as a key gene within a minimal critical region of common HPE-associated cytogenetic re-arrangements, it follows that copy-number variations of the gene should be readily detectable and produce similar biological effects as a more typical loss-of-function allele through crucial coding region alterations. This is indeed the case, since the U. Rennes has identified four independent cases of ZIC2 loss through microdeletion (Bendavid and David, in preparation), the Maastricht team has detected one ZIC2 microdeletion (Paulussen et al., submitted), and our NIH studies have also identified one additional case (∼1.25% of tested probands, Lacbawan and Muenke, in preparation). As new knowledge is gained relating to non-coding regulatory elements essential for ZIC2 expression there are likely to be additional mutational targets identified.
Discussion
Holoprosencephaly is the most common structural malformation of the developing forebrain in humans and its causation is multifarious (including genetic and environmental components) and complex (Muenke and Beachy, 2001; Roessler et al., 2001; Cohen, 2006; Dubourg et al., 2007; Monuki et al., 2007). Its genetic heterogeneity is supported by the fact that nearly 75% of cases are wild-type in their DNA sequence of the coding regions of the nine currently known HPE genes in patients with normal chromosomes. Its characteristic variable expressivity adds additional support for a model of combinatorial interactions among multiple factors (Ming and Muenke, 2002). Although much more needs to be elucidated regarding the potential roles of additional genes, a concerted focus is also required to integrate the potential interactions among the presently known HPE genes and environmental stressors, such as ethanol exposure and maternal diabetes.
The vertebrate system that most closely mirrors genetic susceptibilities for HPE in humans is that of the mouse (reviewed in Krauss et al., 2007). The recent finding that the Kumba mutation (Elms et al., 2003) in murine Zic2 is likely a null suggests that complete absence of this transcription factor causes a mid-gastrulation failure of the axial midline development that can manifest itself as HPE to various degrees (Warr et al., 2008). By contrast, a homozygous hypomorphic Zic2 mutant line (estimated to have ∼20% activity) develops normally through gastrulation and instead displays dorsal forebrain malformations at later stages (Nagai et al., 2001). Mice heterozygous for Zic2 null alleles (50% activity) are phenotypically normal. Therefore, the model that emerges suggests that the cyclopia that follows the loss of both alleles in mice is a consequence of a variable degree of midline arrest, with secondary effects at a later stage on forebrain patterning activities such as Sonic hedgehog. Interestingly, these same investigators could not demonstrate any synergy in the genetic interactions between Zic2 and Shh compound mutants, suggesting that as reflected by these studies that these factors act in parallel, but not directly linked molecular pathways. This is a potentially important observation since the zinc-finger domains of the Zic and Gli factors bind to the identical target sequences (Kinzler et al. 1990; Pavletich and Pabo 1993; Mizugishi et al., 2001) and these facts had suggested that Zic2 was a likely downstream effector of hedgehog signals (Roessler et al., 2001). It now appears likely that the mid-gastrulation functions of Zic2 preceed those of Shh in the mouse (Warr et al., 2008) and that these factors can play important roles at more than one stage of embryonic development.
Our reprise of ZIC2 mutations brings this necessity for integrative gene/environment interactions directly into focus. Heterozygous mutations are the norm in humans. No instances of simultaneous loss of both copies of human ZIC2 have been described to date, despite our clear understanding that this would be a pre-requisite for producing such a phenotype in the mouse model (Warr et al., 2008). Furthermore, no clear pattern of co-morbid genetic changes is apparent among the patients summarized in this report. While ZIC2 screening has been performed for nearly a decade the number of cases of simultaneous genetic lesions involving ZIC2 with other known or candidate HPE genes are anecdotal and infrequent (Ming and Muenke, 2002). Hence, our certitude with respect to the importance and implications of a genetic variant in the ZIC2 gene (as one component of a multiple-hit process) become essential when the second key component of pathogenesis remains obscure. At the present time we can say that the proposed second-hit is not commonly in the SHH gene, or its functionally related pathway factors (Roessler et al., 2005), and may well be one of the poorly understood factors dependent on either direct ZIC2 function or those its downstream targets.
Our structural analysis of these mutations indicates that DNA binding, influenced by the structural integrity of the zinc-fingers and modulated by the COOH-terminus, are the most critical domains detected as mutations among our combined HPE cohorts. For the purposes of genetic counseling, it is important to convey that the expectation of detecting an abnormality in ZIC2 in a new HPE case is high and most of these changes are risk factors for HPE in these families. Undetected sequence variations at other genomic loci, as well as environmental factors and stochastic effects also factor into the extremely variable expression typical of the complex genetic underpinnings of this disorder.
Acknowledgments
The authors wish to thank the patients who participated in this research and the many clinicians who referred them. This research was supported by the Division of Intramural Research of the National Human Genome Research Institute, National Institutes of Health, and by the GIS Intitut des Maladies Rares, COREC (CHU, Facultè de Mèdecine, Rennes, France).
Footnotes
Communicated by Jürgen Horst
References
- Aruga J. The role of Zic genes in neural development. Mol Cell Neurosci. 2004;26:205–221. doi: 10.1016/j.mcn.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Aruga J, Kamiya A, Takahashi H, Fujimi TJ, Shimizu Y, Ohkawa K, Yazawa S, Umesono Y, Noguchi H, Shimizu T, Saitou N, Mikoshiba K, Sakaki Y, Agata K, Tyoda A. A wide-range phylogenetic analysis of Zic proteins: implications for correlations between protein structure conservation and body plan complexity. Genomics. 2006;87:783–792. doi: 10.1016/j.ygeno.2006.02.011. [DOI] [PubMed] [Google Scholar]
- Bendavid C, Haddad BR, Griffin A, Huizing M, Dubourg C, Gicquel I, Cavalli LR, Pasquier L, Long R, Ouspenskaia M, Odent S, Lacbawan F, David V, Muenke M. Multicolor FISH and quantitative PCR can detect submicroscopic deletions in holoprosencephaly patients with a normal karyotype. J Med Genet. 2005a;43:496–500. doi: 10.1136/jmg.2005.037176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendavid C, Dubourg C, Gicquel I, Pasquier L, Saugler-Veber P, Durou MR, Jaillard S, Frebourg T, Haddad BR, Henry C, Odent S, David V. Molecular evaluation of foetuses with holoprosencephaly shows high incidence of microdeletions in the HPE genes. Hum Genet. 2005b;119:1–8. doi: 10.1007/s00439-005-0097-6. [DOI] [PubMed] [Google Scholar]
- Brown S, Russo J, Chitayat D, Warburton D. The 13q- syndrome: the molecular definition of a critical deletion region in band 13q32. Amer J Hum Genet. 1995;57:859–866. [PMC free article] [PubMed] [Google Scholar]
- Brown SA, Warburton D, Brown LY, Yu Cy, Roeder ER, Stengel-Rutkowski S, Hennekam RCM, Muenke M. Holoprosencephaly due to mutations in ZIC2, a homologue of Drosophila odd-paired. Nat Genet. 1998;20:180–183. doi: 10.1038/2484. [DOI] [PubMed] [Google Scholar]
- Brown LY, Odent S, David V, Blayau M, Dubourg C, Apacik C, Delgado MA, Hall BD, Reynolds JF, Sommer A, Wieczorek D, Brown SA, Muenke M. Holoprosencephaly due to mutations in ZIC2; alanine tract expansion mutations may be caused by parental somatic recombination. Hum Mol Genet. 2001;10:791–796. doi: 10.1093/hmg/10.8.791. [DOI] [PubMed] [Google Scholar]
- Brown LY, Hodge SE, Johnson WG, Guy SG, Nye JS, Brown S. Possible association of NTDs with a polyhistidine tract polymorphism in the ZIC2 gene. Amer J Hum Genet. 2002;108:128–131. doi: 10.1002/ajmg.10221. [DOI] [PubMed] [Google Scholar]
- Brown LY, Kottman AH, Brown S. Immunolocalization of Zic2 expression in the mouse forebrain. Gene Expr Patterns. 2003;3:361–367. doi: 10.1016/s1567-133x(03)00043-7. [DOI] [PubMed] [Google Scholar]
- Brown L, Paraso M, Arkell R, Brown S. In vitro analysis of partial loss-of-function ZIC2 mutations in holoprosencephaly: alanine tract expansion modulates DNA binding and transactivation. Hum Mol Genet. 2005;14:411–420. doi: 10.1093/hmg/ddi037. [DOI] [PubMed] [Google Scholar]
- Cheng X, Hsu Cm, Currle DS, Hu JS, Barkovich AJ, Monuki ES. Central roles of the roof plate in telencephalic development and holoprosencephaly. J Neurosci. 2006;26:7640–7649. doi: 10.1523/JNEUROSCI.0714-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MM., Jr Holoprosencephaly: clinical, anatomic, and molecular dimensions. Birth Defects Res Part A Clin Mol Teratol. 2006;76:658–673. doi: 10.1002/bdra.20295. [DOI] [PubMed] [Google Scholar]
- Dubourg C, Bendavid C, Pasquier L, Henry C, Odent S, David V. Holoprosencephaly. Orphant J Rare Dis. 2007;2:8. doi: 10.1186/1750-1172-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubourg C, Lazaro L, Pasquier L, Bendavid C, Blayau M, Le Duff F, Durou MR, Odent S, David V. Molecular screening of SHH, ZIC2, SIX3 and TGIF genes in patients with features of holoprosencephaly spectrum: mutation review and genotype-phenotype correlations. Hum Mut. 2004;24:43–51. doi: 10.1002/humu.20056. [DOI] [PubMed] [Google Scholar]
- Elms P, Siggers P, Napper D, Greenfield, Arkell R. Zic2 is required for neural crest formation and hindbrain patterning during mouse development. Dev Biol. 2003;264:391–406. doi: 10.1016/j.ydbio.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Elms P, Scurry A, Davies J, Willoughy C, Hacker T, Bogani D, Arkell R. Overlapping and distinct expression domains of Zic2 and Zic3 during mouse gastrulation. Gene Expr Patterns. 2004;4:505–511. doi: 10.1016/j.modgep.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Gripp KW, Wotton D, Edwards MC, Roessler E, Ades L, Meinecke P, Richeri-Costa A, Zackai EH, Massague J, Muenke M, Elledge SJ. Mutations in TGIF cause holoprosencephaly and link NODAL signaling to human neural axis determination. Nat Genet. 2000;25:205–208. doi: 10.1038/76074. [DOI] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B. The GLI gene encodes a nuclear protein which binds specific sequences in the human genome. Mol Cell Biol. 1990;10:634–642. doi: 10.1128/mcb.10.2.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauss RS. Holoprosencephaly: new models, new insights. Expert Rev Mol Med. 2007;9:1–17. doi: 10.1017/S1462399407000440. [DOI] [PubMed] [Google Scholar]
- Ming JE, Muenke M. Multiple hits during early embryonic development: digenic disease and holoprosencephaly. Am J Hum Genet. 2002;71:1017–1032. doi: 10.1086/344412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizugishi K, Aruga J, Nakata K, Mikoshiba K. Molecular properties of Zic proteins as transcriptional regulators and their relationship to GLI proteins. J Biol Chem. 2001;276:2180–2188. doi: 10.1074/jbc.M004430200. [DOI] [PubMed] [Google Scholar]
- Monuki ES. The morphogen signaling network in forebrain development and holoprosencephaly. J Neuropath Exp Neurosci. 2007;66:566–575. doi: 10.1097/nen.0b013e3180986e1b. [DOI] [PubMed] [Google Scholar]
- Muenke M, Beachy PA. Holoprosencephaly. In: Scriver CR, et al., editors. The Metabolic & Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2001. pp. 6203–6230. [Google Scholar]
- Orioli IM, Castilla EE, Ming JE, Nazer J, Burle de Aguiar MJ, LLerena JC, Muenke M. Identification of novel mutations in SHH and ZIC2 in a South American (ECLAMC) population with holoprosencephaly. Hum Genet. 2001;109:1–6. doi: 10.1007/s004390100537. [DOI] [PubMed] [Google Scholar]
- Pavletich NP, Pabo CO. Crystal structure of a five-finger GLI-DNA complex: new perspectives on zinc fingers. Science. 1993;261:1701–1707. doi: 10.1126/science.8378770. [DOI] [PubMed] [Google Scholar]
- Nagai T, Aruga J, Minowa O, Sugimoto T, Ohno Y, Noda T, Mikoshiba K. Zic2 regulates the kinetics of neurulation. Proc Natl Acad Sci USA. 2000;97:1618–1623. doi: 10.1073/pnas.97.4.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redemann N, Gaul U, Jäckle H. Disruption of a putative Cys-zinc interaction eliminates the biological activity of the Krüppel finger protein. Nature. 1988;332:90–92. doi: 10.1038/332090a0. [DOI] [PubMed] [Google Scholar]
- Roessler E, Belloni E, Gaudenz K, Jay P, Berta P, Scherer SW, Tsui LC, Muenke M. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat Genet. 1996;14:357–360. doi: 10.1038/ng1196-357. [DOI] [PubMed] [Google Scholar]
- Roessler E, Muenke M. Midline and laterality defects: left and right meet in the middle. BioEssays. 2001;23:888–900. doi: 10.1002/bies.1130. [DOI] [PubMed] [Google Scholar]
- Roessler E, Ermilov AN, Grange DK, Wang A, Grachtchouk M, Dluglosz AA, Muenke M. A previously unidentified amino terminal domain regulates transcriptional activity of wild-type and disease-associated human GLI2. Hum Mol Genet. 2005;14:2181–2188. doi: 10.1093/hmg/ddi222. [DOI] [PubMed] [Google Scholar]
- Wallis DE, Roessler E, Hehr U, Nanni L, Wiltshire T, Richieri-Costa A, Gillessen-Kaesbach G, Zackai EH, Rommens J, Muenke M. Missense mutations in the homeodomain of the human SIX3 gene cause holoprosencephaly. Nat Genet. 1999;22:196–198. doi: 10.1038/9718. [DOI] [PubMed] [Google Scholar]
- Ware SM, Peng J, Zhu L, Fernbach S, Colicos S, Casey B, Towboin J, Belmont JW. Identification and functional analysis of ZIC3 mutations in heterotaxy and related congenital heart defects. Am J Hum Genet. 2004;74:93–105. doi: 10.1086/380998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warr N, Powles-Glover N, Chappell A, Robson J, Norris D, Arkell R. Zic2-associated holoprosencephaly is caused by a transient defect in the organizer region during gastrulation. Hum Mol Genet. 2008 doi: 10.1093/hmg/ddn197. [DOI] [PubMed] [Google Scholar]
- Zhu L, Zhou G, Poole S, Belmont JW. Characterization of the interactions of human ZIC3 mutants with GLI3. Hum Mut. 2008;29:99–105. doi: 10.1002/humu.20606. [DOI] [PubMed] [Google Scholar]