

Introduction
Since the previous review on this subject in this journal [1], significant progress has been made in the characterization of processes that involve the coupling of folding of intrinsically disordered (sometimes termed unstructured) domains with binding to a target molecule, which may itself be more or less disordered. Intrinsically disordered proteins (IDP) are frequently associated with cellular control mechanisms and signaling, and have been identified at the center of “hubs” in protein interaction networks [2]. Interaction with a wide variety of targets therefore appears to be a hallmark of functional disordered proteins. Analyses of correlations between predicted protein disorder and protein function, as assessed by Gene Ontology or Swiss-Prot functional keywords, reveals that disorder is associated with a broad repertoire of biological functions and processes [3–6]. In the present review we survey the most exciting advances that have occurred over the last two years.
Technical Advances
Intrinsically disordered proteins were initially identified experimentally through spectroscopic means [7], as well as theoretically by sequence analysis [8]. Prediction of disordered regions through bioinformatic searches has reached a high degree of sophistication and numerous web servers are available for prediction of disordered regions of protein sequences (reviewed in [9]).
Although information on the conformational propensities of intrinsically disordered domains both free in solution and in complex with targets has been mostly obtained by CD, fluorescence and NMR spectroscopy [10], other experimental methods have recently been employed to provide important insights into the conformational ensemble in the disordered state and into the coupled folding and binding process. These include small angle X-ray scattering (SAXS) [11,12•], paramagnetic spin labeling in combination with ensemble molecular dynamics [13] and single molecule fluorescence [14•]. High speed atomic force microscopy [15] and Raman optical activity [16] have been used to characterize and classify intrinsically disordered proteins and provide insight into the nature of their conformational ensembles and dynamic properties. The combination of NMR spectroscopy and SAXS provides an especially powerful approach to characterize the structural ensemble of intrinsically disordered proteins, as illustrated by the recent determination of the structure of the full length p53 tumor suppressor and its intrinsically disordered transactivation domain [17••]. While X-ray crystallography and NMR remain the primary techniques used to characterize the structures formed by intrinsically disordered proteins when bound to their targets, spectroscopic techniques such as EPR can be applied to study folding of a disordered protein labeled at specific sites with a paramagnetic nitroxide spin probe [18,19].
Coupled Folding and Binding of IDPs
Although it has long been recognized that many intrinsically disordered protein domains fold upon binding to their targets [20], the molecular principles and broad repertoire of interactions are only now becoming understood. While some proteins are completely disordered in the absence of their physiological partner and fold into globular structures only upon binding, most coupled folding and binding events involve relatively short amphipathic motifs contained within longer disordered sequences [1,21]. Indeed, it is frequently possible to identify these folding motifs, which have been termed molecular recognition elements or MoRFs, by bioinformatic analysis of the protein sequence [22•]. These recognition motifs can fold into helix, β-strand, or form irregular structure on binding to a target protein. Binding of disordered linear motifs by β-strand addition has been reviewed recently [23]. A survey of the intermolecular interfaces in IDP complexes indicated that IDPs rely more on hydrophobic interactions than their globular counterparts and form a more intimate fit with their binding partners [24]. An excellent example is the competitive binding of the bacterial toxin colicin to the peptidoglycan-associated lipoprotein (Pal) site of TolB; the intrinsically unfolded colicin can be molded to fit intimately the TolB surface, giving a greater degree of shape complementarity and comparable binding affinity to the much larger and folded Pal [25•]. Colicin mimics key interactions within the TolB-Pal interface, while preventing the conformational change in TolB that is induced by binding of its cognate partner Pal.
A feature of many disordered recognition elements is that they can fold into different structures on binding to different target proteins. Examples include the intrinsically disordered regulatory region near the C-terminus of p53, which folds into helical, β-strand, and extended irregular structures on binding to different protein partners [26], and the C-terminal activation domain of the hypoxia inducible factor HIF-1α which adopts helical and extended structures, respectively, in its complexes with CBP/p300 and the asparagine hydroxylase FIH [21]. Perhaps an even more extreme example is the nuclear coactivator binding domain (or IBiD domain) of CBP/p300, which forms a disordered molten globule state in the absence of a binding partner [27] and folds into helical structures with different topologies upon binding to a p160 nuclear receptor coactivator [28] or the interferon regulatory factor IRF-3 [29] (Figure 1). This structural plasticity, which is displayed by many intrinsically disordered proteins, raises questions about the role of “preformed structural elements” [30] in the recognition process. Even when a disordered protein displays a measurable propensity to populate helical or other secondary structures in the conformational ensemble formed in the unbound state, it would seem unwise to assume that this conformation will necessarily be favored upon binding to a partner protein.
Figure 1.

Structural comparison of the NCBD (IbID) domain of CBP in complex with different partners. A. NCBD in complex with the ACTR domain of p160 [28]. B. NCBD in complex with IRF3 [29].
Mechanism of Coupled Folding and Binding
One of the most intriguing questions related to the coupling of folding and binding of disordered domains is the mechanism of the process, i.e. does folding occur before binding or does binding occur before folding? Two extreme mechanistic possibilities can be envisaged, induced folding or conformational selection (Figure 2). In the first mechanism, the protein associates with its binding partner in a fully disordered state and subsequently folds in association with the target protein; folding is induced by association with the target. In the conformational selection mechanism, the target protein “selects” a conformation closely approximating that of the bound form from the ensemble of conformations populated by the intrinsically disordered protein when free in solution. Clearly, any real system may utilize either one of these extreme mechanisms or some combination of the two; for example, it is to be expected that some degree of conformational adaptation will be required even when a pre-formed structural element in an IDP binds to its target.
Figure 2.
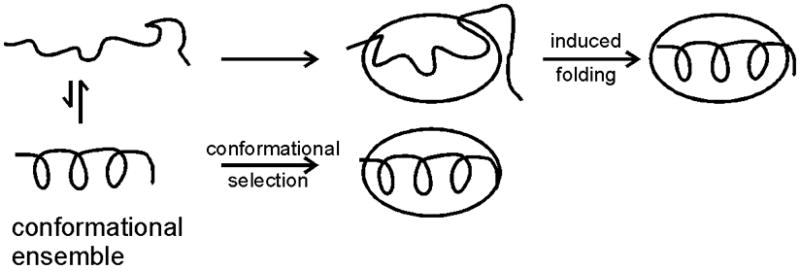
Schematic showing the two possibilities, folding upon binding or conformational selection.
NMR spectroscopy is especially well suited to the elucidation of the mechanisms of coupled folding and binding. Not only do the chemical shifts, residual dipolar couplings, and other NMR parameters give important insights into the ensemble of structures that are sampled by the disordered protein in the absence of its binding partner, but the structure and dynamics of the complex with the target protein can also be determined. Relaxation dispersion NMR spectroscopy provides a particularly powerful approach for investigating millisecond time scale dynamics and protein folding mechanisms [31•]. Relaxation dispersion methods have been applied to investigate the mechanism by which the phosphorylated kinase inducible activation domain (pKID) of the transcription factor CREB folds into a helical structure on binding to its target, the KIX domain of the transcriptional coactivators CBP/p300 [32••]. pKID is largely unstructured in solution [33] and folds upon binding into a pair of helices (αA and βB) that contact a particular face of the folded KIX domain [34]. Upon binding to KIX, pKID forms a weak encounter complex, stabilized by hydrophobic contacts, which exchanges rapidly with the free pKID. The encounter complex is largely unstructured and evolves by way of a partly folded intermediate to the fully bound state, without dissociating from the KIX surface [32••]. This mechanism is summarized in Figure 3. Thus, binding of pKID involves an “induced” folding mechanism.
Figure 3.

Schematic representation of the induced folding mechanism of pKID binding to KIX [32], derived from NMR relaxation dispersion measurements. The conformational ensemble of the free protein contains little propensity for helical structure; the initial encounter complex is heterogeneous and mostly involves the C-terminal end of the pKID peptide. The intermediate structure shows contact between KIX and the entire length of the peptide, but helical structure is not present in either αA or αB. Finally the fully folded complex shows stable helical structure in both regions of pKID. (adapted with permission from Nature NV).
The mechanism by which pKID folds upon binding to KIX has recently been investigated by coarse-grained simulations using a Go-type model [35••]. The simulations are remarkably consistent with the experimental results [32••], indicating that folding is induced after binding of the largely unstructured pKID to the surface of KIX and proceeds by way of a partly folded intermediate. The simulations also provide additional insights into the coupled folding and binding process that are not available from the present experiments. The pKID is largely unfolded in the transition state, and hydrophobic residues in the αB helix of pKID appear to play a crucial role in formation of the transition state and in the association process. Increasing the content of helical structure in the uncomplexed state of pKID leads to a decrease in the rate of association consistent with the “fly-casting” mechanism [36], while formation of transient encounter complexes enhances the association rate without changing the binding mechanism [35]. Taken together, the relaxation dispersion experiments and the simulations provide a remarkably consistent and detailed view of the coupled folding and binding process for binding of pKID to KIX. A similar induced folding mechanism has been revealed by coarse grained molecular dynamics and single molecule spectroscopic studies of binding of the disordered CBD domain of the Wiskott-Aldrich syndrome protein (WASP) to Cdc42 [37]. The unstructured CBD domain first binds to its target and then folds cooperatively into the native structure.
The disordered acidic transactivation domains of c-Myc, Gal4, and VP16 also appear to bind their targets by an induced folding mechanism [38]. However, given the diverse nature and wide ranging conformational propensities of intrinsically disordered proteins, from almost complete lack of structure, through pronounced propensity for formation of local elements of secondary structure, to formation of compact molten globules [21], it is to be expected that such a mechanism will not be universal and that many intrinsically disordered proteins may utilize different mechanisms. Indeed, kinetic studies of ligand-induced folding of a series of mutants of staphylococcal nuclease, considered as a model of an intrinsically disordered protein, demonstrate both folding before binding and binding before folding [39]. Further evidence that conformational selection may play a role in binding of an IDP comes from studies of the retinal phosphodiesterase inhibitory γ-subunit (PDEγ) [40]. The uncomplexed PDEγ is highly disordered but the conformational ensemble includes a loosely folded state that resembles the structure formed by PDEγ when bound to a GTPase-activating protein complex.
In apparent contradiction to the conformational selection mechanism, examples have been reported where increasing the stability of pre-organized secondary structure fails to enhance the binding affinity or even slows the kinetics of complex formation [41,42], suggesting that disorder in the free state may actually be advantageous for the binding process. It seems likely that even in extreme cases where there is a strong propensity for transient formation of the folded conformer in the uncomplexed IDP, a certain amount of structural adaptation will be required when a preformed structural element binds, for which intrinsic flexibility is advantageous. This conclusion is supported by simulations of the interaction of the p21 kinase inhibitor with its target; excessive stabilization of local secondary structural elements in the disordered p21 is detrimental to binding and folding to the native structure [43,44].
Some IDPs remain disordered even after binding to their targets. An example is the interaction between the Smad2 Mad homology domains and the Smad binding domain (SBD) of Smad anchor for receptor activation (SARA). The complex formed between the SBD and Mad homology domain appears to be heterogeneous, with no one region of the SBD making a dominant contribution to the hydrophobic interactions between the proteins [45]. A dynamic complex is also formed between the regulatory (R) region of the cystic fibrosis transmembrane conductance regulator and a nucleotide binding domain (NBD) of the same protein [46•]. Interactions between the nucleotide binding domain and the intrinsically disordered R-region are mediated by multiple sites in R, each of which has helical propensity. The complex is dynamic and all of the helical elements in R interact transiently with the NBD and are stabilized, with no one element being favored over the others. Phosphorylation reduces the helicity of the R-region elements and weakens binding to the NBD. This system represents a variation on the coupled folding and binding scheme, in that the complex formed is highly dynamic and the recognition element in the IDP does not become fully ordered upon binding to the target.
A novel manifestation of coupled folding and binding has been observed in interactions between the Arf tumor suppressor and the ubiquitin E3 ligase Hdm2 [47••]. Arf inhibits the Hdm2-dependent degradation of p53 by binding to Hdm2. The interaction is mediated by short intrinsically disordered domains of Arf and Hdm2, which fold synergistically to bimolecular oligomeric structures that resemble amyloid. This represents an unprecedented example of a molecular complex assembled by folding of small domains from two proteins to form a β-strand-rich oligomer.
Folding and binding kinetics
It has been suggested that the rate of macromolecular association is enhanced by the presence of disorder [36,48]. An unstructured protein has a large capture radius that facilitates the diffusive search for a binding target through the “fly-casting” mechanism [36]. While several theoretical simulations suggest that fly-casting plays an important role in binding and folding of disordered proteins, for example [49], experimental support for this model has been lacking. A recent laser temperature jump analysis of the kinetics of binding of the intrinsically disordered inhibitor IA3 to the yeast protease YPrA [50] indicated a mechanism in which a transient complex is rapidly formed between IA3 and its target, followed by folding into a helical structure within the active site of the enzyme. This result suggested a fly-casting mechanism, in which the initial interaction between the disordered protein and its target facilitates folding and binding without affecting the equilibrium constant.
To date, comparatively few studies of the kinetics of coupled folding and binding processes have been reported. The kinetics of binding of the intrinsically disordered E-cadherin cytoplasmic domain to β-catenin have been investigated using surface plasmon resonance [51]. The association rate is fast (ca. 4 × 105 M−1s−1) and is enhanced more than 10-fold by phosphorylation. Formation of the complex, which involves structuring of nearly 100 residues, is highly disfavored entropically and is driven by a large decrease in enthalpy. The NMR relaxation dispersion analysis of binding of pKID to KIX also revealed a fast association (ca. 6 × 106 M−1s−1) followed by a slower structural rearrangement of the partly folded intermediate [32]. Relaxation dispersion studies have been reported for binding of the intrinsically disordered C-terminal activation domain of hypoxia inducible factor, hydroxylated at Asn803 (HIF-OH), to the TAZ1 domain of the transcriptional coactivator CBP [52]. In this case, the rate of association is diffusion limited (~109 M−1s−1). Thus, disordered proteins appear to display a wide variety of binding rates.
Cooperative binding
It has recently been proposed on theoretical grounds that, in cases where folding accompanies binding, intrinsic disorder can maximize allosteric coupling between binding sites [53]. This suggests a mechanism whereby proteins with intrinsically disordered binding motifs can transmit signals that are triggered by interaction with different types of ligands. The fact that disordered proteins frequently bind to their targets through relatively short amphipathic sequences means that even quite small target proteins may bind more than one IDP simultaneously to form a ternary complex. This characteristic provides the potential for synergy and cross talk between different signaling pathways, mediated by interactions of disordered domains with a common binding partner. For example, the activation domains from the c-Myb and mixed lineage leukemia (MLL) transcription factors bind cooperatively to the KIX domain of CBP to form a ternary complex [54]. Bivalent interactions of the transcription factor CSL with intrinsically disordered (RAM) and partly-folded (ankyrin) domains in the Notch intracellular domain have been described recently [55•]. The RAM domain binds tightly to CSL and enhances ankyrin binding by increasing the effective concentration. A polymer physics model was used to model the binding and suggests that the disordered linker between the RAM and ankyrin domains is of optimal length to enhance the bivalent interaction.
Role of Phosphorylation and Flexibility in Signal Transduction
One of the most common means of signal transduction is through phosphorylation at defined sites within signaling proteins. Analysis of the sequences surrounding known phosphorylation sites reveals a strong propensity towards intrinsic disorder in the neighboring regions of the protein [56]. Phosphorylation can act at different levels, affecting either the conformational propensities in the uncomplexed state or the interactions between the IDP and its partner proteins. Further, phosphorylation can function as a switch or as a “rheostat” to fine-tune the biological response [57].
Perhaps it might be expected that phosphorylation should cause a change in the intrinsic propensity of the protein for structure formation in the free form. Such a change in conformational preference was not observed experimentally for the pKID domain [33], although recent computer simulations appear to suggest that the turn region incorporating the phosphorylated serine residue of pKID may be favored in the uncomplexed state when the phosphoryl group is present [58]. A comprehensive thermodynamic analysis of the pKID/KIX system [42] showed that the effect of the phosphoryl group on the binding of pKID to KIX was primarily enthalpic, an indication that prior formation of secondary structure in the free protein has a negligible effect on the binding interaction.
A variation on the theme of phosphorylation and conformational switching is shown by the Ets-1 transcriptional activator and its interaction with DNA [57]. In the active, unphosphorylated state, the Ets domain binds DNA, an interaction that is allowed by the unfolded and conformationally labile H1 helix. Upon phosphorylation at multiple sites, the H1 helix becomes progressively more highly structured, disfavoring the conformational change that permits the binding of Ets-1 to DNA. The disordered region thus functions as an allosteric effector, essentially a “rheostat” that fine tunes transcription in response to variations in the level of phosphorylation. The cyclin-dependent kinase inhibitor Sic1 is a further example of an intrinsically disordered protein that is regulated by multiple site phosphorylation. In this case, phosphorylation of a threshold number of sites is required for receptor binding, which is driven by cumulative electrostatic interactions with the disordered Sic1 [59•]. A general entropic model has been presented to account for highly cooperative binding driven by multiple site phosphorylation in intrinsically disordered regions [60].
Are these domains disordered in vivo?
For many, the evidence that important transcriptional and signaling domains could be as disordered in vivo as they appear to be in the in vitro experiments described above is hard to countenance. Aside from the issue of stability within the cell, it is has been postulated that the crowded environment within the cell might induce folding of IDPs. This issue has been recently addressed through NMR experiments, with mixed results: while the eukaryotic α-synuclein remains disordered within the cytoplasm in E. coli cells [61], the bacterial protein FlgM appears to gain some structure in the cellular milieu [62].
One of the signature characteristics of a disordered protein is that it is highly susceptible to proteolytic degradation in vitro, and it has been suggested that sensitivity to proteolysis may be important to the signaling function of many IDPs [21]. A recent survey shows that while there is a correlation between protein disorder and short half-life, the correlation is weak suggesting that many disordered proteins are protected from degradation, possibly by complexation with other proteins or nucleic acids [63•]. One of the major functions of chaperone molecules in the cell is to interact with and stabilize incompletely-folded proteins. However, it was recently noted that intrinsically disordered proteins appear to have no preference for interaction with chaperones [64], which argues that IDPs are intrinsically different from the unfolded or mis-folded forms of globular proteins.
CONCLUSIONS
The occurrence of disordered regions in important cellular signaling and regulatory proteins continues to be a common theme as the recently-published genome sequences of many organisms are mined for information. Folding upon binding of these regions to their partners has been a fertile field for the elucidation of the structural bases of a number of physiologically-relevant protein-protein interactions, and will doubtless remain relatively accessible experimentally. Database and computer simulation, together with increasingly sophisticated algorithms for the idenification of sequence motifs characteristic of certain binding interactions, should also provide information on systems that may be too large or heterogeneous for initial experimental characterization, but which can be approached experimentally after they have been parsed in silico.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Dyson HJ, Wright PE. Coupling of folding and binding for unstructured proteins. Curr Opin Struct Biol. 2002;12:54–60. doi: 10.1016/s0959-440x(02)00289-0. [DOI] [PubMed] [Google Scholar]
- 2.Dunker AK, Cortese MS, Romero P, Iakoucheva LM, Uversky VN. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005;272:5129–5148. doi: 10.1111/j.1742-4658.2005.04948.x. [DOI] [PubMed] [Google Scholar]
- 3.Xie HB, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Uversky VN, Obradovic Z. Functional anthology of intrinsic disorder. 1. Biological processes and functions of proteins with long disordered regions. J Proteome Res. 2007;6:1882–1898. doi: 10.1021/pr060392u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vucetic S, Xie HB, Iakoucheva LM, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN. Functional anthology of intrinsic disorder. 2. Cellular components, domains, technical terms, developmental processes, and coding sequence diversities correlated with long disordered regions. J Proteome Res. 2007;6:1899–1916. doi: 10.1021/pr060393m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xie HB, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN. Functional anthology of intrinsic disorder. 3. Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins. J Proteome Res. 2007;6:1917–1932. doi: 10.1021/pr060394e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lobley A, Swindells MB, Orengo CA, Jones DT. Inferring function using patterns of native disorder in proteins. PLoS Comput Biol. 2007;3:e162. doi: 10.1371/journal.pcbi.0030162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kriwacki RW, Hengst L, Tennant L, Reed SI, Wright PE. Structural studies of p21Waf1/Cip1/Sdi1 in the free and Cdk2-bound state: Conformational disorder mediates binding diversity. Proc Natl Acad Sci USA. 1996;93:11504–11509. doi: 10.1073/pnas.93.21.11504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Romero P, Obradovic Z, Kissinger CR, Villafranca JE, Dunker AK. Identifying disordered regions in proteins from amino acid sequences. Proc IEEE International Conference on Neural Networks. 1997;1997:90–95. [Google Scholar]
- 9.Radivojac P, Iakoucheva LM, Oldfield CJ, Obradovic Z, Uversky VN, Dunker AK. Intrinsic Disorder and Functional Proteomics. Biophys J. 2007;92:1439–1456. doi: 10.1529/biophysj.106.094045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dyson HJ, Wright PE. Elucidation of the protein folding landscape by NMR. Methods Enzymol. 2005;394:299–321. doi: 10.1016/S0076-6879(05)94011-1. [DOI] [PubMed] [Google Scholar]
- 11.Bernado P, Blanchard L, Timmins P, Marion D, Ruigrok RW, Blackledge M. A structural model for unfolded proteins from residual dipolar couplings and small-angle x-ray scattering. Proc Natl Acad Sci USA. 2005;102:17002–17007. doi: 10.1073/pnas.0506202102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12•.Bernado P, Mylonas E, Petoukhov MV, Blackledge M, Svergun DI. Structural Characterization of Flexible Proteins Using Small-Angle X-ray Scattering. J Am Chem Soc. 2007;129:5656–5664. doi: 10.1021/ja069124n. SAXS is used as the basis for analysis of the conformational ensemble of flexible proteins using an ensemble optimization method. [DOI] [PubMed] [Google Scholar]
- 13.Dedmon MM, Lindorff-Larsen K, Christodoulou J, Vendruscolo M, Dobson CM. Mapping long-range interactions in alpha-synuclein using spin-label NMR and ensemble molecular dynamics simulations. J Am Chem Soc. 2005;127:476–477. doi: 10.1021/ja044834j. [DOI] [PubMed] [Google Scholar]
- 14•.Mukhopadhyay S, Krishnan R, Lemke EA, Lindquist S, Deniz AA. A natively unfolded yeast prion monomer adopts an ensemble of collapsed and rapidly fluctuating structures. Proc Natl Acad Sci USA. 2007;104:2649–2654. doi: 10.1073/pnas.0611503104. Investigation of the structural ensemble and dynamics of the prion-determining domain of the yeats prion Sup35 using single-molecule fluorescence resonance energy transfer and fluorescence correlation spectroscopy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miyagi A, Tsunaka Y, Uchihashi T, Mayanagi K, Hirose S, Morikawa K, Ando T. Visualization of intrinsically disordered regions of proteins by high-speed atomic force microscopy. ChemPhysChem. 2008;9:1859–1866. doi: 10.1002/cphc.200800210. [DOI] [PubMed] [Google Scholar]
- 16.Zhu F, Tranter GE, Isaacs NW, Hecht L, Barron LD. Delineation of protein structure classes from multivariate analysis of protein Raman optical activity data. J Mol Biol. 2006;363:19–26. doi: 10.1016/j.jmb.2006.08.038. [DOI] [PubMed] [Google Scholar]
- 17••.Wells M, Tidow H, Rutherford TJ, Markwick P, Jensen MR, Mylonas E, Svergun DI, Blackledge M, Fersht AR. Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proc Natl Acad Sci USA. 2008;105:5762–5767. doi: 10.1073/pnas.0801353105. An innovative use of SAXS (see [12] above) and NMR spectroscopy to characterize the solution structure of the full-length p53 and the ensemble structure of the disordered N-terminal transactivation domain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jao CC, Der-Sarkissian A, Chen J, Langen R. Structure of membrane-bound +¦− synuclein studied by site-directed spin labeling. Proc Natl Acad Sci USA. 2004;101:8331–8336. doi: 10.1073/pnas.0400553101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morin B, Bourhis JM, Belle V, Woudstra M, Carriere F, Guigliarelli B, Fournel A, Longhi S. Assessing induced folding of an intrinsically disordered protein by site-directed spin-labeling electron paramagnetic resonance spectroscopy. Journal of Physical Chemistry B. 2006;110:20596–20608. doi: 10.1021/jp063708u. [DOI] [PubMed] [Google Scholar]
- 20.Wright PE, Dyson HJ. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J Mol Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 21.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 22•.Mohan A, Oldfield CJ, Radivojac P, Vacic V, Cortese MS, Dunker AK, Uversky VN. Analysis of Molecular Recognition Features (MoRFs) J Mol Biol. 2006;362:1043–1059. doi: 10.1016/j.jmb.2006.07.087. Describes the identification of sequence motifs within larger intrinsically disordered proteins that exhibit molecular recognition and binding functions. [DOI] [PubMed] [Google Scholar]
- 23.Remaut H, Waksman G. Protein-protein interaction through β-strand addition. Trends Biochem Sci. 2006;31:436–444. doi: 10.1016/j.tibs.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 24.Mészáros B, Tompa P, Simon I, Dosztanyi Z. Molecular Principles of the Interactions of Disordered Proteins. J Mol Biol. 2007;372:549–561. doi: 10.1016/j.jmb.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 25•.Bonsor DA, Grishkovskaya I, Dodson EJ, Kleanthous C. Molecular mimicry enables competitive recruitment by a natively disordered protein. J Am Chem Soc. 2007;129:4800–4807. doi: 10.1021/ja070153n. This study compares the structures of TolB complexes with the globular target protein Pal and with the intrinsically disordered ligand colicin and reveals a novel binding mechanism. [DOI] [PubMed] [Google Scholar]
- 26.Oldfield CJ, Meng J, Yang JY, Yang MQ, Uversky VN, Dunker AK. Flexible nets: disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics. 2008;9 (Suppl 1):S1. doi: 10.1186/1471-2164-9-S1-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Demarest SJ, Deechongkit S, Dyson HJ, Evans RM, Wright PE. Packing, specificity, and mutability at the binding interface between the p160 coactivator and CREB-binding protein. Protein Sci. 2004;13:203–210. doi: 10.1110/ps.03366504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Demarest SJ, Martinez-Yamout M, Chung J, Chen H, Xu W, Dyson HJ, Evans RM, Wright PE. Mutual synergistic folding in recruitment of CBP/p300 by p160 nuclear receptor coactivators. Nature. 2002;415:549–553. doi: 10.1038/415549a. [DOI] [PubMed] [Google Scholar]
- 29.Qin BY, Liu C, Srinath H, Lam SS, Correia JJ, Derynck R, Lin K. Crystal Structure of IRF-3 in Complex with CBP. Structure. 2005;13:1269–1277. doi: 10.1016/j.str.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 30.Fuxreiter M, Simon I, Friedrich P, Tompa P. Preformed structural elements feature in partner recognition by intrinsically unstructured proteins. J Mol Biol. 2004;338:1015–1026. doi: 10.1016/j.jmb.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 31•.Korzhnev DM, Kay LE. Probing Invisible, Low-Populated States of Protein Molecules by Relaxation Dispersion NMR Spectroscopy: An Application to Protein Folding . Acc Chem Res. 2008;41:442–451. doi: 10.1021/ar700189y. A comprehensive review of current methods to characterize low-populated protein folding intermediates using NMR relaxation dispersion. [DOI] [PubMed] [Google Scholar]
- 32••.Sugase K, Dyson HJ, Wright PE. Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature. 2007;447:1021–1025. doi: 10.1038/nature05858. NMR methods, including chemical shift titration and relaxation dispersion, are used to determine the kinetics and mechanism by which the intrinsically disordered pKID activation domain of CREB folds on binding to the KIX domain of CBP. [DOI] [PubMed] [Google Scholar]
- 33.Radhakrishnan I, Pérez-Alvarado GC, Dyson HJ, Wright PE. Conformational preferences in the Ser133-phosphorylated and non-phosphorylated forms of the kinase inducible transactivation domain of CREB. FEBS Letters. 1998;430:317–322. doi: 10.1016/s0014-5793(98)00680-2. [DOI] [PubMed] [Google Scholar]
- 34.Radhakrishnan I, Pérez-Alvarado GC, Parker D, Dyson HJ, Montminy MR, Wright PE. Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: A model for activator:Coactivator interactions. Cell. 1997;91:741–752. doi: 10.1016/s0092-8674(00)80463-8. [DOI] [PubMed] [Google Scholar]
- 35••.Turjanski AG, Gutkind JS, Best RB, Hummer G. Binding-induced folding of a natively unstructured transcription factor. PLoS Comput Biol. 2008;4:e1000060. doi: 10.1371/journal.pcbi.1000060. Simulations using Go-type models to characterize the mechanism by which pKID folds on binding to KIX (see [32] above). An induced-folding mechanism incorporating non-native initial contacts and a partially folded intermediate is consistent with the experimental results. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shoemaker BA, Portman JJ, Wolynes PG. Speeding molecular recognition by using the folding funnel: the fly- casting mechanism. Proc Natl Acad Sci USA. 2000;97:8868–8873. doi: 10.1073/pnas.160259697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu Q, Lu HP, Wang J. Exploring the mechanism of flexible biomolecular recognition with single molecule dynamics. Phys Rev Lett. 2007;98:128105. doi: 10.1103/PhysRevLett.98.128105. [DOI] [PubMed] [Google Scholar]
- 38.Ferreira ME, Hermann S, Prochasson P, Workman JL, Berndt KD, Wright APH. Mechanism of transcription factor recruitment by acidic activators. J Biol Chem. 2005;280:21779–21784. doi: 10.1074/jbc.M502627200. [DOI] [PubMed] [Google Scholar]
- 39.Onitsuka M, Kamikubo H, Yamazaki Y, Kataoka M. Mechanism of induced folding: Both folding before binding and binding before folding can be realized in staphylococcal nuclease mutants. Proteins-Structure Function and Bioinformatics. 2008;72:837–847. doi: 10.1002/prot.21978. [DOI] [PubMed] [Google Scholar]
- 40.Song J, Guo LW, Muradov H, Artemyev NO, Ruoho AE, Markley JL. Intrinsically disordered γ-subunit of cGMP phosphodiesterase encodes functionally relevant transient secondary and tertiary structure. Proc Natl Acad Sci USA. 2008;105:1505–1510. doi: 10.1073/pnas.0709558105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bienkiewicz EA, Adkins JN, Lumb KJ. Functional consequences of preorganized helical structure in the intrinsically disordered cell-cycle inhibitor p27(Kip1) Biochemistry. 2002;41:752–759. doi: 10.1021/bi015763t. [DOI] [PubMed] [Google Scholar]
- 42.Zor T, Mayr BM, Dyson HJ, Montminy MR, Wright PE. Roles of Phosphorylation and Helix Propensity in the Binding of the KIX Domain of CREB-binding Protein by Constitutive (c-Myb) and Inducible (CREB) Activators. J Biol Chem. 2002;277:42241–42248. doi: 10.1074/jbc.M207361200. [DOI] [PubMed] [Google Scholar]
- 43.Verkhivker GM, Bouzida D, Gehlhaar DK, Rejto PA, Freer ST, Rose PW. Simulating disorder-order transitions in molecular recognition of unstructured proteins: where folding meets binding. Proc Natl Acad Sci USA. 2003;100:5148–5153. doi: 10.1073/pnas.0531373100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Verkhivker GM. Protein conformational transitions coupled to binding in molecular recognition of unstructured proteins: deciphering the effect of intermolecular interactions on computational structure prediction of the p27Kip1 protein bound to the cyclin A-cyclin-dependent kinase 2 complex. Proteins. 2005;58:706–716. doi: 10.1002/prot.20351. [DOI] [PubMed] [Google Scholar]
- 45.Chong PA, Ozdamar B, Wrana JL, Forman-Kay JD. Disorder in a target for the Smad2 Mad homology 2 domain and its implications for binding and specificity. J Biol Chem. 2004;279:40707–40714. doi: 10.1074/jbc.M404375200. [DOI] [PubMed] [Google Scholar]
- 46•.Baker JMR, Hudson RP, Kanelis V, Choy WY, Thibodeau PH, Thomas PJ, Forman-Kay JD. CFTR regulatory region interacts with NBD1 predominantly via multiple transient helices. Nature Struct Mol Biol. 2007;14:738–745. doi: 10.1038/nsmb1278. This paper shows that NBD1 forms dynamic and fluctuating complexes with multiple helical motifs in the intrinsically disordered regulatory region of CFTR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47••.Sivakolundu SG, Nourse A, Moshiach S, Bothner B, Ashley C, Satumba J, Lahti J, Kriwacki RW. Intrinsically Unstructured Domains of Arf and Hdm2 Form Bimolecular Oligomeric Structures In Vitro and In Vivo. J Mol Biol. 2008;384:240–254. doi: 10.1016/j.jmb.2008.09.019. This paper reports the novel finding that intrinsically disordered regions in Arf and Hdm2 fold together to form amyloid-like structures that function in the regulation of Hdm2 and p53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pontius BW. Close encounters: why unstructured, polymeric domains can increase rates of specific macromolecular association. Trends Biochem Sci. 1993;18:181–186. doi: 10.1016/0968-0004(93)90111-y. [DOI] [PubMed] [Google Scholar]
- 49.Levy Y, Onuchic JN, Wolynes PG. Fly-casting in protein-DNA binding: frustration between protein folding and electrostatics facilitates target recognition. J Am Chem Soc. 2007;129:738–739. doi: 10.1021/ja065531n. [DOI] [PubMed] [Google Scholar]
- 50.Narayanan R, Ganesh OK, Edison AS, Hagen SJ. Kinetics of Folding and Binding of an Intrinsically Disordered Protein: The Inhibitor of Yeast Aspartic Proteinase YPrA. J Am Chem Soc. 2008;130:11477–11485. doi: 10.1021/ja803221c. [DOI] [PubMed] [Google Scholar]
- 51.Choi HJ, Huber AH, Weis WI. Thermodynamics of β-catenin-ligand Interactions: The roles of the N- and C-terminal tails in modulating binding affinity. J Biol Chem. 2006;281:1027–1038. doi: 10.1074/jbc.M511338200. [DOI] [PubMed] [Google Scholar]
- 52.Sugase K, Lansing JC, Dyson HJ, Wright PE. Tailoring relaxation dispersion experiments for fast-associating protein complexes. J Am Chem Soc. 2007;129:13406–13407. doi: 10.1021/ja0762238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hilser VJ, Thompson EB. Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins. Proc Natl Acad Sci USA. 2007;104:8311–8315. doi: 10.1073/pnas.0700329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.De Guzman RN, Goto NK, Dyson HJ, Wright PE. Structural Basis for Cooperative Transcription Factor Binding to the CBP Coactivator. J Mol Biol. 2006;355:1005–1013. doi: 10.1016/j.jmb.2005.09.059. [DOI] [PubMed] [Google Scholar]
- 55•.Bertagna A, Toptygin D, Brand L, Barrick D. The effects of conformational heterogeneity on the binding of the Notch intracellular domain to effector proteins: a case of biologically tuned disorder. Biochem Soc Trans. 2008;36:157–166. doi: 10.1042/BST0360157. A wormlike chain model is used to show that the disordered region between two domains is of appropriate length to promote recruitment and subsequent complex formation. [DOI] [PubMed] [Google Scholar]
- 56.Iakoucheva LM, Radivojac P, Brown CJ, O’Connor TR, Sikes JG, Obradovic Z, Dunker AK. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004;32:1037–1049. doi: 10.1093/nar/gkh253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pufall MA, Lee GM, Nelson ML, Kang HS, Velyvis A, Kay LE, McIntosh LP, Graves BJ. Variable control of Ets-1 DNA binding by multiple phosphates in an unstructured region. Science. 2005;309:142–145. doi: 10.1126/science.1111915. [DOI] [PubMed] [Google Scholar]
- 58.Solt I, Magyar C, Simon I, Tompa P, Fuxreiter M. Phosphorylation-induced transient intrinsic structure in the kinase-inducible domain of CREB facilitates its recognition by the KIX domain of CBP. Proteins: Structure, Function and Genetics. 2006;64:749–757. doi: 10.1002/prot.21032. [DOI] [PubMed] [Google Scholar]
- 59•.Borg M, Mittag T, Pawson T, Tyers M, Forman-Kay JD, Chan HS. Polyelectrostatic interactions of disordered ligands suggest a physical basis for ultrasensitivity. Proc Natl Acad Sci USA. 2007;104:9650–9655. doi: 10.1073/pnas.0702580104. A mean-field statistical mechanical model is used to predict the threshold number of phosphorylation sites required for the binding of the cyclin-dependent kinase inhibitor Sic1 to its target protein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lenz P, Swain PS. An Entropic Mechanism to Generate Highly Cooperative and Specific Binding from Protein Phosphorylations. Curr Biol. 2006;16:2150–2155. doi: 10.1016/j.cub.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 61.Li C, Charlton LM, Lakkavaram A, Seagle C, Wang G, Young GB, Macdonald JM, Pielak GJ. Differential dynamical effects of macromolecular crowding on an intrinsically disordered protein and a globular protein: implications for in-cell NMR spectroscopy. J Am Chem Soc. 2008 doi: 10.1021/ja801020z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dedmon MM, Patel CN, Young GB, Pielak GJ. FlgM gains structure in living cells. Proc Natl Acad Sci USA. 2002;99:12681–12684. doi: 10.1073/pnas.202331299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63•.Tompa P, Prilusky J, Silman I, Sussman JL. Structural disorder serves as a weak signal for intracellular protein degradation. Proteins. 2008;71:903–909. doi: 10.1002/prot.21773. There appears only a very weak correlation between the lifetimes of intrinsically disordered proteins in the cell and the degree of disorder, and many disordered proteins have surprisingly long half-lives in the cell. [DOI] [PubMed] [Google Scholar]
- 64.Hegyi H, Tompa P. Intrinsically disordered proteins display no preference for chaperone binding in vivo. PLoS Comput Biol. 2008;4:e1000017. doi: 10.1371/journal.pcbi.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]