

Abstract
It has been previously reported that prolonged exposure of an enzyme to organic solvents leads to substantial decrease of activity. This effect was found to be unrelated to the catalysts’ structure or their possible aggregation in organic solvents, and up to the present day the cause for activity loss remains unclear. In the present work, the structural dynamics of the serine protease subtilisin Carlsberg (SC) have been investigated during prolonged exposure to two organic solvents by following hydrogen/deuterium (H/D) exchange of mobile protons. The enzyme, after lyophilization, was incubated in organic solvents at controlled deuteriated water activity for different times and the H/D exchange was allowed to take place. The amount of deuterium exchanged was evaluated by 2H NMR, which in turn gave us a picture of the changing dynamics of our model enzyme during incubation and under different experimental conditions. Our results show that the flexibility of SC decreases during prolonged storage in 1,4-dioxane (Diox) and acetonitrile (ACN) as indicated by the observed 3- to 10-fold decrease in the apparent rate constants of exchange (k) of fast exchangeable protons (FEP) and slow exchangeable protons (SEP) in the protein. Our study also shows that SC is more flexible in ACN than in Diox (k 3−20 times higher in ACN for the FEP and SEP), suggesting that enzyme dynamics are affected by solvent physicochemical properties. Additionally, the enzyme dynamics are also affected by the method of preparation: decreased flexibility (k decreases 3- to 10-fold for FEP and SEP) is observed when the enzyme is chemically modified with poly ethylene glycol (PEGylated) or colyophilized with crown ethers. A possible relationship between activity, enantioselectivity (E), and structural dynamics is discussed, demonstrating that direct correlations, as have been attempted in the past, are hampered by the multi-variable nature and complexity of the system.
Keywords: subtilisin Carlsberg, conformational flexibility, biocatalysis in organic solvents, hydrogen/deuterium exchange
Introduction
The effect of enzyme conformational flexibility is nowadays recognized as a key parameter in the regulation of catalytic process such as enzyme stability, activity and enantioselectivity (E) (Eisenmesser et al., 2002; Henzler-Wildman and Kern, 2007; Lindorff-Larsen et al., 2005; Zavodszky et al., 1998). In the field of nonaqueous biocatalysis it is well recognized that organic solvents affect the activity and also the flexibility of enzymes (Klibanov, 2001). However, the mechanistic reasons for these effects are still under debate, in particular, it is not clear what relationship exists between the flexibility, activity and E value of an enzyme and what, if any, is the effect of prolonged exposure to different solvents and the presence of different additives on the enzyme flexibility.
Most results indicate that lyophilized proteins are less flexible and less active when suspended in nonaqueous systems than in their natural environment (Dempsey, 1995; Desai and Klibanov, 1995). However, an NMR spectroscopic study of cytochrome c suspended in tetrahydrofuran showed that the conformational flexibility of the enzyme in this solvent is greater than in water (Wu and Gorenstein, 1993). Water, on the other hand, has been identified as a lubricant which increases enzyme flexibility and catalytic activity (Affleck et al., 1992b; Broos et al., 1995; Partridge et al., 1998), and the importance of protein hydration in reaching optimum catalytic activity in organic solvents has been shown in several studies (Valivety et al., 1994; Zaks and Klibanov, 1988). Nevertheless various examples state how some enzymes show good activity even when are rigorously dried and supposedly more rigid (Valivety et al., 1992). Contradictory evidence has also been obtained when comparing the solvent physicochemical properties to enzyme flexibility and its relationship to enzyme E. For example, Klibanov et al. proposed that in a highly polar solvent, an enzyme is more flexible, and its active site is more prone to distortion and therefore unable to discriminate between enantiomers, thus associating higher enzyme flexibility (in polar solvents) with lower E value (Fitzpatrick and Klibanov, 1991; Fitzpatrick et al., 1992). A correlation between decreasing E value and increasing solvent dielectric constants was also observed by the same authors. This hypothesis, however, does not apply to all enzyme-substrate systems. In fact Broos et al. (1995) found the opposite to be correct suggesting that high enzyme flexibility is associated with a large numbers of enzyme conformations, enhancing the probability of reaching a conformational state capable of binding and converting an enantiomeric substrate. In support of this work, it was reported that the E value is maximized when the enzyme flexibility is increased (Watanabe et al., 2001). Additives such as crown ether, cyclodextrin, inorganic salts, polyethylene glycol (PEG), on the other hand, have been shown to stabilize, activate and to enhance the E of most enzymes (Fasoli et al., 2006; Secundo et al., 2008), still their effect on protein flexibility remains an issue to be addressed. The understanding of such phenomenon could offer the possibility of optimizing the activity and the E value of enzymes, a particular critical parameter for synthetic purposes. Furthermore, the relationship between enzyme flexibility and stability needs to be addressed. In our lab we have been studying the effect of prolonged (several days) exposure to organic solvents on enzyme's stability, activity and E. A decrease in the catalytic activity of the enzymes studied so far has been observed after prolonged exposure or incubation in organic solvents (Gonzalez et al., 2002). However, the reasons behind this detrimental effect are not clear. A recent paper showed that the loss of activity is not associated with any loss of secondary or tertiary structure (Castillo et al., 2006). The changes in conformational flexibility could be a feasible explanation for the loss in activity.
A growing interest in protein conformational flexibility has led to the development of different analytical techniques to investigate this phenomenon. Mass spectroscopic studies of hydrogen/deuterium (H/D) exchange of the mobile protons in proteins (Wales and Engen, 2006) and NMR (Kay, 2005) have emerged as the most valuable techniques for studying enzyme flexibility in aqueous solution. However, due to some limitations, primarily the insolubility of proteins in nonaqueous system, these techniques are not always applicable to enzyme studies in organic solvents. In such cases, it is possible to focus the attention on the enzyme active site through site specific spin labeling and EPR spectroscopy (Affleck et al., 1992a; Castillo et al., 2006), and time-resolved fluorescence anisotropy measurement and FRET analysis to give a precise picture of the whole enzyme's dynamic process (Broos et al., 1995). Recently H/D exchange studies have been published wherein it has been shown possible to study the kinetics of exchange of amide protons in water (Gekko et al., 2004; Sola and Griebenow, 2006) and organic solvents (Castillo et al., 2008) by FTIR.
NMR spectroscopy is another useful technique since it makes it possible to study both a particular area as well as the global dynamics of a protein. 1H NMR has been used for the purpose of following the deuterium exchange rates of particular buried hydrogen atoms of the enzyme in the presence of organic solvents (Dempsey, 1995; Desai and Klibanov, 1995; Wu and Gorenstein, 1993), while 2H NMR has been used to study the total number of exchanging protons (Hutcheon et al., 2000). Solid state NMR has also been used for the same purpose (Burke et al., 1992).
Few theoretical studies using molecular dynamic simulation have also been done to study enzymes dynamics, showing the importance of hydration to reach nativelike protein flexibility in nonaqueous solvents (Soares et al., 2003).
We undertook this study with the goal of elucidating the effect of prolonged exposure to different organic solvents on the conformational flexibility of subtilisin Carlsberg (SC). We tested different enzyme preparations and solvents to identify the parameters affecting enzyme's rigidity. To address these issues we modified the technique of 2H NMR spectroscopy developed by the group of Moore (Hutcheon et al., 2000) for the qualitative estimation of flexibility of SC in organic solvents by H/D exchange.
Materials and Methods
Enzyme and Reagents
SC (alkaline protease from Bacillus licheniformis, EC 3.4.21.14) was purchased as a lyophilized powder, 18-crown-6, sec-phenethyl alcohol, the solvents (purchased in the Aldrich Sure/Seal bottles, water content below 0.005%), and the inorganic salts were purchased from Sigma-Aldrich (St. Louis, MO). D2O was obtained from Cambridge Isotope Laboratories, Inc. (Andover, MA). Vinyl butyrate was purchased from TCI America Co. (Portland, OR). N-succinimidyl PEG (5 kDa) was obtained from NeKtar Therapeutics (Huntsville, AL).
Enzyme Preparation
SC powder was used as received from the supplier, then dissolved in deionized water (10 mg/mL), frozen in liquid N2 and lyophilized for 24 h. Colyophilization of the enzyme with 18-crown-6 was done the same way, except that the additive was added to the enzyme solution prior to lyophilization at a weight ratio of 1:3.
Enzyme PEGylation
SC was dissolved in sodium 0.1 M borate buffer (pH 9.2) at a concentration of 10 mg/ mL. A threefold (over protein) molar excess of N-succinimidyl PEG (5 kDa) was added and the reaction allowed to proceed with constant stirring for 15 min at 20°C and then for 3 h at 4°C. The reaction was then quenched by reducing the pH to 5.5 with dilute HCl and the solution was dialyzed against 1.5 L of nanopure water (18 MΩ resistance) for 24 h in 25 kDa cutoff dialysis bag. The dialyzed solution was finally lyophilized for 48 h. The degree of PEGylation in terms of number of PEG molecules per molecule of SC was determined by the Habeeb method (Habeeb, 1966). The average PEGylation ratio was 2 mol of PEG per mol of SC.
Calorimetric Analysis
All thermal denaturation experiments were performed using a DSC (Calorimetry Sciences Corp., Lindon, UT) 6100 Nano II differential scanning calorimeter (DSC). Protein samples were prepared at a concentration of 1 mg/mL in deionized water. Heat capacity thermograms were determined at a 1°C/min scan rate. Baselines were run with deionized water. The melting points (Tm) of different protein preparations were determined by digital analysis of the heat capacity thermograms using the program Cp Calc (Calorimetry Sciences Corp.).
Kinetic Measurements
The synthesis of the butyryl ester derivative was performed at a constant water activity (aw) of 0.06 by equilibration with hydrated BaBr2. All the reactants, solvents, and enzyme preparations were individually pre-equilibrated in three separate jars at the desired water activity for 48 h before the reaction. The reactions were then performed as follows: racemic sec-phenethyl alcohol (70 mM) was dissolved in 1.0 mL of solvent. To this solution, the enzyme powder (from 1 to 10 mg, depending on the enzyme preparation) was added. After 5 min n-Butyric acid vinyl ester (200 mM) was added and the reaction was allowed to proceed with constant shaking (200 rpm) at the defined water activity at 25°C. The formation of the ester derivatives was monitored by GC analysis as previously described (Griebenow et al., 1999). The GC instruments (HP 6850, and HP 6890, fitted with Chirasil CB chiral columns, FID detectors, and He as carrier gas) were calibrated with the chiral esters synthesized as reported (Griebenow et al., 1999). All the reactions were performed in duplicate. The enzyme E value was determined from the ratio of the R and S enantiomers of the product, and since a racemic mixture of the alcohol was used, E is equal to the ratio:
H/D Exchange Experiments
Twenty vials of SC (10 mg of enzyme plus additive if needed) were lyophilized, transferred to a desiccator containing phosphorus pentoxide and dried under vacuum for 4 days. The sample vials were then transferred to a glove box and the exchange was initiated by adding 1 mL of the desired organic solvent previously equilibrated for 7 days at a deuteriated water activity (Daw) of 0.78 (KCl gas phase equilibration). After allowing the enzyme to exchange for the desired time (from 2 s to 48 h), the H/D exchange was quenched by quickly flash freezing the sample vials in liquid nitrogen. The organic solvent was removed by lyophilizing the samples for 24 h. The samples were then transferred under N2 atmosphere to a desiccator and kept for 4 days for further drying. These first steps completed the exchange process. The total number of hydrogen atoms that had exchange was determined by the following back exchange procedure: the vials containing the enzyme samples were placed in a glove box and 350 μL of deuterium depleted water was added to each of the vials. The enzyme was denatured by heat shock to expose all exchangeable hydrogens thus allowing them to back exchange with the solvent. Note that the heating was continued for 2 min in order to ensure complete back exchange of unfolded SC. Thermal denaturation of SC was confirmed by DSC analysis. Three hundred microliters of a solution of deuterium depleted water-containing acetone D6 as internal standard (1 mg acetone/mL deuterium depleted water) was added to each vial. The acetone solution was added after heat shock to avoid the evaporation of the internal standard. The denatured enzyme solution was finally transferred to a 5 mm NMR tube, and analyzed by 2H NMR to measure the number of 2H in the solvent.
The relative intensity of the D2O signal from the deuterons exchanged from the protein and that of the acetone D6 signal was measured as shown in Figure 1 and used to calculate the number of hydrogens exchanged as previously reported (Hutcheon et al., 2000). 2H NMR measurements were carried out on a Bruker 400 MHz FT-NMR spectrometer. Two hundred fifty scans were collected using a sweep width of 922 Hz and an acquisition time of 4 s. Data were processed using 1 Hz line broadening and automatic baseline correction.
Figure 1.

Superimposition of three 2H NMR spectra taken after different H/D exchange times: the peak of deuteriated acetone (internal standard) appears at 2.05 ppm. The peak of deuteriated water at 4.8 ppm clearly shows an increase as the exchange proceeds with time. The number of exchanged protons was calculated as the ratio of the two peaks as described in the Materials and Methods Section.
H/D Exchange After 4 days of Incubation
The procedure was repeated as previously described except that the enzyme was incubated for 4 days in 0.2 mL of the desired organic solvent equilibrated at constant water activity with hydrated barium bromide (aw = 0.06). After this point, the procedure was repeated as before: 1 mL of the same solvent previously incubated with potassium chloride in D2O (Daw = 0.78, for at least 7 days) was added to initiate the exchange, and stopped after the desired time by flash freezing. The samples were then lyophilized, dried further in a desiccator for 4 days and the enzyme denatured by heat shock to allow complete back exchange with deuterion depleted water. The samples were analyzed by 2H NMR as before.
Data Analysis
The data of exchanged protons versus time was fitted to the following tri-exponential equation:
where
[NEP], normalized concentration of nonexchanging protons during 48 h of exchanging period; [VFEP], normalized concentration of very fast exchanging protons located on the enzyme surface in direct contact with the solvent; t, time in hours; kVFEP, apparent rate constant for very fast exchanging protons (units: normalized number of protons per hour); [FEP], normalized concentration of fast exchanging protons inside the enzyme; kFEP, apparent rate constant for fast exchanging protons (units: normalized number of protons per hour); [SEP], normalized concentration of slow exchanging protons sequestered in areas of the enzyme where contact with the solvent is retarded; kSEP, apparent rate constant for slow exchanging protons (units: normalized number of protons per hour).
The number of protons were normalized to 1 and plotted as an exponential decay to be in accord with previously published data from this and other laboratories on similar H/D exchange experiments obtained by FTIR spectroscopy, and to facilitate the analysis. The number of hydrogen atoms exchanged was normalized using the relationship:
The total number of exchangeable hydrogens (434) was determined experimentally by heat denaturation. To estimate the actual number of Hydrogen atoms that had exchanged:
The data were fitted using the software SigmaPlot to the equation shown above (f = y0 + a × exp(−b × x) + c × exp(−d × x) + e × exp(−g×x), fit f to y), 100,000 iteractions, a step size of 0.000001 and a tolerance of 1.0 × 10−10.
Results
SC from Bacillus licheniformis has 274 amino acid residues, with which are associated a total of 449 exchangeable protons; 273 are amide protons of the enzyme backbone, 174 of side chains and, 2 of the terminal groups (Smith et al., 1968). Some of these hydrogen atoms are located on the surface of the protein and hence exposed to the solvent molecules while others, due to the supramolecular structure, are buried inside the core of the protein and hence inaccessible to the solvent. These later protons will thus be protected from any exchange with the solvent (protection factor). This distribution of Hs is responsible for the different H/D exchange rates observed. The results from our study show that out of the total 449 theoretically exchangeable protons, an average of 295 protons exchanged after 48 h. With the aim of understanding if this limited number of exchanged protons was due to a limitation of our technique or the protection effect, we repeated the exchange experiment as previously described except that this time the enzyme was denatured trough thermal shock to expose and exchange all exchangeable Hs. Denaturation was confirmed by determining the melting point of the protein through calorimetric analysis. In this case we obtained an average of 434 (±10) exchanged protons suggesting that almost one third of the exchangeable protons are located in the core of the protein, not accessible to the solvent during 48 h and only a few are lost during the experimental procedure, probably due to back exchange.
The protocol developed in our lab permits an appreciation of the fast exchange which takes place during the first few seconds between mobile protons and D2O. In most of the papers published on H/D exchange bi-exponential equations are used and good fits of the data is obtained (Castillo et al., 2008; Gekko et al., 2004; Sola and Griebenow, 2006). However, in our case we found that a better fit is described by a tri-exponential equation as demonstrated for one of the kinetic experiments shown as an example (Fig. 2). The tri-exponential equation describes a population of very fast exchanging protons (VFEP) governed by an apparent rate constant kVFEP. This exchange takes place during the first few seconds of the experiment due to solvent exposed protons, and since we are not able to obtain sufficient data points at this early phase of the exchange the error on kVFEP makes it undistinguishable between ACN days 0 and 4, and dioxane day 4. kVFEP day 0 in dioxane is a bit slower than the rest. It is important to note that actually an increase in this observed rate constant could be an indication of partial denaturation (increased accessible area), and since VFEPs are the external protons not involved in hydrogen bonding, their exchange rate will not reflect on the enzyme flexibility. This hydrogen population cannot be appreciated on a 48 h time scale due to the compression of the data, but it is clearly observed in an expansion of the first part of the graph (insert Fig. 2). The second, third, and fourth populations of hydrogens described by the equation are instead buried into the secondary and tertiary structure of the enzyme. Depending on their respective positions in the enzyme fold they have different rates of exchange and are named as fast exchanging protons (FEP), slow exchanging protons (SEP), and not exchanging protons (NEP). They are related to the apparent rate constants, kFEP and kSEP in the fitting equation.
Figure 2.
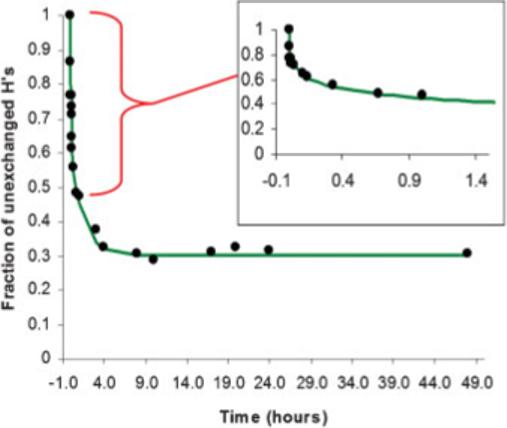
Measurement of protons exchanged versus time. The decay is described by tri-exponential equation. Graph insert shows an expansion of H/D kinetics in the first 1.5 h.
Discussion
As has been discussed in the introduction, prolonged exposure of an enzyme to organic solvents leads to a decrease in its initial activity. This detrimental effect is often more pronounced when the enzyme is colyophilized with additives such as crown ethers, and less evident with the lyophilized powder, probably because the later is already poorly active (Table I). Previous studies have not been able to identify the reason for this activity loss (Castillo et al., 2006; Gonzalez et al., 2002). Thus, we decided to examine the effect of long-term exposure to organic solvents on the global flexibility of SC by H/D exchange/NMR spectroscopy. Incubation of lyophilized SC for a maximum of 96 h (4 days) in 1−4 dioxane (Diox) (a solvent in which this enzyme is most active and enantioselective) showed a decrease in the rate of exchange of both fast (kFEP) and slow (kSEP) exchanging protons, suggesting that incubation in this solvent increases the rigidity of the enzyme, while the relative number of hydrogens in each population remained constant within the experimental error (Fig. 3), suggesting that the structure of the enzyme remains unchanged during incubation. A similar observation was made when this study was repeated in a more polar solvent, acetonitrile (ACN), in which the enzyme is not very active or enantioselective. While the hydrogen population slightly increases at the level of VFEP probably due to a partial denaturation of the enzyme, as in Diox the kFEP and kSEP diminished substantially after 4 days of incubation. This is the first study to show diminution of global enzyme flexibility resulting from incubation in organic solvents that would explain the decrease in enzyme activity previously reported, and it would involve an accepted relationship between activity and structural dynamics: high enzyme flexibility (or dynamics) is required for optimum enzyme activity.
Table I.
Activity and enantioselectivity of subtilisin Carlsberg in organic solvents.
| Prep. | Solvent | Incubationa | Initial activityb | E |
|---|---|---|---|---|
| PEG 5 kDa | Diox | 0 | 264 ± 72 | 21.1 ± 0.2 |
| 4 | 23 ± 13 | 20.3 ± 0.6 | ||
| ACN | 0 | 68 ± 1 | 5.1 ± 0.1 | |
| 4 | 24 ± 1 | 5.2 ± 0.4 | ||
| Lyophilized | Diox | 0 | 18 ± 6 | 19.0 ± 0.6 |
| 4 | 5 ± 2 | 18.9 ± 0.1 | ||
| ACN | 0 | 4 ± 1 | 4.6 ± 0.4 | |
| 4 | 0.1 ± 0.04 | 4.7 ± 0.3 | ||
| Crown ether colyophilized | Diox | 0 | 238 ± 44 | 26.3 ± 0.3 |
| 4 | 5 ± 1 | 25 ± 2 | ||
| ACN | 0 | 60 ± 1 | 5.1 ± 0.7 | |
| 4 | 10 ± 1 | 7 ± 1 |
Data were taken after 0 and 4 days of incubation in organic solvents.
Activity calculated in nmol/mg/min.
Figure 3.
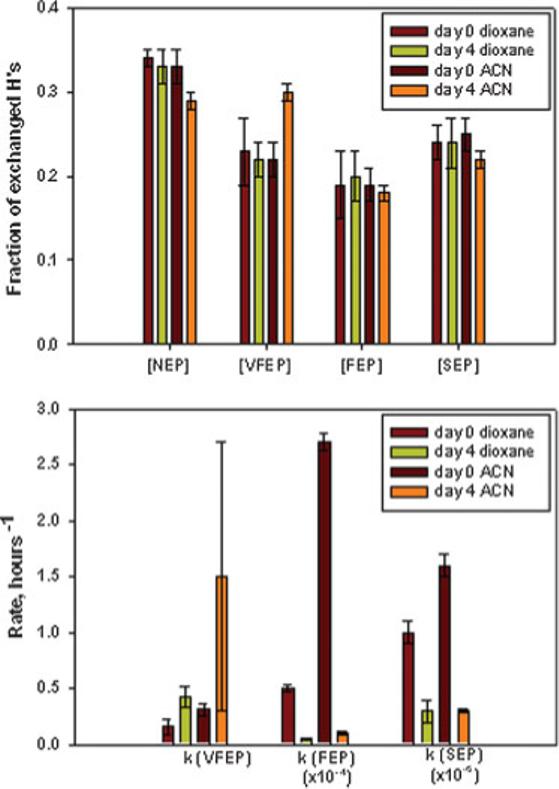
Changes in conformation flexibility of lyophilized subtilisin Carlsberg after 4-days incubation in dioxane and ACN.
With the aim of expanding this finding to a more general system, we decided to evaluate the global dynamics of modified SC. It has been found that adding excipients such as cyclodextrin (Griebenow et al., 1999), crown ether (Secundo et al., 2007; van Unen et al., 2001), PEG (Castillo et al., 2008) and inorganic salts (Morgan and Clark, 2004) consistently improves the activity and the E value of enzymes in organic solvents. This activating effect has been associated with a lyoprotection effect (Fasoli et al., 2006). We thus performed the H/D exchange experiments with two preparations that activate this enzyme: the PEGylated and the crown ether colyophilized in dioxane. To our surprise, we did not observe the same trend between activity and global enzyme flexibility (comparing kSEP and kFEP); PEGylated SC (most active preparation) showed a small decrease in global flexibility at the level of kFEP as compared to the lyophilized (less active preparation), while the crown ether colyophilized enzyme (also more active than the lyophilized) appeared to have significantly less conformational mobility than the lyophilized one (Table II). As previously observed the ratio between the hydrogen populations remains unchanged.
Table II.
Effect of additives on the conformational flexibility of subtilisin Carlsberg in dioxane.
| Lyophilized | Crown ether colyophilized | PEGylated | |
|---|---|---|---|
| [NEP] | 0.34 ± 0.01 | 0.34 ± 0.02 | 0.35 ± 0.01 |
| [VFEP] | 0.23 ± 0.04 | 0.26 ± 0.02 | 0.26 ± 0.02 |
| akVFEP | 0.16 ± 0.07 | 0.38 ± 0.07 | 0.55 ± 0.14 |
| [FEP] | 0.19 ± 0.04 | 0.22 ± 0.01 | 0.18 ± 0.03 |
| a,bkFEP | 5.0 ± 0.3 | 1.1 ± 0.2 | 2.0 ± 0.8 |
| [SEP] | 0.24 ± 0.02 | 0.20 ± 0.02 | 0.21 ± 0.03 |
| a,ckSEP | 10 ± 1 | 1.6 ± 0.1 | 10 ± 1 |
In hours−1.
Values to be multiplied by 10−3.
Values to be multiplied by 10−5.
To convert these values to the actual total number of exchanged hydrogens using the relationship discussed in the Data Analysis Section for instance, Total [NEP]Lyo Dioxane = [(1−0.34) × 434] = 286.4 hydrogens. Likewise, kVFEP for the lyophilized enzyme in dioxane = 433 H × h−1.
One of the most widely accepted hypothesis in nonaqueous enzymology is that organic solvents affect the flexibility of enzymes. It is believed that, as a result of the lower-than-water dielectric constant of organic solvents, an enzyme trapped within this medium should increase its intermolecular interactions and its rigidity should therefore increase (Wescott and Klibanov, 1994). We tried to extend this concept to the rate of exchange of SC suspended in the two solvents used in our study. Conformational flexibility is higher in the polar ACN compared to Diox, as shown by the apparent rate constants kSEP and kFEP. This can be observed in the case of all the three different preparations of the enzyme (Table III). In the case of PEGylated SC in ACN an increased number of VFEP was observed, suggesting partial denaturation of the enzyme. However, the high exchange rate found for the crown ether colyophilized SC and lyophilized SC in ACN compared to Diox could not be due to loss of secondary/tertiary structure, because the number of Hs from each population remains constant within the experimental error. Rather, we believe that the higher exchange rate is due to increased flexibility associated with higher dielectric constant of this solvent. This is in accordance with the notion that solvents with high dielectric constants increase the enzyme flexibility; however, it is in contradiction to the concept of high flexibility high activity, since the enzyme is less active in ACN than in Diox (Table I) for all the preparations used in this study.
Table III.
Effect of solvents on conformational flexibility of subtilisin Carlsberg.
| Lyophilized |
Crown ether colyophilized |
PEGylated |
||||
|---|---|---|---|---|---|---|
| Diox | ACN | Diox | ACN | Diox | ACN | |
| [NEP] | 0.34 ± 0.01 | 0.33 ± 0.02 | 0.34 ± 0.02 | 0.33 ± 0.02 | 0.35 ± 0.01 | 0.34 ± 0.01 |
| [VFEP] | 0.23 ± 0.04 | 0.22 ± 0.02 | 0.26 ± 0.02 | 0.23 ± 0.04 | 0.26 ± 0.02 | 0.37 ± 0.01 |
| akVFEP | 0.16 ± 0.07 | 0.31 ± 0.06 | 0.38 ± 0.07 | 0.64 ± 0.35 | 0.55 ± 0.14 | 0.63 ± 0.07 |
| [FEP] | 0.19 ± 0.04 | 0.19 ± 0.02 | 0.22 ± 0.01 | 0.18 ± 0.03 | 0.18 ± 0.03 | 0.12 ± 0.01 |
| a,bkFEP | 5.0 ± 0.3 | 2.7 ± 0.7 | 1.1 ± 0.2 | 7.4 ± 4.2 | 2.0 ± 0.8 | 4.9 ± 1.5 |
| [SEP] | 0.24 ± 0.02 | 0.25 ± 0.02 | 0.20 ± 0.02 | 0.23 ± 0.02 | 0.21 ± 0.03 | 0.17 ± 0.02 |
| a,ckSEP | 10 ± 1 | 16 ± 1 | 1.6 ± 0.1 | 14 ± 1 | 10 ± 1 | 28 ± 1 |
In hours−1.
Values to be multiplied by 10−3.
Values to be multiplied by 10−5.
So far, in our discussion we have excluded the effect of incubation, preparation and solvents on the enzyme E values. Incubation does not affect the enzyme E (Gonzalez et al., 2002), however the mode of preparation and the solvent used does.
As discussed before, the crown ether preparation of SC showed higher rigidity than the lyophilized powder and PEGylated enzyme (Table II). Interestingly this is also associated with an increased E value for the crown ether preparation (Table I). Furthermore, when comparing between solvents, a high E accompanied by higher rigidity is observed in dioxane for all the enzyme preparations, while ACN makes the SC flexible and less enantioselective (Tables I and III). These data are in accordance with the model proposed by Klibanov where an increased conformational flexibility (due to the polarity of the solvent) is associated with decreased E value. The reason behind this statement is that in a more rigid enzyme, the active site is less prone to distortion and can discriminate better between the two enantiomers. However, we observed that this cannot be taken as a general rule to predict the E value of an enzyme because if it was universally true then an increase in the E value should be seen during prolonged incubation in organic solvents too. In contradiction with Klibanov's hypothesis, the enzyme's E value remains unchanged even after 96 h of incubation, while the flexibility decreases (Table I). These data suggest that additional parameters regulate E value.
Conclusion
We believe that so far this is the first time that changes in protein conformational dynamics upon long time exposure to organic solvents have been studied. It was seen in this study that additives (PEG and crown ether) lead to an increase in rigidity, activity and E value to different degrees. In addition, in case of solvents, in going from a more polar solvent such as ACN to a less polar one such as Diox, an increase in activity, rigidity, and E value was seen. These observations suggest a direct correlation between enzyme activity, E value and conformational rigidity. However, the correlation thus derived does not confirm with the activity losses seen on prolonged exposure of the enzyme to organic solvents, as the decrease in enzyme activity in this case was also accompanied by an increase in rigidity while the E value remains the same. This indicates that the loss of activity observed during incubation in organic solvents is not related only to global enzyme flexibility changes but also to some additional factors operating, perhaps, at the active site of the enzyme.
Acknowledgments
The project described was supported by Grants P20 RR016470, and S06 GM-08216 from the National Center for Research Resources, and the National Institutes of Health (SCORE). The content is solely the responsibility of the authors and does not necessarily represent the official view of the National Center for Research Resources or the National Institutes of Health. We are grateful to Mr Melvin De Jesús for his valuable help with the NMR experiments.
Contract grant sponsor: National Center for Research Resources
Contract grant number: P20 RR016470; S06 GM-08216
Contract grant sponsor: National Institutes of Health (SCORE)
References
- Affleck R, Haynes CA, Clark DS. Solvent dielectric effects on protein dynamics. Proc Natl Acad Sci USA. 1992a;89(11):5167–5170. doi: 10.1073/pnas.89.11.5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Affleck R, Xu ZF, Suzawa V, Focht K, Clark DS, Dordick JS. Enzymatic catalysis and dynamics in low-water environments. Proc Natl Acad Sci USA. 1992b;89(3):1100–1104. doi: 10.1073/pnas.89.3.1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broos J, Visser AJWG, Engbersen JFJ, Verboom W, van Hoek A, Reinhoudt DN. Flexibility of enzymes suspended in organic solvents probed by time-resolved fluorescence anisotropy. Evidence that enzyme activity and enantioselectivity are directly related to enzyme flexibility. J Am Chem Soc. 1995;117(51):12658–12662. [Google Scholar]
- Burke PA, Griffin RG, Klibanov A. Solid-state NMR assessment of enzyme active centers structure under non-aquous conditions. J Biol Chem. 1992;267(28):20057–20064. [PubMed] [Google Scholar]
- Castillo B, Bansal V, Ganesan A, Halling P, Secundo F, Ferrer A, Griebenow K, Barletta G. On the relationship between the activity and structure of PEG-alpha-chymotrypsin conjugates in organic solvents. BMC Biotechnol. 2006;6:51. doi: 10.1002/bit.20863. [DOI] [PubMed] [Google Scholar]
- Castillo B, Sola RJ, Ferrer A, Barletta G, Griebenow K. Effect of PEG modification on subtilisin Carlsberg activity, enantioselectivity, and structural dynamics in 1,4-dioxane. Biotechnol Bioeng. 2008;99(1):9–17. doi: 10.1002/bit.21510. [DOI] [PubMed] [Google Scholar]
- Dempsey CE. Hydrogen bond stabilities in the isolated alamethicin helix: pH-dependent amide exchange measurements in methanol. J Am Chem Soc. 1995;117(28):7526–7536. [Google Scholar]
- Desai UR, Klibanov AM. Assessing the structural integrity of a lyophilized protein in organic solvents. J Am Chem Soc. 1995;117(14):3940–3945. [Google Scholar]
- Eisenmesser EZ, Bosco DA, Akke M, Kern D. Enzyme dynamics during catalysis. Science. 2002;295(5559):1520–1523. doi: 10.1126/science.1066176. [DOI] [PubMed] [Google Scholar]
- Fasoli E, Castillo B, Santos A, Silva E, Ferrer A, Rosario E, Griebenow K, Secundo F, Barletta GL. Activation of subtilisin Carlsberg in organic solvents by methyl-β-cyclodextrin: Lyoprotection versus substrate and product-complex effect. J Mol Catalysis B Enzymatic. 2006;42(1–2):20–26. [Google Scholar]
- Fitzpatrick PA, Klibanov AM. How can the solvent affect enzyme enantioselectivity? J Am Chem Soc. 1991;113(8):3166–3171. [Google Scholar]
- Fitzpatrick PA, Ringe D, Klibanov AM. Computer-assisted modeling of subtilisin enantioselectivity in organic solvents. Biotechnol Bioeng. 1992;40(6):735–742. doi: 10.1002/bit.260400613. [DOI] [PubMed] [Google Scholar]
- Gekko K, Obu N, Li J, Lee JC. A linear correlation between the energetics of allosteric communication and protein flexibility in the Escherichia coli cyclic AMP receptor protein revealed by mutation-induced changes in compressibility and amide hydrogen-deuterium exchange. Biochemistry. 2004;43(13):3844–3852. doi: 10.1021/bi036271e. [DOI] [PubMed] [Google Scholar]
- Gonzalez S, Alvira E, Cordero LV, Ferrer A, Montales CI, Barletta G. High initial activity but low storage stability observed for several preparations of subtilisin Carslberg suspended in organic solvents. Biotechnol Prog. 2002;18:1462–1466. doi: 10.1021/bp025650g. [DOI] [PubMed] [Google Scholar]
- Griebenow K, Laureano YD, Santos AM, Clemente IM, Rodriguez L, Vidal MW, Barletta G. Improved enzyme activity and enantioselectivity in organic solvents by methyl-β-cyclodextrin. J Am Chem Soc. 1999;121(36):8157–8163. [Google Scholar]
- Habeeb AF. Determination of free amino groups in proteins by trinitrobenzenesulfonic acid. Anal Biochem. 1966;14(3):328–336. doi: 10.1016/0003-2697(66)90275-2. [DOI] [PubMed] [Google Scholar]
- Henzler-Wildman K, Kern D. Dynamic personalities of proteins. Nature. 2007;450(7172):964–972. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- Hutcheon GA, Parker MC, Moore BD. Measuring enzyme motility in organic media using novel H-D exchange methodology. Biotechnol Bioeng. 2000;70(3):262–269. [PubMed] [Google Scholar]
- Kay LE. NMR studies of protein structure and dynamics. J Magn Reson. 2005;173(2):193–207. doi: 10.1016/j.jmr.2004.11.021. [DOI] [PubMed] [Google Scholar]
- Klibanov AM. Improving enzymes by using them in organic solvents. Nature. 2001;409(6817):241–246. doi: 10.1038/35051719. [DOI] [PubMed] [Google Scholar]
- Lindorff-Larsen K, Best RB, Depristo MA, Dobson CM, Vendruscolo M. Simultaneous determination of protein structure and dynamics. Nature. 2005;433(7022):128–132. doi: 10.1038/nature03199. [DOI] [PubMed] [Google Scholar]
- Morgan JA, Clark DS. Salt-activation of non-hydrolase enzymes for use in organic solvents. Biotechnol Bioeng. 2004;85:456. doi: 10.1002/bit.10895. [DOI] [PubMed] [Google Scholar]
- Partridge J, Dennison PR, Halling PJ, Moore BD. Subtilisin mobility in hydrophobic organic media measured by proton NMR relaxation: Hydration is more important than solvent dielectric. Biochim Biophys Acta. 1998;1386:79–89. doi: 10.1016/s0167-4838(98)00086-7. [DOI] [PubMed] [Google Scholar]
- Secundo F, Barletta G, Mazzola G. Role of methoxypolyethylen(glycol) on the hydration, activity, conformation and dynamic properties of a lipase in a dry film. Biotechnol Bioeng. 2008;101(2):252–262. doi: 10.1002/bit.21888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Secundo F, Barletta GL, Dumitriu E, Carrea G. Can an inactivating agent increase enzyme activity in organic solvent? Effects of 18-crown-6 on lipase activity, enantioselectivity, and conformation. Biotechnol Bioeng. 2007;97(1):12–18. doi: 10.1002/bit.21223. [DOI] [PubMed] [Google Scholar]
- Smith EL, DeLange RJ, Evans WH, Landon M, Markland FS. Subtilisin Carlsberg. V. The complete sequence; comparison with subtilisin BPN’; evolutionary relationships. J Biol Chem. 1968;243(9):2184–2191. [PubMed] [Google Scholar]
- Soares CM, Teixeira VH, Baptista AM. Protein structure and dynamics in nonaqueous solvents: Insights from molecular dynamics simulation studies. Biophys J. 2003;84(3):1628–1641. doi: 10.1016/S0006-3495(03)74972-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sola RJ, Griebenow K. Chemical glycosylation: New insights on the interrelation between protein structural mobility, thermodynamic stability, and catalysis. FEBS Lett. 2006;580(6):1685–1690. doi: 10.1016/j.febslet.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Valivety RH, Halling PJ, Macrae AR. Rhizomucor Miehei lipase remains highly-active at water activity below 0.001. FEBS Lett. 1992;301(3):258–260. doi: 10.1016/0014-5793(92)80252-c. [DOI] [PubMed] [Google Scholar]
- Valivety RH, Halling PJ, Peilow AD, Macrae AR. Relationship between water activity and catalytic activity of lipases in organic media. Effects of supports, loading and enzyme preparation. Eur J Biochem. 1994;222(2):461–466. doi: 10.1111/j.1432-1033.1994.tb18886.x. [DOI] [PubMed] [Google Scholar]
- van Unen Dirk-Jan, Engbersen JFJ, Reinhoudt DN. Why do crown ethers activate enzymes in organic solvents? Biotechnol Bioeng. 2001;77(3):248–255. doi: 10.1002/bit.10032. [DOI] [PubMed] [Google Scholar]
- Wales TE, Engen JR. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom Rev. 2006;25(1):158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Yoshida T, Ueji S. Optimum conformational flexibility of subtilisin to maximixe the enantioselectivity for subtilisin-catalysed transesterification in an organic solvent with an addition of dimethyl sulfoxide. Chem Commun. 2001;14:1260–1261. [Google Scholar]
- Wescott CR, Klibanov AM. The solvent dependence of enzyme specificity. Biochim Biophys Acta. 1994;1206(1):1–9. doi: 10.1016/0167-4838(94)90065-5. [DOI] [PubMed] [Google Scholar]
- Wu J, Gorenstein DG. Structure and dynamics of cytochrome c in nonaqueous solvents by 2D NH-exchange NMR spectroscopy. J Am Chem Soc. 1993;115(15):6843–6850. [Google Scholar]
- Zaks A, Klibanov AM. The effect of water on enzyme action in organic media. J Biol Chem. 1988;263(17):8017–8021. [PubMed] [Google Scholar]
- Zavodszky P, Kardos J, Svingor A, Petsko GA. Adjustment of conformational flexibility is a key event in the thermal adaptation of proteins. Proc Natl Acad Sci USA. 1998;95(13):7406–7411. doi: 10.1073/pnas.95.13.7406. [DOI] [PMC free article] [PubMed] [Google Scholar]