

Summary
Macrophages are a major source of lipid mediators in the human lung. Expression and contribution of cytosolic (cPLA2) and secreted phospholipases A2 (sPLA2) to the generation of lipid mediators in human macrophages is unclear. We investigated the expression and role of different PLA2s in the production of lipid mediators in primary human lung macrophages. Macrophages express the alpha, but not the zeta isoform of group IV and group VIA cPLA2 (iPLA2). Two structurally-divergent inhibitors of group IV cPLA2 completely block arachidonic acid release by macrophages in response to non-physiological (Ca2+ ionophores and phorbol esters) and physiological agonists (lipopolysaccharide and Mycobacterium protein derivative). These inhibitors also reduce by 70% the synthesis of platelet-activating factor by activated macrophages. Among the full set of human sPLA2s, macrophages express group IIA, IID, IIE, IIF, V, X and XIIA, but not group IB and III enzymes. Me-Indoxam, a potent and cell impermeable inhibitor of several sPLA2s, has no effect on arachidonate release or platelet-activating factor production. Agonist-induced exocytosis is not influenced by cPLA2 inhibitors at concentrations that block arachidonic acid release. Our results indicate that human macrophages express cPLA2-alpha, iPLA2 and several sPLA2s. Cytosolic PLA2-alpha is the major enzyme responsible for lipid mediator production in human macrophages.
Keywords: Arachidonic acid, Platelet Activating Factor, Phospholipase A2, Lung macrophages, Eicosanoids, Inflammation
Introduction
Macrophages are the predominant immunocompetent cells in the lung where they play a pivotal role in immune and inflammatory reactions associated with diseases such as chronic obstructive pulmonary disease [1], bronchial asthma [2], interstitial lung disease [3], infections [4], and cancer [5]. These cells are one of the major sources of arachidonate-derived eicosanoids in the human lung, producing primarily 5-HETE and LTB4 and, to a lesser extent, cysteinyl leukotrienes (LTC4, LTD4 and LTE4) and prostanoids (PGE2, PGF2α and TXA2) [6–8]. In addition, human lung macrophages produce the pleiotropic lipid mediator platelet activating factor (PAF) [9].
The synthesis of eicosanoids and PAF in mammalian cells is biochemically linked [10]. The hydrolysis of 1-alkyl-2-arachidonoyl-sn-glycero-3-phosphocholine, catalyzed by one or more phospholipases A2 (PLA2s), is the initial biosynthetic step to generate free arachidonic acid (AA) and 1-alkyl-2-lyso-sn-glycero-3-phosphocholine, which are the precursors of eicosanoids and PAF, respectively. Different PLA2s have been identified and characterized in mammalian cells [11]. Known enzymes include high molecular weight, calcium-dependent and calcium-independent, cytosolic PLA2s (cPLA2s), and low molecular weight, calcium-dependent, secreted PLA2s (sPLA2s).
The α-isoform of cPLA2 (cPLA2-α, also known as group IVA PLA2, GIVA) is thought to play a critical role in agonist-stimulated AA release leading to eicosanoid production in the majority of mammalian cells [12]. Upon cell activation cPLA2-α is activated by a rise in intracellular calcium, by phosphorylation [13], and possibly by specific phospholipids including phosphatidylinositol-4,5-bisphosphate [14] and ceramide-1-phosphate [15].
Studies on the involvement of cPLA2s in human cells usually rely on pharmacological inhibition of these enzymes. Early studies with the potent cPLA2-α inhibitor AACOCF3 provided evidence that this enzyme was responsible for most of the AA released in thrombin- and collagen-stimulated human platelets [16, 17]. Two new classes of cPLA2-α inhibitors have been recently reported. The pyrrolidine-containing compounds, including pyrrophenone [18] and pyrrolidine-1 [19], have been shown to be highly potent inhibitors of cPLA2-α without influence on the β- and γ-isoforms of cPLA2 [13, 20], on the calcium-independent cytosolic PLA2 (GVIB), and on several sPLA2s [20]. The other class includes 1,3-diphenoxyacetone inhibitors of cPLA2-α that are much more potent than AACOCF3 (compound 22 of [21], referred to as AZ-1 in the present study).
Recent studies with human peripheral blood neutrophils have shown that the cPLA2-α inhibitor pyrrolidine-1 completely blocks AA release and LTB4 production as well as PAF production in response to a variety of physiologically-relevant agonists [22, 23]. Inhibition of lipid mediators by pyrrolidine-1 was virtually identical to that observed in bone marrow derived neutrophils from cPLA2-α-deficient mice [23]. Together, these observations indicate that cPLA2-α is the key enzyme for the generation of lipid mediators in several inflammatory cells. However, there is also evidence that other PLA2s may contribute to AA mobilization when cPLA2-α is blocked [24]. For example, a new GIVF cPLA2 isoform, termed cPLA2-ζ, was recently discovered. This isoform displays similar kinetic properties to cPLA2-α, and show similar sensitivity to the cPLA2-α inhibitors pyrrolidine-1, pyrrophenone, Wyeth-1 [24] and AZ-1 (Leslie, C. and Gelb, M.H., unpublished data). These inhibitors block AA release in serum-stimulated lung fibroblasts that are prepared from cPLA2-α deficient mice, an effect that is likely due to inhibition of cPLA2-ζ [24].
Ten different sPLA2s have been discovered in humans and mice [11]. In contrast to cPLA2s, sPLA2s are usually stored within cytoplasmic granules or pass through the classical secretory pathway to be released in the extracellular environment upon appropriate stimulation of the cell. The possible role of one or more sPLA2s in generating AA for eicosanoid production in mammalian cells is under active investigation. Recent studies with zymosan-stimulated mouse peritoneal macrophages show that disruption of the GV gene leads to an approximate 50% reduction in AA release [25]. In addition, studies with GV- and GX-deficient mice show that these enzymes play a significant role in promoting airway inflammation and eicosanoid production in a mouse model of asthma [26, 27]. This plus the observation that cPLA2-α gene disruption nearly completely eliminates AA release in the same cell/agonist system suggests that some sPLA2s, while not effective per se, can potentiate the action of cPLA2-α. Other studies confirmed this sPLA2-cPLA2-α interaction [28–31], but the molecular basis and directionality of this crosstalk is not understood. Thus, while the central role of cPLA2-α in eicosanoid generation is well established, other cPLA2s and sPLA2s may contribute, under certain circumstances, to provide AA or lyso-PAF available for lipid mediator biosynthesis.
While cPLA2-α appears to be expressed in virtually all mammalian cells, the expression of other members of the PLA2 family is rather heterogeneous [32]. Defining the full set of the PLA2s expressed in inflammatory cells is crucial to understand the role of these enzymes in eicosanoid and PAF metabolism. Data on PLA2 expression in macrophages derive mostly from studies in murine or guinea pig cells. However, macrophages from rodents exhibit marked biochemical and functional differences from human macrophages. For example, activated human lung macrophages synthesize predominantly 5-lipoxygenase metabolites [6, 7], whereas murine macrophages produce both cyclooxygenase and lipoxygenase metabolites [33, 34]. In addition, macrophages isolated from distinct anatomical sites, e.g., pulmonary versus peritoneal, show a different ability to synthesize eicosanoids and PAF [35]. In most cases, studies on AA metabolism in human lung macrophages have been done by using alveolar macrophages obtained from bronchoalveolar lavage fluid [6, 36]. However, the human lung contains different subpopulations of macrophages distinct by morphological, biochemical and functional characteristics [37–39]. In this study we examined the expression of the complete set of cPLA2s and sPLA2s at both the mRNA and protein level in primary macrophages purified from the whole lung tissue. Therefore, the population of macrophages used in this study is representative of all the subsets of macrophages present in the human lung.
Materials and Methods
Reagents
The following were purchased from Sigma: Fatty acid-free HSA and BSA, Percoll®, piperazine–N,N′–bis–2–ethanesulfonic acid (PIPES), Triton X-100, L–glutamine, antibiotic-antimycotic solution (10,000 UI/ml penicillin, 10 mg/ml streptomycin, and 25 μg/ml amphotericin), phenolphthalein glucuronide, lipopolysaccharide (LPS; from E. Coli serotype 026:B6), and phorbol myristate acetate (PMA). RPMI and FCS were from MP Biomedicals Europe. Ca2+ ionophore A23187 is from Calbiochem. [3,6,8,9,11,12,14,15–3H]Aarachidonic acid ([3H]AA; 100 Ci/mmol) and [3H]acetic acid (sodium salt, 4 Ci/mmol) are from Du Pont NEN Products. Peptidoglycan from Staphylococcus Aureus (PGN-SA) is from InvivoGen. Mycobacterium tuberculosis purified protein derivative (PPD) and lipoarabinomannan (LAM) were a generous gift from Chiron S.r.l. A lyophilized stock of 25 mg of PPD and LAM were resuspended in 5 ml of PBS (150 mM NaCl, 3 mM KCl, 10 mM Na2HPO4, 1 mM KH2PO4, pH 7.4) and dialyzed (12,000–14,000 MW cut–off) against PBS overnight at 4°C. The cPLA2-α inhibitors pyrrolidine–1 and AZ–1 were synthesized as described [21, 22]. The sPLA2 inhibitor Me–Indoxam was prepared as described [40]. The cell line HT-29 was purchased from ATCC. Recombinant cPLA2-α was produced as described [41].
Isolation and purification of human lung macrophages
Macrophages were obtained according to human subject research guidelines for the institutions University of Naples Federico II and University of Washington. Macrophages were isolated from the lung parenchyma of patients undergoing thoracic surgery by flotation over Percoll® density gradients, as previously reported [42]. The cells were suspended (106 cells/ml) in RPMI containing 5% FCS, 2 mM L–glutamine, and 1% antibiotic–antimycotic solution and incubated in 24-well plates. After 12 h, the medium was removed and the plates were washed with RPMI. More than 98% of adherent cells were macrophages as assessed by α–naphthylacetate esterase staining [42].
RNA isolation and qPCR
Adherent macrophages were incubated (37°C, 24 hours) in RPMI containing 1% glutamine and 1% antibiotic/antimycotic solution with or without LPS (3 μg/ml). Total RNA extraction from macrophages obtained from 5 donors was performed using the SV total RNA isolation system (Promega). In the final step, the RNA was treated with RNase-free DNase I and was suspended in DEPC treated water. RNA concentrations were determined by OD260 using a Nanodrop apparatus, and RNA quality was evaluated by analysis on an Agilent Bioanalyzer.
First-strand cDNA was synthesized from 5 μg of total RNA using 100 U of MMLV reverse transcriptase (Promega) in a final volume of 50 μl with 500 ng of random primers (Promega). Quantitative PCR (qPCR) was carried out in 96-well ABgene plates using the GENEAMP 5700 sequence Detection System apparatus (Applied Biosystems) with the qPCR Master Mix Plus for SYBRR Green I (Eurogentec). All reactions were performed in a total volume of 16 μl and contained 50 ng of reverse transcribed RNA (based on the initial RNA concentration) and 250 nM of each primer set. The primer sets were designed using the Primer Express program from Applied Biosystems for the following human genes PLA2G2A (NM_000300), PLA2G2D (NM_012400), PLA2G2E (NM_0145891), PLA2G2F (NM_022819), PLA2G3 (NM_015715), PLA2G5 (NM_000929), PLA2G10 (NM_003561), PLA2G12A (BC_017218), PLA2G12B (NM_032562), cPLA2ζ (NM_213600), and iPLA2β (NM_003560). The primer sets were designed to span an intron in order to avoid amplification from potential traces of genomic DNA in the total RNA preparations. No amplification signal was obtained using human genomic DNA as template in the qPCR (not shown). We used QIAGEN commercial primer sets for GIVA cPLA2α (ref QT00085813) and PLA2G1B (ref QT 00000637) genes. The efficiency and specificity of each primer sets were validated using either serial dilutions of cloned human sPLA2 cDNAs or mixed human tissue cDNA for the other genes. Moreover, negative controls without added reverse transcriptase were performed. Finally, it should be mentioned that the primers used in this study detected the respective PLA2 isoforms in human colon tissues [43]. Thermal cycling was performed at 95°C for 10 min, followed by 40 cycles comprising each a denaturation step at 95°C for 15 sec, and an annealing/extension step at 60°C for 1 min. Amplification of the appropriate product was verified by analyzing the dissociation curves that were obtained after PCR with the following steps: 15 sec at 95°C, 20 sec at 60°C, and then a slow ramp of 20 min from 60 to 95°C. The abundance of the mRNA target was calculated relative to the expression of the reference gene GAPDH and is expressed as ΔCt where ΔCt = Ct (gene of interest) − Ct (GAPDH). The Ct values for GAPDH were typically around 19. The data were also validated using TOP1 as a reference gene (not shown).
Additional primer sequences are:
-
hGAPDH-F, CAACGGATTTGGTCGTATTGG;
hGAPDH-R, GCAACAATATCCACTTTACCAGAGTTAA;
-
hGIIA-F, GGCACCAAATTTCTGAGCTACA;
hGIIA-R, TTATCACACTCACACAGTTGACTTCTG;
-
hGIID-F, GCAGAGGCCAACCCAAAGA;
hGIID-R, CAGGTGGTCATAGCAGCAGTCA;
-
hGIIE-F, TGGGCTGTGAGCCCAAA;
hGIIE-R, GCCGGCGCAGAAAATG;
hGIIF-F, GGCCAGCCCAAGGATGA; hGIIF-R, TGGTCAAAGAGTTCCTGGTAGCA;
-
hGIII-F, GGCACAACCTGCTTCAAGCT;
hGIII-R, CGGGCTGACACCCTGATG;
-
hGV-F, AAGGATGGCACCGATTGGT;
hGV-R, CGAATGTTGCAGCCCTTCTC;
-
hGX-F, TGGCAGTGCGTCAATCAGA;
hGX-R, TTGCACAACAGTTCTTGGCATT;
-
hGXIIA-F, TCCACTGTTTGGTGTTCATCTTAAC;
hGXIIA-R, TCATAGCACCTGTCGTGTTGGT;
-
hGXIIB-F, GCGGAGTCTGGGCTTTGTC;
hGXIIB-R, CACGGTGTTGAACACAGTGTCA;
-
hcPLA2zeta-F, AAGAGCTGCAGGTGGAATTTG;
hcPLA2zeta-R, CAGAACCCCGTTGGTGATG;
-
hTOP1-F, CCCTGTACTTCATCGACAAGC;
hTOP1-R, CCACAGTGTCCGCTGTTTC.
cPLA2-α Immunoblotting Analysis
Washed macrophages (3 × 105) were lysed in ice cold lysis buffer (50 mM Hepes, pH 7.4, 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 10% glycerol, 1% Triton X-100 with freshly added protease inhibitors: 10 μg/ml aprotinin, 10 μg/ml leupeptin and 1 mM PMSF) on ice for 30 min. After centrifugation for 20 min at 12,000 × g at 4 °C, the protein concentration in the supernatant was measured using a Bradford-based assay (BioRad). Each lane of an 8% polyacrylamide gel was loaded with 60 μg of cell extract protein. After blotting, cPLA2-α was detected with a polyclonal antiserum [44] at a dilution of 1/1000. This was followed by detection with ECL (GE Biosciences).
Time-resolved fluorescence immunoassay (TRFIA) of sPLA2s
Two ml of antiserum was mixed with 8 ml of binding buffer (20 mM sodium phosphate, pH 7.0, filtered 0.45 μm), and the sample was loaded onto a 1 ml HiTrap protein A HP column (GE Biosciences) at a flow rate of 1 ml/min at room temperature. The column was washed with the same buffer until the OD280 reached a minimum. The IgG fraction was eluted with 10 ml of elution buffer (100 mM glycine, pH 2.7, filtered) with collection of the OD280 peak (about 3 ml) into a tube pre-loaded with 0.3 ml of 1 M Tris, pH 8.0. Purified IgG was stored at −20 °C. Before use it was dialyzed against 4 portions of 10 mM sodium phosphate, pH 7.0 at 4 °C for 1, 2, 5 and 12 h (50 volumes each). The protein concentration was measured with the Bradford assay (BioRad). A portion of sample containing 2 mg of IgG was lyophilized and stored dessicated at −20 °C. The remainder was stored in 0.5 ml aliquots at −20 °C. The lyophilized IgG was dissolved in 0.27 ml of purified water (Milli-Q, Millipore) and 30 μl of 1 M Na2CO3 was added. The pH was checked by spotting a small aliquot onto moistened pH paper (adjusted to 9–9.5 when necessary). A 0.1 ml aliquot of labeling reagent (1 mg of DELFIA Eu-N1 ITC, Perkin Elmer) dissolved in 0.5 ml of purified water, stored −20 °C) was added, and the sample was mixed gently and allowed to sit at 4 °C. The sample was centrifuged at ~12,000 × g for 3 min prior to loading onto a 2×60 cm Superdex 200 column (GE Biosciences) pre-equilibrated in TSA buffer (50 mM Tris, pH 7.8, 0.9% NaCl, 0.05% NaN3) at 1 ml/min at room temperature. Prior to use the column was decontaminated by running 10 mM phthalate, pH 4.1, 0.01% diethylenetriaminepentaacetic acid and then pre-conditioned by running 7.5 mg BSA in TSA buffer. One ml fractions were collected, which were analyzed for europium fluorescence (see below). The protein concentration was measured by a Bradford-based assay, and if less than 0.1 mg/ml, stabilizer (Perkin Elmer, 7.5% heavy metal free BSA) was added to give a final albumin concentration of 0.1%.
One hundred μl of coating solution (10 μg/ml purified, unconjugated IgG, diluted into filtered 0.1 M sodium phosphate, pH 4.9 just prior to use) was added per well of a NUNC 8×12 DELFIA strip plate (Perkin Elmer). The strips were incubated in a sealed container with TSA buffer moistened paper towels for 24 h at room temperature. The solution was removed by aspiration, and 300 μl per well of blocking buffer (50 mM Tris, pH 7.8, 0.9% NaCl, 6% D-sorbitol, 1% BSA, 1 mM CaCl2, filtered) was added, and the strips were incubated overnight at room temperature in the buffer saturated container.
Samples were diluted in assay buffer (50 mM Tris, pH 7.8, 0.9% NaCl, 0.05% NaN3, 0.5% BSA, 0.01% Tween 40, 20 μM diethylenetriaminepentaacetic acid) to give a total volume of 100 μl. Blocking buffer was removed by aspiration from strip wells, and wells were washed twice with 0.3 ml portions of washing solution (50 mM Tris, pH 7.8, 0.9% NaCl, 0.05% NaN3, 0.02% Tween-20). Diluted sample was added to each IgG-coated well followed by incubation for 30 min at room temperature with agitation on a orbital shaker. Liquid was aspirated away, and the well was washed with four 0.3 ml portions of washing solution. Europium conjugated IgG was diluted in assay buffer, diluted solution was added to each well followed by incubation for 30 min at room temperature with agitation on the shaker. Liquid was aspirated away, and the well was washed with another four 0.3 mL portions of washing solution. Enhancement solution (100 μl Perkin Elmer) was added to each well, and incubation on the shaker was continued for 15 min. Time resolved, europium fluorescence was measured with a Victor 1420 Multilabel Counter using the protocol from the manufacturer. To check linearity and sensitivity, standard curves were made using known amounts of recombinant sPLA2 (produced as described [40]) after dilution in assay buffer. Approximate detection limits (pg sPLA2 per assay well) were: GIB, 10; GIIA, 20; GIID, 500; GIIE, 40; GIIF, 300; GIII, 300; GV, 60; GX, 4; GXIIA, 600.
AA Release
Adherent macrophages (1 × 106 cells/well in 24-well plates) were incubated (24 h, 37°C) in 500 μl RPMI supplemented with 5% FCS, 2 mM L–glutamine, 1% antibiotic–antimycotic solution and [3H]AA (1 μCi/106 cells). Previous studies have shown that with this labeling protocol the [3H]AA reaches equilibrium with the endogenous pool of AA [7]. At the end of the labeling procedure the cells were extensively washed with RPMI containing 0.5 mg/ml HSA to remove unincorporated [3H]AA. [3H]AA–labeled macrophages were incubated (1 h, 37°C) with increasing concentrations (0.3–50 μg/ml) of PPD, LAM or PGN-SA. In other experiments, the cells were preincubated (30 min, 37°C) with increasing concentrations (0.01 to 10 μM) of pyrrolidine–1, AZ–1 or Me–Indoxam and then stimulated with the indicated concentrations of PMA, A23187, PPD or LPS. At the end of incubation, supernatants were collected and centrifuged twice (1000 g, 4°C, 5 min) for subsequent determination of released [3H]AA. The cells remaining in the plates were lysed with 2 volumes of methanol. Cell lysate suspensions were mixed with 1 volume of chloroform and 1 volume of high purity water for determination of the total cellular [3H]AA. Both [3H]AA in the supernatant and total [3H]AA in the cell lysates were measured by liquid scintillation counting (2800 TR Liquid Scintillation Analyzer, Perkin Elmer). [3H]AA release was expressed as the percentage of the total cellular [3H]AA determined in cell lysates. Inhibition of [3H]AA release was expressed as percentage of maximum response calculated as (R − Rb)/(Rmax − Rb) × 100, where R is the release in samples treated with the inhibitor, Rb is the release in unstimulated samples and Rmax is the release in samples stimulated in the absence of the inhibitor.
PAF biosynthesis
Freshly isolated macrophages (107 cells/tube) were suspended in 1 ml of PCG buffer containing 0.5 mg/ml HSA, preincubated (30 min, 37°C) with increasing concentrations (0.01 to 10 μM) of pyrrolidine–1 or AZ–1 and then stimulated (10 min, 37°C) with 1 μM A23187 in the presence of 2 nmol of [3H]acetic acid (≈ 7 μCi/tube) [9]. Reactions were stopped by the addition of 2 ml of methanol and 1 ml of chloroform. Unlabeled PAF (50 μg/tube) was added as a carrier immediately before lipid extraction. Lipids were extracted with the technique of Bligh and Dyer and separated by thin layer chromatography on a layer of silica gel G developed in chloroform/methanol/acetic acid/water (50:25:8:4, v/v). In all cases, recovered PAF included both cell–associated and that released in the medium. The PAF standard (1-O-hexadecyl-2-O-acetyl-sn-glycero-3-phosphocholine; C16-PAF; Biomol) was visualized with I2 vapor. Radioactive PAF was detected by scanning the silica plate with a Bioscan system 200 (Canberra Packard), and the corresponding area was isolated and scraped directly into vials to detect radioactivity by liquid scintillation counting. The results were evaluated as cpm of radioactivity incorporated in the area co-migrating with PAF standard. Inhibition of PAF synthesis was expressed as described for AA release.
To confirm that PAF measured by incorporation of [3H]-acetate was truly 1-alkyl-2-[3H]acetyl-glycero-3-phosphocholine, the samples were treated with lipase A1 (from Rhizopus arrhizus, Sigma) as previously described [9]. Briefly, the lipid extract from A23187-stimulated HLM was suspended in 2 ml of diethyl ether and incubated (24 h, 30°C) with 1 ml of borate buffer (0.1 M, pH 6.5) containing 10 mM CaCl2 and 1750 U of lipase A1. At the end of the incubation, the diethyl ether was dried under N2 and lipids were extracted with the technique of Bligh and Dyer. The radioactivity in the organic and aqueous phases was determined by liquid scintillation counting.
β-glucuronidase assay
β-Glucuronidase activity in supernatants and cell pellets was measured by a colorimetric assay, as previously described [42].
Statistical analysis
The data are expressed as the mean ± SE of the indicated number of experiments. p values were determined using Student’s paired t-test.
Results
Characterization of endogenous PLA2s in human lung macrophages
Initial experiments were performed to examine the expression of cPLA2s and sPLA2s in human lung macrophages. To explore the presence of cPLA2-α (GIVA), cPLA2-ζ (GIVF) and iPLA2 (GVIA) in these cells, we carried out quantitative PCR (qPCR) analysis. mRNAs for cPLA2-α and iPLA2 were detected in all 5 preparations of macrophages from different donors (Table 1). Using either the primer set given in “Methods” or those used previously [24], no PCR signal for cPLA2-ζ was detected up to 40 cycles. When these primers were used to analyze, as controls, mRNA from the human colon cell line HT-29 or from a human colon cancer biopsy, the expected size band for cPLA2-ζ was seen after 25 and 24 cycles, respectively (data not shown).
Table I.
Expression of Phospholipases A21 in Human Lung Macrophages
| mRNA Expression (ΔCt)2 |
||||||
|---|---|---|---|---|---|---|
| Cytosolic PLA2s3 | Donor 1 | Donor 2 | Donor 3 | Donor 4 | Donor 5 | Mean |
| GIVA (cPLA2α) | 7.3 | 9.5 | 6.3 | 10.0 | 9.0 | 8.4 |
| GVIA(iPLA2) | 4.7 | 6.8 | 6.8 | 7.3 | 8.0 | 6.7 |
| Secreted PLA2s3 | ||||||
| GIB | 13.3 | 15.7 | Nd | 15.6 | Nd | 14.9 |
| GIIA | 14.1 | 13.8 | 14.8 | 17.3 | 15.0 | 15.0 |
| GIID | 5.8 | 13.0 | 18.8 | 17.7 | 16.4 | 14.3 |
| GX | 15.0 | Nd | 11.2 | 16.2 | 18.6 | 15.3 |
| GXIIA | 7.5 | 9.0 | 9.8 | 9.8 | 8.7 | 9.0 |
The Roman number after the letter G indicates the group, the letter in caps after the number indicates the subgroup (e.g. GIB indicates group IB PLA2).
mRNA Expression is based on qPCR (See “Methods”). Results are expressed as ΔCt calculated as the number of PCR cycles for lift-off for the target mRNA of interest minus the number of PCR cycles for lift-off for GAPDH mRNA (ΔCt = Ct gene of interest – Ct GAPDH). ΔCt < 10 means high to medium expression; ΔCt = 10–15 means medium to low expression, ΔCt > 15 means low expression. Data from 5 different donor are given. Mean was obtained from values of positive donors. Nd: not detected.
No mRNA was detected in any donor for GIVF (cPLA2ζ), GIIE, GIIF, GIII, GV and GXIIB.
Stimulation of macrophages with LPS for 24 hours induced on average a 7-fold increase of cPLA2-α expression (ΔCt in unstimulated macrophages: 8.42 ± 0.70, ΔCt in LPS-treated macrophages: 5.62 ± 0.38, p < 0.01). This time point was selected because preliminary experiments indicated that the effect of LPS on cPLA2-α expression was optimal at 24 hours. An increased expression of cPLA2-α induced by LPS was detectable already after 8 hours of incubation. However, at this time point the effect of LPS was not significant because of great sample variability (data not shown). LPS had no effect on iPLA2 mRNA and did not induced expression of cPLA2-ζ (data not shown). Western blot analysis of cPLA2-α in human lung macrophages confirmed the presence of cPLA2-α protein by revealing a positive signal that co-migrated with the recombinant cPLA2-α (Fig. 1). This analysis also showed a faster migrating band in macrophage lysates whose identity is not known (Fig. 1, lane 1). It should be mentioned, however, that a portion of cPLA2-α into the cells is usually phosphorylated and appears as a band-shift. Thus, it is possible that the two bands represent unphosphorylated and phosphorylated forms of cPLA2-α.
Figure 1. Western blot detection of cPLA2-α in human lung macrophages.
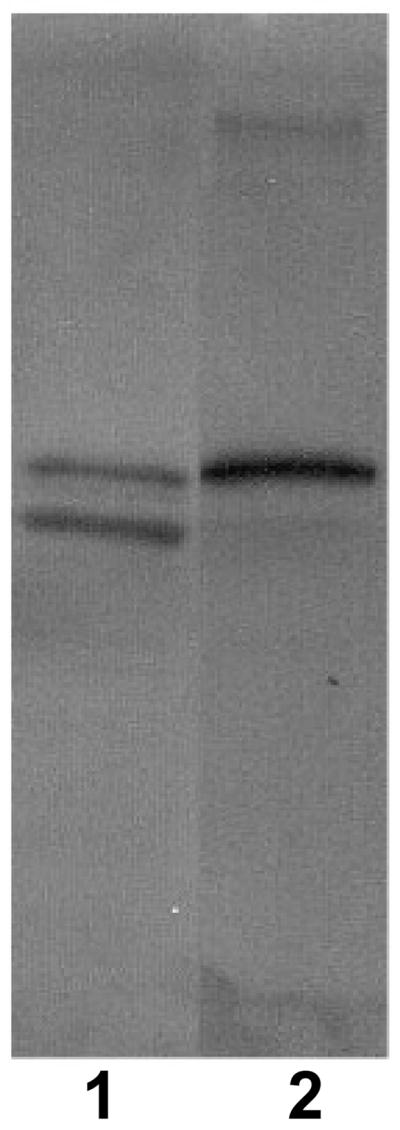
Lane 1 is macrophage lysate, and lane 2 is recombinant cPLA2-α protein. Additional details are given in Experimental Procedures.
We next examined the expression and the release of several forms of sPLA2s in both resting and LPS-activated macrophages. To this aim, we carried out qPCR for sPLA2 mRNAs and a highly sensitive time-resolved fluorescence immunoassay (TRFIA) for protein detection. In fact, western blot analysis of sPLA2s usually does not provide reliable results presumably because of the relatively low abundance of these proteins in mammalian cells. Cell lysates were analyzed for mRNA (Table I) and protein (Fig. 2) and culture medium was analyzed for protein. For human GIB (hGIB), a low level of mRNA was detected in 3 of 5 unstimulated samples. A very low level of hGIB protein was detected only in 1 of 5 unstimulated samples. Expression of this enzyme was not augmented in LPS-stimulated macrophages. A low level of human GIIA (hGIIA) mRNA was detected in all samples of unstimulated cells and it was not further increased by LPS stimulation. Interestingly, hGIIA protein was detected weakly in 2 of 5 unstimulated cells, but it was significantly induced in all samples of LPS-stimulated macrophages (Fig. 2). No release of hGIIA was detected in either unstimulated and LPS-stimulated cells. Human GIID (hGIID) was consistently detected at the mRNA level and hGIID protein was contained in 2 of 5 samples. No differences was observed between unstimulated and LPS-stimulated cells. No mRNA for human GIIE (hGIIE) and human GIIF (hGIIF) was detected in macrophages (Ct values > 40 cycles). However, the relative proteins were detected in, but not consistently released by, about 50% of samples. LPS had no effect on these sPLA2s. All samples examined were negative for human GIII (hGIII) mRNA and protein. Interestingly, mRNA for human GV (hGV) was not detected in any samples, whereas relatively high levels of hGV protein (300–2000 ng per 3×106 cells) were consistently detected in the cell lysates. Three of 5 culture medium samples also contained this enzyme (100–1200 ng per 3×106 cells). Expression and release of this sPLA2 were not influenced by LPS stimulation. Messenger RNA for human GX (hGX) was detected at low level in most samples. Two of the 5 samples contain the protein both in the cell lysates and in the culture medium. LPS had no effect on hGX mRNA expression and protein content. Finally, macrophages showed clearly detectable levels of human GXIIA (hGXIIA) mRNA and protein (500–1400 ng per 3×106 cells), but they did not release it. LPS had no effect on this sPLA2. hGXIIB mRNA was not detected and the protein was not analyzed by TRFIA.
Figure 2. Presence of sPLA2s in human lung macrophages.

Macrophage lysates were analyzed for sPLA2 protein content by time-resolved fluorescence immunoassay (See “Methods”). Approximate detection limits (pg sPLA2 per assay well) were: GIB, 10; GIIA, 20; GIID, 500; GIIE, 40; GIIF, 300; GIII, 300; GV, 60; GX, 4; GXIIA, 600. Data are expressed as ng of sPLA2/3×106 cells and are the mean ± SE of five different donors.
* p < 0.05 vs. respective unstimulated.
Together these results indicate that macrophages contain the following sPLA2s: hGXIIA > hGV > hGIID > hGIIE > hGIIF > hGX >hGIIA. These cells contain little, if any, hGIB and no hGIII. Only hGV and hGX appear to be released to a limited extent. It is interesting to note that hGIIE, hGIIF and hGV are present at the protein level, but no mRNA is detectable. LPS upregulates only the protein content of hGIIA and does not induce or enhance the release of any sPLA2s.
Effect of cPLA2 and sPLA2 inhibitors on AA release induced by non receptor-mediated agonists PMA and Ca2+-ionophore
To evaluate the role of cPLA2-α in catalyzing AA release from primary human lung macrophages, we studied AA release in the presence and absence of two highly-potent, cell permeable, and structurally-divergent inhibitors of cPLA2-α (Pyrrolidine-1 and AZ-1). The indole analog Me-Indoxam was used to study the role of extracellular sPLA2s in macrophage AA release. The structures of these inhibitors are shown in Figure 3. Me-Indoxam is a very potent inhibitor (IC50 in in vitro assays < 100 nM) of hGIIA, hGIIE and hGV [40]. Me-Indoxam is modestly potent (IC50 ~ 200 nM) on hGX and poorly inhibits (IC50 > 2,000 nM) hGIID, hGIIF, hGIII and hGXIIA [40]. Human GIB, GIIA, GIIF, GIII, GV and GX display relatively high specific activity for the hydrolysis of phospholipid vesicles, and thus, one or more of these enzymes may be involved in liberating AA from membrane phospholipids for the biosynthesis of the eicosanoids. On the other hand, hGIID, hGIIE and hGXIIA have orders of magnitude lower specific activity on phospholipid vesicles [40], suggesting that they may not be involved in lipid mediator biosynthesis; their function may be more as ligands for sPLA2 receptors rather than as lipolytic enzymes [32]. The second point about Me-Indoxam is that, unlike the cPLA2-α inhibitors, it cannot cross the plasma membrane of cells and thus, it is able to inhibit the action of the sPLA2 only if the protein is present in the extracellular environment prior to phospholipid hydrolysis [29, 30].
Figure 3.

Structures of the cPLA2-α and sPLA2 inhibitors used in this study.
In the first group of experiments, [3H]AA–labeled human lung macrophages were preincubated with increasing (0.01 μM to 10 μM) concentrations of AZ–1, pyrrolidine–1 or Me–Indoxam and then stimulated (30 min, 37°C) with PMA (1 μM) or A23187 (1 μM). These two stimuli mobilize AA through distinct mechanisms [45]. Under these conditions, the release of AA induced by PMA and A23187 in four experiments was 10.5 ± 1.1% and 12.6 ± 1.1% of total cellular AA, respectively. Figure 4 shows that preincubation with AZ–1 or pyrrolidine–1 dose–dependently inhibited the release of AA from both PMA– and A23187–stimulated human lung macrophages. In contrast, preincubation of macrophages with Me–Indoxam had little, if any effect on AA release, with less than 20% inhibition at the highest dose tested of 10 μM (Fig. 4). None of the inhibitors influenced the spontaneous release of AA from macrophages when tested up to 10 μM (unstimulated: 3.2 ± 0.5 of total cellular AA; AZ–1: 3.4 ± 0.5; pyrrolidine–1: 3.1± 0.4; Me–Indoxam: 3.2 ± 0.6). The inhibitory effect of AZ–1 and pyrrolidine–1 became significant at 0.1 μM and led to an almost complete suppression of AA release at 10 μM. In these experimental conditions, AZ–1 appeared to be more potent than pyrrolidine–1 since: 1) at the maximum concentration (10 μM) AZ–1 caused a more profound inhibition of PMA– and A23187–induced AA release (93.7 ± 0.6% and 94.8 ± 0.7%, respectively) than pyrrolidine–1 (86.3 ± 1.5% and 87.4 ± 1.0%, respectively); and 2) the IC50 values of AZ–1 (330 ± 90 nM and 310 ± 70 nM, on PMA– and A23187–induced release, respectively) were about two–fold lower than those of pyrrolidine–1 (690 ± 210 nM and 510 ± 160 nM, respectively).
Figure 4. Effect of cPLA2 and sPLA2 inhibitors on AA release from PMA-(upper panel) and A23187-stimulated (lower panel) human lung macrophages.

[3H]AA-labeled human lung macrophages were preincubated (30 min, 37°C) with increasing concentrations (0.01–10 μM) of AZ-1 (○), pyrrolidine-1 (●) or Me-Indoxam (■) and then stimulated (30 min, 37°C) with 1 μM PMA (upper panel) or A23187 (lower panel). At the end of the incubation, supernatants were collected and centrifuged twice (1000 g, 4°C, 5 min) for subsequent determination of AA release. Values are the mean ± SE of four different experiments.
* p < 0.05 vs. respective stimulus alone
** p < 0.01 vs. respective stimulus alone
PMA and A23187 have been shown to have a synergistic effect on AA mobilization [46, 47]. We therefore determined whether AZ–1 or pyrrolidine–1 were also effective inhibitors of AA release induced by a combination of the two stimuli. As expected, simultaneous stimulation of macrophages with PMA and A23187 generated a release of AA (19.5 ± 1.8% of total cellular AA) that was almost two–fold higher than that induced by the two stimuli alone. Figure 5 shows that both AZ–1 and pyrrolidine–1 effectively inhibited AA release induced by the combination of PMA and A23187. The IC50 values (280 ± 110 nM and 800 ± 230 nM for AZ–1 and pyrrolidine–1, respectively) were comparable to those obtained in the previous set of experiments when macrophages were stimulated with PMA or A23187 alone, and the results confirmed that AZ–1 was more potent than pyrrolidine–1. Me–Indoxam had no significant effect on AA release induced by PMA and A23187 in combination (Fig. 5). These results indicate that cPLA2-α is largely responsible for AA release induced by PMA and A23187 from human lung macrophages.
Figure 5. Effect of cPLA2 and sPLA2 inhibitors on AA release from PMA + A23187-stimulated human lung macrophages.

[3H]AA-labeled human lung macrophages were preincubated (30 min, 37°C) with increasing concentrations (0.01–10 μM) of AZ-1 (○), pyrrolidine-1 (●) or Me-Indoxam (■) and then stimulated with 1 μM PMA (10 min, 37°C) and subsequently with 1 μM A23187 (30 min, 37°C). At the of the incubation, supernatants were collected and centrifuged twice (1000 g, 4°C, 5 min) for subsequent determination of AA release. Values are the mean ± SE of three different experiments.
* p < 0.05 vs. PMA + A23187
** p < 0.01 vs. PMA + A23187
Effect of cPLA2-α and sPLA2 inhibitors on AA release induced by receptor-mediated agonists PPD and LPS
We next studied the effect of cPLA2-α and sPLA2 inhibitors on AA release induced by two physiological agonists of lung macrophages, PPD and LPS. PPD is the main extracellular protein product of Mycobacterium tuberculosis and it is the major antigenic component eliciting the immune response against this microorganism [48]. PPD is a complex mixture of proteins, polysaccharides, peptidoglycan and lipoarabinomannan that activates cytokine production in human monocytes presumably by interacting with Toll-like receptor-2 (TLR2) [49, 50].
The ability of PPD to induce AA mobilization in human macrophages has not been previously studied. Therefore, we initially examined whether incubation of human lung macrophages with PPD resulted in AA release. Figure 6 shows that PPD (0.3–50 μg/ml) induced a concentration–dependent release of AA from macrophages, an effect that became significant at 3 μg/ml and was maximal at 30 μg/ml (8.1 ± 1.0% of total cellular AA). In addition, since a recent report indicated that peptidoglycan or mannose-based pathogen-associated molecular patterns (PAMPs) induced AA release from human neutrophils [51], we evaluated whether the effect of PPD was due to the presence of peptidoglycan or mannose-based PAMPs. To this purpose, HLM were incubated with increasing concentrations (0.3–50 μg/ml) of PGN from Staphylococcus aureus (PGN-SA) or LAM from Mycobacterium tuberculosis. PGN-SA was used because PGN from Mycobacterium tuberculosis was not available. PGN-SA induced a concentration-dependent release of AA that was comparable to that induced by PPD (Fig. 6). By contrast, LAM did not modify the spontaneous release of AA at all the concentrations examined (Fig. 6). These results indicate that PPD-induced AA release is probably due to peptidoglycan component rather than to mannose-based PAMPs.
Figure 6. Effects of PPD, LAM and PGN-SA on AA release from human lung macrophages.

[3H]AA-labeled human lung macrophages were stimulated (1 h, 37°C) with increasing concentrations (0.3–50 μg/ml) of PPD, LAM, or PGN-SA. At the end of the incubation, supernatants were collected and centrifuged twice (1000 g, 4°C, 5 min) for subsequent determination of AA release. Values are the mean ± SE of three different experiments.
* p < 0.05 vs. unstimulated
** p < 0.01 vs. unstimulated
We next evaluated the effect of the PLA2 inhibitors on the release of AA induced by PPD. Preincubation of macrophages with AZ–1 and pyrrolidine–1 inhibited in a dose–dependent manner the release of AA induced by an optimal concentration of PPD (30 μg/ml) (Fig. 7). In contrast, Me–Indoxam had no effect on PPD–induced AA release (data not shown). Consistent with the results obtained with PMA and A23187, AZ–1 was more potent than pyrrolidine–1 to inhibit PPD–induced AA release (IC50 values of 50 ± 20 nM and 120 ± 40 nM for AZ–1 and pyrrolidine–1, respectively). In these experiments, the highest concentration of AZ–1 used (10 μM) led to a small (≈ 13%) and non significant inhibition of spontaneous AA release. The highest concentration of pyrrolidine-1 and AZ-1 used, 10 μM, led to essentially complete inhibition of AA release (Fig. 7).
Figure 7. Effect of cPLA2 inhibitors on AA release from PPD-stimulated human lung macrophages.

[3H]AA-labeled human lung macrophages were preincubated (30 min, 37°C) with increasing concentrations (0.01–10 μM) of AZ-1 or pyrrolidine-1 and then stimulated (1 h, 37°C) with 30 μg/ml PPD. At the end of the incubation, supernatants were collected and centrifuged twice (1000 g, 4°C, 5 min) for subsequent determination of AA release. Values are the mean ± SE of three different experiments.
§ p < 0.01 vs. unstimulated
* p < 0.05 vs. PPD alone
** p < 0.01 vs. PPD alone
To further explore the role of PLA2s in macrophages activated by physiological agonists, we evaluated the ability of PLA2 inhibitors to block AA release induced by LPS, the main component of the Gram-negative bacterial cell wall. LPS is a well defined stimulus for macrophages, interacting with a receptor complex which includes TLR4 and CD14 [52]. Previous studies have shown that LPS is able to induce AA mobilization from murine and human macrophages [53, 54]. Figure 8 shows that preincubation of macrophages with AZ-1 or pyrrolidine-1 dose-dependently inhibited AA release induced by LPS and completely suppressed it at 10 μM. The IC50 of AZ-1 (90 ± 30 nM) and pyrrolidine-1 (140 ± 30 nM) were comparable to those obtained for inhibition of AA release induced by PPD. The sPLA2 inhibitor Me-Indoxam had no effect on LPS-induced AA release (data not shown). Together these results indicate that the cPLA2-α is the main enzyme involved in AA mobilization induced by two independent, receptor-mediated stimuli in human macrophages.
Figure 8. Effect of cPLA2 inhibitors on AA release from LPS-stimulated human lung macrophages.

[3H]AA-labeled human lung macrophages were preincubated (30 min, 37°C) with increasing concentrations (0.01–10 μM) of AZ-1 or pyrrolidine-1 and then stimulated (2 h, 37°C) with 1 μg/ml LPS. At the end of the incubation, supernatants were collected and centrifuged twice (1000 g, 4°C, 5 min) for subsequent determination of AA release.
Values are the mean ± SE of three different experiments.
§ p < 0.01 vs. unstimulated
** p < 0.01 vs. LPS alone
Effect of cPLA2 inhibitors on PAF synthesis from human lung macrophages
Different pathways can be involved in the synthesis of PAF in macrophages [10]. PAF production may occur through the deacylation-acetylation pathway, which is dependent on PLA2–mediated hydrolysis of 1-alkyl-2-arachidonoyl-sn-glycero-3-phosphocholine, with formation of 1-alkyl-2-lyso-sn-glycero-3-phosphocholine (lyso-PAF), that is in turn acetylated to give PAF. Alternatively, lyso-PAF can be formed by PLA2-mediated hydrolysis of phosphatidylethanolamine (PE) followed by transacylation of lyso-PE by CoA-independent transacylase using 1-alkyl-2-acyl-sn-glycero-phosphocholine as the acyl donor to give lyso-PAF, which is in turn acetylated to give PAF. Finally, PAF can be synthesized by the de novo pathway that involves the transfer of phosphocholine from CDP-choline to 1-alkyl-2-acetylglycerol. The first two pathways, but not the third, are dependent on PLA2 activity.
The above mentioned experimental results show that two, structurally-distinct inhibitors of cPLA2-α blocked essentially all of the AA release in macrophages induced by multiple agonists, and thus we used these inhibitors to study the role of this enzyme in PAF production. We preincubated (30 min, 37°C) human macrophages with increasing concentrations (0.01–10 μM) of AZ-1, pyrrolidine-1 or Me-Indoxam and stimulated (10 min, 37°C) them with A23187 in the presence of [3H]acetic acid. To verify that PAF measured as incorporation of [3H]-acetate was truly 1-alkyl-2-acetyl-sn-glycero-3-phosphocholine the lipid extracts of HLM stimulated with A23187 (1 μM) were hydrolyzed by lipase A1, which cleaves 1-acyl, but not 1-alkyl linkage at the sn-1 position. Radioactivity remaining as phospholipid after lipase A1 hydrolysis was 84 ± 16%, indicating that the majority of PAF produced by A23187-stimulated HLM was 1-alkyl-2-acetyl-sn-glycero-3-phosphocholine. Both AZ–1 and pyrrolidine–1 inhibited in a dose–dependent manner PAF generation in human macrophages (Fig. 9). The sPLA2 inhibitor Me-Indoxam had no effect on A23187-induced PAF production (data not shown). It should be noted that inhibition of PAF synthesis by AZ–1 and pyrrolidine–1 was not complete and reached a maximum of approximately 70% at a concentration of inhibitors of 10 μM. There was no difference in the inhibitory effect of AZ–1 and pyrrolidine–1 on PAF synthesis (Fig. 9). The concentrations required to reach half-maximal PAF inhibition were 275 ± 92 and 290 ± 104 nM for AZ–1 and pyrrolidine–1, respectively. These results indicate that cPLA2-α activity is required for the majority of the PAF produced in response to this agonist, but suggest that about 30% of the PAF in A23187-stimulated macrophages is made by a cPLA2-α-independent process. However, we cannot rule out the possibility that cPLA2-α is required for 100% of the PAF synthesis, but that the cPLA2-α substrates that give rise to lyso-PAF have interfacial KM values for the interaction with cPLA2-α that are lower than the substrates primarily utilized for AA liberation thus leading to a higher concentration of cPLA2-α inhibitor required for complete inhibition. We refrained from using cPLA2-α inhibitor at concentrations > 10 μM because of concern about non-specific effects.
Figure 9. Effect of cPLA2 inhibitors on PAF synthesis from A23187-stimulated human lung macrophages.

Human lung macrophages were preincubated (30 min, 37°C) with increasing concentrations (0.01–10 μM) of AZ–1 (○) or pyrrolidine–1 (●) and then stimulated with 1 μM A23187 (10 min, 37°C) in the presence of [3H]acetic acid. At the end of the incubation, lipid were extracted from both cells and supernatants by the Bligh and Dyer technique. PAF was separated by TLC and quantitated by lipid scintillation counting. Values are the mean ± SE of three different experiments.
** p < 0.01 vs. A23187 alone
Effect of cPLA2-α and sPLA2 inhibitors on exocytosis in human lung macrophages
Activation of endogenous PLA2 and AA mobilization have been suggested to be required for exocytosis in human neutrophils and eosinophils [55, 56]. We therefore tested the PLA2 inhibitors for their effect on the release of β-glucuronidase, a lysosomal enzyme used as a marker of exocytosis, from human lung macrophages. In these experiments, macrophages were preincubated with AZ-1, pyrrolidine-1 or Me-Indoxam before the addition of Ca2+ ionophore A23187. Figure 10 shows that none of the inhibitors, at concentrations up to 10 μM, were able to significantly influence the release of β-glucuronidase either spontaneous or induced by A23187.
Figure 10. Effect of cPLA2 and sPLA2 inhibitors on β-glucuronidase release from A23187-stimulated human lung macrophages.

Human lung macrophages were preincubated (30 min, 37°C) with increasing concentrations (0.01–10 μM) of pyrrolidine-1, AZ-1 or Me-Indoxam and then stimulated with 1 μM A23187 (90 min, 37°C). At the of the incubation, supernatants were collected and centrifuged twice (1000 g, 4°C, 5 min). β-Glucuronidase release was determined by a colorimetric technique. The values are expressed as the percentage of the total cellular content determined in cell aliquots lysed with 0.1% Triton X-100. The data are the mean ± SE of three different experiments. § p < 0.01 vs. unstimulated.
Discussion
In this study we characterized the main isoforms of PLA2s expressed in primary human macrophages. These cells contain cPLA2-α and iPLA2, but not cPLA2-ζ. In addition, macrophages express mRNA and/or contain the protein of several sPLA2s. We also provide strong evidence that cPLA2-α is a critical enzyme for the release of AA which is then available for conversion to eicosanoids. This evidence is based on the use of two structurally-divergent cPLA2-α inhibitors, pyrrolidine-1 and AZ-1, that cause almost complete suppression of AA release induced by various stimuli. Furthermore, both cPLA2-α inhibitors reduce by 70% the generation of PAF from macrophages. In contrast, a cell-impermeable sPLA2 inhibitor, Me-Indoxam, has no effect on AA release and PAF synthesis.
A number of novel observations emerged from this study. First, human macrophages contain a wide profile of sPLA2s which include GIIA, GIID, GIIE, GIIF, GV, GX and GXIIA. Among these sPLA2s, only GIIA is upregulated by LPS, a classical stimulus to induce gene transcription in macrophages. LPS neither enhanced the other sPLA2s constitutively expressed by macrophages (GIID, GIIE, GIIF, GV, GX and GXIIA) nor induced the expression of GIB and GIII, which are undetectable in resting cells. However, LPS significantly upregulated the expression of cPLA2-α. These results are in line with previous observation in rat liver macrophages [57] and in U937 cell line [58].
An intriguing observation of this study was that some sPLA2s were detected at the protein level without measuring any mRNA signal. We previously showed by a novel mass spectrometry method that human lung macrophages contain hGV [59]. This method is unambiguous since it is based on the firm detection of a hGV-derived peptide. It is not clear why mRNA for hGV and other sPLA2s was not detected. Perhaps, the mRNA is rapidly degraded while the protein accumulates in storage granules in macrophages. Another possibility is that some species of sPLA2s, released by other cells in the lung in vivo, can be taken up by macrophages via pino/endocytosis, which is a well known phenomenon occurring in these cells [60]. Indeed, lung surfactant and alveolar fluid contain relatively large quantities of sPLA2s [61, 62]. Thus, it is conceivable that macrophages can internalize these enzymes into phagolysosomes or other intracellular compartments while they are in the lung tissue.
In spite of the large number of sPLA2s expressed in macrophages, these enzymes are poorly released from macrophages. Only GV and GX were detected in the supernatants and the rate of release was not influenced by LPS. This raises the question of the role of these enzymes as intracellular mediators. In this regard, our results, showing a clear role of cPLA2-α in AA release and PAF production, do not rule out a role for one or more sPLA2s in these processes. As noted above, Me-Indoxam is not cell permeable, and thus it is possible that an intracellular action of sPLA2s could be involved in AA release and PAF production. The fact that the cPLA2-α inhibitors block all of the AA release argues that if an intracellularly active sPLA2 is involved, it should act by augmenting the action of cPLA2-α, as shown in other cell systems [25, 28–31]. For example, Satake et al. demonstrated that mouse peritoneal macrophages from GV-deficient mice release 50% less AA in response to opsonized zymosan as compared to cells from wild type mice [25]. In these cells, Me-Indoxam has no effect on AA release (Gelb, M.H. unpublished data). Furthermore, TLR2-dependent activation of cPLA2-α and eicosanoid generation in mast cells from GV-deficient mice were markedly reduced [31]. Indeed, we have shown that the rat gastric mucosal cell line RGM1 uses both cPLA2-α and GIIA in a coordinate way to release AA leading to prostaglandin E2 formation [30]. Similar data were obtained with hGIIA transfected HEK293 cells [29]. In both these cells, GIIA acts prior to secretion into the culture medium and the failure of Me-Indoxam to block the action of this enzyme is due to the inability of the compound to cross cell membranes to come in contact with the sPLA2 in the secretory compartment [29, 30]. We are not aware of any potent sPLA2 inhibitor that is cell permeable, and efforts are underway in the author’s laboratory to remedy this problem. Until such compounds become available, we cannot reliably probe for the involvement of one or more sPLA2 in AA release from human macrophages. Attempts to knockdown GV in human lung macrophages by RNA interference were not performed since no mRNA for this enzyme could be detected by qPCR (Table 1). The present data showing no effect of Me-Indoxam on AA release and PAF production from human lung macrophages suggests that an sPLA2 in the culture medium is probably not involved.
Another novel observation in this study is the ability of PPD to induce AA release from human macrophages. This protein derivative of Mycobacterium tuberculosis has been previously shown to induce cytokine synthesis in human monocytes, the precursors of tissue macrophages [49]. Our data demonstrate that PPD also initiates the AA metabolic cascade in human macrophages and that this effect is mimicked by similar concentrations of PGN from Staphylococcus aureus, but not of LAM from Mycobacterium tuberculosis. These observations are in agreement with a recent study showing that PGN from different bacteria induces AA release from human neutrophils at concentrations of 1–30 μg/ml whereas mannose-based PAMP induced a significant release of AA at concentrations higher than 5 mg/ml [51]. Since PPD and PGN have been shown to interact with TLR2 [50, 63], it is likely that activation of cPLA2-α in macrophages occurs through the engagement of this receptor even though a role for other pattern recognition receptors cannot be ruled out. Previous data have shown that binding of LPS to TLR4 promotes AA release in murine macrophages [54]. Our data, showing that both PPD and LPS induce AA mobilization in human macrophages, indicate that cPLA2-α can be activated by engagement of different TLRs.
Interestingly, neither AZ-1 nor pyrrolidine-1 influenced basal AA release in resting macrophages, suggesting that cPLA2-α is exclusively required for agonist-induced AA release. It is possible that other PLA2s, such as the Ca2+-independent PLA2 (GVI), are primarily involved in the basal turnover of AA in quiescent macrophages [64]. The recent availability of mice deficient in this enzyme may help to resolve this issue [65], at least for murine macrophages.
Activation of cPLA2-α does not seem to be required for exocytosis in human macrophages. Studies with human neutrophils and eosinophils suggest the involvement of one or more PLA2s in degranulation [55, 56], but these studies were carried out with non-specific inhibitors of PLA2 including the highly reactive p-bromo-phenacylbromide. Studies in neutrophils with highly potent and specific PLA2 inhibitors are warranted.
The present study is the first extensive evaluation of PLA2 enzymes in human macrophages purified ex vivo. Our data, showing that the generation of AA can be efficiently suppressed by cPLA2-α inhibitors have important pharmacological implications. First, these compounds are highly effective on primary macrophages which are one of the major cellular source of lipid mediators in the lung. Second, the capacity of the cPLA2-α inhibitors to block or reduce simultaneously the production of the two major classes of lipid mediators, eicosanoids and PAF, potentially endows these molecules with a very potent anti-inflammatory activity. Finally, the observation that AZ-1 and pyrrolidine-1 are effective at submicromolar concentrations when macrophages are stimulated by physiological agonists (PPD and LPS) provides a rationale for the use of these inhibitors in the treatment of inflammatory lung diseases. It should be noted that some eicosanoids derived from cPLA2-α activity, such as PGE2, may exert antinflammatory activities and, therefore, their suppression may results in the exacerbation of lung inflammation. Thus, further studies are necessary to fully evaluate the in vivo effects of cPLA2-α inhibitors such as AZ-1 and pyrrolidine-1.
Acknowledgments
This work was supported by grants from the Ministero dell’Istruzione, dell’Università e della Ricerca (M.T. and G.M.), the Ministero della Salute “Alzheimer Project” (Rome, Italy) (G.M.), the Consiglio Nazionale delle Ricerche (Rome, Italy) (G.M.), the National Institutes of Health (HL50040 and HL36235 to M.H.G.; HL34303 to C. C. L.), the Centre National de la Recherche Scientifique (CNRS) (G.L.) and the Association pour la Recherche sur le Cancer (ARC) (G.L.). We thank Christine Payré for expert assistance in quantitative RT-PCR assays.
Abbreviations
- AA
arachidonic acid
- cPLA2
cytosolic phospholipase A2
- LAM
lipoarabinomannan from Mycobacterium tuberculosis
- LPS
lipopolysaccharide from E. coli
- PAF
platelet activating factor
- PAMPs
pathogen-associated molecular patterns
- PBS
phosphate buffered saline
- PGN-SA
peptidoglycan from Staphylococcus Aureus
- PIPES
piperazine–N,N′–bis–2–ethanesulfonic acid
- PLA2
phospholipase A2*
- PMA
phorbol myristate acetate
- PPD
Mycobacterium tuberculosis purified protein derivative
- qPCR
quantitative PCR
- sPLA2
secreted phospholipase A2
Footnotes
The Roman number after the letter G indicates the group, the letter in caps after the number indicates the subgroup (e.g. GIB indicates group IB PLA2)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Barnes PJ. Alveolar macrophages as orchestrators of COPD. Copd. 2004;1:59–70. doi: 10.1081/COPD-120028701. [DOI] [PubMed] [Google Scholar]
- 2.Peters-Golden M. The alveolar macrophage: the forgotten cell in asthma. Am J Respir Cell Mol Biol. 2004;31:3–7. doi: 10.1165/rcmb.f279. [DOI] [PubMed] [Google Scholar]
- 3.Reynolds HY. Lung inflammation and fibrosis: an alveolar macrophage-centered perspective from the 1970s to 1980s. Am J Respir Crit Care Med. 2005;171:98–102. doi: 10.1164/rccm.200406-788PP. [DOI] [PubMed] [Google Scholar]
- 4.Marriott HM, Dockrell DH. The role of the macrophage in lung disease mediated by bacteria. Exp Lung Res. 2007;33:493–505. doi: 10.1080/01902140701756562. [DOI] [PubMed] [Google Scholar]
- 5.Sica A, Allavena P, Mantovani A. Cancer related inflammation: the macrophage connection. Cancer Lett. 2008;267:204–215. doi: 10.1016/j.canlet.2008.03.028. [DOI] [PubMed] [Google Scholar]
- 6.Balter MS, Eschenbacher WL, Peters-Golden M. Arachidonic acid metabolism in cultured alveolar macrophages from normal, atopic, and asthmatic subjects. Am Rev Respir Dis. 1988;138:1134–1142. doi: 10.1164/ajrccm/138.5.1134. [DOI] [PubMed] [Google Scholar]
- 7.Triggiani M, Oriente A, Marone G. Differential roles for triglyceride and phospholipid pools of arachidonic acid in human lung macrophages. J Immunol. 1994;152:1394–1403. [PubMed] [Google Scholar]
- 8.Peters-Golden M, Henderson WR., Jr Leukotrienes. N Engl J Med. 2007;357:1841–1854. doi: 10.1056/NEJMra071371. [DOI] [PubMed] [Google Scholar]
- 9.Triggiani M, Schleimer RP, Warner JA, Chilton FH. Differential synthesis of 1-acyl-2-acetyl-sn-glycero-3-phosphocholine and platelet-activating factor by human inflammatory cells. J Immunol. 1991;147:660–666. [PubMed] [Google Scholar]
- 10.Prescott SM, Zimmerman GA, Stafforini DM, McIntyre TM. Platelet-activating factor and related lipid mediators. Annu Rev Biochem. 2000;69:419–445. doi: 10.1146/annurev.biochem.69.1.419. [DOI] [PubMed] [Google Scholar]
- 11.Schaloske RH, Dennis EA. The phospholipase A2 superfamily and its group numbering system. Biochim Biophys Acta. 2006;1761:1246–1259. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 12.Leslie CC. Regulation of the specific release of arachidonic acid by cytosolic phospholipase A2. Prostaglandins Leukot Essent Fatty Acids. 2004;70:373–376. doi: 10.1016/j.plefa.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 13.Ghosh M, Tucker DE, Burchett SA, Leslie CC. Properties of the Group IV phospholipase A2 family. Prog Lipid Res. 2006;45:487–510. doi: 10.1016/j.plipres.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 14.Mosior M, Six DA, Dennis EA. Group IV cytosolic phospholipase A2 binds with high affinity and specificity to phosphatidylinositol 4,5-bisphosphate resulting in dramatic increases in activity. J Biol Chem. 1998;273:2184–2191. doi: 10.1074/jbc.273.4.2184. [DOI] [PubMed] [Google Scholar]
- 15.Pettus BJ, Bielawska A, Spiegel S, Roddy P, Hannun YA, Chalfant CE. Ceramide kinase mediates cytokine- and calcium ionophore-induced arachidonic acid release. J Biol Chem. 2003;278:38206–38213. doi: 10.1074/jbc.M304816200. [DOI] [PubMed] [Google Scholar]
- 16.Bartoli F, Lin HK, Ghomashchi F, Gelb MH, Jain MK, Apitz-Castro R. Tight binding inhibitors of 85-kDa phospholipase A2 but not 14-kDa phospholipase A2 inhibit release of free arachidonate in thrombin-stimulated human platelets. J Biol Chem. 1994;269:15625–15630. [PubMed] [Google Scholar]
- 17.Riendeau D, Guay J, Weech PK, Laliberte F, Yergey J, Li C, Desmarais S, Perrier H, Liu S, Nicoll-Griffith D, et al. Arachidonyl trifluoromethyl ketone, a potent inhibitor of 85-kDa phospholipase A2, blocks production of arachidonate and 12-hydroxyeicosatetraenoic acid by calcium ionophore-challenged platelets. J Biol Chem. 1994;269:15619–15624. [PubMed] [Google Scholar]
- 18.Seno K, Okuno T, Nishi K, Murakami Y, Yamada K, Nakamoto S, Ono T. Pyrrolidine inhibitors of human cytosolic phospholipase A2. Part 2: synthesis of potent and crystallized 4-triphenylmethylthio derivative ‘pyrrophenone’. Bioorg Med Chem Lett. 2001;11:587–590. doi: 10.1016/s0960-894x(01)00003-8. [DOI] [PubMed] [Google Scholar]
- 19.Seno K, Okuno T, Nishi K, Murakami Y, Watanabe F, Matsuura T, Wada M, Fujii Y, Yamada M, Ogawa T, Okada T, Hashizume H, Kii M, Hara S, Hagishita S, Nakamoto S, Yamada K, Chikazawa Y, Ueno M, Teshirogi I, Ono T, Ohtani M. Pyrrolidine inhibitors of human cytosolic phospholipase A2. J Med Chem. 2000;43:1041–1044. doi: 10.1021/jm9905155. [DOI] [PubMed] [Google Scholar]
- 20.Ghomashchi F, Stewart A, Hefner Y, Ramanadham S, Turk J, Leslie CC, Gelb MH. A pyrrolidine-based specific inhibitor of cytosolic phospholipase A2 alpha blocks arachidonic acid release in a variety of mammalian cells. Biochim Biophys Acta. 2001;1513:160–166. doi: 10.1016/s0005-2736(01)00349-2. [DOI] [PubMed] [Google Scholar]
- 21.Connolly S, Bennion C, Botterell S, Croshaw PJ, Hallam C, Hardy K, Hartopp P, Jackson CG, King SJ, Lawrence L, Mete A, Murray D, Robinson DH, Smith GM, Stein L, Walters I, Wells E, Withnall WJ. Design and synthesis of a novel and potent series of inhibitors of cytosolic phospholipase A2 based on a 1,3-disubstituted propan-2-one skeleton. J Med Chem. 2002;45:1348–1362. doi: 10.1021/jm011050x. [DOI] [PubMed] [Google Scholar]
- 22.Degousee N, Ghomashchi F, Stefanski E, Singer A, Smart BP, Borregaard N, Reithmeier R, Lindsay TF, Lichtenberger C, Reinisch W, Lambeau G, Arm J, Tischfield J, Gelb MH, Rubin BB. Groups IV, V, and X phospholipases A2s in human neutrophils: role in eicosanoid production and gram-negative bacterial phospholipid hydrolysis. J Biol Chem. 2002;277:5061–5073. doi: 10.1074/jbc.M109083200. [DOI] [PubMed] [Google Scholar]
- 23.Rubin BB, Downey GP, Koh A, Degousee N, Ghomashchi F, Nallan L, Stefanski E, Harkin DW, Sun C, Smart BP, Lindsay TF, Cherepanov V, Vachon E, Kelvin D, Sadilek M, Brown GE, Yaffe MB, Plumb J, Grinstein S, Glogauer M, Gelb MH. Cytosolic phospholipase A2-alpha is necessary for platelet-activating factor biosynthesis, efficient neutrophil-mediated bacterial killing, and the innate immune response to pulmonary infection: cPLA2-alpha does not regulate neutrophil NADPH oxidase activity. J Biol Chem. 2005;280:7519–7529. doi: 10.1074/jbc.M407438200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghosh M, Loper R, Ghomashchi F, Tucker DE, Bonventre JV, Gelb MH, Leslie CC. Function, activity, and membrane targeting of cytosolic phospholipase A2zeta in mouse lung fibroblasts. J Biol Chem. 2007;282:11676–11686. doi: 10.1074/jbc.M608458200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Satake Y, Diaz BL, Balestrieri B, Lam BK, Kanaoka Y, Grusby MJ, Arm JP. Role of group V phospholipase A2 in zymosan-induced eicosanoid generation and vascular permeability revealed by targeted gene disruption. J Biol Chem. 2004;279:16488–16494. doi: 10.1074/jbc.M313748200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henderson WR, Jr, Chi EY, Bollinger JG, Tien YT, Ye X, Castelli L, Rubtsov YP, Singer AG, Chiang GK, Nevalainen T, Rudensky AY, Gelb MH. Importance of group X-secreted phospholipase A2 in allergen-induced airway inflammation and remodeling in a mouse asthma model. J Exp Med. 2007;204:865–877. doi: 10.1084/jem.20070029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Munoz NM, Meliton AY, Arm JP, Bonventre JV, Cho W, Leff AR. Deletion of secretory group V phospholipase A2 attenuates cell migration and airway hyperresponsiveness in immunosensitized mice. J Immunol. 2007;179:4800–4807. doi: 10.4049/jimmunol.179.7.4800. [DOI] [PubMed] [Google Scholar]
- 28.Han WK, Sapirstein A, Hung CC, Alessandrini A, Bonventre JV. Cross-talk between cytosolic phospholipase A2 alpha (cPLA2 alpha) and secretory phospholipase A2 (sPLA2) in hydrogen peroxide-induced arachidonic acid release in murine mesangial cells: sPLA2 regulates cPLA2 alpha activity that is responsible for arachidonic acid release. J Biol Chem. 2003;278:24153–24163. doi: 10.1074/jbc.M300424200. [DOI] [PubMed] [Google Scholar]
- 29.Mounier CM, Ghomashchi F, Lindsay MR, James S, Singer AG, Parton RG, Gelb MH. Arachidonic acid release from mammalian cells transfected with human groups IIA and X secreted phospholipase A2 occurs predominantly during the secretory process and with the involvement of cytosolic phospholipase A2-alpha. J Biol Chem. 2004;279:25024–25038. doi: 10.1074/jbc.M313019200. [DOI] [PubMed] [Google Scholar]
- 30.Ni Z, Okeley NM, Smart BP, Gelb MH. Intracellular actions of group IIA secreted phospholipase A2 and group IVA cytosolic phospholipase A2 contribute to arachidonic acid release and prostaglandin production in rat gastric mucosal cells and transfected human embryonic kidney cells. J Biol Chem. 2006;281:16245–16255. doi: 10.1074/jbc.M513874200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kikawada E, Bonventre JV, Arm JP. Group V secretory PLA2 regulates TLR2-dependent eicosanoid generation in mouse mast cells through amplification of ERK and cPLA2alpha activation. Blood. 2007;110:561–567. doi: 10.1182/blood-2006-10-052258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Valentin E, Lambeau G. Increasing molecular diversity of secreted phospholipases A2 and their receptors and binding proteins. Biochim Biophys Acta. 2000;1488:59–70. doi: 10.1016/s1388-1981(00)00110-4. [DOI] [PubMed] [Google Scholar]
- 33.Humes JL, Sadowski S, Galavage M, Goldenberg M, Subers E, Bonney RJ, Kuehl FA., Jr Evidence for two sources of arachidonic acid for oxidative metabolism by mouse peritoneal macrophages. J Biol Chem. 1982;257:1591–1594. [PubMed] [Google Scholar]
- 34.Peters-Golden M, Shelly C. Inhibitory effect of exogenous arachidonic acid on alveolar macrophage 5-lipoxygenase metabolism. Role of ATP depletion. J Immunol. 1988;140:1958–1966. [PubMed] [Google Scholar]
- 35.Laviolette M, Carreau M, Coulombe R, Cloutier D, Dupont P, Rioux J, Braquet P, Borgeat P. Metabolism of arachidonic acid through the 5-lipoxygenase pathway in normal human peritoneal macrophages. J Immunol. 1988;141:2104–2109. [PubMed] [Google Scholar]
- 36.Elias JA, Ferro TJ, Rossman MD, Greenberg JA, Daniele RP, Schreiber AD, Freundlich B. Differential prostaglandin production by unfractionated and density-fractionated human monocytes and alveolar macrophages. J Leukoc Biol. 1987;42:114–121. doi: 10.1002/jlb.42.2.114. [DOI] [PubMed] [Google Scholar]
- 37.Fathi M, Johansson A, Lundborg M, Orre L, Skold CM, Camner P. Functional and morphological differences between human alveolar and interstitial macrophages. Exp Mol Pathol. 2001;70:77–82. doi: 10.1006/exmp.2000.2344. [DOI] [PubMed] [Google Scholar]
- 38.Ferrari-Lacraz S, Nicod LP, Chicheportiche R, Welgus HG, Dayer JM. Human lung tissue macrophages, but not alveolar macrophages, express matrix metalloproteinases after direct contact with activated T lymphocytes. Am J Respir Cell Mol Biol. 2001;24:442–451. doi: 10.1165/ajrcmb.24.4.4008. [DOI] [PubMed] [Google Scholar]
- 39.Triggiani M, Giannattasio G, Balestrieri B, Loffredo S, Forte V, Granata F. Phenotypical and functional heterogeneity of human lung macrophages. Clin Exp Allergy Rev. 2004;4:129–134. [Google Scholar]
- 40.Singer AG, Ghomashchi F, Le Calvez C, Bollinger J, Bezzine S, Rouault M, Sadilek M, Nguyen E, Lazdunski M, Lambeau G, Gelb MH. Interfacial kinetic and binding properties of the complete set of human and mouse groups I, II, V, X, and XII secreted phospholipases A2. J Biol Chem. 2002;277:48535–48549. doi: 10.1074/jbc.M205855200. [DOI] [PubMed] [Google Scholar]
- 41.Hixon MS, Ball A, Gelb MH. Calcium-dependent and -independent interfacial binding and catalysis of cytosolic group IV phospholipase A2. Biochemistry. 1998;37:8516–8526. doi: 10.1021/bi980416d. [DOI] [PubMed] [Google Scholar]
- 42.Triggiani M, Granata F, Oriente A, De Marino V, Gentile M, Calabrese C, Palumbo C, Marone G. Secretory phospholipases A2 induce β-glucuronidase release and IL-6 production from human lung macrophages. J Immunol. 2000;164:4908–4915. doi: 10.4049/jimmunol.164.9.4908. [DOI] [PubMed] [Google Scholar]
- 43.Mounier CM, Wendum D, Greenspan E, Flejou JF, Rosenberg DW, Lambeau G. Distinct expression pattern of the full set of secreted phospholipases A2 in human colorectal adenocarcinomas: sPLA2-III as a biomarker candidate. Br J Cancer. 2008;98:587–595. doi: 10.1038/sj.bjc.6604184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Glover S, de Carvalho MS, Bayburt T, Jonas M, Chi E, Leslie CC, Gelb MH. Translocation of the 85-kDa phospholipase A2 from cytosol to the nuclear envelope in rat basophilic leukemia cells stimulated with calcium ionophore or IgE/antigen. J Biol Chem. 1995;270:15359–15367. doi: 10.1074/jbc.270.25.15359. [DOI] [PubMed] [Google Scholar]
- 45.Qiu ZH, Gijon MA, de Carvalho MS, Spencer DM, Leslie CC. The role of calcium and phosphorylation of cytosolic phospholipase A2 in regulating arachidonic acid release in macrophages. J Biol Chem. 1998;273:8203–8211. doi: 10.1074/jbc.273.14.8203. [DOI] [PubMed] [Google Scholar]
- 46.Werz O, Klemm J, Samuelsson B, Radmark O. Phorbol ester up-regulates capacities for nuclear translocation and phosphorylation of 5-lipoxygenase in Mono Mac 6 cells and human polymorphonuclear leukocytes. Blood. 2001;97:2487–2495. doi: 10.1182/blood.v97.8.2487. [DOI] [PubMed] [Google Scholar]
- 47.Yoss EB, Spannhake EW, Flynn JT, Fish JE, Peters SP. Arachidonic acid metabolism in normal human alveolar macrophages: stimulus specificity for mediator release and phospholipid metabolism, and pharmacologic modulation in vitro and in vivo. Am J Respir Cell Mol Biol. 1990;2:69–80. doi: 10.1165/ajrcmb/2.1.69. [DOI] [PubMed] [Google Scholar]
- 48.Ellner JJ. Regulation of the human immune response during tuberculosis. J Lab Clin Med. 1997;130:469–475. doi: 10.1016/s0022-2143(97)90123-2. [DOI] [PubMed] [Google Scholar]
- 49.Aung H, Toossi Z, Wisnieski JJ, Wallis RS, Culp LA, Phillips NB, Phillips M, Averill LE, Daniel TM, Ellner JJ. Induction of monocyte expression of tumor necrosis factor alpha by the 30-kD alpha antigen of Mycobacterium tuberculosis and synergism with fibronectin. J Clin Invest. 1996;98:1261–1268. doi: 10.1172/JCI118910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stenger S, Modlin RL. Control of Mycobacterium tuberculosis through mammalian Toll-like receptors. Curr Opin Immunol. 2002;14:452–457. doi: 10.1016/s0952-7915(02)00355-2. [DOI] [PubMed] [Google Scholar]
- 51.Valera I, Vigo AG, Alonso S, Barbolla L, Crespo MS, Fernandez N. Peptidoglycan and mannose-based molecular patterns trigger the arachidonic acid cascade in human polymorphonuclear leukocytes. J Leukoc Biol. 2007;81:925–933. doi: 10.1189/jlb.0706451. [DOI] [PubMed] [Google Scholar]
- 52.Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- 53.Brown GP, Monick MM, Hunninghake GW. Human alveolar macrophage arachidonic acid metabolism. Am J Physiol. 1988;254:C809–815. doi: 10.1152/ajpcell.1988.254.6.C809. [DOI] [PubMed] [Google Scholar]
- 54.Qi HY, Shelhamer JH. Toll-like receptor 4 signaling regulates cytosolic phospholipase A2 activation and lipid generation in lipopolysaccharide-stimulated macrophages. J Biol Chem. 2005;280:38969–38975. doi: 10.1074/jbc.M509352200. [DOI] [PubMed] [Google Scholar]
- 55.Jacobson PB, Schrier DJ. Regulation of CD11b/CD18 expression in human neutrophils by phospholipase A2. J Immunol. 1993;151:5639–5652. [PubMed] [Google Scholar]
- 56.White SR, Strek ME, Kulp GV, Spaethe SM, Burch RA, Neeley SP, Leff AR. Regulation of human eosinophil degranulation and activation by endogenous phospholipase A2. J Clin Invest. 1993;91:2118–2125. doi: 10.1172/JCI116436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dieter P, Kolada A, Kamionka S, Schadow A, Kaszkin M. Lipopolysaccharide-induced release of arachidonic acid and prostaglandins in liver macrophages: regulation by Group IV cytosolic phospholipase A2, but not by Group V and Group IIA secretory phospholipases A2. Cell Signal. 2002;14:199–204. doi: 10.1016/s0898-6568(01)00243-1. [DOI] [PubMed] [Google Scholar]
- 58.Jiang YJ, Lu B, Choy PC, Hatch GM. Regulation of cytosolic phospholipase A2, cyclooxygenase-1 and -2 expression by PMA, TNFalpha, LPS and M-CSF in human monocytes and macrophages. Mol Cell Biochem. 2003;246:31–38. [PubMed] [Google Scholar]
- 59.Lu Y, Bottari P, Turecek F, Aebersold R, Gelb MH. Absolute quantification of specific proteins in complex mixtures using visible isotope-coded affinity tags. Anal Chem. 2004;76:4104–4111. doi: 10.1021/ac049905b. [DOI] [PubMed] [Google Scholar]
- 60.Kaplan J, Ward DM. Movement of receptors and ligands through the endocytic apparatus in alveolar macrophages. Am J Physiol. 1990;258:L263–270. doi: 10.1152/ajplung.1990.258.6.L263. [DOI] [PubMed] [Google Scholar]
- 61.Arbibe L, Koumanov K, Vial D, Rougeot C, Faure G, Havet N, Longacre S, Vargaftig BB, Bereziat G, Voelker DR, Wolf C, Touqui L. Generation of lyso-phospholipids from surfactant in acute lung injury is mediated by type-II phospholipase A2 and inhibited by a direct surfactant protein A-phospholipase A2 protein interaction. J Clin Invest. 1998;102:1152–1160. doi: 10.1172/JCI3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chilton FH, Averill FJ, Hubbard WC, Fonteh AN, Triggiani M, Liu MC. Antigen-induced generation of lyso-phospholipids in human airways. J Exp Med. 1996;183:2235–2245. doi: 10.1084/jem.183.5.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dziarski R, Gupta D. Staphylococcus aureus peptidoglycan is a toll-like receptor 2 activator: a reevaluation. Infect Immun. 2005;73:5212–5216. doi: 10.1128/IAI.73.8.5212-5216.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Balsinde J, Bianco ID, Ackermann EJ, Conde-Frieboes K, Dennis EA. Inhibition of calcium-independent phospholipase A2 prevents arachidonic acid incorporation and phospholipid remodeling in P388D1 macrophages. Proc Natl Acad Sci USA. 1995;92:8527–8531. doi: 10.1073/pnas.92.18.8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bao S, Miller DJ, Ma Z, Wohltmann M, Eng G, Ramanadham S, Moley K, Turk J. Male mice that do not express group VIA phospholipase A2 produce spermatozoa with impaired motility and have greatly reduced fertility. J Biol Chem. 2004;279:38194–38200. doi: 10.1074/jbc.M406489200. [DOI] [PMC free article] [PubMed] [Google Scholar]