

Abstract
Bone morphogenetic proteins (BMPs) are members of the TGF-β superfamily responsible for mediating a diverse array of cellular functions both during embryogenesis and in adult life. Previously, we reported that upregulation of BMP7 in human melanoma correlates with tumor progression. However, melanoma cells are either inhibited by or become resistant to BMP7 as a function of tumor progression, with normal melanocytes being most susceptible. Herein, real-time quantitative reverse transcriptase-polymerase chain reactions and Western blotting revealed that the expression of BMP antagonist, Noggin, correlates with resistance to BMP7 in advanced melanoma cells. To test the hypothesis that coordinated upregulation of Noggin protects advanced melanoma cells from autocrine inhibition by BMP7, functional expression of Noggin in susceptible melanoma cells was achieved by adenoviral gene transfer. The Noggin-overexpressing cells exhibited a growth advantage in response to subsequent BMP7 transduction in vitro under anchorage-dependent and -independent conditions, in three-dimensional skin reconstructs, as well as in vivo in severe combined immune-deficiency mice. In concordance, Noggin knockdown by lentiviral shRNA confers sensitivity to BMP7-induced growth inhibition in advanced melanoma cells. Our findings suggest that, like TGF-β, BMP7 acts as an autocrine growth inhibitor in melanocytic cells, and that advanced melanoma cells may escape from BMP7-induced inhibition through concomitant aberrant expression of Noggin.
Keywords: adenoviral gene transfer, BMP antagonist, BMP inhibitor, skin reconstruct, TGF-β, tumor progression
Bone morphogenetic proteins (BMPs) are pleiotropic cytokines belonging to the TGF-β superfamily. Over 20 members of BMPs have been identified in a wide variety of organisms ranging from insects to mammals.1 Although BMPs were originally shown to induce endochondral bone formation, they are now considered as components of a highly conserved signaling pathway that controls cell growth, differentiation, apoptosis, motility, angiogenesis, and matrix synthesis not only during embryogenesis but also in adult life.2,3 Signaling by BMPs is mediated through both type I and type II transmembrane serine-threonine kinase receptors. Upon ligand binding, the constitutive type II kinase activates the type I receptor and initiates the signal transduction cascade by phosphorylating receptor-regulated “mother against decapentaplegic” (R-Smad) proteins (e.g., Smad 1, 5, and 8). Given the diversity of responses to BMP and the complexity of morphogenic events, their activities are delicately regulated by secretory antagonists (such as Noggin, Chordin, Gremlin, Sclerostin, Follistatin, DAN/Cerberus, and Glypican-3), signaling inhibitors (including SnoN, Smurf 1 and 2, and Smad 6 and 7), and pseudoreceptor BAMBI (BMP and activin membrane-bound inhibitor).4
The discovery that perturbations in BMP pathways are genetically responsible for certain hereditary cancer syndromes (such as familial juvenile polyposis and a subset of Cowden syndrome5,6) has prompted the delineation of their significance in carcinogenesis. Evidence now indicates that various sporadic human cancers also exhibit aberrations in BMP signaling, contributing to tumor development and progression.7-11 It is now clear that the actions of BMPs are cell type-specific, and that the roles of BMPs in carcinogenesis are quite complex, with divergent pro-tumor and anti-tumor effects resulting from both autocrine and paracrine responses.4 However, relatively little is know about BMP signaling in melanoma.
Recently, we4 and others12 independently reported that multiple BMPs, including BMP 2, 4, 6, 7, and 8, are upregulated in melanoma. The expression of BMP7 in particular correlates with tumor progression and disease recurrence 13, but overexpression of BMP7 paradoxically inhibits cell growth to varying degrees through G0-G1 cell cycle arrest and induction of apoptosis. Normal melanocytes are most susceptible to transduced BMP7 whereas melanoma cells are increasingly resistant with tumor progression. The resistance of melanoma cells corresponds to the expression of BMP7 antagonist, Noggin. Using adenoviral transfer, we obtained evidence that forced expression of Noggin in susceptible melanoma cells protects them from BMP7-induced growth inhibition. Furthermore, Noggin overexpressing cells exhibit a growth advantage in response to subsequent BMP7 transduction both in vitro in soft agar and three-dimensional (3-D) skin reconstructs, and in vivo in severe combined immune-deficient (SCID) mice as compared to control green fluorescence protein (GFP)-transduced counterparts. Consistent with these, lentiviral shRNA-mediated Noggin knockdown confers sensitivity to BMP7 in advanced melanoma cells. Our findings suggest that, similar to TGF-β, BMP7 functions as an autocrine growth inhibitor in melanocytic cells, and that advanced melanoma cells may escape BMP7-induced inhibition through coordinated upregulation of Noggin.
Materials and Methods
Cell Culture
The isolation and culture of normal human melanocytes was performed as previously described.14 Isogenic melanoma cell lines derived from the same patient at different disease stages were maintained as described.15,16 These consist of primary vertical growth phase melanoma cell lines WM115 and WM983A, their lymph node metastatic counterparts WM239A and WM983C, respectively. In addition, metastatic/aggressive variants selected in an experimental metastasis model in vivo, such as 1205Lu and C8161, and their parental cell lines WM793 (a vertical growth phase primary human melanoma cell line), and C81-61 (a metastatic human melanoma cell line), respectively, were also included. Normal foreskin keratinocytes and fibroblasts were isolated and propagated as previously described.17,18
Immunohistochemistry
Decoded formalin-fixed and paraffin-embedded melanoma tissue sections were obtained from the archive of the Department of Pathology at the University of Iowa. The sections were dewaxed, quenched, and incubated with a mouse monoclonal antibody raised against human BMP7 (MAB3541, R & D Systems, Minneapolis, MN) at 25 μg/ml overnight at 4 °C. The sections were then washed to remove unbound antibodies and incubated with HRP-conjugated rabbit anti-mouse secondary antibodies (R0260, DakoCytomation, Carpinteria, CA) for 30 min. at room temperature. Visualization was performed using a DAB HRP Substrate-Chromogen 2 liquid component kit (GTX 73338, GeneTex, Inc., San Antonio, TX) with hematoxylin counter stain. As a control, isotype-matched mouse Ig was used in place of the primary antibody. Experiments were repeated twice with consistency.
Semi-quantitative RT-PCR
Total RNAs were extracted from cultured melanoma cells using RNAqueous®-4PCR (Ambion, Austin, TX) according to the manufacturer's instructions. First-strand cDNA was reverse transcribed from 0.1 μg/mL of total RNA with RETROscript (Ambion) using random decamers to prime the reactions. RCR was performed with SuperTaq (Ambion) under conditions described in the package inserts using S-15 as a loading control. The expected sizes of the PCR products, the primer sequences, their annealing temperature, and number of cycles performed are summarized in Table 1. The number of cycles utilized for each set was determined to be within the linear range of amplification of the particular product. Reaction products were analyzed by electrophoresis on 2% ethidium bromide gels and the bands captured by a UVP Biochemi System (Upland, CA) and the expression normalized to the S-15 control.
Table 1.
Sequences of the primer sets and conditions for semi-quantitative RT-PCR.
| Gene | Fwd primer | Rev primer | Product length (bp) |
Annealing temperature (°C) |
Cycle number |
|---|---|---|---|---|---|
| Act-IA (Alk2) | GAGTGATGATTCTTCCTGTGC | TTGGTGGTGATGAGCCCTTCG | 511 | 55 | 25 |
| BMPR-IA (Alk3) |
CCACATCTTGGAGGAGTCGT | TGTGGTTTCTCCCTGGTCAT | 450 | 60 | 25 |
| BMPR-IB (Alk6) |
AAAGGTCGCTATGGGGAAGT | ATTGCTGGTTTGCCTTGAGT | 350 | 62 | 25 |
| BMPR-II | CTGGACAGCAGGACTTCACA | CATGCTCATCAGGACTGGAA | 450 | 62 | 25 |
| ActR-IIA | GCAAAATGAATACGAAGTCTA | GCACCCTCTAATACCTCTGGA | 435 | 60 | 25 |
| ActR-IIB | CAACTTCTGCAACGAGCGCTT | GCGCCCCCGAGCCTTGATCTC | 283 | 60 | 25 |
| Smad1 | AGGAACCAAAACATGGTGC | GTCACTAAGGCATTCGGCAT | 254 | 60 | 25 |
| Smad5 | TGATGAGGAGGAGAAATGGG | AAAGAATGTGGAAACGTGGC | 459 | 62 | 25 |
| Smad8 | ACAACGCCACCTATCCTGAC | CCCAACTCGGTTGTTCAGTT | 341 | 62 | 25 |
| Smad4 | CTGCCAACTTTCCCAACATT | GATCTCCTCCAGAAGGGTCC | 450 | 67 | 30 |
| Noggin | CACTACGACCCAGGATTCAT | CTCCGCAGCTTCTTGCTTAG | 213 | 63 | 25 |
| Chordin | AGGAAATGGCTCCCTGATCT | CTGGTATTGCCTTTCAAAGC | 351 | 53 | 25 |
| Gremlin | TCTGAGGGCAAGAGACCTGT | CTGGTATTGGCCTTTCAAGC | 345 | 53 | 25 |
| DAN/Cerberus | CCTGCCAAGGAATCAAAGAG | CAAGATGACCCCCTGAGAAG | 350 | 60 | 30 |
| Sclerostin | GGAAAGTCCAGGGACTGGTT | TGACCTTGGCACTGAATCAA | 349 | 57 | 35 |
| Follistatin | TGCCACCTGAGAAAGGCTAC | ACAGACAGGCTCATCCGACT | 201 | 63 | 30 |
| Smad7 | GCCCTCTCTGGATATCTTCT | GCTGCATAAACTCGTGGTCA | 320 | 60 | 25 |
| Smurf1 | AGAGGAATCGAAGCCCAGTT | GCGTCTATCAGGTGGATGGT | 350 | 63 | 25 |
| Smurf2 | ATCAACCGCCTCAAAGACAC | CATGTTGCACCATTTGTTCC | 350 | 63 | 25 |
| SnoN | TGAATGGGATGGGAGATGAT | CCAGGAAGAACCTGAGGTGA | 343 | 56 | 25 |
| BAMBI | AGACATCTGCCAAGCCAAAC | AAACGGGAGAGCATCTGTTG | 351 | 57 | 25 |
| Smad6 | TGAATTCTCAGACGCCAGCA | GCTCGAAGTCGAACACCTT | 386 | 50 | 30 |
Real-time quantitative RT-PCR (qRT-PCR)
Total RNA samples from subconfluent cultures of melanoma cells were prepared using RNAqueous®-4PCR (Ambion) according to the manufacturer's protocol. For each cell line, 5 μg of total RNA was reverse transcribed into cDNA in a final reaction mix of 25 μL using SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen). All reagents and probes for real-time RT-PCR were obtained from Applied Biosystems (Foster City, CA). All the probes (except the Noggin probes) used span the intron splice sites, which only detect cDNA. Real-time qRT-PCR was performed on a 7300 Real-time PCR System (Applied Biosystems) in a 25 μL reaction mix containing 1 μL cDNA, 1 x TaqMan Universal PCR Mater Mix and 1x BMP7 (Hs00233477_m1), Noggin (Hs00271352_s1), Sclerostin (Hs00401764_m1), Gremlin (Hs00171951_m1), Glypican-3 (Hs00170471_m1), Chordin (Hs00415315_m1), BAMBI (Hs00180818_m1), Smurf2 (Hs00909284_m1) or GAPDH assay (4352339E). Thermocycling was carried out at 50 °C/2 min., 95 °C/10 min., followed by 40 cycles at 95 °C for 15 sec., and 60 °C for 1 min. All samples were run in triplicate. The relative amounts of BMP inhibitor transcripts were analyzed using the 2−ΔΔCT method.19 Experiments were repeated twice with consistency.
Adenoviral vectors
The adenoviral vectors carrying the green fluorescence protein (Ad/GFP), and human BMP7 (Ad/BMP7) were obtained from the Vector Core, University of Pennsylvania (Philadelphia, PA), and Dr. R. Franceschi at the University of Michigan (Ann Arbor, MI)20, respectively. The recombinant adenovirus expressing mouse Noggin protein (Ad/Noggin) was constructed as follows. The mouse Noggin cDNA (GeneBank #MMU79163) was isolated from the plasmid pMgB950 containing the sequence kindly provided by Dr. R.M. Harland from University of California, Berkeley21,22 using NotI and BamHI digestion. The resulting mouse Noggin cDNA was subcloned into the pacAd5CMVK-NpA recombinant adenoviral vector backbone23 (obtained from the Gene Therapy Center Vector Core at the University of Iowa) to generate a mouse Noggin proviral plasmid pAd.CMV-mNoggin. Recombinant adenovirus was generated and amplified from this proviral plasmid as previously described23 and titered using Adeno-X™ Rapid Titering Kit (Clontech Laboratories, Inc., Mountain View, CA).
Adenoviral Infection of melanoma cells
Subconfluent melanoma cells were transduced with 10 plaque forming units (pfu)/cell of replication deficient adenoviruses for 2 h at 37 °C in a minimum amount of serum-free Dulbecco's modified Eagle's medium sufficient to cover the culture vessels. The optimal multiplicity of infection (M.O.I) was previously determined as the minimum amount of virus required to yield the highest overall gene transfer efficiency without apparent cytotoxicity.18,24 Viral suspensions were then replaced by regular growth medium and cells were incubated overnight. When indicated, subsequent infection with a second adenoviral vector may be performed. After viral infection, cells were allowed to recover for at least 16 h before use. The high efficiency of gene transfer (over 95%)18,24 eliminates the need for selection.
Enzyme-linked Immunosorbant Assay (ELISA)
Twenty-four h after viral transfection of melanocytic cells, the growth medium is replaced with serum-free medium consisting of MCDB153/L15 (v/v: 4/1), CaCl2 (2 mmol/L), and insulin (5 μg/ml) and incubated for 24 h. The supernatant was then collected, volume-measured, and cleared by centrifugation. BMP7 in tissue culture supernatant was quantified in triplicate wells using the human BMP7 DuoSet ELISA Development kit (DY354, R & D Systems) according to the manufacturer's protocol. For VEGF quantification, equal amount of cell lysate (refer to Western blotting for cell lysate preparation) from each sample was added in duplicate wells and ELISA was performed using the human VEGF Quantikine kit (R&D Systems). The results (expressed as amount of secreted BMP7/ml/106 cells/24 h and amount of VEGF/100 μg/ml lysate, respectively) from one representative experiment were shown, however, assays were repeated twice with consistency.
Western blotting
Subconfluent cultures were washed with phosphate buffered saline (PBS), and extracted in lysis buffer containing 1% Triton X-100, 1% deoxycholic acid, 2 mmol/L CaCl2, and protease inhibitors (10 μg/ml leupeptin, 10 μg/ml aprotinin, 1.8 mg/ml iodoacetamide, and 1 mmol/L phenylmethyl sulfonyl fluoride) in PBS. Cell lysates were quantified by a BCA protein assay kit (Pierce, Rockford, IL). An equal amount (50-100 μg) of total protein from each sample was subjected to electrophoresis on NuPAGE 4-12% Bis-Tris gels (Invitrogen), transblotted onto nitrocellulose membranes (Pierce), and probed with primary antibodies, such as anti-phospho Smad 1, 5, 8 (Cell Signaling, Beverly, MA), anti-Noggin (RP57-16) 25,26, anti-bFGF (Abcam, Cambridge, MA), anti-Cripto-1 (R&D Systems), and anti-Nodal (Chemicon, Temecula, CA) antibodies, followed by a peroxidase-conjugated secondary antibody (Pierce). Immunoreactive bands were detected using SuperSignal West Femto Chemiluminescent Substrate (Pierce). Subsequent re-probing using anti-β-actin or anti-tubulin (Abcam) was also performed for internal loading control. Experiments were performed at least twice with consistency.
Conventional anchorage-dependent growth assay
Subconfluent cultures were trypsinized and seeded in 35-mm wells at 1-4 × 105 cells/well. Cells were refed twice weekly. At given intervals, cells in quadruplicate wells were harvested and counted in a Coulter counter (Coulter Electronics, Luton, England). Statistical analyses were performed using the Mann-Whitney U test. Experiments were repeated twice with similar results.
Soft agar assay
Melanoma cells were suspended in MCDB153/L15 medium (v/v: 4/1) supplemented with 25 μg/ml bovine pituitary extract, 2 ng/ml epidermal growth factor (EGF), 2 μg/ml insulin, 4% fetal bovine serum, and 0.25% agar and plated in triplicate at 6 × 104 cells/well in 6-well plates. After 2 weeks, colonies were counted using an inverted microscope. Mann-Whitney U test was used for statistical analyses. Experiments were repeated twice and similar results obtained. Data presented represent results from one experiment.
Cell growth and invasion in 3-D skin reconstructs
Skin reconstructs were prepared as previously described. 17,18,27 Briefly, the growth and invasion of melanoma cells was tested in artificial skin reconstructs, in which human foreskin dermal fibroblasts in rat tail type I collagen were placed on a precast acellular collagen gel. After 6 days, the constricted collagen gels formed a concave surface, serving as a cradle for seeding epidermal cells. Melanoma cells were then mixed with keratinocytes at a 1:5-10 ratio and seeded onto the dermal constructs. After 5 days, cultures were lifted to the air-liquid level for an additional 10 days to allow stratification of epidermal keratinocytes. The reconstructs were then harvested, fixed in paraformaldehyde, embedded in paraffin, sectioned, and stained with hematoxylin and eosin. Apoptosis was evaluated using the Apo-BrdU-IHC™ In Situ DNA Fragmentation Assay Kit (BioVision, Inc., Mountain View, CA). For each condition, triplicate wells were evaluated and experiments were repeated twice with similar results.
Cell cycle analysis
Twenty-four h after viral infection, melanoma cells were maintained at less than 70% confluence for 3 days. Cells were then harvested and fixed with 70% ethanol at 4 °C for 1 h. After washing with PBS, cells were stained with 50 μg/ml of propidium iodide in PBS containing RNase A (0.5 mg/ml), Tris HCl (0.5 mM), and NaCl (0.75 mM) for 30 min. at 4 °C in the dark, and analyzed on flow cytometer at the University of Iowa Flow Cytometry Core Facility. Data shown represent results from one experiment, however, the assay was performed twice with similar results.
Apoptosis detection by anti-phospho histone H2B
Forty-eight h after viral infection, melanoma cells were harvested and stained for 40 min with 20 μg/ml of anti-phospho histone H2B (Upstate, Lake Placid, NY), an early marker of apoptosis, at 4 °C, in triplicates. After removal of excessive primary antibodies, the cells were incubated with Cy3-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) and then analyzed by fluorescence-activated cell sorting (FACS) at the University of Iowa Flow Cytometry Core Facility. Statistical analyses were performed using Mann-Whitney U test. Data shown represent results from one experiment, however, the assay was performed twice with similar results.
In vivo tumorigenicity
Subconfluent melanoma cells were sequentially transduced with different combinations of Ad/GFP, Ad/Nog and Ad/BMP7 at 10 pfu/cell with a 24 h interval between infections. Sixteen h after the second viral infection, cells were harvested and suspended in serum-free medium at a density of 108 cells/ml. One hundred μL of cell suspension were injected subcutaneously in the dorsal skin of each SCID mouse (7 mice/condition). Tumor volume was monitored twice a week and determined as follows: (maximum dimension × minimum dimension)2/2. Statistical analyses were performed using ANNOVA following log transformation. The mice were sacrificed 14 days after injection. Tumors were dissected, weighed, fixed in formalin, and subject to histopathologic examination.
Noggin knockdown in melanoma cells by lentiviral shRNA
Recombinant lentiviral vectors were generated by co-transfecting pLKO.1-Noggin (Sigma), harboring shRNA against human Noggin, or non-target control shRNA (Sigma) with packaging plasmids VSVg and pCMV-ΔR8.2 (Sigma) into 293T packaging cells using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's instructions. Culture supernatants containing recombinant lentiviral particles were used to infect melanoma cells (1205Lu and C8161). Two days after infection, cells were selected with puromycin (1 μg/ml) for a period of 7 d.
Results
BMP7 expression in melanoma correlates with tumor progression
Taking advantage of the isogenic cell lines (Materials and Methods) derived from the same patient at different disease stages as well as aggressive variants selected in an experimental metastasis model in vivo,15 BMP7 mRNA expression was found to correlate with tumor progression using real-time qRT-PCR (Fig. 1A; a normal melanocyte culture (FOM103) was included as baseline control). Cell lines derived from primary melanomas (WM115, WM793, and WM983A) exhibited very low copies of BMP7 transcripts, while their metastatic counterparts (WM239A, 1205Lu, and WM983C) expressed abundant BMP7 mRNA (Fig. 1A). One exceptional metastatic melanoma cell pair, C81-61/C8161, however, displayed low levels of BMP7 transcripts (Fig. 1A). Immunohistochemistry confirmed upregulation of BMP7 protein expression in human melanoma tissue (Fig. 1B-F), using kidney sections as a positive control, since collecting duct tubules have been shown previously to express BMP7.28 Immunoreactivity was detected in primary (Fig. 1B) as well as metastatic melanoma samples, including metastases to lymph node, cutaneous, brain, and bone (Fig. 1C-F). Thus, BMP7 expression correlates with tumor progression and the observed upregulation in aggressive melanoma cells in vitro is biologically relevant and does not represent a tissue culture artifact.
Figure 1.

Expression of BMP7 correlates with melanoma progression in melanoma cell lines by qRT-PCR using normal melanocyte (FOM103) as control (A) and in situ by immunohistochemistry (B-F). Four isogenic cell pairs were analyzed by qRT-PCR, including: WM115 (primary melanoma, ■)/WM239A (metastatic melanoma, □); WM793 (primary melanoma, ■)/1205Lu (metastatic melanoma, □); WM983A (primary melanoma, ■)/WM983C (metastatic melanoma, □); and C81-61 (metastatic melanoma, ■)/C8161 (highly aggressive metastatic melanoma selected in experimental metastasis model in vivo, □). Paraffin sections of primary cutaneous melanoma (Magnification: 200X) and a panel of metastatic lesions in different organ sites (magnification: 400X), including skin (C), bone (D), brain (E), and lymph node (F), were subjected to immunohistochemistry as described in Materials and Methods. Comparing to the corresponding negative controls (inserts), BMP7 expression is evident in melanoma cells.
Adenoviral gene transfer results in functional secretion of biologically active BMP7 transgene product in melanoma cells
To investigate the biological consequences of BMP7 upregulation in melanoma progression, overexpression of the transgene in melanoma cell pairs (WM793/1205Lu and C81-61/C8161) was achieved using recombinant adenovirus. Analysis of the culture supernatants using ELISA revealed that the BMP7-transduced cells produced approximately 1000 ng of BMP7/ml/106 cells/24 h, while their mock- or GFP-transduced counterparts exhibited low/undetectable endogenous levels of BMP7 (Fig. 2A). As expected, BMP7 overexpression resulted in increased R-Smad phosphorylation/activation by Western blotting, comparing to the control GFP-transduced cells, except in the highly aggressive metastatic melanoma cell line C8161 (Fig. 2B). Screening by semi-quantitative RT-PCR demonstrated that melanoma cells express all six known BMP receptors and their downstream signaling machinery, such as Smads (data not shown). The expression of BMPR IB (Alk6) and BMP RII at the protein level was also confirmed by Western blotting (data not shown). These data suggest that virally induced BMP7 is biologically active and functions as an autocrine activator of R-Smad phosphorylation in melanoma cells.
Figure 2.

Biological consequences of functional expression of BMP7 in melanoma cells by adenoviral gene transfer. A. Secretion of transduced BMP7 by ELISA. Culture supernatants of transduced melanoma cells were subjected to ELISA using those of mock- and GFP-transduced cells as control. The levels of BMP7 expression were comparable among different cell lines averaging at ∼1000 ng/ml/106 cells/24 h. B. Transduced BMP7 activates R-Smad phosphorylation as determined by Western blotting. Using an Ab specific for phosphorylated Smads 1, 5, and 8, upregulation of phosphorylated R-Smads following BMP7 transduction in melanoma cells was observed with one exception (C8161 cells). The finding indicates that transduced BMP7 is biologically active and can function as an autocrine in melanoma by eliciting the Smad signaling cascade. C. Transduced BMP7 inhibits melanoma growth to different degrees in vitro in conventional culture. Early/less aggressive melanoma cells (WM793 and C81-61) were susceptible, demonstrating greater than 70% growth retardation in response to BMP7, while their highly aggressive variants (1205Lu and C8161) were relatively resistant with less than 50% inhibition. C8161, in particular, was completely resistant. Asterisks indicate statistically significant differences (p<0.05). D. Propidium iodide stain revealed that BMP7 transduction resulted in an increased percentage of cells resting at G0-G1 (solid column) comparing to that in Ad/GFP- and mock-infected cells. BMP7 drastically induced G0-G1 cell cycle arrest in susceptible melanoma cells (WM793 and C81-61), modestly in relatively resistant 1205Lu melanoma cells, and barely in resistant C8161 cells. E. BMP7 transduction induced early maker of apoptosis, phospho-H2B, in melanoma cells by flow cytometry. Comparing to GFP-transduced melanoma cells, a significant population of BMP7-transduced cells expressed phospho-H2B, particularly in WM793 and C81-61 cells. Asterisks indicate statistically significant differences (p<0.05).
Autocrine effects of BMP7 on melanoma growth, invasion, and motility
Adenoviral gene transfer of BMP7 led to differential growth inhibition in melanoma cells of different stages of tumor progression. Primary (WM793) and less aggressive metastatic (C81-61) melanoma cells were susceptible with greater than 70% growth inhibition (Fig. 2C), whereas their metastatic (1205Lu) and highly aggressive (C8161) counterparts are relatively resistant (Fig. 2C). C8161, in particular, was completely resistant.
To explore the underlying mechanisms of BMP7-mediated growth inhibition in melanoma cells, we performed cell cycle analysis using propidium iodide. We found that BMP7 transduction results in G0-G1 cell cycle arrest. Interestingly, the extent to which BMP7 induces G0-G1 cell cycle arrest correlates with the resistance to growth inhibition by BMP7. In BMP7-sensitive early/less aggressive melanoma cells, such as WM793 and C81-61, the percentage of resting cell population raises drastically from ∼35% in GFP control construct-transduced cells to 70% in BMP7-transduced cells, while the relatively resistant 1205Lu cells exhibit a modest increase (from ∼50% to ∼64%) and the resistant C8161 melanoma cells show no significant difference (Fig. 2D). In addition, when the cells were stained with an early marker for apoptosis, anti-phospho Histone2B, the majority of BMP7-transduced cells undergo apoptosis (Fig. 2E). The degree of BMP7-induced apoptotic cell death also correlates with sensitivity to growth inhibition by BMP7. BMP7-sensitive early/less aggressive variants (WM793 and C81-61) display over 90% positive cells following BMP7 transduction, comparing to ∼30% in GFP-transduced cells, whereas their relatively BMP7-resistant, more aggressive counterparts (1205Lu and C8161) show only ∼40-55% positivity following BMP7 tranduction, comparing to ∼40% in GFP-tranduced cells (Fig. 2E). No changes in phospho Histone 2B expression were observed between the GFP- and BMP7-transfected resistant cell line, C8161 (Fig. 2E).
Since various BMPs have been shown to contribute to tumor progression through stimulating cell motility and invasion,7,29-31 we tested whether BMP7 enhances melanoma migration and invasion in vitro. Using an in vtro scratch migration assay and time-lap video recording, we found no significant difference in cell migration between Ad/BMP7- and Ad/GFP-transduced melanoma cells (data not shown). In addition, the cells behaved similarly in a modified Boyden chamber assay (data not shown).
Characterization of BMP7-transduced melanoma cells in three-dimensional (3D) skin reconstructs
To determine the biological consequences of BMP7 transduction in melanoma cells in an appropriate tissue context, a 3D skin reconstruct model is used to test the invasive capacity as well as the growth characteristics of these cells. This model consists of a dermal compartment containing fibroblasts in a collagen gel separated from an epidermal compartment composed of melanocytic cells and keratinocytes by a naturally deposited basement membrane,27 enabling functional studies of individual genes in a biologically relevant milieu.
Ad/GFP-transduced VGP primary melanoma cells WM793 grow as nests and solitary units within the epidermis and occasionally in the superficial dermis (Fig. 3A), while their BMP7-transduced counterparts display only remnants of small clusters as well as single cells at the dermal-epidermal junction and superficial dermis. Free 3′-OH ends resulting from DNA fragmentation and indicative of apoptotic cell death, are detected in these cells using the Apo-BrdU-IHC™ In Situ DNA Fragmentation Assay Kit (BioVision; Fig 3F). Control GFP vector transduced aggressive 1205Lu melanoma cells traverse the basement membrane and grow deeply into the dermis, forming invasive tumor nests (Fig. 3C), whereas, their Ad/BMP7-infected counterparts show dermal tumor nesting with morphological evidence of apoptosis (Fig. 3D and E), such as nuclear condensation and formation of apoptotic bodies. Similar results were obtained using C81-61 metastatic melanoma cells (Fig. 3G and H). However, when we incorporate highly aggressive C8161 metastatic melanoma cells, which are shown to be resistant to BMP7-mediated autocrine inhibition in the traditional two-dimensional culture (Fig. 2C), both the control Ad/GFP- and Ad/BMP7-infected cells grow aggressively into the dermis and eventually partially replace the epidermis (Fig. 3I and J). These data suggested that consistent with the results obtained from the conventional monolayer culture, BMP7 is growth-inhibitory in melanoma cells and that advanced/aggressive melanoma cells are progressively resistant.
Figure 3.
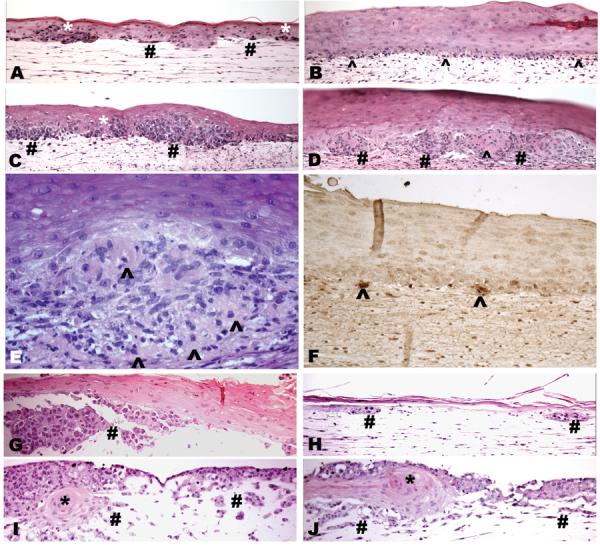
Effects of transduced BMP7 on melanoma growth and invasion in 3D skin reconstructs. In susceptible primary melanoma cell line WM793, Ad/GFP-infected cells grow as large nests and single cells within the epidermis (*) and superficial dermis (#; A). Upon BMP7-transduction, WM793 cells show small clusters as well as single units at the dermal epidermal junction (B, ^) reactive to TUNNEL stain (F, ^). GFP tranfectants of the relative resistant isogenic cell line 1205Lu grow aggressively forming large tumor nests in the deep dermis (C, #), while the BMP7-transfectants (D, #) displayed extensive cell death (^) with morphological hallmarks of apoptosis (D and E, ^). Control GFP-transfected susceptible metastatic C81-61 melanoma cells grow aggressively as large nests and nodules within the dermis (G, #), whereas their BMP7-transduced counterparts only show small remnants of dermal tumor nests (H, #). In the resistant, highly aggressive C8161 melanoma cells, both Ad/GFP- (I) and Ad/BMP7-infected cells (J) grow aggressively into the dermis (#) and eventually also replace the epidermis with rare residual keratinocyte islands (I, J, *).
Advanced/aggressive melanoma cells escape growth inhibition by BMP7 through concomitant upregulation of BMP antagonist, Noggin
Using semi-quantitative RT-PCR, initial screening indicate that the resistance to induced BMP7 in advanced/aggressive melanoma cells correlates with upregulation of BMP antagonist, Noggin, but not Dan/Cerberus, Follistatin, Sclerostin, Gremlin, Chordin, Glypican-3 (GPC3), Smurf 1 and 2, SnoN, Smad6, Smad7, or BAMBI). 4 Real-time qRT-PCR (Fig. 4A) and Western blotting (Fig. 4B) further confirmed these observations. It is worth noting that BMP7 transduction did not induce Smad phosphorylation in the highly aggressive metastatic melanoma cell line C8161 (Fig. 2B), which exhibited abundant Noggin transcripts (∼104 fold increase comparing to its less aggressive parental cell line C81-61, Fig. 4A), consistent with the known antagonist function of Noggin.
Figure 4.

Noggin mediates resistance to BMP7. Noggin upregulation correlates with melanoma progression and resistance to BMP7 by qRT-PCR (A) and Western blotting (B). Isogenic cell pairs: 1. WM115 (primary melanoma, ■)/WM239A (metastatic melanoma, □); 2. WM793 (primary melanoma, ■)/1205Lu (metastatic melanoma, □); 3. WM983A (primary melanoma, ■)/WM983C (metastatic melanoma, □); 4. C81-61 (metastatic melanoma, ■)/C8161 (highly aggressive metastatic melanoma, □). C. Infection of susceptible melanoma cells with Noggin-expressing adenovirus and analysis by Western blotting (G: Ad/GFP-infected cells, N: Ad/Noggin-infected cells). D. Noggin expression blocks BMP7-induced phosphorylation of R-Smads. (G: Ad/GFP-infected cells, B: Ad/BMP7-infected cells, N+G: Ad/Noggin and Ad/GFP double infected cells, N+B: Ad/Noggin and Ad/BMP7 double infected cells.)
To test the hypothesis that advanced/aggressive melanoma cells escape growth inhibition by BMP7 through coordinated upregulation of Noggin, we first overexpressed Noggin in susceptible melanoma cells in an attempt to rescue them from BMP7-mediated growth inhibition. Forced functional expression of Noggin was achieved via adenoviral gene transfer. Western blotting confirmed the presence of the transgene product at the protein level (Fig. 4C) and the transduced Noggin was effective in blocking BMP7-induced Smad signaling (Fig. 4D). Conventional growth assays revealed that pre-infection with Ad/Nog protects susceptible melanoma cells from subsequently induced BMP7 (Fig. 5A).
Figure 5.

Noggin expression confers BMP7 resistance in susceptible melanoma cells in vitro. A. Conventional growth assay. Isogenic melanoma cell pairs (WM793/ 1205Lu, and C81-61/C8161) were sequentially infected with different combinations of adenoviral vectors and analyzed for growth rate in conventional culture conditions. BMP7 expression in control Ad/GFP-infected susceptible melanoma cell lines (WM793, and C81-61) results in marked growth inhibition, while in their Ad/Noggin-infected counterparts, cell growth is restored to the same rate as control Ad/GFP and Ad/GFP- double infected cells. Similar rescuing effect of Noggin is also observed in the relatively resistant 1205Lu cells. The resistant, highly aggressive melanoma cell line, C8161 remains resistant regardless of Noggin rescue. Asterisks indicate statistical significance (p<0.05). B. Colony formation in soft agar. Twenty-four h following double infection with different combinations of adenoviral vectors, melanoma cells were resuspended in 0.25% agar and seeded in triplicate wells at 6 × 104 cells/well. Two weeks later, the colony-forming efficiency was determined as the percentage of cells forming colonies containing four or more cells. Ten random fields were examined for each condition. Data represent mean ± SD. Asterisks indicate significant difference (p<0.05). Representative micrographs of individual colonies of 1205Lu and C8161 are also shown. C. 3D skin reconstructs. Twenty-four h following double infection using various combinations of adenoviral vectors, susceptible WM793 primary melanoma cells were incorporated into 3D skin reconstructs. While Ad/GFP and Ad/BMP7-double infected cells result in scattered apoptotic cells at the dermal-epidermal junction and superficial dermis (^), the Ad/Noggin and Ad/BMP7-double infected cells grow as large tumor nests (*) within the epidermis and focally in the superficial dermis, a pattern similar to those of Ad/GFP and Ad/GFP-, and Ad/Noggin and Ad/GFP-infeected control.
In soft agar assays, Noggin transduction in BMP7-susceptible melanoma cells restores colony formation (Fig. 5B). In 3D skin reconstructs, Noggin transduction rescues WM793 primary vertical growth phase melanoma cells from BMP7-induced apoptotic cell death, leading to tumor growth in the superficial dermis, at the dermal-epidermal junction, and within the epidermis (Fig. 5C). Similar rescue is also observed in other melanoma cell lines (WM164, 1205Lu, and C81-61, data not shown). In addition, in tumorigenicity assays in SCID mice, Noggin transduction, as expected, protects melanoma cells from BMP7-mediated growth inhibition (Fig. 6). At 17 days post subcutaneous injection, the tumors derived from Nog/BMP7-transduced 1205Lu melanoma cells measure 4 times larger in size and weigh twice as much as those from GFP- and BMP7-double transduced counterparts (Fig. 6A and B). Routine histology examination of the tumors revealed that the Ad/GFP- and Ad/BMP7-double infected cells induce ectopic bone formation at the periphery of the tumors (Fig. 6C, left panel), consistent with the known osteogenic function of BMP7, while the Nog/BMP7-infected cells grow as large, partially encapsulated, subcutaneous nodules without evidence of heterotropic ossification (Fig. 6C, right panel).
Figure 6.

Ectopic Noggin expression rescues tumorigenicity of Ad/BMP7-infected melanoma cells in vivo. Sixteen h after double infection, 1205Lu melanoma cells were injected subcutaneously in to SCID mice. Tumor sizes were monitored for 17 days (A) before the tumors were harvested, weighed (B), and processed for routine histology (C). Hematoxylin and Eosin stained slides show that the Ad/GFP and Ad//BMP7-double infected cells induce ectopic bone formation at the periphery of the tumor with central necrosis (*), while the Ad/Noggin and Ad/BMP7-double infected cells grow as large cutaneous nodules in delicate fibrous capsules (arrow heads) without evidence of heterotropic ossification.
To test the hypothesis that Noggin knockdown in advanced/resistant melanoma cells confers sensitivity to BMP7-induced growth inhibition, we generated stable Noggin knockdown 1205Lu and C8161 cell lines using the lentiviral shRNA approach. As shown in Fig. 7A by Western blotting, over 75% knockdown efficiency was achieved (using cells infected by lentiviral vector containing control shRNA as control). In conventional monolayer growth assays, both 1205Lu and C8161 Noggin knockdown variants exhibit increased sensitivity to BMP7, compared to their non-target control shRNA counterparts (Fig. 7B).
Figure 7.

Noggin knockdown in advanced melanoma cells confers sensitivity to BMP7-induced growth inhibition. A. Noggin knockdown by lentiviral shRNA. Stable cell lines expressing lentiviral shRNA against human Noggin and non-target control were subjected to Western blotting using tubulin as internal loading control. Above 75% Noggin knockdown efficiency is achieved as determined by densitometry in advanced 1205Lu and C8161 melanoma cells. B. Noggin knockdown renders advanced melanoma cells more sensitive to BMP7-induced growth inhibition. Upon infection by Ad/BMP7, 1205Lu and C8161 Noggin knockdown melanoma cells (□) show greater degrees of growth retardation compared to the their non-target control counterparts (○). Asterisks indicate statistically significant difference.
To explore the possibility that Noggin may restore growth in BMP7-transduced melanoma cells indirectly through induction of other growth factors, we examined the expression of potential melanoma growth-promoting factors, such as bFGF, Nodal, Cripto-1 (by Western blotting), and VEGF (by ELISA) following Noggin overexpression (Fig. 8). We found that Noggin overexpression upregulates Nodal and VEGF in one (WM793/1205Lu) but not the other isogenic melanoma cell pairs. This suggests that Noggin rescue of melanoma growth in response to BMP7 may in part be attributed to induction of Nodal and VEGF in some but not all melanoma cell lines.
Figure 8.
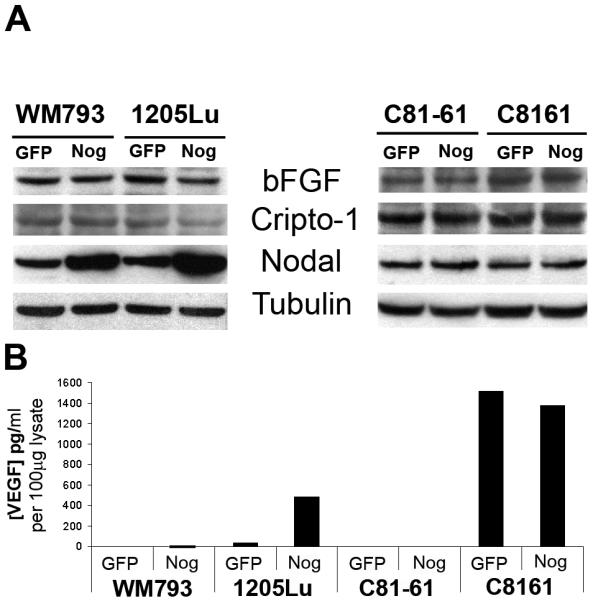
Effects of Noggin on melanoma growth promoting factors by Western blotting (A) and ELISA (B). Noggin transduction upregulates Nodal and VEGF in one isogenic melanoma cell pair WM793/1205Lu but not the other C81-61/C8161.
Discussion
Although originally identified by their capacity to induce endochondral bone formation, BMP signaling pathways have now been shown to play critical roles in a diverse array of non-osteogenic processes.4 The discovery that perturbations in BMP signaling are genetically responsible for certain familial cancer syndromes, such as familial juvenile polyposis, has stimulated active interests in delineating the functional significance of BMPs in tumor development and progression. Consistent with this, reactivation of developmental/morphogenetic signaling pathways such as Hedgehog, Wnt, and Notch, has also recently been implicated in tumorigenesis. It is postulated that such activations may lead to or result from tumor dedifferentiation towards a stem cell-like phenotype.32
Despite the substantial progress achieved during the past years, relatively little is known about BMP signaling in melanocytic cells. Using semi-quantitative RT-PCR, we previously screened expression of BMPs in melanocytic cells4 and reported that the expression of BMP7, in particular, correlates with tumor aggressiveness in vitro. We have now verified our previous observation by real-time qRT-PCR (Fig. 1A). The biological relevance of BMP7 upregulation was further confirmed in situ by immunohistochemistry on melanoma tissue sections (Fig. 1B-F). Aberrant BMP7 expression during tumor progression is not unique to melanoma. High levels of BMP7 have also been detected in bone metastasis of prostate cancer.33 Studies have demonstrated that prostate carcinoma cells produce increasing amounts of BMPs as they progress to a more aggressive phenotype and that the upregulation of BMP7 expression in metastatic cells is a critical component of developing osteoblastic lesions. In addition, BMP7 is also upregulated in breast carcinoma and the expression paradoxically correlates with differentiation markers, such as estrogen and progesterone receptors.34 On the contrary, nephroblastoma cells exhibit downregulation of BMP7.35 The apparent divergent regulation of BMP7 in different human cancers may reflect the cell type-specific actions of individual BMPs.
BMP7-mediated growth regulation has been extensively studied in carcinogenesis. The findings have been conflicting with divergent effects of both growth stimulation and inhibition. 36,37,38 To investigate the biological consequences of BMP7 in human melanoma, we overexpressed the transgene using adenoviral gene transfer, which led to differential growth inhibition in traditional monolayer culture (Fig. 2C), as well as in 3D skin reconstructs (Fig. 3). When comparing isogenic cell lines, biologically advanced, aggressive melanoma cells (1205Lu, and C8161) are less responsive, whereas their biologically early (WM793) or less aggressive counterparts (C81-61) appear more sensitive. Although the level of transduced BMP7 by adenoviral vectors may be superphysiologic, it allow for augmentation of the biological read-out to gain insights into BMP7 function in melanoma as the actions of cytokines can be subtle and difficult to appreciate.39
While not a main focus of our study, we found expression of all 6 known BMP receptors as well as their signal transduction proteins in melanoma cells using semi-quantitative RT-PCR. In addition, gene sequencing also revealed no evidence of mutations or deletions in BMP receptors (data not shown). Taken together, these results indicate an intact and functional autocrine BMP pathway in melanoma cells, with the manifestation of growth suppression in biologically early cells, but resistance in late aggressive cells.
We further explored the mechanisms through which BMP7 expression exerts its growth inhibitory activities in susceptible melanoma cells. Cell cycle analyses revealed that ectopic expression of BMP7 resulted in G0-G1 cell cycle arrest (Fig. 2D). In addition, a significant portion of BMP7-transduced cells expressed phospho-Histone2B, an early marker for apoptotic cells, by flow cytometry (Fig. 2E). Apoptotic death in BMP7-transduced cells was further confirmed by a TUNEL stain (Fig. 3F). Consistent with our findings, involvement of BMP7 in cell cycle control and apoptosis regulation has been previously described. BMP7 has been shown to induce apoptosis in myeloma cells 36 and to inhibit proliferation of androgen-insensitive prostate cancer cells, e.g. PC-3 and DU-145, both in vitro and in vivo through G0-G1 cell cycle arrest.40 In contrast, BMP7 promotes cell survival under serum deprivation in androgen-sensitive LNCaP and C4-2B prostate cancer cell lines.41 The seemingly disparate observations in prostate cancer suggest that the biological responses to BMP7 in a given cell type may depend on cross-talk with other signaling pathways.42
In contrast to what has been previously reported, where BMP4 and BMP7 were shown to enhance cell invasion and migration in melanoma,12 and prostate as well as colon cancer cell lines,30,31,41 respectively, we did not observe similar activities in BMP7-transduced melanoma cells in in vitro scratch migration and modified Boyden chamber assays. Consistent with this, BMP7-transduced susceptible melanoma cells retained their ability to invade the artificial dermis in our 3D skin reconstruct model even though the cells exhibited evidence of apoptosis (Fig. 3B and D). This is not surprising given that the biological responses of individual BMPs are cell- and tissue context-specific.43
One possible mechanism through which melanoma cells may bypass the antiproliferative effect elicited by BMP7 is the disruption of BMP signaling through BMP inhibitors. Using semi-quantitative RT-PCR with confirmation by real-time qRT-PCR, it appears that the resistance to induced BMP7 in advanced/aggressive melanoma correlates with upregulation of BMP antagonist, Noggin4 (Fig. 4A). To test the hypothesis that concurrent upregulation of Noggin protects advanced/aggressive melanoma cells from growth retardation by BMP7, we investigated the consequences of Noggin overexpression in susceptible melanoma cells, as well as those of Noggin knockdown in resistant melanoma cells, in response to induced BMP7. We found that overexpression of Noggin conferred BMP7 resistance in susceptible melanoma cells not only in vitro in conventional monolayer growth assays (Fig. 5A), soft agar clonogenicity assays (Fig. 5B), and 3D skin reconstructs (Fig. 5C), but also in vivo in experimental animals (Fig. 6). In conventional monolayer cultures, Noggin knockdown confers sensitivity to BMP7 in resistant melanoma cells (Fig. 7). Using Western blotting and ELISA, we also found that Noggin upregulates melanoma growth –promoting factors, such as Nodal and VEGF in a subset of but not all melanoma cell lines (Fig. 8). These suggest that the observed restoration of growth by Noggin may in part be attributed to the indirect effect of Nodal and VEGF induction.
There are ample examples in which tumor cells harbor aberrant expression of BMP signaling inhibitors that contribute to tumorigenesis and progression. For instance, Chordin, which reduces the motility of the tumor cells, is downregulated in ovarian cancer cells.44 In esophageal squamous cell carcinoma, Smurf2 expression correlates with poor prognosis.45 Loss of GPC3 was also noted in a significant portion of ovarian and breast cancers46. Furthermore, its restoration inhibited colony-forming potential suggesting that GPC3 acts as a negative growth regulator in these tumors.47 In contrast, overexpression of GPC3 was demonstrated in embryonal tumors,48 colon cancer,49 hepatocellular carcinoma,50 and melanoma.51,52 Analogous to Noggin counteracting the autocrine inhibition of BMP7 in melanoma, upregulation of GPC3 in hepatocellular carcinoma has also been shown to modulate the growth inhibitory effect of BMP7.37 However, unlike Noggin, GPC3 expression does not correlate with melanoma progression.51
In summary, two key events associated with BMP7 signaling take place during melanoma development and progression: 1) the acquisition of the ability to express increased levels of BMP7 and 2) the development of resistance to the autocrine inhibition by BMP7 through concomitant upregulation of antagonist, Noggin. Given that BMP7 is growth inhibitory in human melanoma, it remains puzzling as to why the malignant cells secrete such a factor without apparent autocrine benefits. There are a few possible explanations. First, the degree of growth suppression by endogenous BMP7 may be moderate and thus easily overcome by other intrinsic/extrinsic pro-proliferative signals. Second, interactions with other signaling pathways may modulate the growth inhibitory action of BMP7 in melanoma. 53-55 Finally, BMP7 may offer paracrine stimulation for melanoma cells in the tumor microenvironment. Indeed, recent studies indicated that BMPs may contribute to tumor progression through stromal induction, such as promoting angiogenesis.43,56,57 Thus, like TGF-β, an accepted “double-edged sword” in tumorigenesis, BMPs may function both as oncogenes and tumor suppressors depending on the relative dosage and disease stage.4 Moreover, it remains to be determined whether inhibition of Noggin upregulation may unmask the antiproliferative effects of BMP7, a potentially novel translational strategy for melanoma therapy. It is for this reason that further investigation is warranted to bridge the gap between our current knowledge of BMP7 signaling in melanoma and its potential as a therapeutic target.
Supplementary Material
Acknowledgments
We are grateful to Drs. Meenhard Herlyn (Wistar Institute, Phliadelphia, PA) and Mary J.C. Hendrix (Northwestern University, Chicago, IL) for their generous gifts of melanoma cell lines. Dr. Renny T. Franceschi (University of Michigan, Ann Arbor, MI) is acknowledged for providing Ad/BMP7. This work is supported by start-up funds from Department of Pathology and the Holden Comprehensive Cancer Center, University of Iowa, Iowa City, IA, Department of Pathology, Brigham and Women's Hospital, Boston (M-Y. H), MA, and NIH grants, DK047967 (J.F.E.), and HL084815, CA93683, and AR42689 (G.F.M.).
Abbreviations
- 3-D
three-dimensional
- Ad
Adenovirus
- BMP
bone morphogenetic protein
- ELISA
enzyme-linked immunosorbant assay
- GFP
green fluorescence protein
- GPC3
glypican-3
- HRP
horse reddish peroxidase
- Ig
immunoglobulin
- PFU
plaque forming unit
- qRT-PCR
quantitative reverse transcriptase-polymerase chain reaction
- SCID
severe combined immune deficiency
- VGP
vertical growth phase
Footnotes
The authors have no duality of interest to declare.
Reference
- 1.Balemans W, Van Hul W. Extracellular regulation of BMP signaling in vertebrates: a cocktail of modulators. Dev Biol. 2002;250:231–250. [PubMed] [Google Scholar]
- 2.Whitman M. Smads and early developmental signaling by the TGFbeta superfamily. Genes Dev. 1998;12:2445–2462. doi: 10.1101/gad.12.16.2445. [DOI] [PubMed] [Google Scholar]
- 3.Botchkarev VA. Bone morphogenetic proteins and their antagonists in skin and hair follicle biology. J Invest Dermatol. 2003;120:36–47. doi: 10.1046/j.1523-1747.2003.12002.x. [DOI] [PubMed] [Google Scholar]
- 4.Hsu MY, Rovinsky S, Penmatcha S, et al. Bone morphogenetic proteins in melanoma: angel or devil? Cancer Metastasis Rev. 2005;24:251–263. doi: 10.1007/s10555-005-1575-y. [DOI] [PubMed] [Google Scholar]
- 5.Howe JR, Sayed MG, Ahmed AF, et al. The prevalence of MADH4 and BMPR1A mutations in juvenile polyposis and absence of BMPR2, BMPR1B, and ACVR1 mutations. J Med Genet. 2004;41:484–491. doi: 10.1136/jmg.2004.018598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Waite KA, Eng C. From developmental disorder to heritable cancer: it's all in the BMP/TGF-beta family. Nat Rev Genet. 2003;4:763–773. doi: 10.1038/nrg1178. [DOI] [PubMed] [Google Scholar]
- 7.Langenfeld EM, Calvano SE, Abou-Nukta F, et al. The mature bone morphogenetic protein-2 is aberrantly expressed in non-small cell lung carcinomas and stimulates tumor growth of A549 cells. Carcinogenesis. 2003;24:1445–1454. doi: 10.1093/carcin/bgg100. [DOI] [PubMed] [Google Scholar]
- 8.Kim IY, Lee DH, Lee DK, et al. Restoration of bone morphogenetic protein receptor type II expression leads to a decreased rate of tumor growth in bladder transitional cell carcinoma cell line TSU-Pr1. Cancer Res. 2004;64:7355–7360. doi: 10.1158/0008-5472.CAN-04-0154. [DOI] [PubMed] [Google Scholar]
- 9.Horvath LG, Henshall SM, Kench JG, et al. Loss of BMP2, Smad8, and Smad4 expression in prostate cancer progression. Prostate. 2004;59:234–242. doi: 10.1002/pros.10361. [DOI] [PubMed] [Google Scholar]
- 10.Wen XZ, Miyake S, Akiyama Y, et al. BMP-2 modulates the proliferation and differentiation of normal and cancerous gastric cells. Biochem Biophys Res Commun. 2004;316:100–106. doi: 10.1016/j.bbrc.2004.02.016. [DOI] [PubMed] [Google Scholar]
- 11.Helms MW, Packeisen J, August C, et al. First evidence supporting a potential role for the BMP/SMAD pathway in the progression of oestrogen receptor-positive breast cancer. J Pathol. 2005;206:366–376. doi: 10.1002/path.1785. [DOI] [PubMed] [Google Scholar]
- 12.Rothhammer T, Poser I, Soncin F, et al. Bone morphogenic proteins are overexpressed in malignant melanoma and promote cell invasion and migration. Cancer Res. 2005;65:448–456. [PubMed] [Google Scholar]
- 13.Rothhammer T, Wild PJ, Meyer S, et al. Bone morphogenetic protein 7 (BMP7) expression is a potential noval prognostic marker for recurrence in patients with primary melanoma. Cancer Biomark. 2007;3:111–117. doi: 10.3233/cbm-2007-3205. [DOI] [PubMed] [Google Scholar]
- 14.Hsu M-Y, Li L, Herlyn M. Cultivation of normal human epidermal melanocytes in the absence of phorbol esters. In: Picot J, Totowa NJ, editors. Human Cell Culture Protocols. Humana Press; 2004. pp. 13–28. [DOI] [PubMed] [Google Scholar]
- 15.Hsu M-Y, Elder DE, Herlyn M. Melanoma: the Wistar melanoma (WM) cell lines. In: Masters JRW, Palsson B, editors. Human Cell Culture. 1st ed. Kluwer Academic Publishers; Nowell, MA: 1999. pp. 259–274. [Google Scholar]
- 16.van der Schaft DW, Seftor RE, Seftor EA, et al. Effects of angiogenesis inhibitors on vascular network formation by human endothelial and melanoma cells. J Natl Cancer Inst. 2004;96:1473–1477. doi: 10.1093/jnci/djh267. [DOI] [PubMed] [Google Scholar]
- 17.Hsu MY, Shih DT, Meier FE, et al. Adenoviral gene transfer of beta3 integrin subunit induces conversion from radial to vertical growth phase in primary human melanoma. Am J Pathol. 1998;153:1435–1442. doi: 10.1016/s0002-9440(10)65730-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsu MY, Meier FE, Nesbit M, et al. E-cadherin expression in melanoma cells restores keratinocyte-mediated growth control and down-regulates expression of invasion-related adhesion receptors. Am J Pathol. 2000;156:1515–1525. doi: 10.1016/S0002-9440(10)65023-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 20.Franceschi RT, Wang D, Krebsbach PH, et al. Gene therapy for bone formation: in vitro and in vivo osteogenic activity of an adenovirus expressing BMP7. J Cell Biochem. 2000;78:476–486. doi: 10.1002/1097-4644(20000901)78:3<476::aid-jcb12>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 21.Smith WC, Harland RM. Expression cloning of noggin, a new dorsalizing factor localized to the Spemann organizer in Xenopus embryos. Cell. 1992;70:829–840. doi: 10.1016/0092-8674(92)90316-5. [DOI] [PubMed] [Google Scholar]
- 22.Zimmerman LB, De Jesus-Escobar JM, Harland RM. The Spemann organizer signal noggin binds and inactivates bone morphogenetic protein 4. Cell. 1996;86:599–606. doi: 10.1016/s0092-8674(00)80133-6. [DOI] [PubMed] [Google Scholar]
- 23.Anderson RD, Haskell RE, Xia H, et al. A simple method for the rapid generation of recombinant adenovirus vectors. Gene Ther. 2000;7:1034–1038. doi: 10.1038/sj.gt.3301197. [DOI] [PubMed] [Google Scholar]
- 24.Hsu M, Andl T, Li G, et al. Cadherin repertoire determines partner-specific gap junctional communication during melanoma progression. J Cell Sci. 2000;113:1535–1542. doi: 10.1242/jcs.113.9.1535. [DOI] [PubMed] [Google Scholar]
- 25.Valenzuela DM, Economides AN, Rojas E, et al. Identification of mammalian noggin and its expression in the adult nervous system. J Neurosci. 1995;15:6077–6084. doi: 10.1523/JNEUROSCI.15-09-06077.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marcelino J, Sciortino CM, Romero MF, et al. Human disease-causing NOG missense mutations: effects on noggin secretion, dimer formation, and bone morphogenetic protein binding. Proc Natl Acad Sci U S A. 2001;98:11353–11358. doi: 10.1073/pnas.201367598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meier F, Nesbit M, Hsu MY, et al. Human melanoma progression in skin reconstructs : biological significance of bFGF. Am J Pathol. 2000;156:193–200. doi: 10.1016/S0002-9440(10)64719-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang SN, Lapage J, Hirschberg R. Loss of tubular bone morphogenetic protein-7 in diabetic nephropathy. J Am Soc Nephrol. 2001;12:2392–2399. doi: 10.1681/ASN.V12112392. [DOI] [PubMed] [Google Scholar]
- 29.Scherberich A, Tucker RP, Degen M, et al. Tenascin-W is found in malignant mammary tumors, promotes alpha8 integrin-dependent motility and requires p38MAPK activity for BMP-2 and TNF-alpha induced expression in vitro. Oncogene. 2005;24:1525–1532. doi: 10.1038/sj.onc.1208342. [DOI] [PubMed] [Google Scholar]
- 30.Ye L, Lewis-Russell JM, Kynaston H, et al. Endogenous bone morphogenetic protein-7 controls the motility of prostate cancer cells through regulation of bone morphogenetic protein antagonists. J Urol. 2007;178:1086–1091. doi: 10.1016/j.juro.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 31.Grijelmo C, Rodrigue C, Svrcek M, et al. Proinvasive activity of BMP-7 through SMAD4/src-independent and ERK/Rac/JNK-dependent signaling pathways in colon cancer cells. Cell Signal. 2007;19:1722–1732. doi: 10.1016/j.cellsig.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 32.Taipale J, Beachy PA. The Hedgehog and Wnt signalling pathways in cancer. Nature. 2001;411:349–354. doi: 10.1038/35077219. [DOI] [PubMed] [Google Scholar]
- 33.Masuda H, Fukabori Y, Nakano K, et al. Increased expression of bone morphogenetic protein-7 in bone metastatic prostate cancer. Prostate. 2003;54:268–274. doi: 10.1002/pros.10193. [DOI] [PubMed] [Google Scholar]
- 34.Alarmo EL, Rauta J, Kauraniemi P, et al. Bone morphogenetic protein 7 is widely overexpressed in primary breast cancer. Genes Chromosomes Cancer. 2006;45:411–419. doi: 10.1002/gcc.20307. [DOI] [PubMed] [Google Scholar]
- 35.Higinbotham KG, Karavanova ID, Diwan BA, et al. Deficient expression of mRNA for the putative inductive factor bone morphogenetic protein-7 in chemically initiated rat nephroblastomas. Mol Carcinog. 1998;23:53–61. doi: 10.1002/(sici)1098-2744(199810)23:2<53::aid-mc1>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 36.Ro TB, Holt RU, Brenne AT, et al. Bone morphogenetic protein-5, -6 and -7 inhibit growth and induce apoptosis in human myeloma cells. Oncogene. 2004;23:3024–3032. doi: 10.1038/sj.onc.1207386. [DOI] [PubMed] [Google Scholar]
- 37.Midorikawa Y, Ishikawa S, Iwanari H, et al. Glypican-3, overexpressed in hepatocellular carcinoma, modulates FGF2 and BMP-7 signaling. Int J Cancer. 2003;103:455–465. doi: 10.1002/ijc.10856. [DOI] [PubMed] [Google Scholar]
- 38.Pouliot F, Blais A, Labrie C. Overexpression of a dominant negative type II bone morphogenetic protein receptor inhibits the growth of human breast cancer cells. Cancer Res. 2003;63:277–281. [PubMed] [Google Scholar]
- 39.Valtieri M, Tocci A, Gabbianelli M, et al. Enforced TAL-1 expression stimultaes primitive, erythroid and megakaryocytic progenitors but blocks the granulopoeitic differentiation program. Cancer Res. 1998;58:562–569. [PubMed] [Google Scholar]
- 40.Miyazaki H, Watabe T, Kitamura T, et al. BMP signals inhibit proliferation and in vivo tumor growth of androgen-insensitive prostate carcinoma cells. Oncogene. 2004;23:9326–9335. doi: 10.1038/sj.onc.1208127. [DOI] [PubMed] [Google Scholar]
- 41.Yang S, Zhong C, Frenkel B, et al. Diverse biological effect and Smad signaling of bone morphogenetic protein 7 in prostate tumor cells. Cancer Res. 2005;65:5769–5777. doi: 10.1158/0008-5472.CAN-05-0289. [DOI] [PubMed] [Google Scholar]
- 42.Ide H, Yoshida T, Matsumoto N, et al. Growth regulation of human prostate cancer cells by bone morphogenetic protein-2. Cancer Res. 1997;57:5022–5027. [PubMed] [Google Scholar]
- 43.Langenfeld EM, Langenfeld J. Bone morphogenetic protein-2 stimulates angiogenesis in developing tumors. Mol Cancer Res. 2004;2:141–149. [PubMed] [Google Scholar]
- 44.Moll F, Millet C, Noel D, et al. Chordin is underexpressed in ovarian tumors and reduces tumor cell motility. Faseb J. 2006;20:240–250. doi: 10.1096/fj.05-4126com. [DOI] [PubMed] [Google Scholar]
- 45.Fukuchi M, Fukai Y, Masuda N, et al. High-level expression of the Smad ubiquitin ligase Smurf2 correlates with poor prognosis in patients with esophageal squamous cell carcinoma. Cancer Res. 2002;62:7162–7165. [PubMed] [Google Scholar]
- 46.Xiang YY, Ladeda V, Filmus J. Glypican-3 expression is silenced in human breast cancer. Oncogene. 2001;20:7408–7412. doi: 10.1038/sj.onc.1204925. [DOI] [PubMed] [Google Scholar]
- 47.Lin H, Huber R, Schlessinger D, et al. Frequent silencing of the GPC3 gene in ovarian cancer cell lines. Cancer Res. 1999;59:807–810. [PubMed] [Google Scholar]
- 48.Saikali Z, Sinnett D. Expression of glypican 3 (GPC3) in embryonal tumors. Int J Cancer. 2000;89:418–422. [PubMed] [Google Scholar]
- 49.Lage H, Dietel M, Froschle G, et al. Expression of the novel mitoxantrone resistance associated gene MXR7 in colorectal malignancies. Int J Clin Pharmacol Ther. 1998;36:58–60. [PubMed] [Google Scholar]
- 50.Man XB, Tang L, Zhang BH, et al. Upregulation of Glypican-3 expression in hepatocellular carcinoma but downregulation in cholangiocarcinoma indicates its differential diagnosis value in primary liver cancers. Liver Int. 2005;25:962–966. doi: 10.1111/j.1478-3231.2005.01100.x. [DOI] [PubMed] [Google Scholar]
- 51.Nakatsura T, Kageshita T, Ito S, et al. Identification of glypican-3 as a novel tumor marker for melanoma. Clin Cancer Res. 2004;10:6612–6621. doi: 10.1158/1078-0432.CCR-04-0348. [DOI] [PubMed] [Google Scholar]
- 52.Motomura Y, Senju S, Nakatsura T, et al. Embryonic stem cell-derived dendritic cells expressing glypican-3, a recently identified oncofetal antigen, induce protective immunity against highly metastatic mouse melanoma, B16-F10. Cancer Res. 2006;66:2414–2422. doi: 10.1158/0008-5472.CAN-05-2090. [DOI] [PubMed] [Google Scholar]
- 53.Kraunz KS, Nelson HH, Liu M, et al. Interaction between the bone morphogenetic proteins and Ras/MAP-kinase signalling pathways in lung cancer. Br J Cancer. 2005;93:949–952. doi: 10.1038/sj.bjc.6602790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakashima A, Katagiri T, Tamura M. Cross-talk between Wnt and bone morphogenetic protein 2 (BMP-2) signaling in differentiation pathway of C2C12 myoblasts. J Biol Chem. 2005;280:37660–37668. doi: 10.1074/jbc.M504612200. [DOI] [PubMed] [Google Scholar]
- 55.Litsiou A, Hanson S, Streit A. A balance of FGF, BMP and WNT signalling positions the future placode territory in the head. Development. 2005;132:4051–4062. doi: 10.1242/dev.01964. [DOI] [PubMed] [Google Scholar]
- 56.Dai J, Kitagawa Y, Zhang J, et al. Vascular endothelial growth factor contributes to the prostate cancer-induced osteoblast differentiation mediated by bone morphogenetic protein. Cancer Res. 2004;64:994–999. doi: 10.1158/0008-5472.can-03-1382. [DOI] [PubMed] [Google Scholar]
- 57.Raida M, Clement JH, Leek RD, et al. Bone morphogenetic protein 2 (BMP-2) and induction of tumor angiogenesis. J Cancer Res Clin Oncol. 2005;131:741–750. doi: 10.1007/s00432-005-0024-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.