

Abstract
The active site electronic structure of the azide complex of substrate-bound human heme oxygenase-1, (hHO) has been investigated by 1H NMR spectroscopy to shed light on the orbital/spin ground state as an indicator of the unique distal pocket environment of the enzyme. 2D 1H NMR assignments of the substrate and substrate-contact residue signals reveal a pattern of substrate methyl contact shifts, that places the lone iron π-spin in the dxz orbital, rather than the dyz orbital found in the cyanide complex. Comparison of iron spin relaxivity, magnetic anisotropy and magnetic susceptibilities argues for a low-spin, (dxy)2(dyz,dxz)3, ground state in both azide and cyanide complexes. The switch from singly-occupied dyz for the cyanide to dxz for the azide complex of hHO is shown to be consistent with the orbital hole determined by the azide π-plane in the latter complex, which is ∼90° in-plane rotated from that of the imidazole π-plane. The induction of the altered orbital ground state in the azide relative to the cyanide hHO complex, as well as the mean low-field bias of methyl hyperfine shifts and their paramagnetic relaxivity relative to those in globins, indicate that azide exerts a stronger ligand field in hHO than in the globins, or that the distal H-bonding to azide is weaker in hHO than in globins. The Asp140 → Ala hHO mutant that abolishes activity retains the unusual WT azide complex spin/orbital ground state. The relevance of our findings for other HO complexes and the HO mechanism is discussed.
Keywords: Hyperfine shift, spin ground state, orbital ground state, heme oxygenase, H-bonding
Heme oxygenase, (1-3) HO1, is a non-metal enzyme that uses protoheme, PH, as both substrate and cofactor to generate biliverdin, iron and CO via three intermediates, mesohydroxyheme, verdoheme and iron(III)-biliverdin, as shown in Figure 1. The biliverdin product of the majority of HOs is further reduced to bilirubin by biliverdin reductase. In mammals, the ∼300 residue enzyme is membrane bound, and the three products have key roles, respectively as precursors to the antioxidant, bilirubin (4), in iron homeostasis (5), and as potential neural messenger (6). A smaller (∼200 residue) soluble HO is found in plants and photosynthetic bacteria (7) where the product biliverdin is converted to light-harvesting pigments, and in numerous pathogenic bacteria, (3, 8-10) where its prime role is to secure iron from the host. While the HOs exhibit variable sequence homology, (9) their structures exhibit a new, but remarkably conserved, α-helical fold. (11-16) The distal HO pocket is unique in possessing a cluster of ordered water molecules organized within an extended H-bond network with some much stronger than usual H-bonds. (12, 13, 16-22) Two prominent features of the HO reaction are that the ring cleavage is highly stereospecific, (3, 10, 23) and that the ferryl species resulting from heterolytic O-O bond cleavage, which is responsible for oxidation by cytochromes P450 and peroxidases, is catalytically incompetent in HOs (23-25).
Figure 1.
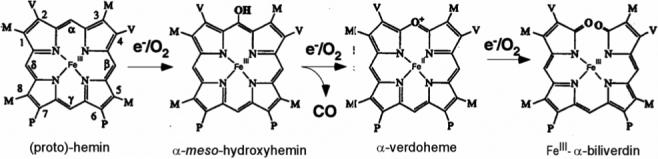
Sequence of intermediates in the heme oxygenase mechanism.
Control of heme stereoselectivity is rationalized by steric blocking by the distal helix of three of the meso-positions, and steric tilt/orientation of the exogenous ligand toward the fourth, unblocked position; differential stereoselectivity is determined primarily by differential heme seating in the active site. (2, 3, 10-16, 26, 27) While the majority of HOs are α-meso-selective, the HO from the opportunistic pathogeneic bacterium Pseudomonas aeruginosa, (28) PaHO, is unusual in that the product is a mixture of β- and δ-biliverdin. Initial mechanistic studies favored electrophilic attack on a meso position by Fe+3-OOH (3, 23-25), but computational results suggested that this would involve an insurmountably high energy barrier and instead proposed (29-31)a tethered free-radical mechanism where the heme-bound O-O bond is homolytically cleaved. The destabilization of heterolytic O-O bond cleavage in HO has been attributed to H-bonding to the iron ligand by a set of conserved, ordered water molecules. (3, 12, 13, 16, 17, 29-31) In the mammalian HO, these water molecules are H-bonded to distal Asp140 whose mutagenesis (32, 33) perturbs the ordered water molecule arrangement and abolishes HO activity, yielding the inactive ferryl unit.
The preference for homolytic over heterolytic O-O bond cleavage is unique to HOs and early efforts were directed to searching for a unique spectroscopic signature of the active site of HOs that could be related to an active site environment distinct from those of the more common heme proteins. However, uv-visible, resonance Raman, epr and, until recently, (34, 35) 1H NMR spectra of HO complexes all were comparable to those for the same derivatives of a globin. (2, 36-42) NMR spectra of paramagnetic hemoprotein derivatives provide unique information through their hyperfine shifts (42, 43) that reflect on functionally relevant structural information not readily gleaned from either crystal structures or other spectroscopic methods. Low-spin ferrihemoprotein complexes exhibit substrate contact shift patterns that reflect the pattern of delocalized spin density. (42, 43) For the majority of S = 1/2 ferrihemoproteins with relatively planar hemin, the lone spin resides in the π-bonding dyz or dxz orbital, which, for the geometry depicted in Figure 2A, can be delocalized into the symmetry-adapted 3eπ(yz) (spin distribution in Figure 3A) or 3eπ(xz) (spin distribution in Figure 3B) porphyrin molecular orbitals. (42, 44, 45) For the extensively investigated His/CN−-ligated ferrihemoproteins, the orbital ground state (singly occupied dxz or dyz) is determined by the axial His imidazole plane orientation, (44) as defined by the angle φH relative to the porphyrin based x-axis (Figure 2A). For α-meso-selective2 HOs (i.e., with the substrate orientation depicted in Figure 2A), φH ∼0, and the lone spin in dyz delocalizes into 3eπ(yz) with large predicted, (44) and observed (18, 19, 22, 41, 46) contact shifts for porphyrin positions 2−, 3−, 6−, and 7 (Figure 3).
Figure 2.
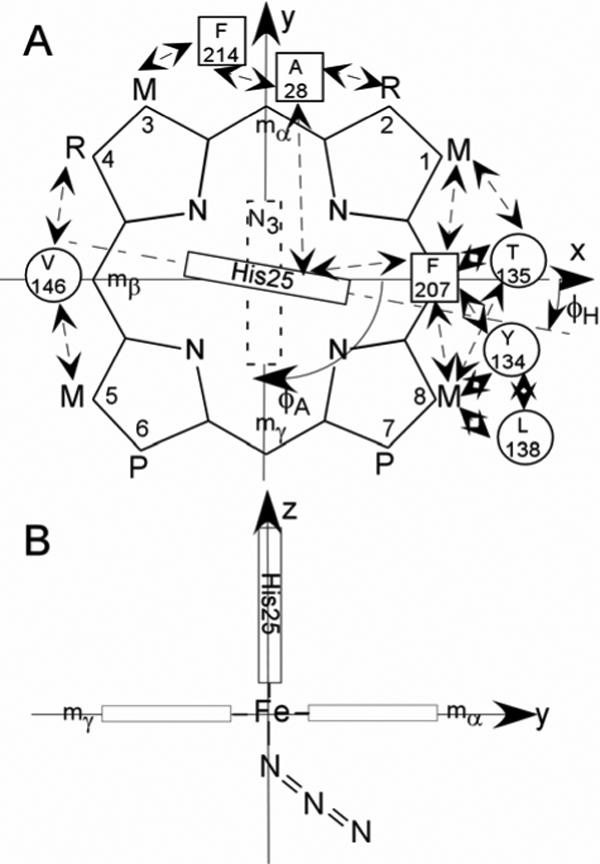
Structure of the (A) substrate, native protohemin, (PH; R = vinyl), and two-fold symmetric 2,4-dimethyldeuterohemin (DMDH; R = CH3). The coordinate system for the iron d orbitals has the x and y-axes passing through the β-,δ-meso positions (and His25 imidazole plane) and α-,γ-meso positions (azide π-plane), respectively, for the substrate orientation of the α-meso-selective hHO. The substrate contact residues are shown as squares and circles for proximal and distal residues, respectively. The orientations of the proximal His imidazole and distal azide π-planes are shown as solid and dashed rectangles, respectively; the angles between the x-axis and the proximal His imidazole and distal azide π-planes are φH and φA, respectively. Contacts expected from the crystal structures, (11, 13) and observed by 1H NMR for WT hHO-DMDH-N3, are shown by double-sided arrows; (B) edge-on view from the positive x-axis that depicts the expected Fe-N3 orientation, observed in the complex of rat HO. (62)
Figure 3.
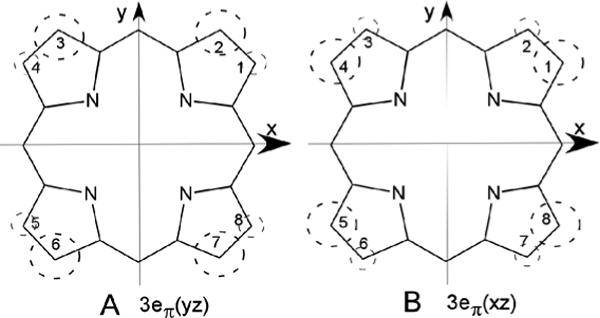
Symmetry-adapted substrate 3eπ molecular orbitals that illustrate the position of significant delocalized π-spin density on the substrate resulting from singly-occupied metal-porphyrin π-bonding. The magnitude of the spin density, ρπ , is shown by the size of the dashed circle. (A) 3eπ(yz), which interacts only with dyz and results in large low-field methyl contact shifts for positions 2, 3, 6 and 7, as observed (18) for the cyanide complexes of α-meso selective hHO, and (B) 3eπ(xz), which interacts only with dxz and results in large low-field methyl contact shifts at positions 1, 4, 5 and 8, as observed in the azide complexes of α-meso selective hHO. The orbitals in (A) and (B) are similarly occupied in all α-meso selective HO cyanide (18, 19, 22, 40, 41, 46) and azide (34, 35) complexes, respectively.
Azide and cyanide complexes of ferri-globins exhibit very similar substrate contact shift patterns, and hence similar orbital ground states. (42) It was therefore, surprising that the azide complexes of both PaHO and the HO from the pathogenic bacterium Neisseria meningitidis, NmHO, exhibited (34) a switch between the two alternate orbital ground states (i.e., molecular orbitals in Figure 3), upon substituting azide for cyanide (22, 28), even though the axial His imidazole orientation can be expected (12, 14) to be unchanged. Based on the failure of the correlation between the contact shift pattern and the His orientation in the azide complex, and supported by somewhat larger magnetic moments for the azide relative to cyanide complex, a novel S = 3/2, (dyz)2(dxy)1(dxz)1(dz2)1, rather than the common S = 1/2, (dxy)2(dxz dyz)3 ground state, was proposed (34) for HO azide complexes. This unprecedented intermediate-spin ground state was attributed to a weaker than “normal” (i.e., globins) axial field strength for the azide complex. Since H-bond donation to the azide will weaken its ligand field, it was concluded (34) that the distal H-bonding to the ligand by the ordered water molecules in HOs is stronger than the H-bonding by distal basic side chains in globins. The interchange between the singly occupied dxz and dyz (with spin delocalization into 3eπ(xz) and 3eπ(yz), respectively) between azide and cyanide complexes of HOs was suggested (34) as a feature diagnostic of a catalytically competent heme oxygenase active site. While a spectroscopic signature for HO activity would be very useful, it remains to be determined whether this proposed unique property, a switch in orbital ground states between the azide and cyanide complexes, (34) is exhibited by all HOs, and if so, exactly what molecular/electronic structural feature is perturbed relative to other ferrihemoproteins with ostensibly conserved His/CN− and chromophores, and how it relates to HO activity.
For the purpose of attempting to resolve some of these questions herein we extend our 1H NMR studies to the azide complex of human heme oxygenase-1, hHO. Sequence homology between NmHO and PaHO is stronger (9) than either with hHO, particularly in the distal H-bond network, and it is of interest to compare the NMR properties of the azide complexes of hHO with those of NmHO (27, 34) and PaHO. (34) Unfortunately, hHO in different complexes with the native substrate, protohemin, PH (R = vinyl in Figure 2A), exhibits molecular heterogeneity (40, 41, 47) with respect to the orientation of PH about the α-,γ-meso axis that reduces sensitivity and increases spectra overlap, which severely complicates assignments. Instead we employ the two-fold symmetric heme, 2,4-dimethyldeuterohemin, DMDH (R = CH3 in Figure 2A), which was found (18) to generate a single, homogeneous species in solution with a molecular structure essentially indistinguishable from that of the PH complex. (20, 47, 48) To assess whether any NMR spectroscopic anomalies are related to the distinctive HO distal pocket environment that stabilizes hemolytic over heterolytic O-O bond scission, we include 1H NMR data on the Asp140Ala point mutant, D140A-hHO-PH-N3, which abolishes mammalian HO activity, (32, 33) and instead favors heterolytic O-O bond cleavage.
We demonstrate that the switch between the alternate orbital ground states for the azide and cyanide complexes is apparently a general HO characteristic, but that this orbital/spin state is retained in the inactive D140A-hHO-PH-N3 mutant, indicating that this unusual property is not uniquely correlated with HO activity. These results allow us to formulate an alternate orbital/spin ground state for HO azide complexes that is consistent with differences in the π bonding capability of and CN− and argue, contrary to previous proposals, (10, 34, 49) for stronger, rather than weaker, azide field strength, and hence weaker rather than stronger H-bonding to the exogenous ligands in HOs than in globins.
EXPERIMENTAL
Protein sample
Solubilized, wild-type hHO, and the point mutant D140A-hHO were prepared as reported previously (24, 32) 2,4-dimethylprotoporphyrin-IX, DMDH (Figure 2A with R = methyl), was purchased from Mid-Century Chemicals and the iron inserted by standard procedures. (50) The 1:1 molar equivalents of DMDH was titrated into apo-hHO in 2H2O in the presence of 80 mM KN3 buffered by 50 mM phosphate at pH ∼7.1.
NMR spectroscopy
1H NMR data were collected on Bruker AVANCE 500 and 600 spectrometers operating at 500 and 600 MHz, respectively. Reference spectra were collected in 2H2O over the temperature range 15−35 °C at both a repetition rate of 1 s−1 over 40 ppm spectral width and at 5 s−1 over a 200 ppm spectral width. Chemical shifts are referenced to 2,2-dimethyl-2-silapentane-5-sulfonate (DSS) through the water resonance calibrated at each temperature. Non-selective T1s were determined by the standard inversion-recovery pulse sequence and estimated from the null point. 600 MHz NOESY spectra (51) (mixing time 40 ms; repetition rate 1−2.5 s−1) and 500 MHz Clean-TOCSY spectra (to suppress ROESY response (52); spin lock 25 ms; repetition rate 1−2 s−1) were recorded over a bandwidth of 25 KHz (NOESY) and 12 KHz (TOCSY) using 512 t1 blocks of 128 and 256 scans each consisting of 2048 t2 points. 2D data sets were processed using Bruker XWIN software on a Silicon Graphics Indigo workstation, and consisted of 30°- or 45°-sine-squared-bell-apodization in both dimensions, zero-filling to 2048 × 2048 data points prior to Fourier transformation.
Paramagnetic susceptibility comparison
Solution 1H NMR (53, 54) was used to determine the difference in paramagnetic susceptibility, ΔχM,
| (1) |
between the accepted S = 1/2 hHO-DMDH-CN complex and the hHO-DMDH-N3 complex, in order to assess the potential population of a S > 1/2 ground state for the latter complex. A 2.0 mM hHO-DMDH-H2O solution 50 mM in phosphate, pH 7.1 (concentration determined by ε = 140 mM−1 cm−1, to which was added 10 mM dioxane,3 was split into two samples. The first solution was diluted by 10% by volume with phosphate buffered, pH 7.1 1H2O solution 400 mM in KN3 to yield a 1.8 mM hHO-DMDH-N3 solution in 1H2O. The second solution was diluted by 10% by volume with phosphate buffered pH 7.1, 50%:50% v:v 1H2O:2H2O, 400 mM in KCN, to yield a 1.8 mM hHO-DMDH-CN solution in 95% 1H2O, 5% 2H2O. The equimolar (1.8 mM) hHO-DMDH-CN and hHO-DMDH-N3 solutions at identical ionic strength were placed in the center tube and annulus of a concentric 5 mm NMR tube and the chemical shift difference, Δδ , measured on a 600 MHz spectrometer at 25° and 35°C. This difference in chemical shift is related to the difference in paramagnetic susceptibility between the cyanide and azide complex as given by: (53, 54)
| (2) |
Using the essentially invariant paramagnetic susceptibility for the established low-spin cyanide complex of ferric HO (34), χM(cyanide complex) ∼ 3.6 × 10−8 m3/mol, χM(hHO-DMDH-N3 can be estimated, and the effective magnetic moment, μeff for the azide complex can be determined by:
| (3) |
RESULTS
Spectral comparison of and CN− complexes
The strongly low-field, contact-shifted methyl resonances indicative of large π-spin density are resolved in the low-field portion of the 1H NMR spectrum. The low-field resolved portion of the 600 MHz 1H NMR reference spectrum of hHO-DMDH-N3 in 2H2O is shown in Figure 4A, and can be compared to that of hHO-DMDH-CN (18) in 2H2O in Figure 4A'. Assignments for the substrate resonances are given in the Fisher notation, and for amino acid residues, by residue number and position. Noteworthy is the observation (18) that the CN− complex exhibits two resolved methyls, while the WT azide complex displays four methyls in this window. The non-selective T1s (not shown) for the resolved methyls of hHO-DMDH-N3 are ∼85 msec, which is less than a factor ∼2 shorter than for the cyanide complex. (18, 41) The 1CH3 and 8CH3 peaks of hHO-DMDH-N3 exhibit line-broadening over those of the 4CH3 and 5CH3 peaks (in spite of similar T1s), which arises from rapid exchange (55) between alternate micro-environments at the substrate periphery. The two remaining methyls will be shown to appear within the diamagnetic envelope 0−10 ppm. The DMDH methyl peaks to low-field of ∼10 ppm, and to the high-field of ∼10 ppm, in hHO-DMDH-N3 exhibit very weak anti-Curie, and markedly stronger anti-Curie behavior, respectively (not shown; see Supporting Information; Figure S1).
Figure 4.
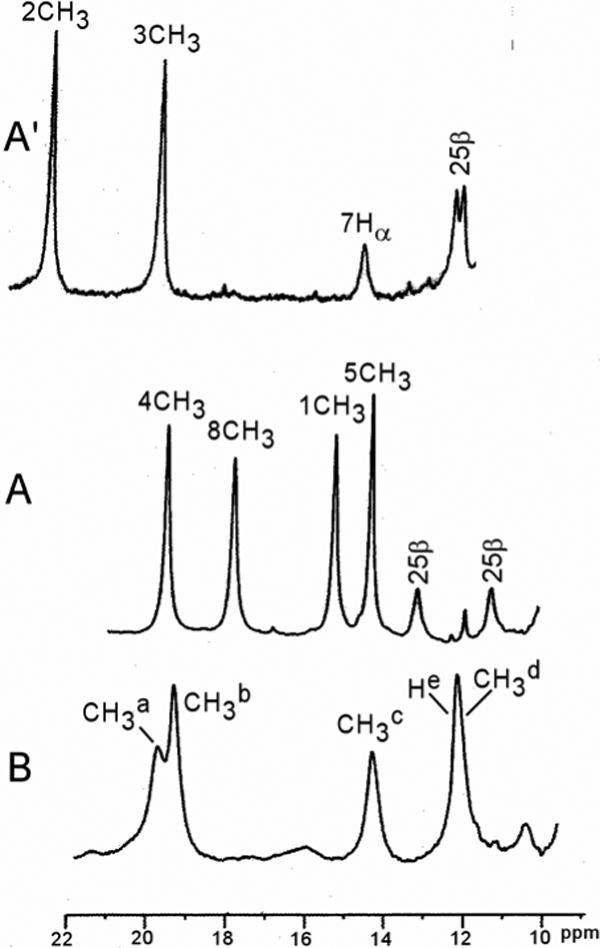
Resolved portions of the 600 MHz 1H NMR spectra of: (A) hHO-DMDH-N3; (B) D140A-hHO-DMDH-N3; and (A') Reproduces the same spectral portions for hHO-DMDH-CN (18). All samples are at 30°C in 2H2O, 50 mM in phosphate at pH 7.4. DMDH peaks are labeled by the Fisher notation (see Figure 3), and residue peaks are labeled by the residue number and its position within the residue.
The low-field resolved portion of the NMR spectrum of D140A-hHO-DMDH-N3 in 2H2O is shown in Figure 4B. Spectra over a wider range of temperature (not shown; see Supporting Information; Figure S2) reveal four methyls, labeled CH3a-CH3d (intensity ∼3 protons), and a single proton peak, He, to the low-field of ∼10 ppm similar3 to that for hHO-DMDH-N3. The T1s for the methyl resonances are similar (∼80 ms) to those for the WT complex. The methyl peaks exhibit significant and differentially broadened resonances at 30°C, which broaden further at lower temperature (not shown; see Supporting Information; Figure S2). This behavior indicates an exchange phenomenon, as observed for WT, but with slower rate. (55) Weak anti-Curie behavior is displayed by the low-field methyls in D140A-hHO-DMDH-N3, similar to that observed for WT (not shown; see Supporting Information; Figure S2).
Assignment protocols
We focus here only on the electronic structure of the chromophore, and hence the only resonances of interest are those for DMDH and sufficient active site residues in contact with DMDH to associate each pyrrole with a specific protein environment in the heme pocket, as defined in the crystal structure. (13) These limited, but crucial assignments were optimally pursued in a 2H2O solution of hHO-DMDH-N3. While the hHO azide crystal structure has not been reported, comparison of the rat HO azide crystal structure (12) with those of rat HO having numerous other ligands, including cyanide, (14), show that heme contact residues are unchanged in the various derivatives. Our working hypothesis is that this is also true for hHO.
The DMDH assignments are assigned very straight forwardly by the pattern of inter-methyl and methyl-propionate CαH NOESY cross peaks about the substrate periphery, as reported for hHO-DMDH-CN (18) and NmHO-DMDH-N3 (46) We note that DMDH, because of its two-fold symmetry, does not possess unique labeling for each pyrrole substituent. However, we adopt the convention of numbering positions (as reported for the hHO-DMDH-CN complex (18)), which parallels that for the major solution isomer of hHO-PH, (41) as depicted in Figure 2A. Active site residues are assigned by TOCSY-detection of the spin-connectivity that uniquely identifies residue type, and by the NOESY contacts among these residues and between these residues and DMDH, as predicted by the crystal structure. (13) The uniqueness of both scalar and dipolar connectivities are confirmed on the basis of 2D experiments over a range of temperatures, where the natural temperature dependence of hyperfine shifts serves as a “third dimension” for editing the spectra. The expected, and observed, active site residue-substrate contacts are depicted schematically in Figure 2A. The assignments were greatly aided by the observation of very similar dipolar shifts for heme contact residues in the azide and the previously reported (18) cyanide complex. Moreover, hHO possesses an extended aromatic cluster on the distal side, conserved in both substrate and substrate-free hHO (13, 56), that predicts, and allows the observation of, (18) highly characteristic NOESY cross peak patterns among TOCSY-detected aromatic rings with characteristic low-field shifts, in the pattern: Phe95-Trp96-Phe47-Phe167-Leu164-Phe166-Tyr58.
Assignment of WT hHO-DMDH-N3
Portions of the 600 MHz 1H NMR NOESY spectrum of hHO-DMDH-N3 in 2H2O illustrating the key contacts about the DMDH periphery in the order CαH-CH3-CH3-CH3-CH3-CH3-CH3-CαH, with weak cross peaks between the outer and central methyl pairs and moderate cross peaks between the other two methyl pairs, are shown by arrows in Figure 5. Key DMDH-residue and inter-residue dipolar contacts are illustrated in both Figures 5 and 6. A pair of strongly low-field shifted and relaxed geminal protons can arise only from the remaining heme ligand, His25 CβHs (Figures 6C, 6D, 6F).
Figure 5.
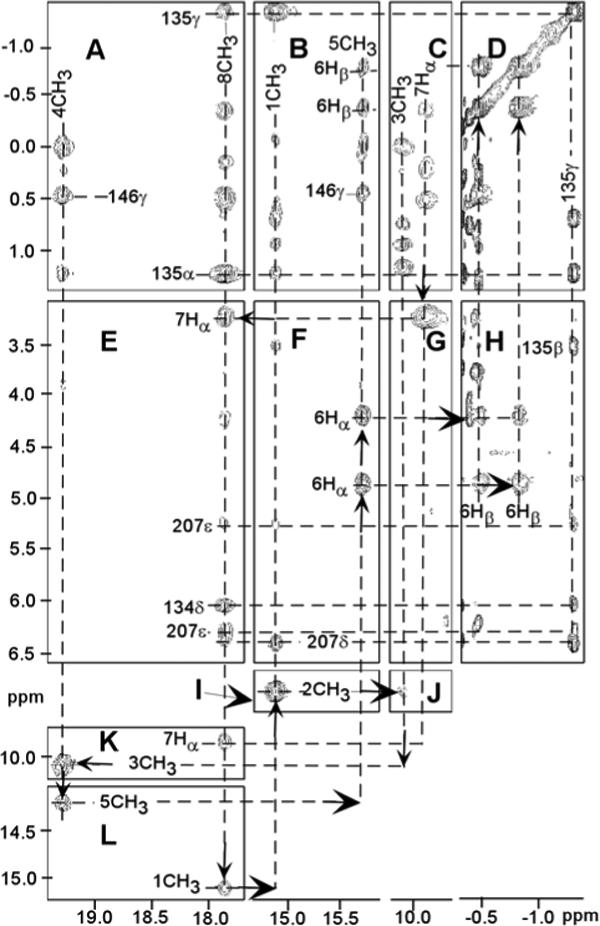
Portions of the 600 MHz 1H NMR NOESY spectrum (mixing time 40 ms; repetition rate 2.5 s−1; sweep width 35 ppm) of hHO-DMDH-N3 in 2H2O, 50 mM in phosphate, pH 7.4, at 30°C, illustrating the dipolar contacts about the DMDH perimeter, as shown by arrows (A-L). Key residue-DMDH and inter-residue contacts are: Val146 to 4CH3 (A) and 5CH3 (B), Thr135 to 8CH3 (A) and 1CH3 (B), Phe207 to 8CH3 (E) and 1CH3 (F), Tyr134 ring to 8CH3 (E) and Thr135 CγH3 (H).
Figure 6.
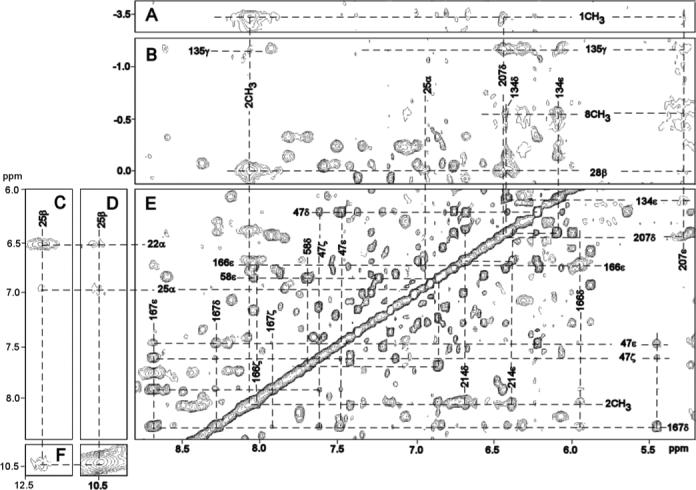
Portions of the 600 MHZ 1H NMR NOESY spectrum (mixing time 40 ms, repetition rate 1.5 s−1; sweep width 17 ppm) illustrating key DMDH-residue and inter-residue contacts: (A) 1CH3 to Phe207 ring; (B) 8CH3 to Tyr134 ring, Ala28 CβH3 to Phe207 ring and His25 CαH; (C, D, F) His25 intra-residue; (E) 2CH3 to Phe214 ring; and (E) inter-aromatic and aromatic-aliphatic contacts involving Phe47, Tyr58, Tyr134, Phe166, Phe167 and Leu164 in the distal aromatic cluster. Note that in panel (B), 1CH3 and 8CH3 are folded-in.
The NOESY cross peak of an Ala methyl to the His25 CαH (Figure 6B), together with Ala methyl NOESY cross peask to an upfield DMDH methyl (Figure 6B), uniquely identify Ala28 and assign the 2CH3 and 3CH3. Ala28 CβH3 NOESY cross peaks to a Phe ring and to 2CH3 (Figure 6B) uniquely identify Phe214, while an Ala28 CβH3 cross peak to another aromatic ring (Figure 6B) that exhibits cross peaks to 1CH3 (Figure 6A) and 8CH3 (Figure 5E), uniquely locates the ring of Phe207. NOESY cross peaks of a Thr to both 8CH3 (Figure 5A) and 1CH3 (Figure 5B), and a cross peak between an aromatic ring and both 8CH3 (Figure 5E) and Phe207 (Figure 6E), uniquely locate Thr135 and Tyr134. The methyls of a Val exhibit contacts to both 4CH3 (Figure 5A) and 5CH3 (Figure 5B) that can only arise from Val146. The expected and observed heme pocket contacts are depicted schematically in Figure 2A and chemical shifts of DMDH and heme contact residues are provided in Table 1. Lastly, characteristic NOESY cross peaks among weak to moderately dipolar-shifted rings in the aromatic cluster identify an additional six aromatic (Phes 47, 95, 166, 167; Tyr58, Trp96), and part of one aliphatic (Leu164) residues with shifts listed in Supporting Information.
Table 1.
Chemical shifts for DMDH and key active site residues for DMDH-azide complexes of WT hHO and D140A-hHO
| WT hHO | D140A-hHO | |||
|---|---|---|---|---|
| Residue | Proton | Azidea | Cyanideb | Azidea |
| DMDH | 1CH3 | 15.09 | 8.95 | 19.9 |
| 2CH3 | 7.71 | 21.37 | ||
| 3CH3 | 10.07 | 18.25 | ||
| 4CH3 | 19.34 | 8.51 | 19.4/14.5c | |
| 5CH3 | 14.27 | 9.78 | 14.5/19.4c | |
| 6CαH | 4.20 | 11.02 | ||
| 6CαH' | 4.85 | 10.24 | ||
| 6CβH | −3.90 | −0.05 | ||
| 6CβH' | −7.50 | 0.12 | ||
| 7CαH | 3.24 | 13.13 | ||
| 7CαH' | 9.84 | 7.02 | ||
| 8CH3 | 17.78 | 8.29 | 12.4 | |
| Arg22 | CαH | 6.67 | 6.27 | 6.21 |
| His25 | Cβ1H | 10.78 | 8.95 | 9.3 |
| Cβ2H | 12.68 | 10.75 | 12.3 | |
| Ala28 | CαH | 3.40 | 2.42 | |
| CβH3 | −0.07 | −2.14 | ||
| Tyr134 | CδHs | 6.37 | 6.56 | 6.55 |
| CεHs | 6.05 | 6.25 | 6.13 | |
| Thr135 | CαH | 1.23 | 2.39 | 2.36 |
| CβH | 3.75 | 3.35 | 3.55 | |
| CδH3 | −1.31 | −0.10 | 0.07 | |
| Leu138 | CαH | 3.76 | 3.12 | - |
| CβHs | −0.35, 0.47 | −0.40, 0.55 | - | |
| CδH | 0.48 | 0.55 | - | |
| Val146 | CαH | 3.88 | 3.76 | |
| CβH | 2.26 | 2.32 | ||
| CγH3 | 0.51 | 0.56 | ||
| CγH3' | 0.47 | 0.71 | ||
| Phe207 | CδHs | 6.43 | 6.43 | - |
| CεHs | 5.25 | 5.82 | - | |
| Phe214 | CδHs | 6.80 | 6.67 | - |
| CεHs | 6.35 | 6.35 | - | |
| CζH | 6.27 | 6.45 | - | |
Chemical shift, in ppm referenced to DSS via the residual solvent signals, in 2H2O, 50 mM in phosphate, pH 7.1 at 30°.
Chemical shift in ppm, referenced to DSS, in 1H2O, 50 mM in phosphate. Data taken from Li et al (18).
The 4CH3 and 5CH3 assignments could not be differentiated.
The large hyperfine shifts for the hHO-DMDH-N3 methyls at positions 1−, 4−, 5− and 8− dictate single occupancy of dxz, with spin delocalized into 3eπ(xz), (Figure 3A) as observed (34) for NmHO-PH-N3. (34) The active site residues, on the other hand, exhibit dipolar shifts very similar to those of the CN− complex (18) whose dyz (with spin delocalization into 3eπ(yz); Figure 3B) is singly occupied (see Table 1 and Supporting Information, S1).
Assignments in D140A-hHO-DMDH-N3
The methyls on a given pyrrole exhibit very sizable NOESY cross peaks and numerous common cross peaks to pyrrole contact residues. Two methyls adjacent to the same meso-H exhibit only a very small inter-methyl cross peak but still exhibit common cross peaks to the same heme contact residue. Variable temperature NMR spectra of hHO-DMDH-N3 identify four resolved methyl peaks and one resolved single proton peak He (not shown, see Figure S2 Supporting Information). The much larger linewidths, lower concentration (∼1.5 mM), and hence strongly reduced sensitivity relative to the WT complex, preclude the detection of the weak NOESY cross peak within the methyl pairs on adjacent pyrroles, 1CH3/8CH3 and 4CH3/5CH3, but should readily allow the detection of the moderate intensity cross peak within methyl pairs on the same pyrrole, 1CH3/2CH3 and 3CH3/4CH3. The excessive linewidth, moreover, precludes the detection of the intra-residue TOCSY cross peaks for DMDH contact residues. However, one qualitative conclusion is that there are still four low-field DMDH methyls, as in WT hHO-DMDH-N3 complex,3 and not two, as in hHO-DMDH-CN (18) indicating that the singly occupied dxz is retained in the mutant azide complex.
Portions of the NOESY spectrum of D140A-hHO-DMDH-N3 in 2H2O containing the low-field methyl resonances are shown in Figure 7. The absence of cross peaks between well-separated low-field methyl peaks excludes any of these pairs arising from methyls of the same pyrrole. The near-degeneracy of CH3a and CH3b prevents detection of even a moderate intensity NOESY cross peak between them; however, the absence of any common cross peaks to CH3a and CH3b over a range of temperatures similarly discounts their positions either on the same pyrrole or adjacent to the same meso position. Methyl peaks CH3a (Figure 7A) and CH3d (Figure 7C) exhibit common NOESY cross peaks to an aliphatic and aromatic residue indicating aliphatic-aromatic residue contacts (Figures 7D, 7F) that are very similar in pattern to those observed in hHO-DMDH-N3 for 1CH3 and 8CH3 with Thr135, Tyr134, respectively, and are so assigned. Methyl peaks CH3b (Figure 7A) and CH3c (Figure 7B) exhibit a common NOESY cross peak to an upfield-shifted methyl in a manner similar to that observed in hHO-DMDH-N3 for 4CH3 (Figure 5A) and 5CH3 (Figure 5B) with Val146, and are so assigned; the two methyls could not be differentiated. Thus, the same four methyl peaks appear in the low-field resolved spectral window for both the WT azide and D140A mutant azide complexes. The intense NOESY cross peak for peak He is consistent with it arising from the His25 CβHs (Figure 7G). The logical conclusion from this NOESY pattern similarity between the mutant and WT proteins is that they display highly conserved active site structures. This is further supported by the observation of very similar NOESY cross peaks among TOCSY-detected rings in the distal aromatic cluster of both proteins (Not shown; see Supporting Information).
Figure 7.
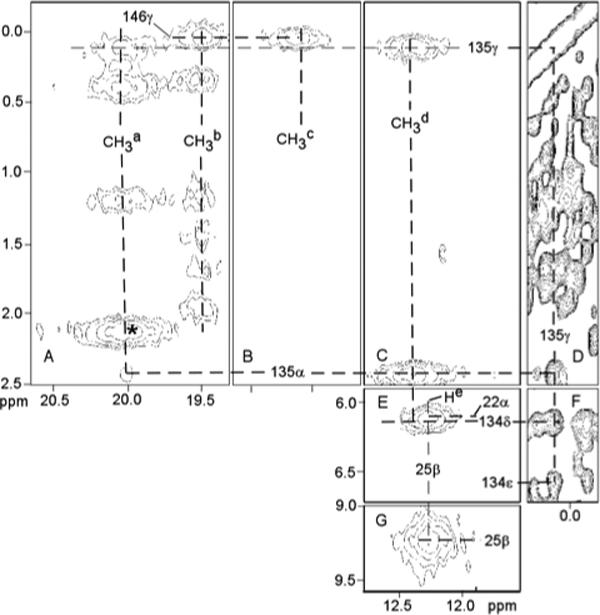
Portion of the 600 MHZ 1H NMR NOESY spectrum (mixing time 40 ms; repetition rate 25 s−1) of D140A-hHO-DMDH-N3 in 2H2O, 100 mM in phosphate, pH 7.1 at 30°C illustrating the DMDH contacts to the four low-field resolved methyls labeled Ma-Md to key active site residues (A-C, E, F); as well as key intra Thr135 (D), Thr135 to Tyr134 (F) and both inter His25 (G) and His25 to Lys22 contacts (E). The peak marked * possibly arises from 2CH3.
Comparison of magnetic susceptibilities of hHO-DMDH-N3 and hHO-DMDH-CN
The splitting of the dioxane signal, Δδ, is measured as 2.8 ± 0.1 Hz at 25°C and 3.1 ± 0.1 Hz at 35°C, with the azide complex exhibiting the larger χM. The Δδ at 25°C leads to ΔχM = 0.81 ×10−8 m3/mol via Eqs. (1) and (2). The χM value for the low-spin, cyanide complex of HO (34) indicate χM(hHO-DMDH-CN) ∼ 3.6 × 10−8 m3/mol, such that we obtain χM(hHO-DMDH-N3) ∼ 4.4 × 10−8 m3/mol. Eq. (3) yields μeff(hHO-DMDH-CN) ∼2.6 BM and μeff(hHO-DMDH-N3) ∼2.9 BM at 25°C. Clearly, χM and μeff for the azide complex are only very modestly increased (∼20% and ∼10%, respectively) over those for the cyanide complex.
DISCUSSION
Active site molecular structure of the wild-type azide complex
The observation of all of the contacts between active site residues and the individual methyls of DMDH expected on the basis of both crystal structure (13) and solution structure of the cyanide complex, (18) confirm an essentially conserved active site structure for the WT azide complex. This is further supported by the observation of the dipolar contacts among numerous rings in the aromatic cluster of the distal side of the substrate. Crystal structures of the azide and cyanide complexes of the highly homologous rat HO complex have already demonstrated (12, 14) that the active site structure is highly conserved.
The pattern of DMDH hyperfine shifts has clearly shown(18) that dyz (with spin delocalization into 3eπ(yz); Figure 3A) is singly occupied for hHO-DMDH-CN. The pattern of DMDH methyl hyperfine shifts for hHO-DMDH-N3, large low-field contact-dominated hyperfine shifts for 1CH3, 4CH3, 5CH3 and 8CH3, are consistent only with single spin occupation of dxz (with spin delocalized into 3eπ(xz); Figure 3B). This further confirms the original observation (34) of the switch of the orbital grounds states between the azide and cyanide complexes of PaHO and NmHO, and supports the notion that this switch may be diagnostic of the active site structure of HOs. For both the PaHO and NmHO complexes, it was proposed, (34) based on the assumption that only the axial His orientation can control the orbital ground state, (44) that the singly-occupied dxz [dyz], for α-meso- [β-,δ-meso-] selective HOs can arise only with the unusual S = 3/2, (dyz)2(dxz)1(dxy)1(dz2)1 [(dxz)2(dyz)1(dxy)1(dz2)1] ground state, respectively. In order to determine the orbital/spin state consistent with the present 1H NMR data on the DMDH azide complex of α-meso selective hHO, we will consider separately the spin and orbital parts.
hHO-DMDH-N3 possesses an S = 1/2, rather than S = 3/2, ground state
An S = 3/2 ground state was postulated (34) for bacterial HOs primarily on the basis of a switch between dyz and dxz single-spin occupation in azide and cyanide complexes of PaHO and NmHO that is completely inconsistent with the expected conserved axial His orientation, and the finding that the magnetic susceptibility in PaHO-PH-N3 was twice that for the S = 1/2 cyanide complex. In contrast, we conclude that the spin ground state for hHO-DMDH-N3 is S = 1/2, as in hHO-DMDH-CN, (18) for four reasons: 1) Nuclear T1s, and hence electronic T1s, (57) are within a factor of 2 for azide and cyanide complexes and highly characteristic of S = 1/2 ferric hemoproteins; (42, 43, 57) 2) The dispersion of chemical shifts for non-ligated residues, that arises from dipolar shifts due to anisotropy of the paramagnetic susceptibility tensor, (42, 43) is very large and essentially the same in hHO-DMDH-N3 and hHO-DMDH-CN, as evidenced by the very similar shift magnitude and shift pattern for non-ligated residues in the cyanide and azide complexes (Table 1); 3) Most directly, the magnetic susceptibility difference between the azide and cyanide complexes of hHO is only ∼0.8 × 10−8 m3/mol, which, with the reported χM ∼3.6 × 10−8 m3/mol for an HO cyanide complex (34) indicates an only ∼20% increase in χM for the azide over the cyanide complex and this difference is a factor ∼5 smaller than the reported difference in χM for the same pair of PaHO complexes. (34) The small increase of μeff of the azide, ∼2.9 BM, over that of the cyanide (∼2.6 BM) is much smaller than expected even for a spin-only S = 3/2 ground state. The very modest increase in ΔχM at elevated temperature, as well as the weak anti-Curie behavior of the hHO-DMDH-N3 methyl hyperfine shifts, (not shown, see Supporting Information, Figure S1) are in complete accord with an S = 1/2 ground state and a very modest thermal population of the S = 5/2 excited state, as is commonly found in globin azide derivatives. (42, 58, 59) 4) Lastly, and very importantly, as shown below, a robust interpretation can be offered for the switch of the singly-occupied dπ-orbital of dyz in hHO-DMDH-CN to dxz in hHO-DMDH-N3 complex within the S = 1/2 ground state that is completely consistent with the expected conservation of the axial His orientation and the π-bonding properties of ligated azide. It is noted that EPR data at cryogenic temperatures on the azide complex of rat HO have also been interpreted as arising from a low-spin state with eclipsed axial ligand planes. (60)
The axial field strength is stronger in hHO than globin azide complexes
1H NMR studies of azide complexes of globins have been interpreted (42, 59) on the basis of an S = 1/2 ↔ S = 5/2 equilibrium, (58) with the low-field bias of the methyl shifts and the degree of anti-Curie behavior indicative of the degree of high-spin population. The azide complexes of globins are qualitatively differentiated from those of HOs in that their low-field methyl shifts are greater and the degree of their anti-Curie behavior is larger. Hence, the lower S = 5/2 population for hHO than globins dictates a stronger axial field strength in the azide complexes of hHO than of the globins. Magnetic resonance studies have shown that the axial anisotropy increases, and the electronic T1 decreases (nuclear T1 increases), with increasing axial field strength in an S = 1/2 ferri-hemin or ferri-hemoprotein. (42, 45) The methyl T1s and dipolar shift dispersion that relate to the magnitude of the magnetic anisotropy, are also much larger in hHO than in globin azide complexes, confirming a stronger axial field in the HO complexes. The nature of the Fe-His bonding in HOs has been concluded (37, 39, 61) to be “normal” and not detectably differentiated from that in globins. Hence the greater azide axial field strength in hHO than globin complexes must result from a stronger bonding of the azide ligand in hHO than in globins.
The (dxy)2(dxz)2(dxz) orbital ground state in hHO-DMDH-N3 is determined by the azide orientation
The pattern of large low-field DMDH 1CH3, 4CH3, 5CH3 and 8CH3 contact shifts dictates a singly occupied dxz that yields the delocalized spin in 3eπ(xz) (shown in Figure 3B), and it remains to be shown that this is not only consistent with the expected molecular structure (62) of the azide complex, but can be reasonably anticipated with the presently established larger azide axial field strength in HO than globin complexes. The pattern of DMDH methyl hyperfine shifts as a function of the axial His orientation, given by the angle, φΗ, (shown in Figure 2A) has been empirically predicted (44) and gives rise to the pattern illustrated (27) in Figure 8. However, this plot can be generalized to represent the DMDH methyl shifts as a function of the angle, φ, to the unique π-plane of whichever axial ligand serves as the stronger π-donor to the iron. For His/CN− ligation, the axial symmetry of the ligated cyanide necessarily leads to control of the orbital ground state by the axial histidine plane (φH in Figure 2A). (42, 44) The methyl shift pattern for the hHO-DMDH-CN complex has been reported (18) with contact shift magnitudes in the order: 3CH3 ∼2CH3 >8CH3 ∼5CH3 ∼4CH3 ∼1CH3, which dictates φ ∼0° in Figure 8 (vertical arrow under CN−), in reasonable agreement with the crystallographic φH ∼0 (Figure 2A) (13)
Figure 8.
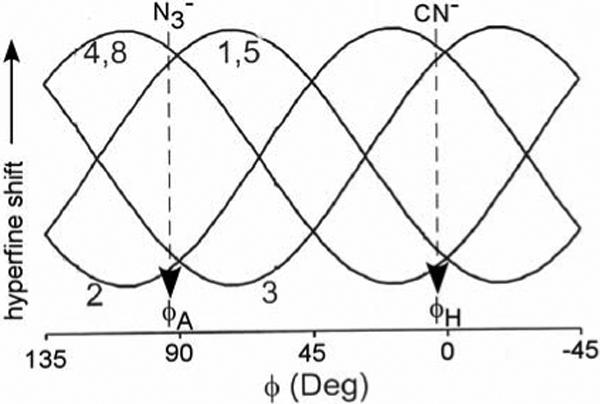
Empirical plot of DMDH methyl shifts as a function of the angle φ between the unique π-plane of the axial ligand that serves as the stronger π-donor to the iron in a S = 1/2, (dxy)2(dxz,dyz)3 ferrihemoprotein. (27, 44) The angle, φ, is defined in Figure 2A. The pattern of the observed DMDH methyl shifts, and the corresponding φ, are indicated by vertical arrows under CN− (for the cyanide complex) and under (for the azide complex).
When there are two non-axially symmetric ligands, the orbital ground state will depend on which of the two ligands serves as the stronger π-donor to the iron. In formulating (34) the S = 3/2 ground state for the azide complex of the NmHO and PaHO to account for the switch between the two π orbitals in Figure 3 for the azide and cyanide complexes, the possible contribution of azide π-bonding to the orbital ground state was discounted on the basis of the axial symmetry of the azide ion. However, while the azide is axially symmetric, upon coordination to iron, the axial symmetry is broken and contributes a single π-system with well-defined orientation relative to the heme. An orientation of the azide terminus towards the α-meso position (i.e., as depicted in Figure 2B) in hHO can be reasonably expected since the bound NO in hHO-PH-NO is sterically constrained to orient towards the α-meso position (13) as part of the stereoselectivity mechanism. The crystal structure of the azide complex reported for the highly homologous rat HO complex (12) shows the bent Fe-N3 unit directed towards the α-meso position as depicted in Figure 2B. The presently observed methyl shift pattern for hHO-DMDH-N3 is: 1CH3 ∼4CH3 ∼5CH3 ∼8CH3 >2CH3 ∼3CH3 > indicates φ ∼90° from Figure 8 (vertical arrow under ). This φ is precisely that expected for the orientation of the azide π-system (φA in Figure 2). Thus, it is the azide π-bonding that determines the orbital ground state in the azide complex of hHO, and the logical conclusion that follows is that the azide must be a better π-donor (stronger field) than the His imidazole in hHO.
Comparison of the axial field in HO and globin azide complexes
The question arises whether the azide axial field strength is generally stronger than for the His imidazole, or whether this only occurs in HOs. The heme methyl 1H NMR assignments of both the cyanide and azide complexes have been reported for globins, (42) and, while the low-field bias of the heme methyls at the same ambient temperature is always larger in the azide than cyanide complexes, the pattern of shifts for the cyanide and azide complexes is very similar, with low-field shift magnitudes 5CH3 > 1CH3 > 8CH3 > 3CH3. This suggests that the imidazole π bonding controls the orbital ground state in both cyanide and azide complexes of globins, or that azide is a weaker π donor than the axial His imidazole in globins. It should be noted that, once it is recognized that the orbital ground state in hHO-DMDH-N3 is determined by the azide, rather than the imidazole π-plane orientation, as in globins, the conclusion that azide exert a stronger ligand field in HOs than globins, follows directly.
Correlation between azide ligand field strength and distal H-bonding
The ligand field strength of azide can be modulated by distal H-bonding, with proton donation to either N1 or N3 resulting in weaker Fe-N1 bonding (axial field strength). Hence, the stronger ligand field of the azide in HO than globins indicates that the distal H-bonding to the azide is weaker in hHO than globins. The key distal H-bond interactions that involve the exogenous ligand, Tyr58, Thr135, Arg136, Gly143, and Asp140 and two ordered water molecules, water#1 (magenta) and water#2 (dark green), in WT hHO-PH-NO (13), are depicted in Figure 9. Since water#1 must serve as donor to the strong Asp140 carboxylate acceptor, it is unlikely to serve as a significant H-bond donor to the iron ligand. A similar pocket structure can be assumed for the azide hHO complex. Thus, water#1 and Gly143 NH appear to provide much weaker H-bonds to the axial ligand than provided by the generally basic residues in the distal pocket of globins. (63)
Figure 9.
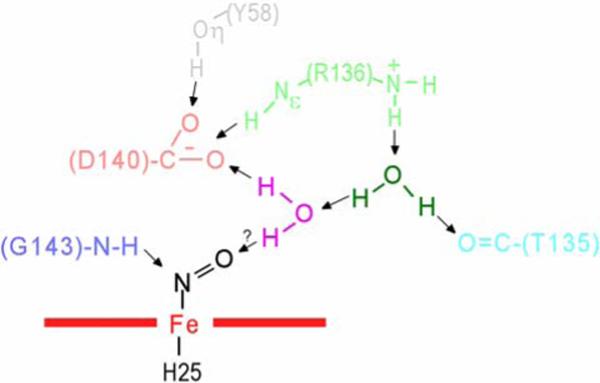
Schematic of active site structural features for the crystal structures (13) of WT hHO-PH-NO, illustrating the relative positions of four key residues in the distal H-bonding network, Tyr58 (grey), Thr135(aqua), Arg136(light green), Asp140(pink), Gly143(light blue), as well as two non-ligated, ordered water molecules, water#1(magenta) and water#2(dark green). (13) The most significant structural difference in the D140A-hHO mutant is the replacement of the Asp140 carboxylate with a new water molecule. H-bonds are shown by arrows.
Relevance to other studies
The ground states for PaHO [NmHO] azide complexes have been proposed (34) as S = 3/2, (dyz)2(dxy)1(dxz)1(dz2)1, [S = 3/2, (dxz)2(dxy)1(dyz)1(dz2)1] on the basis of the failure of the substrate methyl contact shifts to correlate with the axial His imidazole orientation and supported by a significant elevation of χM of the azide over the cyanide complexes. The arguments presented above obviate the first point. The fact that both NmHO and PaHO exhibit a switch between the alternate singly occupied dxz and dyz for the azide (34) and cyanide (22, 28) complexes in a manner very similar to that presently observed for hHO suggests a common S = 1/2 ground state for both azide and cyanide complexes of HOs in general, and dictates that the orbital ground state is similarly controlled in the azide complexes of all HOs. The larger increase in χM in the azide over the cyanide complex (34) for the bacterial HOs than hHO complexes likely arises from a larger population of the excited S = 5/2 state in the bacterial HOs. Consistent with this conclusion, the mean low-field methyl hyperfine shifts in NmHO and PaHO azide complexes are larger than in the hHO complex, and the methyl T1 values are larger in the hHO (∼85 ms) than bacterial HO (∼40 ms (35)) azide complexes. Hence, the weaker azide field strength in bacterial relative to mammalian HO indicates stronger distal H-bonding to the azide in either PaHO or NmHO than in hHO.
Implications for the HO mechanism
Based on the fact that hydrogen peroxide and ethyl peroxide yield the α-meso-hydroxy- and α-meso-ethoxy-hemin, respectively, (24, 25) and the ENDOR-detection (64) of the catalytically competent Fe+3-OOH at cryo-temperatures upon radiolysis of the oxy-complex, the mechanism (2, 3) of reaction was proposed to result from electrophilic attack on the α-meso carbon by the intact Fe+3-OOH. Computational results, (29) and interpretation of NMR data (10) have argued for a free-radical mechanism, where the hydroxyl radical resulting from homolytic O-O bond scission is tethered to the ordered water molecule cluster over the meso-position that is attacked. (31) A radical mechanism is supported by recent studies of electrophilic versus free-radical reactions with heme in horseradish peroxidase. (65)
An argument for the free-radical mechanism has been advanced (10, 34, 49, 66) on the basis of a proposed substrate “self-activation” for free-radical attack at a meso position due to single-spin population of the dxy orbital that leads to ruffling of the porphyrin and significant π-spin delocalization to the meso positions. The rationale for proposing the single-spin population of dxy was based on the premise that the exogenous ligand (directly proposed for azide (34) and hydroxide, (49) and inferred for hydroperoxide (10)) exerts a significantly weakened axial field in HOs relative to that in globins and that this weakened axial field in HOs results from stronger distal H-bonding to the ligand in HOs than in globins. The weaker than “normal” (i.e., globins) axial field strength of hydroxide and azide in HO was attributed to strong distal H-bond donation by ordered water molecules. The present 1H NMR data on hHO do not directly bear on the viability of the free-radical mechanism, but do not confirm the premise that the axial field strength of the exogenous ligand is weaker in HOs than globins. In order to disfavor the O-O cleavage in the Fe+3-OOH complexes of the globins, a weaker distal H-bond to azide in HOs than in globins might be reasonably expected, and is confirmed here. Consistent with the present findings, EPR data on the hydroperoxy complex of rat HO have been interpreted on the basis of the conventional dπ ground state (60) rather than the dxy ground state predicted (10, 49) on the basis of strong distal H-bonding.
Influence of the Asp140 → Ala mutation
The switch in the S = 1/2 orbital ground state for cyanide and azide complexes may be diagnostic of the stronger-than-“normal” azide ligand field in HOs in general, but it clearly does not correlate with HO activity, inasmuch as the azide complex of the inactive (32) D140A-hHO similarly exhibits4 four low-field methyls with T1 values similar to those in hHO-DMDH-N3, and similar weak anti-Curie behavior indicative of single occupation of the dxz orbital as for hHO-DMDH-N3. The crystal structure of D140A-hHO-PH-NO has shown (13) that the most significant structural difference between the WT and the mutant is that a new ordered water molecule (water#3) replaces the carboxylate group of the Asp140. Most importantly, the position of water#1 relative to the ligand, remains about the same. However, the replacement of the strong H-bond acceptor carboxylate of Asp140 in the D140A-hHO mutant, with a water molecule (water#3) makes water#1 a better H-bond donor to the ligand. The greater H-bond donation to the ligand by water#1 in the mutant is sufficient to accelerate O-O bond cleavage, but insufficient to make the ligated azide a poorer H-bond donor than the His imidazole, hence the retention of the S = 1/2, (dxy)2(dyz)2(dxz)1 ground state.
The very delicate balance between the donor strength of the ordered water molecule (water#1) in the discrimination between the rate of homolytic versus heterolytic O-O bond cleavage is indicated by a recent study of HO from Corynebacteria diphtheriae, CdHO (67) CdHO exhibits significant sequence (9) and structural (13, 16) homology to hHO, in particular in the distal helix and the H-bond/ordered water molecule network, (18, 19) with Tyr53, Arg132, Asp136, and Gly139 replacing Tyr58, Arg136, Asp140, and Gly143 respectively, and an ordered water molecule (water#1) similarly bridging the Asp136 carboxylate and exogenous ligand. However, the D136A-CdHO mutant retains about ∼50% activity. (67) Preliminary NMR evidence (not shown) suggests that the coupling between the exogenous ligand and homologous distal H-bond/ordered water network differ significantly between hHO and CdHO; a more detailed comparative investigation of the hHO and CdHO enzymes is in progress.
The present NMR results on hHO DMDH-N3 indicate that the complex is primarily low-spin with a (dxy)2(dyz)2(dxz)1 orbital ground state which is determined by the orientation of the ligated azide ion. The similarity of the substrate methyl contact shift patterns argue for a conserved S = 1/2 ground state (with variable thermal occupation of the S = 5/2 state) for the azide complexes of the pathogenic bacterial HOs, (34) with the orbital ground state determined by the proximal His ring orientation in the cyanide complexes (22, 28) and distal azide orientation in the azide complexes. Control of the orbital ground state in HOs by the azide ligand rather than by the axial His, is attributed to a stronger ligand field by the azide in HOs than globin complexes. This stronger azide axial field strength in HOs than in globins indicates weaker distal H-bonding to the ligand in HOs than globins. The fact that this unusual orbital ground state is conserved in the inactive Asp140 → Ala mutant of hHO indicates that, while the novel orbital ground state may be characteristic of an orbital hole switch between the azide and cyanide all HO complexes, it simply reflects the relatively weak distal H-bonding to the ligand by an ordered water molecule, and does not correlate with HO activity. Lastly, the proposed (10, 34, 49) weaker axial ligand field strength in azide-ligated HOs compared to azide-ligated globins that would stabilize a singly occupied dxy, which favors porphyrin ruffling that is key to the proposed free-radical mechanism, is not supported by NMR data for HOs.
Supplementary Material
Footnotes
This research was supported by grants from the National Institutes of Health, GM62830 (GNL) and DK30297 (PROM).
Abbreviations use: HO, heme oxygenase; hHO, human heme oxygenase #1; PaHO, Pseudomonas aeruginosa heme oxygenase; NmHO, Neisseria meningitidis, CdHO, Corynebacterium diphtheriae; PH, protohemin; DMDH, 2,4-dimethyldeuterohemin; DSS, 2,2-dimethyl-2-silapentane-5-sulfonate; NOESY, two-dimensional nuclear Overhauser spectroscopy; TOCSY, two-dimensional total correlation spectroscopy.
In the case of β-(or δ-)-meso-selective PaHO, the substrate is rotated in-plane (15, 49) ∼90° relative to the conserved protein matrix and imidazole orientation compared to that depicted in Figure 2A, such that the unpaired dxz spin is delocalized into 3eπ(xz) in PaHO-PH-CN, with predicted (44) and observed (49) large contact shifts for positions 1, 4, 5, and 8.
Both dioxane and t-butanol were used as susceptibility markers. However, addition of t-butanol significantly perturbed the hyperfine shift pattern of the azide complex, indicating specific interactions near the active site. Addition of dioxane left the 1H NMR spectra of both the azide and cyanide complexes unperturbed.
D140A-hHO-DMDH-CN yields chemical shifts very similar to those of WT hHO-DMDH-CN,(18) with 1CH3, 2CH3, 3CH3 , 4CH3, 5CH3, 8CH3, shifts at 25°C of 8.3, 20.0, 17.6, 7.6, 10.6, 9.4 ppm for the former and 9.0, 21.3, 18.2, 8.5, 9.8, 8.3 ppm for the latter complex, respectively.
SUPPORTING INFORMATION PARAGRAPH
One table and six figures (Curie plots for DMDH methyls in hHO-DMDH-N3, and D140A-hHO DMDH-N3, D140A-hHO DMDH-N3 spectra as a function of temperature, TOCSY spectra of paramagnetic and aliphatic residues in hHO-DMDH-N3, and NOESY and TOCSY spectra for aromatic rings in D140A-hHO-DMDH-N3), 8 total pages. This material is available free of charge via the internet at http://pubs.acs.org.
REFERENCES
- 1.Tenhunen R, Marver HS, Schmid R. Microsomal heme oxygenase. Characterization of the enzyme. J. Biol. Chem. 1969;244:6388–6394. [PubMed] [Google Scholar]
- 2.Ortiz de Montellano PR, Auclair K. Heme Oxygenase Structure and Mechanism. In: Kadish KM, Smith KM, Guilard R, editors. The Porphryin Handbook. Elsevier Science; San Diego, CA: 2003. pp. 175–202. [Google Scholar]
- 3.Unno M, Matsui T, Ikeda-Saito M. Structure and catalytic mechanism of heme oxygenase. Nat. Prod. Rep. 2007;24:553–570. doi: 10.1039/b604180a. [DOI] [PubMed] [Google Scholar]
- 4.Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Biliruben is an Antioxidant of Possible Physiological Importance. Science. 1987;235:1043–1046. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- 5.Uzel C, Conrad ME. Absorption of heme iron. Seminars in Hematology. 1998;35:27–34. [PubMed] [Google Scholar]
- 6.Baranano DE, Snyder SH. Neural roles for heme oxygenase: Contrasts to nitric oxide synthase. Proc. Natl. Acad. Sci. (USA) 2001;98:10996–11002. doi: 10.1073/pnas.191351298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beale SI. Biosynthesis of open-chain tetrapyrroles in plants, algae, and cyanobacteria. Ciba Foundation Symposium. 1994;180:156–168. doi: 10.1002/9780470514535.ch9. [DOI] [PubMed] [Google Scholar]
- 8.Wilks A. Heme Oxygenase: Evolution, Structure, and Mechanism. Antioxidants Redox Signal. 2002;4:603–614. doi: 10.1089/15230860260220102. [DOI] [PubMed] [Google Scholar]
- 9.Frankenberg-Dinkel N. Bacterial Heme Oxygenases. Antoxidants Redox Signal. 2004;6:825–834. doi: 10.1089/ars.2004.6.825. [DOI] [PubMed] [Google Scholar]
- 10.Rivera M, Zeng Y. Heme Oxygenase, steering dioxygen activation toward heme hydroxylation. J. Inorg. Biochem. 2005;99:337–354. doi: 10.1016/j.jinorgbio.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 11.Schuller DJ, Wilks A, Ortiz de Montellano PR, Poulos TL. Crystal structure of human heme oxygenase-1. Nature Struct. Biol. 1999;6:860–867. doi: 10.1038/12319. [DOI] [PubMed] [Google Scholar]
- 12.Sugishima M, Sakamoto H, Higashimoto Y, Omata Y, Hayashi S, Noguchi M, Fukuyama K. Crystal structure of rat heme oxygenase-1 in complex with heme bound to azide: Implication for regiospecific hydroxylation of heme at the α-meso carbon. J. Biol. Chem. 2002:45086–45090. doi: 10.1074/jbc.M207267200. [DOI] [PubMed] [Google Scholar]
- 13.Lad L, Wang J, Li H, Friedman J, Bhaskar B, Ortiz de Montellano PR, Poulos TL. Crystal Structures of the Ferric, Ferrous and Ferrous-NO Forms of the Asp140Ala Mutant of Human Heme Oxygenase-1: Catalytic Implications. J. Mol. Biol. 2003;330:527–538. doi: 10.1016/s0022-2836(03)00578-3. [DOI] [PubMed] [Google Scholar]
- 14.Sugishima M, Sakamoto H, Noguchi M, Fukuyama K. Crystal Structures of CO-, CN-, and NO-Bound Forms of Rat Heme Oxygenase-1 (HO-1) in Complex with Heme: Structural Implications for Discrimination between CO and O2 in HO-1. Biochemistry. 2003;42:9898–9905. doi: 10.1021/bi027268i. [DOI] [PubMed] [Google Scholar]
- 15.Friedman J, Lad L, Li H, Wilks A, Poulos TL. Structural Basis for Novel δ-Regioselective Heme Oxygenation in the Opportunistic Pathogen Pseudomonas aeruginosa. Biochemistry. 2004;43:5239–5245. doi: 10.1021/bi049687g. [DOI] [PubMed] [Google Scholar]
- 16.Unno M, Matsui T, Chu GC, Coutoure M, Yoshida T, Rousseau DL, Olson JS, Ikeda-Saito M. Crystal Structure of the Dioxygen-bound Heme Oxygenase from Corynebacterium diphtheriae. J. Biol. Chem. 2004;279:21055–21061. doi: 10.1074/jbc.M400491200. [DOI] [PubMed] [Google Scholar]
- 17.Friedman JM, Lad L, Deshmukh R, Li HY, Wilks A, Poulos TL. Crystal structures of the NO- and CO-bound heme oxygenase from Neisseriae meningitidis - Implications for O2 activation. J. Biol. Chem. 2003;278:34654–34659. doi: 10.1074/jbc.M302985200. [DOI] [PubMed] [Google Scholar]
- 18.Li Y, Syvitski RT, Auclair K, Wilks A, Ortiz de Montellano PR, La Mar GN. Solution NMR characterization of an unusual distal H-bond network in the active site of the cyanide-inhibited, human heme oxygenase complex of the symmetric substrate, 2,4-dimethyldeuterohemin. J. Biol. Chem. 2002;277:33018–33031. doi: 10.1074/jbc.M204216200. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Syvitski RT, Chu GC, Ikeda-Saito M, La Mar GN. Solution 1H NMR investigation of the active site molecular and electronic structures of the substrate-bound, cyanide-inhibited bacterial heme oxygenase from C. diphtheriae. J. Biol. Chem. 2003;279:6651–6663. doi: 10.1074/jbc.M211249200. [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Syvitski RT, Auclair K, Ortiz de Montellano PR, La Mar GN. Solution 1H, 15N NMR spectroscopic characterization of substrate-bound cyanide-inhibited, human heme oxygenase: water occupation of the distal cavity. J. Am. Chem. Soc. 2003;125:13392–13403. doi: 10.1021/ja036176t. [DOI] [PubMed] [Google Scholar]
- 21.Syvitski RT, Li Y, Auclair K, Ortiz de Montellano PR, La Mar GN. 1H NMR detection of immobilized water molecules within a strong hydrogen-bonding network in the distal side of substrate-bound human heme oxygenase. J. Am. Chem. Soc. 2002;124:14296–14297. doi: 10.1021/ja028108x. [DOI] [PubMed] [Google Scholar]
- 22.Liu Y, Zhang X, Yoshida T, La Mar GN. 1H NMR characterization of the solution active site structure of substrate-bound, cyanide-inhibited heme oxygenase from Neisseria meningitidis; Comparison to crystal structures. Biochemistry. 2004;43:10112–10126. doi: 10.1021/bi049438s. [DOI] [PubMed] [Google Scholar]
- 23.Ortiz de Montellano PR. The mechanism of heme oxygenase. Curr. Opin. Chem. Biol. 2000;4:221–227. doi: 10.1016/s1367-5931(99)00079-4. [DOI] [PubMed] [Google Scholar]
- 24.Wilks A, Demontellano PRO. Rat Liver Heme Oxygenase - High Level Expression Of a Truncated Soluble Form and Nature Of the Meso-Hydroxylating Species. J. Biol. Chem. 1993;268:22357–22362. [PubMed] [Google Scholar]
- 25.Wilks A, Torpey J, Ortiz de Montellano PR. Evidence for the electrophilic oxygen addition to the porphyrin ring in the formation of α-meso-hydroxyheme. J. Biol. Chem. 1994;269:29553–29556. [PubMed] [Google Scholar]
- 26.Wang J, Evans JP, Ogura H, La Mar GN, Ortiz de Montellano PR. Alteration of the Regiospecificity of Human Heme Oxygenase-1 by Unseating of the Heme but not Disruption of the Distal Hydrogen Bonding Network. Biochemistry. 2006;45:61–73. doi: 10.1021/bi051645k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ogura H, Evans JP, Ortiz de Montellano PR, La Mar GN. Implication for Using Heme Methyl Hyperfine Shifts as Indicators of Heme Seating as Related to Stereoselectivity in the Catabolism of Heme by Heme Oxygenase: In-Plane Heme versus Axial His Rotation. Biochemistry. 2008;47:421–430. doi: 10.1021/bi7017333. [DOI] [PubMed] [Google Scholar]
- 28.Caignan GA, Deshmukh R, Wilks A, Zeng Y, Huang H.-w., Moenne-Loccoz P, Bunce RA, Eastman MA, Rivera M. Oxidation of heme to β- and δ-biliverdin by Pseudomonas aeruginosa Heme Oxygenase as a Consequence of an Unusual Seating of the Heme. J. Am. Chem. Soc. 2002;124:14879–14892. doi: 10.1021/ja0274960. [DOI] [PubMed] [Google Scholar]
- 29.Sharma PK, Kevorkiants R, de Visser SP, Kumar D, Shaik S. Porphyrin Traps Its Terminator! Concerted and Stepwise Porphyrin Degradation Mechanisms Induced by Heme-Oxygenase and Cytochrome P450. Angew. Chem. Int. Ed. 2004;43:1129–1132. doi: 10.1002/anie.200352943. [DOI] [PubMed] [Google Scholar]
- 30.Kumar D, de Visser SP, Shaik S. Theory Favors a Stepwise Mechanism of Porphyrin Degradation by a Ferric Hydroperoxide Model fo the Active Species of Heme Oxygenase. J. Am. Chem. Soc. 2005;127:8204–8213. doi: 10.1021/ja0446956. [DOI] [PubMed] [Google Scholar]
- 31.Chen H, Moreau Y, Derat E, Shaik s. Quantum Mechanical/Molecular Mechanical Study of Mechanisms of Heme Degradation by the Enzyme Heme Oxygenase: The Strategic Function of the Water Cluster. J. Am. Chem. Soc. 2008;130:1953–1965. doi: 10.1021/ja076679p. [DOI] [PubMed] [Google Scholar]
- 32.Koenigs Lightning L, Huang H-W, Moënne-Loccoz P, Loehr TM, Schuller DJ, Poulos TL, Ortiz de Montellano PR. Disruption of an active site hydrogen bond converts human heme oxygenase-1 into a peroxidase. J. Biol. Chem. 2001;276:10612–10619. doi: 10.1074/jbc.M010349200. [DOI] [PubMed] [Google Scholar]
- 33.Fujii H, Zhang X, Tomita T, Ikeda-Saito M, Yoshida T. A role for highly conserved carboxylate, Aspartate-140, in oxygen activation and heme degradation by heme oxygenase-1. J. Am. Chem. Soc. 2001;123:6475–6484. doi: 10.1021/ja010490a. [DOI] [PubMed] [Google Scholar]
- 34.Zeng Y, Caignan GA, Bunce RA, Rodriguez JC, Wilks A, Rivera M. Azide-inhibited Bacterial Heme Oxygenases Exhibit an S=3/1 (dxz,dyz)3(dxy)1(dz2)1 Spin State: Mechanistic Implications for Heme Oxidation. J. Am. Chem. Soc. 2005;127:9794–9807. doi: 10.1021/ja0425987. [DOI] [PubMed] [Google Scholar]
- 35.Ma L-H, Liu Y, Zhang X, Yoshida T, La Mar GN. 1H NMR study of the effect of variable ligand on heme oxygenase electronic and molecular structure. J. Inorg. Biochem. 2009;103:10–19. doi: 10.1016/j.jinorgbio.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoshida T, Noguchi M, Kikuchi G. Oxygenated Form of Heme-Heme Oxygenase Complex and Requirement for Second Electron to Initiate Heme Degradation from the Oxygenated Complex. J. Biol. Chem. 1980;255:4418–4420. [PubMed] [Google Scholar]
- 37.Sun J, Wilks A, Ortiz de Montellano PR, Loehr TM. Resonance Raman and EPR Spectroscopic Studies on Heme Heme Oxygenase Complexes. Biochemistry. 1993;32:14151–14157. doi: 10.1021/bi00214a012. [DOI] [PubMed] [Google Scholar]
- 38.Takahashi S, Wang JL, Rousseau DL, Ishikawa K, Yoshida T, Takeuchi N, Ikeda-Saito M. Heme-Heme Oxygenase Complex - Structure and Properties of the Catalytic Site from Resonance Raman Scattering. Biochemistry. 1994;33:5531–5538. doi: 10.1021/bi00184a023. [DOI] [PubMed] [Google Scholar]
- 39.Chu GC, Tomita T, Sönnichsen FD, Yoshida T, Ikeda-Saito M. The heme complex of Hmu O, a bacterial heme degradation enzyme from Corynebacterium diphtheriae. J. Biol. Chem. 1999;274:24490–24496. doi: 10.1074/jbc.274.35.24490. [DOI] [PubMed] [Google Scholar]
- 40.Hernández G, Wilks A, Paolesse R, Smith KM, Ortiz de Montellano PR, La Mar GN. Proton NMR Investigation of Substrate-bound Heme Oxygenase: Evidence for Electronic and Steric Contributions to Stereoselective Heme Cleavage. Biochemistry. 1994;33:6631–6641. doi: 10.1021/bi00187a033. [DOI] [PubMed] [Google Scholar]
- 41.Gorst CM, Wilks A, Yeh DC, Ortiz de Montellano PR, La Mar GN. Solution 1H NMR investigation of the molecular and electronic structure of the active site of substrate-bound human heme oxygenase: the nature of the distal hydrogen bond donor to bound ligands. J. Am. Chem. Soc. 1998;120:8875–8884. [Google Scholar]
- 42.La Mar GN, Satterlee JD, de Ropp JS. NMR of Hemoproteins. In: Kadish KM, Smith KM, Guilard R, editors. The Porphyrins Handbook. Academic Press; San Diego: 2000. pp. 185–298. [Google Scholar]
- 43.Bertini I, Luchinat C. NMR of Paramagnetic Substances. Coord. Chem. Rev. 1996;150:1–296. [Google Scholar]
- 44.Shokhirev NV, Walker FA. The Effect of Axial Ligand Plane Orientation on the Contact and Pseudocontact Shifts of Low-spin Ferriheme Proteins. J. Biol. Inorg. Chem. 1998;3:581–594. [Google Scholar]
- 45.Walker FA. Proton NMR and EPR Spectroscopy of Paramagnetic Metalloporphyrin. In: Kadish KM, Smith KM, Guilard R, editors. The Porphyrin Handbook. Academic Press; Boston: 2000. pp. 1–183. [Google Scholar]
- 46.Liu Y, Ma L-H, Zhang X, Yoshida T, Satterlee JD, La Mar GN. 1H NMR study of the influence of hemin vinyl-methyl substitution on the interaction between the C-terminus and substrate and the ”aging” of the heme oxygenase from N. meningitidis. Induction of active site structural heterogeneity by a two-fold symmetric hemin. Biochemistry. 2006;45:13875–13888. doi: 10.1021/bi061747q. [DOI] [PubMed] [Google Scholar]
- 47.Zhu W, Li Y, Wang J, Ortiz de Montellano PR, La Mar GN. Solution NMR study of environmental effects on substrate seating in human heme oxygenase; Influence of polypeptide truncation, substrate modification and axial ligand. J. Inorg. Biochem. 2006;100:97–107. doi: 10.1016/j.jinorgbio.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 48.La Mar GN, Asokan A, Espiritu B, Yeh DC, Auclair K, Ortiz de Montellano PR. Solution 1H NMR of the active site of substrate-bound, cyanide-ligated, human heme oxygenase. Comparison to the crystal structure of the water-ligated form. J. Biol. Chem. 2001;276:15676–15687. doi: 10.1074/jbc.M009974200. [DOI] [PubMed] [Google Scholar]
- 49.Caignan GA, Deshmukh R, Zeng Y, Wilks A, Bunce RA, Rivera M. The Hydroxide Complex of Pseudomonas aeruginosa Heme Oxygenase as a Model of the Low-Spin Iron(III) Hydroperoxide Intermediate in Heme Catabolism: 13C NMR Spectroscopic Studies Suggest the Active Participation of the Heme in Macrocycle Hydroxylation. J. Am. Chem. Soc. 2003;125:11842–11852. doi: 10.1021/ja036147i. [DOI] [PubMed] [Google Scholar]
- 50.Smith KM, Kehres LA. Syntheses of Methyl Devinylporphyrins Related to Protoporphyrin-IX. Initial Studies on the Mechanism of the Copper (II) Catalyzed Cyclizations of 1',8'-Dimethyl-a,c-biladienes. J. Chem. Soc., Perkins Trans. 1983;1:2329–2335. [Google Scholar]
- 51.Jeener J, Meier BH, Bachmann P, Ernst RR. Investigation of Exchange Processes by Two Dimensional NMR Spectroscopy. J. Chem. Phys. 1979;71:4546–4553. [Google Scholar]
- 52.Griesinger C, Otting G, Wüthrich K, Ernst RR. Clean TOCSY for 1H Spin System Identification in Macromolecules. J. Am. Chem. Soc. 1988;110:7870–7872. [Google Scholar]
- 53.Evans DF. The determination of the paramagnetic susceptibility of substances in solution by nuclear magnetic resonance. J. Chem. Soc. 1959:2003–2005. [Google Scholar]
- 54.Bertini I, Luchinat C, Turano P, Battaini G, Casella L. The Magnetic Properties of Myoglobin as Studied by NMR Spectroscopy. Chem. Eur. J. 2003;9:2316–2322. doi: 10.1002/chem.200204562. [DOI] [PubMed] [Google Scholar]
- 55.Sandström J. Dynamic NMR Spectroscopy. Academic Press; New York: 1982. [Google Scholar]
- 56.Lad L, Schuller DJ, Shimizu H, Friedman J, Li H, Ortiz de Montellano PR, Poulos TL. Comparison of the Heme-free and -bound Crystal Structures of Human Heme Oxygenase-1. J. Biol. Chem. 2003;278:7834–7843. doi: 10.1074/jbc.M211450200. [DOI] [PubMed] [Google Scholar]
- 57.Banci L, Bertini I, Luchinat C. Nuclear and electronic relaxation. VCH; Weinheim: 1991. [Google Scholar]
- 58.Iizuka T, Kotani M. Analysis of thermal equilibrium between high-spin and low-spin stsates in ferrimyoglobin complexes. Biochim. Biophys. Acta. 1969;181:275–286. doi: 10.1016/0005-2795(69)90250-5. [DOI] [PubMed] [Google Scholar]
- 59.Iizuka T, Morishima I. 220 MHz Proton NMR Studies of Hemoproteins High-spin - Low-spin Equilibrium in Ferric Myoglobin and Hemoglobin Derivatives. Biochim. Biophys. Acta. 1974;371:1–13. doi: 10.1016/0005-2795(74)90148-2. [DOI] [PubMed] [Google Scholar]
- 60.Ikeda-Saito M, Fujii H. EPR Characterization of the Heme Oxygenase Reaction Intermediates and Its Implication for the Catalytic Mechanism. In: Tesler J, editor. Paramagnetic Resonance of Metallobiomolecules. Oxford University Press; New York: 2003. pp. 97–112. [Google Scholar]
- 61.Takahashi S, Wang J, Rousseau DL, Ishikawa K, Yoshida T, Host JR, Ikeda-Saito M. Heme-Heme Oxygenase Complex Structure of the Catalytic Site and Its Implication for Oxygen Activation. J. Biol. Chem. 1994;269:1010–1014. [PubMed] [Google Scholar]
- 62.Sugishima M, Sakamoto H, Kakuta Y, Omata Y, Hayashi S, Noguchi M, Fukuyama K. Crystal Structue of Rat Apo-Heme Oxygenase-1 (HO-1): Mechanism of Heme Binding in HO-1 Inferred from Structural Comparison of the Apo and Heme Complex Forms. Biochemistry. 2002;41:7293–7300. doi: 10.1021/bi025662a. [DOI] [PubMed] [Google Scholar]
- 63.Springer BA, Sligar SG, Olson JS, Phillips GN. Mechanisms Of Ligand Recognition In Myoglobin. Chem. Rev. 1994;94:699–714. [Google Scholar]
- 64.Davydov RM, Yoshida T, Ikeda-Saito M, Hoffman BM. Hydroperoxy-Heme Oxygenase Generated by Cryoreduction Catalyzes the Formation of α-meso-Hydroxyheme as Detected by EPR and ENDOR. J. Am. Chem. Soc. 1999;121:10656–10657. [Google Scholar]
- 65.Wojciechowski G, Ortiz de Montellano PR. Radical Energies and the Regiochemistry of Addition to Heme Groups. Methylperoxy and Nitrite Radical Additions to the Heme of Horseradish Peroxidase. J. Am. Chem. Soc. 2007;129:1663–1672. doi: 10.1021/ja067067s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rivera M, Caignan GA, Astashkin AV, Raitsimring AM, Shokhireva TK, Walker FA. Models of the Low-Spin Iron(III) Hydroperoxide Intermediate of Heme Oxygenase: Magnetic Resonance Evidence for Thermodynamic Stabilization of the dxy Electronic State at Ambient Temperature. J. Am. Chem. Soc. 2002;124:6077–6089. doi: 10.1021/ja017334o. [DOI] [PubMed] [Google Scholar]
- 67.Matsui T, Furukawa M, Unno M, Tomita T, Ikeda-Saito M. Roles of Distal Asp in Heme Oxygenase from Corynebacterium diphtheriae, HmuO. J. Biol. Chem. 2005;280:2981–2989. doi: 10.1074/jbc.M410263200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.