

The selective oxidation of primary amines to hydroxylamines is a synthetic transformation that currently lacks a straightforward solution. While oxidations of primary amines to nitro groups, imines, or oximes with both organic and metal-mediated oxidants are well known,i there is no general method for interrupting these oxidations at the more reactive and oxidatively labile hydroxylamine stage.ii Direct methods for the preparation of NOBz derivatives, while convenient for simple substrates, fail with most functionalized amines and lead to imine formation with α-amino acid derivatives.iii In this communication, we disclose a reagent that makes possible the conversion of primary amines of α-peptides to nitrones and, by hydrolysis, to chiral hydroxylamines via an operationally simple protocol and with retention of stereochemistry (Scheme 1).
Scheme 1.
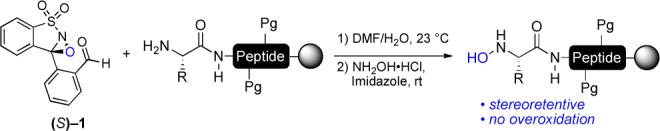
Preparation of N-terminal peptide hydroxylamines with sulfonyloxaziridine reagent 1.
Our interest in the synthesis of hydroxylamines from the corresponding primary amines emerged from our recent discovery of the α-ketoacid–hydroxylamine amide-forming ligation reaction, which allows two unprotected molecules to be chemoselectively conjugated via an amide bond. iv, v While the ligation reaction itself is straightforward, the preparation of peptide chains containing the requisite functional groups at the C-and N-termini presents an unmet synthetic challenge. We have communicated an approach to peptide α-ketoacids,vi but the only viable route we found to enantiopure N-terminal hydroxylamines is a three step protocol based on a report by Fukuyama.vii, viii Although very effective for simple amines, it was almost unworkable for complex peptides and could not be extended to solid phase peptide synthesis. We have also found that this method can lead to erosion of the amine stereochemistry if not carefully executed.
The Fukuyama method, as well as its antecedents reported by Polovoski and others,ix effects the oxidation of an imine to a nitrone or oxaziridine as the key N–O bond forming reaction. We reasoned that a single reagent effecting both imine formation and oxidation would simplify the overall process and avoid prolonged intermediacy of the stereochemically labile imine. Inspired by the work of Davis on selective oxidizing agents,x we designed and synthesized N-sulfonyloxaziridine 1 bearing a pendant aldehyde moiety via a high-yielding sequence (Scheme 2). While Davis reagents are known to overoxidize primary amines,xi we hoped that rapid, prior imine formation would prevent side reactions.
Scheme 2.
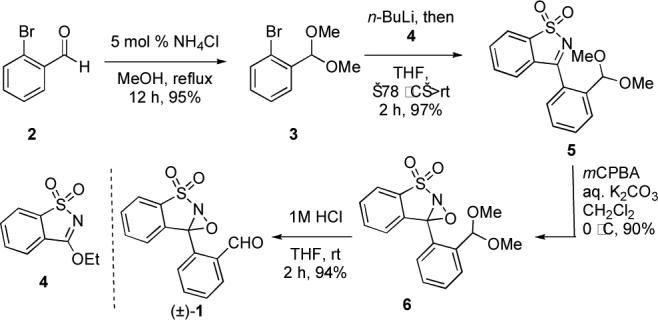
Synthesis of (±)–1.
Preliminary solution phase studies with simple amines confirmed the ability of reagent (±)–1 to convert a primary amine to a nitrone and revealed reaction conditions consisting of aqueous DMF with a small amount of added acid as preferred conditions. We immediately turned to the more challenging and relevant application of (±)-1 for the synthesis of solid supported N-terminal peptide hydroxylamines. Using tripeptide 7 as a model substrate, we examined the utility of reagent (±)-1 to effect the formation of the N-terminal hydroxylamine. In our initial efforts, we elected to evaluate the effectiveness of the reaction by a three-step sequence involving nitrone formation, hydrolysis to the hydroxylamine, and ligation with a peptide α-ketoacid 9 to afford a tetrapeptide 10 (Scheme 3). The purity of the resulting product and its approximate chemical yield were assayed following cleavage from Rink amide MBHA linked resin.
Scheme 3.
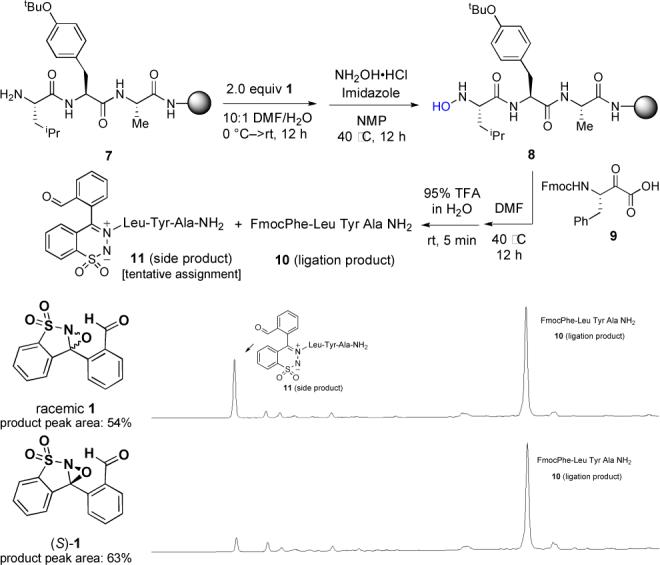
Selective oxidation of tripeptide model 7 with 1.
Our studies using tripeptide 7 were consistently plagued by the formation of a side product with the identical mass as the expected intermediate nitrone, xii and which we have tentative assigned as azomethine ylide 11 arising from N-amidation of the amine.xiii We hypothesized that it resulted from a stereoelectronic mismatch between the (S)-configured peptide amine and one of the two enantiomers of the chiral, but racemic, oxaziridine reagent 1. To test this we independently prepared the two enantiomers of 1 for the oxidation and subsequent ligation of tripeptide 7 (See Supporting Information for the synthesis of enantiopure (S)-1 and (R)-1). The matched, (S)-configured reagent provided the desired ligation product 10 cleanly almost without trace of the undesired side product 11. In contrast the (R)-configured reagent increased formation of the side product (see Supporting Information for HPLC data). The availability and disparate reactivity of both enantiomers of 1 provide a lead for future investigations.
Peptide hydroxylamines are configurationally stable, but there is potential for epimerization at the nitrone stage. A key component of our investigations was to demonstrate that the overall process did not erode the stereochemistry of the α-position. This was accomplished by preparing both epimers of tripeptide 7 at the leucine residue and comparing the stereochemical outcomes following oxidation by (±)-1, hydrolysis, and ligation with Fmoc-Phe-ketoacid 9. Analyses of the resulting tetrapeptides (See Supporting Information) confirmed that the procedures occurred with preservation of stereochemistry.
We envision that the greatest utility of reagent 1 for the preparation of peptide hydroxylamine will be for the rapid synthesis and screening of potential ligation sites for the preparation of larger peptides. Once a suitable ligation site is identified, we anticipate that the hydroxylamine can be introduced via more involved, but higher yielding, approaches. xiv We therefore explored the substrate scope of reagent 1 with respect to the identity of the N-terminal amine residue. Table 1 shows the application of (S)-1 to 4 different N-terminal amines in an otherwise constant pentapeptide (entries 1−4). Both racemic and (S)-configured 1 were viable (entries 1,2), however yields and crude purities of the resulting ligation products were improved with the use of the enantiopure reagent. A completely different sequence (entry 6) was also tested to confirm the general application to N-terminal peptide hydroxylamine synthesis.
Table 1.
Selective oxidations of solid supported N-terminal peptide amines with 1 and subsequent ligations.a
 | |||||
|---|---|---|---|---|---|
| Entry | AA1 = | Ligation product | Product peak areac / % | Mass yield (from resin) | Isolated yieldd / % |
| 1 | Ser | Fmoc-Phe–Ser-Asp-Tyr-Lys-Ala-NH2 | 55% (220 nm) | 4.8 mg from 49.4 mg 12a | 22 |
| 2b | Ser | Fmoc-Phe–Ser-Asp-Tyr-Lys-Ala-NH2 | 37% (220 nm) | not determined | not determined |
| 3 | Glu | Fmoc-Phe–Glu-Asp-Tyr-Lys-Ala-NH2 | 41% (220 nm) | 6.8 mg from 6.2 mg 12b | 25 |
| 4 | Phe | Fmoc-Phe–Phe-Asp-Tyr-Lys-Ala-NH2 | 42% (220 nm) | 5.1 mg from 64.6 mg 12c | 17 |
| 5 | Ala | Fmoc-Phe–Ala-Asp-Tyr-Lys-Ala-NH2 | 47% (220 nm) | 2.9 mg from 32.9 mg 12d | 18 |
| 6 | Leu | Fmoc-Phe–Leu-Lys-Ser-Glu-Tyr-NH2 | 31% (220 nm) | 3.4 mg from 38.7 mg 12e | 20 |
See Supporting Information for further experimental details.
(±)-1 was used.
Determined by HPLC analyses of unpurified peptides following cleavage.
Approximate yields over entire sequence based on mass recovery of the ligated peptide following preparative HPLC.
We believe that the selectivity of this reagent arises from the rapid formation of N-terminal imine II that effects an intramolecular oxygen atom transfer to provide nitrone IV (Figure 1). The beneficial effect of a small amount of water on the rate of this reaction suggests that the actual atom transfer step may occur onto hemiaminal I, rather than onto imine II directly. We believe that rapid precomplexation of the peptide and 1 reduces competing, and possibly more rapid, oxidation reactions at other sites of the peptide substrate. To test this hypothesis, we repeated the 4-step sequence with tripeptide standard 7 and an N-sulfonyloxaziridine that lacks the aldehyde moiety. With either this reagent alone or in combination with benzaldehyde, only a trace amount of the ligation product was obtained and the major product was a peptide oxime arising from oxidation of the N-terminal amine to a ketone (see Supporting Information).
Figure 1.
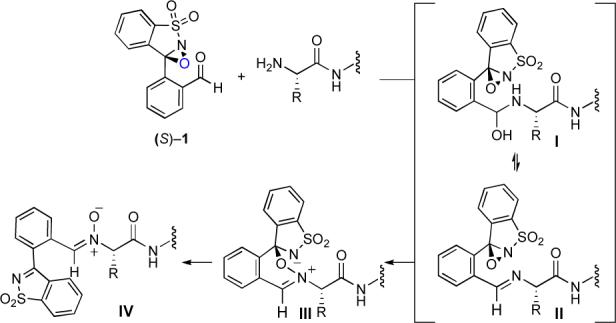
Postulated directed oxidation of amines by 1.
Reagent 1 offers a direct, convenient approach to the selective synthesis of hydroxylamines from primary amines. Our preliminary studies have focused on its application to the synthesis of N-terminal peptide hydroxylamine for chemoselective ligation; however, we expect this reagent and its design principles will find application in other directed oxidations.
Supplementary Material
Acknowledgments
We are grateful to the NIH (GM-076320), the Packard Foundation and generous gifts from Bristol Myers Squibb, Eli Lilly, and Roche for the support of this research program. T.F. was a fellow of the Uehara Foundation. J.W.B is a fellow of the Packard Foundation and the Beckman Foundation.
Footnotes
Supporting Information Available. Experimental procedures and characterization data for all new compounds. This material is available free of charge via the internet at http://pubs.acs.org.
References
- i.Rosenblatt DH, Burrows EP. In: Supplement F: The Chemistry of Amino, Nitroso, and Nitro Compounds and Their Derivatives. Part 2. Patai S, editor. Wiley and Sons; Chichester: 1982. pp. 1085–1149. [Google Scholar]
- ii.a Field JD, Kropp PJ. J. Org. Chem. 2000;65:5937–5941. doi: 10.1021/jo0002083. [DOI] [PubMed] [Google Scholar]; b Wittman MD, Halcomb RL, Danishefsky SJ. J. Org. Chem. 1990;55:1981–1983. [Google Scholar]
- iii.a Fennell KA, Miller MJ. J. Org. Chem. 1999;64:7451–7458. [Google Scholar]; b John ORS, Killeen NM, Knowles DA, Yau SC, Bagley MC, Tomkinson NCO. Org. Lett. 2007;9:4009–4012. doi: 10.1021/ol701774y. [DOI] [PubMed] [Google Scholar]
- iv.Bode JW, Fox RM, Baucom KD. Angew. Chem. Int. Ed. 2006;45:1248–1252. doi: 10.1002/anie.200503991. [DOI] [PubMed] [Google Scholar]
- v.Hackenberger CPR, Schwarzer D. Angew. Chem. Int. Ed. 2008;47:10030–10074. doi: 10.1002/anie.200801313. [DOI] [PubMed] [Google Scholar]
- vi.Ju L, Lippert AR, Bode JW. J. Am. Chem. Soc. 2008;130:4253–5255. doi: 10.1021/ja800053t. [DOI] [PubMed] [Google Scholar]
- vii.Fukuyama Org. Syn. Tokuyama H, Kuboyama T, Fukuyama T. Org. Syn. 2003;80:207–218. [Google Scholar]
- viii.Ottenheijm HCJ, Herscheid JDM. Chem. Rev. 1986;86:697–707. [Google Scholar]
- ix.a Polonski T, Chimiak A. Tetrahedron Lett. 1974;15:2453–2453. [Google Scholar]; b Grundke G, Keese W, Rimpler M. Synthesis. 1987;12:1115–1116. [PubMed] [Google Scholar]; c Millet P, Lusinchi X. Tetrahedron Lett. 1985;26:3791–3794. [Google Scholar]
- x.Davis FA, Sheppard AC. Tetrahedron. 1989;18:5703–5742. [Google Scholar]
- xi.Zajac WW, Walters TR, Darcy MG. J. Org. Chem. 1988;53:5856–5860. [Google Scholar]
- xii.We initially hypothesized that this product was the saccharine-derived amide; independent synthesis of this product proved otherwise.
- xiii.Vidal J, Damestoy S, Guy L, Hannachi J-C, Aubry A, Collet A. Chem. Eur. J. 1997;3:1691–1709. [Google Scholar]
- xiv.Procedures for the asymmetric synthesis and coupling of suitably protected peptide hydroxylamines will be reported separately.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.