

Abstract
Background:
The myotonic dystrophies (DM1, DM2) are the most common adult muscle diseases and are characterized by multisystem involvement. DM1 has been described in diverse populations, whereas DM2 seems to occur primarily in European Caucasians. Both are caused by the expression of expanded microsatellite repeats. In DM1, there is a reservoir of premutation alleles; however, there have been no reported premutation alleles for DM2. The (CCTG)DM2 expansion is part of a complex polymorphic repeat tract of the form (TG)n(TCTG)n(CCTG)n(NCTG)n(CCTG)n. Expansions are as large as 40 kb, with the expanded (CCTG)n motif uninterrupted. Reported normal alleles have up to (CCTG)26 with one or more interruptions.
Methods:
To identify and characterize potential DM2 premutation alleles, we cloned and sequenced 43 alleles from 23 individuals. Uninterrupted alleles were identified, and their instability was confirmed by small-pool PCR. We determined the genotype of a nearby single nucleotide polymorphism (rs1871922) known to be in linkage disequilibrium with the DM2 mutation.
Results:
We identified three classes of large non-DM2 repeat alleles: 1) up to (CCTG)24 with two interruptions, 2) up to (CCTG)32 with up to four interruptions, and 3) uninterrupted (CCTG)22–33. Large non-DM2 alleles were more common in African Americans than in European Caucasians. Uninterrupted alleles were significantly more unstable than interrupted alleles (p = 10−4 to 10−7). Genotypes at rs1871922 were consistent with the hypothesis that all large alleles occur on the same haplotype as the DM2 expansion.
Conclusions:
We conclude that unstable uninterrupted (CCTG)22-33 alleles represent a premutation allele pool for DM2 full mutations.
GLOSSARY
- AfrAm
= African American;
- DM
= myotonic dystrophy;
- EurCauc
= European Caucasian;
- FISH
= fluorescent in situ hybridization;
- MIM
= Mendelian Inheritance in Man;
- NA
= not available;
- SNP
= single nucleotide polymorphism;
- SP-PCR
= small-pool PCR.
Myotonic dystrophy type 1 (DM1, Mendelian Inheritance in Man [MIM] 160900) and type 2 (DM2, MIM 602668) are the most common adult-onset muscle diseases, characterized by autosomal dominant inheritance, muscular dystrophy, myotonia, and multisystem involvement.1,2 DM is caused by unstable microsatellite repeat expansions—in DM1 a (CTG)n expansion in the 3′ UTR of DMPK in chromosome 19q13.3,3–5 and in DM2 a (CCTG)n expansion in intron 1 of ZNF9 in 3q21.3.6 The principal features of the DMs are progressive distal muscle weakness and atrophy in DM1 and proximal muscle weakness in DM2. Muscle stiffness, myotonia, cataracts, cardiac conduction disturbances, endocrinologic abnormalities (including insulin resistance, male hypogonadism frontal balding), and elevated γ-glutamyl-transferase are common findings. However, in any individual patient, some of these symptoms may be absent, and in DM2, myotonia may be variable over time. Although DM1 and DM2 share the same core features, very apparent differences exist, and to the clinician these are clearly different diseases. Similar to DM1,7 DM2 has been reported with rare exception in populations of European descent.8–11 In populations where prevalence has been estimated, the occurrence of DM2 is at least as high as that of DM1 and may be even higher.8 DM2 expansions are generally much larger than those in DM1, with expansions commonly as large as 40 kb. The smallest reported expansion had an uninterrupted mosaic (CCTG)75 as estimated by Southern blot.6 For DM1, there is a reservoir of premutation alleles in the population.12 However, there have been no reports of premutation alleles for DM2, and the minimum size of a pathogenic expansion is not known.
The repeat tract within intron 1 of ZNF9 is a complex array with several polymorphic elements, first described as (TG)n(TCTG)n (CCTG)n.6 In most individuals, there are two or more stretches of (CCTG)n interrupted by one or more (TCTG) or (GCTG) motifs. The most common interruption seems to be of the form (GCTG)1(CCTG)1(TCTG)1. It has been suggested that the loss of these cryptic repeats always accompanies expansion.6 We hypothesized that some larger alleles may represent a source of new expansion mutations. If premutations exist, we further predicted that they would have long uninterrupted (CCTG)n tracts, be somatically unstable, and occur on the same conserved haplotype as DM2 mutations. To determine the existence of DM2 premutation alleles, we cloned and sequenced a number of unusually large alleles along with short alleles and alleles from a DM2 family with an amplifiable expansion as controls.
METHODS
Samples.
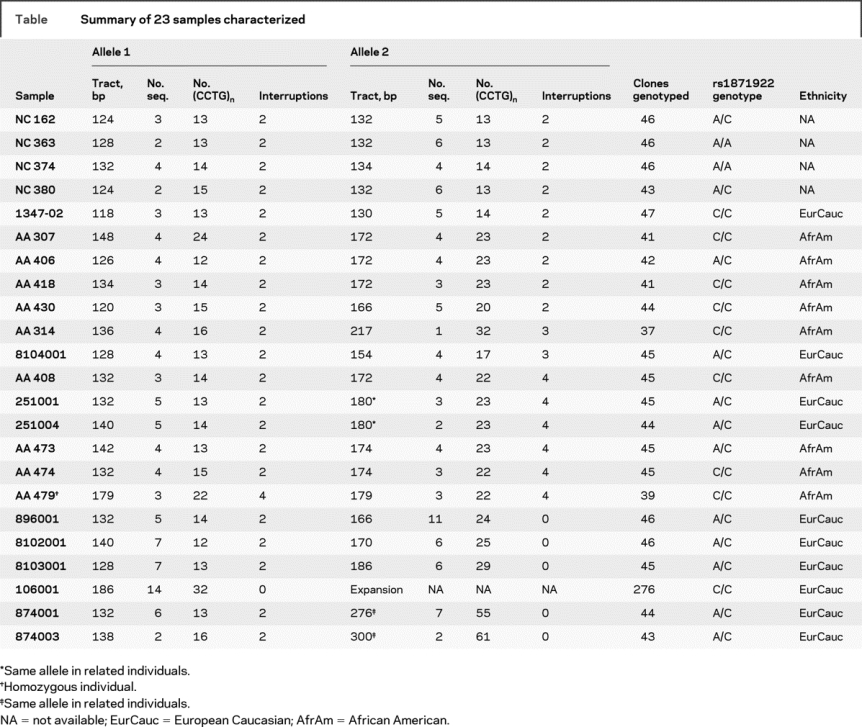
Enrollment of participants for this study was approved by the respective institutional review boards in accordance with the Declaration of Helsinki. After molecular genetic diagnosis, participants were recruited by their attending physicians. After obtaining informed consent, blood was drawn, and DNA was prepared using GenePure (Gentra). Three groups of population samples were used for genotyping. Group1 consisted of European Caucasian individuals, most of whom were normal relatives of DM2 patients. This group also included individuals with a molecular diagnosis of DM2, whose normal allele was counted in the population study. In all, group 1 contained 169 independent normal chromosomes. Sample group 2 consisted of 90 African American individuals (180 independent normal chromosomes). Group 3 was composed of 1,062 unrelated normal individuals from Finland (2,124 independent chromosomes). With regard to the Caucasian samples, approximately 14% of the samples were anonymous, and no demographic information was available. Of the remainder, 48% were male and 52% were female. We do not have age information for the majority of the samples; however, all were adults. All the African American samples and Finnish samples were anonymous; therefore, only information on ethnicity is available. Altogether, DM2-repeat alleles of 23 samples were selected from the above populations, cloned, and sequenced, and 43 independent amplifiable alleles were characterized as summarized in the table. One sample was homozygous for a large allele, two samples (251001 and 251004) were related and shared their large allele, one sample (106001) had a large DM2 expansion as the second allele, and two related samples (874001 and 874003) had small expansion mutations.
Table Summary of 23 samples characterized
Determination of allele size.
DM2-repeat alleles were genotyped by PCR across the repeat tract. Three-primer amplification using a fluorescent-labeled universal primer was performed on an ABI 9700 thermocycler, using a Qiagen Hot Start Master Mix as previously described.9 Products were analyzed on an ABI 3100 Genetic Analyzer. For sizing of products larger than 350 bp, we used the Mapmarker 1000 standard (Bioventures, Murfreesboro, TN).
Amplification, cloning, genotyping, and sequencing.
For cloning, alleles were amplified using unlabeled primers and the conditions described above in a two primer reaction. Products were cloned using the StrataClone PCR cloning kit according to the manufacturer’s instructions. For each sample, at least 45 colonies were amplified directly for genotyping. DNA from at least eight independent clones of representative alleles was prepared using a QIAprep Spin Miniprep kit. Plasmid DNA was sequenced directly using Big Dye terminator version 3 (ABI) and was visualized by capillary electrophoresis on an ABI 3100 Genetic Analyzer. Sequences were assembled using Sequencher software (Gene Codes, Ann Arbor, MI).
Single genome equivalent amplification.
Samples were subjected to single genome equivalent amplification (small-pool PCR [SP-PCR]) as previously described.13 Briefly, DNA samples were diluted to approximately single diploid genome levels (6 pg). Multiple heminested SP-PCRs were conducted using the three-primer reaction described above. At least 120 PCR replicates per sample were amplified to ensure counting at least 100 independent alleles. A Primus-96 plus thermocycler (MWG Biotech, High Point, NC) was used for PCR setup and amplification. Cycling conditions were 95°C/7 minutes, followed by 42 cycles of (95°C/45 seconds, 62°C/30 seconds, 72°C/30 seconds), and a final extension step of 72°C/7 minutes. Labeled amplification products were subjected to capillary electrophoresis as described above.
Data analysis.
After ABI GeneScan analysis, the data were scored for allele counts and variants. A model in which the number of alleles in replicate pools follows a Poisson distribution and in which particular allele frequencies constituted a fixed proportion of the total has been described.14 Maximum likelihood estimates of the mean number of alleles in each pool and the frequencies of each allele were derived accordingly. The mutant frequencies were compared between groups for significance using the arc-sin transformed mutant frequencies and a bootstrap standard error.14
Genotyping of SNP rs1871922.
Genotyping of single nucleotide polymorphism (SNP) rs1871922 was previously described.9 The polymorphism alters a HaeIII restriction site such that the C allele produces a fragment of 88 bp, whereas the A allele produces a fragment of 125 bp. The locus was PCR amplified; PCR products were digested with HaeIII (NEB), electrophoresed through 4% MetaPhor agarose gels (FMC), and visualized by ethidium bromide staining.
RESULTS
Occurrence of larger-than-average alleles is more common in African American samples.
Normal Caucasian chromosomes show a unimodal distribution of alleles (mean tract length 132 bp) with most differing by 2 bp, indicating that the (TG)n motif is contributing significantly to the polymorphism at this locus. Alleles in the tail of the distribution were 4 bp apart, indicating additional variation in the (CCTG)n or (TCTG)n motifs. In contrast, a survey of African American alleles yielded a bimodal distribution with a secondary peak at around 174 bp (figure 1). Alleles with repeat tracts ≥160 bp are relatively rare in populations of European Caucasian descent. We identified only 17 such alleles among 973 non-DM2 chromosomes (1.8%). However, among 176 African American chromosomes, we identified 15 such alleles (8.5%), suggesting that large alleles are more common in the sub-Saharan populations (p = 1.769 × 10−6), consistent with the ethnic diversity of sub-Saharan Africans.

Figure 1 Distribution of normal repeat track sizes
(A) Alleles with a repeat track ≥160 bp are rare in European Caucasians (17 of 973 non-DM2 chromosomes, 1.8%). (B) Such large alleles occur more frequently in the African American population (8 of 180 chromosomes, 8.3%; p = 1.769 × 10−6).
Sequence structure of large alleles at the DM2 locus.
To investigate the structure of these large alleles, we selected representative individuals from both European Caucasian and African American populations having alleles with repeat tracts ≥160 bp. Alleles were amplified, cloned, genotyped, and sequenced along with a number of smaller alleles with repeat tracts <160 bp. In all, 23 samples were characterized as summarized in the table. At least 45 clones were genotyped for each sample, and at least 8 of the clones from each sample were sequenced. In all, 41 independent non–disease-causing DM2-repeat alleles were characterized along with amplifiable DM2 pathogenic expansions from two brothers. All alleles, regardless of size, showed hypervariability of cloned amplification products. Sequencing revealed that, for interrupted alleles, this variability was entirely confined to the (TG)n tract. We identified four individuals with uninterrupted (CCTG)24–32 tracts, and these alleles showed hypervariability of the (CCTG)n motif as well as the (TG)n. We observed three types of interrupted alleles: 1) small alleles with (CCTG)12–16 containing two interruptions, 2) medium to large alleles with (CCTG)20–24 and two interruptions, and 3) large alleles with (CCTG)17–32 containing four (or more rarely three) interruptions. Figure 2 shows the various repeat tract structures observed.

Figure 2 Repeat tract structures of the DM2 locus observed in 44 sequenced alleles
We observed three classes of repeat tracts at the DM2 locus: 1) interrupted short alleles with up to (CCTG)24 and two interruptions, 2) interrupted long alleles with up to (CCTG)32 and up to four interruptions, and 3) uninterrupted (CCTG)22–33. Repeat tracts ≥160 bp often showed duplication of the (GCTG)-(CCTG)-(TCTG)-(CCTG)n portion of the tract. Occasional variations in the interruption sequence were also observed. Repeat tract length in bp and number of alleles observed are indicated for each variant (n = 44). The most common short allele (A) is also found in the Rhesus monkey (NW_001096632), suggesting that it is ancestral. Variants B, D, and F were only seen in the African American samples. Uninterrupted alleles (G and H) were seen only in Caucasians.
Large alleles likely occur on the same haplotype as DM2 mutations.
We previously identified a single shared haplotype extending across at least 132 kb among DM2 patients from different European populations.9 With the exception of the (CCTG)n expansion, the DM2 mutant haplotype was identical to the most common haplotype (47.2%) in normal individuals, a situation reminiscent of that seen in DM1.12,15,16 Taken together, these data suggested a single founding mutation in DM2 patients of European origin, occurring between 4,000 and 12,000 years ago.9 The majority of the known SNP markers in the region are monomorphic in the Caucasian population, and those that are not have minor allele frequencies <5%. The single highly informative marker in this haplotype is rs1871922, located in intron 1 of ZNF9, approximately 12 kb from the repeat tract, with approximately equal allele frequencies in European Caucasians and the C allele in linkage disequilibrium with the DM2 expansion. This allele is monomorphic in the chimpanzee (UCSC Browser, dbSNP), indicating that this allele is ancestral. For the four premutation alleles, all individuals had at least one C allele at rs1871922 and one was homozygous for C, consistent with the hypothesis that the premutation allele occurs on the same haplotype as the DM2 expansion.
Among the 88 African American samples genotyped, 69 were C/C, 2 were A/A, and 17 were A/C. These findings did not deviate significantly from Hardy–Weinberg equilibrium. This locus was not typed as part of the HapMap project, and no frequency information was available at this locus for sub-Saharan populations, but the C allele should be very common, based on our results. It is even possible that the C allele is monomorphic in sub-Saharan populations and that the presence of the A allele in our African American chromosomes is due to population admixture. Of the 10 independent large interrupted alleles sequenced, 7 are in individuals homozygous for the C allele at rs1871922. The remaining 4 individuals were heterozygous. No individual with a large allele was homozygous for the A allele. Thus, it is altogether possible that there is a single haplotype associated with all large alleles, of both uninterrupted and multiple-interruption types. However, lacking specific phase information for the African American samples, it is impossible to demonstrate this relationship conclusively. All SNP genotypes are included in the table.
Large uninterrupted alleles are unstable.
We observed that the entire locus was surprisingly unstable in cloning, irrespective of allele size or presence of interruptions. Allele distributions observed on cloning are presented in figure e-1 on the Neurology® Web site at www.neurology.org. Sequencing showed that all alleles had many clones with deletions in the (TG)n tract. On the other hand, only uninterrupted alleles showed variability in the (CCTG)n tract. To distinguish between inherent (CCTG)n instability and possible cloning artifacts, we performed single genome equivalent amplification (SP-PCR). Results of the SP-PCR are shown in figure 3. Interrupted alleles, both small and large, had average mutation frequencies of 12% to 14% and were considerably more stable than uninterrupted alleles, with average mutation frequency of approximately 40%. This difference was highly significant for all four uninterrupted alleles tested. The p values for the individual samples were 6.5 × 10−7 (896001), 4.3 × 10−4 (8103001), 1.4 × 10−4 (8102001), and 3.9 × 10−7 (106001). A muscle biopsy from an individual with a large interrupted allele [tract length of 180 bp or (CCTG)23 with four interruptions] was examined by RNA-fluorescent in situ hybridization (FISH) and was found not to form ribonuclear inclusions characteristic of uninterrupted disease-causing repeats (data not shown).
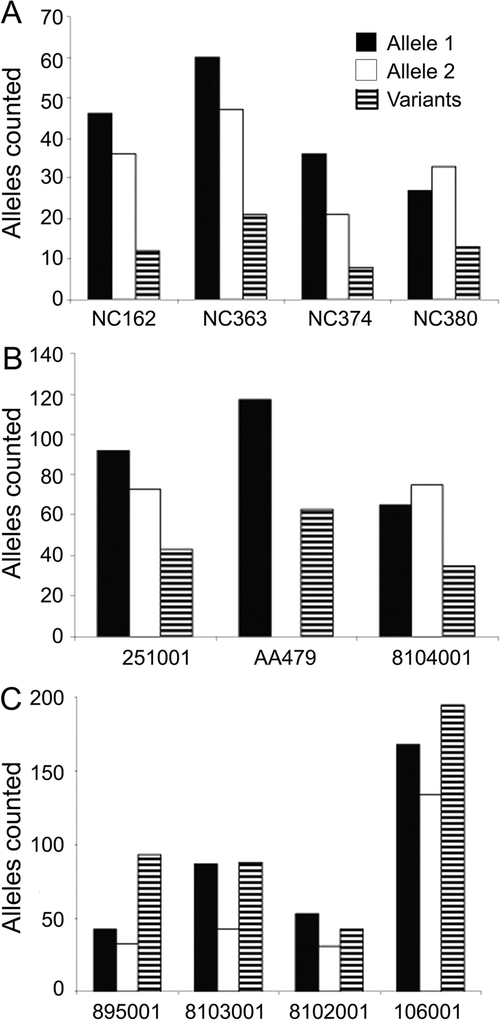
Figure 3 Results of small-pool PCR experiment
The DM2 locus was constitutively more unstable than other microsatellite loci commonly used to assess instability. Sequencing revealed that this instability is confined to the (TG)n portion of the tract. Bars represent number of alleles counted in single genome equivalent amplification (small-pool PCR). Solid black and white bars represent the progenitor alleles and striped bars represent novel alleles identified. Both small (A) and large (B) interrupted alleles had average mutation frequencies of 12% to 14%. Uninterrupted alleles (C) had mutation frequencies ranging from 30% to 53%, with an average of 39.4%. These alleles showed instability of the (CCTG)n tract as well as the (TG)n. The mutation frequencies for uninterrupted alleles were highly significantly different from interrupted controls. The p values for the four premutation alleles were 6.5 × 10−7 (896001), 4.3 × 10−4 (8103001), 1.4 × 10−4 (8102001), and 3.9 × 10−7 (106001).
DISCUSSION
We investigated the structure and stability of large repeat alleles at the DM2 locus. Such alleles are rare in the European Caucasian population (<2%) but significantly more common among African Americans (8.5% in our sample; p = 1.769 × 10−6). We cloned and sequenced large alleles from nine independent African American samples. These alleles either had two interruptions with unusually long (CCTG)n or (TCTG)n tracts, or they had average (CCTG)n tract lengths with three or four interruptions. Four of the five large Caucasian alleles had uninterrupted (CCTG)23–33 tracts. The remaining large Caucasian allele was of the multiple interruption type. The four uninterrupted alleles were shown to be extremely unstable by SP-PCR when compared with alleles with interruptions. Thus, the instability of the (CCTG)n tract seemed to be preexistent in the somatic DNA of these individuals and was not an artifact of cloning. Studies of repeat instability, primarily in the trinucleotide expansion diseases DM1 and Huntington disease, have suggested that there exists a threshold for repeat expansion of between 100 and 200 bp. Uninterrupted repeats of less than this length are stable, whereas larger repeats expand with frequencies of 80% to 100%.17,18 The unstable (CCTG)23–33 alleles described here, with lengths of 92 to 132 bp, seem to support the threshold hypothesis and extend it to include tetranucleotide repeats. Interestingly, the mutation frequency for normal interrupted alleles at the DM2 locus by SP-PCR (12%–14%) is extraordinarily high. Other microsatellite repeats used to assess instability have typical mutation frequencies <4%.13 This observation is consistent with the instability observed in the clones. It is possible that the reason for this high mutation frequency is related to the tendency of the (TG)n tract to form Z-DNA.19
We identified a DM2 family with nonmendelian segregation of a small amplifiable DM2 expansion. Although the exact size of the mutant expansion was not determined, the proband and his brother had predominant alleles in their peripheral blood of (CCTG)55 and (CCTG)61, respectively. Clones of the four premutation alleles exhibited instability of their (CCTG)n tracts comparable with those observed in these two small expansion alleles.
Although phase could not be determined, we observed that all the large alleles occurred coincident with the C allele at rs1871922. This observation suggested a scenario by which such large alleles might have evolved from small alleles by unequal crossing over (figure 4A). The conversion of a normal stable allele to an expandable premutation allele at the DM2 locus would require both the loss of the stabilizing interruption(s) and an increase of the repeat length. Several mechanisms have been proposed by which stabilizing interruptions could be lost. It has been previously shown that loss of mismatch repair activity predisposes to loss of stabilizing interruptions in yeast.20 Alterations in replication origin through activation of cryptic initiation sites, insertion of repetitive elements, or epigenetic events have likewise been suggested.18 Although unequal meiotic recombination is unlikely to account for repeat expansion per se, it remains a possible mechanism by which interruptions could be lost.18 The reciprocal products of such an unequal crossover between two short normal alleles would be one long allele with four interruptions and another with no interruptions. If instability is a function of contiguous (CCTG)n length, over time such an uninterrupted allele could reach a threshold at which expansion accelerates. Figure 4B proposes an evolutionary model for the DM2 mutation. Most probably, the first large alleles arose in Africa, possibly by unequal meiotic crossing over. Although other mechanisms are certainly possible, the unequal crossing-over model is attractive, because both uninterrupted and multiply interrupted alleles would arise in the germ line from the same event. This event would have occurred while the C allele predominated in the population, as seems to be the case in individuals of sub-Saharan descent. Over time, the most common alleles, both large and small, acquired rare variants, involving mainly alternative interruptions, whereas the uninterrupted alleles continued to either expand or die out. There is no way to know whether selection has acted over time to keep the frequency of these alleles low.

Figure 4 Model for evolution of the DM2 mutation
(A) Mis-pairing and unequal crossing over of DM2 repeat tracts (A1) can result in large alleles. Reciprocal crossover products can have either multiple interruptions (A2) or none (A3). These uninterrupted alleles are inherently unstable and eventually expand to pathogenic range (A4). (B) Evolutionary model for the DM2 locus. The ancestral hominid allele probably had a short (CCTG)n tract with two interruptions in cis with the C allele at rs1871922, the same as the most common modern allele (green line). Based on the higher frequency of large alleles in our African American samples, the first large alleles probably arose in Africa, possibly by unequal crossing over. Long alleles with multiple interruptions (orange line) accumulated variants, whereas uninterrupted alleles (pink line) gradually expanded, eventually reaching a pathogenic threshold (red line). The most parsimonious scenario is that the A allele at rs1871922 arose after the exodus from Africa on the same chromosome as a small two-interruption allele (blue line). The apparent rapid increase in frequency of the A allele could be due to selection at some nearby locus. (C) The Tajima D statistic21 indicates probable positive selection (D <−2) in Europeans in the region approximately 100 kb centromeric to ZNF9.
Based on current population frequencies, the predominant alleles in the population that migrated out of Africa were of the two-interruption variety. However, some uninterrupted alleles were also present. It is likely that the A allele at rs1871922 arose after the exodus from Africa on the same chromosome as a two-interruption allele. However, this haplotype seems to have rapidly increased in frequency, perhaps because of selection at some nearby locus. This inference is supported by the Tajima D statistic21 (UCSC Genome Browser) shown in figure 4C, which indicates positive selection in Europeans in the region approximately 100 kb centromeric to ZNF9.
Although this scenario is attractive and ties together a number of different observations, it also raises some interesting questions. For example, if large alleles actually arose by unequal crossing over, we would expect to find uninterrupted alleles in Africa. Did these alleles exist and subsequently disappear? If so, why? If not, why is there no DM2 in sub-Saharan Africans? Symptoms of DM2 are generally milder than DM1 and usually self-reported. Therefore, one explanation could simply be the unavailability of diagnostic opportunities. Larger population-based studies would be needed to determine whether uninterrupted alleles exist in sub-Saharan populations.
Of primary importance is the frequency of premutation alleles among Caucasians of European descent, the population where DM2 occurs. In one set of 973 European Caucasian nondisease alleles (169 were independent chromosomes), we observed a single premutation allele (0.6%). In a second set of 1,062 Finns (2,124 independent chromosomes), we identified three additional premutation alleles (0.1%). Taken together, these findings would suggest a premutation frequency of approximately 0.1% to 0.6%, although for accurate assessment a more systematically ascertained sample set would be needed. Nevertheless, these results strongly suggest that DM2 premutation alleles are present in the European Caucasian population at an appreciable frequency. It is not yet known whether such alleles may have any clinical or phenotypic consequence.
ACKNOWLEDGMENT
The authors thank the participating families for their cooperation. The authors thank Charles A. Thornton (University of Rochester) for contribution of DNA samples and Wolfram Kress (University of Würzburg) for sharing of molecular diagnostic data. The authors acknowledge Tamer Ahmed and Vlad Codrea for technical assistance with the genotyping, cloning, and sequencing; Marzena Wojciechowska for the RNA-FISH; and Keith A. Baggerly for statistical assistance. They also wish to thank the European Neuro-Muscular Centre for their continued support of the International Working Group on DM2/PROMM and Other Myotonic Dystrophies.
Supplementary Material
Address correspondence and reprint requests to Dr. Ralf Krahe, Department of Cancer Genetics, Unit 1010, University of Texas M.D. Anderson Cancer Center, 1515 Holcombe Blvd., Houston, TX 77030-4009 rkrahe@mdanderson.org
Supplemental data at www.neurology.org
Editorial, page 484
e-Pub ahead of print on November 19, 2008, at www.neurology.org.
R.K. was supported in part by NIH grant AR48171 and MDA. B.U. was supported by Medicinska understödsföreningen Liv och Hälsa r.f., the Tampere University Hospital Research Funds, and the Folkhälsan Institute of Genetics.
Disclosure: The authors report no disclosures.
Received April 21, 2008. Accepted in final form July 22, 2008.
REFERENCES
- 1.Harper PS. Myotonic Dystrophy, 3rd ed. London, UK: W.B. Saunders; 2001. [Google Scholar]
- 2.Krahe R, Bachinski LL, Udd B. Myotonic dystrophy type 2: clinical and genetic aspects. In: Wells RD, Ashizawa T, eds. Genetic Instabilities and Neurological Diseases. Amsterdam, Boston: Academic Press/Elsevier; 2006:131–150. [Google Scholar]
- 3.Brook JD, et al. Correction: Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 1992;69:385. [DOI] [PubMed] [Google Scholar]
- 4.Fu YH, Pizzuti A, Fenwick RG Jr, et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 1992;255:1256–1258. [DOI] [PubMed] [Google Scholar]
- 5.Mahadevan M, Tsilfidis C, Sabourin L, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science 1992;255:1253–1255. [DOI] [PubMed] [Google Scholar]
- 6.Liquori CL, Ricker K, Moseley ML, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 2001;293:864–867. [DOI] [PubMed] [Google Scholar]
- 7.Ashizawa T, Epstein HF. Ethnic distribution of myotonic dystrophy gene. Lancet 1991;338:642–643. [DOI] [PubMed] [Google Scholar]
- 8.Udd B, Meola G, Krahe R, et al. Report of the 115th ENMC workshop: DM2/PROMM and other myotonic dystrophies. 3rd Workshop, 14–16 February 2003, Naarden, The Netherlands. Neuromuscul Disord 2003;13:589–596. [DOI] [PubMed] [Google Scholar]
- 9.Bachinski LL, Udd B, Meola G, et al. Confirmation of the type 2 myotonic dystrophy (CCTG)n expansion mutation in patients with proximal myotonic myopathy/proximal myotonic dystrophy of different European origins: a single shared haplotype indicates an ancestral founder effect. Am J Hum Genet 2003;73:835–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liquori CL, Ikeda Y, Weatherspoon M, et al. Myotonic dystrophy type 2: human founder haplotype and evolutionary conservation of the repeat tract. Am J Hum Genet 2003;73:849–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saito T, Amakusa Y, Kimura T, et al. Myotonic dystrophy type 2 in Japan: ancestral origin distinct from Caucasian families. Neurogenetics 2008;9:61–63. [DOI] [PubMed] [Google Scholar]
- 12.Imbert G, Kretz C, Johnson K, Mandel JL. Origin of the expansion mutation in myotonic dystrophy. Nat Genet 1993;4:72–76. [DOI] [PubMed] [Google Scholar]
- 13.Coolbaugh-Murphy MI, Xu J, Ramagli LS, Brown BW, Siciliano MJ. Microsatellite instability (MSI) increases with age in normal somatic cells. Mech Ageing Dev 2005;126:1051–1059. [DOI] [PubMed] [Google Scholar]
- 14.Coolbaugh-Murphy M, Maleki A, Ramagli L, et al. Estimating mutant microsatellite allele frequencies in somatic cells by small-pool PCR. Genomics 2004;84:419–430. [DOI] [PubMed] [Google Scholar]
- 15.Neville CE, Mahadevan MS, Barcelo JM, Korneluk RG. High resolution genetic analysis suggests one ancestral predisposing haplotype for the origin of the myotonic dystrophy mutation. Hum Mol Genet 1994;3:45–51. [DOI] [PubMed] [Google Scholar]
- 16.Yamagata H, Miki T, Nakagawa M, Johnson K, Deka R, Ogihara T. Association of CTG repeats and the 1-kb Alu insertion/deletion polymorphism at the myotonin protein kinase gene in the Japanese population suggests a common Eurasian origin of the myotonic dystrophy mutation. Hum Genet 1996;97:145–147. [DOI] [PubMed] [Google Scholar]
- 17.Gatchel JR, Zoghbi HY. Diseases of unstable repeat expansion: mechanisms and common principles. Nat Rev Genet 2005;6:743–755. [DOI] [PubMed] [Google Scholar]
- 18.Mirkin SM. Expandable DNA repeats and human disease. Nature 2007;447:932–940. [DOI] [PubMed] [Google Scholar]
- 19.Sinden RR. Molecular biology: DNA twists and flips. Nature 2005;437:1097–1098. [DOI] [PubMed] [Google Scholar]
- 20.Rolfsmeier ML, Lahue RS. Stabilizing effects of interruptions on trinucleotide repeat expansions in Saccharomyces cerevisiae. Mol Cell Biol 2000;20:173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989;123:585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.