

Abstract
Arsenic (As) is a well-known environmental toxicant and carcinogen as well as an effective chemotherapeutic agent. The underlying mechanism of this dual capability, however, is not fully understood. Tumor suppressor gene p53, a pivotal cell cycle checkpoint signaling protein, has been hypothesized to play a possible role in mediating As-induced toxicity and therapeutic efficiency. In this study, we found that arsenite (As3+) induced apoptosis and cell cycle arrest in a dose-dependent manner in both p53+/+ and p53−/− mouse embryonic fibroblasts (MEFs). There was, however, a distinction between genotypes in the apoptotic response, with a more prominent induction of caspase-3 in the p53−/− cells than in the p53+/+ cells. To examine this difference further, a systems-based genomic analysis was conducted comparing the critical molecular mechanisms between the p53 genotypes in response to As3+. A significant alteration in the Nrf2-mediated oxidative stress response pathway were found in both genotypes. In p53+/+ MEFs, As3+ induced p53-dependent gene expression alterations in DNA damage and cell cycle regulation genes. However, in the p53−/− MEFs, As3+ induced a significant up-regulation of pro-apoptotic genes (Noxa) and down-regulation of genes in immune modulation. Our findings demonstrate that As-induced cell death occurs through a p53-independent pathway in p53 deficient cells while apoptosis induction occurs through p53-dependent pathway in normal tissue. This difference in the mechanism of apoptotic responses between the genotypes provides important information regarding the apparent dichotomy of arsenic’s dual mechanisms, and potentially leads to further advancement of its utility as a chemotherapeutic agent.
Keywords: Arsenite/Arsenic, gene expression profiling, p53, cell cycle regulation, apoptosis and oxidative stress
Introduction
Arsenic (As) is a well-known environmental toxicant that has been associated with numerous human health impacts including dermatosis, diabetes mellitus, cardiovascular disorders, and cancer (Tapio and Grosche, 2006). Within the last decade, arsenic has also been used as an effective chemotherapeutic agent for acute promyelocytic leukemia (APL) (Chen et al., 1997; Miller et al., 2002) and has been shown to induce apoptosis in a relatively wide spectrum of tumors such as lung, ovarian, gastric, neuroblastoma, and breast cancer (Bode and Dong, 2002). Despite its well-known toxicity and carcinogenicity, arsenic trioxide is approved by the Food and Drug Administration as a chemotherapeutic agent for the treatment of APL (FDA, 2000). Currently, there are over 40 clinical trials underway to extend arsenic’s therapeutic application(Litzow, 2008). The underlying mechanism of arsenic’s paradoxical capability to act both as a carcinogen and as a chemotherapeutic agent, however, is unclear. Understanding this dichotomy has the potential to improve the ability to effectively use As3+ as a chemotherapeutic agent as well as protect against its toxicity.
Arsenic-induced cell cycle arrest and apoptosis is commonly associated with an increased generation of reactive oxygen species (ROS), depletion of the cellular antioxidant system, inhibition of DNA repair and DNA methylation, and decreased mitochondrial membrane potential accompanied by cytochrome c release and caspase activation (Miller et al., 2002). Research data imply that arsenic significantly affects specific signal transduction molecules that are involved in mediating cellular proliferation or apoptosis, including MAPKs, p53, AP-1, and NFκB. These changes in cellular signaling pathways have been associated with both arsenic’s carcinogenicity and cancer-therapeutic effect (Kitchin, 2001; Qian et al., 2003).
The tumor suppressor gene p53, for example, has been linked to the DNA damage, cell cycle perturbations, and apoptosis that is seen with arsenic (Park et al., 2000; Yih and Lee, 2000). P53 is crucial in maintaining genome integrity through the induction of cell cycle arrest, allowing for the DNA repair or apoptosis for cells with irreparable damage (Vogelstein et al., 2000). In the presence of functional p53, exposure to 5 μM arsenite has been found to induce DNA strand breaks, increasing the p53 phosphorylation (Yih and Lee, 2000; Yih et al., 2002). Arsenic trioxide was also shown to inhibit proliferation of the human B lymphoma MBC-1 by up-regulating p53 expression and inducing apoptosis (Shen et al., 2000).
However, a number of alternate studies have demonstrated that As3+ also disrupts mitosis and induces apoptotic cell death in p53 deficient cells such as HeLa S3 and U937 cells (Huang and Lee, 1998; McCabe et al., 2000). In addition, P53 deficient myeloma cells were shown to be more sensitive to As3+ treatment exhibiting a higher level of apoptosis than p53+/+ cells (Liu et al., 2003). In an earlier study exploring the induction of p53 in cell lines with different genetic backgrounds including C-33A with mutations of the p53 gene, HeLa, Jurkat, and a transformed lymphoblast cell line, Salazar et al. found that C-33A cells showed the higher sensitivity to arsenic treatment while HeLa, Jurkat, and LCL-EBV cells showed similar cytotoxicity curves (Salazar et al., 1997). In addition, transfected Jurkat cells and human lymphocytes with mutated p53 genes showed increased sensitivity to arsenic cytotoxicity. Since treatment with arsenite or arsenate resulted in apoptosis in both wild-type p53 (p53+/+) and p53-deficient (p53−/−) cells, Bode and Dong suggested that p53 signaling pathway is not involved in arsenic-induced apoptosis (Huang et al., 1999; Bode and Dong, 2002). The use of cell lines to characterize the cellular response to arsenic has provided some clues but due to the complexity of the genetic background of these cell lines, there remains no firm consensus on the overall role of p53 in mediating As-induced toxicity. In this study, we applied mouse embryonal fibroblasts (MEFs) isolated from p53+/+ and p53−/− mouse to examine the role of p53 in As3+-induced changes in signaling pathways. The ease of isolation of MEFs from mid-gestation mouse embryos and the ability to derive these cells from mice harboring various genetic alterations has been made the MEFs ideal model system for examining signaling pathways changes under different conditions (Vengellur et al., 2003). We have applied this culture system to investigate the molecular mechanism of metal-induced cell cycle arrest and apoptosis, as well as the role of p53 (Gribble et al., 2005).
High throughput gene expression profiling from mRNA expression microarray has become a powerful tool in the characterizations of biological processes, disease states, and response to drugs, as well as response to genetic perturbations (Lamb et al., 2006). In this study, we applied this approach to testify our hypothesis that although arsenic induces cell cycle arrest and apoptosis in both p53+/+ and p53−/− cells, the underlying molecular mechanisms or gene pathways are different. A systems-biology based examination of differential signaling pathways from the microarray study in the normal cells (p53 wildtype) and genetic abnormal MEFs (p53 knockout) may provide important information regarding the apparent dichotomy of arsenic’s dual mechanisms potentially leading to further advancement of its utility as a chemotherapeutic agent.
Material and Methods
Cell culture
Mouse embryonal fibroblasts (MEFs) were isolated following a modified protocol (Gribble et al., 2005). Briefly, embryos were separated from the uteri of pregnant females on day 14 of gestation. The torso and limbs were dissected to isolate fibroblasts. DNA obtained from the tail was used for PCR genotyping. Single cell suspensions were plated in 100 mm dishes in DMEM F-12 with 10% v/v Nu-Serum. The ease of isolation and the ability to derive these cells from mice harboring various genetic alterations has made the MEFs an ideal model system for studying aspects of cell growth control and functional genetics (Vengellur et al., 2003).
Arsenic treatments, cell cytotoxicity and apoptosis
MEFs, at either passage 6 or 7, were grown to near-confluency (85–90%) and switched to lower serum (containing 5% Nu-serum) DMEM F-12 culture medium for 24h. MEFs were treated with sodium arsenite (As3+) at concentrations of 0–20 μM for 24 h. All cultures were monitored with an inverted phase-contrast microscope during culture to assess their general appearance as previously described (Yu et al., 2005). Cells were harvested and total protein and mRNA were extracted as described previously (Yu et al., 2005). Cell viability was determined using the neutral red uptake assay (Borenfreund and Puerner, 1985), based on lysosomal accumulation of neutral red in viable cells. Apoptosis-associated endpoints were determined either by quantitatively measuring the activities of caspase 3/7 and 8 with a fluorometric assay using DEVD-AMC and IETD-AMC (Axxora, San Diego, CA) as the respective caspase-specific substrates or semi-quantitatively detecting the cleaved form of caspase-3 with Western blot analysis using an anti-caspase-3 antibody (Cell Signaling, Inc).
Cell cycle analysis
Cell cycle kinetics were examined using bivariate BrdU-Hoechst flow cytometry according to our established protocol, which allows for the dynamic change of cell cycle progression to be monitored (Sidhu et al., 2005). Briefly, cells were cultured with 5-bromo-2′-deoxyuridine (BrdU) to incorporate BrdU into their DNA. After incubation, cells were harvested and re-suspended in Hoechst 33258 buffer and propidium iodide (PI) with RNase (0.1%). Cells were analyzed using a Coulter Epics Elite flow cytometer (Beckman Coulter). MPLUS 5.0 software (Phoenix Flow Systems) was used to analyze the data. Analysis was conducted to determine the proportion of cells in each cell cycle phase (G0/G1, S, G2/M) from successive rounds of cell division.
RNA Isolation and Microarray Hybridization
For the microarray-based gene expression analysis, treatments were restricted to one time point (24 h) and a single concentration (5 μM). The concentration selected was based on minimal impacts on morphology, cell viability and caspase 3/7 activity after 24 h treatment. Total RNA from three biological samples for each group was isolated using the RNeasy Mini Kit (Qiagen, Valencia, CA) and quality was assayed on the Agilent 2100 Bioanalyzer (Agilent, Palo Alto, CA). Array analysis was conducted in the NIEHS Center for Ecogenetic and Environmental Health (CEEH), the Functional Genomics Laboratory following the manufacturer’s protocols for the technical aspects associated with the determination of gene expression using the CodeLink Mouse UniSet 10K oligonucleotide microarray. Three independent samples were collected per treatment. The first step in preparing samples for hybridization was the synthesis of cDNA from 5 μg total RNA using Superscript II. The resulting cDNA was column purified using the QIAquick purification kit for PCR products (Qiagen, Valencia, CA). Lastly, cRNA was produced from the second strand of DNA using the In Vitro Transcription Kit (Amersham Biosciences, Piscataway, NJ). In addition to the nucleotide mix, 10 mM Biotin-11-UTP was added (Perkin Elmer Life Sciences, Boston, MA). The amplified product was purified with the RNeasy Mini Kit (Qiagen, Valencia, CA). cRNA was quantified by its absorbance at 260 nm on a UV spectrophotometer, while the quality of the cRNA was assessed on the Agilent 2100 Bioanalyzer. cRNA was stored at −80°C until used for array hybridization. Hybridization was carried out for 18 h on an orbital shaker set at 300 rpm and 37°C. After removing the hybridization chamber, arrays were washed with 0.75× TNT for 1 h at 46°C. Incubation for 30 min with AlexaFlour 647-streptavidin (Molecular Probes, Inc., Eugene, OR) was followed by four 5 min washes in 1× TNT and two vigorous rinses in 0.05% Tween-20. Slides were dried by centrifugation at 2000 rpm for 3 min and arrays were scanned on an Axon GenePix 4000 Scanner (Axon Instruments, Union City, CA) set to a wavelength of 635 nm, a PMT voltage of 600 V, and 10 μM resolutions. CodeLink array data was first run through accompanying software from Amersham Biosciences. Raw data extracted from the images are deposited in the NCBI GEO data base. These values were imported to BRB Array Tools v3.0 and a log base 2 transformation was applied and normalized each array by using the median intensity over entire arrays (Wright and Simon, 2003).
A systems-based GO-Quant functional GO and pathway analysis of microarray data
Microarray analysis was conducted based on our recently established GO-Quant approach (Yu et al., 2006). Briefly, a class comparison was conducted in each treatment by using the Randomized Variance Model (Wright and Simon, 2003). Three hundred and eight-four genes significantly altered in either genotype (p53+/+ or p53−/−) at p ≤0.001 were selected and ratios were derived by dividing each normalized gene value by the average value of the controls. Two-class unpaired comparison of fold changes of these significant changed genes between the two genotypes after treatment was conducted using SAM (Significant Microarray Analysis) program (Tusher et al., 2001), which is built in MeV (MultiExperiment Viewer) software (Delta 1.8 and 90% percentile false discovery rate set at 0.05%) (Saeed et al., 2003). Further hierarchical clustering analysis of each group was conducted in MeV.
To establish the association between the treatment and the affected gene ontology (GO) terms, we applied the MAPPfinder to identify enriched biological themes, particularly GO terms at p ≤ 0.001 (Dahlquist et al., 2002). Z-score and permutation p-value was used to rank these terms of biological significance (Dahlquist et al., 2002). The output of the MAPPfinder calculation was input into the GO-Quant program and the average ratio of each GO item was calculated, which allows for the quantitative comparison between the two genotypes after As3+ treatment (Yu et al., 2006). In addition, a canonical pathway-based analysis was conducted with Ingenuity Pathway Analysis (IPA; Ingenuity® Systems, www.ingenuity.com) for either genotype to identify the genes significantly associated with canonical pathways in the Ingenuity Pathways Knowledge Base. IPA 6-1202 includes more than 100 curated canonical pathways incorporating more than 6000 discreet gene concepts. The significance of the association between the significantly changed genes (p≤0.01 and fold change ≥1.5) and the canonical pathway was determined based on a p-value calculated with Fisher’s exact test. Further comparison of pathways changed with As3+ treatment between p53+/+ and p53−/− MEFs was conducted.
The results of quantitative data on cell viability, caspase activity assays, and densitometry analyses of western blot data of cleaved caspase 3 are expressed as mean ± standard error of the mean (SEM). Differences between dose of treatments and genotypes were examined for statistical significance by two-way ANOVA followed by Tukey HSD pair-wise comparison.
Quantitative RT-PCR (qRT-PCR) analysis
MEFs were treated with As3+ under the same conditions as in the microarray study, and total RNA were isolated following 8 and 24 h treatment. We conducted quantitative gene expression analysis of cell cycle regulatory genes including Cyclin B1, Cdc25c, Uchl1, and Ube2c using Taqman Universal PCR Master Mix and 6-FAM dye-labeled Taqman assays (Applied Biosystems) (Luderer et al., 2003) and antioxidant glutathione S-transferase Gsta2 gene using SYBR green PCR Master Mix (Applied Biosystems). GAPDH was utilized as an internal control to normalize the data. RNA was collected using TRIZOL (Invitrogen) and purified with the RNAeasy kit (Qiagen). cDNA synthesis was generated using Oligo(dT)12–18 and Superscript II Reverse Transcriptase (Gibco). For gene expression of Cyclin B1, Cdc25c, Uchl1, Ube2c, and GAPDH, 4 uL of cDNA were included in a PCR reaction that also consisted of the appropriate forward and reverse primers at 360 nM each, 80nM TaqMan probe and TaqMan Universal PCR Master Mix, No Amperase UNG (Applied Biosystems Inc., Foster City, CA). The PCR reaction for Gsta2 differed in consisting of 120 nM of each primer and SYBR green PCR Master Mix (Applied Biosystems Inc., Foster City, CA). The PCR primers and the dual-labeled probes (6-carboxy-fluorescein (FAM) and 6-carboxy-tetramethyl-rhodamine (TAMRA) for all genes were designed using ABI Primer Express v.1.5 software (Applied Biosystems Inc., Foster City, CA). All oligo sequences are listed in Table 1. Amplification and detection of PCR amplicons were performed with the ABI PRISM 7700 system (Applied Biosystems Inc., Foster City, CA) with the following PCR reaction profile: 1 cycle of 95°C for 10 min., 40 cycles of 95°C for 30sec, and 62°C for 1 min. GAPDH amplification plots derived from serial dilutions of an established reference sample (mouse liver RNA) were used to create a linear regression formula in order to calculate Cyclin B1, Cdc25c, Uchl1, Ube2c, and Gsta2 expression levels. GAPDH gene expression levels were utilized as an internal control to normalize the data. Reported fold changes in expression are the ratios of treatment over control values.
Table 1.
Sequences for PCR primers and probes
| Gene | Forward primer (FOR) | Probe |
|---|---|---|
| Reverse primer (REV) | ||
| Cyclin B1 | FOR: 5′-TGAAGACTCCCTGCTTCCTGTTATG-3′ | 5′-FAM-TCACAAAGCACATGACTGTCAAGAACAAGTATGC A-TAMRA-3′ |
| REV: 5′-TCAGCTGTGCCAGCGTGC-3′ | ||
| Cdc25c | FOR: 5′-GCATTATATTACCCTGAGCTATATATCCTCAA-3′ | 5′-FAM-TCACACAGCTCCATATACTCTGGAAAGAAATCTC TGTAGC-TAMRA-3′ |
| REV: 5′-GAAGCATGGGACAGTAGCTCTGTG-3′ | ||
| Uchl1 | FOR: 5′-TGCCCTTTCCAGTGAACCATG-3′ | 5′-FAM-TGGCAGCATCCTGCAGCAGAGAGTCC-TAMRA-3′ |
| REV: 5′-CAGAGAAGCGGACCTCCCC-3′ | ||
| Ube2c | FOR: 5′-TCTATCCAGAGCCTGCTAGGAGAAC-3′ | 5′-FAM-AAAACCCCACAGCATTTAAGAAATACCTGCAAGA AAC-TAMRA-3′ |
| REV: 5′-CTTGGCTGGAGACCTGCTTTG-3′ | ||
| Gapdh | FOR: 5′-TCCTGCACCACCAACTGCTT-3′ | 5′-FAM-CACTCATGACCACAGTCCATGCCATCAC-TAMRA-3′ |
| REV: 5′-GAGGGGCCATCCACAGTCTT-3′ | ||
| Gsta2 | FOR: 5′-GTATTATGTCCCCCAGACCAAAGAG-3′ | |
| REV: 5′-CTGTTGCCCACAAGGTAGTCTTGT-3′ |
Results
As3+ induced cell death and cell cycle arrest in both p53-wildtype and knockout MEFs
In this study, arsenic-induced cytotoxicity and cell cycle arrest were examined in both p53+/+ and p53−/− MEFs. As shown in Figure 1, As3+ treatment resulted in a consistent concentration-dependent compromise to morphological integrity. No significant morphological changes were observed with 5 μM in either genotype (Fig. 1A). Significant morphological changes were observed in p53−/− cell at 10 μM including irregular cell shape and condensation. Significant disruptions of morphology such as round-up, and detachment were observed in both genotypes at the high concentration of 20 μM. As3+ treatment reduced cell viability as assessed by NR dye uptake assay in a dose- dependent manner (Fig. 1B). Significant decreases in NR dye uptake were observed at 5 and 10 μM with greater decreases in p53−/− cells than that in the p53+/+ cells. NR dye uptake rates were sharply reduced to less than 15% at 20 μM in both p53+/+ and p53−/− (Fig. 1B). Furthermore, there was a dose-dependent induction of apoptosis as evidenced by up-regulation of cleaved caspase 3 (Fig. 1C) and increases in caspase 3/7 (Fig.1D), and caspase 8 activities (Fig.1E) in both genotypes, but were significantly higher in the p53−/− cells compared to the p53+/+ cells.
Figure 1. Arsenic (As3+) exposure induced changes in cell morphology (A), viability assessed by neutral red dye (NR) uptake assay (B), and apoptosis (C–E).
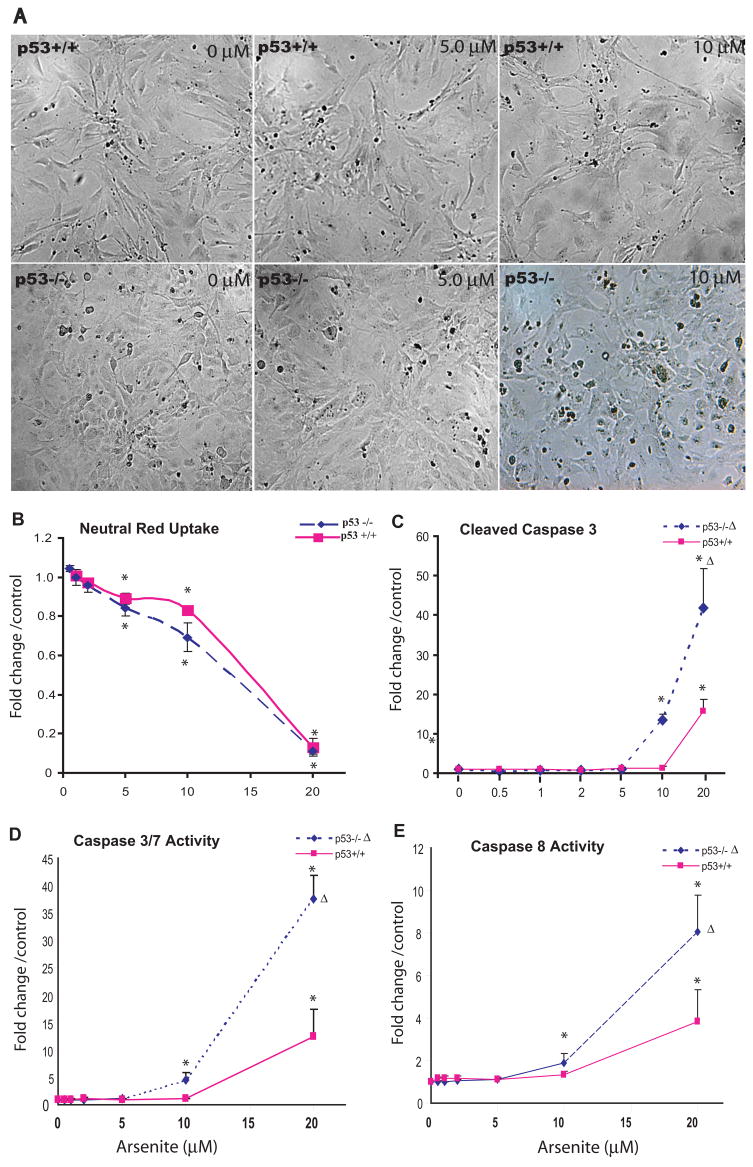
Cultures of p53 wildtype (p53+/+) and knockout (p53−/−) MEF cells were exposed to various concentrations of As3+ (μM) for 24h. Following the exposure, cell morphological changes were monitored with an inverted phase-contrast microscope (A). Cell viability was assessed utilizing the NR uptake assay, which indirectly reflects the uptake function of the cells (B). Apoptosis was monitored by measuring cleaved caspase-3 with Western blot analysis (C) as well as caspase 3/7 (D) and caspase 8 (E) activities. Quantitative analysis of band intensities and area were achieved using the “Quantity One” software (Bio-Rad Laboratories). Statistical analysis of cell viability, caspase activities was conducted using two-way analysis of variance (ANOVA, p ≤ 0.001 (Δ)) followed by Dunnett’s method to the control, with a significance level of p ≤ 0.05 (*). Data presented as mean ± SE, n ≥3. Morphological impacts including irregular cell shape, condensation are evident after 24 h exposure at an As3+ ≥ 10 μM. Dose-dependent decreases in NR uptake were observed, significantly from the concentration ≥ of 5 μM in both genotypes. The induction of caspase 3/7 and caspase 8 activities as well as cleaved caspase-3 increased in a dose-dependent manner in both genotypes and was significantly higher in the p53−/− than those in the p53+/+ cells (ANOVA, p ≤ 0.001).
As3+ treatment resulted in a dose-dependent inhibition of cell cycling that was observed as an increase in all phases of the original cell population (G0/G10, G2/M0) and a decrease in any cells taking up BrdU (new cell cycle) at concentrations ≥2 μM (24 h), independent of genotype (Fig. 2A). In the p53+/+ cells, a significant increase of the cell population in G2/M0 phase was observed at concentrations ≥5 μM (Fig. 2B), leading to fewer cells progressing to the next round of G2/M phase (G2/M1) at concentrations ≥2 μM (Fig. 2B). In the p53−/− cells, we observed a concentration-dependent increase of cells in G2/M0 at concentrations ≥5.0 μM (Fig. 2B) and a significant decrease in second round of G2/M (G2/M1) phase at concentrations ≥5 μM. We also observed a significant decrease in the third round of G2/M (G2/M2) at concentrations ≥2 μM (Fig. 2B).
Figure 2. Arsenic (As3+) causes a concentration-dependent cell cycle disruption.

Decease in the proportion of cells reaching new round of cell cycle (A) and G2/M cell cycle inhibition (B). Cultures of p53 wildtype (p53+/+) and knockout (p53−/−) MEF cells were exposed to various concentrations of As3+ (μM) for 24h. Evaluation of bivariate Hoechst-PI flow cytograms was conducted to determine the total number of cells, and the proportion of cells in the first and successive rounds of cell division. Statistical analysis was conducted using ANOVA followed by Dunnett’s method to the control, with a significance level of p ≤ 0.05 (*). Data presented as mean ± SE, n ≥3. Dose-dependent inhibitions of cell cycle were observed, significantly at concentration ≥ 2 μM in both genotypes (A). As3+ exposure causes a concentration dependent increase in the number of cells in G2/M0 in both p53 genotypes (B), with significant inhibitions at 5 and 10 μM of As3+ in both at 24h. Cell cycle progression inhibition can be observed by the lack of new cells in the second round of cell division G2/M1, significant at concentration ≥ 2 μM As3+ in p53+/+ cells at 24h. In the p53−/− cells, As3+ caused decrease in the number of cells in the second round of G2/M1 at concentration ≥ 5 and in the third round of G2/M2 at concentration ≥ 2 μM.
Differential gene expression between p53+/+ and p53−/− MEFs
Without functional p53, the p53−/− cells in our study proliferated faster than p53+/+ cells as illustrated by the progression of cell cycle (Fig. 2). Knocking down the p53 gene induced dramatic differential gene expression. We identified 817 genes that were differentially expressed between the two genotypes at a cut-off of p ≤ 0.001 (Fig. 3). 53 dependent genes, such as p53 (Trp53), p21 (Cdkn1a), Ccng1, Bax, Perp, Pmaip1, Ccng1, and Ccne1, were significantly down-regulated in the p53−/− MEFs, while other cell cycle regulatory genes, such as Cdkn2a, Rhou, Nmyc1, Ccni, and Cdkn1c, were up-regulated. The functional canonical pathway analysis shows that gene expression of 38 genes within apoptosis regulation and 30 genes in cell cycle regulation were altered (Supplementary Table 1). The altered gene expression in p53- knockout MEFs confirms its critical role in regulating cell cycle and apoptosis, and the identification of p53 dependent signaling pathway by the knockout of p53 further validates this approach as a tool to characterize potential signaling pathways.
Figure 3. Volcano plot illustrating the differential gene expression between p53+/+ and p53 −/− MEF cells.

A microarray based gene expression analysis was conducted and a randomized variance t test (Wright and Simon, 2003) was used to select the significantly down-regulated genes (green) and the up-regulated genes (red) at p ≤ 0.001 (n = 3). A total of 817 genes were differently expressed in the p53−/− MEFs as compared to the p53+/+ MEFs. In the p53−/− cells there was a significant up-regulation of chemokines and cytokines such as Ccl5, Ccl8, Cxcl5, Cxcl10, Cxcl12, Cxcl14, Cxcl15, Cxcl16 and growth factor genes such as Igf1, Igf2, Fgf18, Fgf9, Fgf13, Fgf21, Gdf5, Bmp2 and Bmp6. Genes involved in the regulation of cell proliferation, cell death and apoptosis such as Bax, Mgmt, p53 (Trp53), p21 (Cdkn1a), Ccng1, Pmaip1 were down-regulated in the p53−/− cells.
Gene expression changes in response to As3+ in p53+/+ and p53−/− MEF cells
A total of 186 genes were found to be responsive to As3+ at a cut-off of p ≤ 0.001 in p53+/+. Among these genes, 37 genes were significantly up-regulated and 149 genes were down-regulated. The top ten up-regulated genes (based on the p value of the statistical analysis) were Gsta2, Gsta3, Gsta4, Hmox1, 2310005E10Rik, Abca8b, Ubd, Enpp2, Zfp91, and Apoh (Supplement Table 2). The top ten down-regulated genes were 2010317E24Rik, Calmbp1, Ccnb2, Cdkn3, Ccnb1, Kif20a, Gzme, Cdc20, 2310007D09Rik, Cdca3, Hmmr, Mcpt8, and Ube2c. In the p53−/− genotype, a total of 229 genes were differentially expressed when treated with As3+ for 24 h. Among these genes, 93 genes were up-regulated and 136 genes were down-regulated at cutoff p ≤ 0.001. The top ten up-regulated genes (based on the p value of the statistical analysis) include Gsta2, Gsta3, and Gsta4, Pmaip1, Fetub, Hmox1, 1700016M24Rik, Gstm7, Krt1-13 and Nqo1(Supplement Table 2). The top ten down-regulated genes include Hp, Rsad2, Serpina3n, Ccl5, Rac3, Lum, Sod3, Mmp3, Gck and Zbp1.
The comparison between the genotypes after As3+ treatment is shown in Figure 4. Within these 384 significantly changed genes either in p53+/+ or p53−/− cells, we found 149 genes whose expression levels were either significantly up or down-regulated in both p53+/+ and p53−/− cells after As3+ treatment (A) based on SAM two-unpaired group comparison with Delta value of 1.8 and 90% percentile false discovery rate of 0.05%. This group included up-regulated genes in antioxidant and phase II enzymes such as Gsta2, Gsta3, Gsta4, and hmox1, as well as down-regulated genes such as penk1, Rsad2, Tgtp, and Has2. Significant lower expression levels in p53+/+ cells than those in the p53−/− cell are listed in Fig. 4B. Within these 126 genes, 103 genes were significantly down-regulated in p53+/+ cells such as Ccnb1 (Cyclin B1), Ccnb2 (Cyclin B2), Ube2c, and Cdca3, and 23 genes were significantly up-regulated in p53−/− cells such as Fetub, Pmaip, NCf2, and Star. Fig. 4C shows significantly down-regulated genes (90 genes) in p53−/−, including a number of genes in immune responses such as Cxcl10, Zbp1, Mmp3, Lum, Ifit1, Ifitm7, and Ccl5.
Figure 4. Differential gene expression in p53+/+ and p53−/− cells after As3+ treatment.

Synchronous cultures of p53 wildtype (p53+/+) and knockout (p53−/−) MEF cells were exposed to As3+ (5 μM) for 24h. A microarray based gene expression analysis was conducted as described in Experimental Procedures. A minimum of 3 arrays was analyzed for each experimental condition. A randomized variance t test (Wright and Simon, 2003) was used to select the significant down-regulated genes and up-regulated genes at p ≤ 0.001. A total of 186 and 229 genes were found to be responsive to As3+ at the cut-off of p ≤ 0.001 in p53+/+ and p53−/− MEF, respectively. Further comparison of these significantly changed genes (384 genes) after As3+ treatment between two genotypes (p53+/+ and p53−/−) was conducted using SAM program (Significant Microarray Analysis) (Tusher et al., 2001), which is built in MeV (MultiExperiment Viewer) software (Saeed et al., 2003). Three groups, no significant difference (A), significant lower expression levels (B) or higher (C) in p53+/+ cells than those in the p53−/− cell were identified with Delta value of 1.8 and 90% percentile false discovery rate of 0.05%. Fig. 3A shows 149 genes, whose expression levels were either significantly up or down-regulated in both p53+/+ and p53−/− cells after As treatment. The cluster included up-regulated genes in antioxidant and phase II enzymes such as Gsta2, Gsta3, Gsta4, and hmox1 as well as down-regulated genes such as penk1, Rsad2, Tgtp, and Has2. Fig. 3B shows significantly down-regulated genes (126 genes) in p53+/+ cells such as Ccnb1 (Cyclin B1), Ccnb2 (Cyclin B2), and Cdca3 or up-regulated genes (23 genes) in p53−/− cells such as Fetub, Pmaip, NCf2 and Star. Fig. 3C showed significantly down-regulated genes (90 genes) in the p53−/−, including a number of genes in immune responses such as Cxcl10, Zbp1, Mmp3, Lum, Ifit1, Ifitm7, Mmp3, and Ccl5.
Figure 5 summarizes GO-Quant-based quantitative functional comparative analysis between the p53+/+ and p53−/− cells. Significantly altered GO terms as identified by MAPPFinder were listed with a GO hierarchic structure (path) and quantitative calculation of average fold changed in each GO term by GO-Quant was visualized proportional to fold changes in colors. Common alteration of gene expression in both p53 genotypes were up-regulation of glutathione transferase activity (7), down-regulation in stimulus response (2), immune response (2), steroid metabolism (2), and protein catabolism (4). In the p53+/+ cells, significant down-regulation of gene terms occurred in cell cycle (1), especially genes in M and S phase, and DNA metabolism including DNA replication and initiation in p53+/+ (3 and 8). In the p53−/− cells, significant down-regulation in G-protein-coupled receptor binding and chemokine receptor binding and activity were observed in the p53−/− cells after As3+ treatment (5).
Figure 5. GO-Quant-based quantitative functional comparison of gene expression alteration in p53+/+ and p53−/− cells after As3+ treatment.

GO-Quant based quantitative functional Gene Ontological comparison between the p53+/+ and p53−/− after As3+ treatment was conducted according to our previous report (Yu et al., 2006). MAPPFinder (Doniger et al., 2003) was used to link the gene expression data to the GO hierarchy (Ashburner et al., 2000) and establish associations between treatment and the affected GO terms. MAPPFinder calculates a Z score as well as a permutation test p value for the genes that are significantly altered after treatment (p ≤ 0.001) in each genotype. The right side of Figure 4 listed the significantly altered GO terms (p ≤ 0.05) for the significantly changed genes either in p53+/+ and p53−/− at p 0.001 and the left heatmap illustrates the quantitative result of the average fold changes in each GO term by GO-Quant program. The brightness of color is proportional to the fold changes of gene expression level, and color indicates up-regulation (red) or down-regulation (green) genes. Path (Gene ontology hierarch path), GOID (gene ontology identification number), N (the number of gene changed), Z (the Z score) and p (permutation p value) and GO Term (Gene Ontology term), were listed. Common alteration of gene expression in both p53 genotypes were up-regulation of glutathione transferase activity, down-regulation of steroid metabolism and protein catabolism. Significant down-regulation of genes terms in cell cycle in the p53+/+ and significant down-regulation in G-protein-coupled receptor binding and chemokine receptor binding and activity in the p53−/− cells were observed after As3+ treatment.
Comparison of these significantly associated pathways changed with As3+ in the p53+/+ cells and p53−/− cells by the IPA is listed in Fig. 6.
Figure 6.

Canonical pathway-based analyses of gene expression data from p53+/+ and p53−/− MEFs treated with As3+. Ingenuity Pathway Analysis (IPA; Ingenuity® Systems, www.ingenuity.com) was used to identify the genes significantly associated with canonical pathways in the Ingenuity Pathways Knowledge Base. IPA 6-1202 includes more than 100 curated canonical pathways incorporating more than 6000 discreet gene concepts. The significance of the association between the significantly changed genes (p ≤ 0.01 and fold change ≥ 1.5) and the canonical pathway was determined based on a p value calculated with Fisher’s exact test. Further comparison of pathways changed with As3+ treatment between p53+/+ and p53−/− MEF was conducted. As3+ treatment induced alteration of NRf2-mediated oxidative stress response, glutathione metabolism, biosynthesis of steroids, and aryl hydrocarbon receptor signaling pathways in both p53+/+ and p53−/− cells. In the p53+/+ cells, significant alteration in cell cycle, especially G2/M DNA damage checkpoint regulation, and pyrimidine metabolism were observed after As3+ treatment. In the p53−/− cells, As3+ treatment induced significant pathways changed mainly related to immune responses including interferon, IL-6, NFκB, toll-like receptor, leukocyte extravasation, antigen presentation acute phase response, LPS/IL-1 mediated inhibition of RXR function, complement system and IL-10 signaling pathways.
NRf2-mediated oxidative stress response
Independent of genotype, As3+ induced significant alterations in NRf2-mediated oxidative stress response pathway (Fig. 6 and 7A). These genes are also related to glutathione metabolism and xenobiotics cytochrome p450 metabolism, as well as Aryl hydrocarbon receptor signaling. As shown in Fig. 6, a detailed pathway comparison between p53+/+ and p53−/− cells, As3+ treatment induced significant up-regulation of Gsta2, Gsta3, Gsta4, Gsto1, Gstm1, Gstm3, Gclm, Noq1, Hmox1, Ephx1, Ephx2, and Cat in both genotypes. We further explored kinetic transcriptional changes of Gsta2 gene using quantitative RT-PCR, and observed an increase in Gsta2 at 8h implying that defense mechanisms in response to As3+-induced oxidative stress precede cell cycle inhibition and cell death as expected (Fig. 8A).
Figure 7. Nrf2-mediated oxidative stress response pathway (A), cycle regulations (B), and IL6 and NFκB signaling pathways (C) in response to As3+ in p53 wildtype (p53+/+) and knockout (p53−/−) MEF cells.



Cell cycle regulation pathway was modified from KEGG and a knowledge based pathways about NRf2-mediated oxidative stress response and IL6 and NFκB were built in GenMAPP program (www.gennmap.org) (Dahlquist et al., 2002). Differential gene expression was based on treatment versus control expression change (1.5-fold and p<0.05, t test). Red indicates a higher level of expression in the treatment while green indicates decreased level of expression in the treatments. Grey indicates that the selection criteria were not met but the gene is represented on the array. As3+ induced significant alterations in NRf2-mediated oxidative stress response pathway in both p53+/+ and p53−/− cells (Fig 6A). P53-dependent down-regulation of cell cycle regulatory genes in p53+/+ such as Cdc25B, Cdc25C, Ccnb1, Top2A, Ccnb2, Plk1, Cdc2, Chek2 were observed (Fig. 6B). Activation of Gadd45a and Gadd45g, Chk1, Topa/b in p53−/− cells were observed. As3+ induced a significant down-regulation in canonical NFκB signaling and IL6 pathways in the p53−/− cells (C).
Figure 8. Quantitative Realtime PCR (qRT-PCR) measurement of specific gene expressions in As3+ treated p53 MEF cells.


Figure A shows the correlation of the gene expressions measured from the Codelink microarray and qRT-PCR and Figure B and C shows the time-dependent gene expression alterations of five selected genes. Quantitative Realtime PCR (qRT-PCR) gene expression measurements for Uchl1, Ube2c, Ccnb1, Cdc25c and GST2a in three biological samples exposed to As3+ (5 μM) for 8 and 24h in the same condition as microarray study were conducted as described in Experimental Procedure. Correlation of the gene expression patterns (in Log 2 fold) between the microarray and qRT-PCR at 24 h was performed (A). For all of the five selected genes, we found consistent results between the Codelink microarray and the qRT-PCR methods (r = 0.94, p ≤ 0.001). For the time-dependent alterations in gene expression measured by qRT-PCR, statistical analysis was conducted using ANOVA followed by Dunnett’s method to the control, with a significance level of p ≤ 0.05 (*). Data presented as mean ± SE, n =3.
DNA damage and cell cycle regulatory genes
Although As3+ induced similar effects on cell cycle arrest and cell death in both p53−/− and p53+/+ cells, changes in gene expression associated with DNA damage and cell cycle regulation were different between the two genotypes. We observed that As3+ induced significant changes in p53 canonical pathway in p53+/+ cells, however, in the p53−/− cells, we also observed significant genes changed previously found to be p53-dependent, such as Gadd45a, Gadd45g, Perp, and Pmip1 (Figure 6). In the p53+/+ cells, we observed that As3+ induced a significant down-regulation of DNA replication and cell cycle regulatory genes which have been previously identified as p53-dependent target genes (Fig. 5B, Fig. 6, and 7B). Figure 7B, a modified cell cycle gene pathway from the KEGG database (Ogata et al., 1999), shows p53 mediates stress by regulating transcription of genes involved in executing cell cycle arrest and apoptosis. As3+ treatment induced a p53-dependent alteration in the cell cycle regulatory genes in p53+/+ cells. Specifically, within the p53+/+ cells, As3+ treatment resulted in a significant up-regulation of Gadd45a, Gadd45g, Mdm2, and Cyclin A1, and a significant decrease in PIK1, p18, p19, Cyclin A2, Cdc2a in G1/S phase and Chek2, Cdc25b/c, Cyclin B1/B2, Top2a, Cdkn2d, and Cdc20 in the G2/M phase. In the p53−/− cells, we observed up-regulation of Gadd45a, Gadd45g, Chek1 and Top2a/b genes (Fig. 7B).
Unique gene expression alterations in immune responses network in p53−/− MEFs
In the p53−/− cells, As3+ treatment induced significant changes in pathways related to immune response network such as interferon, IL-6, NFκB, toll-like receptor, leukocyte extravasation, antigen presentation, acute phase response, LPS/IL-1mediated inhibition of RXR function, complement system, and IL-10 signaling pathways (Fig. 6). Fig 7C is a modified canonical NFκB pathway with the interaction of cytokine IL-6 pathway. Significant down-regulation of genes involved in both NFκB and IL-6 signaling pathways was observed in p53−/− cells.
Temporal changes of genes of Gsta2, Ccnb1, Cdc25c, Uchl1, and Ube2c by quantitative reverse transcription-PCR (qRT-PCR)
Quantitative analysis of gene expression in the antioxidant pathway, Gsta2, and cell cycle regulation of Ccnb1, Cdc25c, Uchl1 and Ube2c was conducted using qRT-PCR. A significant correlation between the gene expression results in the microarray analyses and the qRT-PCR analysis for these genes was observed (r = 0.94, P ≤ 0.001) (Fig. 8A). This result validate this subset of expression patterns from the microarray experiment. In addition, the time-dependent examinations of these genes demonstrate that As3+ treatment at 5 μM significantly increase the Gsta1 antioxidant response at both 8 and 24 h in both genotypes (Fig. 8B). Significant decreases in Cyclin B1, Cdc25c, and Ube2c were observed in the p53+/+ cells both at 8h and 24h. While in p53−/−, significant decreases in Cyclin B1 and Cdc25c were only observed at 8h and no difference at 24 h. (Fig. 8B). Significant increase in Uchl1 was also observed in both genotypes at both time-points.
Discussion
Our study demonstrates that As3+ induced dose-dependent cell cycle arrest and apoptosis in both p53+/+ and p53−/− cells, supporting findings of previous reports (Huang et al., 1999; Bode and Dong, 2002). Induced cell death in p53−/− cells appears to be unique to As3+, whereas this response is not seen in other metals such cadmium and methylmercury (Gribble et al., 2005), where less cytotoxicity were observed in p53−/− cells versus p53+/+ cells. Previous studies have demonstrated that p53-deficient cells have a greater sensitivity to As3+-mediated cytotoxicity, cell cycle arrest, and apoptosis (Huang and Lee, 1998; McCabe et al., 2000; Liu et al., 2003). This unique response in genetically modified p53−/− cells may be associated with arsenic’s unique ability to act as a chemotherapeutic agent for cancer cells since p53 mutations have been linked with more than 60% of all human cancers (Olivier et al., 2002). A majority of the literature currently available supports the premise that many chemotherapeutic agents cause DNA damage and activate the p53 pathway to induce growth arrest and apoptosis. However, p53 function is often inactivated or suppressed in human cancers (Olivier et al., 2002). Compared to the vast amount of literature available about the p53-dependent pathway, only a few studies have reported p53-independent pathways leading to cell cycle arrest and apoptosis.. Therefore, the understanding of a p53-independent pathway may provide important information for the development of new therapies or novel biological targets for cancer treatment.
The gene knockout approach has been widely used as an efficient approach in identifying critical signaling pathway. The differences in the phenotype such as toxicity and cell cycle arrest between the wildtype and knockout are used to draw conclusions as to whether the specific gene is involved in or not. For example, methylmercury and cadmium have been shown to induce significantly more apoptosis and cell cycle arrest in p53+/+ versus p53−/− cells, which suggests that p53+/+ is critical in methylmercury-and cadmium-induced toxicity (Gribble et al., 2005). If there is no difference between the two genotypes, it is generally concluded that p53 is not a critical genes. Our current study, as well as the previous study, demonstrates that As3+ induces cytotoxicity within p53+/+ and p53−/− cells in a similar dose-dependent manner (Huang et al., 1999; Bode and Dong, 2002). This observation seems to suggest that p53 is not involved in As-induced cell death as proposed by previous studies (Huang et al., 1999; Bode and Dong, 2002). However, our systems-based genomic analysis demonstrated that arsenic induced differential gene pathways leading to cell cycle arrest and apoptosis in p53+/+ cells versus p53−/− cells. In p53+/+ MEFs, As3+ induced p53-dependent gene expression alterations in DNA damage and cell cycle regulation genes, while in the p53−/− MEFs, As3+ induced a significant up-regulation of apoptosis-related genes (Noxa) and down-regulation of genes involved in the regulation of immune modulation. Our current study illustrates the need for careful interpretation when using the gene knockout approach for identification of critical gene.
The As3+-mediated induction of antioxidant and phase II detoxification enzyme genes from our microarray analysis highlights these potential cellular pathways as contributors to As3+-induced cell death in both p53-proficient and-deficient cells (Figure 6). Antioxidant and phase II detoxification enzymes have been shown to act as important defense mechanisms against As3+ treatment (Miller et al., 2002). As3+ was found to generate oxidative stress by inhibiting mitochondrial respiratory function leading to an increase in ROS and depleted ATP (Pelicano et al., 2003). The cellular and molecular adaptive responses to oxidative stress involve increased expression of antioxidant enzymes, phase II detoxification enzymes, and stress-inducible cytoprotective genes aimed at reversing the oxidant imbalance and achieving cellular homeostasis. Recent research revealed that Nrf2 is essential for the coordinate transcriptional induction of various antioxidant and phase II detoxifying enzymes through the antioxidant response element (ARE) (Moi et al., 1994). ARE sequences have been characterized within the proximal regulatory sequences of genes encoding the antioxidant enzymes glutathione GST, NQO1, heme oxygenase-1 (HO-1), and γ-GCS and it regulates a wide range of metabolic responses to oxidative stress caused by ROS (Prestera et al., 1995). As a consequence of inadequate induction of these molecules, Nrf2-deficient mice are sensitive to high oxidative stress and drug-induced stress (Chan et al., 2001; Enomoto et al., 2001). Ramos-Gomez et al. showed that Nrf2 was central to the constitutive and inducible expression of phase II enzymes in vivo and dramatically influences susceptibility to carcinogens (Ramos-Gomez et al., 2003). Nrf2-mediated oxidative stress including GSTs, heme oxygenase, and metallothionein has been reported after As3+ treatment (Miller et al., 2002). A recent study shows that elevation of ROS levels by Emodin increased As2O3-induced apoptosis (Yi et al., 2004).
The p53-dependent regulations of cell cycle alteration observed in this study (Figure 6 & 7) are consistent with previously published gene targets that are regulated by p53 (Kho et al., 2004). In addition, the p53-dependent gene profile alterations by As3+ are quite similar to that of other metals, such as cadmium and methylmercury (Yu et al., 2007). Our results indicate p53 dependency in As3+-induced cell cycle arrest, and these transcriptional changes may contribute to As3+-induced inhibition of cell proliferation in the functional p53 cells. The above results suggest that cells with a functioning p53 protein utilize this pathway to mediate cellular defense and thereby mitigate damage. Impairment of this checkpoint mechanism causes rearrangement, amplification, and loss of chromosomes, events that are causally associated with cancer. Alteration of p53-dependent cell cycle regulatory genes, therefore, might play an important role in As3+-induced toxicity as well as carcinogenesis.
Our microarray results are consistent with previous reports that Gadd45 is induced by genotoxic stress through both p53-dependent and-independent pathways (O’Reilly et al., 2000). Gadd45 has been considered a p53 target gene whose expression is dependent on the activation of p53. Gadd45 also binds and synergizes with p21 to elicit growth arrest (Vairapandi et al., 2000). In addition to its ability to inhibit proliferation and stimulate DNA repair, Gadd45 can also induce apoptosis when over-expressed in primary human fibroblasts in vitro (Wang et al., 1999). Both up-regulation in the p53+/+ and p53−/− MEF cells suggests that the induction of Gadd45 by As3+ is also through a p53-independent pathway. Arsenite-induced G2/M cell cycle arrest was also found partially through the p53-independent but c-Jun-N-terminal kinase-dependent induction of Gadd45a (Chen et al., 2001). Our current study suggests the important role of Gadd45 in the response to DNA damage by As3+, especially in the p53−/− cells. In the p53−/− cells, As3+ also induced up-regulation of Top2a, while this gene is found to be down-regulated in p53+/+ cells. The relationship between the induction of Gadd45 and other genes such as Top2a and cell cycle arrest in p53−/− cells still needs to be studied.
In the p53−/− cells, we identified 118 unique gene expression alterations in response to As3+. Activations of apoptosis pathway genes, Perp and Pmaip1, previously known as Noxa (Attardi et al., 2000; Villunger et al., 2003) were observed, highlighting the possible molecular targets induced by As3+ in the treatment of cancer cells. The Perp gene is highly expressed in cells undergoing p53-dependent apoptosis (Attardi et al., 2000). Until now, Perp was found to be a direct p53 target gene, and its over-expression sufficient to induce cell death in fibroblasts (Attardi et al., 2000). Cellular stress leads to DNA damage and activation of the intrinsic mitochondria-initiated apoptotic pathway. Mitochondria-initiated apoptosis is tightly regulated by BCL-2 family proteins. Anti-apoptotic members such as BCL-2 and BCL-X contain four Bcl-2 homology (BH) domains; the multi-BH domain pro-apoptotic members BAX and BAK contain BH domains 1–3, and a diverse group of loosely related pro-apoptotic proteins such as BID, BAD, BIM, BIK, PUMA, and NOXA contain only BH domain 3 (BH3) (Puthalakath and Strasser, 2002). BH3-only BCL-2 family proteins Noxa and Puma have been found to be important downstream genes of p53 to induce apoptosis (Villunger et al., 2003). Noxa is essential for the release of cytochrome c from mitochondria and mediate stress responses, both dependent and independent of p53 (Villunger et al., 2003). These responses are activated both by cytokine deprivation, cytotoxic drugs, and radiation (Villunger et al., 2003). The efficacy of chemotherapeutic agents on tumor cells has been shown to be modulated by p53 and its target genes such as Bcl-2 family members. However, various chemotherapeutic agents can induce cell death in tumor cells that do not express the functional p53, suggesting that some chemotherapeutic agents may induce cell death in a p53-independent pathway. Liang et al recently demonstrated a p53-independent mechanism for induction of Noxa by another anti-cancer drug, GSI (Liang et al., 2004). Treatment of melanoma cells with GSI can activate apoptotic machinery via Noxa in the absence of p53. Moreover, Noxa−/− MEFs exhibited modest but significant resistance to etoposide-induced apoptosis (Villunger et al., 2003). Our current study suggests that As3+ induces the apoptotic process in p53−/− MEF cells through the activation of p53-independent Noxa signaling pathway and As3+-induced significant up-regulation of BCL-2 BH3 only is tightly related to its anticancer capability.
In the p53−/− cells, significant alterations in genes linked to the immune responses involving multiple pathways such as IL6, IL10, Toll-like receptor, and NFκB signaling pathways were observed. Cytokine deregulation such as elevation of serum IL-6 has been shown to participate in the development or evolution of the malignant process (Kurzrock, 2001). Serum IL-6 levels are elevated in both relapsed and newly-diagnosed Hodgkin’s and non-Hodgkin’s lymphoma (Heinrich et al., 1990), and these levels correlate with established prognostic features (Negrier et al., 2004). Cytokines can be used to decrease apoptosis in normal cells and inhibition of cytokine activity may improve cancer therapy by enhancing apoptosis in cancer cells. Apoptosis can be induced not only by cytokine withdrawal from cytokine-dependent normal and cancer cells, but also by various DNA damaging and other cytotoxic agents. An early study showed that development and aging of the immune system were accelerated in p53-deficient mice (Ohkusu-Tsukada et al., 1999). A recent study by Komarova et al suggested that p53 inhibits inflammation through suppression of NFκB (Komarova et al., 2005). A systems-based comparative gene expression study in head and neck cancer cell lines also identified NFκB regulons are differentially expressed in cancer subsets with distinct p53 status (Yan et al., 2008). The concerted alterations in gene expression patterns reflect cross-talk among NFκB and p53 signaling pathways. The unique regulation in cytokine responses by As3+ in the p53-deficient cells might play a critical role in induction of apoptosis. Further clarification of the role of the immune response and NFκB signaling pathway of As3+ in tumor cells will help add to the understanding of the molecular mechanism of As3+ in cancer treatment.
In summary, although our results demonstrated that As3+ induced concentration-dependent apoptosis and cell cycle arrest in both wildtype and p53-knockout MEFs, our gene expression profiling revealed both common and unique gene pathways involved between the two genotypes. As3+ induces ROS, leading to alterations in antioxidant potentials such as glutathione transferase, heme oxygenase 1, and induces DNA damage in both p53+/+ and p53−/− cells. However, in the presence of functional p53, p53 is activated by several post-transcriptional modifications in response to DNA damage, which in turn activate its downstream targets such as p21, Gadd45, and cyclin B to cause cell cycle arrest and Noxa and Perp to induce apoptosis. In contrast to effects of As3+ mediated via p53, which trigger only slight apoptosis, As3+ triggers prominent apoptosis in p53−/− cells through the p53-independent pathway. In addition to production of ROS and induction of antioxidant genes, treatment with As3+ in p53−/− cells uniquely induces alterations in the modulation of immune responses, leading to the activation of the p53-independent cell cycle regulatory genes and pro-apoptotic machinery including Noxa and Perp.
The observed differential mechanism of cell death between p53+/+ and p53−/− cells might be directly link to As’s unique ability to act as both a carcinogen and a chemotherapeutic agent. Our finding of the unique response to As3+ in p53 genotype cells provides a potential new direction for exploring the role of p53 in carcinogenesis as well as p53-independent apoptosis in cancer treatment with arsenic. The understanding of the different mode of action of arsenic in the normal tissues and p53-deficient cancer cells may provide critical information in developing new clinical applications and, in particular, for designing “cocktails” for enhanced chemotherapy. This approach has the potential to be useful in the treatment of certain human cancers, especially for “drug-resistant” cancers, the effectiveness of which are through p53-dependent apoptosis induced by most of the clinically used chemotherapy agents.
Supplementary Material
Acknowledgments
This work was supported by NIEHS (U10ES11387, R01ES10613, 1PO1ES09601 and P30ES07033) and EPA (R826886). We thank Sean Quigley and Hannah Viernes in the Biomarker Lab for their excellent assistance with the microarray and qRT-PCR analyses.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Attardi LD, Reczek EE, Cosmas C, Demicco EG, McCurrach ME, Lowe SW, Jacks T. PERP, an apoptosis-associated target of p53, is a novel member of the PMP-22/gas3 family. Genes Dev. 2000;14:704–718. [PMC free article] [PubMed] [Google Scholar]
- 3.Bode AM, Dong Z. The paradox of arsenic: molecular mechanisms of cell transformation and chemotherapeutic effects. Crit Rev Oncol Hematol. 2002;42:5–24. doi: 10.1016/s1040-8428(01)00215-3. [DOI] [PubMed] [Google Scholar]
- 4.Borenfreund E, Puerner J. Toxicity determined in vitro by morphological alterations and neurtral red absorption. Toxicol Lett. 1985;24:119–124. doi: 10.1016/0378-4274(85)90046-3. [DOI] [PubMed] [Google Scholar]
- 5.Chan K, Han XD, Kan YW. An important function of Nrf2 in combating oxidative stress: detoxification of acetaminophen. Proc Natl Acad Sci U S A. 2001;98:4611–4616. doi: 10.1073/pnas.081082098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen F, Lu Y, Zhang Z, Vallyathan V, Ding M, Castranova V, Shi X. Opposite effect of NF-kappa B and c-Jun N-terminal kinase on p53-independent GADD45 induction by arsenite. J Biol Chem. 2001;276:11414–11419. doi: 10.1074/jbc.M011682200. [DOI] [PubMed] [Google Scholar]
- 7.Chen GQ, Shi XG, Tang W, Xiong SM, Zhu J, Cai X, Han ZG, Ni JH, Shi GY, Jia PM, Liu MM, He KL, Niu C, Ma J, Zhang P, Zhang TD, Paul P, Naoe T, Kitamura K, Miller W, Waxman S, Wang ZY, de The H, Chen SJ, Chen Z. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): I. As2O3 exerts dose-dependent dual effects on APL cells. Blood. 1997;89:3345–3353. [PubMed] [Google Scholar]
- 8.Dahlquist KD, Salomonis N, Vranizan K, Lawlor SC, Conklin BR. GenMAPP, a new tool for viewing and analyzing microarray data on biological pathways. Nat Genet. 2002;31:19–20. doi: 10.1038/ng0502-19. [DOI] [PubMed] [Google Scholar]
- 9.Doniger SW, Salomonis N, Dahlquist KD, Vranizan K, Lawlor SC, Conklin BR. MAPPFinder: using Gene Ontology and GenMAPP to create a global gene-expression profile from microarray data. Genome Biol. 2003;4:R7. doi: 10.1186/gb-2003-4-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Enomoto A, Itoh K, Nagayoshi E, Haruta J, Kimura T, O’Connor T, Harada T, Yamamoto M. High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE-regulated drug metabolizing enzymes and antioxidant genes. Toxicol Sci. 2001;59:169–177. doi: 10.1093/toxsci/59.1.169. [DOI] [PubMed] [Google Scholar]
- 11.FDA. FDA APPROVES ARSENIC TRIOXIDE FOR LEUKEMIA TREATMENT IN RECORD TIME FOR A CANCER DRUG DEVELOPMENT PROGRAM. Food and Drug Administration U.S. Department of Health and Human Services Public Health Service; 5600 Fishers Lane Rockville, MD 20857: 2000. http://www.fda.gov/bbs/topics/ANSWERS/ANS01040.html. [Google Scholar]
- 12.Gribble EJ, Hong SW, Faustman EM. The magnitude of methylmercury-induced cytotoxicity and cell cycle arrest is p53-dependent. Birth Defects Res Part A-Clin Mol Teratol. 2005;73:29–38. doi: 10.1002/bdra.20104. [DOI] [PubMed] [Google Scholar]
- 13.Heinrich PC, Castell JV, Andus T. Interleukin-6 and the acute phase response. Biochem J. 1990;265:621–636. doi: 10.1042/bj2650621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang C, Ma WY, Li J, Dong Z. Arsenic induces apoptosis through a c-Jun NH2-terminal kinase-dependent, p53-independent pathway. Cancer Res. 1999;59:3053–3058. [PubMed] [Google Scholar]
- 15.Huang SC, Lee TC. Arsenite inhibits mitotic division and perturbs spindle dynamics in HeLa S3 cells. Carcinogenesis. 1998;19:889–896. doi: 10.1093/carcin/19.5.889. [DOI] [PubMed] [Google Scholar]
- 16.Kho PS, Wang Z, Zhuang L, Li Y, Chew JL, Ng HH, Liu ET, Yu Q. p53-regulated transcriptional program associated with genotoxic stress-induced apoptosis. J Biol Chem. 2004;279:21183–21192. doi: 10.1074/jbc.M311912200. [DOI] [PubMed] [Google Scholar]
- 17.Kitchin KT. Recent advances in arsenic carcinogenesis: modes of action, animal model systems, and methylated arsenic metabolites. Toxicol Appl Pharmacol. 2001;172:249–261. doi: 10.1006/taap.2001.9157. [DOI] [PubMed] [Google Scholar]
- 18.Komarova EA, Krivokrysenko V, Wang K, Neznanov N, Chernov MV, Komarov PG, Brennan ML, Golovkina TV, Rokhlin OW, Kuprash DV, Nedospasov SA, Hazen SL, Feinstein E, Gudkov AV. p53 is a suppressor of inflammatory response in mice. Faseb J. 2005;19:1030–1032. doi: 10.1096/fj.04-3213fje. [DOI] [PubMed] [Google Scholar]
- 19.Kurzrock R. Cytokine deregulation in cancer. Biomed Pharmacother. 2001;55:543–547. doi: 10.1016/s0753-3322(01)00140-8. [DOI] [PubMed] [Google Scholar]
- 20.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, Reich M, Hieronymus H, Wei G, Armstrong SA, Haggarty SJ, Clemons PA, Wei R, Carr SA, Lander ES, Golub TR. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 21.Liang XQ, Cao EH, Zhang Y, Qin JF. A P53 target gene, PIG11, contributes to chemosensitivity of cells to arsenic trioxide. FEBS Lett. 2004;569:94–98. doi: 10.1016/j.febslet.2004.05.057. [DOI] [PubMed] [Google Scholar]
- 22.Litzow MR. Arsenic trioxide. Expert Opin Pharmacother. 2008;9:1773–1785. doi: 10.1517/14656566.9.10.1773. [DOI] [PubMed] [Google Scholar]
- 23.Liu Q, Hilsenbeck S, Gazitt Y. Arsenic trioxide-induced apoptosis in myeloma cells: p53-dependent G1 or G2/M cell cycle arrest, activation of caspase-8 or caspase-9, and synergy with APO2/TRAIL. Blood. 2003;101:4078–4087. doi: 10.1182/blood-2002-10-3231. [DOI] [PubMed] [Google Scholar]
- 24.Luderer U, Diaz D, Faustman EM, Kavanagh TJ. Localization of glutamate cysteine ligase subunit mRNA within the rat ovary and relationship to follicular apoptosis. Mol Reprod Dev. 2003;65:254–261. doi: 10.1002/mrd.10298. [DOI] [PubMed] [Google Scholar]
- 25.McCabe MJ, Jr, Singh KP, Reddy SA, Chelladurai B, Pounds JG, Reiners JJ, Jr, States JC. Sensitivity of myelomonocytic leukemia cells to arsenite-induced cell cycle disruption, apoptosis, and enhanced differentiation is dependent on the inter-relationship between arsenic concentration, duration of treatment, and cell cycle phase. J Pharmacol Exp Ther. 2000;295:724–733. [PubMed] [Google Scholar]
- 26.Miller WH, Jr, Schipper HM, Lee JS, Singer J, Waxman S. Mechanisms of action of arsenic trioxide. Cancer Res. 2002;62:3893–3903. [PubMed] [Google Scholar]
- 27.Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci U S A. 1994;91:9926–9930. doi: 10.1073/pnas.91.21.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Negrier S, Perol D, Menetrier-Caux C, Escudier B, Pallardy M, Ravaud A, Douillard JY, Chevreau C, Lasset C, Blay JY. Interleukin-6, interleukin-10, and vascular endothelial growth factor in metastatic renal cell carcinoma: prognostic value of interleukin-6--from the Groupe Francais d’Immunotherapie. J Clin Oncol. 2004;22:2371–2378. doi: 10.1200/JCO.2004.06.121. [DOI] [PubMed] [Google Scholar]
- 29.O’Reilly MA, Staversky RJ, Watkins RH, Maniscalco WM, Keng PC. p53-independent induction of GADD45 and GADD153 in mouse lungs exposed to hyperoxia. Am J Physiol Lung Cell Mol Physiol. 2000;278:L552–559. doi: 10.1152/ajplung.2000.278.3.L552. [DOI] [PubMed] [Google Scholar]
- 30.Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999;27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohkusu-Tsukada K, Tsukada T, Isobe K. Accelerated development and aging of the immune system in p53-deficient mice. J Immunol. 1999;163:1966–1972. [PubMed] [Google Scholar]
- 32.Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, Hainaut P. The IARC TP53 database: new online mutation analysis and recommendations to users. Hum Mutat. 2002;19:607–614. doi: 10.1002/humu.10081. [DOI] [PubMed] [Google Scholar]
- 33.Park WH, Seol JG, Kim ES, Hyun JM, Jung CW, Lee CC, Kim BK, Lee YY. Arsenic trioxide-mediated growth inhibition in MC/CAR myeloma cells via cell cycle arrest in association with induction of cyclin-dependent kinase inhibitor, p21, and apoptosis. Cancer Res. 2000;60:3065–3071. [PubMed] [Google Scholar]
- 34.Pelicano H, Feng L, Zhou Y, Carew JS, Hileman EO, Plunkett W, Keating MJ, Huang P. Inhibition of mitochondrial respiration: a novel strategy to enhance drug-induced apoptosis in human leukemia cells by a reactive oxygen species-mediated mechanism. J Biol Chem. 2003;278:37832–37839. doi: 10.1074/jbc.M301546200. [DOI] [PubMed] [Google Scholar]
- 35.Prestera T, Talalay P, Alam J, Ahn YI, Lee PJ, Choi AM. Parallel induction of heme oxygenase-1 and chemoprotective phase 2 enzymes by electrophiles and antioxidants: regulation by upstream antioxidant-responsive elements (ARE) Mol Med. 1995;1:827–837. [PMC free article] [PubMed] [Google Scholar]
- 36.Puthalakath H, Strasser A. Keeping killers on a tight leash: transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins. Cell Death Differ. 2002;9:505–512. doi: 10.1038/sj.cdd.4400998. [DOI] [PubMed] [Google Scholar]
- 37.Qian Y, Castranova V, Shi X. New perspectives in arsenic-induced cell signal transduction. J Inorg Biochem. 2003;96:271–278. doi: 10.1016/s0162-0134(03)00235-6. [DOI] [PubMed] [Google Scholar]
- 38.Ramos-Gomez M, Dolan PM, Itoh K, Yamamoto M, Kensler TW. Interactive effects of nrf2 genotype and oltipraz on benzo[a]pyrene-DNA adducts and tumor yield in mice. Carcinogenesis. 2003;24:461–467. doi: 10.1093/carcin/24.3.461. [DOI] [PubMed] [Google Scholar]
- 39.Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- 40.Salazar AM, Ostrosky-Wegman P, Menendez D, Miranda E, Garcia-Carranca A, Rojas E. Induction of p53 protein expression by sodium arsenite. Mutat Res. 1997;381:259–265. doi: 10.1016/s0027-5107(97)00207-8. [DOI] [PubMed] [Google Scholar]
- 41.Shen L, Chen TX, Wang YP, Lin Z, Zhao HJ, Zu YZ, Wu G, Ying DM. As2O3 induces apoptosis of the human B lymphoma cell line MBC-1. J Biol Regul Homeost Agents. 2000;14:116–119. [PubMed] [Google Scholar]
- 42.Sidhu JS, Ponce RA, Vredevoogd MA, Yu X, Gribble E, Hong SW, Schneider E, Faustman EM. Cell Cycle Inhibition by Sodium Arsenite in Primary Embryonic Rat Midbrain Neuroepithelial Cells. Toxicol Sci. 2005;89:475–484. doi: 10.1093/toxsci/kfj032. [DOI] [PubMed] [Google Scholar]
- 43.Tapio S, Grosche B. Arsenic in the aetiology of cancer. Mutat Res. 2006;612:215–246. doi: 10.1016/j.mrrev.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 44.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vairapandi M, Azam N, Balliet AG, Hoffman B, Liebermann DA. Characterization of MyD118, Gadd45, and proliferating cell nuclear antigen (PCNA) interacting domains. PCNA impedes MyD118 AND Gadd45-mediated negative growth control. J Biol Chem. 2000;275:16810–16819. doi: 10.1074/jbc.275.22.16810. [DOI] [PubMed] [Google Scholar]
- 46.Vengellur A, Woods BG, Ryan HE, Johnson RS, LaPres JJ. Gene expression profiling of the hypoxia signaling pathway in hypoxia-inducible factor 1alpha null mouse embryonic fibroblasts. Gene Expr. 2003;11:181–197. doi: 10.3727/000000003108749062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, Adams JM, Strasser A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–1038. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- 48.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 49.Wang XW, Zhan Q, Coursen JD, Khan MA, Kontny HU, Yu L, Hollander MC, O’Connor PM, Fornace AJ, Jr, Harris CC. GADD45 induction of a G2/M cell cycle checkpoint. Proc Natl Acad Sci U S A. 1999;96:3706–3711. doi: 10.1073/pnas.96.7.3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wright GW, Simon RM. A random variance model for detection of differential gene expression in small microarray experiments. Bioinformatics. 2003;19:2448–2455. doi: 10.1093/bioinformatics/btg345. [DOI] [PubMed] [Google Scholar]
- 51.Yan B, Chen G, Saigal K, Yang X, Jensen ST, Van Waes C, Stoeckert CJ, Chen Z. Systems biology-defined NF-kappaB regulons, interacting signal pathways and networks are implicated in the malignant phenotype of head and neck cancer cell lines differing in p53 status. Genome Biol. 2008;9:R53. doi: 10.1186/gb-2008-9-3-r53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yi J, Yang J, He R, Gao F, Sang H, Tang X, Ye RD. Emodin enhances arsenic trioxide-induced apoptosis via generation of reactive oxygen species and inhibition of survival signaling. Cancer Res. 2004;64:108–116. doi: 10.1158/0008-5472.can-2820-2. [DOI] [PubMed] [Google Scholar]
- 53.Yih LH, Lee TC. Arsenite induces p53 accumulation through an ATM-dependent pathway in human fibroblasts. Cancer Res. 2000;60:6346–6352. [PubMed] [Google Scholar]
- 54.Yih LH, Peck K, Lee TC. Changes in gene expression profiles of human fibroblasts in response to sodium arsenite treatment. Carcinogenesis. 2002;23:867–876. doi: 10.1093/carcin/23.5.867. [DOI] [PubMed] [Google Scholar]
- 55.Yu X, Griffith WC, Hanspers K, Dillman JF, 3rd, Ong H, Vredevoogd MA, Faustman EM. A system-based approach to interpret dose- and time-dependent microarray data: quantitative integration of gene ontology analysis for risk assessment. Toxicol Sci. 2006;92:560–577. doi: 10.1093/toxsci/kfj184. [DOI] [PubMed] [Google Scholar]
- 56.Yu X, Robinson J, Sidhu J, Hong S, Faustman E. Disruption of Ubiquitin-Proteasome System and Altered Cell Cycle Regulation Involved in Metal-induced cytotoxicity in Mouse Embryonic Fibroblast (MEF): a microarray-based gene expression analysis. Environmental Perspective. 2007 Submitted. [Google Scholar]
- 57.Yu X, Sidhu JS, Hong S, Faustman EM. Essential role of extracellular matrix (ECM) overlay in establishing the functional integrity of primary neonatal rat Sertoli cell/gonocyte co-cultures: an improved in vitro model for assessment of male reproductive toxicity. Toxicol Sci. 2005;84:378–393. doi: 10.1093/toxsci/kfi085. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.