

Abstract
Gametes carry the DNA that will direct the development of the next generation. By compromising genetic integrity, DNA damage and mutagenesis threaten the ability of gametes to fulfill their biological function. DNA repair pathways function in germ cells and serve to ameliorate much DNA damage and prevent mutagenesis. High base excision repair (BER) activity is documented for spermatogenic cells. DNA polymerase-beta (POLB) is required for the short-patch BER pathway. Because mice homozygous null for the Polb gene die soon after birth, mice heterozygous for Polb were used to examine the extent to which POLB contributes to maintaining spermatogenic genomic integrity in vivo. POLB protein levels were reduced only in mixed spermatogenic cells. In vitro short-patch BER activity assays revealed that spermatogenic cell nuclear extracts obtained from Polb heterozygous mice had one third the BER activity of age-matched control mice. Polb heterozygosity had no effect on the BER activities of somatic tissues tested. The Polb heterozygous mouse line was crossed with the lacI transgenic Big Blue mouse line to assess mutant frequency. The spontaneous mutant frequency for mixed spermatogenic cells prepared from Polb heterozygous mice was 2-fold greater than that of wild-type controls, but no significant effect was found among the somatic tissues tested. These results demonstrate that normal POLB abundance is necessary for normal BER activity, which is critical in maintaining a low germline mutant frequency. Notably, spermatogenic cells respond differently than somatic cells to Polb haploinsufficiency..
Keywords: base excision repair, DNA repair, gamete biology, gametogenesis, lacI, mutagenesis, POLB, spermatogenesis, spermatogenic cells
Heterozygosity for Polb results in elevated mutagenesis in spermatogenic cells, indicating the importance of base excision repair in maintaining germline mutant frequencies.
INTRODUCTION
The chemistry of DNA makes it susceptible to many forms of damaging agents. The base excision repair (BER) pathway is the main guardian against damage due to cellular metabolism [1–6]. It is estimated that the BER pathway excises about 1 million lesions per cell per day based on the levels of endogenous base lesions that mammalian cells constantly experience [7]. Two major subpathways of BER are recognized, namely, a short-patch pathway that results in the replacement of one nucleotide and a long-patch pathway that results in the replacement of two to 12 nucleotides [1–6, 8]. DNA polymerase-β (POLB) can function in both subpathways and is essential for the short-patch subpathway [9–13].
Short-patch BER activity varies among mammalian tissues and was found to be greatest in spermatogenic cell nuclear extracts compared with tested somatic tissues [14–17]. Consistent with high levels of BER activity, the spontaneous mutant frequency of spermatogenic cells was found to be significantly lower than that in somatic tissues tested [18, 19]. These data suggest an inverse correlation between BER activity and spontaneous mutagenesis. Consistent with this putative correlation, short-patch BER activity was significantly reduced in spermatogenic cell and somatic tissue nuclear extracts obtained from mice heterozygous for Apex1 (also known as Ape1) [15, 20], an enzyme that functions in short- and long-patch BER [21, 22], with a corresponding increase in spontaneous mutant frequency by 9 mo of age [23]. Notably, the spontaneous mutant frequency for liver and spleen was elevated in 4- to 6-mo-old mice [23]. Together, these data suggest that BER has a major role in maintaining a low spontaneous mutant frequency in a variety of cells and tissues.
Although data from heterozygous Apex1 mice suggest that BER is involved in maintaining a low spontaneous mutant frequency in vivo, it is important to consider that APEX1 is multifunctional, acting as 5′ endonuclease, 3′ phosphodiesterase, 3′ phosphatase, 3′ to 5′ exonuclease, strong p53 activator [24], and redox activator of multiple transcription factors [25–28], as well as having RNase H activities [29]. POLB, however, functions preferentially in the nucleus to fill short gaps in DNA with 5′-phosphate termini [30]. POLB lacks proofreading exonuclease activity and is a distributive polymerase except that short gaps with 5′-phosphate termini are filled processively [31]. Although POLD or POLE can substitute for the DNA synthesis capability of POLB [32], it has been shown that there is no backup for its 5′dRPase activity [33]. The deficiency in 5′dRPase activity is believed to be the cause of neurotoxic effects leading to neonatal death in POLB-deficient mice [33, 34]. Because homozygous null mice for Polb die at 18.5 days post coitum or soon after birth [34, 35], Polb heterozygous (Polb+/−) mice were used for the present study.
It has been speculated that BER proteins must be tightly regulated because imbalances in BER lead to genomic instability [36]. Changes in POLB expression are found in approximately one third of human tumors [37]. Therefore, the abundances of APEX1, POLB, XRCC1, and DNA ligase III were examined in the Polb+/− mice to determine if deficiency of POLB affected other BER proteins. In vitro short-patch BER activity and the effect on mutagenesis were assessed for liver, brain, and spermatogenic cells in these Polb+/− mice as a direct test of the role of POLB and short-patch BER in germline mutagenesis. Finally, because deficiency of DNA repair proteins has been shown to increase apoptosis [23, 38], the prevalence of TUNEL-positive spermatogenic cells was examined to determine if cell death was elevated among POLB-deficient cells in the face of spontaneous DNA damage.
MATERIALS AND METHODS
Polb Heterozygous Animals
lacI Transgenic mice (C57BL/6J) were obtained from Stratagene or from in-house breeding regimens. Mice heterozygous for Polb [35] were mated with homozygous lacI transgenic mice to generate Polb(+/−),lacI+ progeny. The Polb promoter and first exon were deleted in the construction of these mice. The Polb+/− mice were backcrossed at least 10 generations with C57BL/6J mice before these experiments. Genotyping by PCR amplification was performed on DNA obtained from tail biopsy specimens [39, 40] from potential Polb(+/−),lacI+ animals. The primers for the amplification of the Polb(−) allele were MBEX2 and MBFOR2 with the corresponding sequences of 5′CTGGCTCACGTTCTTCTCAAAGTTTGCGAG′3 and 5′AAGGACGGAA GGTGGAGGGAGAGCTAATGC′3, respectively. The amplimer was visualized as a 420-bp band. The primers for the amplification of the lacI transgene were T7LacI and LacI5 with the corresponding sequences of 5′TAATACGACTCACTATAGGGACACCATCGAA TGGTGCAAAAC′3 and 5′ATTTAGGTGACACTATAGGAGAACTTAATGGGCCCG′3, respectively, and produced a 680-bp amplimer. Primers for exon IV of the Apex1 gene were used for an internal control. The sequence for sense Apex1 was 5′GTGATTGTGGCTGAATT TGA′3 and 5′GTCTAAAGGAAACCGGAAGT′3 for antisense Apex1. This primer set produced a 650-bp amplimer [23]. The PCR consisted of 30 cycles, including denaturation at 94°C for 1 min, annealing at 54°C for 1.5 min, and extension at 72°C for 1 min. The PCR products were subjected to electrophoresis using 2% agarose gel electrophoresis.
All animals were housed in an American Association for the Accreditation of Laboratory Animal Care-accredited facility and were maintained on standard food and water ad libitum. Before embarking on experiments, all animal manipulations were approved by the Institutional Animal Care and Use Committee. The mice were specific pathogen free, anesthetized with isoflurane, and humanely euthanized by cervical dislocation at age 10 days or at age 4–7 mo. Liver and brain were rapidly removed and used for preparations of nuclear extracts or high-molecular-weight DNA isolation. Testes were removed and fixed in formalin for subsequent histological analysis or used immediately for mixed germ cell preparations (i.e., all spermatogenic cell types in the testis essentially free of somatic cells). Afterward, nuclear extracts or high-molecular-weight DNAs were prepared from mixed germ cell preparations.
Mixed Germ Cell Preparations
Preparation of mixed germ cells was conducted as described previously [41, 42]. Testes harvested from mice were decapsulated by making a small incision in the tunica albuginea and gently squeezing out the seminiferous tubules. The decapsulated testes were placed in Krebs solution [41, 42] previously brought to pH 7.3 with aeration for 20 min of a gas mixture of 5% carbon dioxide and 95% air. Collagenase was added to a final concentration of 0.5 mg/ml, and the tubules were incubated for 15 min at 33°C in a shaking water bath. The seminiferous tubules were washed once with Krebs solution to maximize removal of interstitial cells and blood cells. Trypsin was added to the tubules to a final concentration of 0.5 mg/ml in Krebs solution after washing to disaggregate the germ cells. A total of 5 μg of DNase was also added. The tubules were incubated for 12 min at 33°C in a shaking water bath under increased agitation compared with the collagenase treatment. The germ cells were dissociated using a wide-bore pipette by pipetting up and down until homogeneous, yielding a suspension of single cells. The germ cell suspension was filtered through a 100-μm nylon mesh membrane and pelleted at 1100× g at 4°C for 15 min. The supernatant was discarded, and the germ cell pellet was washed once with Krebs solution. Before the final centrifugation, the cells were counted on a hemacytometer.
Nuclear Extract Preparations
Spermatogenic cell, liver, and brain crude nuclear extracts were prepared as described previously [30] and as modified by Intano et al. [14, 15, 43]. Further modification included the addition of Complete Mini protease inhibitor cocktail tablets from Roche to the homogenization and lysis buffers at 1 tablet/10 ml of solution. Just before lysis, 2.5 ng/μl of luciferase per milliliter of lysis buffer was added for a total of 2500 ng for each mixed germ cell sample and 5000 ng for liver and brain. Protein concentrations were determined by the Bradford assay [44] using immunoglobulin as the protein standard. Aliquots of nuclear extracts were flash frozen in liquid nitrogen and stored at −80°C.
Western Blot Analysis
Mixed germ cell, liver, and brain nuclear extracts (75 μg) were separated on a 12% discontinuous SDS-PAGE (30% acrylamide:bis solution, 29:1) gel and transferred onto a nitrocellulose membrane via electroblotting. After transfer, each blot was cut into three strips to separate regions containing 1) DNA ligase IIIα, 2) XRCC1, and 3) POLB and APEX1. Each section was hybridized with primary and then secondary antibodies before realigning the sections for chemiluminescent exposure. Accordingly, the relative proportions of each protein within a tissue were determined using the same gel, membrane, exposure, and antibody incubation conditions. For each tissue, at least one BER protein did not vary by genotype, and it was used as an internal loading control. Rabbit polyclonal anti-hAPEX1 antibody (Novus Biologicals), mouse monoclonal POLB antibody (NeoMarkers), rabbit polyclonal anti-hXRCC1 antisera (Serotec), and mouse monoclonal DNA ligase IIIα antibody (Genetex) were used to detect APEX1, POLB, XRCC1, and DNA ligase IIIα proteins, respectively. Incubation with primary antibodies at 1:500 dilution for APEX1 and DNA ligase IIIα and at 1:1000 for POLB and XRCC1 was followed by incubation with horseradish peroxidase-conjugated goat anti-rabbit or goat anti-mouse secondary antibodies (Pierce) at 1:250 000 dilution and by signal generated-enhanced chemiluminescence (Pierce). Intensity of the bands generated was quantified using a ChemiImager 4400 (Alpha Innotech) as integrated density value (IDV). Data collected as IDV per microgram of protein from the Polb heterozygotes were normalized against wild-type littermate controls. BenchMark Prestained Protein Ladder (Invitrogen) and purified hAPEX1 and hPOLB proteins (Trevigen) were included as molecular mass standards.
BER Assay
Short-patch BER activity assays were conducted as previously described [11, 14, 15, 43]. Single-stranded oligonucleotides purified via PAGE and HPLC were purchased from Integrated DNA Technologies, Inc. The lesion is a uracil at position 22 in the 51-mer sequence 5′56FAM/GCTTGCATGCCTGCAGGTCGAUTCTAGAGGATCCCCGGGTACCGAGCTA3′. The 56-FAM denotes the 5′ fluorescein end label on the U-containing strand. The complementary strand was annealed to the U-containing oligonucleotide in 5 times molar excess, producing a double-stranded oligonucleotide or guanine:uracil22 (G:U22) 51-mer at 1 pmol/μl by incubation at 95°C followed by overnight incubation in a heating block that gradually reached room temperature. Nuclear extracts were incubated with 3 pmol of double-stranded G:U22 51-mer, 2.5 μl of salt buffer (1 M Tris-HCl [pH 7.5], 50 mM MgCl2, 10 mM dithiothreitol [DTT], and 1 mM EDTA), 2.5 μl of energy buffer (20 mM ATP, 2 mM NAD, 50 mM ditrisphosphocreatine, 200 μM dATP, 200 μM dTTP, 200 μM dGTP, and 200 nM dCTP), 2.5 μl of nontransgenic C57BL/6J genomic DNA, 2.5 μl of 4 U/μl of creatine phosphokinase, 2.0 μl of [α33P] dCTP, and buffer B [14, 15, 43] brought to a 25-μl total reaction volume. Samples were incubated at 37°C for 10 min, and the reaction was immediately stopped by placement on ice and the addition of 4.5 μl of loading dye (80% formamide, 10 mg/ml of xylene cyanol, 10 mg/ml of bromophenol blue, 50 mM EDTA, and 0.3 M sodium chloride). Samples were heated to 65°C for 3 min, immediately cooled on ice, and loaded on a 12% polyacrylamide gel containing 7 M urea in Tris/borate/EDTA buffer (89.2 mM Trizma base, 89 mM boric acid, and 2 mM EDTA [pH 8.0]). Known amounts of G:U22 51-mer were loaded on the same gel as the nuclear extract samples, and the resulting fluorescence was used to determine oligonucleotide recovery for experimental samples. Afterward, the gel was exposed on screens that were scanned on a BioRad Molecular Imager FX for radioactivity quantitation and on an Alpha Innotech ChemiImager 4400 for fluorescence quantification [14].
Protein amounts were tested to identify the range that would yield linear results. Although the actual limits of linearity were not identified, 10–50 μg of mixed germ cell nuclear extracts, 40–320 μg of liver nuclear extracts, and 80–400 μg of brain nuclear extracts resulted in linear increases in oligonucleotide repair. Consequently, 40 μg of mixed germ cell nuclear extracts and 160 μg of liver and brain nuclear extracts were chosen for the assays.
Repaired G:U22 51-mer was calculated as follows: counts of bands × G:U22 51-mer oligonucleotide added (in picomoles) × amount of luciferase added (in nanograms) / amount of nuclear extracts (in micrograms) × activity × exposure time of autoradiography (in minutes) × G:U22 51-mer oligonucleotide recovered (in picomoles) × luciferase recovered (in nanograms). The machine count per minute for 1 fmol of [α33P] dCTP was empirically determined to be 243.
A luciferase assay was performed on the nuclear extracts the same day the BER activity assays were performed as described previously [14, 15, 43]. The luciferase activity was used to normalize BER activity to protein recovery during the nuclear extract preparation procedure. One microliter of nuclear extract sample was diluted 1:200 in assay buffer (60 mM DTT, 0.075% BSA, and 5 mM ATP in luciferase buffer [60 mM Tris-acetate, pH 7.5, 2.5 mM EDTA, and 12 mM magnesium acetate]). Two microliters of diluted nuclear extract was mixed with 38 μl of luciferase buffer and injected with luciferin-assay buffer mixture. Based on the relative light units, the amount of luciferase recovered by the nuclear extracts was determined with Berthold's Lumat LB 9501 using a standard curve generated from luciferase standards [45, 46].
Big Blue Mouse Mutant Assay
High-molecular-weight genomic DNA was prepared using the RecoverEase DNA isolation kit from Stratagene according to the manufacturer's recommendations with one modification: DNA isolation from brain was modified by using a loose-fitting pestle for homogenization. Mixed germ cells were isolated, flash frozen in liquid nitrogen, and stored at −80°C. The germ cell pellet was homogenized by pipetting up and down in lysis buffer until homogeneous. The homogenate was not filtered, and the pellet was incubated with proteinase K for 3 h. Otherwise, the aforementioned protocol was followed.
The λ transgenic shuttle vector recovery was performed as described in Stratagene's Transpack Packaging Extracts, producing packaged DNA samples. The packaged DNA was incubated with SCS-8 cells, then mixed with agar that contained the chromogenic substrate X-gal (5-bromo-4-chloro-e-indolyl-β-D-galactopyranoside) and poured on assay trays. The assay trays were allowed to solidify and were incubated at 37°C overnight. Putative mutants, identified as blue-appearing plaques, were cored and replated to determine if the mutant was likely mouse derived as described previously [23]. The mutant frequency was calculated by the ratio of confirmed mutants to the total number of plaque-forming units (pfu).
Terminal Deoxynucleotidyl TUNEL
Testes from 10-day-old and 4- to 7-mo-old mice were harvested and fixed in 10% formalin solution. The tissues were paraffin embedded, and TUNEL assays [47] and hematoxylin-eosin (H&E) staining were performed on 4-μm testis sections by the San Antonio Cancer Institute Pathology Core Shared Resource. For 10-day-old animals, sections from two different depths of the testis, one near the surface and one approximately at the midpoint of the testis, were stained to ensure that nonadjacent sections were analyzed. Sections from four different depths, evenly distributed across the testis, were stained and examined for adult animals. The number of seminiferous tubules with at least one TUNEL-positive cell was counted, so that at least 230 seminiferous tubule cross-sections were counted per animal in the 10-day-old group and at least 580 seminiferous tubule cross-sections were counted per animal in the adult group. The percentage of seminiferous tubule cross-sections displaying one or more apoptotic cells was quantified using four microscopic fields at 16× magnification for 10-day-old mice and using two microscopic fields at 6.3× magnification for adults. Because of the considerably larger seminiferous tubules in adults compared with 10-day-olds, lower magnification was used in the adults to visualize a large number of different seminiferous tubules. For each genotype, at least 320 apoptotic spermatogonia were counted for 10-day-olds, and at least 90 apoptotic spermatogonia were counted for young adults. The ratio of total number of apoptotic germ cell types to total number of tubules counted per animal was analyzed in both age groups.
Statistical Analysis
Western blot, BER, and TUNEL data were analyzed by ANOVA, and comparisons among means were Bonferroni adjusted. Mutant frequency data were analyzed using chi-square test.
RESULTS
Western Blot Analysis
In mixed germ cell nuclear extracts, two cross-reacting bands for anti-hAPEX1 were found, a 37-kDa and a 34-kDa band (Fig. 1A), whereas only a 37-kDa band was observed in the somatic tissues tested (Fig. 1, B and C). The level of APEX1 protein was significantly lowered to 58%, 75%, and 49% in mixed spermatogenic cells, liver, and brain, respectively, obtained from Polb+/− samples vs. wild-type controls (P ≤ 0.05) (Fig. 1D). POLB and DNA ligase IIIα levels were also decreased to 58% and 80%, respectively, of those of wild-type controls in mixed germ cells, whereas XRCC1 levels were only decreased in the somatic tissues of Polb heterozygotes (P ≤ 0.05). XRCC1 protein levels were decreased to 78% and 77% in liver and brain, respectively (P ≤ 0.05).
FIG. 1.

Western blot analysis. Western blot analysis of nuclear extracts prepared from mixed germ cells (MGC) (A), liver (B), and brain (C) obtained from wild-type control and Polb+/− animals. Each tissue was analyzed separately from other tissues. Alongside samples, lanes were also loaded with purified APEX1 and POLB as standards. APEX1 protein is distinct as two bands on MGC Western blots at 37 kDa and at 34 kDa. POLB is at 39 kDa, XRCC1 is at 69 kDa, and DNA ligase IIIα is at 93 kDa. D) Summary of Western blot quantification using nuclear extracts prepared from Polb+/− and wild-type mice. Data are given as mean ± SEM after normalization to values for wild-type controls. At least four animals were used per genotype. Bands were quantified as IDV per microgram of protein. Solid black bars, MGC; solid gray bars, liver; hatched bars, brain; and a, significantly different from wild-type control in same tissue.
BER Activity Assays
To examine the effect of Polb heterozygosity on BER activity, BER activity assays were performed using nuclear extracts obtained from select tissues of 4- to 7-mo-old adult Polb heterozygotes. Various protein amounts were tested to identify the range that would yield linear results. For a given amount of protein, there was consistently greater BER activity for mixed germ cells compared with somatic tissues (P ≤ 0.05) (Fig. 2A). Mixed germ cell nuclear extracts obtained from Polb heterozygous mice showed a striking 60% lower BER activity vs. wild-type controls (i.e., mean ± SEM of 0.39 ± 0.063 vs. 0.13 ± 0.036 fmol/μg of protein; P ≤ 0.05) (Fig. 2, B and E). Notably, liver and brain nuclear extract BER activities were similar between Polb heterozygous and wild-type control mice (Fig. 2, C–E). Liver nuclear extracts exhibited approximately 3-fold higher activity than brain nuclear extracts regardless of genotype (P ≤ 0.05). Also regardless of genotype, mixed germ cell nuclear extracts displayed significantly greater BER activity compared with liver and brain nuclear extracts (P ≤ 0.05 for both). For wild-type controls, mixed germ cell nuclear extracts displayed 14-fold greater BER activity than liver nuclear extracts and 50-fold greater BER activity than brain nuclear extracts (P ≤ 0.05 for both). For Polb heterozygous mice, mixed germ cell nuclear extracts displayed 4-fold greater BER activity than liver nuclear extracts and 17-fold greater BER activity than brain nuclear extracts (P ≤ 0.05 for both).
FIG. 2.

BER activity assay results. A) Linearity determinations for mixed germ cells (MGC), liver, and brain BER activities relative to protein concentration. The best-fit line is shown for wild-type control MGC (solid line), liver (long dashed line), and brain (short dashed line). a, significantly different from liver and brain; b, significantly different from brain. B–D) BER activities for nuclear extracts prepared from MGC (B), liver (C), and brain (D) obtained from wild-type (WT) control and Polb+/− animals. Forty micrograms of MGC nuclear extracts and 160 μg of liver and brain nuclear extracts were used. Results are given as mean ± SEM (E). Four animals were used per genotype for MGC and liver and three for brain. Results are from at least two replicate assays for each sample. Solid bars, control; hatched bars, Polb+/−; a, significantly different from wild-type control in same tissue; b, significantly different from MGC and brain same of genotype; and c, significantly different from MGC and liver of same genotype.
Mutant Frequency Assays
To determine the effects of Polb heterozygosity and BER deficiency on spontaneous mutagenesis, Polb heterozygous mice were crossed with lacI transgenic mice, and spontaneous mutant frequencies were measured from mixed germ cells, liver, and brain obtained from 4- to 7-mo-old mice. At least 2 million pfu per genotype were counted for mixed germ cells, and at least 1 million pfu were counted for liver and brain each (Table 1). Consistent with previously published results [18, 19, 23], the spontaneous mutant frequency of mixed spermatogenic cells was significantly lower than somatic tissue mutant frequencies (P ≤ 0.036) regardless of genotype. In an inverse correlation to the BER activity findings, the spontaneous mutant frequency for Polb(+/−),lacI+ mixed germ cells (mean ± SEM, 2.41 ± 0.34 × 10−5) was significantly greater than that for wild-type controls (1.44 ± 0.26 × 10−5) (P ≤ 0.022) (Fig. 3). Although the spontaneous mutant frequency for Polb(+/−),lacI+ liver was reduced compared with wild-type controls, it did not reach statistical significance (P ≤ 0.064). The spontaneous mutant frequency for Polb(+/−),lacI+ brain was similar to that of wild-type controls (P ≤ 0.996).
TABLE 1.
Spontaneous mutant frequencies for MGC, liver, and brain.

FIG. 3.

Spontaneous mutant frequencies for MGC and somatic tissues obtained from wild-type control (solid bars) and Polb(+/−),lacI+ (hatched bars) animals. Results are given as mean ± SEM. Five animals per genotype were used minimally, and at least two replicate assays were performed on DNA obtained from each animal. a, significantly different from wild-type control in same tissue; b, significantly different from liver of same genotype; and c, significantly different from brain of same genotype.
Prevalence of Apoptosis
To examine the potential role of apoptosis on germline mutant frequency, TUNEL assays were performed on formalin-fixed testes sections obtained from 10-day-old and 4- to 7-mo-old mice (Fig. 4). Testes cross-sections were also stained with H&E to further examine the morphology of the testes. An approximate 2-fold decline was found in the percentage of seminiferous tubules with apoptotic cells from young adults compared with 10-day-olds regardless of genotype (P ≤ 0.05) (Table 2). The ratio of apoptotic spermatogonia to seminiferous tubules was also analyzed. The number of apoptotic spermatogonia per seminiferous tubules declined 7-fold in young adults compared with that in 10-day-old mice for each genotype (P ≤ 0.05). In conclusion, no significant difference was found between wild-type controls and Polb heterozygous mice when the number of apoptotic cells and the number of apoptotic spermatogonia per seminiferous tubules were analyzed.
FIG. 4.
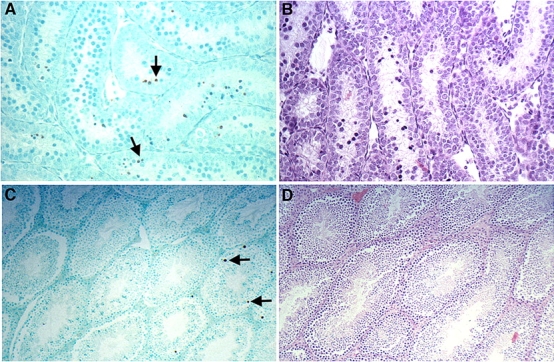
TUNEL staining of testes cross-sections obtained from 10-day-old and young adult wild-type control mice. A) TUNEL-stained cross-section from a 10-day-old mouse. B) An H&E-stained testis from a 10-day-old mouse. C) TUNEL-stained young adult testis. D) An H&E-stained young adult testis. Brown precipitate on TUNEL-stained slides indicates TUNEL-positive nuclei, indicative of cells scored positive for apoptosis. Prevalence of apoptosis among the testis decreases approximately by half as the animal ages from 10 days to adulthood in wild-type controls and in Polb+/− mice. Examples of apoptotic nuclei are designated by black arrows. Original magnification A and B ×16; C and D ×6.3.
TABLE 2.
Prevalence of apoptosis in testis.

DISCUSSION
To determine the effect of a functional Polb allele on BER activity in various tissues, short-patch BER activities were assessed using nuclear extracts obtained from mice heterozygous for Polb. A major finding in the present study was the 2-fold increased spontaneous mutagenesis in spermatogenic cells obtained from young adult Polb+/− mice. The greater short-patch BER activity in mixed spermatogenic cell nuclear extracts compared with somatic tissues tested is consistent with previous results for wild-type mice [14–17]. Notably, many short-patch BER genes display the highest levels of expression in the testis of adult mice. For instance, POLB, XRCC1, and DNA ligase IIIα display high expression in pachytene spermatocytes and round spermatids [14, 16, 48–53]. BER activity was found to be significantly lower in the male germ cells of Polb heterozygous mice compared with wild-type controls. Cabelof et al. [54] detected reduced POLB and BER activity in testis but did not report a spontaneous mutant frequency for spermatogenic cells.
The Western blot analyses and in vitro BER activity data presented herein indicate that Polb haploinsufficiency causes a deficit in BER in spermatogenic cells. It is unclear at present which POLB activities are involved in mediating the elevated mutant frequency. It is tempting to speculate that unrepaired lesions (POLB substrates) act as substrates for error-prone translesion synthesis DNA polymerases such as Rev1, resulting in increased mutagenesis. It was found that methyl methanesulfonate-induced increase in mutations in Polb null mouse embryonic fibroblasts (MEFs) was mediated by REV1 [55].
In contrast to the results for spermatogenic cells, mutant frequency is decreased in the brain of Polb homozygous null embryos [56]. Apoptosis of neuronal cells with damaged DNA may have contributed to the decreased spontaneous mutant frequency found in the brain of Polb homozygous null embryos. Extensive apoptosis is observed in the developing nervous systems of POLB-deficient mice [34]. It has also been shown that POLB deficiency activates p53-dependent apoptosis [57] and that POLB deficiency in cells is associated with hypersensitivity to damaging agents due to induction of necrotic cell death [58] and apoptosis [59, 60]. The elevated mutant frequency in spermatogenic cells indicated an elevated level of DNA damage in spermatogenic cells presumably due to decreased DNA repair. Based on findings by Niimi et al. [56] that suggested an increased apoptotic response to decreased POLB, apoptosis was assessed in testis because mixed germ cells displayed reduced POLB abundance. Consistent with previous findings [61], a greater prevalence of apoptosis was found among both genotypes at age 10 days, corresponding to a time in the first wave of spermatogenesis, compared with young adults. However, Polb+/− mice did not display more apoptosis at either age compared with age-matched controls. These results indicate that apoptosis did not affect the link between Polb heterozygosity and spontaneous mutant frequency in the testes and suggest that there may be tissue-specific differences in response to Polb haploinsufficiency. In a previous study [23], an increased prevalence of seminiferous tubules displaying TUNEL-positive cells was detected among the testes of Apex1 heterozygous mice, suggesting that deficiencies in different BER proteins may elicit different responses within the same tissue.
A second major finding of the present study is the different response of somatic cells to Polb haploinsufficiency. Nuclear extracts prepared from liver and brain showed normal levels of BER proteins and normal BER activity compared with previously published results [14, 20, 43]. Only nuclear extracts prepared from mixed spermatogenic cells obtained from Polb heterozygotes showed perturbed levels of POLB. Wild-type animals show tissue-specific differences in expression of BER proteins and BER activity. For example, all BER genes tested show the greatest expression in testis or spermatogenic cells, and short-patch BER activity is greatest in spermatogenic cells [14–17]. It perhaps should not be surprising that somatic cells respond differently to Polb haploinsufficiency than germ cells in light of the already established differences in normal BER gene expression among tissues. The unchanged abundance of POLB among somatic tissues of Polb heterozygous mice suggests that somatic cells can maintain normal POLB levels through transcriptional or posttranscriptional mechanisms. Recently, BER proteins not in complex with DNA damage were shown to be ubiquitinated and degraded [62] as a mechanism for coordinated expression. Other mechanisms that have not yet been identified may exist to regulate BER protein abundances. Presumably, these same mechanisms do not function or are inadequate to compensate for the normally greater abundance of POLB in spermatogenic cells, as POLB abundance is reduced relative to other BER proteins in spermatogenic cells obtained from Polb heterozygous mice. Tissue-specific differences in response to deficiencies in DNA repair genes are well known. For example, deficiencies in nucleotide excision repair genes lead to xeroderma pigmentosum and a greatly elevated incidence of skin cancer. Deficiencies in mismatch repair gene can lead to hereditary nonpolyposis colorectal cancer. Therefore, in response to DNA repair protein deficiencies, effects can be found in one tissue, while the consequences are more subtle or nondetectable in another tissue.
Consistent with BER assay results, Polb heterozygosity does not significantly affect liver and brain spontaneous mutant frequencies in young adult mice. The results observed in the present study for liver are in agreement with previously published results [54] and are consistent with the similar spontaneous mutant frequency observed for Polb null MEFs compared with wild-type controls [63]. Somewhat surprisingly, the spontaneous mutant frequency for liver obtained from Polb heterozygous mice was slightly reduced (P = 0.064). Although not statistically significant at age 4–7 mo, it is possible that a significant difference might be realized as the animals age. Spontaneous mutant frequencies for brain obtained from Polb heterozygous adult mice have not been previously reported (to our knowledge), although Niimi et al. [56] detected a decrease in brain spontaneous mutant frequency of Polb homozygous null embryos. The results herein are consistent with previously published reports for wild-type brain [18, 64, 65].
The data for POLB abundance and short-patch BER activity in somatic tissues in the present study contrast with results by Cabelof et al. [54], who found a decrease in POLB and short-patch BER activity in all tissues tested. The reasons for the discrepancies in results are unclear and are not easily resolved. The preparation of nuclear extracts by Cabelof et al. had slight modifications, which may account partially for the difference in results. They did not include internal standards to normalize experimental data, and this may account for some of the difference, although the corrections for recovery based on the internal controls did not alter the pattern of results in the present experiments compared with the data without recovery corrections. It seems more likely that differences in environmental conditions could contribute to the differences in results. The consequences of different diets, exposure to endocrine disrupters such as bisphenol A, or epigenetic reprogramming may influence repair gene expression and mutagenesis. Although Cabelof et al. reported decreased POLB abundance and BER activity in select somatic tissues, they did not report a difference in spontaneous mutagenesis. Indeed, the spontaneous mutant frequencies were similar for wild-type and Polb+/− liver. The similarity in somatic tissue spontaneous mutagenesis is inconsistent with the 50% reduction in POLB abundance and BER activity but is consistent with the data reported in the present study. Most important, regardless of the source of discrepancies, the combined studies clearly demonstrate that haploinsufficiency results in reduced POLB and reduced BER in testis or spermatogenic cells. This is consistent with the increased spontaneous mutagenesis in spermatogenic cells detected in the present study.
POLB has been shown to interact with APEX1 [9], XRCC1/DNA ligase IIIα heterodimer [66, 67], and DNA ligase I [68]. The interaction with multiple BER proteins has led to the suggestion that POLB participates in the sequential coordination of the BER pathway [69]. Our study found decreased levels of APEX1 in liver, brain, and mixed germ cells and DNA ligase IIIα of mixed germ cells from Polb heterozygotes. XRCC1 levels were also significantly reduced in the somatic tissues tested. XRCC1 has other protein partners and functions in addition to partnering with ligase IIIα [70, 71]. Changes in XRCC1 can be observed without changes in ligase IIIα. A decrease in ligase IIIα would only be expected if XRCC1 levels decrease below an equimolar level of ligase IIIα. Ligase IIIα seems also to have roles unrelated to XRCC1. For example, ligase IIIα is detected in mitochondria, but XRCC1 is not [72, 73]. It is not clear how reduced abundance of POLB in spermatogenic cells mediates a decline in APEX1 and DNA ligase IIIα, although the recent finding of increased degradation of BER proteins not complexed with DNA damage may be explanatory [62]. Potentially less APEX1 and DNA ligase IIIα are complexed with DNA damage when POLB abundance is reduced. More perplexing is the reduced abundances of APEX1 and XRCC1 in liver and brain of Polb heterozygous mice, while the abundance of POLB was apparently not altered in these tissues. A speculative explanation is that the antibodies for APEX1 and XRCC1 have a lower affinity for the respective proteins than the antibody generated against POLB. Therefore, modest changes in POLB would be less readily detected over the normal variability found with Western blots, while the lower affinity of the other antibodies would result in a more readily detected modest decrease. It seems likely that as-yet undetected mechanisms contribute to regulation of BER protein abundances.
In summary, heterozygosity for Polb elicited different effects in male mixed germ cells compared with somatic tissues of young adult mice. The results indicate that heterozygosity for Polb in the germline results in decreased abundance of the protein, reduced BER activity, and elevated spontaneous mutagenesis. Inactivation of a Polb allele in the somatic tissues tested, liver and brain, had no detectable effect on in vitro BER activity or in vivo spontaneous mutagenesis in young adult mice. That is, genetic instability was not altered significantly in these somatic tissues. Therefore, the abundance and activity of POLB normally present in spermatogenic cells are necessary for retaining a low spontaneous mutant frequency for the male germline. Together, the data indicate tissue-specific responses to Polb heterozygosity. Although Polb heterozygosity is tolerated or compensated for in somatic tissues, it has profound effects in spermatogenic cells on BER activity and spontaneous mutagenesis, thereby demonstrating the importance of short-patch BER in sustaining germline genetic integrity.
Footnotes
1Supported by grant P20 CA103730 from the Elsa U. Pardee Foundation and the National Institutes of Health to R.W.S. and by grants 1 R01 AG24364-01 and AG21163 from the National Institutes of Health to C.A.W. This research was supported in part by the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences. The contents are solely the responsibility of the authors and do not necessarily reflect the views of the funding agencies.
REFERENCES
- Norbury CJ, Hickson ID.Cellular responses to DNA damage. Annu Rev Pharmacol Toxicol 2001; 41: 367–401.. [DOI] [PubMed] [Google Scholar]
- Wood RD.DNA repair in eukaryotes. Annu Rev Biochem 1996; 65: 135–167.. [DOI] [PubMed] [Google Scholar]
- Nielsen H, Krokan HE.Base excision repair in a network of defense and tolerance. Carcinogenesis 2001; 22: 987–998.. [DOI] [PubMed] [Google Scholar]
- Lindahl T, Wood RD.Quality control by DNA repair. Science 1999; 286: 1897–1905.. [DOI] [PubMed] [Google Scholar]
- Lindahl T.Suppression of spontaneous mutagenesis in human cells by DNA base excision-repair. Mutat Res 2000; 462: 129–135.. [DOI] [PubMed] [Google Scholar]
- Fortini P, Pascucci E, Parlanti E, D'Errico MD, Simonelli V, Dogliotti E.The base excision repair: mechanisms and its relevance for cancer susceptibility. Biochimie 2003; 85: 1053–1057.. [DOI] [PubMed] [Google Scholar]
- Holmquist GP.Endogenous lesions, S-phase-independent spontaneous mutations, and evolutionary strategies for base excision repair. Mutat Res 1998; 400: 59–68.. [DOI] [PubMed] [Google Scholar]
- Frosina G, Fortini G, Rossi O, Carrozzino F, Raspaglio G, Cox LS, Lane DP, Abbondandodo A, Dogliotti E.Two pathways for base excision repair in mammalian cells. J Biol Chem 1996; 271: 9573–9578.. [DOI] [PubMed] [Google Scholar]
- Bennett RAO, Wilson DM, III, Wong D, Demple B.Interaction of human apurinic endonuclease and DNA polymerase β in the base excision repair pathway. Proc Natl Acad Sci U S A 1997; 94: 7166–7169.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto Y, Kim K.Excision of deoxyribose phosphate residues by DNA polymerase β during DNA repair. Science 1995; 269: 699–702.. [DOI] [PubMed] [Google Scholar]
- Singhal RK, Prasad R, Wilson SH.DNA polymerase β conducts the gap-filling step in uracil-initiated base excision repair in a bovine testis nuclear extract. J Biol Chem 1995; 270: 949–957.. [DOI] [PubMed] [Google Scholar]
- Liu Y, Beard WA, Shock DD, Prasad R, Hou EW.DNA polymerase β and flap endonuclease 1 enzymatic specificities sustain DNA synthesis for long patch base excision repair. J Biol Chem 2005; 280: 3665–3674.. [DOI] [PubMed] [Google Scholar]
- Prasad R, Dianov GL, Bohr VA, Wilson SH.Fen1 stimulation of DNA polymerase β mediates an excision step in mammalian long patch base excision repair. J Biol Chem 2000; 275: 4460–4466.. [DOI] [PubMed] [Google Scholar]
- Intano GW, McMahan CA, Walter RB, McCarrey JR, Walter CA.Mixed spermatogenic germ cell nuclear extracts exhibit high base excision repair activity. Nucleic Acids Res 2001; 29: 1366–1372.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intano GW, McMahan CA, McCarrey JR, Walter RB, McKenna AE, Matsumoto Y, MacInnes MA, Chen DJ, Walter CA.Base excision repair is limited by different proteins in male germ cell nuclear extracts prepared from young and old mice. Mol Cell Biol 2002; 22: 2410–2418.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen A, Bjortuft H, Wiger R, Holme JA, Seeberg EC, Bjoras M, Brunborg G.Highly efficient base excision repair (BER) in human and rat male germ cells. Nucleic Acids Res 2001; 29: 1781–1790.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabelof DC, Raffoul JJ, Yanamadala S, Ganir C, Guo Z, Heydari AR.Attenuation of DNA polymerase β-dependent base excision repair and increased DMS-induced mutagenicity in aged mice. Mutat Res 2002; 500: 135–145.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter CA, Intano GW, McCarrey JR, McMahan CA, Walter RB.Mutation frequency declines during spermatogenesis in young mice but increases in old mice. Proc Natl Acad Sci U S A 1998; 95: 10015–10019.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler SW, Provost GS, Fieck A, Kretz PL, Bullock WO, Putman DL, Sorge JA, Short JM.Analysis of spontaneous and induced mutations in transgenic mice using a lambda ZAP/lacI shuttle vector. Environ Mol Mutagen 1991; 18: 316–321.. [DOI] [PubMed] [Google Scholar]
- Raffoul JJ, Cabelof DC, Nakamura J, Meira LB, Friedberg EC, Heydari AR.Apurinic/apyrimidinic endonuclease (APE/REF-1) haploinsufficient mice display tissue-specific differences in DNA polymerase β base excision repair. J Biol Chem 2004; 279: 18425–18433.. [DOI] [PubMed] [Google Scholar]
- Linsley WS, Penhoet EE, Linn S.Human endonuclease specific for apurinic/apyrimidinic sites in DNA. J Biol Chem 1977; 252: 1235–1242.. [PubMed] [Google Scholar]
- Ranalli TA, Tom S, Bambara RA.AP endonuclease 1 coordinates flap endonuclease 1 and DNA ligase I activity in long patch base excision repair. J Biol Chem 2002; 277: 41714–41724.. [DOI] [PubMed] [Google Scholar]
- Huamani J, McMahan CA, Herbert DC, Reddick R, McCarey JR, MacInnes MI, Chen DJ, Walter CA.Spontaneous mutagenesis is enhanced in Apex heterozygous mice. Mol Cell Biol 2004; 24: 8145–8153.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman LJ, Murthy KGK, Zhu C, Curran T, Xanthoudakis S, Prives C.Identification of redox/repair protein Ref-1 as a potent activator of p53. Genes Dev 1997; 11: 558–570.. [DOI] [PubMed] [Google Scholar]
- Walker LJ, Robson CN, Black E, Gillespie D, Hickson ID.Identification of residues in the human DNA repair enzyme HAP1 (Ref-1) that are essential for redox regulation of Jun DNA binding. Mol Cell Biol 1993; 13: 5370–5376.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xanthoudakis S, Curran T.Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA binding activity. EMBO J 1992; 11: 653–665.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xanthoudakis S, Miao G, Wang F, Pan YC, Curran T.Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J 1992; 11: 3323–3335.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xanthoudakis S, Miao G, Curran T.The redox and DNA repair activities of Ref-1 are encoded by nonoverlapping domains. Proc Natl Acad Sci U S A 1994; 91: 23–27.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans AR, Limp-Foster M, Kelley MR.Going APE over ref-1. Mutat Res 2000; 461: 183–108.. [DOI] [PubMed] [Google Scholar]
- Prasad R, Beard WA, Wilson SH.Studies of gapped DNA substrate binding by mammalian DNA polymerase β. J Biol Chem 1994; 269: 18096–18101.. [PubMed] [Google Scholar]
- Singhal RK, Wilson SH.Short gap-filling synthesis by DNA polymerase β is processive. J Biol Chem 1993; 268: 15906–15911.. [PubMed] [Google Scholar]
- Fortini P, Pascucci B, Parlanti E, Sobol RW, Wilson SH, Dogliotti E.Different DNA polymerases are involved in the short-and long-patch base excision repair in mammalian cells. Biochemistry 1998; 37: 3575–3580.. [DOI] [PubMed] [Google Scholar]
- Sobol RW, Prasad R, Evenski A, Baker A, Yang X, Horton JK, Wilson SH.The lyase activity of the DNA repair protein β-polymerase protects from DNA-damage-induced cytotoxicity. Nature 2000; 405: 807–810.. [DOI] [PubMed] [Google Scholar]
- Sugo N, Aratani Y, Nagashima Y, Kubota Y, Koyama H.Neonatal lethality with abnormal neurogenesis in mice deficient in DNA polymerase β. EMBO J 2000; 19: 1397–1404.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Marth JD, Orban PC, Mossmann H, Rajewsky K.Deletion of a DNA polymerase β gene segment in T cells using cell type-specific gene targeting. Science 1994; 26: 103–106.. [DOI] [PubMed] [Google Scholar]
- Chan KKL, Zhang ZM, Dianov GL.Base excision repair fidelity in normal and cancer cells. Mutagen 2006; 21: 173–178.. [DOI] [PubMed] [Google Scholar]
- Albertella MR, Lau A, O'Connor MJ.The overexpression of specialized DNA polymerases in cancer. DNA Repair (Amst) 2005; 4: 583–593.. [DOI] [PubMed] [Google Scholar]
- Trivedi RM, Almeida KH, Fornsaglio JL, Schamus S, Sobol RW.The role of base excision repair in the sensitivity and resistance to temozolomide-mediated cell death. Cancer Res 2005; 65: 6394–6400.. [DOI] [PubMed] [Google Scholar]
- Walter CA, Nasr-Schirf D, Luna VJ.Identification of transgenic mice carrying the CAT gene with PCR amplification. Biotechniques 1989; 7: 1065–1070.. [PubMed] [Google Scholar]
- Walter CA.Transgenic manipulation of the mouse genome. Yu BP.Methods in Aging Research, New York:CRC;1999: 38–415.. [Google Scholar]
- Bellve AR, Cavicchia JC, Millette CF, O'Brien DA, Bhatnagar YM, Dym M.Spermatogenic cells of the prepubertal mouse. J Cell Biol 1977; 74: 68–85.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellve AR.Purification, culture, and fractionation of spermatogenic cells. Methods Enzymol 1993; 225: 84–113.. [DOI] [PubMed] [Google Scholar]
- Intano GW, Cho EJ, McMahan CA, Walter CA.Age-related base excision repair activity in mouse brain and liver nuclear extracts. J Gerontol A Biol Sci Med Sci 2003; 58: 205–211.. [DOI] [PubMed] [Google Scholar]
- Bradford MM.A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976; 72: 248–254.. [DOI] [PubMed] [Google Scholar]
- De Wet JR, Wood KV, Helinski DR, DeLuca M.Cloning of firefly luciferase cDNA and the expression of active luciferase in Escherichia coli. Proc Natl Acad Sci U S A 1985; 82: 7870–7873.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Wet JR, Wood KV, DeLuca M, Helinski DR, Subramani S.Firefly luciferase gene: structure and expression in mammalian cells. Mol Cell Biol 1987; 7: 725–737.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrieli Y, Sherman Y, Ben-Sasson B.Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol 1992; 119: 493–501.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose F, Hotta Y, Yamaguchi M, Matsukage A.Difference in the expression level of DNA polymerase β among mouse tissues: high expression in the pachytene spermatocyte. Exp Cell Res 1989; 181: 169–180.. [DOI] [PubMed] [Google Scholar]
- Alcivar AA, Hake LE, Hecht NB.DNA polymerase-β and poly(ADP)ribose polymerase mRNAs are differentially expressed during the development of male germinal cells. Biol Reprod 1992; 46: 201–207.. [DOI] [PubMed] [Google Scholar]
- Walter CA, Lu J, Bhakta M, Zhou ZQ, Thompson LH, McCarrey JR.Testis and somatic Xrcc-1 DNA repair gene expression. Somat Cell Mol Genet 1994; 20: 451–561.. [DOI] [PubMed] [Google Scholar]
- Walter CA, Trolian DA, McFarland MB, Street KA, Gurram GR, McCarrey JR.Xrcc-1 expression during male meiosis in the mouse. Biol Reprod 1996; 55: 630–635.. [DOI] [PubMed] [Google Scholar]
- Chen J, Tomkinson AE, Ramos W, Mackey ZB, Danehower S, Walter CA, Schultz RA, Besterman JM, Husain I.Mammalian DNA ligase III: molecular cloning, chromosomal localization, and expression in spermatocytes undergoing meiotic recombination. Mol Cell Biol 1995; 15: 5412–5422.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackey ZB, Ramos W, Levin DS, Walter CA, McCarrey JR, Tomkinson AE.An alternative splicing event which occurs in mouse pachytene spermatocytes generates a form of DNA ligase III with distinct biochemical properties that may function in meiotic recombination. Mol Cell Biol 1997; 17: 989–998.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabelof DC, Guo Z, Raffoul JJ, Sobol RW, Wilson SH, Richardson R, Heydari AR.Base excision repair deficiency caused by polymerase β-haploinsufficiency: accelerated DNA damage and increased mutational response to carcinogens. Cancer Res 2003; 63: 5799–5807.. [PubMed] [Google Scholar]
- Poltoratsky V, Horton JK, Prasad R, Wilson SH.REV1 mediated mutagenesis in base excision repair deficient mouse fibroblast. DNA Repair (Amst) 2005; 4: 1182–1188.. [DOI] [PubMed] [Google Scholar]
- Niimi N, Sugo N, Aratani Y, Gondo Y, Katsuki M, Koyama H.Decreased mutant frequency in embryonic brain of DNA polymerase β null mice. Mutagen 2006; 21: 55–59.. [DOI] [PubMed] [Google Scholar]
- Sugo N, Niimi N, Aratani Y, Takiguchi-Hayashi K, Koyama H.p53 Deficiency rescues neuronal apoptosis but not differentiation in DNA polymerase β-deficient mice. Mol Cell Biol 2004; 24: 9470–9477.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JK, Stefanick DF, Wilson SH.Involvement of poly(ADP-ribose) polymerase activity in regulating Chk1-dependent apoptotic cell death. DNA Repair (Amst) 2005; 4: 1111–1120.. [DOI] [PubMed] [Google Scholar]
- Ochs K, Sobol RW, Wilson SH, Kaina B.Cells deficient in DNA polymerase β are hypersensitive to alkylating agent-induced apoptosis and chromosomal breakage. Cancer Res 1999; 59: 1544–1551.. [PubMed] [Google Scholar]
- Frechet M, Canitrot Y, Cazaux C, Hoffman J.DNA polymerase β imbalance increases apoptosis and mutagenesis induced by oxidative stress. FEBS Lett 2001; 505: 229–232.. [DOI] [PubMed] [Google Scholar]
- Mori C, Nakamura N, Dix DJ, Fujioka M, Nakagawa S, Shiota K, Eddy EM.Morphological analysis of germ cell apoptosis during postnatal testis development in normal and Hsp70–2 knockout mice. Dev Dyn 1997; 208: 125–136.. [DOI] [PubMed] [Google Scholar]
- Parsons JL, Tait PS, Finch D, Dianova II, Allinson SL, Dianov GL.CHIP-mediated degradation and DNA damage-dependent stabilization regulate base excision repair proteins. Mol Cell 2008; 29: 477–487.. [DOI] [PubMed] [Google Scholar]
- Sobol RW, Watson DE, Nakamura J, Yakes FM, Hou E, Horton JK, Ladapo J, Van Houten B, Swenberg JA, Tindall KR, Samson LD, Wilson SH.Mutations associated with base excision repair efficiency and methylation-induced genotoxic stress. Proc Natl Acad Sci U S A 2002; 99: 6860–6865.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart GR, Oda Y, de Boer JG, Glickman BW.Mutation frequency and specificity with age in liver, bladder and brain of lacI transgenic mice. Genetics 2000; 154: 1291–1300.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provost GS, Kretz PL, Hamner RT, Matthews CD, Rogers BJ, Lundberg KS, Dycaico MJ, Short JM.Transgenic systems for in vivo mutation analysis. Mutat Res 1993; 288: 133–149.. [DOI] [PubMed] [Google Scholar]
- Caldecott KW, Aoufouchi S, Johnson P, Shall S.XRCC1 polypeptide interacts with DNA polymerase nontransgenic control and heterozygotes β and possibly poly (ADP-ribose) polymerase, and DNA ligase III is a novel molecular ‘nick-sensor' in vitro. Nucleic Acids Res 1996; 24: 4387–4394.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota Y, Nash RA, Klungland A, Schar P, Barnes DE, Lindahl T.Reconstitution of DNA base excision-repair with purified human proteins: interaction between DNA polymerase β and the XRCC1 protein. EMBO J 1996; 15: 6662–6670.. [PMC free article] [PubMed] [Google Scholar]
- Prasad R, Singhal RK, Srivastava DK, Molina JT, Tomkison AE, Wilson SH.Specific interaction of DNA polymerase β and DNA ligase I in a multiprotein base excision repair complex from bovine testis. J Biol Chem 1996; 271: 16000–16007.. [DOI] [PubMed] [Google Scholar]
- Wilson SH, Kunkel TA.Passing the baton in base excision repair. Nature 2000; 7: 176–178.. [DOI] [PubMed] [Google Scholar]
- Levy N, Martz A, Bresson A, Spenlehauer C, de Murcia G, Menissier-de Murcia J.XRCC1 is phosphorylated by DNA-dependent protein kinase in response to DNA damage. Nucleic Acids Res 2006; 34: 32–41.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E, Keil C, Oei SL.Roles of DNA ligase III and XRCC1 in regulating the switch between short patch and long patch BER. DNA Repair (Amst) 2006; 5: 544–555.. [DOI] [PubMed] [Google Scholar]
- Pinz KG, Bogenhagen DF.Efficient repair of abasic sites in DNA by mitochondrial enzymes. Mol Cell Biol 1998; 18: 1257–1265.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshmipathy U, Campbell C.Mitochondrial DNA ligase III function is independent of Xrcc1. Nucleic Acids Res 2000; 28: 3880–3886.. [DOI] [PMC free article] [PubMed] [Google Scholar]