

Abstract
Many risk genes interact synergistically to produce schizophrenia and many neurotransmitter interactions have been implicated. We have developed a circuit-based framework for understanding gene and neurotransmitter interactions. NMDAR hypofunction has been implicated in schizophrenia because NMDAR antagonists reproduce symptoms of the disease. One action of antagonists is to reduce the excitation of fast-spiking interneurons, resulting in disinhibition of pyramidal cells. Overactive pyramidal cells, notably those in the hippocampus, can drive a hyperdopaminergic state that produces psychosis. Additional aspects of interneuron function can be understood in this framework, as follows. (i) In animal models, NMDAR antagonists reduce parvalbumin and GAD67, as found in schizophrenia. These changes produce further disinhibition and can be viewed as the aberrant response of a homeostatic system having a faulty activity sensor (the NMDAR). (ii) Disinhibition decreases the power of gamma oscillation and might thereby produce negative and cognitive symptoms. (iii) Nicotine enhances the output of interneurons, and might thereby contribute to its therapeutic effect in schizophrenia.
Introduction
Schizophrenia affects nearly 1% of the population [1]. Clinically, the disorder is characterized by positive symptoms (psychosis, hallucinations and paranoia), negative symptoms (flat affect, poor attention, lack of motivation and deficits in social function) and cognitive deficits. Population, family and twin studies indicate that schizophrenia is highly heritable, but no single gene has a strong effect. Rather, the disorder is due to the synergistic interaction of multiple genes and environmental factors [2]. Recent association and linkage studies have identified over a dozen risk genes for schizophrenia [3]. Another line of research has focused on neurotransmitter systems and, again, the evidence, rather than identifying a single factor, points to abnormalities in multiple systems: glutamate, GABA, dopamine and acetylcholine have all been implicated. There is therefore a strong need for an integrative approach to explain how multiple genes and neurotransmitters can interact in a synergistic way to produce the disorder. In this review, we will describe neural circuitry that provides a framework for understanding many of these interactions. Our description builds on several previous integrative approaches [4,5] but extends that work in several ways, notably by suggesting a systems-level explanation for the changes in GABAergic interneurons that occur in schizophrenia.
GABA hypofunction
Studies of postmortem brain tissue have provided strong evidence that the GABAergic system is impaired in schizophrenia (this is termed hypofunction). These studies showed reductions in the concentration of cortical GABA [6] and the activity of glutamate decarboxylase (GAD) [7], the enzyme that synthesizes GABA. These observations were confirmed and extended in subsequent studies showing alteration in several presynaptic components of the GABAergic system [8–14]. The GABA deficit does not affect all classes of cortical GABAergic interneurons equally [15], but is restricted mainly to the basket and chandelier type of interneurons [16,17]. These two types have fast-spiking properties, contain the Ca2+-binding protein parvalbumin, and synapse on the perisomatic region of pyramidal cells. Because they target the spike-initiating region of neurons, fast-spiking interneurons are thought to have a key role in controlling the overall firing properties of brain networks. Although there might be a modest reduction in the number of interneurons, the major changes are in the concentration of particular proteins, notably GAD67 and parvalbumin [18,19]. Such changes are found in many cortical regions [20] and in the hippocampus [12,17], particularly in CA2/3 and the stratum oriens of CA1 [21]. A reduction in GABA synthesis and release would be expected to produce a compensatory upregulation of postsynaptic GABA receptors, and there is now clear evidence for such compensation [15,22–25].
The existence of GABAergic deficits in schizophrenia is supported by experiments using noninvasive methods. GABA can be measured in the human brain by magnetic resonance spectroscopy and has been shown to be reduced in schizophrenia [26]. Furthermore, inhibitory action, as measured by transcranial magnetic stimulation [27], is reduced. These experiments, taken together with the pathophysiology, strongly suggest that the GABA system is compromised in schizophrenia. We will return later to the origin and functional consequences of these changes.
NMDA hypofunction
The NMDA hypofunction theory of schizophrenia (reduced NMDA channel function) is based on two findings: (i) dissociative anesthetics (PCP, MK801 and ketamine) are antagonists of NMDA receptors, and (ii) when abused, these drugs induce a condition that resembles schizophrenia [28]. In laboratory experiments, a subanesthetic dose of ketamine given to normal volunteers induces the symptoms of schizophrenia more effectively than any other known drug [29–32]. NMDA antagonists reproduce both negative and positive symptoms, as well as many of the cognitive deficits associated with the disease. By contrast, amphetamine, a drug that increases dopamine release, induces only positive symptoms. In patients with schizophrenia, ketamine strongly exacerbates their symptoms [28,33].
More recent work has used genetic methods to produce NMDA hypofunction in rodent models. A genetically induced reduction of the NR1 subunit of the NMDA channel [34] resulted in deficits in attention, impaired social behavior and cognitive symptoms consistent with those in schizophrenia. Similar results were obtained by altering the glycine binding site on the NR1 subunit, a site that must be occupied by glycine or D-serine for the NMDA channel to open [35].
Direct evidence for altered NMDA function in schizophrenia comes from two lines of investigation. An evoked potential generated in the supra-granular layer of primary auditory cortex called mismatch negativity [36] is reduced in schizophrenia [37]. Source-sink analysis of monkey cortex shows that this potential is caused by current through NMDA channels. Thus, the reduction in mismatch negativity is an indication of NMDA hypofunction. Other work using double in situ hybridization on postmortem tissue shows a reduction in the NR2A subunit on parvalbumin interneurons [13]. Although no functional conclusions can be drawn from this result, it provides the clearest evidence to date for molecular changes in the NMDA channel.
The causes of NMDAR hypofunction in schizophrenia are probably varied, as would be expected for a disorder involving many genes. The possibilities include reduction in the concentrations of the co-agonists, glycine [38,39] and D-serine levels [40], elevated levels of endogenous antagonists (NAAG/kynurenic acid) [41,42], alterations in the redox state of the NMDA channel [43] or reduced channel expression or trafficking [13,44].
If NMDAR hypofunction contributes to the symptoms of schizophrenia, treatment with agonists of the glycine site should reduce these symptoms (this site is normally not fully occupied [45]). During the last decade, over a dozen placebo-controlled clinical trials with glycine site agonists including D-cycloserine, glycine and D-serine have been carried out in patients with schizophrenia who were receiving concurrent antipsychotic medications. With the exception of one study, negative symptoms were reduced [46]. Treatment with the endogenous glycine transport inhibitor sarcosine has also been reported to reduce negative symptoms, improve cognition and further reduce positive symptoms in schizophrenic patients receiving concurrent antipsychotics [47,48].
The importance of glutamatergic transmission in schizophrenia is underscored by the recent report that an mGlu2/3 agonist is effective, by itself, in treating the disease [49]. This is the first successful treatment not based on direct antagonism of dopamine D2 receptors. The first indication of the therapeutic potential of this drug came from animal models showing that mGlu2/3 agonist reduces the overactivity produced by NMDA antagonist [50,51]. Such reduction might occur as a result of presynaptic reduction in excitation, but a recent report raises the possibility of postsynaptic enhancement of the NMDAR function [52].
NMDA/GABA interaction: disinhibition
In pyramidal cells, excitatory postsynaptic potential (EPSPs) are generated primarily by AMPA channels; the main role of NMDA channels in these cells is in the synaptic plasticity that underlies learning. It was therefore unclear why administration of NMDA antagonist to humans should have large effects on mental processes not related to learning. A key finding [53] was the discovery that NMDA channels contribute strongly to the EPSP in interneurons and that acute inhibition of these channels reduces inhibitory output (see also Refs [54–56]). There is an enormous diversity of interneuron types [57]; it is thus important to note that large NMDA-mediated EPSPs have been found in the parvalbumin-containing basket cells [54,58] that mediate the feedback inhibition (Figure 1) discussed later in this review. It is also important to note that in addition to reducing the EPSP, NMDA antagonists hyperpolarize neurons by blocking the effect of ambient glutamate [59]; this would also make it more difficult to excite interneurons.
Figure 1.
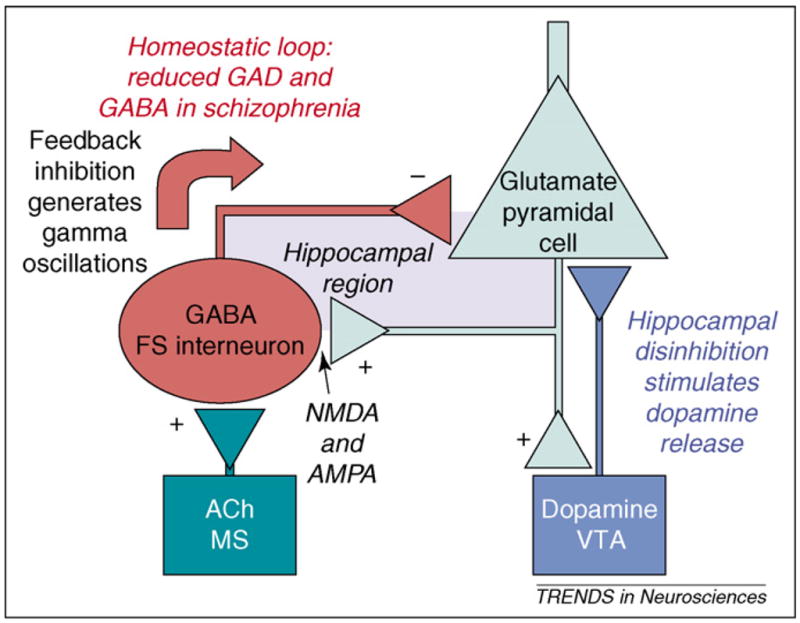
Simplified circuitry that provides a framework for understanding the actions of neurotransmitters and risk genes in schizophrenia. Two loops are shown: (i) the reciprocal interactions between pyramidal cells and fast-spiking interneurons and (ii) the hippocampal ventral tegmental area (VTA) loop (note that the connection between the hippocampus and VTA is shown as monosynaptic for simplicity, but is actually polysynaptic through the striatum and ventral pallidum). The effect of dopamine on the hippocampus is probably excitatory [109], raising the possibility that the hippocampal-VTA loop could go into positive feedback, thereby generating the sudden onset of psychosis. The reciprocal relationship of pyramidal cells and fast-spiking interneurons is ubiquitous in the hippocampus and cortex. This loop is responsible for homeostasis of firing of pyramidal cells and for the generation of gamma frequency oscillations. Abnormalities in these oscillations might underlie cognitive and negative symptoms. Cholinergic input to fast-spiking (FS) interneurons is from the medial septal region (MS). ACh = acetylcholine.
If the output of interneurons is reduced by NMDA antagonists, pyramidal cell activity should increase. This has been observed in rodents by electrophysiological methods [60], metabolic imaging methods [61,62] and measurements of glutamate release [63]. The prolonged overactivity of pyramidal cells could have deleterious consequences; indeed, this is the likely explanation of the fact that prolonged inhibition of NMDARs produces swelling of pyramidal cells and other signs of cellular stress [64]. Taken together, these experiments strongly argue that a major effect of NMDA antagonists is to produce disinhibition of pyramidal cells.
If disinhibition occurs in schizophrenia, there should be an increase in brain metabolic activity. This has been observed using functional imaging. Importantly, the increase in activity correlates with the severity of psychopathology [65,66] and is predictive of cognitive abnormalities [67]. Recent work used newly developed methods to quantitatively measure basal blood flow at very high spatial resolution [68,69]. The results showed elevated blood flow in the hippocampus of schizophrenia patients, particularly in the CA1 region. This activity showed a strong correlation with psychosis, particularly symptoms of delusion.
The disinhibition produced by NMDA antagonists is only partial because AMPA-mediated excitation of interneurons remains. Thus, normal interaction of pyramidal cells in interneurons might be affected by this form of disinhibition without causing the large-scale epileptic activity produced by complete block of inhibition. There is, however, an overlap of schizophrenia and epilepsy. NMDA antagonists produce an EEG signature similar to some forms of epilepsy [70], and there is a substantially increased risk of schizophrenia in patients with epilepsy [71]. Moreover, temporal lobe epilepsy can often produce symptoms related to those in schizophrenia [72]. Still, it remains to be resolved why agents other than NMDA antagonists that reduce inhibition in humans do not produce symptoms of schizophrenia. One possibility is that such drugs reduce both the inhibitory output of interneurons and the inhibitory input onto interneurons; these effects might cancel each other. NMDA antagonists, by selectively reducing the excitation of interneurons, might have a more specific effect. The situation is further complicated by regional variability in the sensitivity of interneurons to NMDA antagonist. A recent study found that such antagonists affected inhibitory circuits more strongly in the entorhinal cortex than in the hippocampus [73] (see also Ref. [74]). Other work has pointed to the thalamus as a region particularly sensitive to antagonist [61,70]. The basis of this regional variability remains to be determined.
NMDARs on interneurons as a sensor for homeostatic regulation of pyramidal cell firing
The existence of an NMDAR-mediated component of the EPSP in interneurons helps to connect the NMDA and GABA hypotheses, but does not explain the decreased expression of GAD and parvalbumin. Here we propose a novel explanation of this decrease. Our starting point is the idea that a major function of the fast-spiking interneurons is the homeostatic regulation of overall pyramidal cell firing. These interneurons sum the responses from hundreds of pyramidal cells and then inhibit these cells, thus providing negative feedback control of the summed firing level [75]. Acute application of an NMDA antagonist will immediately interfere with this homeostatic function by reducing the gain of negative feedback. There is now good evidence for a second, slower mechanism that further reduces the efficacy of inhibition. In vivo treatment with NMDAR antagonists for several days produces a reduction in cortical GAD67 and parvalbumin mRNA [76,77], much like that seen in schizophrenia (see above). The reduction in GAD67 would be expected to reduce GABA levels and therefore decrease inhibition. Although this prediction has not been directly tested after NMDA antagonist application, it has been tested in another model of schizophrenia that has reduced hippocampal GAD67. Physiological recordings from pyramidal cells in this model show reduced evoked inhibition and reduced miniature inhibitory postsynaptic current amplitude [78]. Remarkably, a study [79] shows that the deficits in GAD67 produced by NMDAR antagonist can be observed in cell cultures of pyramidal cells and interneurons. The fact that this cardinal feature of the human pathological data can be reproduced in such a simple system indicates that it must arise through a local circuit mechanism. The experiments also showed that the reduction in GAD67 is specific to parvalbumin-containing interneurons, that cell death is not involved, that the effect depends on blocking NR2A receptors and that it is the Ca2+ entry through these channels [80] that triggers the change in protein levels. A recent study provides insight into some of the biochemical mechanisms involved [81].
It at first seems counterintuitive that application of an NMDAR antagonist should produce an acute reduction of inhibition followed by a further, slower, reduction of inhibition – most slow changes are compensatory rather than reinforcing. We suggest a simple explanation. As noted above, the homeostatic function of these interneurons is to stabilize overall pyramidal cell firing, and it would make sense if slow biochemical changes served this function. It appears (see above) that interneurons use NMDARs to sense pyramidal cell activity. It follows that NMDAR hypofunction would be falsely ‘interpreted’ as inactivity of pyramidal cells. The interneuron, as part of its homeostatic function, would then attempt to compensate for the apparent inactivity by reducing its inhibitory output. This would be done by lowering GAD67 levels and therefore GABA.
Interestingly, recent biophysical results raise the possibility that the reduction in parvalbumin that occurs in schizophrenia (and in response to NMDAR antagonist in rodents) might be part of the same homeostatic mechanism. Parvalbumin is a protein that buffers Ca2+. During action potentials, it binds the Ca2+ that enters through voltage-dependent Ca2+ channels. Because the buffer is loaded with Ca2+, it will act to maintain free Ca2+ at a level higher than the resting level well after the action potential’s end. This ‘tail’ of Ca2+ elevation triggers what is termed ‘delayed’ GABA release. It has recently been found that such delayed release is cumulatively larger than the synchronous release that occurs at the time of the action potential [82]. Thus, the net effect of reducing parvalbumin levels is to decrease inhibitory output.
In summary, the reduction of GAD67 and parvalbumin appear to serve a common homeostatic function carried out by fast-spiking interneurons. The biochemical machinery that controls this homeostasis depends on the NMDA channel as a sensor for pyramidal cell activity. If the sensor malfunctions, indicating low pyramidal cell activity, the interneuron may synthesize less GABA and parvalbumin in an attempt to restore pyramidal cell activity to the correct level. These homeostatic compensations are maladaptive in this context and unfortunately produce overactivity of pyramidal cells that could trigger further problems, as discussed in the next section. More needs to be learned about this homeostatic loop. Interestingly, there are indications that GAD67 reduction occurs in sensory cortex during sensory deprivation, which would also be expected to reduce glutamatergic input to fast-spiking interneurons [83,84].
The hyperdopaminergic state and the role of the hippocampus
Dopamine was the first neurotransmitter system to be strongly implicated in schizophrenia. Antagonists of the D2 receptor reduce the positive symptoms of the disorder [85,86], and standard drug treatments of schizophrenia remain based on this antagonism. By implication, it would seem that the disease might be due to a hyperdopaminergic state (excess dopamine). Consistent with this, increasing dopamine release with amphetamine produces positive symptoms in normal subjects [87]. Direct evidence for a dopaminergic abnormality in schizophrenia comes from studies that measured the ability of endogenous dopamine to displace dopamine receptor radioligands in the striatum. Such studies showed that dopamine release is hyper-responsive to amphetamine in schizophrenia patients and that responsiveness correlates with the exacerbation of psychosis [88].
An important advance in understanding neurotransmitter interactions in schizophrenia was the finding that the hyperdopaminergic state can be a consequence of NMDAR hypofunction [89,90]. This was supported by the finding that acute application of NMDAR antagonist stimulates dopamine release in animal models [91] and humans [92,93] (but see Ref. [94]).
Progress has been made in understanding which brain regions are critical for the effect of NMDA antagonists (and the resulting disinhibition) on the dopamine system. Because recurrent inhibition is a fundamental feature of cortical circuitry, blockade of NMDARs will likely cause disinhibition in many brain regions. Consistent with this, in schizophrenia there are abnormalities in sensory processes mediated by sensory cortex [95–97], as well as in high-level functions (working memory) carried out in prefrontal cortex [98]. However, there appears to be a special role of disinhibition in the hippocampal region in stimulating the hyperdopaminergic state (and the consequent psychosis). The hippocampal region has been implicated in schizophrenia and in forms of psychosis not related to schizophrenia [99,100]. Importantly, artificially activating the subiculum, an output structure of the hippocampus, is sufficient to increase the population activity of dopamine neurons in the ventral tegmental area (VTA) [101] and to release dopamine [102]. Other studies utilized an animal model for schizophrenia [103] to investigate the causal role of the hippocampus. In this model, interneurons are preferentially reduced by treatment with a mitogen late in gestation [104]. This results in elevated VTA activity and hyper-responsiveness to amphetamine in adults (as occurs in schizophrenia). Importantly, these effects could be acutely reversed [105] by inactivating the subiculum, indicating that the hippocampal region is necessary for producing the hyperdopaminergic state. This kind of circuit analysis is powerful and it will be important to determine whether similar results can be obtained with other models of schizophrenia.
There is increased understanding of the special relationship of the hippocampus and VTA in normal memory function. The hippocampus is a memory store, one function of which is to detect novelty (by comparison of input to stored information); this detection appears to trigger the novelty-dependent firing of the VTA [106,107]. The dopaminergic cells of the VTA project to many regions, including the hippocampus. The resulting dopamine release in the hippocampus appears to have several effects on neurons. It is important for the consolidation of long-term potentiation, and thus the entry of information into long-term memory [106]. Furthermore, dopamine can alter synaptic transmission [108], and the net effect is to produce further disinhibition [109] (raising the possibility of a positive feedback process). The changes in the hippocampus-VTA loop appear to have functional consequences: in schizophrenia patients, there is a failure of the hippocampal fMRI signal to habituate with repeated presentation of emotional faces; thus, everything is novel [110]. Without habituation processes that allow gating (filtering) of sensory stimuli, sensory processes can become overloaded [111]. Hyperactivation of the dopamine system is also likely to affect other cognitive systems, notably the working memory processes of prefrontal cortex (reviewed in Ref. [112]).
Disinhibition might produce some cognitive symptoms by reducing gamma oscillations
In the above section, we explored how malfunction of the feedback loop between pyramidal cells and fast-spiking interneurons could affect the dopamine system. In addition, this loop is directly involved in the generation of gamma oscillations and there are now strong reasons for believing that abnormalities in these oscillations could contribute to some of the symptoms of schizophrenia. Gamma frequency (30–100 Hz) oscillations are present in the local field potential and EEG, reflecting the synchronized firing of groups of pyramidal cells. It has been found that gamma oscillations are reduced in schizophrenia and that the degree of this reduction correlates with the severity of negative symptoms [113]. Because the power of gamma oscillations varies dramatically with behavioral state, there is concern that the reduced gamma power in schizophrenia might be a result of an alteration in behavioral state rather than a specific change in the gamma-producing circuitry. This issue cannot yet be resolved with certainty, but it is noteworthy that reductions in gamma power are seen during tasks that do not involve attention and can be observed in unmedicated patients [114].
Gamma oscillations arise through negative feedback inhibition of pyramidal cells by fast-spiking interneurons [74,115], the same interneuron type we discussed above. Because NMDA channels contribute to the excitation of fast-spiking interneurons, NMDAR antagonists should reduce gamma oscillations. This has now been directly demonstrated in slices of the entorhinal cortex [73].
Testing the role of gamma oscillations in cognitive processes is difficult, but a recent study has made progress in this direction. In this study, GluR1 or GluR4 were knocked out of parvalbumin interneurons [115]. Because these receptors contribute to the excitation of interneurons, their removal reduced feedback inhibition and would thus be expected to reduce gamma power. This reduction was observed, notably in the hippocampus, where most of the experiments were conducted. Because the neural code organized by gamma oscillations is critical for effective communication between brain structures, abnormalities in gamma rhythm could interfere with cognitive processes. Specific support for a role of gamma in memory comes from computational models [116] showing how interference with hippocampal gamma would compromise memory function. Consistent with these models, reducing gamma power by knocking out GluR1/GluR4 in interneurons produced deficits in hippocampal memory tasks [115]. Taken together, these experiments suggest how NMDAR hypofunction, acting through both acute and slower biochemical mechanisms, could reduce gamma oscillations and thereby produce memory deficits in schizophrenia. The extent to which other negative symptoms of schizophrenia can be attributed to abnormalities in gamma oscillations remains to be examined, but it would seem unlikely that all symptoms can be related to gamma. Indeed, there is evidence that hypofunction of the NMDAR-medicated excitation of magnocellular pyramidal cells could account for deficits in early visual processing [117].
The cholinergic system: reversing disinhibition and cognitive deficits
The disinhibition model described above is also useful in understanding the role of the cholinergic system in schizophrenia [118]. One hint of the relevance of the cholinergic system to schizophrenia is that the prevalence of smoking among individuals with schizophrenia exceeds 70%, 2- to 4-fold higher than in the general population [119]. This heavy use of nicotine is believed to be an attempt at self-medication [120]. In controlled experiments on schizophrenia patients, nicotine has been found to enhance cognitive function [121–123].
Analysis of circuit function is beginning to provide insight into how these cholinergic effects might arise. α7 nicotinic receptors are concentrated on interneurons [124], can depolarize interneurons and can thereby enhance GABA release [125]. Recent studies show that there is a second and more surprising way that nicotine enhances GABA release: nicotine inhibits the inhibitory synapses onto interneurons [126]. Through these synergistic actions, nicotine can enhance the excitation of interneurons and thereby enhance inhibitory output. Consistent with this, nicotine increases the gamma oscillations that are dependent on interneuron function [127].
Toward a circuit-based explanation of synergistic gene action
The goal of systems biology is to understand how genes work together in biochemical and cellular networks to produce function. Such an integrated understanding is of special importance in schizophrenia research because the disorder results from the synergistic interaction of many risk genes, none of which has a large effect. To determine whether genes act synergistically, it is necessary to have a circuit-based model. This is illustrated by analysis of NMDAR function; these receptors are present both on pyramidal cells and interneurons. It is only because of the physiological experiments indicating the special importance of NMDARs in the excitation of interneurons (see above) that NMDAR hypofunction (which reduces GABA release) can be seen as synergistic with other factors that also reduce GABA release (e.g. the mutation in GAD67).
Many of the genes that have been identified as risk genes for schizophrenia relate to glutamatergic transmission (Box 1). Indeed, this association is substantially greater than chance [128]. In some cases (e.g. G72, DAOO and serine racemase), the available evidence strongly suggests that NMDAR hypofunction could result. In other cases, for instance the metabotropic glutamate receptor 3 (GRM3), glutamatergic transmission is implicated [129], but it is not known how NMDAR function is affected. These considerations emphasize the need for physiological analysis of risk genes.
Box 1. Schizophrenia risk genes that affect transmission at glutamatergic, nicotinic and GABAergic synapses
DAOA: The gene, also termed G72, is of recent evolutionary appearance. It encodes a protein that activates DAAO, the enzyme that catabolizes D-serine [130]. DAAO appears to be the critical determinant of D-serine levels, as its activity correlates inversely with D-serine levels both regionally and developmentally [40,131]. As D-serine acts as a co-agonist with glutamate for NMDAR, reduced availability of D-serine would lead to NMDAR hypofunction [132]. Since G72 was first proposed as a risk gene for schizophrenia [130], over a dozen studies have supported this association (for a review, see Ref. [133]). One study reported increased expression of G72 in prefrontal cortex [134]. The impressive replications of the association of G72 with the risk for schizophrenia is all the more intriguing, given recently replicated findings that (i) D-serine reduces negative symptoms, improves cognition and reduces positive symptoms in patients with chronic schizophrenia who are receiving concurrent typical antipsychotic medications [135,136] and (ii) that serum and cerebrospinal fluid levels of D-serine are reduced in schizophrenic subjects [40,131].
DAAO: The gene encoding D-amino acid oxidase, the enzyme that degrades D-serine, has also been linked to the risk of schizophrenia in several studies [137]. The enzymatic consequences of the DAAO mutation are not known, but a postmortem study revealed elevated levels of DAAO in the hippocampus of patients with schizophrenia that correlated with the duration of illness [138]. This would account for the observed reduction of D-serine levels, a reduction that would produce NMDAR hypofunction [139].
Serine racemase: This enzyme produces D-serine from L-serine. There is a single-nucleotide polymorphism in the 5′ promoter region in the gene encoding serine racemase that is linked to schizophrenia [140]. This results in reduced expression of the protein. It would be expected that this would reduce D-serine levels and produce NMDAR hypofunction. PICK1, a protein that interacts with serine racemase, has been identified as a risk gene for schizophrenia [141].
GRM3: GRM3 encodes for mGluR3, for which N-acetyl-aspartyl glutamate (NAAG) is a potent and specific agonist [142]. mGluR3 downregulates the release of glutamate and thereby could cause NMDAR hypofunction. Research suggests that the mGluR3 agonist NAAG might be increased in corticolimbic regions in schizophrenia as a result of downregulation of its catabolic enzyme, glutamate carboxypetidase II [41,143,144].
DTNP1: Dysbindin (DTNP1; 6p24-22) has emerged as another promising risk gene for schizophrenia [145]. Dysbindin is concentrated in the presynaptic glutamatergic terminals where it interacts with SNAP and synapsin 1 and modulates vesicular release of glutamate [146]. The expression of dysbindin is reduced in prefrontal cortex and hippocampus in schizophrenia [147]. Notably, the dysbindin genotype has been inversely associated with general cognitive ability and poor premorbid function in schizophrenia [148,149].
NRG1: The association of the gene encoding neuregulin with the risk for schizophrenia is also particularly robust [150]. Neuregulin is a component of the ErbB signaling pathway. Mice with a null mutation of its gene express lower levels of NR1 [151], and have altered tyrosine phosphorylation of the channel [152]. Consistent with this downregulation, reducing presynaptic neuregulin with RNAi reduces the NMDAR component of transmission [153]. Importantly, there is a lowered level of neuregulin in the synaptic complex isolated from the brain of schizophrenics [152,154]. The effects of neuregulin are not likely to be exclusively on glutamatergic transmission, as nicotinic and GABAergic transmission are also affected [155–157].
GAD1: Glutamic acid decarboxylase (67 kDa) is responsible for the bulk of GABA synthesized in neurons. Single-nucleotide polymorphisms in the 5′ promoter region of its gene, GAD1, are associated with childhood onset of schizophrenia and cortical gray matter loss [158].
CHNRA7: The α7 nicotinic receptor is expressed by interneurons and acts to excite them. Their deficit would thus lead to disinhibition of pyramidal cells. This gene is contained in a region that shows strong linkage to schizophrenia and affects gating deficits associated with the disease [159].
The circuitry on which we have focused provides a framework for integrating results on the glutamate, GABA, dopamine and cholinergic neurotransmitter systems, and a significant number of risk genes can be seen as working synergistically within this circuit (Box 1). These results are encouraging, but we emphasize that the development of circuit-based models is at a very early stage and that models will undoubtedly have to undergo substantial revision. What we hope this article has made clear is that a circuit-based approach is possible, that some progress has been made in this direction and that this approach is the correct strategy for understanding a disease that produces its devastating consequences through synergistic gene action.
Acknowledgments
The authors would like to thank Gordon Fain for advice on the manuscript and Margarita Behrens and Claudia Racca for useful discussions. J.E.L., J.T.C., D.C.J. and R.W.G. are grateful for the support of NIH Conte Center grant P50 MH060450. F.M.B. has been supported by NIH grants MH42261, MH31862 and MH60450. S.H. has been supported by MH067999 and MH070560. R.W.G. gratefully acknowledges support from the Department of Veterans Affairs.
References
- 1.Perala J, et al. Lifetime prevalence of psychotic and bipolar I disorders in a general population. Arch Gen Psychiatry. 2007;64:19–28. doi: 10.1001/archpsyc.64.1.19. [DOI] [PubMed] [Google Scholar]
- 2.Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005;10:40–68. doi: 10.1038/sj.mp.4001558. [DOI] [PubMed] [Google Scholar]
- 3.Kirov G, et al. Finding schizophrenia genes. J Clin Invest. 2005;115:1440–1448. doi: 10.1172/JCI24759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lewis DA, Moghaddam B. Cognitive dysfunction in schizophrenia: convergence of γ-aminobutyric acid and glutamate alterations. Arch Neurol. 2006;63:1372–1376. doi: 10.1001/archneur.63.10.1372. [DOI] [PubMed] [Google Scholar]
- 5.Carlsson A. The neurochemical circuitry of schizophrenia. Pharmacopsychiatry. 2006;39(Suppl 1):S10–S14. doi: 10.1055/s-2006-931483. [DOI] [PubMed] [Google Scholar]
- 6.Perry TL, et al. γ-Aminobutyric-acid deficiency in brain of schizophrenic patients. Lancet. 1979;1:237–239. doi: 10.1016/s0140-6736(79)90767-0. [DOI] [PubMed] [Google Scholar]
- 7.Bird JM. Computed tomographic brain studies and treatment response in schizophrenia. Can J Psychiatry. 1985;30:251–254. doi: 10.1177/070674378503000407. [DOI] [PubMed] [Google Scholar]
- 8.Simpson MD, et al. Reduced GABA uptake sites in the temporal lobe in schizophrenia. Neurosci Lett. 1989;107:211–215. doi: 10.1016/0304-3940(89)90819-7. [DOI] [PubMed] [Google Scholar]
- 9.Akbarian S, et al. Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch Gen Psychiatry. 1995;52:258–266. doi: 10.1001/archpsyc.1995.03950160008002. [DOI] [PubMed] [Google Scholar]
- 10.Guidotti A, et al. Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch Gen Psychiatry. 2000;57:1061–1069. doi: 10.1001/archpsyc.57.11.1061. [DOI] [PubMed] [Google Scholar]
- 11.Volk DW, et al. Decreased glutamic acid decarboxylase67 messenger RNA expression in a subset of prefrontal cortical γ-aminobutyric acid neurons in subjects with schizophrenia. Arch Gen Psychiatry. 2000;57:237–245. doi: 10.1001/archpsyc.57.3.237. [DOI] [PubMed] [Google Scholar]
- 12.Heckers S, et al. Differential hippocampal expression of glutamic acid decarboxylase 65 and 67 messenger RNA in bipolar disorder and schizophrenia. Arch Gen Psychiatry. 2002;59:521–529. doi: 10.1001/archpsyc.59.6.521. [DOI] [PubMed] [Google Scholar]
- 13.Woo TU, et al. Density of glutamic acid decarboxylase 67 messenger RNA-containing neurons that express the N-methyl-D-aspartate receptor subunit NR2A in the anterior cingulate cortex in schizophrenia and bipolar disorder. Arch Gen Psychiatry. 2004;61:649–657. doi: 10.1001/archpsyc.61.7.649. [DOI] [PubMed] [Google Scholar]
- 14.Volk D, et al. GABA transporter-1 mRNA in the prefrontal cortex in schizophrenia: decreased expression in a subset of neurons. Am J Psychiatry. 2001;158:256–265. doi: 10.1176/appi.ajp.158.2.256. [DOI] [PubMed] [Google Scholar]
- 15.Benes FM, et al. Deficits in small interneurons in prefrontal and cingulate cortices of schizophrenic and schizoaffective patients. Arch Gen Psychiatry. 1991;48:996–1001. doi: 10.1001/archpsyc.1991.01810350036005. [DOI] [PubMed] [Google Scholar]
- 16.Woo TU, et al. A subclass of prefrontal γ-aminobutyric acid axon terminals are selectively altered in schizophrenia. Proc Natl Acad Sci U S A. 1998;95:5341–5346. doi: 10.1073/pnas.95.9.5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang ZJ, Reynolds GP. A selective decrease in the relative density of parvalbumin-immunoreactive neurons in the hippocampus in schizophrenia. Schizophr Res. 2002;55:1–10. doi: 10.1016/s0920-9964(01)00188-8. [DOI] [PubMed] [Google Scholar]
- 18.Woo TU, et al. Schizophrenia and the parvalbumin-containing class of cortical local circuit neurons. Am J Psychiatry. 1997;154:1013–1015. doi: 10.1176/ajp.154.7.1013. [DOI] [PubMed] [Google Scholar]
- 19.Lewis DA, et al. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- 20.Hashimoto T, et al. Regional survey of GABA-related gene expression in the neocortex of subjects with schizophrenia. Schizophr Bull. 2007;33:203–631. [Google Scholar]
- 21.Benes FM, et al. Regulation of the GABA cell phenotype in hippocampus of schizophrenics and bipolars. Proc Natl Acad Sci U S A. 2007;104:10164–10169. doi: 10.1073/pnas.0703806104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Benes FM, et al. Differences in the subregional and cellular distribution of GABAA receptor binding in the hippocampal formation of schizophrenic brain. Synapse. 1996;22:338–349. doi: 10.1002/(SICI)1098-2396(199604)22:4<338::AID-SYN5>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 23.Newell KA, et al. Alterations of muscarinic and GABA receptor binding in the posterior cingulate cortex in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:225–233. doi: 10.1016/j.pnpbp.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 24.Ohnuma T, et al. Measurement of GABAergic parameters in the prefrontal cortex in schizophrenia: focus on GABA content, GABA(A) receptor α-1 subunit messenger RNA and human GABA transporter-1 (HGAT-1) messenger RNA expression. Neuroscience. 1999;93:441–448. doi: 10.1016/s0306-4522(99)00189-x. [DOI] [PubMed] [Google Scholar]
- 25.Volk DW, et al. Reciprocal alterations in pre- and postsynaptic inhibitory markers at chandelier cell inputs to pyramidal neurons in schizophrenia. Cereb Cortex. 2002;12:1063–1070. doi: 10.1093/cercor/12.10.1063. [DOI] [PubMed] [Google Scholar]
- 26.Rosso IM, et al. Cingulate cortex GABA concentration in schizophrenia: a two-dimensional proton magnetic resonance spectroscopy study. Biol Psychiatry. 2006;59(Suppl):1515. [Google Scholar]
- 27.Daskalakis ZJ, et al. Evidence for impaired cortical inhibition in schizophrenia using transcranial magnetic stimulation. Arch Gen Psychiatry. 2002;59:347–354. doi: 10.1001/archpsyc.59.4.347. [DOI] [PubMed] [Google Scholar]
- 28.Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–1308. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- 29.Krystal JH, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- 30.Umbricht D, et al. Ketamine-induced deficits in auditory and visual context-dependent processing in healthy volunteers: implications for models of cognitive deficits in schizophrenia. Arch Gen Psychiatry. 2000;57:1139–1147. doi: 10.1001/archpsyc.57.12.1139. [DOI] [PubMed] [Google Scholar]
- 31.Parwani A, et al. The effects of a subanesthetic dose of ketamine on verbal memory in normal volunteers. Psychopharmacology (Berl) 2005;183:265–274. doi: 10.1007/s00213-005-0177-2. [DOI] [PubMed] [Google Scholar]
- 32.Saykin AJ, et al. Neuropsychological function in schizophrenia. Selective impairment in memory and learning. Arch Gen Psychiatry. 1991;48:618–624. doi: 10.1001/archpsyc.1991.01810310036007. [DOI] [PubMed] [Google Scholar]
- 33.Lahti AC, et al. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology. 1995;13:9–19. doi: 10.1016/0893-133X(94)00131-I. [DOI] [PubMed] [Google Scholar]
- 34.Mohn AR, et al. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell. 1999;98:427–436. doi: 10.1016/s0092-8674(00)81972-8. [DOI] [PubMed] [Google Scholar]
- 35.Ballard TM, et al. Severe impairment of NMDA receptor function in mice carrying targeted point mutations in the glycine binding site results in drug-resistant nonhabituating hyperactivity. J Neurosci. 2002;22:6713–6723. doi: 10.1523/JNEUROSCI.22-15-06713.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Javitt DC, et al. Role of cortical N-methyl-D-aspartate receptors in auditory sensory memory and mismatch negativity generation: implications for schizophrenia. Proc Natl Acad Sci U S A. 1996;93:11962–11967. doi: 10.1073/pnas.93.21.11962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shelley AM, et al. Mismatch negativity: an index of a preattentive processing deficit in schizophrenia. Biol Psychiatry. 1991;30:1059–1062. doi: 10.1016/0006-3223(91)90126-7. [DOI] [PubMed] [Google Scholar]
- 38.Sumiyoshi T, et al. Plasma glycine and serine levels in schizophrenia compared to normal controls and major depression: relation to negative symptoms. Int J Neuropsychopharmacol. 2004;7:1–8. doi: 10.1017/S1461145703003900. [DOI] [PubMed] [Google Scholar]
- 39.Neeman G, et al. Relation of plasma glycine, serine, and homocysteine levels to schizophrenia symptoms and medication type. Am J Psychiatry. 2005;162:1738–1740. doi: 10.1176/appi.ajp.162.9.1738. [DOI] [PubMed] [Google Scholar]
- 40.Hashimoto K, et al. Decreased serum levels of D-serine in patients with schizophrenia: evidence in support of the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia. Arch Gen Psychiatry. 2003;60:572–576. doi: 10.1001/archpsyc.60.6.572. [DOI] [PubMed] [Google Scholar]
- 41.Tsai G, et al. Abnormal excitatory neurotransmitter metabolism in schizophrenic brains. Arch Gen Psychiatry. 1995;52:829–836. doi: 10.1001/archpsyc.1995.03950220039008. [DOI] [PubMed] [Google Scholar]
- 42.Erhardt S, et al. Kynurenic acid and schizophrenia. Adv Exp Med Biol. 2003;527:155–165. doi: 10.1007/978-1-4615-0135-0_18. [DOI] [PubMed] [Google Scholar]
- 43.Choi YB, Lipton SA. Redox modulation of the NMDA receptor. Cell Mol Life Sci. 2000;57:1535–1541. doi: 10.1007/PL00000638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci. 2007;8:413–426. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- 45.Chen L, et al. Glycine transporter-1 blockade potentiates NMDA-mediated responses in rat prefrontal cortical neurons in vitro and in vivo. J Neurophysiol. 2003;89:691–703. doi: 10.1152/jn.00680.2002. [DOI] [PubMed] [Google Scholar]
- 46.Coyle JT, Tsai G. The NMDA receptor glycine modulatory site: a therapeutic target for improving cognition and reducing negative symptoms in schizophrenia. Psychopharmacology (Berl) 2004;174:32–38. doi: 10.1007/s00213-003-1709-2. [DOI] [PubMed] [Google Scholar]
- 47.Lane HY, et al. Glycine transporter I inhibitor, N-methylglycine (sarcosine), added to clozapine for the treatment of schizophrenia. Biol Psychiatry. 2006;60:645–649. doi: 10.1016/j.biopsych.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 48.Tsai G, et al. Glycine transporter I inhibitor, N-methylglycine (sarcosine), added to antipsychotics for the treatment of schizophrenia. Biol Psychiatry. 2004;55:452–456. doi: 10.1016/j.biopsych.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 49.Patil ST, et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat Med. 2007;13:1102–1107. doi: 10.1038/nm1632. [DOI] [PubMed] [Google Scholar]
- 50.Moghaddam B, Adams BW. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science. 1998;281:1349–1352. doi: 10.1126/science.281.5381.1349. [DOI] [PubMed] [Google Scholar]
- 51.Homayoun H, et al. Activation of metabotropic glutamate 2/3 receptors reverses the effects of NMDA receptor hypofunction on prefrontal cortex unit activity in awake rats. J Neurophysiol. 2005;93:1989–2001. doi: 10.1152/jn.00875.2004. [DOI] [PubMed] [Google Scholar]
- 52.Tyszkiewicz JP, et al. Group II metabotropic glutamate receptors enhance NMDA receptor currents via a protein kinase C-dependent mechanism in pyramidal neurones of rat prefrontal cortex. J Physiol. 2004;554:765–777. doi: 10.1113/jphysiol.2003.056812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grunze HC, et al. NMDA-dependent modulation of CA1 local circuit inhibition. J Neurosci. 1996;16:2034–2043. doi: 10.1523/JNEUROSCI.16-06-02034.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jones RS, Buhl EH. Basket-like interneurones in layer II of the entorhinal cortex exhibit a powerful NMDA-mediated synaptic excitation. Neurosci Lett. 1993;149:35–39. doi: 10.1016/0304-3940(93)90341-h. [DOI] [PubMed] [Google Scholar]
- 55.Lei S, McBain CJ. Distinct NMDA receptors provide differential modes of transmission at mossy fiber-interneuron synapses. Neuron. 2002;33:921–933. doi: 10.1016/s0896-6273(02)00608-6. [DOI] [PubMed] [Google Scholar]
- 56.Maccaferri G, Dingledine R. Control of feedforward dendritic inhibition by NMDA receptor-dependent spike timing in hippocampal interneurons. J Neurosci. 2002;22:5462–5472. doi: 10.1523/JNEUROSCI.22-13-05462.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Somogyi P, Klausberger T. Defined types of cortical interneurone structure space and spike timing in the hippocampus. J Physiol. 2005;562:9–26. doi: 10.1113/jphysiol.2004.078915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Buhl EH, et al. Physiological properties of anatomically identified basket and bistratified cells in the CA1 area of the rat hippocampus in vitro. Hippocampus. 1996;6:294–305. doi: 10.1002/(SICI)1098-1063(1996)6:3<294::AID-HIPO7>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 59.Binshtok AM, et al. NMDA receptors in layer 4 spiny stellate cells of the mouse barrel cortex contain the NR2C subunit. J Neurosci. 2006;26:708–715. doi: 10.1523/JNEUROSCI.4409-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jackson ME, et al. NMDA receptor hypofunction produces concomitant firing rate potentiation and burst activity reduction in the prefrontal cortex. Proc Natl Acad Sci U S A. 2004;101:8467–8472. doi: 10.1073/pnas.0308455101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vaisanen J, et al. Effects of NMDA-receptor antagonist treatment on c-fos expression in rat brain areas implicated in schizophrenia. Cell Mol Neurobiol. 2004;24:769–780. doi: 10.1007/s10571-004-6918-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sharp FR, et al. Psychosis: pathological activation of limbic thalamocortical circuits by psychomimetics and schizophrenia? Trends Neurosci. 2001;24:330–334. doi: 10.1016/s0166-2236(00)01817-8. [DOI] [PubMed] [Google Scholar]
- 63.Moghaddam B, et al. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17:2921–2927. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Olney JW, et al. NMDA receptor hypofunction model of schizophrenia. J Psychiatr Res. 1999;33:523–533. doi: 10.1016/s0022-3956(99)00029-1. [DOI] [PubMed] [Google Scholar]
- 65.Friston KJ, et al. The left medial temporal region and schizophrenia. A PET study. Brain. 1992;115:367–382. doi: 10.1093/brain/115.2.367. [DOI] [PubMed] [Google Scholar]
- 66.Malaspina D, et al. Resting neural activity distinguishes subgroups of schizophrenia patients. Biol Psychiatry. 2004;56:931–937. doi: 10.1016/j.biopsych.2004.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Heckers S, et al. Impaired recruitment of the hippocampus during conscious recollection in schizophrenia. Nat Neurosci. 1998;1:318–323. doi: 10.1038/1137. [DOI] [PubMed] [Google Scholar]
- 68.Lewandowski NM, et al. Isolating hippocampal subregions most vulnerable to schizophrenia. 2005 Annual Society for Neuroscience Meeting; Washington, DC. 2005. Poster/Program 443.1. [Google Scholar]
- 69.Schobel SA, et al. The CA1 subfield is a primary site of hippocampal dysfunction associated with schizophrenia. 2007 Annual Society for Neuroscience Meeting; San Diego, CA. 2007. Abstract/Program 806.9/V27. [Google Scholar]
- 70.Sharp FR, Hendren RL. Psychosis: atypical limbic epilepsy versus limbic hyperexcitability with onset at puberty? Epilepsy Behav. 2007;10:515–520. doi: 10.1016/j.yebeh.2007.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Qin P, et al. Risk for schizophrenia and schizophrenia-like psychosis among patients with epilepsy: population based cohort study. BMJ. 2005;331:23. doi: 10.1136/bmj.38488.462037.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ounsted C, Lindsay J. Epilepsy and Psychiatry. Churchill Livingstone; 1981. [Google Scholar]
- 73.Cunningham MO, et al. Region-specific reduction in entorhinal gamma oscillations and parvalbumin-immunoreactive neurons in animal models of psychiatric illness. J Neurosci. 2006;26:2767–2776. doi: 10.1523/JNEUROSCI.5054-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fisahn A, et al. Cholinergic induction of network oscillations at 40 Hz in the hippocampus in vitro. Nature. 1998;394:186–189. doi: 10.1038/28179. [DOI] [PubMed] [Google Scholar]
- 75.Bartos M, et al. Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat Rev Neurosci. 2007;8:45–56. doi: 10.1038/nrn2044. [DOI] [PubMed] [Google Scholar]
- 76.Cochran SM, et al. Induction of metabolic hypofunction and neurochemical deficits after chronic intermittent exposure to phencyclidine: differential modulation by antipsychotic drugs. Neuropsychopharmacology. 2003;28:265–275. doi: 10.1038/sj.npp.1300031. [DOI] [PubMed] [Google Scholar]
- 77.Keilhoff G, et al. Repeated application of ketamine to rats induces changes in the hippocampal expression of parvalbumin, neuronal nitric oxide synthase and cFOS similar to those found in human schizophrenia. Neuroscience. 2004;126:591–598. doi: 10.1016/j.neuroscience.2004.03.039. [DOI] [PubMed] [Google Scholar]
- 78.Gisabella B, et al. Regulation of synaptic plasticity in a schizophrenia model. Proc Natl Acad Sci U S A. 2005;102:13301–13306. doi: 10.1073/pnas.0506034102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kinney JW, et al. A specific role for NR2A-containing NMDA receptors in the maintenance of parvalbumin and GAD67 immunoreactivity in cultured interneurons. J Neurosci. 2006;26:1604–1615. doi: 10.1523/JNEUROSCI.4722-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Goldberg JH, et al. Ca2+ imaging of mouse neocortical interneurone dendrites: contribution of Ca2+-permeable AMPA and NMDA receptors to subthreshold Ca2+ dynamics. J Physiol. 2003;551:67–78. doi: 10.1113/jphysiol.2003.042598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Behrens MM, et al. Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science. 2007;318:1645–1647. doi: 10.1126/science.1148045. [DOI] [PubMed] [Google Scholar]
- 82.Collin T, et al. Developmental changes in parvalbumin regulate presynaptic Ca2+ signaling. J Neurosci. 2005;25:96–107. doi: 10.1523/JNEUROSCI.3748-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hendry SH, Jones EG. Reduction in number of immunostained GABAergic neurones in deprived-eye dominance columns of monkey area 17. Nature. 1986;320:750–753. doi: 10.1038/320750a0. [DOI] [PubMed] [Google Scholar]
- 84.Jiao Y, et al. Major effects of sensory experiences on the neocortical inhibitory circuits. J Neurosci. 2006;26:8691–8701. doi: 10.1523/JNEUROSCI.2478-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Seeman P. Dopamine receptors and the dopamine hypothesis of schizophrenia. Synapse. 1987;1:133–152. doi: 10.1002/syn.890010203. [DOI] [PubMed] [Google Scholar]
- 86.Snyder SH. The dopamine hypothesis of schizophrenia: focus on the dopamine receptor. Am J Psychiatry. 1976;133:197–202. doi: 10.1176/ajp.133.2.197. [DOI] [PubMed] [Google Scholar]
- 87.Angrist BM. Amphetamine psychosis: clinical variations of the syndrome. In: Cho AK, Segal DS, editors. Amphetamine and Its Analogs. Academic Press; 1994. pp. 387–414. [Google Scholar]
- 88.Laruelle M, et al. Single photon emission computerized tomography imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects. Proc Natl Acad Sci U S A. 1996;93:9235–9240. doi: 10.1073/pnas.93.17.9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Carlsson M, Carlsson A. Interactions between glutamatergic and monoaminergic systems within the basal ganglia—implications for schizophrenia and Parkinson’s disease. Trends Neurosci. 1990;13:272–276. doi: 10.1016/0166-2236(90)90108-m. [DOI] [PubMed] [Google Scholar]
- 90.Grace AA. Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: a hypothesis for the etiology of schizophrenia. Neuroscience. 1991;41:1–24. doi: 10.1016/0306-4522(91)90196-u. [DOI] [PubMed] [Google Scholar]
- 91.Pawlowski L, et al. Phencyclidine activates rat A10 dopamine neurons but reduces burst activity and causes regularization of firing. Acta Physiol Scand. 1990;139:529–530. doi: 10.1111/j.1748-1716.1990.tb08955.x. [DOI] [PubMed] [Google Scholar]
- 92.Vollenweider FX, et al. Effects of (S)-ketamine on striatal dopamine: a 11Craclopride PET study of a model psychosis in humans. J Psychiatr Res. 2000;34:35–43. doi: 10.1016/s0022-3956(99)00031-x. [DOI] [PubMed] [Google Scholar]
- 93.Aalto S, et al. Cortical glutamate-dopamine interaction and ketamine-induced psychotic symptoms in man. Psychopharmacology (Berl) 2005;182:375–383. doi: 10.1007/s00213-005-0092-6. [DOI] [PubMed] [Google Scholar]
- 94.Kegeles LS, et al. NMDA antagonist effects on striatal dopamine release: positron emission tomography studies in humans. Synapse. 2002;43:19–29. doi: 10.1002/syn.10010. [DOI] [PubMed] [Google Scholar]
- 95.Javitt DC, et al. Impaired mismatch negativity generation reflects widespread dysfunction of working memory in schizophrenia. Arch Gen Psychiatry. 1995;52:550–558. doi: 10.1001/archpsyc.1995.03950190032005. [DOI] [PubMed] [Google Scholar]
- 96.Leitman DI, et al. The neural substrates of impaired prosodic detection in schizophrenia and its sensorial antecedents. Am J Psychiatry. 2007;164:474–482. doi: 10.1176/ajp.2007.164.3.474. [DOI] [PubMed] [Google Scholar]
- 97.Butler PD, et al. Subcortical visual dysfunction in schizophrenia drives secondary cortical impairments. Brain. 2007;130:417–430. doi: 10.1093/brain/awl233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Goldman-Rakic PS. Cellular basis of working memory. Neuron. 1995;14:477–485. doi: 10.1016/0896-6273(95)90304-6. [DOI] [PubMed] [Google Scholar]
- 99.Suzuki K, et al. Auditory hallucinations and cognitive impairment in a patient with a lesion restricted to the hippocampus. Schizophr Res. 2003;64:87–89. doi: 10.1016/s0920-9964(02)00386-9. [DOI] [PubMed] [Google Scholar]
- 100.Takebayashi H, et al. Unilateral auditory hallucinations in schizophrenia after damage to the right hippocampus. Schizophr Res. 2002;58:329–331. doi: 10.1016/s0920-9964(01)00399-1. [DOI] [PubMed] [Google Scholar]
- 101.Floresco SB, et al. Glutamatergic afferents from the hippocampus to the nucleus accumbens regulate activity of ventral tegmental area dopamine neurons. J Neurosci. 2001;21:4915–4922. doi: 10.1523/JNEUROSCI.21-13-04915.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Legault M, et al. Chemical stimulation of the ventral hippocampus elevates nucleus accumbens dopamine by activating dopaminergic neurons of the ventral tegmental area. J Neurosci. 2000;20:1635–1642. doi: 10.1523/JNEUROSCI.20-04-01635.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Moore H, et al. A neurobehavioral systems analysis of adult rats exposed to methylazoxymethanol acetate on E17: implications for the neuropathology of schizophrenia. Biol Psychiatry. 2006;60:253–264. doi: 10.1016/j.biopsych.2006.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Johnston MV, et al. Methylazoxymethanol treatment of fetal rats results in abnormally dense noradrenergic innervation of neocortex. Science. 1979;203:369–371. doi: 10.1126/science.32620. [DOI] [PubMed] [Google Scholar]
- 105.Lodge DJ, Grace AA. Aberrant hippocampal activity underlies the dopamine dysregulation in an animal model of schizophrenia. J Neurosci. 2007;27:11424–11430. doi: 10.1523/JNEUROSCI.2847-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lisman JE, Grace AA. The hippocampal-VTA loop: controlling the entry of information into long-term memory. Neuron. 2005;46:703–713. doi: 10.1016/j.neuron.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 107.Bunzeck N, Duzel E. Absolute coding of stimulus novelty in the human substantia nigra/VTA. Neuron. 2006;51:369–379. doi: 10.1016/j.neuron.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 108.Lisman JE, Otmakhova NA. Storage, recall, and novelty detection of sequences by the hippocampus: elaborating on the SOCRATIC model to account for normal and aberrant effects of dopamine. Hippocampus. 2001;11:551–568. doi: 10.1002/hipo.1071. [DOI] [PubMed] [Google Scholar]
- 109.Hammad H, Wagner JJ. Dopamine-mediated disinhibition in the CA1 region of rat hippocampus via D3 receptor activation. J Pharmacol Exp Ther. 2006;316:113–120. doi: 10.1124/jpet.105.091579. [DOI] [PubMed] [Google Scholar]
- 110.Holt DJ, et al. Sustained activation of the hippocampus in response to fearful faces in schizophrenia. Biol Psychiatry. 2005;57:1011–1019. doi: 10.1016/j.biopsych.2005.01.033. [DOI] [PubMed] [Google Scholar]
- 111.Braff DL, et al. Impact of prepulse characteristics on the detection of sensorimotor gating deficits in schizophrenia. Schizophr Res. 2001;49:171–178. doi: 10.1016/s0920-9964(00)00139-0. [DOI] [PubMed] [Google Scholar]
- 112.Seamans JK, Yang CR. The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol. 2004;74:1–58. doi: 10.1016/j.pneurobio.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 113.Lee KH, et al. Synchronous gamma activity: a review and contribution to an integrative neuroscience model of schizophrenia. Brain Res Brain Res Rev. 2003;41:57–78. doi: 10.1016/s0165-0173(02)00220-5. [DOI] [PubMed] [Google Scholar]
- 114.Gallinat J, et al. Reduced oscillatory gamma-band responses in unmedicated schizophrenic patients indicate impaired frontal network processing. Clin Neurophysiol. 2004;115:1863–1874. doi: 10.1016/j.clinph.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 115.Fuchs EC, et al. Recruitment of parvalbumin-positive interneurons determines hippocampal function and associated behavior. Neuron. 2007;53:591–604. doi: 10.1016/j.neuron.2007.01.031. [DOI] [PubMed] [Google Scholar]
- 116.Lisman J. The theta/gamma discrete phase code occuring during the hippocampal phase precession may be a more general brain coding scheme. Hippocampus. 2005;15:913–922. doi: 10.1002/hipo.20121. [DOI] [PubMed] [Google Scholar]
- 117.Butler PD, et al. Early-stage visual processing and cortical amplification deficits in schizophrenia. Arch Gen Psychiatry. 2005;62:495–504. doi: 10.1001/archpsyc.62.5.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Martin LF, Freedman R. Schizophrenia and the α7 nicotinic acetylcholine receptor. Int Rev Neurobiol. 2007;78:225–246. doi: 10.1016/S0074-7742(06)78008-4. [DOI] [PubMed] [Google Scholar]
- 119.de Leon J. Smoking and vulnerability for schizophrenia. Schizophr Bull. 1996;22:405–409. doi: 10.1093/schbul/22.3.405. [DOI] [PubMed] [Google Scholar]
- 120.Kumari V, Postma P. Nicotine use in schizophrenia: the self medication hypotheses. Neurosci Biobehav Rev. 2005;29:1021–1034. doi: 10.1016/j.neubiorev.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 121.Barr RS, et al. The effects of transdermal nicotine on cognition in nonsmokers with schizophrenia and nonpsychiatric controls. Neuropsychopharmacology. 2008;33:480–490. doi: 10.1038/sj.npp.1301423. [DOI] [PubMed] [Google Scholar]
- 122.Jacobsen LK, et al. Nicotine effects on brain function and functional connectivity in schizophrenia. Biol Psychiatry. 2004;55:850–858. doi: 10.1016/j.biopsych.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 123.Smith RC, et al. Effects of nicotine nasal spray on cognitive function in schizophrenia. Neuropsychopharmacology. 2006;31:637–643. doi: 10.1038/sj.npp.1300881. [DOI] [PubMed] [Google Scholar]
- 124.Krenz I, et al. Parvalbumin-containing interneurons of the human cerebral cortex express nicotinic acetylcholine receptor proteins. J Chem Neuroanat. 2001;21:239–246. doi: 10.1016/s0891-0618(01)00112-0. [DOI] [PubMed] [Google Scholar]
- 125.Hulo S, Muller D. Tetrodotoxin-sensitive enhancement of inhibition in CA1 pyramidal neurones by nicotine. Neuroreport. 2001;12:1351–1354. doi: 10.1097/00001756-200105250-00012. [DOI] [PubMed] [Google Scholar]
- 126.Wanaverbecq N, et al. Cholinergic axons modulate GABAergic signaling among hippocampal interneurons via postsynaptic α 7 nicotinic receptors. J Neurosci. 2007;27:5683–5693. doi: 10.1523/JNEUROSCI.1732-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Song C, et al. Role of α7-nicotinic acetylcholine receptors in tetanic stimulation-induced gamma oscillations in rat hippocampal slices. Neuropharmacology. 2005;48:869–880. doi: 10.1016/j.neuropharm.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 128.Harrison PJ, West VA. Six degrees of separation: on the prior probability that schizophrenia susceptibility genes converge on synapses, glutamate and NMDA receptors. Mol Psychiatry. 2006;11:981–983. doi: 10.1038/sj.mp.4001886. [DOI] [PubMed] [Google Scholar]
- 129.Gupta DS, et al. Metabotropic glutamate receptor protein expression in the prefrontal cortex and striatum in schizophrenia. Synapse. 2005;57:123–131. doi: 10.1002/syn.20164. [DOI] [PubMed] [Google Scholar]
- 130.Chumakov I, et al. Genetic and physiological data implicating the new human gene G72 and the gene for D-amino acid oxidase in schizophrenia. Proc Natl Acad Sci U S A. 2002;99:13675–13680. doi: 10.1073/pnas.182412499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hashimoto K, et al. Reduced D-serine to total serine ratio in the cerebrospinal fluid of drug naive schizophrenic patients. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:767–769. doi: 10.1016/j.pnpbp.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 132.Stevens ER, et al. D-serine and serine racemase are present in the vertebrate retina and contribute to the physiological activation of NMDA receptors. Proc Natl Acad Sci U S A. 2003;100:6789–6794. doi: 10.1073/pnas.1237052100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Detera-Wadleigh SD, McMahon FJ. G72/G30 in schizophrenia and bipolar disorder: review and meta-analysis. Biol Psychiatry. 2006;60:106–114. doi: 10.1016/j.biopsych.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 134.Hattori E, et al. Polymorphisms at the G72/G30 gene locus, on 13q33, are associated with bipolar disorder in two independent pedigree series. Am J Hum Genet. 2003;72:1131–1140. doi: 10.1086/374822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tsai G, et al. D-serine added to antipsychotics for the treatment of schizophrenia. Biol Psychiatry. 1998;44:1081–1089. doi: 10.1016/s0006-3223(98)00279-0. [DOI] [PubMed] [Google Scholar]
- 136.Heresco-Levy U, et al. Placebo-controlled trial of D-cycloserine added to conventional neuroleptics, olanzapine, or risperidone in schizophrenia. Am J Psychiatry. 2002;159:480–482. doi: 10.1176/appi.ajp.159.3.480. [DOI] [PubMed] [Google Scholar]
- 137.Corvin A, et al. Evidence for association and epistasis at the DAOA/G30 and D-amino acid oxidase loci in an Irish schizophrenia sample. Am J Med Genet B Neuropsychiatr Genet. 2007;144:949–953. doi: 10.1002/ajmg.b.30452. [DOI] [PubMed] [Google Scholar]
- 138.Bendikov I, et al. A CSF and postmortem brain study of D-serine metabolic parameters in schizophrenia. Schizophr Res. 2007;90:41–51. doi: 10.1016/j.schres.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 139.Shinkai T, et al. Association analyses of the DAOA/G30 and D-amino-acid oxidase genes in schizophrenia: further evidence for a role in schizophrenia. Neuromolecular Med. 2007;9:169–177. doi: 10.1007/BF02685890. [DOI] [PubMed] [Google Scholar]
- 140.Morita Y, et al. A genetic variant of the serine racemase gene is associated with schizophrenia. Biol Psychiatry. 2007;61:1200–1203. doi: 10.1016/j.biopsych.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 141.Fujii K, et al. Serine racemase binds to PICK1: potential relevance to schizophrenia. Mol Psychiatry. 2006;11:150–157. doi: 10.1038/sj.mp.4001776. [DOI] [PubMed] [Google Scholar]
- 142.Wroblewska B, et al. N-acetylaspartylglutamate selectively activates mGluR3 receptors in transfected cells. J Neurochem. 1997;69:174–181. doi: 10.1046/j.1471-4159.1997.69010174.x. [DOI] [PubMed] [Google Scholar]
- 143.Hakak Y, et al. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc Natl Acad Sci U S A. 2001;98:4746–4751. doi: 10.1073/pnas.081071198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Huffacker S, et al. Large scale gene chip analysis of postmortem brains from schizophrenia and bipolar affect in disorder patients. 2003 Annual Society for Neuroscience Meeting; New Orleans, LA. 2003. Abstract/Program 312.16. [Google Scholar]
- 145.Williams NM, et al. Is the dysbindin gene (DTNBP1) a susceptibility gene for schizophrenia? Schizophr Bull. 2005;31:800–805. doi: 10.1093/schbul/sbi061. [DOI] [PubMed] [Google Scholar]
- 146.Numakawa T, et al. Evidence of novel neuronal functions of dysbindin, a susceptibility gene for schizophrenia. Hum Mol Genet. 2004;13:2699–2708. doi: 10.1093/hmg/ddh280. [DOI] [PubMed] [Google Scholar]
- 147.Talbot K, et al. Dysbindin-1 is reduced in intrinsic, glutamatergic terminals of the hippocampal formation in schizophrenia. J Clin Invest. 2004;113:1353–1363. doi: 10.1172/JCI20425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Burdick KE, et al. DTNBP1 genotype influences cognitive decline in schizophrenia. Schizophr Res. 2007;89:169–172. doi: 10.1016/j.schres.2006.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Gornick MC, et al. Dysbindin (DTNBP1, 6p22.3) is associated with childhood-onset psychosis and endophenotypes measured by the Premorbid Adjustment Scale (PAS) J Autism Dev Disord. 2005;35:831–838. doi: 10.1007/s10803-005-0028-3. [DOI] [PubMed] [Google Scholar]
- 150.Petryshen TL, et al. Support for involvement of neuregulin 1 in schizophrenia pathophysiology. Mol Psychiatry. 2005;10:366–374. doi: 10.1038/sj.mp.4001608. [DOI] [PubMed] [Google Scholar]
- 151.Falls DL. Neuregulins: functions, forms, and signaling strategies. Exp Cell Res. 2003;284:14–30. doi: 10.1016/s0014-4827(02)00102-7. [DOI] [PubMed] [Google Scholar]
- 152.Bjarnadottir M, et al. Neuregulin1 (NRG1) signaling through Fyn modulates NMDA receptor phosphorylation: differential synaptic function in NRG1+/− knock-outs compared with wild-type mice. J Neurosci. 2007;27:4519–4529. doi: 10.1523/JNEUROSCI.4314-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Li B, et al. The neuregulin-1 receptor erbB4 controls glutamatergic synapse maturation and plasticity. Neuron. 2007;54:583–597. doi: 10.1016/j.neuron.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hahn CG, et al. Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nat Med. 2006;12:824–828. doi: 10.1038/nm1418. [DOI] [PubMed] [Google Scholar]
- 155.Yang X, et al. A cysteine-rich isoform of neuregulin controls the level of expression of neuronal nicotinic receptor channels during synaptogenesis. Neuron. 1998;20:255–270. doi: 10.1016/s0896-6273(00)80454-7. [DOI] [PubMed] [Google Scholar]
- 156.Chang Q, Fischbach GD. An acute effect of neuregulin 1 β to suppress α 7-containing nicotinic acetylcholine receptors in hippocampal interneurons. J Neurosci. 2006;26:11295–11303. doi: 10.1523/JNEUROSCI.1794-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Woo RS, et al. Neuregulin-1 enhances depolarization-induced GABA release. Neuron. 2007;54:599–610. doi: 10.1016/j.neuron.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 158.Addington AM, et al. GAD1 (2q31.1), which encodes glutamic acid decarboxylase (GAD67), is associated with childhood-onset schizophrenia and cortical gray matter volume loss. Mol Psychiatry. 2005;10:581–588. doi: 10.1038/sj.mp.4001599. [DOI] [PubMed] [Google Scholar]
- 159.Leonard S, Freedman R. Genetics of chromosome 15q13-q14 in schizophrenia. Biol Psychiatry. 2006;60:115–122. doi: 10.1016/j.biopsych.2006.03.054. [DOI] [PubMed] [Google Scholar]