

Abstract
Cytochrome P450 (P450) enzymes typically require the presence of at least cytochrome P450 reductase (CPR) and NADPH to carry out the metabolism of xenobiotics. To address whether the need for redox transfer proteins and the NADPH cofactor protein could be obviated, CYP2C9 was bonded to a gold electrode through an 11-mercaptoundecanoic acid and octanethiol self-assembled monolayer (SAM) through which a current could be applied. Cyclic voltammetry demonstrated direct electrochemistry of the CYP2C9 enzyme bonded to the electrode and fast electron transfer between the heme iron and the gold electrode. To determine whether this system could metabolize warfarin analogous to microsomal or expressed enzyme systems containing CYP2C9, warfarin was incubated with the CYP2C9-SAM-gold electrode and a controlled potential was applied. The expected 7-hydroxywarfarin metabolite was observed, analogous to expressed CYP2C9 systems, wherein this is the predominant metabolite. Current-concentration data generated with increasing concentrations of warfarin were used to determine the Michaelis-Menten constant (Km) for the hydroxylation of warfarin (3 μM), which is in good agreement with previous literature regarding Km values for this reaction. In summary, the CYP2C9-SAM-gold electrode system was able to carry out the metabolism of warfarin only after application of an electrical potential, but in the absence of either CPR or NADPH. Furthermore, this system may provide a unique platform for both studying P450 enzyme electrochemistry and as a bioreactor to produce metabolites without the need for expensive redox transfer proteins and cofactors.
Cytochrome P450 (P450) enzymes are important for drug metabolism in humans, accounting for ∼75% metabolism of drugs (Williams et al., 2004). Numerous factors affect the P450 metabolism rate and the resulting metabolite structure including electron transfer, protein-protein interactions, concentration and structure of the substrate, and the source and specific means whereby the P450 was prepared (Guengerich, 2007). In practice, it is difficult to isolate the individual contributions of these factors because they are often inter-dependent. Thus, it is of interest to devise model platforms that can be used to independently control these parameters to monitor their respective effects on metabolism.
P450 catalysis requires a constant supply of NADPH as the electron source and cytochrome P450 reductase (CPR) to deliver the electrons. In attempts to obviate the requirement of these redox partners/cofactors for catalysis, P450 enzymes have been immobilized on electrodes so that the electrode supplies electrons to drive the P450 catalytic cycle (Estabrook et al., 1996; Reipa et al., 1997; Gilardi et al., 2002) with effective electrical communication between the electrode and the enzyme being critical (Yang et al., 2008). Direct adsorption of enzymes on bare electrodes, such as gold, platinum, and graphite, results in diminished biocatalytic activity (Habermüller et al., 2000; Joseph et al., 2003; Shumyantseva et al., 2005). Complicating these systems, the active redox site is often deeply embedded in the interior of the enzyme, insulating it from the electrode and impeding electron transfer between the electrode and the redox site of enzyme. Finally, immobilization by direct attachment may impede or prohibit substrate binding or metabolite release through steric constraints (Habermüller et al., 2000; Joseph et al., 2003; Shumyantseva et al., 2005).
Various methods for enzyme immobilization have been explored to solve the problems that were caused by direct attachment (Reipa et al., 1997; Gilardi et al., 2002; Joseph et al., 2003; Johnson et al., 2005; Shumyantseva et al., 2005). These methods include immobilizing the enzyme in a polyion film (Masuda and Fukuda, 1995), entrapping the enzyme in a sol-gel film (Masuda et al., 2000), incorporating the enzyme into conducting polymer films (Funk et al., 2005), modifying the enzyme with an electrical relay (Hahm and Lieber, 2003), functionalizing the electrode with a biomembrane-like surfactant (Ohldag et al., 2006), and supporting the enzyme in a clay nanoparticle modified electrode (Shumyantseva et al., 2004). However, because these methods do not control enzyme aggregation on the electrode surface, the efficiency of electron transfer to the active site of the enzyme may be reduced (Habermüller et al., 2000; Gilardi et al., 2001; Gilardi and Fantuzzi, 2001; Chen et al., 2002), potentially through altered enzyme conformation.
Enzyme orientation and concentration at the electrode surface are critical to the sensitivity of enzymatic devices (Habermüller et al., 2000; Gilardi et al., 2001; Gilardi and Fantuzzi, 2001), and, thus, regioselectively bonding the enzyme to a preformed, self-assembled monolayer (SAM) has been used to control these factors. For example, alkane-thiol or other thiol-terminated chains covalently bonded to a noble-metal surface and functionalized at the terminus with a group that binds to a specific site of the protein, which have been used to control enzyme binding orientation (Collinson and Bowden, 1992; Pardo-Yissar et al., 2000; Chen et al., 2002). CYP2E1 has been bonded through thiols, both directly or via a cysteine-maleimide linker, to a gold surface although enzyme orientation remained ambiguous (Fantuzzi et al., 2004). Bonding to the N terminus of enzymes provides a more specific alternative than bonding to surface cystines that can result in undesirable orientations of the enzyme. Molecular modeling of CYP2C9, based on its crystal structure (Wester et al., 2004), suggests the presence of lysine residues on the surface, some of which are located near the substrate access channel or purported reductase binding site and thus a less than optimal site for attachment. However, it is hypothesized that attachment of CYP2C9 to a gold surface through the N terminus lysine is more likely based on pKa and steric constraints.
In a previously study (Gannett et al., 2006), we bonded CYP2C9 molecules via the N terminus to the carboxylic acid group of 11-mercaptoundecanoic acid (MUA), resulting in a system with catalytic activity toward substrate. However, this system was still dependent on the presence of NADPH and CPR, both of which are expensive, require continual addition upon extended incubations, and can confound isolation and purification of metabolites. To address these issues, this work has now been extended to realize the direct electrochemistry of the surface-bonded CYP2C9 enzyme and evaluate the electrocatalytic activity of CYP2C9 (absent of both NADPH and CPR) toward the hydroxylation of the CYP2C9 substrate warfarin.
Materials and Methods
Chemicals and Reagents. All chemicals were of analytical reagent grade and were used without further purification. MUA, 1-octanethiol (OT), N-((3-dimethylamino)-propyl)-N-ethyl carbodiimide hydrochloride (EDC), N-hydroxysulfosuccinimide (NHS), flurbiprofen, dapsone, NADPH, and warfarin were purchased from Sigma-Aldrich (St. Louis, MO). CYP2C9 was prepared by expression in an Escherichia coli system, isolated, and purified as described previously (Gannett et al., 2006). Cytochrome P450 reductase was purchased from BD Biosciences (San Jose, CA). All electrochemical measurements were made in potassium phosphate-buffered solution [(PBS) 40 mM, pH 7.4] containing 154 mM NaCl in Milli-Q water (18.2 MΩ.cm) unless otherwise indicated. Gold-coated glass slides (25 mm × 75 mm) with a 4-nm chromium layer and a 100-nm-thick gold top layer were purchased from EMF Corp. (Ithaca, NY).
Immobilization of CYP2C9 Enzyme on Gold Electrode. Gold-coated glass slides were sonicated in acetone for 5 min, rinsed with deionized water, and then soaked in Piranha solution (98% H2SO4: 30% H2O2 = 3:1 v/v) at 80°C for 15 min. (Caution: Piranha solution reacts violently with organic chemicals.) The gold-coated glass slides were then rinsed with deionized water and ethanol and then soaked in an ethanolic solution of 8-octanethiol (75 mM) and 11-mercaptoundecanoic acid (25 mM) for 18 h to allow the SAM to attach to the gold surface. Excess physisorbed thiols were removed by rinsing the SAM-bearing, gold-coated glass slides with absolute ethanol and PBS, three times for each slide. To bond the CYP2C9 to the SAM, the SAM-coated slides were covered with PBS containing 2.0 mM EDC and 5.0 mM NHS for 2 h. The treated slides were then removed from the EDC/NHS solution and immersed in a PBS solution containing 50 nM CYP2C9, 40 μM flurbiprofen, and 40 μM dapsone for 24 h. All processes were performed under argon at room temperature. Herein, the gold-coated slide functionalized with a MUA/OT monolayer is abbreviated as MUA/OT-Au, and the MUA/OT-Au sample with attached CYP2C9 is abbreviated as CYP2C9-MUA/OT-Au.
X-Ray Photoelectron Spectroscopy Characterization of Immobilized CYP2C9 Enzyme. X-ray photoelectron spectroscopy (XPS) analysis was performed on the SAM-bearing, gold-coated slides with and without immobilized CYP2C9. The XPS measurement was carried out with an Omicron ESCA Probe (Taunusstein, Germany) equipped with an EA125 energy analyzer. Photoemission was stimulated by monochromatic aluminum K-α radiation (1486.6 eV) with an operating power of 300 W. A low-energy electron flood gun was used for charge neutralization. Survey and high-resolution scans were collected using the pass energies of 50 and 25 eV, respectively. Binding energies of the spectra were referenced to the carbon 1s electron (C 1s) binding energy (284.8 eV).
Electrochemical Measurements. The electrochemical measurements were carried out in a three-electrode electrochemical cell comprised of a platinum wire (1 mm o.d.) counter electrode, an Ag AgCl (3M KCl) reference electrode, and the CYP2C9-bonded gold electrode (CYP2C9-MUA/OT-Au) as the working electrode. The working electrode surface area was approximately 7 mm2. Different CYP2C9-MUA/OT-Au working electrodes were prepared, and all yielded comparable results. Likewise, CYP2C9 from several enzyme preparations were used for electrode construction and had no effect on the experimental data. All electrochemical measurements were carried out in an aqueous solution (154 mM NaCl, 40 mM PBS, pH 7.4). Cyclic voltammetry (CV) and amperometry measurements were performed using a potentiostat (Gamry Reference 600; Gamry Instruments, Warminster, PA). For anaerobic experiments, solutions were deoxygenated by purging with nitrogen gas for 60 min before measurements and then blanked under a nitrogen atmosphere during measurement. For the aerobic experiments, the solution was purged with oxygen for 1 min before initial measurements. Oxygen saturation was maintained during measurement. All electrochemical experiments were conducted at ambient temperature (25 ± 1°C).
Determination of Enzyme Surface Coverage and Electron Transfer Rate. For redox-active molecules immobilized on an electrode surface, the peak current (ip) is given by the Laviron equation (Laviron, 1979) (eq. 1):
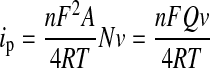 |
(1) |
where n is the number of electrons transferred, F is Faraday's constant, A is the area of the electrode, N is the number of redox active sites on the surface, ν is the scan rate, Q is the charge passing through the electrode in columbs, R is the gas constant, and T is the temperature. Equation 1 was used to calculate n (0.72) by obtaining the slope of the linearly fitted curve of ip versus v. Furthermore, given n = 0.72, the surface coverage of the CYP2C9 molecules can be calculated (8.0 × 10–13 or 4.8 × 1011 mol/cm2). This number is within a factor of 2 of our previous estimate based on atomic force microscopy (Gannett et al., 2006).
For the immobilized CYP2C9, the electron-transfer rate constant (ks) can be obtained according to eq. 2 in the case of nΔEp < 200 mV (Laviron, 1979), where α is the electron transfer coefficient and the other variables are as defined previously.
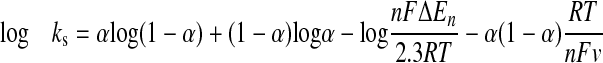 |
(2) |
Electrochemical Determination of CYP2C9-Mediated Kinetics Warfarin Hydroxylation. Cyclic voltammetry measurements were made with the Au-SAM-CYP2C9 electrode in solutions containing warfarin (1, 2.5, 5, 10, 15, 20, 30, and 40 μM) and the potential swept cathodically and anodically. The peak reduction current at each concentration was then fit. The apparent Michaelis-Menten constant (K appm) and imax (functionally equivalent to Vmax) were used to evaluate the kinetics of warfarin (to 7-OH-warfain) catalysis by the bonded CYP2C9 enzyme. These parameters were estimated by the Michaelis-Menten equation (eq. 3), where C is the warfarin concentration, i is the current, and imax is the maximum current.
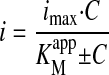 |
(3) |
Controlled Potential Electrolysis of Warfarin and Liquid Chromatography Analysis. Electrocatalytic metabolism was performed on the CYP2C9-MUA/OT-Au electrodes in solutions containing 50 μM warfarin dissolved in 154 mM NaCl and 40 mM PBS (pH 7.4) at a fixed potential of –0.38 V for 2 h. The metabolites were isolated by concentration to dryness of the electrolysis solution, dissolving the resulting solid in 2 ml acetonitrile, filtering, and then concentration to dryness. The resulting solid was dissolved in 200 μl of PBS and spiked with 7-ethoxycoumarin (50 μl of acetonitrile containing 1 μg/ml) as an internal standard. Samples were centrifuged (5 min at 13,000g), and 200 μl of the supernatant was loaded into liquid chromatography (LC) vials. LC was performed with a Waters Alliance 2965 separations module using a Waters Symmetry column (150 × 4.6 mm, 3.6-μm particle size; Waters, Milford, MA), eluting with a 60:40 mixture of 0.5% aqueous phosphoric acid/acetonitrile (1 ml/min), with fluorescence detection (Waters 2475, λex = 320 nm, λem = 415 nm) and an injection volume of 10 μl. All chromatographic analysis was performed using Empower 2 software (Waters). The peak corresponding to 7-hydroxywarfarin was determined by comparison to a 7-hydroxywarfarin standard obtained from Sigma-Aldrich.
Reconstituted CYP2C9 Enzyme Incubation. Reconstituted CYP2C9 enzyme incubations with warfarin were conducted using enzymes prepared from insect cells infected with recombinant baculovirus containing a human CYP2C9*1 cDNA and rabbit NADPH-P450 reductase. For these incubations, 50 μM warfarin was incubated with 125 nM CYP2C9 in 40 mM potassium phosphate buffer (pH 7.4), 1.6 mM phospholipid in THAM, and 250 nm CPR in total volume of 200 μl. Samples were preincubated for 3 min at 37°C before the addition of 1 mM NADPH (final concentration). Incubations were conducted for 20 min at 37°C and quenched by the addition of acetonitrile containing 7-ethoxycoumarin (50 μl of a 1 μg/ml in acetonitrile) as the internal standard. The incubation mixture was then centrifuged (15 min at 13000g) to pellet proteins. A total of 200 μl of the supernatant was then loaded into LC vials for analysis. Chromatographic detection parameters used were the same as those described above.
Results
Characterization of CYP2C9-Immobilized Gold Electrode. During XPS measurement, the energy from X-ray photons of an incident beam are absorbed by an atom in a molecule or solid, leading to ionization and the emission of a core (inner shell) electron in the s, p, d, and f levels. The binding energies of these core-level electrons are heavily dependent upon the chemical states of the atoms, which chemically shift with the coordination environment. Therefore, the binding energies of the core-level electrons are used for analysis of the chemical structure. Figure 1a shows the XPS spectrum of the S 2p core level that was obtained from the MUA/OT-Au sample. The S 2p3/2 peak at 162 eV is ascribed to the thiol covalently bonded to the gold surface (Nuzzo et al., 1987). Figure 1b shows the C 1s core level spectrum of the MUA/OT-Au sample. The peak at 284.8 eV is characteristic of the aliphatic carbons present in MUA. The peak component at 289.5 eV is ascribed to the carbonyl carbon of the carboxylic acid group of MUA (Wu et al., 2004; Lim et al., 2007). No peaks were observed in the MUA/OT-Au sample that would be expected for an amide nitrogen or the carbonyl carbon of an amide.
Fig. 1.

a, XPS spectrum of the sulfur 2p (S 2p3/2) core level electrons obtained from the MUA/OT-Au sample corresponding to the S-Au bond (162.0 eV). b, XPS spectrum of the C 1s electrons obtained from the MUA/OT-Au sample characteristic of C-C bonds (284.8 eV) and –C = OOH bonds (289.5 eV). c, XPS spectrum of the N 1s obtained from the CYP2C9-MUA/OT-Au sample ascribed to N-C = O bonds (400.3 eV). d, XPS spectrum of the C 1s electrons attributed to C-C bonds (284.8 eV), C-N peptide bonds (286.1 eV), and the N-C = O bonds (288.0 eV) from the CYP2C9-MUA/OT-Au sample.
After the MUA/OT-Au sample was modified with the CYP2C9 (CYP2C9-MUA/OT-Au), XPS peaks consistent with the presence of the protein were observed in the CYP2C9-MUA/OT-Au sample. The N 1s peak at 400.3 eV in Fig. 1c corresponds to the amide linkages that are expected for the CYP2C9 peptide bonds and to the amide bonds between MUA and CYP2C9. The peak at 286.1 eV in Fig. 1d is assigned to the C-N peptide bonds of CYP2C9 and the MUA-CYP2C9 amide bonds. The C 1s peak at 288.0 eV is due to the O = C-N bond (Song et al., 1993). It is noteworthy that the peak at 289.5 eV, which was present in MUA/OT-Au before enzyme attachment (Fig. 1b), is not observed after attachment of CYP2C9 (Fig. 1d), suggesting amide bond formation between CYP2C9 and the MUA. Thus, the XPS spectra are consistent with the proposal that the CYP2C9 is covalently bonded to the MUA-functionalized gold surface.
Direct Electrochemistry of Immobilized CYP2C9 Enzyme in the Absence of Oxygen. Figure 2 shows the cyclic voltammograms obtained from a bare gold electrode (Fig. 2, curve a) and the MUA/OT-Au electrode (Fig. 2, curve b) in PBS containing 1 mM K3[Fe(CN)6]3 and 1 mM K4[Fe(CN)6]4 under anaerobic conditions. Scans began at 0.6 V, and, as the potential was driven negative, Fe(III) was reduced to Fe(II) with a maximum negative current at 0.16 V. At a potential of –0.10 V, the sweep was reversed, and, as the potential became more positive, Fe(II) was re-oxidized to Fe(III) with a maximum positive current at 0.28 V. The reduction potential (Ere) for the bare gold electrode was 0.16 V, and the oxidation potential (Eox) was 0.28 V. The formal potential, Ef, which is defined as Ef = (Eox + Ere)/2, was 0.22 V (versus Ag AgCl, 3M KCl). In contrast, after the gold electrode was covered by the SAM, no redox current above the baseline was observed at any potential, which indicated that the SAM acted as an insulator between the gold electrode and the bulk solution. Furthermore, the SAM must be compact and uniform to efficiently block electron transfer (Diao et al., 1999). The CV of the MUA/OT-Au electrode under aerobic conditions was also obtained (data not shown) and was identical to the CV for the MUA/OT-Au electrode under the anaerobic condition (Fig. 2, curve b).
Fig. 2.

Cyclic voltammograms obtained in 40 mM PBS containing 1 mM K3Fe(CN)6, 1 mM K3/4Fe(CN)6, and 154 mM NaCl at a scan rate of 10 mV/s under nitrogen-saturated conditions with curve a corresponding to a bare Au electrode and curve b corresponding to a MUA/OT-Au gold electrode. Both scans started at a potential of 0.6 V, went down to 0.15 V, and then the scan was reversed back to 0.6 V.
Cyclic voltammograms obtained from the electrode modified with the CYP2C9 enzyme under anaerobic conditions caused a well defined redox couple as shown in Fig. 3a. The data indicate direct electrochemistry of the covalently coupled CYP2C9 to the SAM-modified electrode. The CV exhibited a formal potential Ef [Ef = (Eox + Ere)/2, Eox oxidation potential, Ere reduction potential] at –0.45 V (versus Ag/AgCl, 3M KCl). The peak currents (those above the background current) observed for the oxidation (–0.42 V) and reduction (–0.49 V) peaks were nearly identical. The peak difference (ΔEp) between Eox and Ere is 70 mV (scan rate 1.6 V/s), which indicates a quasireversible single-electron redox reaction and is consistent with the reversible conversion of heme iron shown in eq. 4. This phenomenon has also been observed with other P450 isoenzymes such as CYP2B4 (Shumyantseva et al., 2004), CYP2E1 (Fantuzzi et al., 2004), P450 BM3 (Fantuzzi et al., 2006), CYP2C9 (Johnson et al., 2005), and CYP3A4 (Joseph et al., 2003).
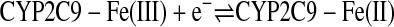 |
(4) |
Both the anodic and cathodic peak currents linearly increase with increasing scan rate from 0.5 to 1.6 V/s (Fig. 3b), suggesting that the voltammetric response is due to the surface-confined adsorbed redox molecules without any influence of protein diffusion away from or to the electrode surface (Bard and Faulkner, 2001; Myland and Oldham, 2005). It is notable that the peak width at a scan rate of 1.6 V/s (Fig. 3a) is close to the theoretical value of 90 mV for a typical single-electron wave of an adsorbed protein (Shukla et al., 2005). This peak width result is most likely reflective of the homogeneity in the conformation or orientation of the bonded CYP2C9 across the modified electrode surface, leading to an identical microscopic redox potential.
Fig. 3.

a, cyclic voltammograms obtained from a CYP2C9-MUA/OT-Au electrode in 40 mM PBS solution containing 154 mM NaCl at a scan rate of 1.6 V/s under the nitrogen-saturated condition (dashed lines indicate the background current). b, the cathodic peak current (negative slope) and the anodic peak current (positive slope) as a function of scan rate from 500 to 1600 mV/s.
Determination of Enzyme Surface Coverage and Electron Transfer Rate. For redox-active molecules immobilized on an electrode surface, the peak current (ip) is proportional to the number of redox-active sites (N) and the number of electrons transferred (n). The values of n and N can be determined by fitting the oxidation or reduction peak current as a function of scan rate (Fig. 3b) (Laviron, 1979). Using eq. 1, n was determined to be 0.72 and indicates that a single-electron-transfer reaction occurs in the immobilized CYP2C9 under anaerobic conditions (Bard and Faulkner, 2001). Furthermore, given n = 0.72, the surface coverage of active CYP2C9 molecules can be calculated and the result was 8.0 × 10–13 mol/cm2 (equivalent to 4.8 × 1011 molecules/cm2). This number is within a factor of 2 of our previous estimate based on atomic force microscopy measurement (Gannett et al., 2006). In addition, n can also be used to calculate the electron transfer rate constant (ks; eq. 2) and is in the range of 6 to 31 s–1 with the scan rates from 0.5 to 1.6 V/s (Laviron, 1979). This range is comparable with the data obtained from cytochrome P450cam absorbed on clay-bridged glassy carbon electrode (Lei et al., 2000).
Cyclic Voltammetry of Immobilized CYP2C9 Enzyme in the Presence of Oxygen. Figure 4 shows the CV data obtained with the CYP2C9-MUA/OT-Au electrode under anaerobic (Fig. 4, curve a) and aerobic conditions (Fig. 4, curve b), respectively. The data used for Fig. 4, curve a, is identical to that shown in Fig. 3a. In the absence of oxygen and warfarin, the catalytic cycle stalls after the reduction of Fe(III) to Fe(II) of CYP2C9, and a reversible CV of CYP2C9 is observed with an apparent formal potential of –0.45 V (eq. 4). Under anaerobic conditions, no variation in redox potential or the observed oxidation/reduction currents were observed in the presence of warfarin (data not shown). In the presence of oxygen and without the warfarin (Fig. 4, curve b), the oxidation peak is no longer observed and the current of the reduction peak, at approximately –0.49 V, increased. Under aerobic conditions, the addition of warfarin leads to an increase in the reduction current at –0.49 V (Fig. 4, curve c). Subsequent experiments revealed that this increase in reduction current was dependent on warfarin concentration.
Fig. 4.

Cyclic voltammograms obtained from the CYP2C9-MUA/OT-Au electrodes in 40 mM PBS solution containing 154 mM NaCl at a scan rate of 1.6 V/s; curve a was obtained in the absence of warfarin under nitrogen-saturated conditions, curve b was obtained in the absence of warfarin under oxygen-saturated conditions, and curve c was obtained in the presence of 0.5 μM warfarin under oxygensaturated conditions.
Warfarin Metabolism Electrocatalyzed by Immobilized CYP2C9. The electrochemical behavior of this system suggests that warfarin was being oxidized by the CYP2C9, possibly to the expected metabolite, 7-hdroxywarfarin. To confirm the electrocatalytical metabolism of warfarin at the CYP2C9-MUA/OT-Au electrode, a constant potential (–0.38 V) electrolysis was performed with the CYP2C9-MUA/OT-Au electrode in a solution containing warfarin (50 μM) and under aerobic conditions. After 2 h of controlled potential electrolysis, the solution was analyzed by high-performance liquid chromatography (HPLC) (Fig. 5, curve b). For comparison, CYP2C9-mediated warfarin metabolism was also performed in a solution containing NADPH, CPR, phospholipids, and CYP2C9. The chromatogram shown in Fig. 5, curve a, was obtained from the incubation of CYP2C9 and warfarin in solution. Comparison of the traces in Fig. 5 shows that both methods led to the formation of 7-hydroxywarfarin. It is noteworthy that the key major metabolites, observed under both conditions, are similar. Metabolite peaks from the solution-based incubations are larger than those obtained from the controlled potential electrolysis. This is probably due to the fact that the amount of CYP2C9 in solution (25 pmol) is considerably greater than that on the Au-SAM-CYP2C9 electrode (0.06 pmol, more than 400 times less than in solution).
Fig. 5.

HPLC traces obtained from CYP2C9-mediated conversion of warfarin to the 7-hydroxy metabolite (inset). Curve a wash obtained from solution-based incubation containing NADPH and CPR, and curve b by a controlled potential electrolysis (–0.38 V) at a Au-CYP2C9 electrode in the absence of NADPH and CPR. The dashed traces, offset above curves a and b, are the controls and were obtained on incubation mixtures that lacked NADPH (above curve a) or no current was applied (above curve b).
Electrochemically Driven CYP2C9-Mediated Enzyme Kinetics of Warfarin. Figure 6 depicts the response of the reduction peak currents as a function of the warfarin concentration under oxygen-saturated conditions. The peak current increased with increasing warfarin concentrations, indicating good electrocatalytic activity of the immobilized CYP2C9. The peak current was linearly proportional to the warfarin concentration from 0 to 5 μM with a regression correlation coefficient (R2) of 0.97. In the linear range, the sensitivity of the current response to concentration of warfarin was estimated to be 64 μA · cm–2 · μM–1.
Fig. 6.

The current of the cathodic peak (at –0.45 V) as a function of warfarin concentration under nitrogen-saturated conditions (open squares) and under oxygensaturated conditions (closed circles). The solid line is the nonlinear regression fit of the oxygen saturation data, and the dashed line is the linear regression line of the nitrogen saturation data (saturation was not observed with the nitrogen saturation data).
The apparent Michaelis-Menten constant (Kmapp) and imax (functionally equivalent to Vmax) were used to evaluate the kinetics of warfarin catalysis by the bonded CYP2C9 enzyme. By using the Michaelis-Menten Equation (eq. 3), the calculated Kmapp is 3 μM for the immobilized CYP2C9 on the electrode. The value for metabolism of warfarin in the present work is in the range estimated in the literature of Km ∼ 4 μM, obtained by the incubation of CYP2C9 and warfarin in the solution containing NADPH (Land and Böcker, 1995).
Discussion
The use of expressed, purified enzymes for both mechanistic and kinetic studies of P450 metabolism is common and can offer some advantages over the use of liver microsomes. At the same time, reconstituted enzyme systems (e.g., in artificial membranes of phospholipid vesicles) are a departure from the native membrane-bound environment and may permit interactions, such as P450 aggregation, that may not normally occur (Backes and Kelley, 2003). It is unfortunate that solution-based experiments do not allow easy control over these interactions and are not readily amenable to the direct study of electron transfer processes. To address the issue of controlling P450 reinsertion into artificial membranes, protein-protein interactions and the resulting effects, nanodiscs, were developed (Bayburt and Sligar, 2002). Nanodiscs have been used to colocate CYP3A4 and CPR and mediate metabolism using NADPH. This approach permits study of isolated P450s but does not, as of this time, permit study of the effect of P450 aggregation (Denisov et al., 2007).
To overcome this issue of P450 catalysis requirements (NADPH/CPR or peroxides), allow for control of protein-protein interactions, and permit simultaneous control of electron transfer, an alternative approach using P450 enzymes immobilized to a gold surface through a SAM has been developed (Gannett et al., 2006). This system is unique in that the bonded P450 (CYP2C9) is able to function with NADPH and CPR as the electron source and can be modified to study P450-P450 aggregation effects. Although this system can be developed for several applications including bioreactors for metabolite synthesis and biosensors for monitoring drug concentrations, the current work explores its use with an electrochemical means source for the potential study of electron transfer processes (Chen et al., 2002) (minus the NADPH/CPR components) and enzyme kinetics.
Previous work in other laboratories has involved P450s bound to a SAM electrostatically or immobilized in a matrix. In the current work, it was observed that bonding of the enzyme to a gold surface was required for electron transfer. The bonding probably occurs via the N terminus of the P450, although there are numerous surface lysine residues distributed over the entire surface of the CYP2C9 (Gannett et al., 2006). Previous work in our laboratory suggests that the surface lysines are not very accessible for bonding and that the N terminus is the most likely site for bonding to occur (Gannett et al., 2006). The redox behavior observed in the current work supports this proposal because it is consistent with homogeneous attachment, which requires each enzyme molecule to be attached in the same manner. If both surface lysines and the terminal residue were involved, very different redox behavior would be expected (Shukla et al., 2005).
It is interesting to note the redox potential observed for the bonded CYP2C9. For P450s in solution, the redox potential generally falls in the range of –0.4 to –0.28 V (Johnson et al., 2005). However, the redox potential typically shifts anodically when the P450 is immobilized. Shifts in redox potential can range as large as 0.4 V. In the present case, the observed redox potential is more similar to P450s in solution than immobilized. This suggests that the CYP2C9 is behaving as though it was free in solution and oriented as shown in Fig. 7. In addition, and by analogy to cytochrome c, the electron transfer rate measured (in the range of 6–31 s–1) is more consistent with this orientation than if CYP2C9 were laying on the SAM (Yue et al., 2008).
Fig. 7.

Model of CYP2C9 bonded to the Gold-SAM electrode. The porphyrin system is shown as sticks, and the iron atom is depicted as a sphere. The gold surface/SAM to iron atom separation is shown.
As shown in Fig. 8, when electrons transfer between the electrode and heme iron, reduction of the heme iron occurs (step 2) and the P450 enters the catalytic cycle. In the absence of oxygen, only reduction from Fe(III) to Fe(II), as well as reoxidation (Fe(II)→Fe(III)), should be possible (Fig. 8, step 2 and the reverse). In the presence of oxygen, reduction to Fe(II) will result in oxygen binding (Fig. 8, step 3). If no substrate is present, one of the three possible shunt processes (Fig. 8, step 3a, 4a, and/or 5a) will occur. Under these circumstances, the iron is returned to the Fe(III) state and an oxidation current will not be detected. Because some shunt processes require more than one electron (Fig. 8, step 4a or 5a), the reduction current should increase relative to the CV data obtained in the absence of oxygen (Fig. 4, curve b). When both oxygen and a suitable substrate are present, metabolite production should be observed and electron consumption should further increase (Fig. 4, curve b) because both shunt (Fig. 8, steps 3a–5a) and metabolite formation (Fig. 8, steps 1–7) can now occur. The experimental observations are in complete agreement with these predictions, suggesting that the CYP2C9-MUA/OT-Au system and, with electrochemical provision of electrons, the CYP2C9-MUA/OT-Au electrode behave in a way that is similar to CYP2C9 in solution (metabolism with CPR and NADPH).
Fig. 8.

The P450 catalytic cycle. Steps 1 to 7 refer to normal P450-mediated metabolism of a substrate (RH) resulting in metabolite (ROH) production. Steps 3a to 5a are shunt processes resulting in the production of superoxide anion, hydrogen peroxide, or water, respectively.
It is notable that the controlled potential electrolysis of warfarin resulted in formation of the expected 7-hydroxywarfain metabolite. The amount of enzyme on the surface of the electrode was far less than is typically used for solution-based incubations; in the present case, approximately 0.06 pmol of enzyme was bonded to the surface. In contrast, the comparator solution incubations contained 25 pmol of enzyme, more than 400 times the amount on the gold-SAM electrode. The HPLC profiles of the solution and electrochemical incubation mixtures were not identical, and additional species are observed in the electrochemical incubations. Nevertheless, 7-hydroxywarfarin and a second metabolite with a slightly longer retention time are observed under either set of incubation conditions. There are many possibilities (e.g., salt concentration) that may be responsible for these differences, and elucidating the reason for these differences will be addressed in future work. Finally, after accounting for the relative amounts of CYP2C9 present and difference in incubation times, production of the 7-hydroxywarfarin metabolite in the electrochemical system is estimated, based on the amount of CYP2C9 and incubation time, to be 35 times greater than the solution system.
A final test of this electrochemical-CYP2C9-mediated enzyme metabolism and how well it mimics normal solution-based systems is the measurement of the kinetics of warfarin metabolism. We expect some difference in enzyme kinetic parameters, if for no other reason than a difference in the state of enzyme aggregation. Nevertheless, we find in our system a Kmapp of 3 μM, a value that is in excellent agreement with the literature value of Kmapp of ∼4 μM (Land and Böcker, 1995). Analogous results were reported for CYP2E1 bonded directly to a gold electrode using surface cysteines, but it was not reported whether the CYP2E1 could metabolize the substrate using NADPH/CPR as the electron source (Fantuzzi et al., 2004).
In theory, bonding to the SAM may provide a pathway for electron passage from the outer surface and through the enzyme. Modeling of the enzyme bonded to the SAM via the N terminus suggests that the heme group of CYP2C9 is oriented in a fashion similar to bound cytochrome c (Yue et al., 2008), and, in the latter case, bonding is not required for electron transfer (Fig. 7). Therefore, if electron transfer is solely dependent upon the heme orientation, bonding may only serve to constrain the CYP2C9 to the proper orientation or keep the heme within a distance that is amenable to electron transfer with a gold-heme separation of approximately 5.3 nm (Fig. 7). Alternatively, the flexible nature of the linkage between the CYP2C9 and the SAM may permit the CYP2C9 to lay on the surface of the SAM. At physiological pH, the surface lysines would be protonated and the SAM carboxylic acid groups deprotonated, hence favoring an electrostatic interaction for this arrangement. If this brought the CPR binding site into contact with the SAM surface, it would reduce the gold-heme distance to approximately 3 nm and may increase the rate of electron transfer if distance is the sole determinant. However, if orientation of the heme relative to gold is of significant importance, it may inhibit or block electron transfer.
In summary, direct electrochemistry of substrate (warfarin) was observed when CYP2C9 was bonded to a gold electrode through a self-assembled monolayer in the presence of oxygen. It is noteworthy that whereas the endogenous electron donor NADPH/CPR can serve as the source of electrons, the system also functions with the electrochemical provision of electrons. Metabolite production and kinetic measurements were consistent with solution-based incubations. Although purely speculative, the observation that electrons can transfer to the P450 from outside the protein from the gold surface suggests the possibility of multiple pathways (i.e., in addition to the putative CPR binding site) for electrons to move through the P450 and reduce the heme iron. The ability to control electron transfer, independent of P450 reductase, provides opportunities to potentially assist in identification of P450 cycle intermediates by controlling the progress of the P450 cycle, to evaluate the impact of P450 reductase binding on P450 conformation and cycle kinetics, and begin to address issues related to the contributions of cytochrome b5 to P450 conformation and electron transfer.
Acknowledgments
The primary laboratory of origin was that of Professor Nianqiang Wu.
This work was supported in part by the National Institutes of Health National Institute of General Medical Sciences [Grant GM063215]; the National Science Foundation Research Infrastructure Improvement [Grant EPS0554328]; the West Virginia University Research Corporation; and the West Virginia Experimental Program to Stimulate Competitive Research.
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.108.025452.
ABBREVIATIONS: P450, cytochrome P450; CPR, cytochrome P450 reductase; SAM, self-assembled monolayer; MUA, 11-mercaptoundecanoic acid; OT, 1-octanethiol; EDC, N-((3-dimethylamino)-propyl)-N-ethyl carbodiimide hydrochloride; NHS, N-hydroxysulfosuccinimide; PBS, phosphate-buffered solution; XPS, X-ray photoelectron spectroscopy; C 1s, carbon 1s electron; CV, cyclic voltammetry; LC, liquid chromatography; S 2p, sulfur 2p electron; N 1s, nitrogen 1s electron; HPLC, high-performance liquid chromatography.
References
- Backes WL and Kelley RW (2003) Organization of multiple cytochrome P450s with NADPH-cytochrome P450 reductase in membranes. Pharmacol Ther 98 221–233. [DOI] [PubMed] [Google Scholar]
- Bard AJ and Faulkner LR (2001) Electrochemical Methods, Fundamentals, and Applications. Wiley, New York.
- Bayburt TH and Sligar SG (2002) Single-molecule height measurements on microsomal cytochrome P450 in nanometer-scale phospholipid bilayer disks. Proc Natl Acad Sci U S A 99 6725–6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Ferrigno R, Yang J, and Whitesides GM (2002) Redox properties of cytochrome c absorbed on self-assembled monolayers: a probe for protein conformation and orientation. Langmuir 18 7009–7015. [Google Scholar]
- Collinson M and Bowden EF (1992) Voltammetry of covalently immobilized cytochrome c on self-assembled monolayer electrodes. Langmuir 8 1247–1250. [Google Scholar]
- Denisov IG, Baas BJ, Grinkova YV, and Sligar SG (2007) Cooperativity in cytochrome P450 3A4: linkages in substrate binding, spin-state, uncoupling, and product formation. J Biol Chem 282 7066–7076. [DOI] [PubMed] [Google Scholar]
- Diao P, Jiang D, Cui X, Gu D, Tong R, and Zhong B (1999) Studies of structural disorder of self-assembled thiol monolayers on gold by cyclic voltammetry and AC impedance. J Electroanal Chem 464 61–67. [Google Scholar]
- Estabrook RW, Faulkner KM, Shet MS, and Fisher CW (1996) Application of electrochemistry for P450-catalyzed reactions. Meth Enzymol 272 44–51. [DOI] [PubMed] [Google Scholar]
- Fantuzzi A, Fairhead M, and Gilardi G (2004) Direct electrochemistry of immobilized human cytochrome P450 2E1. J Am Chem Soc 126 5040–5041. [DOI] [PubMed] [Google Scholar]
- Fantuzzi A, Meharenna YT, Briscoe PB, Sassone C, Borgia B, and Gilardi G (2006) Improving catalytic properties of P450 BM3 haem domain electrodes by molecular Lego. Chem Commun (Camb) 1289–1291. [DOI] [PubMed]
- Funk T, Deb A, George SJ, Wang H, and Cramer SP (2005) X-Ray magnetic circular dichroism–a high energy probe of magnetic properties. Coord Chem Rev 249 3–30. [Google Scholar]
- Gannett PM, Kabulski J, Perez FA, Liu Z, Lederman D, Locuson CW, Ayscue RR, Thomsen NM, and Tracy TS (2006) Preparation, characterization, and substrate metabolism of gold-immobilized cytochrome P450 2C9. J Am Chem Soc 128 8374–8375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilardi G and Fantuzzi A (2001) Manipulating redox systems: application to nanotechnology. Trends Biotechnol 19 468–476. [DOI] [PubMed] [Google Scholar]
- Gilardi G, Fantuzzi A, and Sadeghi SJ (2001) Engineering and design in the bioelectrochemistry of metalloproteins. Curr Opin Struct Biol 11 491–499. [DOI] [PubMed] [Google Scholar]
- Gilardi G, Meharenna YT, Tsotsou GE, Sadeghi SJ, Fairhead M, and Giannini S (2002) Molecular Lego: design of molecular assemblies of P450 enzymes for nanobiotechnology. Biosens Bioelectron 17 133–145. [DOI] [PubMed] [Google Scholar]
- Guengerich FP (2007) Mechanisms of cytochrome P450 substrate oxidation: MiniReview. J Biochem Mol Toxicol 21 163–168. [DOI] [PubMed] [Google Scholar]
- Habermüller K, Mosbach M, and Schuhmann W (2000) Electron-transfer mechanisms in amperometric biosensors. Fresenius J Anal Chem 366 560–568. [DOI] [PubMed] [Google Scholar]
- Hahm J-I and Lieber CM (2003) Direct ultrasensitive electrical detection of DNA and DNA sequence variations using nanowire nanosensors. Nano Letters 4 51–54. [Google Scholar]
- Johnson DL, Lewis BC, Elliot DJ, Miners JO, and Martin LL (2005) Electrochemical characterisation of the human cytochrome P450 CYP2C9. Biochem Pharmacol 69 1533–1541. [DOI] [PubMed] [Google Scholar]
- Joseph S, Rusling JF, Lvov YM, Friedberg T, and Fuhr U (2003) An amperometric biosensor with human CYP3A4 as a novel drug screening tool. Biochem Pharmacol 65 1817–1826. [DOI] [PubMed] [Google Scholar]
- Land D and Böcker R (1995) Highly sensitive and specific high-performance liquid chromatographic analysis of 7-hydroxywarfarin, a marker for human cytochrome P4502C9 activity. J Chromatogr B Biomed Appl 672 305–309. [DOI] [PubMed] [Google Scholar]
- Laviron E (1979) The use of linear potential sweep voltammetry and of AC voltammetry for the study of the surface electrochemical reaction of strong adsorbed systems and of redox modified electrodes. J Electroanal Chem 100 263–270. [Google Scholar]
- Lei C, Wollenberger U, Jung C, and Scheller FW (2000) Clay-bridged electron transfer between cytochrome P450cam and electrode. Biochem Biophys Res Commun 268 740–744. [DOI] [PubMed] [Google Scholar]
- Lim MS, Feng K, Chen X, Wu N, Raman A, Nightingale J, Gawalt ES, Korakakis D, Hornak LA, and Timperman AT (2007) Adsorption and desorption of stearic acid self-assembled monolayer on aluminum oxide. Langmuir 23 2444–2453. [DOI] [PubMed] [Google Scholar]
- Masuda H and Fukuda K (1995) Ordered metal nanohole arrays made by a two-step replication of honeycomb structures of anodic alumina. Science 268 1466–1468. [DOI] [PubMed] [Google Scholar]
- Masuda H, Yasui K, and Nishio K (2000) Fabrication of ordered arrays of multiple nanodots using anodic porous alumina as an evaporation mask. Adv Materials 12 1031–1033. [Google Scholar]
- Myland JC and Oldham KB (2005) Quasireversible cyclic voltammetry of a surface confined redox system: a mathematical treatment. Electrochem Commun 7 282–287. [Google Scholar]
- Nuzzo RG, Zegarski BR, and Dubois LH (1987) Fundamental Studies of the chemisorption of organosulfur compounds on Au(111). Implications for molecular self-assembly on gold surfaces. J Am Chem Soc 109 733–740. [Google Scholar]
- Ohldag H, Shi H, Arenholz E, Stöhr J, and Lederman D (2006) Parallel versus antiparallel interfacial coupling in exchange biased Co/FeF2. Phys Rev Lett 96 027203. [DOI] [PubMed] [Google Scholar]
- Pardo-Yissar V, Katz E, Willner I, Kotlyar AB, Sanders C, and Lill H (2000) Biomaterial engineered electrodes for bioelectronics. Faraday Discuss 119–134; discussion 171–190. [DOI] [PubMed]
- Reipa V, Mayhew MP, and Vilker VL (1997) A direct electrode-driven P450 cycle for biocatalysis. Proc Natl Acad Sci U S A 94 13554–13558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla A, Gillam EM, Mitchell DJ, and Bernhardt PV (2005) Direct electrochemistry of enzymes from the cytochrome P450 2C family. Electrochem Commun 7 437–442. [Google Scholar]
- Shumyantseva VV, Carrara S, Bavastrello V, Jason Riley D, Bulko TV, Skryabin KG, Archakov AI, and Nicolini C (2005) Direct electron transfer between cytochrome P450scc and gold nanoparticles on screen-printed rhodium–graphite electrodes. Biosens Bioelectron 21 217–222. [DOI] [PubMed] [Google Scholar]
- Shumyantseva VV, Ivanov YD, Bistolas N, Scheller FW, Archakov AI, and Wollenberger U (2004) Direct electron transfer of cytochrome P450 2B4 at electrodes modified with nonionic detergent and colloidal clay nanoparticles. Anal Chem 76 6046–6052. [DOI] [PubMed] [Google Scholar]
- Song S, Clark R, and Bowden EF (1993) Characterization of cytochrome-C alkanethiolate structures prepared by self-assembly on gold. J Phys Chem 97 6564–6572. [Google Scholar]
- Wester MR, Yano JK, Schoch GA, Yang C, Griffin KJ, Stout CD, and Johnson EF (2004) The structure of human cytochrome P450 2C9 complexed with flurbiprofen at 2.0 A resolution. J Biol Chem 279 35630–35637. [DOI] [PubMed] [Google Scholar]
- Williams JA, Hyland R, Jones BC, Smith DA, Hurst S, Goosen TC, Peterkin V, Koup JR, and Ball SE (2004) Drug-drug interactions for UDP-glucuronosyltransferase substrates: a pharmacokinetic explanation for typically observed low exposure (AUC1/AUC) ratios. Drug Metab Dispos 32 1201–1208. [DOI] [PubMed] [Google Scholar]
- Wu NQ, Fu L, Su M, Aslam M, Aslam M, Wong KC, and Dravid VP (2004) Interaction of fatty acid monolayers with cobalt nanoparticles. Nano Letters 4 383–386. [Google Scholar]
- Yang M, Wang J, Li H, Zheng J-G, and Wu NQ (2008) A lactate electrochemical biosensor with a titanate nanotube as direct electron transfer promoter. Nanotechnology 19 075502. [DOI] [PubMed] [Google Scholar]
- Yue H, Waldeck DH, Schrock K, Kirby D, Knorr K, Switzer S, Rosmus J, and Clark RA (2008) Multiple sites for electron transfer tunneling between cytochrome c and mixed self-assembled monolayers. J Phys Chem C 112 2514–2521. [Google Scholar]