

Abstract
Mechanical stress is known to modulate postnatal skeletal growth and development. However, the mechanisms underlying the mechanotransduction are not fully understood. Polycystin-1 (PC1) is a promising candidate among proteins that may play a role in the process as it has been shown to function as a flow sensor in renal epithelium and it is known to be important for skeletal development. To investigate whether PC1 is involved in mechanotransduction in skeletal tissues, mice with a conditional deficiency for PC1 in neural crest cells, osteoblasts or chondrocytes were subjected to midpalatal suture expansion. Dynamic bone labeling revealed that new bone formation in response to expansion was significantly reduced in Wnt1Cre;Pkd1 mice, as the suture area containing new bone was 14.0±3.4% in mutant mice versus 65.0±3.8% in control mice at 2 weeks (p<0.001). In contrast, stress-induced new bone formation was not affected in OsxCre;Pkd1 mice. The increase in cell proliferation and differentiation into osteoblasts, seen in wild-type mice 1 day after force delivery, was not observed until 14 days in Wnt1Cre;Pkd1 mice. TUNEL labeling showed a significant increase in apoptotic suture cells at days 1 and 3 (from 7.0±0.5% to 13.5±1.4% at day 1 and from 4.6±1.1% to 10.5±1.7% at day 3, p<0.05). Abnormal ossification of nasal cartilage of Wnt1Cre;Pkd1 mice was accelerated upon suture expansion. Such ossification was also observed, but to a lesser extent in Col2a1-ER*Cre;Pkd1 mice. Transcript levels of Runx2 and MMP13 were significantly increased in the nasal cartilage of Wnt1Cre;Pkd1 mice compared to controls (p<0.05 and p<0.001, respectively), and in mutant mice with expansion versus without expansion (p<0.05 and p<0.001, respectively). Lack of PC1 in chondroprogenitor cells also resulted in increased cell apoptosis and an altered arrangement of chondrocytes in nasal cartilage. These results indicate that PC1 plays a critical role in the response of osteochondroprogenitor cells to the mechanical tissue stress induced by midpalatal suture expansion. They also suggest that the combination of an in vivo mechanical model, such as midpalatal suture expansion, with conditional deficiency for proteins that play a role in mechanotransduction, represents a powerful experimental strategy to explore underlying mechanisms.
Keywords: suture, mechanical force, polycystin 1, osteoblast, chondrocyte
Introduction
It is well known that postnatal skeletal development and growth are modulated by mechanical loading. Regular exercise and physical loading have been shown to increase bone mineral density while disuse or low gravity will lead to osteoporosis [1-3]. The ability of skeletal tissue to respond to mechanical stress has also been used in orthopedic and orthodontic treatment to induce bone modeling/remodeling, eg. distraction osteogenesis, orthodontic tooth movement and midpalatal suture expansion. Understanding the cellular mechanisms by which skeletal tissues sense and respond to mechanical stress would greatly increase our knowledge of skeletal biology and help to improve clinical practice in orthopedics and orthodontics.
It has long been recognized that osteoblasts, osteocytes and chondrocytes can all respond to mechanical stress, and their interaction is essential in generating tissue responses. For instance, application of fluid flow to osteoblasts in vitro inhibits cell apoptosis via induction of insulin-like growth factor-I (IGF-I) signaling [4]. In addition, osteoblasts subjected to intermittent cell stretching may upregulate the expression of receptor activator of nuclear factor-kappaB ligand (RANKL), which could influence osteoclast recruitment [5]. For osteocytes, it has been demonstrated that fluid shear stress promotes their survival, while disuse promotes apoptosis [6]. Moreover, osteocytes subjected to oscillatory fluid flow may inhibit osteoclastogenesis via release of soluble factors [7]. Although in vitro mechanical models provide relatively simple systems to study changes in gene expression and cell behavior upon mechanical stimulation, it is impossible to recapitulate the in vivo biomechanical environment and study tissue responses in such models. Therefore, any model of mechanical modulation of the skeleton must ultimately be tested in vivo, as only the physiological environment in vivo would allow conclusions about the functional adaptation of bone.
Previously, we developed a mouse midpalatal suture expansion model to study the biological responses of midpalatal suture cells to mechanical loading in vivo [8]. We have shown that an expansive force across the suture will induce new bone formation in the suture within an experimental period of two weeks. Further analysis has demonstrated that increased periosteal cell proliferation and differentiation into osteoblasts, starting as early as day 1, are the major contributions to new bone formation observed later. Thus, this model provides a unique opportunity to study mechanical modulation of bone from the early cellular changes to the later process of new bone formation. In addition, the model makes it possible to test candidate genes that play a role in mechanotransduction in vivo by using genetically modified mouse strains.
Polycystin-1 (PC1) is a promising candidate for mechanotransduction as it spans the cell membrane and connects extracelluar matrix components with cytoskeleton and intracellular signaling pathways [9-13]. Recently, it has been shown that PC1 functions as a fluid flow sensor in renal epithelium and that dysfunction of PC1 leads to the abnormal tissue morphogenesis observed in polycystic kidney disease [14].
In addition to its role in kidney development and morphogenesis, PC1 may participate in the growth and development of the skeleton. Pkd1 transcripts were detected in neural crest derivatives and prechondrogenic tissue, as well as in MC3T3-E1 osteoblasts and MLO-Y4 osteocytes [15,16]. Furthermore, skeletal abnormalities have been reported in several Pkd1-deficient animals [17, 18]. However, it is unclear whether PC1 plays a role in skeletal tissue responses to mechanical stress in vivo. To address this question, midpalatal suture expansion was studied in PC1-deficient mice. In order to elucidate the function of PC1 in different cell types and at different differentiation stages, PC1 was inactivated in neural crest cells, osteoblasts and chondrocytes by crossing mice with floxed alleles of Pkd1with Wnt1Cre, OsxCre and Col2a1-Cre ER* deleter strains, respectively. Our data suggest that PC1 plays a significant role in the responses of osteochondroprogenitor cells to mechanical stress induced by midpalatal suture expansion.
Materials and Methods
Animals
Mice carrying floxed alleles of the Pkd1 gene were reported previously [19]. Wnt1-Cre and Osx-Cre deleter strains were kindly provided by Dr. Andy McMahon. Standard PCR genotyping was performed using primers specific for the Cre recombinase coding sequence: Cre5’ (TGC TCT GTC CGT TTG CCG) and Cre3’ (ACT GTG TCC AGA CCA GGC). Col2a1-Cre ER* transgenic mice were described previously [20]. To induce Cre recombinase activity, 10-day-old mice were injected with tamoxifen (1mg/10g) (Sigma-Aldrich, St. Louis, MO) in corn oil intraperitoneally every other day for a total of 10 times. Mice injected with corn oil alone served as control. All procedures performed in the experiments were approved by the Institutional Animal Care and Use Committee of Harvard Medical School where the animals were raised and studied.
Expansion of midpalatal suture
Six-week-old male mice from each genotype (Cre positive floxed Pkd1 homozygotes, Cre positive floxed Pkd1 heterozygotes, and Cre negative floxed Pkd1 homozygotes) were subjected to midpalatal suture expansion for 1, 3, 7, and 14 days. After anesthesia with intraperitoneal injection of ketamine (87mg/kg) and xylazine (13mg/kg), the mice were subjected to midpalatal suture expansion according to the method of our previous study [8]. Briefly, an opening loop made of 0.014-in. stainless steel orthodontic wire (GAC International Inc., Bohemia, NY) was bonded to first and second maxillary molars on both sides using a light cured adhesive (3M Unitek, Monrovia, CA). The initial force magnitude used in the experiments was 0.56N. Since we have shown that sham operation has no effect on the midpalatal suture [8], mice without operation were used as non-expansion control in this study. In addition, since no overall difference was observed between Cre positive floxed Pkd1 heterozygous mice and Cre negative floxed Pkd1 homozygous mice, they were grouped together as wild-type controls. In general, at least 3 mice were used for each genotype, time point and sample processing method including Osteo-Bed embedding, paraffin embedding, OCT embedding and RNA extraction.
Fluorescence labeling and sample processing
Fluorescence labeling was performed according to the procedures described previously [8]. Briefly, mice were injected with alizarin complexone (60mg/kg) on the day the opening loop was applied and calcein (6mg/kg) on day 14. Control mice were injected with the fluorescent dyes at the same times. Mice were sacrificed 2 days after calcein injection, and the maxillae were dissected for analysis. After fixation with 4% (w/v) paraformaldehyde and dehydration with a series of graded alcohols, samples were embedded in Osteo-Bed resin (Polysciences, Inc., Warrington, PA) following manufacturer’s instruction. Frontal sections of 140μm were cut using a low speed saw (Buehler, Lake Bluff, IL) and mounted on slides using EUKITT mounting medium (Electron Microscopy Sciences, Hatfield, PA). The sections were examined using a fluorescence microscope (80i Upright Microscope, Nikon, Japan). Images were obtained and analyzed using NIH image (developed at the U.S. National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nih-image/). The distance between the outlines of alizarin complexone labels was measured at the midpoint of oral third, middle third and nasal third, and the average of the measurements was calculated as suture width. The distance between the borders of the palatal bone on the nasal and oral side was measured at the midpoint of left third, middle third and right third, and the average of the measurements was calculated as suture thickness. New bone formation was measured as the area of bone labeled with calcein in the suture, and total suture area was measured as the area between the outlines of alizarin complexone labels, a method modified from the one described by Cowan et al. [21].
Histology, immunohistochemistry, in situ hybridization and TUNEL
The maxillae including the midpalatal suture were dissected out from all the control and experimental mice. Samples were fixed in 4% (w/v) paraformaldehyde overnight and demineralized in 0.5M ethylenediaminetetra-acetic acid (EDTA) for 14 days at 4°C before they were embedded in paraffin. Serial frontal sections between first and second molar levels were cut at 5 μm for subsequent analysis.
For morphological observation, paraffin-embedded sections were stained with hematoxylin and eosin and then microphotographs were taken. Suture width was measured on these microphotographs using Adobe Photoshop. For each suture, the distances between the edges of palatal bones were measured at the midpoint of the oral third, mid third and nasal third. Suture width was calculated for each sample as the average of the measurements and subjected to statistical analysis. The expression of Ki67, MMP13 and cleaved collagen type II was examined by immunohistochemistry using anti-mouse Ki67 monoclonal antibody (clone TEC-3) (DakoCytomation, Glostrup, Denmark), anti-MMP13 polyclonal antibody (Chemicon International, Inc., Temecula, CA) and Col2 3/4C short polyclonal antibody (IBEX Pharmaceuticals Inc., Quebec, Canada) according to the manufacturer’s recommendation. To count the Ki67-positive cells, a rectangle was drawn to include the midpalatal suture within the edges of the palatal bones and the periosteum on the oral and nasal sides. Ki67-positive cells and total cells in the rectangle were counted manually on all sections. The percentage of Ki67-positive cells were calculated for each sample and subjected to statistical analysis. Using digoxigenin-11-UTP-labeled murine Col1a1 antisense probe, fluorescent in situ hybridization with tyramide signal amplification was performed as described previously [8].
Apoptotic cells were detected using DeadEnd™ Fluorometric TUNEL system (Promega Corporation, Madison, WI) following manufacturer’s instruction. In the midpalatal suture, TUNEL-positive cells and total cells in the suture were counted on all sections. The percentage of TUNEL-positive cells were calculated for each sample and subjected to statistical analysis. In the nasal cartilage, the number of TUNEL-positive chondrocytes per section was counted, and chondrocytes were distinguished from surrounding perichondrium and cells in bone marrow based on morphology.
β-galactosidase staining
To visualize the Cre efficiency, we crossed Wnt1Cre, OsxCre and Col2a1-CreER* mice with the conditional LacZ reporter mouse strain Rosa 26R, in which β-galactosidase expression is activated upon Cre activation [22]. Wnt1Cre;R26R and OsxCre;R26R mice were euthanized at postnatal day 2 and the maxillae including the midpalatal suture were dissected out.Col2a1-CreER*;R26R mice were injected with tamoxifen (1mg/10g) or corn oil using the same procedure as described for Col2a1-Cre;ER*Pkd1 mice. Mice were euthanized at 6 weeks and the maxillae including the midpalatal suture were dissected out. Samples were fixed in 4% (w/v) paraformaldehyde overnight and demineralized in 0.5M ethylenediaminetetra-acetic acid (EDTA) for 14 days at 4°C before they were embedded in Tissue-Tek OCT compound (Miles Laboratories, Naperville, USA). Serial frontal sections between first and second molar levels were cut at 8 μm. Detection of β-galactosidase activity was accomplished by incubation in 1% X-gal for several hours at 37°C.
Real-time quantitative PCR
Nasal septal cartilages from non-expansion mice and mice subjected to midpalatal suture expansion for 3 days were dissected and total RNA was rapidly isolated from cartilage using TRIzol reagent (Invitrogen, Carlsbad, CA) according to manufacturer’s instruction. cDNA was then synthesized from total RNA using iScript cDNA Synthesis Kit (BioRad, Hercules, CA) and subjected to real-time quantitative PCR for Runx2, VEGFA, MMP13 and GAPDH. Primer sets were purchased from Superarray Biosciences (Frederick, MD) and were prevalidated to generate single amplicons. PCR conditions were: 35 cycles of denaturation for 30s at 95°C, annealing for 30s at 55°C, extension for 30s at 72°C and was performed using iCycler (BioRad).
Statistical analysis
Results were presented as mean±SD. Cell count differences were compared using the unpaired Student’s t-test. Real-time quantitative PCR results were analyzed using Mann Whitney’s U test. Differences were considered significant at p<0.05.
Results
Deficient new bone formation in response to expansive force in Wnt1Cre;Pkd1 mice
It has been demonstrated that an expansive force across the midpalatal suture results in new bone formation within a 2-week period as shown by dynamic bone labeling [8]. As expected, control mice in this study subjected to expansive force for 2 weeks had significant amounts of calcein labeled newly formed bone in the expanded suture area. The thickness and width of the midpalatal suture in Wnt1Cre;Pkd1 mice were comparable to those of the control mice (Suppl. fig. 1). However, there was significantly less new bone formation in the mutant mice subjected to expansive force during the same experimental period. The suture area containing new bone was 14.0±3.4% in Wnt1Cre;Pkd1 mice versus 65.0±3.8% in control mice at day 14 (p<0.001).
To determine whether Pkd1 is required for new bone formation in response to midpalatal suture expansion at a later differentiated osteoblast stage, expansive force across the midpalatal suture was applied to OsxCre;Pkd1 mice with a conditional deficiency for PC1 in osteoblasts. Although the thicknesses of the suture and palatal bones of these OsxCre;Pkd1 mice are about 2/3 of those of controls (Suppl. fig. 1) (the thicknesses of the suture and palatal bones of OsxCre positive floxed Pkd1 heterozygotes are comparable to those of controls), new bone formation was observed in OsxCre;Pkd1 mice, and there was no significant difference of the amount of new bone formation compared to controls as indicated by the percent suture area containing new bone (63.7±6.3% vs. 65.0±3.8%, p>0.05) (Fig. 1). The efficacy of our gene inactivation by Wnt1Cre and OsxCre deleter strains was confirmed by examining the expression of β-galactosidase in the midpalatal suture of Wnt1Cre;R26R and OsxCre;R26R mice (Suppl. fig.2). Wnt1Cre was expressed by multiple cell lineages except epithelial cells. While the expression pattern of OsxCre is more restricted than that of Wnt1Cre, it was expressed by periosteal cells, osteocytes and some chondrocytes.
Figure 1.
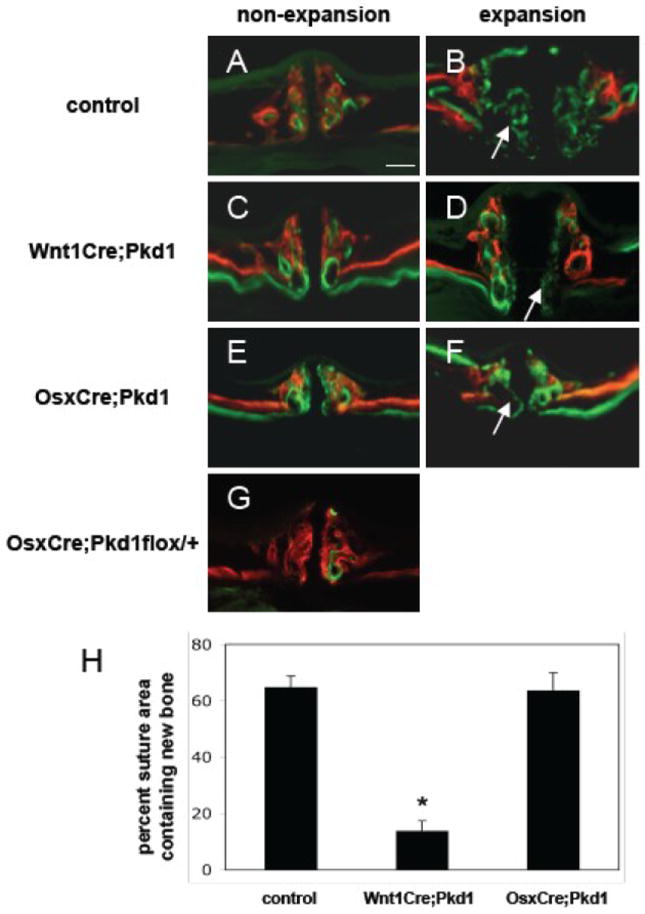
Deficient new bone formation in response to expansive force in Wnt1Cre;Pkd1 mice. All mice were injected with alizarin complexone (red) and calcein (green) 15 days and 1 day before euthanasia, respectively. Frontal sections of maxillae of control (A and B), Wnt1Cre;Pkd1 (C and D), and OsxCre;Pkd1 (E and F) mice with (B, D and F) or without (A, C and E) application of the opening loops. (G) The thickness of midpalatal suture and palatal bones of OsxCre;Pkd1flox/+ mice is comparable to that of controls. (H) Comparative analysis of percent suture area containing new bone in control group, Wnt1Cre;Pkd1 group and OsxCre;Pkd1 group at day 14 (*p<0.001). (B, D and F) Arrows point to new bone formed in the expanded suture area during the experiment period of 2 weeks. Scale bar (A): 100μm.
Changes in suture morphology in Wnt1Cre;Pkd1 mice in response to expansive force
To study the biological processes underlying the deficient new bone formation of the midpalatal suture of Wnt1Cre;Pkd1 mice in response to midpalatal suture expansion, histological analysis was performed on control mice and Wnt1Cre;Pkd1 mice subjected to the procedure for 1, 3, 7 and 14 days.
Histologically, the midpalatal suture of non-expansion mice in the control group had two cartilaginous masses separated by a layer of fibrous tissue. The border of the suture was covered by a continuation of the fibrous and cellular periosteum around the margins of adjacent bones (Fig. 2A). Briefly, the midpalatal suture was expanded and the width of the suture continued to increase during the experimental period. At day 14, the suture was filled with cellular fibrous tissue, and newly formed bone was evident at both the oral and nasal margins of the suture (Fig. 2B-E).
Figure 2.
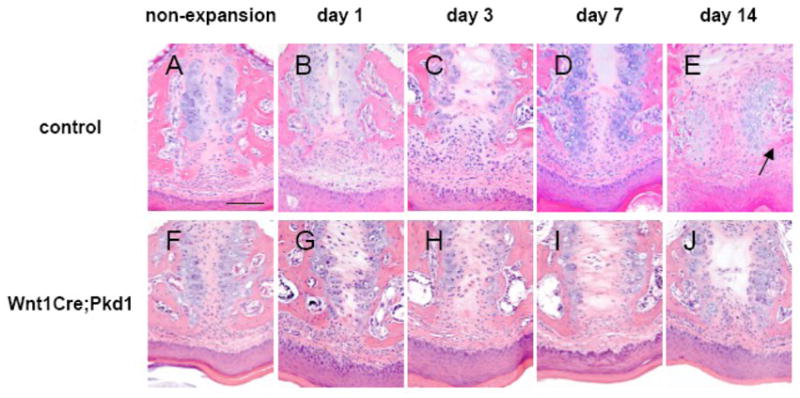
Effects of midpalatal suture expansion on control and Wnt1Cre;Pkd1 mice. Hematoxylin and eosin staining of frontal sections of midpalatal sutures of control (A-E) and Wnt1Cre;Pkd1 (F-J) animals without expansion (A and F) and with expansion at days 1 (B and G), 3 (C and H), 7 (D and I) and 14 (E and J). (E) Arrow points to the area of new bone formation at the edges of palatal bones. Scale bar (A): 100μm.
The midpalatal suture of non-expansion mice in the Wnt1Cre;Pkd1 group had a similar structure to that of the control group (Fig. 2F). After the placement of the opening loops, the midpalatal suture was expanded and the two masses of cartilage were laterally displaced at day 1. The suture continued to be expanded for the following days until day 7. However, new bone formation was not evident at the end of the 2-week experimental period by hematoxylin and eosin staining (Fig. 2G-J).
As there is a linear relationship between the stress and the strain within a range of extension, widths of the sutures of control and Wnt1Cre;Pkd1 mice were measured to assess the mechanical stress applied to the suture. At day 0, there was no significant difference in suture width between control and Wnt1Cre;Pkd1 mice. The suture width was increased significantly in both groups one day after the placement of the opening loop, while there was no significant difference in suture width between Wnt1Cre;Pkd1 and control mice. The suture width reached the maximum at day 14 for both control and Wnt1Cre;Pkd1 mice, however, the suture width of Wnt1Cre;Pkd1 mice was significantly narrower compared to the suture width of control mice (p>0.05) (Suppl. fig. 2).
Defective periosteal cell proliferation and differentiation in Wnt1Cre;Pkd1 mice in response to expansive force
To understand the mechanisms underlying the significantly reduced bone formation observed in Wnt1Cre;Pkd1 mice following the placement of the opening loop, proliferation and differentiation of periosteal cells was evaluated using Ki67 immunohistochemistry and Col1a1 in situ hybridization. In the control group, cell proliferation rate was low in non-expansion mice with Ki67 positive cells accounting for 2.5±0.9% of total suture cells. The proliferation rates were significantly increased following the placement of the opening loop compared to the non-expansion mice, ranging from 11.1±2.7% to 17.9±2.4% (p<0.05 for all). The Ki67 positive cells were mainly observed in the region of periosteal cells at days 1 and 3, and scattered throughout the suture at days 7 and 14. In the Wnt1Cre;Pkd1 group, the number of Ki67 positive cells represented 1.3±0.6% of total suture cells in non-expansion mice and from 1.5±0.5% to 2.3±0.3% in expansion mice. Although there was no significant difference between expansion and non-expansion mice, Ki67 positive cells were detected in the region of periosteal cells at day 14 (Fig. 3 and data not shown).
Figure 3.
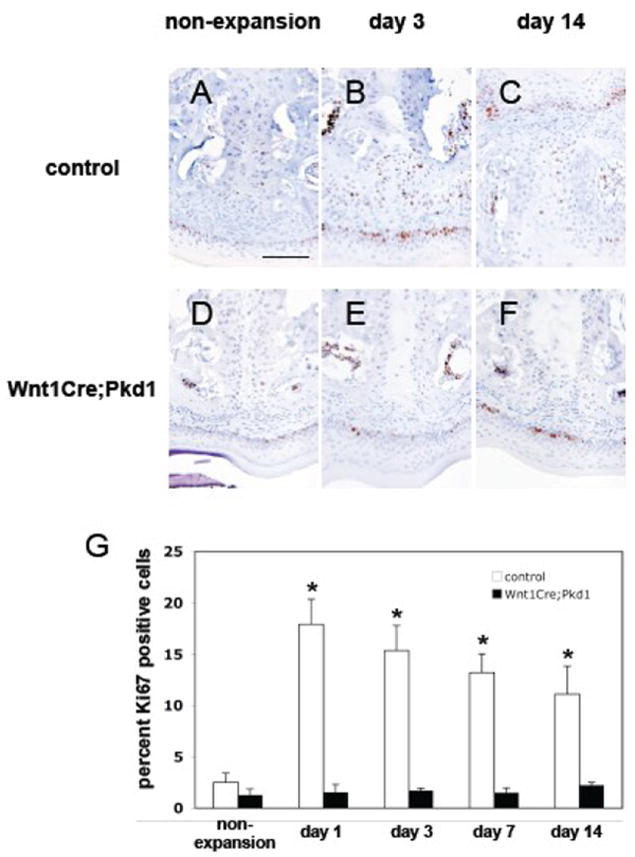
Deficient periosteal cell proliferation in Wnt1Cre;Pkd1 mice in response to expansive force. Ki67 immunohistochemical staining of frontal sections of midpalatal sutures of control (A-C) and Wnt1Cre;Pkd1 (D-F) animals without expansion (A and D) and with expansion at days 3 (B and E) and 14 (C and F). (G) Comparative analysis of Ki67 expression in control groups and Wnt1Cre;Pkd1 groups without expansion and with expansion at days 1, 3, 7 and 14 (*p<0.05). (B) Ki67-positive cells (brown) are located in the periosteal region. (C) Ki67-positive cell are distributed within the expanded suture. (F) Ki67-positive cells are located in the periosteal region. Scale bar (A): 100μm.
These periosteal cells were further evaluated for their differentiation status using in situ hybridization for type I collagen. In the control group, the expression level of type I collagen mRNA was very low in non-expansion mice and significantly upregulated in expansion mice, with Col1a1 mRNA-positive cells detected in the region of periosteal cells at days 1 and 3, and distributed along the medial surface of newly formed bone and cartilage at days 7 and 14 (Fig. 4A-E). In contrast, the expression level of type I collagen mRNA was low in non-expansion and expansion mice in the Wnt1Cre;Pkd1 group, except that stronger signals were observed in the periosteal cells close to palatal bones at day 14 (Fig. 4F-J).
Figure 4.
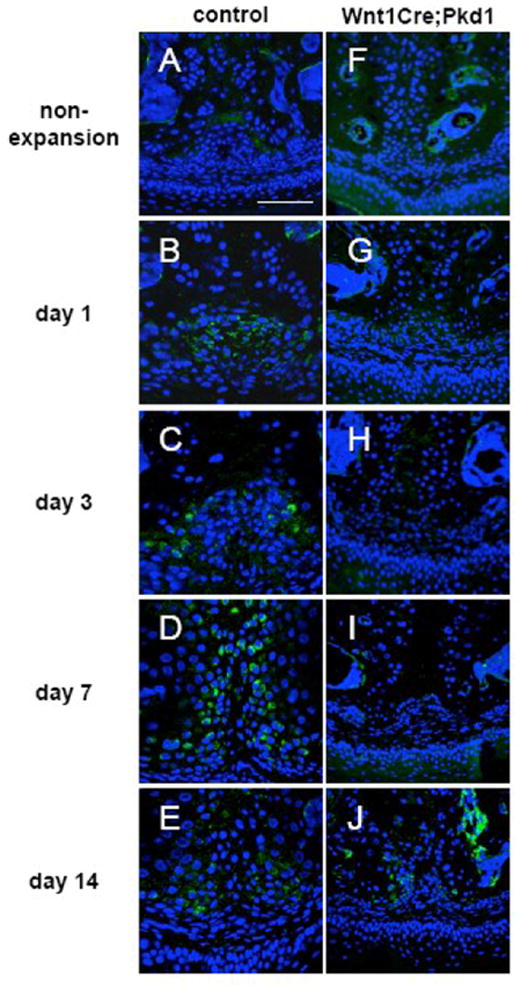
Defective differentiation of periosteal cells into osteoblasts in Wnt1Cre;Pkd1 mice in response to expansive force. Col1a1 in situ hybridization of frontal sections of midpalatal sutures of control (A-E) and Wnt1Cre;Pkd1 (F-J) animals without expansion (A and F) and with expansion at days 1 (B and G), 3 (C and H), 7 (D and I) and 14 (E and J). (B and C) Increased expression for Col1a1 (green) in the periosteal region. (D) Col1a1-positive cells are distributed in the suture. (J) Col1a1-positive cells are located in the perioteal region. Scale bar (A): 100μm.
Increased periosteal cell apoptosis in Wnt1Cre;Pkd1 mice in response to expansive force
Since tissue remodeling requires coordinate regulation of cell proliferation, differentiation and cell death/apoptosis, we studied the role of apoptosis in the response of periosteal cells to midpalatal suture expansion. In the non-expansion mice of both control and Wnt1Cre;Pkd1 groups, TUNEL-positive cells were mainly detected in the region of chondrocytes, accounting for 4.8±1.6% and 5.1±1.5% of total suture cells, respectively. After the application of the opening loop, TUNEL-positive cells were observed in the regions of periosteal cells and chondrocytes at days 1 and 3 (Fig. 5), and mainly in the region of chondrocytes at days 7 and 14 (not shown). In the control mice, TUNEL-positive cells account for 7.0±0.5% and 4.6±1.1% of total suture cells at days 1 and 3 respectively. In the Wnt1Cre;Pkd1 mice, TUNEL-positive periosteal cells were observed in the zone of proliferating cells and aligned along the direction of the stretched collagen fibers. The number of TUNEL-positive cells represented 13.5±1.4% and 10.5±1.7% of total suture cells and were significantly higher than those of controls at days 1 and 3 (p<0.05 for both) (Fig. 5).
Figure 5.
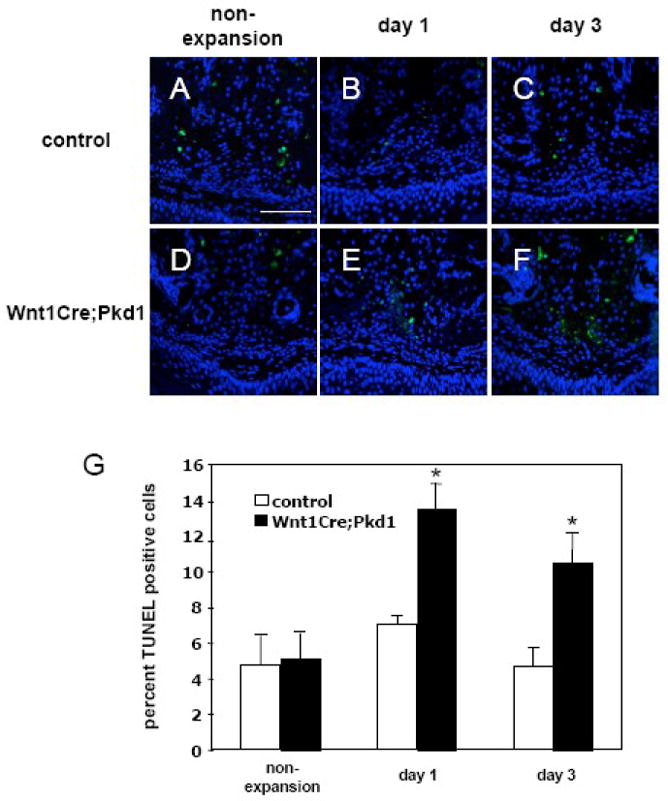
Increased periosteal cell apoptosis in Wnt1Cre;Pkd1 mice in response to expansive force. TUNEL staining of frontal sections of midpalatal sutures of control (A-C) and Wnt1Cre;Pkd1 (D-F) animals without expansion (A and D) and with expansion at days 1 (B and E) and 3 (C and F). (G) Comparative analysis of TUNEL-positive cells between control and Wnt1Cre;Pkd1 groups without expansion and with expansion at days 1 and 3 (*p<0.05). (B, C, E and F) TUNEL positive cells (green) in the periosteal region. Scale bar (A): 100μm.
Changes in nasal cartilage morphology in young adult mice
While the histological appearance of the midpalatal suture of Wnt1Cre;Pkd1 mice was similar to that of control mice, the nasal septal cartilage of the conditional knockout mice exhibited a different postnatal morphology from that of their control counterparts. Histologically, the nasal septum of control mice was covered by perichondrium and consisted of pale round mature chondrocytes in cluster-like arrangements toward the center of the septum and smaller, more darkly stained and more elongated chondrocytes at the periphery. The histological appearance of the nasal septum was not changed over the course of aging and was consistent from the anterior to the posterior end (Fig. 6A-F).
Figure 6.
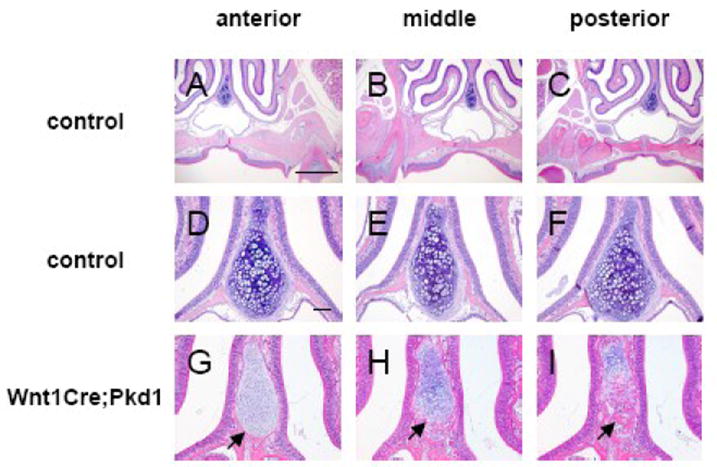
Abnormal ossification in the nasal cartilage in Wnt1Cre;Pkd1 mice. Hematoxylin and eosin staining of frontal sections of nasal cartilages of 3-month-old control (A-F) and Wnt1Cre;Pkd1 (G-I) animals. Serial sections were cut between first and second maxillary molars and representative sections were shown from the anterior (A, D and G), middle (B, E and H) and posterior (C, F, and I) regions. (G, H and I) Arrows point to the ossification areas in the nasal cartilage. Scale bar (A and D): 1mm and 100μm, respectively.
In contrast, nasal septal cartilage of Wnt1Cre;Pkd1 mice showed a disorganized arrangement of chondrocytes where round mature chondrocytes were mixed with smaller, more darkly stained chondrocytes in the middle region of the cartilage. Moreover, nasal septal cartilage of Wnt1Cre;Pkd1 mice showed signs of ossification around the ages of 8-12 weeks (Fig. 6G-I). Although there were some variations in the extent of ossification in terms of the onset and starting position along the anterior-posterior axis, ossification was observed in all of the mutant mice. Typically, the ossification started at the very edge of the inferior septal border and then extended further into the cartilaginous septum (Fig. 6, arrows). Along the anterior-posterior axis, the ossification was more evident and extensive at the posterior end than the anterior end.
Accelerated ossification of nasal cartilage upon midpalatal suture expansion in Wnt1Cre;Pkd1 mice and Col2a1-Cre ER*; Pkd1 mice
As natural activity such as mastication can exert mechanical stress on the nasal septum, it is unclear to what extent force has contributed to the process of nasal cartilage ossification observed in adult mutant mice. Since previous studies have shown that midpalatal suture expansion can exert various stresses on different structures of the skull including nasal septum [23], the nasal septum of Wnt1Cre;Pkd1 mice with or without midpalatal suture expansion were analyzed to investigate the possibility that mechanical stress facilitates ossification in Wnt1Cre;Pkd1 mice. While the histological appearance of the nasal septum of control mice was unchanged upon the placement of the opening loop, the nasal septum of Wnt1Cre;Pkd1 mice displayed accelerated ossification in response to suture expansion. As shown in Fig. 7, after one day of suture expansion, histological characteristics of ossification were observed at the inferior border of the nasal septum at the first molar level where the septum was completely cartilaginous in non-expansion Wnt1Cre;Pkd1 mice. During the following days, the area of ossification, containing trabecular bone with bone marrow cavities, increased and extended upward into the nasal septum. The cartilaginous tissue had completely disappeared and was replaced by compact bone at day 14 (Fig. 7A-E and data not shown). The process was also observed in Col2a1-Cre ER*; Pkd1 mice but to a much lesser extent. At day 7, ossification appeared at the inferior border of the nasal cartilage in some of the mutant mice subjected to midpalatal suture expansion. The chondrocytes exhibited an abnormal arrangement with hypertrophic chondrocytes located in peripheral regions adjacent to ossification sites, and separated from the hypertrophic chondrocytes in the middle by a zone of small, darkly stained chondrocytes. This altered arrangement seemed to be associated with the ossification process as it was not observed in non-expansion mutant mice or expansion mice without the abnormal ossification (Fig. 7F and G). The efficacy of our gene inactivation protocol was confirmed by examining the expression of β-galactosidase in the nasal cartilage of Col2a1-CreER*;R26R mice (Fig. 7H).
Figure 7.
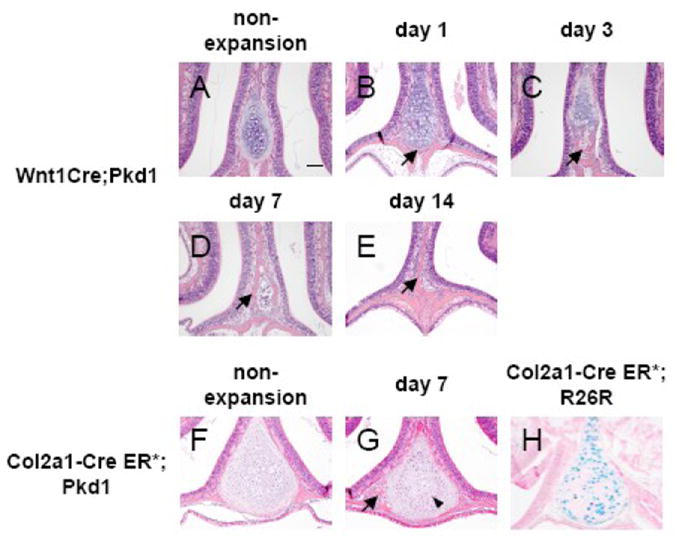
Abnormal ossification in the nasal cartilage in Wnt1Cre;Pkd1 mice and Col2a1-Cre ER*; Pkd1 mice in response to expansive force. Hematoxylin and eosin staining of frontal sections of nasal cartilages of Wnt1Cre;Pkd1 (A-E) and Col2a1-Cre ER*; Pkd1 (F and G) animals without expansion (A and F) and with expansion at days 1 (B), 3 (C), 7 (D and G) and 14 (E). (H) β-galactosidase staining of nasal cartilage of Col2a1-Cre ER*;Rosa26R reveals that Cre recombinase is expressed by the majority of the chondrocytes. (B, C, D, E and G) Arrows point to the ossification area in the nasal cartilage. (G) Arrowhead points to the zone of small, darkly stained chondrocytes located between two zones of hypertrophic chondrocytes. Scale bar (A): 100μm.
Altered expression of regulators of chondrocyte differentiation, matrix degradation and vascular invasion in Wnt1Cre;Pkd1 mice
To further investigate the mechanisms underlying the abnormal ossification observed in the nasal cartilage of Wnt1Cre;Pkd1 mice, we examined the expression of several factors, including RUNX2, MMP13 and VEGFA, involved in regulating hypertrophic chondrocyte differentiation, matrix degradation and angiogenesis during endochondral ossification. Transcript levels of Runx2 and Mmp13 were significantly increased in the nasal cartilage isolated from Wnt1Cre;Pkd1 mice compared to control mice (P<0.05 and P<0.001, respectively), while there was no significant change in the transcript levels of Vegfa between these two groups. Upon application of the opening loop, expression of the Runx2 was significantly upregulated in Wnt1Cre;Pkd1 mice (P<0.05) but not in control mice, while expression of the Mmp13 was significantly upregulated in both Wnt1Cre;Pkd1 mice (P<0.001) and control mice (P<0.05). Expression of Vegfa was not altered in response to the mechanical stress (Fig. 8A).
Figure 8.
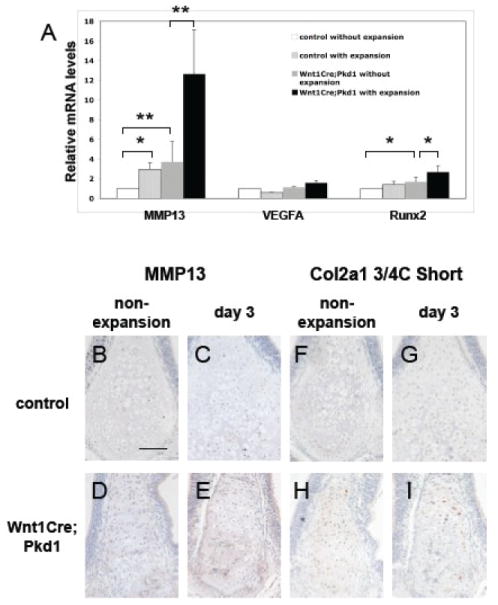
Expression of Mmp13, Vegfa, Runx2 and cleaved collagen type II in the nasal cartilages in Wnt1Cre Pkd1 mice. (A) Real-time quantitative PCR analysis of Mmp13, Vegfa and Runx2 in control and Wnt1Cre;Pkd1 groups without expansion and with expansion at day 3 (*p<0.05 and ** p<0.001). Immunohistochemical staining of Mmp13 (B-E) and cleaved collagen type II (F-I) of frontal sections of nasal cartilages of control (B, C, F and G) and Wnt1Cre;Pkd1 (D, E, H and I) animals without expansion (B, D, F and H) and with expansion at day 3 (C, E, G and I). Scale bar (B): 100μm.
Using immunohistochemistry, we also examined the expression level of Mmp13 protein. In control mice, Mmp13 positive staining was detected in hypertrophic chondrocytes in nasal cartilage with or without force. In Wnt1Cre;Pkd1 mice, Mmp13 positive staining was observed in hypertrophic as well as in prehypertrophic chondrocytes. At the same time, the intensity of Mmp13 staining was increased compared with control mice. In Wnt1Cre;Pkd1 mice subjected to midpalatal suture expansion, Mmp13 staining was also detected in some of the cells in bone marrow in addition to chondrocytes (Fig. 8B-E). Since a major function of Mmp13 is to cleave type II collagen, which is the major fibrillar extracellular matrix component of cartilage, we used an antibody that detects a neoepitope at the cleavage site of the 3/4 fragment of type II collagen following cleavage by Mmp13 to assess the state of collagen remodeling. In the control group, immunostaining for cleaved collagen was barely detectable in both non-expansion and expansion mice. In contrast, cleaved collagens were present around both hypertrophic and prehypertrophic chondrocytes in the nasal cartilage in Wnt1Cre;Pkd1 group. The intensity of the immunostaining for cleaved collagen was increased following application of the opening loop (Fig. 8F-I).
Increased chondrocyte apoptosis in nasal cartilage in Wnt1Cre;Pkd1 mice
During endochondral ossification, hypertrophic chondrocytes undergo programmed cell death before the cartilage is invaded by blood vessels, osteoblasts, osteoclasts and mesenchymal precursor cells. To investigate the relationship between the ossification of nasal cartilage in Wnt1Cre;Pkd1 mice and apoptosis, TUNEL labeling of nasal cartilage was performed to identify end-stage apoptotic chondrocytes. In control mice, no TUNEL-positive chondrocytes in the nasal cartilage was observed regardless of the presence of mechanical stress. In contrast, TUNEL-positive staining was detected in peripheral chondrocytes in the regions where the initial ossification occurred in the non-expansion mice of the Wnt1Cre;Pkd1 group. The number of TUNEL-positive chondrocytes varied between two and four per section. In the expansion mice of the Wnt1Cre;Pkd1 group, TUNEL-positive staining was observed in chondrocytes close to the ossification sites. Here the number of TUNEL-positive chondrocytes varied between four and eight per section. The numbers of TUNEL-positive cells in Wnt1Cre;Pkd1 group were significantly higher that those of controls with or without expansion (P<0.05 for both) (Fig. 9).
Figure 9.
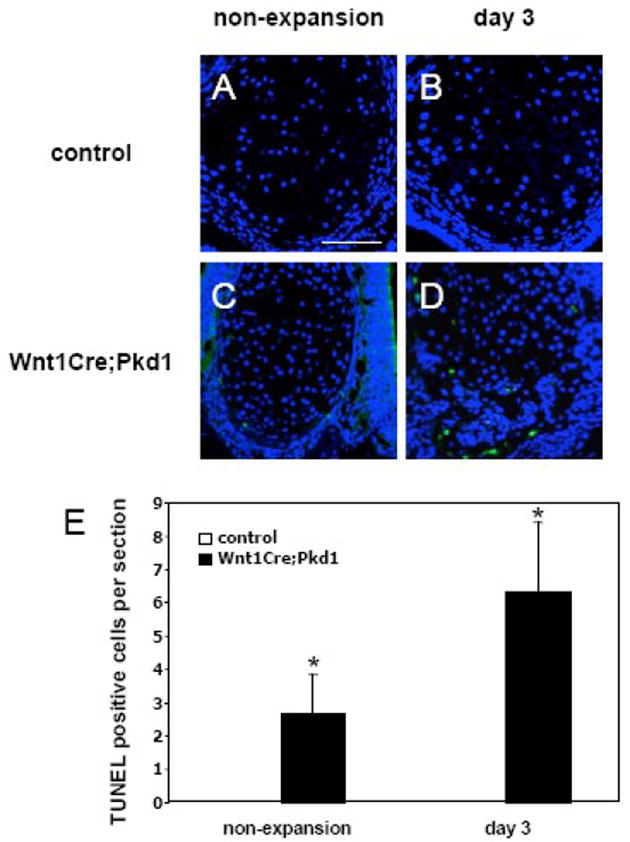
Increased chondrocyte apoptosis in nasal cartilage in Wnt1Cre;Pkd1 mice. TUNEL staining of the nasal cartilages in control (A and B) and Wnt1Cre;Pkd1 (C and D) animals without expansion (A and C) and with expansion at day 3 (B and D). (E) Comparative analysis of TUNEL-positive cells in control and Wnt1Cre;Pkd1 groups without expansion and with expansion at day 3 (*p<0.05). (C) TUNEL-positive cells (green) are located in the peripheral regions of nasal cartilage. (D) TUNEL-positive cells are scattered in nasal cartilage. Scale bar (A): 100μm.
Discussion
In this study, we have shown that Pkd1 plays a critical role in the response of craniofacial cells to mechanical tissue stress. Using the mouse midpalatal suture expansion model [8], we provide evidence that Pkd1-deficient mice exhibit a significantly reduced osteogenic response to tensile stress across the suture. This is due to reduced proliferation, delayed differentiation and increased apoptosis of osteochondroprogenitor cells. Furthermore, nasal cartilage of Pkd1-deficient mice undergoes unusual postnatal endochondral ossification and this process is accelerated upon application of expansive force across the midpalatal suture.
It is well accepted that mechanical stimuli are essential in regulating skeletal growth and adaptation. The appendicular skeleton responds to mechanical stress by periosteal and endocortical bone apposition and resorption. In the craniofacial skeleton, sutures and synchondroses are believed to transmit mechanical stress and enable bone growth at their margins [24-26]. Based on the results of the present studies, we conclude that this function of craniofacial sutures requires Pkd1. Although Wnt1Cre;Pkd1 mice exhibited premature ossification of the presphenoid synchondrosis at the skull base and retarded postnatal growth of the anterior craniofacial complex [27], the midpalatal suture and palatal bones in these mice appeared histologically normal, except that the distance between the first maxillary molars was increased. To account for this difference between wild-type and conditional mutant mice, we modified the size of the opening loop to ensure that the same magnitude of initial force was applied to both Wnt1Cre;Pkd1 and control mice. As a result, there was no significant difference in the suture width between the control mice and Wnt1Cre;Pkd1 mice at day 0 and day 1, indicating a similar tensile stress produced by the opening loop on the midpalatal suture. Thus, the different responses we observed in control versus Wnt1Cre;Pkd1 mice are likely due to the inactivation of Pkd1 in cranial neural crest cells rather than a difference in the mechanical stress introduced by suture expansion.
The protein product of Pkd1, PC1, seems to have multiple functions in cells of the osteoblastic cell lineage depending on the stage of differentiation. First, PC1 is required at an early osteoprogenitor stage for the proliferative response to supraphysiological levels of mechanical tissue stimulation, as the relative amount of stress-induced new bone was significantly reduced in Wnt1Cre;Pkd1, but not in OsxCre;Pkd1 mice that are conditionally deficient for PC1 in differentiated osteoblasts. In contrast, OsxCre;Pkd1, but not Wnt1Cre;Pkd1 mice, exhibited thinner palatal bones post puberty (Fig. 1A, C and E), indicating that PC1 might be important for bone formation under physiological conditions. While our studies do not allow conclusions about the specific mechanistic role of PC1 in the response of cells to mechanical stress applied to the whole suture tissue, one hypothetical model is that PC1 functions as a mechanosensor in osteoprogenitor cells to high levels of mechanical stress, mediating cell proliferation and differentiation into osteoblasts. At levels of mechanical stress within a lower, physiological range, PC1 exerts a negative effect on the differentiation of osteoprogenitor cells to osteoblasts, and a positive effect on more mature osteoblasts to facilitate their differentiation and matrix production. In this model, inactivation of PC1 in osteoblasts results in reduced bone formation as reflected by the decreased thickness of the palatal bones, while inactivation of PC1 in osteoprogenitor cells has no effect on palatal bone thickness. As the expression level and subcellular localization of PC1 have been shown to be developmentally regulated in kidney epithelial cells [15, 28, 29], it is possible that PC1 is expressed at different levels and locations in osteoprogenitor cells and differentiated osteoblasts, thus activating distinct downstream signaling pathways. It has also been reported that cellular responses may vary dependent on mechanical force magnitude, frequency or duration [5, 30, 31], as well as the stage of cellular differentiation. For instance, stretch applied to immature osteoblasts in culture results in decreased cell numbers as cells are stimulated to undergo apoptosis, while stretch has no effect on apoptosis in more mature osteoblasts in culture [32]. These data are consistent with the hypothesis that stimulation of a mechanosensor, such as PC1, elicits different cellular responses to mechanical stress depending on stress levels and cell differentiation.
Recent studies have shown that PC1 associated with the primary cilium of renal epithelia can function as a molecular sensor of fluid flow, thereby regulating tissue morphogenesis [14]. Further work has revealed that PC1 can regulate the cell cycle via upregulation of p21, a cyclin-dependent kinase inhibitor, and that loss of PC1 function will lead to increased cell proliferation and the renal cysts seen in autosomal dominant polycystic kidney disease (ADPKD) patients [33, 34]. However, our data indicate that PC1 is a positive cell cycle regulator in osteoprogenitor cells as the upregulated proliferation of sutural cells in response to midpalatal suture expansion is severely diminished in Wnt1Cre;Pkd1 mice. Compared to control mice in which significantly upregulated cell proliferation was observed as early as day 1, Ki67 positive cells were not detected until day 14 in Wnt1Cre;Pkd1 mice. Consistent with the notion that the role of PC1 in regulating the cell cycle in skeletal cells might be that of a stimulator, Kolpakova-Hart et al. observed reduced proliferative activity in chondrocytes and osteochondroprogenitor cells in Pkd1-deficient mice during embryonic and postnatal development [27].
Since tissue remodeling usually requires a coordinated regulation of cell proliferation and apoptosis, the effects of mechanical stress on cell apoptosis have also been studied in a variety of systems. Apoptotic cells have been detected at sites of bone formation in distraction osteogenesis [35]. During orthodontic tooth movement, apoptotic cells have been observed in dental pulp, periodontal ligaments and periapical areas within 1 to 3 days after application of mechanical force [36, 37]. Similarly, we have found that midpalatal suture expansion induces a low level of cell apoptosis of periosteal cells at days 1 and 3 in control mice. The induction of apoptosis occurs at the time when these periosteal cells undergo rapid proliferation, suggesting that a certain level of apoptosis is associated with the stress induced by suture expansion. In contrast, while the expansive force failed to induce a proliferative response in periosteal cells until day 14 in Wnt1Cre;Pkd1 mice, it resulted in a significantly higher number of apoptotic periosteal cells in these mutant mice. As these periosteal cells provide a major source of osteoblastic cells for new bone formation in the expanded suture area, the increased apoptotic activity combined with the deficient proliferative response of periosteal cells to midpalatal suture expansion would result in a smaller pool of progenitor cells, and significantly less bone formation in Wnt1Cre;Pkd1 mice. Taken together, these data has suggested that PC1 plays a positive role on cell proliferation and differentiation, as well as a negative role on cell apoptosis in the response of periosteal cells to midpalatal suture expansion.
An interesting finding of this study was that the nasal cartilage of Wnt1Cre;Pkd1 mice underwent abnormal ossification during postnatal development and in response to midpalatal suture expansion. Ossification of nasal cartilage was also observed to a lesser extent in Col2a1-Cre ER*; Pkd1 mice upon mechanical stimulation. The histological changes in the nasal cartilage of mutant mice are similar to those typical of endochondral ossification, a process involving chondrocyte hypertrophy and apoptosis, cartilage matrix remodeling, vascular invasion and trabecular bone formation [38-40]. In wild-type mice with non-ossifying nasal cartilage, hypertrophic chondrocytes were located in the center with resting condrocytes in the peripheral regions. In nasal cartilage exhibiting abnormal ossification, chondrocytes appeared to be rearranged with hypertrophic chondrocytes located next to the ossification sites around the periphery of the cartilage and proliferative chondrocytes located inside these hypertrophic chondrocytes. This rearrangement was most obvious in Col2a1-Cre ER*; Pkd1 mice subjected to midpalatal suture expansion, as the process was proceeding at a slower pace in these mice than in Wnt1Cre;Pkd1 mice and could therefore be “captured” by histology in its initial stages. Moreover, real-time quantitative PCR revealed that the chondrocytes from mutant nasal cartilage expressed higher levels of Runx2 and Mmp13 transcripts compared to chondrocytes from controls. Although the expression of Runx2 and Mmp13 was upregulated when comparing the expansion versus non-expansion mice in the control group, the upregulation of Runx2 and Mmp13 was significantly higher in the Wnt1Cre;Pkd1 group following mechanical stimulation. In addition to its role in regulating osteoblastic differentiation, Runx2 is a positive regulator of chondrocyte maturation, while Mmp13, a downstream target of Runx2, is essential for degradation of the extracellular matrix in cartilage at the ossification front [41-43]. Thus, the abnormal ossification in nasal cartilage of Wnt1Cre;Pkd1 mice mimics the process of endochondral ossification in endochondral bones and is likely a result of accelerated chondrocyte hypertrophy and excessive cartilage matrix breakdown, mediated by upregulated expression of Runx2 and Mmp13. Recent studies have shown that up-regulation of Runx2 in chondrocytes may contribute to pathogenesis of osteoarthritis in mice [44] and in humans [45,46]. Moreover, MMP13 is also up-regulated in osteoarthritis cartilage [47]. It is interesting to note that the pathological manifestations of PC1 inactivation in the cranial cartilage are reminiscent of those in osteoarthritis [27]. However, the effect of PC1 deficiency on Runx2 expression in osteoblasts was different from its effect in chondrocytes. Previous studies have shown that PC1 deficiency results in Runx2 down-regulation in osteoblasts [16, 48]. Similarly, delayed ossification was observed in the calvarial and other cranial bones in PC1-deficient mice indicating a possible down-regulation of Runx2 [27]. Therefore, it is possible that different cell types respond differently to a similar signaling “trigger”.
TUNEL positive cells were observed in the peripheral regions of nasal cartilage in Wnt1Cre;Pkd1 mice. Positive TUNEL labeling was also observed in the perichondrium of the presphenoid sychondroses in these mice as reported by Kolpakova-Hart et al. [27]. It is possible that Pkd1 is required for survival of chondrocytes and chondroprogenitor cells by providing survival signals and/or regulating anti-apoptotic signals [49]. As apoptotic cells may secrete cytokines to attract osteoclasts/chondroclasts, thus initiating bone formation/remodeling [50], the abnormal ossification may also be partly attributed to the activity of these apoptotic cells at the periphery of nasal cartilage in Wnt1Cre;Pkd1 mice.
In addition to the effects of Pkd1 inactivation, the fact that the abnormal ossification of nasal cartilage started around postnatal week 8 to 12 suggests that there might be other age-dependent factors contributing to this process. It is well known that mechanical force generated by the masticatory muscles can affect the growth pattern of the craniofacial skeleton [51]. Since the abnormal ossification of nasal cartilage was observed long after the mice were weaned and fed with regular solid food, it is possible that mechanical stress induced by mastication contributes to the replacement of cartilage by bone. As the nasal septum is exposed to high stress during midpalatal suture expansion [23], this hypothesis is supported by the observation that the ossification of nasal cartilage was initiated at an earlier time point (one day after the application of the opening loop) and at a more anterior site in Wnt1Cre;Pkd1 mice subjected to suture expansion than in non-expansion Wnt1Cre;Pkd1 controls.
It should be noted that when PC1 was inactivated predominantly in Col2a1 positive chondrocytes using the Col2a1-Cre ER* deleter strain, the process of ossification of the nasal cartilage was greatly reduced as it was observed only in some of the mutant mice subjected to suture expansion and at a much later time point compared to the Wnt1Cre;Pkd1 mice. As Col2a1 is not only expressed by differentiated chondrocytes, but also by chondroprogenitor cells, it is possible that PC1 is required primarily in the chondroprogenitor cells to mediate mechanical cues and regulate nasal cartilage morphogenesis. However, it is also possible that ossification of nasal cartilage is a paracrine result of inactivation of PC1 in non-chondrocytes such as perichondrial cells. However, we believe that the altered arrangement of chondrocytes in the nasal cartilage of Wnt1Cre;Pkd1 and Col2a1-Cre ER*;Pkd1 mice is more likely to be cell autonomous. Taken together, these data suggest that Pkd1 is required in chondroprogenitor cells for the survival, maturation and organization of chondrocytes and therefore the integrity of nasal cartilage. Perhaps chondrocytes without functional PC1 are unable to detect the mechanical cues that regulate tissue morphogenesis, and this results in cartilage destruction and replacement with osseous tissue upon mechanical loading.
In vitro studies have implicated several transmembrane molecules in mechanotransduction, including integrins and stretch-activated channels (SACs) [52-55]. One question is how PC1 function is integrated with these putative mechanosensors in cells. It has been reported that PC1 forms multiprotein complexes with integrins and other focal adhesion proteins such as paxillin, talin, tensin, focal adhesion kinase and c-src in renal epithelial cells [56, 57]. In addition, integrins can physically and functionally interact with some classes of ion channels to mediate responses to mechanical stimuli. For instance, activation of α5β3 integrin enhances the volume-sensitive calcium influx pathway which is mediated by stretch-activated cation channels (SA-Cat) [58]. Therefore, it is possible that PC1, integrins, SACs and other potential mechanosensors form a macromolecular complex at mechanical transducer sites such as focal adhesions and produce a coordinated response to various extracellular mechanical environments. The crosstalk between these transmembrane molecules may also happen at intracellular levels so that activation of one signaling pathway may regulate the status of other signaling pathways. The specific transmembrane molecule or signaling pathway to be activated may depend on their availability in cells, which vary with cell differentiation state and developmental stage.
In conclusion, our data provide evidence for a novel role of Pkd1 in the responses of skeletal tissues to mechanical stress. Using the mouse midpalatal suture expansion model, we have demonstrated for the first time that Pkd1 is required for the proliferation, differentiation and survival of periosteal osteochondroprogenitor cells upon mechanical stimulation of the suture tissue. Pkd1 is also important for normal nasal cartilage development and inactivation of Pkd1 results in abnormal ossification of nasal cartilage in response to mechanical midpalatal suture stress. Although the signaling pathways upstream and downstream of Pkd1 in skeletal cells remain to be elucidated, our study provides a starting point for future investigations on the role of Pkd1 in skeletal tissue responses to mechanical stress.
Supplementary Material
Acknowledgments
The authors wish to thank Dr. Jennifer Waters and Lara Petrak at the Nikon Imaging Center at Harvard Medical School for their assistance with microscopy. The authors thank Sofiya Plotkina for technical support and Yulia Pittel for the clerical assistance. This work was supported by research grants R01AR036810 and R21AR053143 from the National Institutes of Health (to B.R.O.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Turner CH, Pavalko FM. Mechanotransduction and functional response of the skeleton to physical stress: the mechanisms and mechanics of bone adaptation. J Orthop Sci. 1998;3(6):346–55. doi: 10.1007/s007760050064. [DOI] [PubMed] [Google Scholar]
- 2.Martin RB. Toward a unifying theory of bone remodeling. Bone. 2000;26(1):1–6. doi: 10.1016/s8756-3282(99)00241-0. [DOI] [PubMed] [Google Scholar]
- 3.Turner CH, Robling AG. Mechanisms by which exercise improves bone strength. J Bone Miner Metab. 2005;23(Suppl):16–22. doi: 10.1007/BF03026318. [DOI] [PubMed] [Google Scholar]
- 4.Triplett JW, O’Riley R, Tekulve K, Norvell SM, Pavalko FM. Mechanical loading by fluid shear stress enhances IGF-1 receptor signaling in osteoblasts in a PKCzeta-dependent manner. Mol Cell Biomech. 2007;4(1):13–25. [PubMed] [Google Scholar]
- 5.Kreja L, Liedert A, Hasni S, Claes L, Ignatius A. Mechanical regulation of osteoclastic genes in human osteoblasts. Biochem Biophys Res Commun. 2008;368(3):582–7. doi: 10.1016/j.bbrc.2008.01.106. [DOI] [PubMed] [Google Scholar]
- 6.Bakker A, Klein-Nulend J, Burger E. Shear stress inhibits while disuse promotes osteocyte apoptosis. Biochem Biophys Res Commun. 2004;320(4):1163–8. doi: 10.1016/j.bbrc.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 7.You L, Temiyasathit S, Lee P, Kim CH, Tummala P, Yao W, et al. Osteocytes as mechanosensors in the inhibition of bone resorption due to mechanical loading. Bone. 2008;42(1):172–9. doi: 10.1016/j.bone.2007.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hou B, Fukai N, Olsen BR. Mechanical force-induced midpalatal suture remodeling in mice. Bone. 2007;40(6):1483–93. doi: 10.1016/j.bone.2007.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hughes J, Ward CJ, Peral B, Aspinwall R, Clark K, San Millán JL, et al. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet. 1995;10(2):151–60. doi: 10.1038/ng0695-151. [DOI] [PubMed] [Google Scholar]
- 10.Qian F, Germino FJ, Cai Y, Zhang X, Somlo S, Germino GG. PKD1 interacts with PKD2 through a probable coiled-coil domain. Nat Genet. 1997 Jun;16(2):179–83. doi: 10.1038/ng0697-179. [DOI] [PubMed] [Google Scholar]
- 11.Sandford R, Sgotto B, Aparicio S, Brenner S, Vaudin M, Wilson RK, et al. Comparative analysis of the polycystic kidney disease 1 (PKD1) gene reveals an integral membrane glycoprotein with multiple evolutionary conserved domains. Hum Mol Genet. 1997;6(9):1483–9. doi: 10.1093/hmg/6.9.1483. [DOI] [PubMed] [Google Scholar]
- 12.Xu GM, Sikaneta T, Sullivan BM, Zhang Q, Andreucci M, Stehle T, et al. Polycystin-1 interacts with intermediate filaments. J Biol Chem. 2001 Dec 7;276(49):46544–52. doi: 10.1074/jbc.M107828200. [DOI] [PubMed] [Google Scholar]
- 13.Boca M, D’Amato L, Distefano G, Polishchuk RS, Germino GG, Boletta A. Polycystin-1 induces cell migration by regulating phosphatidylinositol 3-kinase-dependent cytoskeletal rearrangements and GSK3beta-dependent cell cell mechanical adhesion. Mol Biol Cell. 2007;18(10):4050–61. doi: 10.1091/mbc.E07-02-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33(2):129–37. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 15.Guillaume R, D’Agati V, Daoust M, Trudel M. Murine Pkd1 is a developmentally regulated gene from morula to adulthood: role in tissue condensation and patterning. Dev Dyn. 1999;214(4):337–48. doi: 10.1002/(SICI)1097-0177(199904)214:4<337::AID-AJA6>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 16.Xiao Z, Zhang S, Mahlios J, Zhou G, Magenheimer BS, Guo D, et al. Cilia-like structures and polycystin-1 in osteoblasts/osteocytes and associated abnormalities in skeletogenesis and Runx2 expression. J Biol Chem. 2006;281(41):30884–95. doi: 10.1074/jbc.M604772200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boulter C, Mulroy S, Webb S, Fleming S, Brindle K, Sandford R. Cardiovascular, skeletal, and renal defects in mice with a targeted disruption of the Pkd1 gene. Proc Natl Acad Sci U S A. 2001;98(21):12174–9. doi: 10.1073/pnas.211191098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu W, Shen X, Pavlova A, Lakkis M, Ward CJ, Pritchard L, et al. Comparison of Pkd1-targeted mutants reveals that loss of polycystin-1 causes cystogenesis and bone defects. Hum Mol Genet. 2001;10(21):2385–96. doi: 10.1093/hmg/10.21.2385. [DOI] [PubMed] [Google Scholar]
- 19.Starremans PG, Li X, Finnerty PE, Guo L, Takakura A, Neilson EG, et al. A mouse model for polycystic kidney disease through a somatic in-frame deletion in the 5’ end of Pkd1. Kidney Int. 2008;73(12):1394–405. doi: 10.1038/ki.2008.111. [DOI] [PubMed] [Google Scholar]
- 20.Maeda Y, Nakamura E, Nguyen MT, Suva LJ, Swain FL, Razzaque MS, et al. Indian Hedgehog produced by postnatal chondrocytes is essential for maintaining a growth plate and trabecular bone. Proc Natl Acad Sci U S A. 2007;104(15):6382–7. doi: 10.1073/pnas.0608449104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cowan CM, Cheng S, Ting K, Soo C, Walder B, Wu B, et al. Nell-1 induced bone formation within the distracted intermaxillary suture. Bone. 2006;38(1):48–58. doi: 10.1016/j.bone.2005.06.023. [DOI] [PubMed] [Google Scholar]
- 22.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21(1):70–1. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 23.Jafari A, Shetty KS, Kumar M. Study of stress distribution and displacement of various craniofacial structures following application of transverse orthopedic forces--a three-dimensional FEM study. Angle Orthod. 2003;73(1):12–20. doi: 10.1043/0003-3219(2003)073<0012:SOSDAD>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 24.Opperman LA. Cranial sutures as intramembranous bone growth sites. Dev Dyn. 2000;219(4):472–85. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1073>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 25.Mao JJ. Mechanobiology of craniofacial sutures. J Dent Res. 2002;81(12):810–6. doi: 10.1177/154405910208101203. [DOI] [PubMed] [Google Scholar]
- 26.Nie X, Luukko K, Kettunen P. FGF signalling in craniofacial development and developmental disorders. Oral Dis. 2006;12(2):102–11. doi: 10.1111/j.1601-0825.2005.01176.x. [DOI] [PubMed] [Google Scholar]
- 27.Kolpakova-Hart E, McBratney-Owen B, Hou B, Fukai N, Nicolae C, Zhou J, et al. Growth of cranial synchondroses and sutures requires polycystin-1. Dev Biol. 2008;321(2):407–19. doi: 10.1016/j.ydbio.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Geng L, Segal Y, Peissel B, Deng N, Pei Y, Carone F, et al. Identification and localization of polycystin, the PKD1 gene product. J Clin Invest. 1996;98(12):2674–82. doi: 10.1172/JCI119090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ward CJ, Turley H, Ong AC, Comley M, Biddolph S, Chetty R, et al. Polycystin, the polycystic kidney disease 1 protein, is expressed by epithelial cells in fetal, adult, and polycystic kidney. Proc Natl Acad Sci U S A. 1996;93(4):1524–8. doi: 10.1073/pnas.93.4.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen YJ, Huang CH, Lee IC, Lee YT, Chen MH, Young TH. Effects of cyclic mechanical stretching on the mRNA expression of tendon/ligament-related and osteoblast-specific genes in human mesenchymal stem cells. Connect Tissue Res. 2008;49(1):7–14. doi: 10.1080/03008200701818561. [DOI] [PubMed] [Google Scholar]
- 31.Koyama Y, Mitsui N, Suzuki N, Yanagisawa M, Sanuki R, Isokawa K, et al. Effect of compressive force on the expression of inflammatory cytokines and their receptors in osteoblastic Saos-2 cells. Arch Oral Biol. 2008;53(5):488–96. doi: 10.1016/j.archoralbio.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 32.Weyts FA, Bosmans B, Niesing R, van Leeuwen JP, Weinans H. Mechanical control of human osteoblast apoptosis and proliferation in relation to differentiation. Calcif Tissue Int. 2003;72(4):505–12. doi: 10.1007/s00223-002-2027-0. [DOI] [PubMed] [Google Scholar]
- 33.Bhunia AK, Piontek K, Boletta A, Liu L, Qian F, Xu PN, et al. PKD1 induces p21(waf1) and regulation of the cell cycle via direct activation of the JAK-STAT signaling pathway in a process requiring PKD2. Cell. 2002 Apr 19;109(2):157–68. doi: 10.1016/s0092-8674(02)00716-x. [DOI] [PubMed] [Google Scholar]
- 34.Li X, Luo Y, Starremans PG, McNamara CA, Pei Y, Zhou J. Polycystin-1 and polycystin-2 regulate the cell cycle through the helix-loop-helix inhibitor Id2. Nat Cell Biol. 2005;7(12):1202–12. doi: 10.1038/ncb1326. [DOI] [PubMed] [Google Scholar]
- 35.Meyer T, Meyer U, Stratmann U, Wiesmann HP, Joos U. Identification of apoptotic cell death in distraction osteogenesis. Cell Biol Int. 1999;23(6):439–46. doi: 10.1006/cbir.1999.0381. [DOI] [PubMed] [Google Scholar]
- 36.Rana MW, Pothisiri V, Killiany DM, Xu XM. Detection of apoptosis during orthodontic tooth movement in rats. Am J Orthod Dentofacial Orthop. 2001;119(5):516–21. doi: 10.1067/mod.2001.113654. [DOI] [PubMed] [Google Scholar]
- 37.Hamaya M, Mizoguchi I, Sakakura Y, Yajima T, Abiko Y. Cell death of osteocytes occurs in rat alveolar bone during experimental tooth movement. Calcif Tissue Int. 2002;70(2):117–26. doi: 10.1007/s002230010021. [DOI] [PubMed] [Google Scholar]
- 38.Olsen BR, Reginato AM, Wang W. Bone development. Annu Rev Cell Dev Biol. 2000;16:191–220. doi: 10.1146/annurev.cellbio.16.1.191. [DOI] [PubMed] [Google Scholar]
- 39.Karsenty G, Wagner EF. Reaching a genetic and molecular understanding of skeletal development. Dev Cell. 2002;2(4):389–406. doi: 10.1016/s1534-5807(02)00157-0. [DOI] [PubMed] [Google Scholar]
- 40.Ortega N, Behonick DJ, Werb Z. Matrix remodeling during endochondral ossification. Trends Cell Biol. 2004;14(2):86–93. doi: 10.1016/j.tcb.2003.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Knäuper V, López-Otin C, Smith B, Knight G, Murphy G. Biochemical characterization of human collagenase-3. J Biol Chem. 1996;271(3):1544–50. doi: 10.1074/jbc.271.3.1544. [DOI] [PubMed] [Google Scholar]
- 42.Jiménez MJ, Balbín M, López JM, Alvarez J, Komori T, López-Otín C. Collagenase 3 is a target of Cbfa1, a transcription factor of the runt gene family involved in bone formation. Mol Cell Biol. 1999;19(6):4431–42. doi: 10.1128/mcb.19.6.4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Enomoto H, Enomoto-Iwamoto M, Iwamoto M, Nomura S, Himeno M, Kitamura Y, et al. Cbfa1 is a positive regulatory factor in chondrocyte maturation. J Biol Chem. 2000;275(12):8695–702. doi: 10.1074/jbc.275.12.8695. [DOI] [PubMed] [Google Scholar]
- 44.Kamekura S, Kawasaki Y, Hoshi K, Shimoaka T, Chikuda H, Maruyama Z, et al. Contribution of runt-related transcription factor 2 to the pathogenesis of osteoarthritis in mice after induction of knee joint instability. Arthritis Rheum. 2006;54(8):2462–70. doi: 10.1002/art.22041. [DOI] [PubMed] [Google Scholar]
- 45.Sato S, Kimura A, Ozdemir J, Asou Y, Miyazaki M, Jinno T, et al. The distinct role of the Runx proteins in chondrocyte differentiation and intervertebral disc degeneration: Findings in murine models and in human disease. Arthritis Rheum. 2008;58(9):2764–75. doi: 10.1002/art.23805. [DOI] [PubMed] [Google Scholar]
- 46.Hopwood B, Tsykin A, Findlay DM, Fazzalari NL. Microarray gene expression profiling of osteoarthritic bone suggests altered bone remodelling, WNT and transforming growth factor-beta/bone morphogenic protein signalling. Arthritis Res Ther. 2007;9(5):R100. doi: 10.1186/ar2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Appleton CT, Pitelka V, Henry J, Beier F. Global analyses of gene expression in early experimental osteoarthritis. Arthritis Rheum. 2007 Jun;56(6):1854–68. doi: 10.1002/art.22711. [DOI] [PubMed] [Google Scholar]
- 48.Xiao Z, Zhang S, Magenheimer BS, Luo J, Quarles LD. Polycystin-1 regulates skeletogenesis through stimulation of the osteoblast-specific transcription factor RUNX2-II. J Biol Chem. 2008;283(18):12624–34. doi: 10.1074/jbc.M710407200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tare RS, Townsend PA, Packham GK, Inglis S, Oreffo RO. Bcl-2-associated athanogene-1 (BAG-1): a transcriptional regulator mediating chondrocyte survival and differentiation during endochondral ossification. Bone. 2008;42(1):113–28. doi: 10.1016/j.bone.2007.08.032. [DOI] [PubMed] [Google Scholar]
- 50.Bronckers AL, Goei W, van Heerde WL, Dumont EA, Reutelingsperger CP, van den Eijnde SM. Phagocytosis of dying chondrocytes by osteoclasts in the mouse growth plate as demonstrated by annexin-V labelling. Cell Tissue Res. 2000;301(2):267–72. doi: 10.1007/s004410000238. [DOI] [PubMed] [Google Scholar]
- 51.Yamamoto S. The effects of food consistency on maxillary growth in rats. Eur J Orthod. 1996;18(6):601–15. doi: 10.1093/ejo/18.6.601. [DOI] [PubMed] [Google Scholar]
- 52.Sakai K, Mohtai M, Iwamoto Y. Fluid shear stress increases transforming growth factor beta 1 expression in human osteoblast-like cells: modulation by cation channel blockades. Calcif Tissue Int. 1998;63(6):515–20. doi: 10.1007/s002239900567. [DOI] [PubMed] [Google Scholar]
- 53.Kapur S, Mohan S, Baylink DJ, Lau KH. Fluid shear stress synergizes with insulin-like growth factor-I (IGF-I) on osteoblast proliferation through integrin-dependent activation of IGF-I mitogenic signaling pathway. J Biol Chem. 2005;280(20):20163–70. doi: 10.1074/jbc.M501460200. [DOI] [PubMed] [Google Scholar]
- 54.Motokawa M, Kaku M, Tohma Y, Kawata T, Fujita T, Kohno S, et al. Effects of cyclic tensile forces on the expression of vascular endothelial growth factor (VEGF) and macrophage-colony-stimulating factor (M-CSF) in murine osteoblastic MC3T3-E1 cells. J Dent Res. 2005;84(5):422–7. doi: 10.1177/154405910508400505. [DOI] [PubMed] [Google Scholar]
- 55.Plotkin LI, Mathov I, Aguirre JI, Parfitt AM, Manolagas SC, Bellido T. Mechanical stimulation prevents osteocyte apoptosis: requirement of integrins, Src kinases, and ERKs. Am J Physiol Cell Physiol. 2005;289(3):C633–43. doi: 10.1152/ajpcell.00278.2004. [DOI] [PubMed] [Google Scholar]
- 56.Wilson PD, Geng L, Li X, Burrow CR. The PKD1 gene product, “polycystin-1,” is a tyrosine-phosphorylated protein that colocalizes with alpha2beta1-integrin in focal clusters in adherent renal epithelia. Lab Invest. 1999;79(10):1311–23. [PubMed] [Google Scholar]
- 57.Geng L, Burrow CR, Li HP, Wilson PD. Modification of the composition of polycystin-1 multiprotein complexes by calcium and tyrosine phosphorylation. Biochim Biophys Acta. 2000;1535(1):21–35. doi: 10.1016/s0925-4439(00)00079-x. [DOI] [PubMed] [Google Scholar]
- 58.Miyauchi A, Gotoh M, Kamioka H, Notoya K, Sekiya H, Takagi Y, et al. AlphaVbeta3 integrin ligands enhance volume-sensitive calcium influx in mechanically stretched osteocytes. J Bone Miner Metab. 2006;24(6):498–504. doi: 10.1007/s00774-006-0716-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.