

Abstract
The MRE11/RAD50/NBN (MRN) complex plays a key role in recognizing and signaling DNA double-strand breaks (DSBs). Hypomorphic mutations in NBN (previously known as NBS1) and MRE11A give rise to the autosomal-recessive diseases Nijmegen breakage syndrome (NBS) and ataxia-telangiectasia-like disorder (ATLD), respectively. To date, no disease due to RAD50 deficiency has been described. Here, we report on a patient previously diagnosed as probably having NBS, with microcephaly, mental retardation, ‘bird-like’ face, and short stature. At variance with this diagnosis, she never had severe infections, had normal immunoglobulin levels, and did not develop lymphoid malignancy up to age 23 years. We found that she is compound heterozygous for mutations in the RAD50 gene that give rise to low levels of unstable RAD50 protein. Cells from the patient were characterized by chromosomal instability; radiosensitivity; failure to form DNA damage-induced MRN foci; and impaired radiation-induced activation of and downstream signaling through the ATM protein, which is defective in the human genetic disorder ataxia-telangiectasia. These cells were also impaired in G1/S cell-cycle-checkpoint activation and displayed radioresistant DNA synthesis and G2-phase accumulation. The defective cellular phenotype was rescued by wild-type RAD50. In conclusion, we have identified and characterized a patient with a RAD50 deficiency that results in a clinical phenotype that can be classified as an NBS-like disorder (NBSLD).
Introduction
Deficiencies in DNA double-strand break (DSB) signaling and repair are hallmarks of distinctive neurodegenerative syndromes.1,2 Germ-line mutations in the ATM and MRE11A genes underlie the autosomal-recessive cerebellar ataxia syndromes ataxia-telangiectasia (A-T [MIM 208900])3 and ataxia-telangiectasia-like disorder (ATLD [MIM 604391]),4 whereas germ-line mutations in the NBN (previously termed NBS1) gene are responsible for the Nijmegen breakage syndrome (NBS [MIM 251260]), an autosomal-recessive disorder characterized by microcephaly, growth retardation, immunodeficiency, radiosensitivity, and cancer predisposition.5,6 The MRE11/RAD50/NBN (MRN) complex is a highly conserved protein complex implicated in both homologous recombination repair (HRR) and nonhomologous end joining (NHEJ) of DNA DSBs,7,8 in telomere maintenance, and in DNA replication.9,10 This protein complex acts as a sensor of DSBs and recruits the ataxia telangiectasia mutated (ATM) protein to sites of the breaks, where it is activated.11–13
The MRN complex is rapidly relocalized to nuclear foci in response to irradiation of cells.14 It was shown that ATM, which phosphorylates NBN for activation of the S-phase checkpoint, was not required for association of the MRN complex with sites of DNA damage.15 The complex also binds tightly to chromatin during normal S phase.16 The initial event in recognizing and responding to DSBs is the binding of a RAD50/MRE11 heterotetramer (R/M complex) to DNA, tethering broken ends of DSBs.17 This binding is achieved through the two DNA-binding motifs of MRE11 and is arranged as a globular domain with RAD50 Walker A and B motifs (ATPase domains). The bridging of DNA molecules is achieved through CXXC sequences in the middle part of RAD50.18 These sequences occur at the ends of a 960-aa-long heptad repeat coiled-coil region and appear to dimerize by the coordination of a Zn2+ ion.19 The dynamic architecture of the MRN complex is altered for parallel orientation of the coiled coils of RAD50, favoring intercomplex association.20 The exonuclease and endonuclease activities of MRE11 are stimulated upon association with RAD50.21,22 NBN also stimulates its endonuclease activity.23 The complete complex can partly unwind or dissociate a short DNA duplex with a 3′ overhang, and this activity is stimulated by ATP.23
MRN is required for full activation of ATM at the DSB.24 As part of its engagement, ATM is recruited to DNA flanking the DSB, where it is activated by autophosphorylation.25,26 Conversion of an inactive dimer to an active monomer is part of this process. The likely sequence of events has recently been elucidated at defined endogenous DSBs.27,28 The initial activation of ATM appears to occur by relaxation of chromatin, followed by local disruption of the nucleosome structure, recruitment of ATM to the MRN complex at the break, and subsequent admittance of DNA-repair proteins. Once activated, ATM can then phosphorylate a multitude of downstream substrates involved in DNA repair, cell-cycle control, and regulation of transcription.29,30 NBN is one of these substrates and plays a role as adaptor for the phosphorylation of downstream substrates involved in regulating S-phase checkpoints.31 No disorder has been associated with biallelic mutations in RAD50, thus far. Here, we report on a patient with a variant form of NBS, who revealed RAD50 protein deficiency due to two germline mutations in the RAD50 gene.
Material and Methods
Cell Cultures and Treatments
Blood samples and skin biopsies were obtained with informed consent of the patients and parents. The procedures followed the ethical standards of institutional committees. Whole blood was stimulated by 2.5% phytohemagglutinin. Lymphoblast cell lines (LCLs) were established from Ficoll-isolated blood lymphocytes via transformation by Epstein-Barr virus. Primary fibroblast-like cells were grown from skin explants. The RAD50-deficient fibroblasts, but not those of the heterozygous parents, displayed prematurely senescent morphology and behavior, with diminished growth rates, even at early passages. Suspension cell cultures were maintained in RPMI 1640, adherent cultures in Earle's MEM, both with GlutaMAX (Invitrogen, Karlsruhe, Germany), supplemented with 15% heat-inactivated FBS (Sigma, Taufkirchen, Germany), in a humidified atmosphere with 5% CO2 at 37°C. Ionizing radiation (IR) employed either a 60Co- or an X-ray source (dose rates approximately 1.5 Gy/min), both controlled by dosimetry.
For assessment of univariate cell-cycle distributions, lymphoblasts were harvested 48 hr after IR, lymphocytes and fibroblasts 72 hr after IR. The cells were permeabilized, and nuclei were stained with 4 μg/ml DAPI for 30 min in the dark. Flow histograms were recorded on an analytical dual-laser-equipped flow cytometer (LSR1, BD Biosciences, Heidelberg, Germany). Curves were fitted and quantified in the MPLUS AV software package (Phoenix Flow Systems, San Diego, CA).
Genomic Workup
Total RNA and genomic DNA were isolated from peripheral white blood cells, lymphoblasts, and fibroblasts. RNA was reverse-transcribed with the use of random hexamers (Amersham Bioscience, Munich, Germany) for priming of first-strand cDNA synthesis. The entire RAD50 coding region was PCR amplified from the patient́s samples and sequenced in ten overlapping fragments with primers derived from the published cDNA sequence. For the amplification of genomic DNA, we constructed primers based on exon-flanking intron sequences of the RAD50 gene. Primer sequences and genomic PCR conditions are provided in Table S1, available online.
Sequencing of the purified PCR products required dye-terminator chemistry on ABI 310 instruments (Applied Biosystems, Darmstadt, Germany) with reagents supplied by the manufacturer. Nucleotide sequence changes are numbered according to the sequence of the major RAD50 mRNA (transcript variant 1, GenBank NM_005732.2), starting with the first nucleotide of the initiation codon; likewise, changes of the protein sequence were described according to the recommendations of the Human Genome Variation Society. For further genotype analysis, we used restriction-enzyme digestion of exon 21 and exon 25 PCR products, followed by electrophoresis of the fragments on 2% agarose gels. The c.3277C→T substitution destroyed a TaqI site of the wild-type (WT) exon 21 PCR product (185 and 146 bp fragments) and left the PCR product of 331 bp from the mutant allele uncleaved. The c.3939A→T substitution created a new recognition site for PsiI that resulted in fragments of 285 and 52 bp from the mutant allele, compared to a single WT exon 25 PCR product of 337 bp. Restriction enzymes were purchased from New England BioLabs (Ipswich, MA).
Protein Analysis
Subcultures of lymphoblast or fibroblast lines were left untreated or exposed to 6 Gy of ionizing radiation and harvested 30 min after irradiation. Extracts were prepared by resuspending the cells in lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 2 mM EGTA, 2 mM EDTA, 25 mM NaF, 25 mM β-glycerophosphate, 0.1 mM ortho-vanadate, 0.1 mM phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin, 1 μg/ml aprotinin, 0.2% Triton X-100, and 0.3% nonidet P-40), followed by incubation on ice for 30 min. Supernatants were collected after centrifugation at 13,000 × g for 10 min. For the determination of relative RAD50, NBN, and MRE11 protein levels and the extent of radiation-induced NBN phosphorylation, whole-cell extracts were separated by SDS-PAGE at 20 μg total protein per lane. The gels were blotted onto Hybond-C (Amersham Pharmacia, Munich, Germany) nitrocellulose membranes at 35 V for 3 to 4 hr with the use of carbonate buffer (10 mM NaHCO3, 3 mM Na2CO3, pH 9.9) with 20% methanol. Alternatively, 7% Tris-Acetate Gels (Invitrogen, Karlsruhe, Germany) and buffers supplied by the manufacturer were used. After blocking with 5% skim milk and 0.05% Tween 20 (Sigma, Taufkirchen, Germany) in PBS, membranes were probed with appropriate antibodies. These included mouse monoclonal anti-RAD50 (RAD50-13B3, diluted 1:5000, GeneTex, San Antonio, TX), rabbit polyclonal NBN antiserum (anti-p95, diluted 1:5000, Novus Biologicals, Littleton, CO) or mouse monoclonal anti-MRE11 (MRE11-12D7, diluted 1:400, GeneTex, San Antonio, TX); mouse monoclonal anti-phospho p53 (serine-15) (diluted 1:1000, New England Biolabs, Ipswich, MA); rabbit polyclonal anti-CHEK1 pSer317 (1:1000, Cell Signaling Technology, Danvers, MA); rabbit polyclonal anti-ATM Ser1981 (1:2000, Rockland, Gibertsville, PA); mouse monoclonal anti-ATM (1:2000, GeneTex, San Antonio, TX); mouse monoclonal anti-SMC1 pSer957 (1:2000 Upstate, Charlottesville, VA), and rabbit polyclonal anti-SMC1 (1:2000, Novus, Littleton, CO). For comparison with a protein of uniform abundance, blots were reprobed with monoclonal anti-β-actin (diluted 1:5000, Sigma, Taufkirchen, Germany). We used horseradish peroxidase-conjugated rabbit anti-mouse and goat anti-rabbit IgG secondary antibodies (dilution 1:5000, Sigma, Taufkirchen, Germany) and enhanced chemiluminescence (ECL, Pierce/Thermo Fisher Scientific, Rockford, IL, or Amersham/GE Healthcare, Munich, Germany) for visualization and autoradiography. Immunoblot signals were quantitated densitometrically.
For immunoprecipitation (IP), 220 μg (controls) or 440 μg (HA239) protein were mixed after preclearance with primary antibody (rabbit anti-NBN [Novus, Littleton, CO] or rabbit anti-MRE11 [Novus, Littleton, CO]) for 1 hr and were immunoprecipitated overnight (4°C) by the addition of protein G-Sepharose (Amersham/GE Healthcare, Munich, Germany). Immunoprecipitates were washed extensively and separated by denaturating gel electrophoresis. For immunodetection, various mouse anti-NBN, -MRE11, or -RAD50 were used (all provided by GeneTex, San Antonio, TX).
Immunofluorescence
Normal control and RAD50-deficient fibroblasts were seeded onto coverslips in 12-well plates, grown for 24 hr, and exposed to 12 Gy of ionizing radiation. The cells were incubated for an additional 14 hr with fresh growth media, followed by fixation and permeabilisation in 4% paraformaldehyde and 0.1% Triton X-100 for 15 min. After washing with PBS, coverslips were blocked in PBS with 5% goat serum (Jackson ImmunoResearch, West Grove, PA) at room temperature (RT) for 1 hr. The cell monolayers were covered with rabbit polyclonal NBN antiserum (Novus, Littleton, CO) at 1:100 dilution, rabbit polyclonal MRE11 antiserum (Novus, Littleton, CO) at 1:200 dilution, or mouse monoclonal anti-RAD50 (GeneTex, San Antonio, TX) at 1:200 dilution and were incubated at RT for 1 hr. After washing with PBS, the slides were incubated with Alexa Fluor 594 goat anti-rabbit IgG or Alexa Fluor 594 goat anti-mouse conjugate (Molecular Probes), respectively, at 1:200 dilution, for 1 hr at RT. In the case of double-staining, Alexa Fluor 488 goat anti-rabbit IgG was used. Nuclei were counterstained with DAPI at 1 ng/ml for 2 min. After three final washing steps, the coverslips were mounted in Vectashield mounting medium (Vector Laboratories, Burlingame, CA). They were examined by fluorescence microscopy on an Axiophot microscope (Zeiss, Oberkochen, Germany). Images of the same nuclei were captured with the use of Texas red, FITC, and DAPI filters and an Applied Imaging System with EasyFish version 1.2 software.
Complementation Assays
Human RAD50 cDNA in pFLAG-1 vector (Kodak Scientific Imaging Systems, New Haven, CT) was a generous gift of K. K. Kim, Kwangju, South Korea. The coding region was amplified by PCR and subcloned into the vector pREP4 (Invitrogen, Karlsruhe, Germany) with the use of BamHI and NotI restriction sites. Transient transfection of lymphoblastoid cells was achieved by lipofection with FuGene (Roche, Mannheim, Germany) in accordance with the manufacturer's protocol. Telomerase immortalization of RAD50-deficient fibroblasts was accomplished by infection of primary skin fibroblasts with a retroviral vector encoding hTERT (pBABE-puro-hTERT; Addgene, Cambridge, MA) as previously described.32 RAD50 cDNA was also subcloned into the pCLNXS retroviral plasmid (Imgenex, San Diego, CA) with the use of EcoRI and BamHI sites. hTERT-immortalized RAD50(−/−) cells (YS8) were infected with packaged viral particles and selected with G418. Expression of RAD50 was tested with 13B3 Ab from GeneTex (San Antonio, TX) via immunoblotting and IF. The results indicated that about 30% of the cells initially expressed the recombinant protein.
Cell Survival
Clonogenic cell survival after exposure to IR was carried out as described previously.33 Control and F239 RAD50-deficient cells (fibroblasts) were resuspended at 2 × 105/ml in cell-culture medium. Cells were irradiated with 1, 2, 3, 4, or 5 Gy and plated out for the determination of survival via counting of surviving colonies. The A-T fibroblast line AT4BI was included as a radiosensitive control. Survival data were derived from three separate experiments.
Cytogenetic Analysis
Both blood lymphocytes and LCLs were examined for spontaneous and radiation-induced chromosomal instability. Exponentially growing cells were irradiated in the G2 phase of the cell cycle with doses of 0.5 or 1 Gy, respectively. Colcemid (0.06 μg/ml) was added to the cultures 2 hr after irradiation, and the cells were harvested 2 hr later. Metaphases were GTG banded via pancreatin and Giemsa staining. Chromosome aberrations were scored according to ISCN nomenclature. Chromosome painting was performed via standard hybridization procedures with whole-chromosome paint (WCP) probes for chromosomes 1, 2, 4, 7, and 14 (Vector Laboratories, Burlingame, CA).
DNA-Replication Labeling and DNA-Fiber Spreads
Cells were labeled consecutively with 15 μM Chlorodeoxyuridine (CldrU) for 20 min, including a mock or 5 Gy treatment, then a further 20 min with 15 μM Iododeoxyuridine (IdrU). DNA fibers were prepared as described elsewhere.34 In brief, 2 μl of cells (1 × 106 cells/mL) were mixed with 6 μl lysis solution (0.5% SDS, 200 mM Tris-HCl pH 7.4 and 50 mM EDTA), and after 10 min, slides were tilted to 20° to spread the DNA fibers. Slides were air dried both before and after fixation in methanol:acetic acid (3:1). DNA fibers were denatured in 2.5 M HCl for 60 min and immunolabelled with rat monoclonal anti-bromodeoxyuridine (BrdrU) (BU1/75, Abcam) and anti-rat Alexa 488-conjugated secondary antibody (Molecular Probes) for labeling of CldrU, followed by mouse monoclonal anti-BrdrU (Becton Dickinson) and donkey anti-mouse Alexa 594-conjugated secondary antibody (Molecular Probes) for labeling of IdrU as previously described.35 DNA tracks were scored (300–500) for each cell line for mock and 5 Gy treatments. The percentage of new DNA tracks was determined (new initiations/continuing tracks × 100), and the percentage of inhibition of DNA synthesis was computed as [(irradiated new initiations/continuing) / (unirradiated new initiations/continuing)] × 100.
Results
Patient Phenotype
Previously, a four-year-old girl of nonconsanguineous German descent had been reported as having an NBS-like condition.36 Her diagnosis was based on typical congenital and laboratory findings, including microcephaly, growth retardation, spontaneous chromosomal instability including characteristic translocations between chromosomes 7 and 14, cellular hypersensitivity to ionizing radiation, and radioresistant DNA synthesis. By age 15, she presented with mild to moderate retardation of psychomotor development, mild spasticity, and very modestly impaired sensomotor coordination, which appeared as a subtle unsteadiness in straight-line walking, resembling a slight and nonprogressive ataxia. Physical examination also included recognition of a “bird”-like face and revealed height (130 cm), weight (29 kg), and head circumference (43 cm) well below the third percentile. Puberty and secondary sexual characteristics appeared normal. Areas of hyper- and hypopigmentation were noticed all over her body, and she was suffering from a massive hyperopia. A photograph and a summary of clinical findings are provided in Figure S1 and Table S2. Of note, the girl had never experienced severe or recurrent infections. Laboratory studies performed when she was 18 years old showed normal serum levels of immunoglobulin (Ig) classes G, A, M, D, as well as of IgG subclasses, as had been observed previously, when she was a child.36 Lymphocyte counts, subset distributions, and lymphocyte response to mitogen were within normal limits, as was the level of serum alpha-fetoprotein. There was no evidence of lymphoid malignancy such as frequently occurs over time in individuals with an NBS phenotype. At her current age of 23 years, the patient is living in her own flat in an assisted-living facility.
Mutations in the RAD50 Gene
The initial sequencing of the NBN gene did not reveal a disease-causing mutation. Heterozygosity for a rare amino acid substitution (p.R169H, c.506G→A in exon 5), which could not explain a NBS phenotype, was noted. Immunoblot analysis of patient-derived lymphoblasts (HA239) failed to reveal any radiation-induced phosphorylation of NBN(Ser343). This observation prompted studies of NBN interactors. Among them, RAD50 protein was undetectable, in contrast to that of classic NBS patients (Figures 1A and 1B). However, traces of residual RAD50 protein were detected in LCLs after extended exposure of X-ray films, indicating a weak, normal-sized RAD50 protein band and an isoform with an estimated 5-10 kDa increase in its molecular weight (Figure 1C). Residual levels of RAD50 protein were below 5% of WT. Although RAD50 protein was not detected at normal exposures of the autoradiographs (Figures 1A and 1B), the other components of the MRN complex were easily detected (Figure 1B). Protein levels of the other components of the MRN complex varied from 20%–40% of WT for NBN and 30%–70% of WT for MRE11, in different experiments (Figure 1B). Primary fibroblasts from the same patient also had markedly reduced levels of RAD50 (data not shown). Sequence analysis of the entire coding region of RAD50 at cDNA (LCL) and gDNA levels for both lymphocytes and fibroblasts revealed compound heterozygosity for mutations that are predicted to be deleterious (Figure 1D). The maternally inherited mutation (c.3277C→T; p.R1093X) is located in exon 21 and creates a premature termination codon (Figure 1D, upper panel). The paternally inherited mutation (c.3939A→T), located in exon 25, affects the RAD50 stop codon by changing the ochre triplet at codon 1313 into a tyrosine codon (Figure 1D, middle). Conceptual translation of the paternal allele predicts a larger polypeptide than WT, extended by 66 additional amino acids at the carboxy terminal (p.X1313YextX∗66). The prediction of a larger-size polypeptide produced by the aberrant paternal allele is consistent with the detection of a higher-molecular-size protein band (Figure 1C). The very low amounts of the larger polypeptide could be consequence of protein instability and/or of an unstable nonstop mRNA. Yet, this allele was predominating; sequence analysis of cDNA showed quasi-hemizygous expression of the paternal allele, c.3939A→T, in lymphoblast mRNA, implying nonsense-mediated mRNA decay for the maternal allele (Figure 1D, bottom panel). Thus, most of the residual RAD50 protein appeared to be derived from the paternal p.X1313YextX∗66 allele. The existence of alternatively spliced RAD50 mRNA as specified in the NCBI database may explain the observed traces of normal-sized protein, though additional studies were precluded by the extremely low amounts of protein in the RAD50-deficient cells. Screening of 350 chromosomes from a German random population sample failed to reveal either of the two mutations noted in the patient, likely excluding the possibility that these are polymorphic variants.37
Figure 1.
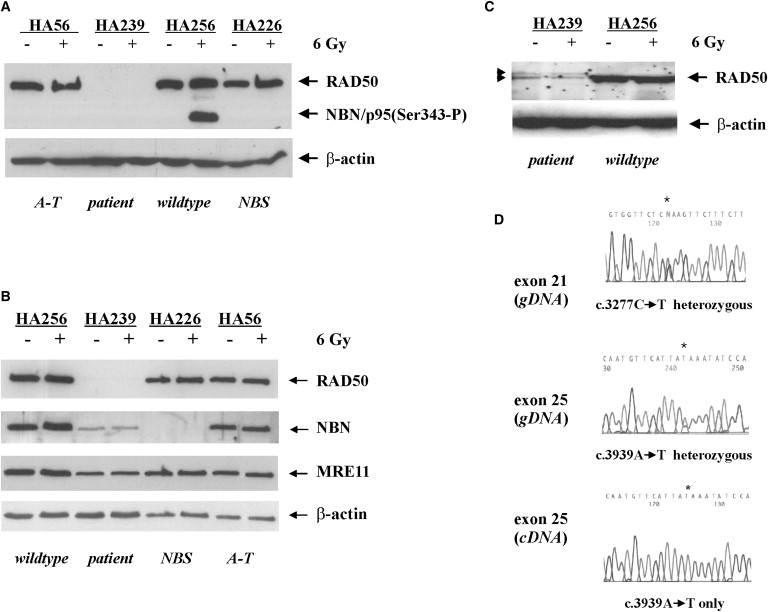
RAD50 Deficiency Associated with Two Germ-Line Mutations
(A–C) Immunoblot analysis of NBN, MRE11, and RAD50 in whole-cell extracts of either untreated (−) or 6 Gy irradiated (+) patient-derived lymphoblasts (HA239) (A and B) compared to normal control (WT, HA256) and A-T or NBS lymphoblasts (HA56, HA226) (B). Immunoblot analysis of RAD50 in the patient-derived LCLs after strong overexposure of the blots, arrows pointing to weak bands consistent with a normal-sized and a larger RAD50 variant (C).
(D) Analysis of the RAD50 sequence. An asterisk indicates two heteroallelic base substitutions, c.3277C→T (p.R1093X) and c.3939A→T (p.X1313YextX∗66) in the genomic RAD50 sequence (upper and middle panel). Only the allele carrying the mutation c.3939A→T was observed in the RAD50 cDNA sequence (bottom panel).
Defect in Localization of RAD50 to Sites of DNA Damage
Given that the human RAD50, MRE11, and NBN proteins colocalize in large nuclear repair foci after irradiation,38 we investigated whether the patient's fibroblasts would still be able to form irradiation-induced nuclear foci (IRIF). As shown in Figure 2, NBN and RAD50 were diffusely distributed throughout the nucleus in normal control fibroblasts and relocated to distinct foci after irradiation (Figure 2A; a–d, q–t). Predominantly nuclear staining for NBN was also apparent in the patient's fibroblasts, but there were no IRIFs (Figure 2A; e–h). Immunofluorescence failed to detect RAD50 in the patient's fibroblasts, irrespective of whether the cells were irradiated or not (Figure 2A; u–x). Immunostaining of MRE11 showed diffuse and predominantly nuclear distribution in control fibroblasts. After IR, MRE11 was seen to accumulate within nuclear IRIF (Figure 2A; i–l). In RAD50-deficient cells, however, MRE11 staining was predominantly cytoplasmic. This did not change after IR, and there was no induction of MRE11-specific IRIFs (Figure 2A; m–p). Retroviral transduction of RAD50 cDNA into hTERT-immortalized patient-derived fibroblasts restored both nuclear relocation of MRE11 and IRIF formation of RAD50 and MRE11 in response to IR (Figure 2B). These results indicated that intact RAD50 is essential for nuclear localization of MRE11 and for DNA-damage-induced recruitment of NBN and MRE11 into nuclear foci. In normal lymphoblasts, MRE11 and RAD50 proteins could be coimmunoprecipitated with antibodies against NBN (Figure S2) and vice versa (data not shown). In patient cells, the amount of MRE11 coimmunoprecipitating with NBN was markedly reduced as compared to controls, indicating a stabilizing function of RAD50 on the MRN complex (Figure S2).
Figure 2.
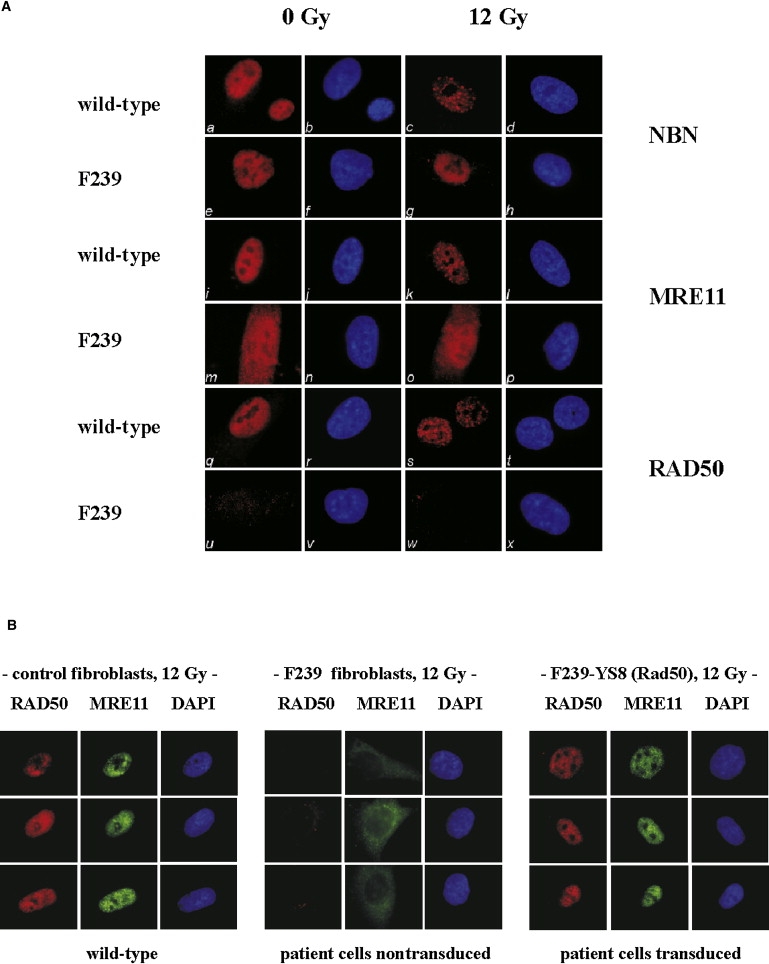
Irradiation-Induced Interactions of NBN, MRE11, and RAD50
(A) Immunofluorescence of primary fibroblasts with mutant RAD50 (F239) compared with WT control fibroblasts assessed for their ability to form ionizing radiation-induced foci (IRIF) for NBN, MRE11, and RAD50 in response to a radiation dose of 12 Gy; a–d, control fibroblasts unirradiated and irradiated, stained with anti-NBN and DAPI nuclear stain, respectively; e–h, corresponding data for RAD50-deficient cells, F239; i–l, control fibroblasts unirradiated and irradiated, stained with anti-MRE11 and DAPI, respectively; m–p, corresponding analysis for F239 cells; q–t, control fibroblasts unirradiated and irradiated, stained with anti-RAD50 and DAPI, respectively; u–x, corresponding analysis for F239 cells.
(B) Nuclear localization and foci formation of MRE11 and RAD50 is restored after transduction of RAD50 cDNA. RAD50-deficient fibroblasts were immortalized with hTERT. Stable cell lines expressing WT RAD50 were established by transduction with a retroviral RAD50 vector (YS8) and selection in G418. Cells were exposed to 12 Gy and IRIF for RAD50 and MRE11 determined in control fibroblasts (left panel), uncomplemented F239 cells (middle panel), and RAD50-transduced F239 cells (right panel).
ATM Activation and Signaling Is Defective in RAD50-Deficient Cells
IR-induced activation of ATM appears to be impaired in NBN-deficient cells.24 A commonly used biological marker for ATM activation is its autophosphorylation at serine residue 1981.25 In RAD50-deficient fibroblasts, DNA-damage-induced ATM autophosphorylation was markedly impaired after 5 Gy irradiation (Figure 3A). In addition, there was little, if any, detectable phosphorylation of either SMC1 on Ser957 or CHEK2 on Thr68, and radiation-induced ATM-dependent phosphorylation of p53 on Ser15 was reduced by approximately 50% relative to control cells (Figure 3A and data not shown). Similarly, in the RAD50-deficient LCL HA239, DNA-damage-induced ATM autophosphorylation was impaired after 10 Gy irradiation (Figure 3B). Additionally, there was very little detectable phosphorylation of either SMC1 on Ser957 or NBN on Ser343, and the radiation-induced ATM-dependent phosphorylation of p53 on Ser15 was reduced by ∼50% relative to control cells (Figure 3B and data not shown). Transient transfection of RAD50 cDNA into RAD50-deficient lymphoblasts followed by irradiation restored phosphorylation of p53(Ser15) and ATM(Ser1981), as well as of NBN(Ser343) (Figure 3C). The effects of RAD50 deficiency were not limited to the cellular ionizing radiation response, however; p53 phosphorylation on Ser15 was also diminished after hydroxyurea treatment of RAD50-deficient cells (Figure 3D). Phosphorylation of the ATR substrate CHEK1(Ser317) was similarly reduced after exposure to hydroxyurea, to an extent comparable with classic NBS cells, suggesting that, by analogy with NBN39 and as reported for a RAD50-depleted colon cancer cell line,40 RAD50 deficiency may also impair ATR-dependent signaling pathways after replication fork stalling (Figure 3E).
Figure 3.
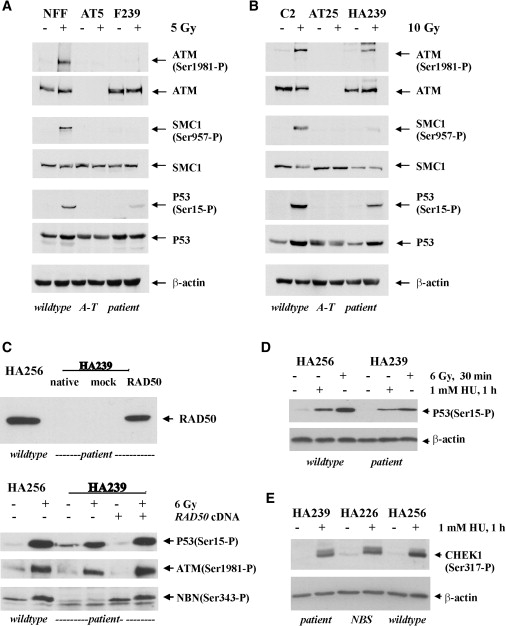
Role of RAD50 in ATM- and ATR-Dependent Signaling
(A) Role of RAD50 in ATM-dependent IR-induced signaling in fibroblasts. Impairment of IR-induced ATM autophosphorylation and signaling to ATM substrates in both A-T (AT5) and RAD50-deficient (F239) fibroblasts compared with control fibroblasts (NFF).
(B) Role of RAD50 in ATM-dependent IR-induced signaling in lymphoblastoid cells. Impairment of the IR-induced signaling to ATM substrates in both A-T- (AT25ABR) and RAD50-deficient (HA239) LCL compared with control cells (C2ABR).
(C) Restoration of radiation-induced phosphorylation of NBN (Ser343), as well as of phosphorylation of p53 (Ser15) and ATM autophosphorylation at Ser1981, after transient transfection of RAD50 cDNA into HA239 LCL.
(D) Comparative immunoblot analysis of p53(Ser15) phosphorylation after treatment of RAD50-deficient or control lymphoblastoid cells with either hydroxyurea (HU, 1 mM, 1 hr) or ionizing radiation (6 Gy, 30 min), consistent with reduced p53 phosphorylation in RAD50- deficient cells.
(E) Immunoblot analysis of ATR-dependent signaling to CHEK1 after hydroxyurea (HU) treatment (1 mM, 1 hr) of RAD50-deficient lymphoblastoid cells (HA239) in comparison to NBS cells homozygous for the NBN c.657del5 mutation (HA226) and to WT control cells (HA256), consistent with reduced CHEK1 phosphorylation in both NBN- and RAD50-deficient cells.
RAD50-Deficient Cells Exhibit Increased Cellular and Chromosomal Radiosensitivity
A characteristic feature of cells with defective ATM activation is their hypersensitivity to IR. To address this in the patient with RAD50 deficiency, we measured the clonogenic survival of RAD50-deficient fibroblasts after irradiation with different doses in between 1 and 5 Gy. As is shown in Figure 4A, RAD50-deficient fibroblasts displayed a significantly lower proportion of surviving colonies than did normal control fibroblasts, although the extent of radiosensitivity was somewhat lower than that in A-T fibroblasts (Figure 4A). We also investigated the level of chromosomal radiosensitivity and were able to show that RAD50-deficient blood lymphocytes and lymphoblasts showed an increased level of IR-induced chromosomal damage similar that of to A-T and NBS cells (Table S3).
Figure 4.
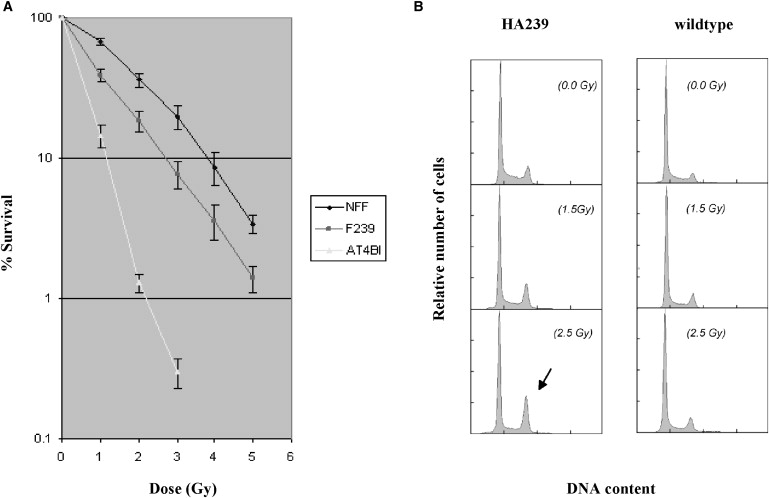
Clonogenic Survival and Cell-Cycle Checkpoints
(A) Radiosensitivity in RAD50-deficient fibroblasts (F239) was measured by counting of colonies surviving radiation doses of 1–5 Gy. Colony survival was expressed as a percentage of irradiated/unirradiated cells. Error bars represent SEM, and the data are derived from three separate experiments. NFF, control; F239, RAD50-deficient cells; AT4B1, A-T cells.
(B) Univariate flow histograms of asynchronous RAD50-deficient LCL (HA239). Cells were irradiated at the indicated dosage levels and harvested 48 hr later. G2-phase cell accumulation is indicated by an arrow. Quantitative analysis is shown in Table 1.
RAD50 deficiency was also associated with an increased spontaneous chromosomal instability, as has been suggested by the initial cytogenetic studies in this patient.36 Interestingly, RAD50-deficient cells showed a particularly high frequency of spontaneous chromatid exchanges (Figure S3A, chte); 7.6 chte per 100 metaphases in lymphocytes and 2.7 chte per 100 metaphases in LCLs, detected neither in A-T nor in NBS cells. All chte figures (multiradial interchanges) involved nonhomologous chromosomes. Further analyses by both G banding and WCP assays revealed spontaneous translocations of chromosomes 7 and 14 in 1.5%–4% of the patient lymphocytes, a frequency lower than that observed in lymphocytes of NBS or A-T patients (∼10%–20%, refs. 5,41,42 and M.S., unpublished data). Extended WCP assays including probes for chromosomes 1, 2, and 4 additionally disclosed a high rate of spontaneous translocations (23/1000 metaphases) (Figure S3B), in this case at a frequency comparable to that observed in NBS or A-T cells43.In summary, RAD50-deficient cells showed a similar level of IR-induced chromosomal damage compared with A-T or NBS cells. However, aberrations spontaneously occurring in RAD50-deficient cells appear to include fewer translocations involving chromosomes 7 and 14. In contrast, the predominance of chromatid interchanges (multiradials) that are due to successful but inaccurate repair of chromatid-type lesions (DSBs) supposedly reflects a role for RAD50 in homology-directed recombinational DNA repair.
RAD50-Deficient Cells Reveal G1/S and Intra-S Checkpoint Defects and Accumulation in G2 after Irradiation
NBS and A-T cells exhibit cell-cycle abnormalities that can be assessed by flow cytometric analysis.44,45 An early study of the present patient suggests that this holds true, in similar form, for RAD50-deficient cells.36 When we increased levels of irradiation from 0 to 8 Gy, S-phase fractions of HA239 LCL remained grossly unchanged, whereas they decreased in a dose-dependent manner in normal control lymphoblasts (Figure 4B and Table 1). Likewise, with increasing dosage levels of irradiation from 0 to 8 Gy, the G2-phase fractions of HA239 lymphoblasts sharply increased in a dose-dependent manner, whereas the G2-phase fractions showed only slight increases within the same dosage range in control lymphoblasts. These findings suggest that HA239 lymphoblasts are uninhibited by irradiation in entering S phase (their S-phase fractions remain grossly unchanged) and in their progress through S phase (their G2-phase fractions strongly increase). These data provide direct and indirect evidence that the G1/S and intra-S checkpoints in RAD50-deficient lymphoblasts are compromised and that the cells accumulate at the G2/M checkpoint at later times after irradiation, similar to what has previously been reported for A-T cells.44 Akin to lymphoblasts, RAD50-deficient lymphocytes and fibroblasts showed comparable G1 and S cell-cycle-checkpoint defects and accumulated in G2 phase in a dose-dependent manner after IR.
Table 1.
Cell-Cycle Distributions of Lymphoblast Cultures 48 Hr after Irradiation
| IR Dosage (Gy) | HA239 G1 S G2 (%) | Wild-Type Control (CON) G1 S G2 (%) |
|---|---|---|
| 0.0 | 66.0 20.6 13.4 | 56.8 31.1 12.1 |
| 2.5 | 53.9 22.0 22.0 | 67.8 15.9 16.3 |
| 4.0 | 41.9 20.2 37.9 | 71.2 11.4 17.4 |
| 8.0 | 34.0 20.7 45.2 | 77.3 7.4 15.3 |
Proportions were calculated by curve fits of the histograms shown in Figure 4B.
Failure to inhibit DNA synthesis in response to IR treatment to the same extent as that of controls is a hallmark of A-T, NBS, and ATLD cells.46–48 This is referred to as radioresistant DNA synthesis (RDS) and is used as a measure of the intra-S-phase checkpoint. RDS is normally determined by incorporation of radioactive nucleotides into DNA, which is subject to considerable variability. Previous observations suggest that both DNA-chain initiation and elongation are resistant to IR-induced damage introduced into DNA of A-T cells.44 To address this, we have developed a DNA-replication assay to differentiate between chain elongation and new initiations, with a view to increasing the sensitivity of the assay (A.W.K., unpublished data). The assay involves a short pulse with CldrU for labeling of existing chains, followed by radiation exposure and a pulse with IudrU for labeling of new initiations. The extent of DNA synthesis is expressed as the ratio of new initiations to total elongations. Under these conditions, WT fibroblasts (NFF) showed marked inhibition of DNA synthesis after radiation, whereas RAD50-deficient cells (F239) displayed RDS (Table 2). Thus, consistent with data from a different approach in an early report of this patient,36 radioresistant DNA synthesis is an inherent feature of the patient′s cells and might contribute to the genomic instability and radiosensitivity in this NBS-like disorder.
Table 2.
Radioresistant DNA Synthesis in RAD50-Deficient (F239) Fibroblasts
| New DNA Initiations (%)a |
|||
|---|---|---|---|
| Cell Line | Unirradiated | Irradiated | Inhibition (%) |
| NFF | 7.6 (7.2, 8.0)b | 1.5 (1.3, 1.6) | 80 |
| F239 | 10.1 (10.7, 9.4) | 4.8 (5.5, 4.0) | 52 |
New DNA replication initiations were determined by sequential labeling of cells with Cld Urd (existing chains) and Id Urd (new initiations), with or without exposure to radiation between pulse labeling. Initiations are standardized to total elongation in the fields of view.
Average values for two experiments. NFF is the normal control fibroblast and F239 is the RAD50 mutant. Between 80 and 155 fibers were scored for the different samples.
Discussion
This study identifies and further characterizes RAD50 mutations as the underlying cause of a disease entity, RAD50 deficiency, that shows features of a Nijmegen Breakage Syndrome-like disorder (NBSLD). To our knowledge, this disease entity has not been previously published. The cellular and clinical phenotype of RAD50 deficiency adds an important piece of evidence to our knowledge of how defects in DNA-damage signaling may relate to radiosensitivity, neurodevelopmental disease, and immunodeficiency. Because null mutations in RAD50—like those in MRE11A and NBN—cause embryonic lethality in mice,49–51 most of what we know about the developmental functions of the MRN complex is presently based on hypomorphic inactivation of NBN and MRE11A.4,52 The biallelic mutations in the present case individual with RAD50 deficiency might also result in a hypomorphic combination, given the evidence for subtle amounts of residual RAD50 protein in her lymphoblastoid cells. This condition is associated with a clinical phenotype resembling NBS rather than ATLD, suggesting potential differences in the roles for MRE11 and RAD50 or NBN in neuronal development. The description of human RAD50 deficiency in this study complements the series of reports on defects in the MRN complex,4,38,52 important for its bilateral interaction with ATM and for downstream signaling in the DSB-repair pathways. A-T and NBS represent the clinically most devastating and divergent phenotypes of the corresponding gene defects, whereas MRE11 deficiency (ATLD) and RAD50 deficiency (NBSLD) can be regarded as attenuated disorders of the former or latter, respectively. They constitute variant forms without immunodeficiency and malignancies (also with normal serum alpha-fetoprotein levels), such that affected individuals reach higher mean ages than do individuals suffering from A-T or NBS. This is also observed in the pattern of radiosensitivity in which RAD50-deficient cells (F239) are intermediate in their sensitivity between A-T and WT control cells and closely resemble ATLD in this characteristic.4 Cells from patients with classical NBS have a pattern of sensitivity more similar to that of A-T.38
Like the NBN protein, MRE11 and RAD50 are regarded as DSB sensors that are involved in the activation of ATM at early time points after low doses of IR.13 RAD50 deficiency was found to be associated with reduced ATM autophosphorylation and impaired radiation-induced phosphorylation of ATM target proteins, a finding consistent with the proposed role of RAD50 in sensing IR-induced DNA damage and in tethering DSBs.17,18 RAD50 deficiency was also found to be associated with RDS and with impaired function of G1- and S-phase cell-cycle checkpoints; this may be a consequence of reduced ATM-mediated phosphorylation of cell-cycle regulators such as p53 or SMC1.29–31 However, the role of RAD50 could also be indirect, given that RAD50 deficiency impaired the nuclear localization of MRE11 and its interaction with NBN. In addition, mutational inactivation of RAD50 caused some reduction in the expression levels of the remaining components of the complex, similar to what was observed for MRE11A mutations.4 But whereas RAD50 was not detected at normal exposure conditions of autoradiographs, the other components of the MRN complex were easily detected at protein levels that are close to those expected for ATLD or NBS heterozygotes. Because ATLD or NBS heterozygotes do not exhibit such marked cellular phenotypes as described for the RAD50-deficient cells, we consider it unlikely that the downregulation of the NBN and MRE11 proteins alone would be responsible for the observed features. It may nevertheless be noteworthy that the overall levels of MRE11 protein appeared more stable than those of NBN in RAD50-deficient cells, paralleling the closer clinical relationship of RAD50 deficiency with NBS than with ATLD (MRE11 deficiency). In both NBS- and RAD50-deficient cells, MRE11 is mainly detected in the cytoplasm, suggesting that both proteins are required for the nuclear localization of MRE11.38,53–55 It remains possible that extranuclear functions of MRE11 can prevent to some extent the cerebellar ataxia that is characteristic for A-T and ATLD but is not observed in NBS and RAD50 deficiency. On the other hand, specific roles of NBN and RAD50 in ATR-mediated signaling pathways could contribute to the microcephaly that has been correlated with defects in ATR function56–58 but is not observed in patients with A-T or ATLD. Interestingly, RAD50 can form complexes with NBN and bind to DNA in the absence of MRE11 in vitro.59 Perhaps the closer clinical phenotype between NBS and RAD50 deficiency might reflect a more critical role for NBN and RAD50 in downstream signaling to specific intermediates or pathways.31,60 Specific modifications to those downstream molecules, such as phosphorylation, might also influence their activities at sites of DSBs or may impinge on functions unrelated to DNA-damage sensing.
In summary, we describe the first patient with RAD50 deficiency, a very rare neurodegenerative disorder due to biallelic RAD50 mutations. Our data corroborate the clinical and molecular relationship among the three genes encoding the components of the MRN complex, but they also suggest potential differences, as apparent by microcephaly associated with NBN and RAD50 deficiency in contrast to MRE11A mutations. These findings and the availability of patient-derived RAD50-deficient cell lines provide the basis for the further delineation of the biological roles of RAD50 as an early participant in cellular DNA-damage responses.
Supplemental Data
Supplemental Data include three figures and three tables and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Human Genome Variation Society, http://www.hgvs.org/mutnomen/recs.htm
NCBI GenBank, http://www.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Acknowledgments
The authors thank the patient, her parents, and clinicians in charge for their participation in this study. We are indebted to G. Barbi for cell lines; B. Plückthun, B. Bütow and R. Drange for technical assistance; R. Friedl for flow cytometry; N. Turmanov for mutational screening; P. Heidari for additional cDNA analyses; P. Chen for cell survival data; T. Liehr and A. Küchler for FISH probes; K. K. Kim for RAD50 cDNA; and C. Sohn, P. Hillemanns, J. H. Karstens, P. Wieacker, H. Hoehn, and K. Sperling for providing technical equipment and funding.
Contributor Information
Detlev Schindler, Email: schindler@biozentrum.uni-wuerzburg.de.
Thilo Dörk, Email: doerk.thilo@mh-hannover.de.
References
- 1.McKinnon P.J., Caldecott K. DNA Strand Break Repair and Human Genetic Disease. Annu. Rev. Genomics Hum. Genet. 2007;8:37–55. doi: 10.1146/annurev.genom.7.080505.115648. [DOI] [PubMed] [Google Scholar]
- 2.Kulkarni A., Wilson D.M. The involvement of DNA-damage and -repair defects in neurological dysfunction. Am. J. Hum. Genet. 2008;82:539–566. doi: 10.1016/j.ajhg.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Savitsky K., Bar-Shira A., Gilad S., Rotman G., Ziv Y., Vanagaite L., Tagle D.A., Smith S., Uziel T., Sfez S. A single ataxia-telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 4.Stewart G.S., Maser R.S., Stankovic T., Bressan D.A., Kaplan M.I., Jaspers N.G., Raams A., Byrd P.J., Petrini J.H., Taylor A.M. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell. 1999;99:577–587. doi: 10.1016/s0092-8674(00)81547-0. [DOI] [PubMed] [Google Scholar]
- 5.van der Burgt I., Chrzanowska K.H., Smeets D., Weemaes C. Nijmegen breakage syndrome. J. Med. Genet. 1996;33:153–156. doi: 10.1136/jmg.33.2.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Digweed M., Sperling K. Nijmegen breakage syndrome: clinical manifestation of defective response to DNA double-strand breaks. DNA Repair (Amst.) 2004;3:1207–1214. doi: 10.1016/j.dnarep.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 7.Kobayashi J., Antoccia A., Tauchi H., Matsuura S., Komatsu K. NBS1 and its functional role in the DNA damage response. DNA Repair (Amst.) 2004;3:855–861. doi: 10.1016/j.dnarep.2004.03.023. [DOI] [PubMed] [Google Scholar]
- 8.Yang Y.G., Saidi A., Frappart P.O., Min W., Barrucand C., Dumon-Jones V., Michelon J., Herceg Z., Wang Z.Q. Conditional deletion of Nbs1 in murine cells reveals its role in branching repair pathways of DNA double-strand breaks. EMBO J. 2006;25:5527–5538. doi: 10.1038/sj.emboj.7601411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ciapponi L., Cenci G., Ducau J., Flores C., Johnson-Schlitz D., Gorski M.M., Engels W.R., Gatti M. The Drosophila Mre11/Rad50 complex is required to prevent both telomeric fusion and chromosome breakage. Curr. Biol. 2004;14:1360–1366. doi: 10.1016/j.cub.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 10.Maser R.S., Mirzoeva O.K., Wells J., Olivares H., Williams B.R., Zinkel R.A., Farnham P.J., Petrini J.H. Mre11 complex and DNA replication: linkage to E2F and sites of DNA synthesis. Mol. Cell. Biol. 2001;21:6006–6016. doi: 10.1128/MCB.21.17.6006-6016.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petrini J.H. The mammalian Mre11-Rad50-Nbs1 protein complex: integration of functions in the cellular DNA-damage response. Am. J. Hum. Genet. 1999;64:1264–1269. doi: 10.1086/302391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paull T.T., Lee J.H. The MRE11/RAD50/NBN complex and its role as a DNA double-strand break sensor for ATM. Cell Cycle. 2005;4:737–740. doi: 10.4161/cc.4.6.1715. [DOI] [PubMed] [Google Scholar]
- 13.Lavin M.F. ATM and the Mre11 complex combine to recognize and signal DNA double-strand breaks. Oncogene. 2007;26:7749–7758. doi: 10.1038/sj.onc.1210880. [DOI] [PubMed] [Google Scholar]
- 14.Maser R.S., Monsen K., Nelms B.E., Petrini J.H. hMre11 and hRad50 nuclear foci are induced during the normal cellular response to DNA double-strand breaks. Mol. Cell. Biol. 1997;17:6087–6096. doi: 10.1128/mcb.17.10.6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mirzoeva O.K., Petrini J.H. DNA damage-dependent nuclear dynamics of the Mre11 complex. Mol. Cell. Biol. 2001;21:281–288. doi: 10.1128/MCB.21.1.281-288.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mirzoeva O.K., Petrini J.H. DNA replication-dependent nuclear dynamics of the Mre11 complex. Mol. Cancer Res. 2003;1:207–218. [PubMed] [Google Scholar]
- 17.De Jager M., van Noort J., van Gent D.C., Dekker C., Kanaar R., Wyman C. Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol. Cell. 2001;8:1129–1135. doi: 10.1016/s1097-2765(01)00381-1. [DOI] [PubMed] [Google Scholar]
- 18.Van den Bosch M., Bree R.T., Lowndes N.F. The MRN complex: coordinating and mediating the response to broken chromosomes. EMBO Rep. 2003;4:844–849. doi: 10.1038/sj.embor.embor925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hopfer U. A Maxwell's Demon type of membrane transport: possibility for active transport by ABC-type transporters? J. Theor. Biol. 2002;214:539–547. doi: 10.1006/jtbi.2001.2479. [DOI] [PubMed] [Google Scholar]
- 20.Moreno-Herrero F., de Jager M., Dekker N.H., Kanaar R., Wyman C., Dekker C. Mesoscale conformational changes in the DNA-repair complex Rad50/Mre11/Nbs1 upon binding DNA. Nature. 2005;437:440–443. doi: 10.1038/nature03927. [DOI] [PubMed] [Google Scholar]
- 21.Paull T.T., Gellert M. The 3′ to 5′ exonuclease activity of Mre11 facilitates repair of DNA double-strand breaks. Mol. Cell. 1998;1:969–979. doi: 10.1016/s1097-2765(00)80097-0. [DOI] [PubMed] [Google Scholar]
- 22.Williams R.S., Williams J.S., Tainer J.A. Mre11-Rad50-Nbs1 is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template. Biochem. Cell Biol. 2007;85:509–520. doi: 10.1139/O07-069. [DOI] [PubMed] [Google Scholar]
- 23.Paull T.T., Gellert M. Nbs1 potentiates ATP-driven DNA unwinding and endonuclease cleavage by the Mre11/Rad50 complex. Genes Dev. 1999;13:1276–1288. doi: 10.1101/gad.13.10.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uziel T., Lerenthal Y., Moyal L., Andegeko Y., Mittelman L., Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003;22:5612–5621. doi: 10.1093/emboj/cdg541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bakkenist C.J., Kastan M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 26.Kozlov S.V., Graham M.E., Peng C., Chen P., Robinson P.J., Lavin M.F. Involvement of novel autophosphorylation sites in ATM activation. EMBO J. 2006;25:3504–3514. doi: 10.1038/sj.emboj.7601231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berkovich E., Monnat R.J., Kastan M.B. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat. Cell Biol. 2007;9:683–690. doi: 10.1038/ncb1599. [DOI] [PubMed] [Google Scholar]
- 28.You Z., Ballis J.M., Johnson S.A., Dilworth S.M., Hunter T. Rapid activation of ATM on DNA flanking double-strand breaks. Nat. Cell Biol. 2007;9:1311–1318. doi: 10.1038/ncb1651. [DOI] [PubMed] [Google Scholar]
- 29.Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer. 2003;3:155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- 30.Kastan M.B. DNA Damage Responses: Mechanisms and Roles in Human Disease. Mol. Cancer Res. 2008;6:517–524. doi: 10.1158/1541-7786.MCR-08-0020. [DOI] [PubMed] [Google Scholar]
- 31.Kitigawa R., Bakkenist C.J., McKinnon P.J., Kastan M.B. Phosphorylation of SMC1 is a critical downstream event in the ATM-NBS1-BRCA1 pathway. Genes Dev. 2004;18:1423–1438. doi: 10.1101/gad.1200304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bodnar A.G., Ouelette M., Frolkis M., Holt S.E., Chiu C.P., Morin G.B., Harley C.B., Shay J.W., Lichtsteiner S., Wright W.E. Extension of life-span by introduction of telomerase into normal tissue human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 33.Chen P.C., Lavin M.F., Kidson C., Moss D. Identification of ataxia telangiectasia heterozygotes, a cancer prone population. Nature. 1978;274:484–486. doi: 10.1038/274484a0. [DOI] [PubMed] [Google Scholar]
- 34.Parra I., Windle B. High resolution visual mapping of stretched DNA by fluorescent hybridization. Nat. Genet. 1993;5:17–21. doi: 10.1038/ng0993-17. [DOI] [PubMed] [Google Scholar]
- 35.Aten J.A., Bakker P.J., Stap J., Boschman G.A., Veenhof C.H. DNA double labelling with IdUrd and CldUrd for spatial and temporal analysis of cell proliferation and DNA replication. Histochem. J. 1992;24:251–259. doi: 10.1007/BF01046839. [DOI] [PubMed] [Google Scholar]
- 36.Barbi G., Scheres J.M., Schindler D., Taalman R.D., Rodens K., Mehnert K., Müller M., Seyschab H. Chromosome instability and X-ray hypersensitivity in a microcephalic and growth-retarded child. Am. J. Med. Genet. 1991;40:44–50. doi: 10.1002/ajmg.1320400109. [DOI] [PubMed] [Google Scholar]
- 37.Collins J.S., Schwartz C.E. Detecting Polymorphisms and Mutations in Candidate Genes. Am. J. Hum. Genet. 2002;71:1251–1252. doi: 10.1086/344344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carney J.P., Maser R.S., Olivares H., Davis E.M., Le Beau M., Yates J.R., Hays L., Morgan W.F., Petrini J.H. The hMRE11/hRAD50 protein complex and Nijmegen breakage syndrome: Linkage of double-strand break repair to the cellular DNA damage response. Cell. 1998;93:477–486. doi: 10.1016/s0092-8674(00)81175-7. [DOI] [PubMed] [Google Scholar]
- 39.Stiff T., Reis C., Alderton G.K., Woodbine L., O'Driscoll M., Jeggo P.A. Nbs1 is required for ATR-dependent phosphorylation events. EMBO J. 2005;24:199–208. doi: 10.1038/sj.emboj.7600504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhong H., Bryson A., Eckersdorff M., Ferguson D.O. Rad50 depletion impacts upon ATR-dependent DNA damage responses. Hum. Mol. Genet. 2005;14:2685–2693. doi: 10.1093/hmg/ddi302. [DOI] [PubMed] [Google Scholar]
- 41.Aurias A., Dutrillaux B., Buriot B., Lejeune J. High frequency of inversions and translocations of chromosomes 7 and 14 in ataxia-telangiectasia. Mutat. Res. 1980;69:369–374. doi: 10.1016/0027-5107(80)90101-3. [DOI] [PubMed] [Google Scholar]
- 42.Taylor A.M., Oxford J.M., Metcalfe J.A. Spontaneous cytogenetic abnormalities in lymphocytes from thirteen patients with ataxia telangiectasia. Int. J. Cancer. 1981;27:311–319. doi: 10.1002/ijc.2910270309. [DOI] [PubMed] [Google Scholar]
- 43.Stumm M., Neubauer S., Keindorff S., Wegner R.D., Wieacker P., Sauer R. High frequency of spontaneous translocations revealed by FISH in cells from patients with the cancer-prone syndromes ataxia telangiectasia and Nijmegen breakage syndrome. Cytogenet. Cell Genet. 2001;92:186–191. doi: 10.1159/000056900. [DOI] [PubMed] [Google Scholar]
- 44.Beamish H., Lavin M.F. Radiosensitivity in ataxia-telangiectasia: anomalies in radiation-induced cell cycle delay. Int. J. Radiat. Biol. 1994;65:175–184. doi: 10.1080/09553009414550211. [DOI] [PubMed] [Google Scholar]
- 45.Seyschab H., Schindler D., Friedl R., Barbi G., Boltshauser E., Fryns J.P., Hanefeld F., Korinthenberg R., Krägeloh-Mann I., Scheres J.M. Simultaneous measurement, using flow cytometry, of radiosensitivity and defective mitogen response in ataxia telangiectasia and related syndromes. Eur. J. Pediatr. 1992;151:756–760. doi: 10.1007/BF01959085. [DOI] [PubMed] [Google Scholar]
- 46.Houldsworth J., Lavin M.F. Effect of ionizing radiation on DNA synthesis in ataxia telangiectasia cells. Nucleic Acids Res. 1980;8:3709–3720. doi: 10.1093/nar/8.16.3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Painter R.B., Young B.R. Radiosensitivity in ataxia-telangiectasia: a new explanation. Proc. Natl. Acad. Sci. USA. 1980;77:7315–7317. doi: 10.1073/pnas.77.12.7315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ford M.D., Houldsworth J., Lavin M.F. Ataxia telangiectasia: An anomaly in DNA replication after irradiation. Nucleic Acids Res. 1981;9:1395–1404. doi: 10.1093/nar/9.6.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luo G., Yao M.S., Bender C.F., Mills M., Bladl A.R., Bradley A., Petrini J.H. Disruption of mRAD50 causes embryonic stem cell lethality, abnormal embryonic development, and sensitivity to ionizing radiation. Proc. Natl. Acad. Sci. USA. 1999;96:7376–7381. doi: 10.1073/pnas.96.13.7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Petrini J.H., Theunissen J.W. Double strand break metabolism and cancer susceptibility: lessons from Mre11 complex. Cell Cycle. 2004;3:541–542. doi: 10.4161/cc.3.5.835. [DOI] [PubMed] [Google Scholar]
- 51.Zhu J., Petersen S., Tessarollo L., Nussenzweig A. Targeted disruption of the Nijmegen breakage syndrome gene NBS1 leads to early embryonic lethality in mice. Curr. Biol. 2001;11:105–109. doi: 10.1016/s0960-9822(01)00019-7. [DOI] [PubMed] [Google Scholar]
- 52.Varon R., Vissinga C., Platzer M., Cerosaletti K.M., Chrzanowska K.H., Saar K., Beckmann G., Seemanová E., Cooper P.R., Nowak N.J. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell. 1998;93:467–476. doi: 10.1016/s0092-8674(00)81174-5. [DOI] [PubMed] [Google Scholar]
- 53.Difilippantonio S., Celeste A., Fernandez-Capetillo O., Chen H.T., Reina San Martin B., Van Laethem F., Yang Y.P., Petukhova G.V., Eckhaus M., Feigenbaum L. Role of Nbs1 in the activation of the Atm kinase revealed in humanized mouse models. Nat. Cell Biol. 2005;7:675–685. doi: 10.1038/ncb1270. [DOI] [PubMed] [Google Scholar]
- 54.Desai-Mehta A., Cerosaletti K.M., Concannon P. Distinct functional domains of nibrin mediate Mre11 binding, focus formation and nuclear localization. Mol. Cell. Biol. 2001;21:2184–2191. doi: 10.1128/MCB.21.6.2184-2191.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cerosaletti K., Wright J., Concannon P. Active role for nibrin in the kinetics of ATM activation. Mol. Cell. Biol. 2006;26:1691–1699. doi: 10.1128/MCB.26.5.1691-1699.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.O'Driscoll M., Ruiz-Perez V.L., Woods C.G., Jeggo P.A., Goodship J.A. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat. Genet. 2003;33:497–501. doi: 10.1038/ng1129. [DOI] [PubMed] [Google Scholar]
- 57.O'Driscoll M., Dobyns W.B., van Hagen J.M., Jeggo P.A. Cellular and clinical impact of haploinsufficiency for genes involved in ATR signaling. Am. J. Hum. Genet. 2007;81:77–86. doi: 10.1086/518696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Griffith E., Walker S., Martin C.A., Vagnarelli P., Stiff T., Vernay B., Al Sanna N., Saggar A., Hamel B., Earnshaw W.C. Mutations in pericentrin cause Seckel syndrome with defective ATR-dependent DNA damage signaling. Nat. Genet. 2008;40:232–236. doi: 10.1038/ng.2007.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van der Linden E., Sanchez H., Kinoshita E., Kanaar R., Wyman C. RAD50 and NBS1 form a stable complex functional in DNA binding and tethering. Nucleic Acids Res. 2009 doi: 10.1093/nar/gkn1072. published online Jan. 16, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shull E.R., Lee Y., Nakane H., Stracker T.H., Zhao J., Russell H.R., Petrini J.H., McKinnon P.J. Differential DNA damage signaling accounts for distinct neural apoptotic responses in ATLD and NBS. Genes Dev. 2009;23:171–180. doi: 10.1101/gad.1746609. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.