

Summary
MicroRNAs (miRNAs) are small non-coding regulatory RNAs that reduce stability and/or translation of fully or partially sequence-complementary target mRNAs. In order to identify miRNAs and to assess their expression patterns, we sequenced over 250 small RNA libraries from 26 different organ systems and cell types of human and rodents, enriched in neuronal as well as normal and malignant hematopoietic cells and tissues. We present expression profiles derived from clone count data and provide novel computational tools for their analysis. Unexpectedly, a relatively small set of miRNAs, many of which are ubiquitously expressed, account for most of the difference in miRNA profiles between cell lineages and tissues. This broad survey also provides detailed and accurate information about mature sequences, precursors, genome locations, maturation processes, inferred transcriptional units and conservation patterns. We also propose a subclassification scheme for miRNAs for assisting future experimental and computational functional analyses.
Introduction
MicroRNAs (miRNAs) are small (~22-nucleotide) non-coding regulatory RNA molecules encoded by plants, animals and some viruses (reviewed in Bartel, 2004; Berezikov and Plasterk, 2005; Cullen, 2006; Mallory and Vaucheret, 2006). They were first discovered in Caenorhabditis elegans and were shown to regulate expression of partially complementary mRNAs (Lee et al., 1993; Wightman et al., 1993; Moss et al., 1997). Most miRNAs are evolutionary conserved in related species and some even show conservation between invertebrates and vertebrates (Pasquinelli et al., 2000; Lagos-Quintana et al., 2001; Lau et al., 2001; Lee and Ambros, 2001). Many miRNAs have well-defined developmental and cell-type specific expression patterns (reviewed in Wienholds and Plasterk, 2005). However, for most mammalian miRNAs the relative abundance and specificity of expression remain to be investigated.
miRNAs regulate a variety of developmental and physiological processes (reviewed in Cao et al., 2006; Plasterk, 2006; Shivdasani, 2006). The analysis of miRNA function in animals is either performed genetically or by delivery of synthetic miRNA precursors or antisense oligonucleotides (antagomirs) (reviewed recently in Krützfeldt et al., 2006). Such analysis revealed that 100 to 200 target mRNAs are repressed and destabilized by a single miRNA (Krützfeldt et al., 2005; Lim et al., 2005; Linsley et al., 2007). Other mRNAs appear to be under selective pressure to avoid complementarity to co-expressed highly-abundant miRNAs (Farh et al., 2005; Stark et al., 2005; Sood et al., 2006). Many computational studies have been conducted to define miRNA regulatory networks (reviewed in Rajewsky, 2006), yet most molecular targets of miRNAs remain experimentally undefined.
Posttranscriptional editing of some double-stranded precursor miRNAs by adenosine deamination (Luciano et al., 2004; Pfeffer et al., 2005; Blow et al., 2006; Kawahara et al., 2007) can further control targeting specificity as well as modulate the stability and processing of miRNA precursor transcripts (Gottwein et al., 2006; Yang et al., 2006). Polymorphic sequence variation identified in some other pre-miRNA sequences, in contrast, had no effect on miRNA processing (Iwai and Naraba, 2005; Diederichs and Haber, 2006). Regulated processing of miRNA precursor transcripts has also been reported in the context of cell-type and stage-specific expression (Obernosterer et al., 2006; Thomson et al., 2006).
The increasing number of studies addressing the role of miRNAs in development and in various diseases including cancer emphasizes the need for a comprehensive catalogue of accurate sequence, expression and conservation information for the large number of recently proposed miRNAs. Here we present a database and analysis of over 250 small RNA cDNA libraries obtained by cloning and sequencing. We have developed interactive analysis tools, and illustrate their utility in discovering miRNA expression changes associated with hematopoietic and nervous system differentiation and malignant transformation.
Results
miRNA profiling by sequence analysis of small RNA clone libraries
We cloned and sequenced more then 330,000 independent small RNA sequences from 256 small RNA libraries prepared from 26 distinct organ systems and cell types of human and/or rodents (Table S1), and also re-analyzed some previously described small RNA libraries (Lagos-Quintana et al., 2002; Lagos-Quintana et al., 2003; Poy et al., 2004). Each library was covered by about 1,300 clones and contained on average 65% miRNA sequences representing 70 to 75 distinct mature miRNAs (Tables S2–4). Our small RNA annotation procedure and miRNA profile analysis (Figure S1) kept track of small RNA clones that mapped equally well to more than one miRNA precursor (Tables S5–8). About one third of all miRNA clones mapped to multicopy miRNA genes with indistinguishable mature sequences while another 1% mapped to two or more paralogs with related, but not identical mature miRNA sequence. Clone counts of miRNAs were collected in two distinct table formats either distributing the clones between miRNA genes or precursors (Tables S9–11) or collecting them as unique mature sequences (Tables S12–14). In the annotation process, we identified 33 new miRNAs (Table 1), and we confirmed the expression of many evolutionary conserved miRNAs previously only cloned in other species.
Table 1.
miRNAs identified in this study and not listed in miRBase (release 9.1)
| Human miRNA | Sequence | Clone # | Mouse miRNA | Sequence | Clone # | Rat miRNA | Sequence | clone # |
|---|---|---|---|---|---|---|---|---|
| miR-872__FM | AAGGUUACUUGUUAGUUCAGG | 11 | miR-872 | AAGGUUACUUGUUAGUUCAGG | 394 | miR-872 | AAGGUUACUUGUUAGUUCAGG | 373 |
| miR-879 | AGAGGCUUAUAGCUCUAAGCC | 33 | miR-879 | AGAGGCUUAUAGCUCUAAGCC | 5 | |||
| miR-8822 | AGGAGAGAGUUAGCGCAUUAGU | 1 | miR-882a6 | AUGGGAAUGUUAGAGCAUUAGC | 0 | |||
| miR-882b | AUGGCAAUGUUAGAGCAUUAGC | 0 | ||||||
| miR-874 | CUGCCCUGGCCCGAGGGACCGA | 17 | miR-874 | CUGCCCUGGCCCGAGGGACCGA | 2 | miR-874 | CUGCCCUGGCCCGAGGGACCGA | 0 |
| miR-877 | GUAGAGGAGAUGGCGCAGGG | 40 | miR-877 | GUAGAGGAGAUGGCGCAGGG | 6 | miR-877 | GUAGAGGAGAUGGCGCAGGG | 2 |
| miR-297c3 | AUGUAUGUGUGCAUGUACAUGU | 16 | ||||||
| miR-743b4 | GAAAGACAUCAUGCUGAAUAGA | 8 | miR-743b7 | GAAAGACACCAUACUGAAUAGA | 0 | |||
| miR-883a | UAACUGCAACAGCUCUCAGUAU | 6 | ||||||
| miR-883b | UAACUGCAACAUCUCUCAGUAU | 1 | miR-883 | UAACUGCAACAUCUCUCAGUAU | 0 | |||
| miR-880 | UACUCCAUCCUCUCUGAGUAGA | 119 | miR-880 | UACUCCAUUCAUUCUGAGUAGA | 3 | |||
| miR-878 | GCAUGACACCACACUGGGUAGA | 22 | miR-878 | GCAUGACACCAUACUGGGUAGA | 1 | |||
| miR-874 | AACUGUGUCUUUUCUGAAUAGA | 60 | miR-881 | AACUGUGGCAUUUCUGAAUAGA | 0 | |||
| miR-871__FM | UAUUCAGAUUAGUGCCAGUCAUG | 2 | miR-871 | UAUUCAGAUUAGUGCCAGUCAUG | 11 | miR-871 | UAUUCAGAUUGGUGCCGGUCACA | 0 |
| miR-8901 | UACUUGGAAAGGCAUCAGUUG | 10 | ||||||
| miR-888 | UACUCAAAAAGCUGUCAGUCA | 16 | ||||||
| miR-465d | UAUUUAGAAUGGUACUGAUGUG | 0 | ||||||
| miR-892b | CACUGGCUCCUUUCUGGGUAGA | 14 | miR-465c | UAUUUAGAAUGGCGCUGAUCUG | 42 | |||
| miR-891b | UGCAACUUACCUGAGUCAUUGA | 4 | ||||||
| miR-891a | UGCAACGAACCUGAGCCACUGA | 24 | ||||||
| miR-466f5 | UACGUGUGUGUGCAUGUGCAUG | 7 | ||||||
| miR-466e | GAUGUGUGUGUACAUGUACAUA | 1.5 | ||||||
| miR-466b | GAUGUGUGUGUACAUGUACAUG | 7 | ||||||
| miR-466b8 | UAUGUGUGUGUGUAUGUCCAUG | 1 | ||||||
| miR-466c | GAUGUGUGUGUGCAUGUACAUA | 11 | ||||||
| miR-466c | UGUGAUGUGUGCAUGUACAUG | 2 | ||||||
| miR-467e | AUAAGUGUGAGCAUGUAUAUGU | 125 | ||||||
| miR-466d | UGUGUGUGCGUACAUGUACAUG | 4 | ||||||
| miR-466g | AUACAGACACAUGCACACACA | 2 | ||||||
| miR-466h | UGUGUGCAUGUGCUUGUGUGUA | 4 | ||||||
| miR-885 | UCCAUUACACUACCCUGCCUCU | 5 | ||||||
| miR-887 | GUGAACGGGCGCCAUCCCGAGG | 3 | ||||||
| miR-886 | CGCGGGUGCUUACUGACCCUU | 16 |
The predominantly cloned sequence and orthologs are indicated. Bars beside the name indicate clustered miRNAs.
part of the cluster-hsa-mir-888(6), epididymis-specific expression;
part of the cluster-mmu-mir-134(33), enriched in embryonic tissue;
part of the cluster-mmu-mir-297a-1(46);
part of the cluster-mmu-mir-463(19), epididymis (testis) enriched expression;
part of the cluster-mmu-mir-297a-3(43);
part of the cluster-rno-mir-134(41);
part of the cluster-rno-mir-463(12);
part of the cluster-rno-mir-297c(4); Abbr.: FM: “from mouse”; the same miRNA sequence than in mouse was cloned, but could not be found in the current human assembly.
We provide evidence for expression of 340, 303 and 205 distinct mature miRNAs from human, mouse and rat; these are encoded by 395, 363 and 231 different miRNA genes, respectively. When we include orthology relationships for cloned miRNAs (Table S15), we obtained a final list of 416, 386 and 325 miRNA genes present in human, mouse, and rat, contained in 214, 190 and 168 transcription units, respectively. The 8.2 release of miRBase includes 84, 7, and 13 additional miRNA gene candidates for human, mouse, and rat, respectively, for which we have not found supporting evidence in this study. About 80% of these miRNA candidates were initially identified at very low or single clone counts in single libraries or after sequence-based enrichment procedures (discussed in the Supplemental Experimental Procedures).
The relative cloning frequencies of miRNAs represent a measure of miRNA expression. We implemented three distinct ways of visualizing miRNA expression patterns. (1) We represent each mature miRNA sequence independently. (2) Assuming that similar miRNAs regulate similar targets, we defined “sequence groups” of mature miRNAs based on sequence similarity (Table S16, Figure S3). (3) We grouped miRNA precursors that either share a mature form (multicopy miRNAs) or are clustered in the genome in “precursor clusters” (Tables S17–19). Available cDNA and EST expression evidence support most of our spatially linked precursor clusters and pairwise comparison of the relative expression of clustered mature miRNAs across tissues yielded higher correlation coefficients than for non-clustered miRNAs (r=0.49 for mouse and human vs. r=0.11 and 0.15 for mouse and human, respectively; Tables S20–22). We named precursor clusters according to the name of the lowest numbered cluster member and added in brackets the number of members in the cluster, e.g. expression from the hsa-mir-15a/mir-16-1 genes and hsa-mir-15b/mir-16-2 genes was summarized in the precursor cluster-hsa-mir-15a(4); miRNA sequence families were named with similar convention. For the comparison of miRNA profiles and clustering of samples, we used a Bayesian probability framework that takes into account the sampling noise affecting the relative frequencies of miRNAs. A web-accessible database (http://www.mirz.unibas.ch/smiRNAdb) allows users to perform sequence searches and to visualize and analyze the miRNA expression profiles across individual samples or groups of samples.
Technical and biological replicates indicated that miRNA cloning frequencies are reproducible (Figure S3). We then compared the cloning frequencies of five of our samples with signal intensities from miRNA microarrays using the same input total RNA (Figures S4, S5) and observed a strong and significant correlation of expression ranks within each sample. Furthermore, our cloning profiles correlated with most liver and brain profiles reported by many other laboratories (Figure S6). In order to evaluate how miRNA cloning frequencies correlate with the actual miRNA concentration across samples, we determined the absolute amount of several miRNAs in a subset of our samples by quantitative Northern blotting. We found a significant correlation between miRNA clone frequencies and concentrations measured by Northern blotting (Figures S7–11). To assess how well cloning frequencies reflect relative concentrations within a sample, and to identify potential systematic biases in our library preparation protocol, we prepared a pool of 816 synthetic 5′ phosphorylated oligoribonucleotides with known relative concentrations containing the predominantly cloned miRNA sequences. We sequenced a total of 7498 miRNA clones from two independently prepared libraries (Table S23). Although the miRNA clone counts correlated significantly with their concentrations in the pool, the correlation coefficient was rather low. To a large extent, this is expected for the relatively small sample sizes, as shown by resampling tests (Figure S12, Supplemental Experimental Procedures). However, because the clone counts of the two libraries correlated better with each other than two resampled samples did, we conjectured that a systematic bias must also be present in the cloning protocol. The most likely source of this bias is the secondary structure of the different small RNAs that affect the adapter ligation efficiency. Such a RNA self-structure dependent bias would also explain the robust correlation of miRNA profiles across samples and to miRNA array platforms.
Sequence variations in mature miRNA sequences
Approximately 20% of miRNA clone sequences in human, mouse and rat contained mismatches with respect to their genomic locus that were caused by post-transcriptional modification and/or PCR and sequencing errors. We identified sequence variations for 143 different human and 109 mouse miRNA genes above the sequencing error background (Figure 1, Tables S24 and S25). The frequency of sequence alteration (Table S26) was dependent on the specific nucleotide and its position with the miRNA and was sometimes also tissue-dependent (Tables S27, S28). The prevailing modifications were caused by A-to-I editing (identified as A-to-G changes) and 3′ terminal A and U additions. The A-to-G transitions (Figure 1C) occurred at 4-fold lower frequency in mouse than in human (0.5% in mouse and 2.2% overall in human), in agreement with recent reports of substantially higher A-to-I editing in primates (Kim et al., 2004; Eisenberg et al., 2005).
Figure 1. Nucleotide 3′ additions and editing events in mature miRNA sequences.

Examples for 3′ A additions (A), U additions (B), and A to G nucleotide exchanges due to possible deamination events (C) are given. Nucleotides of sequenced miRNAs that do not match their genomic sequence are colored. The black bar underneath each sequence logo indicates the predominantly cloned sequence. A list of all modifications and their occurrence in the different libraries is given in Tables S24 and S25.
Classifying miRNAs to guide miRNA functional analysis
The most important criteria for annotating a cloned sequence as a miRNA are their characteristic length (~22-nucleotide) and a compact pre-miRNA fold-back structure (Ambros et al., 2003). Furthermore, miRNAs generally adhere to additional properties including precise 5′ end processing, asymmetric strand accumulation, and sequence conservation. Surprisingly, not all miRNA sequences complied with these criteria when examining the large number of miRNA clones obtained in our study. Therefore we subdivided miRNAs into four categories (Tables S29–31, for details see Supplemental Experimental Procedures). We termed the miRNAs in those categories “prototypical”, “repeat-clustered”, “repeat-derived” and “unclassified” miRNAs.
miRNAs were considered “prototypical” if they met defined criteria regarding their 5′ end processing, lack of repetitiveness and cross-species conservation. More than 97% of our miRNA clones originated from “prototypical” miRNA genes. Overall, 59% of all miRNA genes were classified as protoypical.
“Repeat-clustered” and “repeat-derived” miRNAs originate from highly repetitive genomic sequences, either clustered or dispersed (Smalheiser and Torvik, 2005). In human and mouse, we identified only a single “repeat-cluster”. The human cluster contains 40 sequence-related precursors that are co-expressed in placenta (see also Bentwich et al., 2005) whereas the mouse cluster consists of 43 precursors preferentially expressed in neuronal tissues. In contrast to the human repeat-clustered miRNAs, the mouse repeat-clustered miRNAs had additional matches to nearly 2000 dispersed regions in the mouse genome, and we therefore called them also “repeat-derived”. In contrast to prototypical miRNAs, precursors of repeat-clustered miRNAs did not preserve the strand asymmetry between sequence-related precursors. Finally, from the dispersed repeat miRNA category, we identified 75 and 74 miRNA genes in human and mouse, respectively. The overall cloning frequency of all repeat-associated miRNA sequences was 2.6%, though the number of genes in this category represented more than 23% of all miRNA genes.
The remaining 28% of miRNA genes were termed “unclassified”, and their clone counts only amount to 0.4% of all miRNA clones in our study. We found evidence of expression for 42.3% of these genes, and their products showed irregularities in processing, and/or unusual sequence variations including deletions and variation in the seed sequence between human and rodent orthologous.
In order to assess if the our classification scheme of miRNAs has functional implications, we computed the number of conserved 3′ UTR sequence segments complementary to position 1 to 8 (seed) of conserved miRNAs (the “signal”) and for shuffled sequences (the “noise”) (Lewis et al., 2005). Prototypical miRNAs showed an enrichment for putative target sites (average signal-to-noise ratio=3.2:1), consistent with previous analysis of smaller sets of predominantly prototypical miRNAs (Lewis et al., 2003; Lewis et al., 2005; Stark et al., 2005). Repeat-derived miRNAs showed an average ratio of 1.2:1, while unclassified miRNAs had an average ratio of 1.0:1, suggesting that non-prototypical miRNAs either have fewer conserved mRNA targets and/or different requirements for mRNA targeting.
Specificity of miRNA expression in human, mouse and rat organs and cell types
Understanding cell-type specificity of miRNA expression is an important step in elucidating miRNA function. We defined this in terms of information content of the distribution of the relative cloning frequencies in each individual tissue type (see Experimental Procedures and Tables S32–34). The miRNA expression varied from highly specific to ubiquitous and, for conserved miRNAs, was comparable between rodents and human. Surprisingly, very few miRNAs were exclusively found in individual tissues or cell types and only a third of the analyzed miRNAs were expressed with a higher degree of tissue specificity (Figure 2A). When we examined the overall miRNA cloning frequency, we noticed that several abundant miRNAs were ubiquitously expressed, even in embryonic derived cell lines and tissues (Figure 2B). Most notably, miR-16, which is encoded by the mir-15a/16-1 and the mir-15b/16-2 polycistrons, was always the most abundantly cloned miRNAs in each of the over 250 samples analyzed. miR-21 was detected in the majority of samples, but in contrast to miR-16, its relative cloning frequency varied greatly and was higher in tumor samples and cell lines compared to normal tissues.
Figure 2. Tissue specificity of miRNA expression.

(A) The 51 most specific miRNA precursor clusters for human and mouse are depicted. The total height of each bar represents the information content reflecting tissue specificity, while the relative heights for each of the tissues are proportional to the clones of a miRNA precursor cluster in a given tissue type relative to all tissue types. The enrichment of miR-124 in the endocrine tissue in mouse is due to high expression of this miRNA in the MIN-6 beta-cell line, but miR-124 is not expressed in primary β-cells residing in mouse or human islets. (B) The 26 most expressed miRNA precursor clusters for human and mouse are depicted. The mean clone count for each tissue type (striped bars) and the tissue specificity (white bars) are indicated. The most abundant miRNA clusters are ubiquitously expressed and show little tissue specificity.
The expression patterns of miRNAs were recently examined by in situ hybridization analysis during development of zebrafish and mouse, and in human brain samples (Wienholds et al., 2005; Kloosterman et al., 2006a; Kloosterman et al., 2006b; Nelson et al., 2006). The derived expression patterns largely overlap with our cloning data for miRNA precursor clusters analyzed in both studies (Table S32). However, for some miRNAs (e.g. mir-200a(3), mir-203(1), hsa-mir-141(2)), we find enriched expression in other tissues besides those noted in zebrafish.
Analysis of miRNA expression in normal and malignant cells of the hematopoietic system
Distinct cell populations of the hematopoietic system accumulate in acute leukemia and lymphoma making it an interesting model system for studying basic mechanisms of stem cell maintenance, differentiation, and malignant transformation. Regulation of a subset of miRNAs during hematopoiesis has been studied in mouse and human by microarray analysis and Northern blotting (recently reviewed by Kluiver et al., 2006), but a comprehensive analysis of the human hematopoietic system and its associated diseases is lacking. We prepared 98 different small RNA libraries from the human hematopoietic system including sorted cell populations from healthy donors, whole bone marrow and tumor biopsies collected from patients with hematopoietic malignancies and various cell lines (Figure 3). Comparison of miRNA expression in the hematopoietic system to all other organ systems examined in this study indicates that only 5 miRNAs are highly specific for hematopoietic cells: miR-142, miR-144, miR-150, miR-155, and miR-223.
Figure 3. miRNA expression in hematopoietic lineage differentiation.
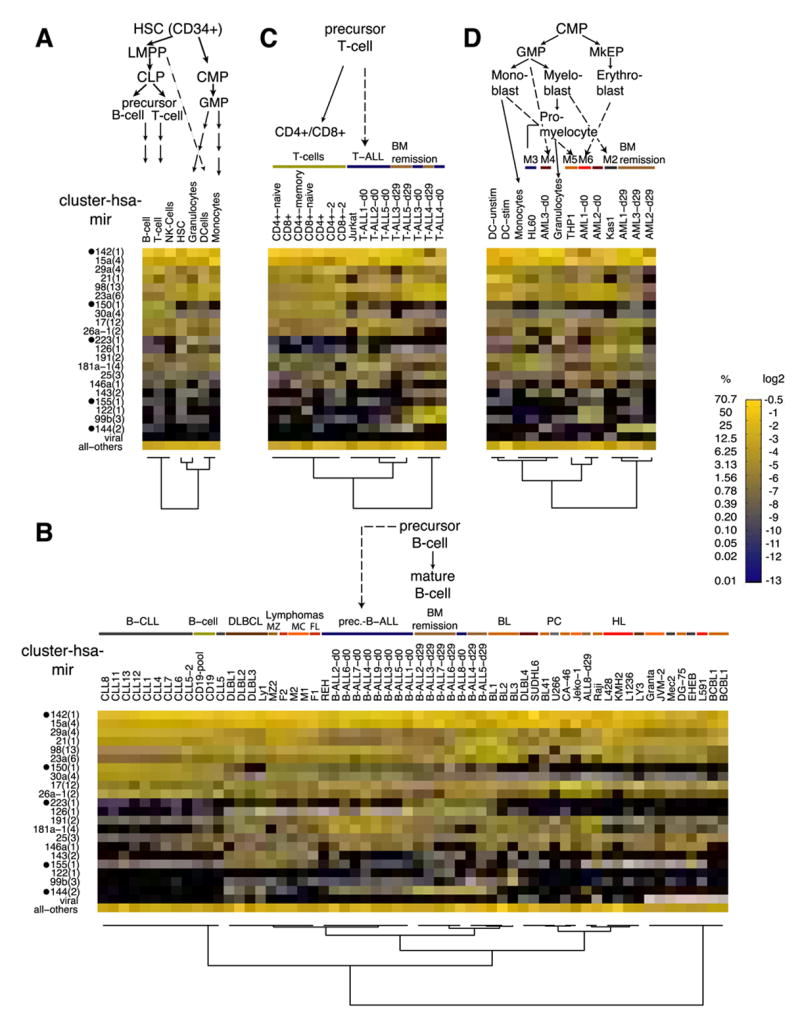
miRNA precursor clusters with at least 5% relative cloning frequency in at least one of the samples are depicted individually in a color code. All other miRNAs and viral miRNAs were pooled and are displayed together. Undetected miRNAs are displayed in black. Hematopoietic-specific miRNA clusters are marked by black dots. B-, T- and dendritic cell (DC) samples were combined and their composite profiles are shown in panel (A) while panel B, C, and D, respectively, show the individual samples. The lineage is indicated above each figure. Different disease entities and normal cells are indicated with color bars. Patient-derived samples are labeled with the disease name and patient number, while for cell lines the respective names are given. The FAB classification is given for the AML samples and the progenitor cells are indicated. Abbreviations: LMPP, lymphoid primed multipotent progenitor; CLP, common lymphoid progenitor; CMP, common myeloid progenitor; GMP, granulocyte/macrophage progenitor; MkEP, megakaryocyte/erythrocyte progenitor; BM remission, bone marrow in remission; DLBCL, diffuse large B-cell lymphoma; MZ, marginal zone; MC, mantle cell; FL, follicular lymphoma; BL, Burkitt’s lymphoma; PC, plasmocytoma; HL, Hodgkin lymphoma.
Hierarchical clustering (See Supplementary methods) shows that lymphoid cells cluster separately from stem cells and from cells derived from the myeloid lineage (Figure 3A). Patient samples and cell lines of the B-lymphoid lineage clustered in distinct subgroups mostly consistent with the diagnosis (Figure 3B). This was caused by variation in the expression of a small number of miRNAs including the mir-181a-1(4) cluster, absent from B-cell chronic lymphocytic leukemia (B-CLL) patient samples and mature B-cells, and miR-126, strongly expressed in precursor B-cell acute lymphoblastic leukemia (pre-B-ALL) samples, bone marrow from patients in remission and most germinal center-derived lymphoma (5.8% average relative cloning frequency), but not in the germinal center-derived Burkitt’s lymphoma and several lymphoma cell lines. In Burkitt’s and diffuse large B-cell lymphoma (DLBCL) miR-150 expression was reduced about 20-fold in comparison to other closely related germinal center malignancy. In the T-lymphoid lineage the miRNA expression profiles of sorted different mature T-cell types were similar and clustered together (Figure 3C). In contrast to patient samples for pre-B-ALL, the miRNA profiles from bone marrow samples of T-ALL patients before and after remission were not clearly distinct whereas in the myeloid lineage disease bone marrow samples clustered separately from samples of the patients in remission (Figure 3D). We did not find any striking differences in the miRNA expression profile of cell lines derived from different myeloid sublineages, except for the induction of expression of the granulocyte/monocyte-specific miR-223.
miRNA profiles of tissues and cells of the nervous system in human and rodents
To assess whether differential miRNA expression in the nervous system contributes to its complex cell type composition, we investigated expression of miRNAs in different brain regions in human, mouse and rat (Figure 4A, B and C, respectively). miR-9, miR-124, miR-128a and miR-128b were highly and specifically expressed in brain regions (38.6% relative cloning frequency) except for pituitary gland, that showed abundant expression of miR-7, miR-375, and the clusters of mir-141(2) and mir-200a(3) (19.8%) (Figure 4A). Cluster analysis based on the expression of orthologous miRNAs separates the human adult brain regions from those of adult rat brain, with the exception of the hippocampus region.
Figure 4. miRNA expression in the nervous system.
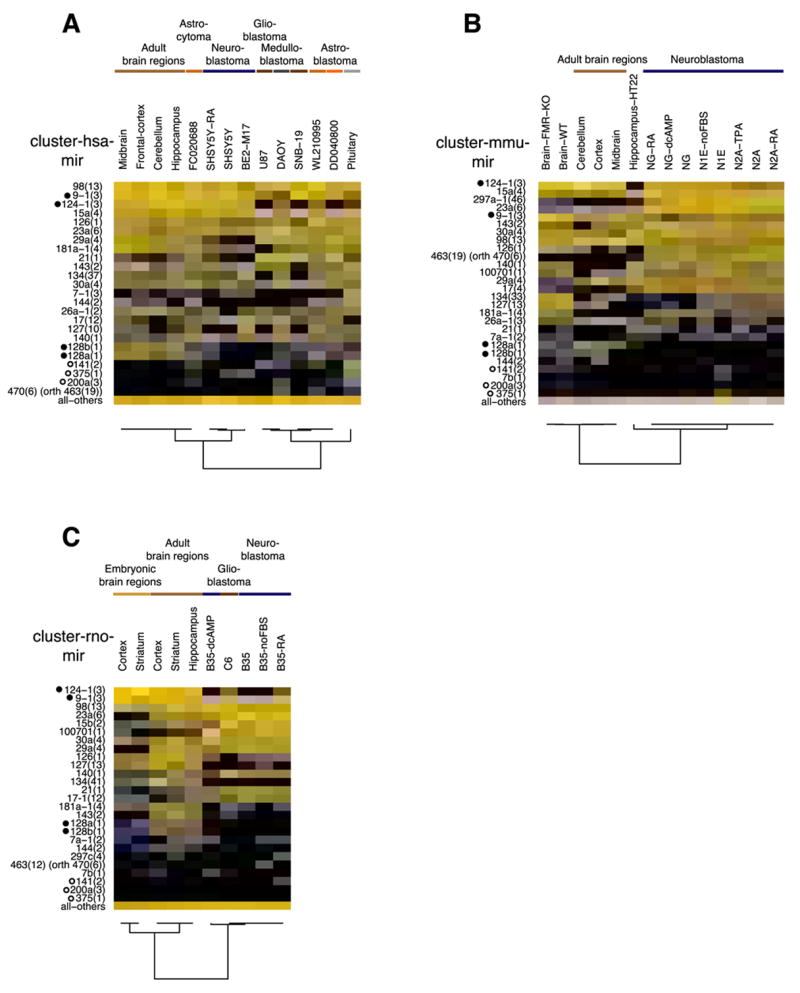
Expression profiles of miRNA precursor clusters with at least 5% relative expression in one of the samples as well as the neuronal-specific miRNA clusters (marked with black dot) are depicted individually for human (A), mouse (B) and rat (C) brain sections, brain tumor samples (FC0020688, WL210995, DD040800) and cell lines as indicated (color code of expression values see Figure 3). The open circles indicate miRNAs enriched in endocrine tissue including the pituitary gland. All panels show orthologous miRNA precursor clusters (if present). Abbreviations: dcAMP, dibutyryl cAMP; FBS, fetal bovine serum; TPA, trans-phorbol ester; RA, retinoic acid; orth, indicates that the hsa-mir-470 cluster is orthologous to the rodent mir-463 clusters.
To assess whether miRNA expression changes during embryonic development of the brain, we investigated miRNA profiles of embryonic and adult striatum and cortex in rat (Figure 4C). Adult and embryonic brain tissues clustered separately, due to expression of the mir-29a(4) cluster, which is absent in the embryonic tissues, but expressed at high frequency in the adult cortex (5.6%) and adult striatum (8.8%).
Analysis of miRNA profiles of cell lines and patient material derived from the central or the sympathetic nervous system revealed expression differences according to the glial or neuronal cellular origin of the tumor. The most striking difference was the near absence of the neuron-specific miR-124 in the glial tumors, and its presence in neuroblastoma derived from sympathetic neurons. Consistent with previous reports (Chan et al., 2005), miR-21 was 5- to 32-fold upregulated in all glial derived tumors or cell lines compared to midbrain (0.3%). The miRNA profiles of mouse neuroblastoma were consistent with their human counterpart, except for the presence of the mouse-specific cluster of mir-297a-1(46) (12.3% average cloning frequency), which was not expressed in normal mouse brain. Unexpectedly, the neuroblastoma cell line B35 from rat did not express miR-124 and its miRNA profile was very similar to the rat glioblastoma cell line C6.
Finally, in order to evaluate the role of Fragile X mental retardation protein (FMRP), which was shown to interact with Dicer and Ago protein in the miRNA pathway (Caudy et al., 2002; Jin et al., 2004; Plante et al., 2006), the miRNA profile of the whole brain of 4-day-old FMRP knockout mice and homozygous wild-type littermate were compared, but no differences in miRNA expression were observed (Figure 4B). FMRP apparently does not affect miRNA maturation or miRNP assembly and may at most play a role in downstream mRNA target recognition.
miRNA expression changes in cell differentiation experiments
As model systems for neuronal differentiation, we treated neuroblastoma (B35, SH-SY5Y, NG, N1E, and N2A) and embryonic carcinoma (NTera2) cell lines with dibutyryl cyclic AMP (cAMP), retinoic acid (RA), and FBS-deficient media until we observed the characteristic morphologic changes indicative of neuronal differentiation (Figure 5A). The most significant change in miRNA expression was observed after dibutyryl cAMP treatment of the rodent neuroblastoma cell line B35, when the frequency of the rat mir-16 cluster (mir-15b(2)) drops from 13.1% to 1.2%. The behavior of the human Ntera2 embryonic carcinoma cell line was unique in that RA-treatment turned on the expression of the miR-21 (8.9%), mir-141(2) (9.6%) and mir-371(46) (9.4%) clusters, while the neuronal miR-124 was only detected at low levels (1.4%) and was not induced.
Figure 5. miRNA expression in cell differentiation experiments.
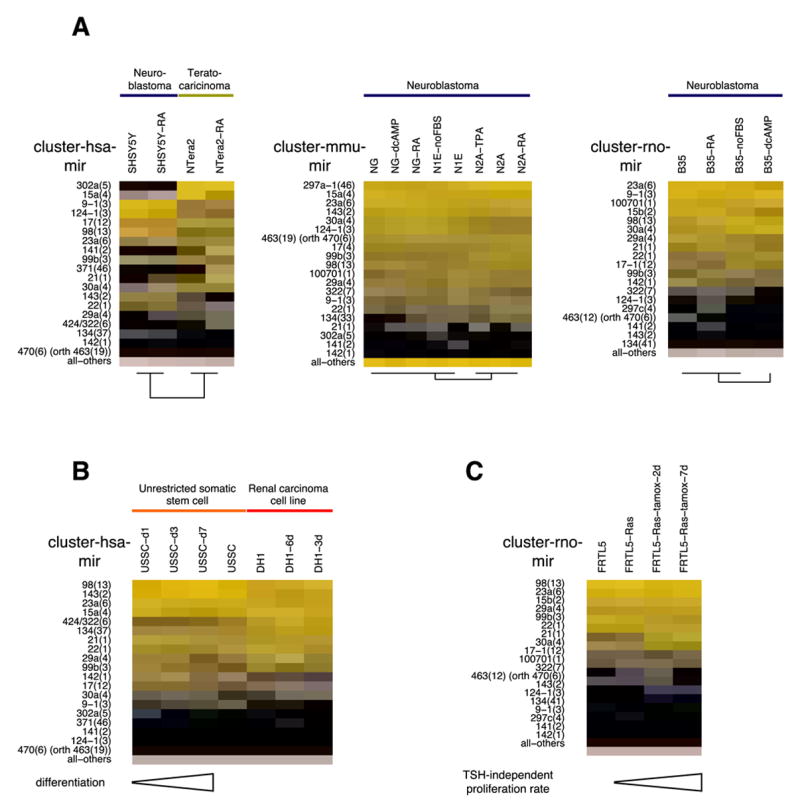
Cell lines were treated and differentiated into neurons (A) and osteoblasts (B). (C) A thyroid stimulating hormone (TSH)-dependent rat cell line FRTL5 was dedifferentiated by induction of a chimeric RAS protein. Expression profiles for orthologous miRNA precursor clusters with 5% or more relative cloning frequency in at least one sample are depicted in a color-coded manner (see Figure 3 for details). Abbreviations: dcAMP, dibutyryl cAMP; FBS, fetal bovine serum; TPA, trans-phorbol ester; RA, retinoic acid; USSC, human unrestricted somatic stem cell; orth, indicates that the hsa-mir-470 cluster is orthologous to the rodent mir-463 clusters.
To assess miRNA involvement in osteoblast differentiation, we differentiated unrestricted somatic stem cells (USSCs) obtained from umbilical cord blood (Koegler et al., 2004) as well as the Wilms tumor cell line (DH-1) for 1, 3, and 7 days and for 3 and 6 days, respectively (Figure 5B). The expression of the hematopoietic-specific miR-142 decreased from 5.0% to 0.3% after 7 days in USSCs while the let-7 clusters (mir-98(13)) increased 2-3 fold after 7 days in USSCs and after 3 days in the DH-1 cell line to 26.3% and 33%, respectively. In the latter there was a significant degree of apoptosis following differentiation, and the trend observed after day 3 was not followed beyond day 6.
We also examined a restricted thyroid cell line FRTL-5 that depends on the presence of thyroid-stimulating hormone (TSH) or after induction of a stable, ectopic tamoxifen-regulated RAS oncogene (De Vita et al., 2005). Ras induction leads to de-differentiation and to TSH-independent proliferation of the thyroid cells and to upregulation of miR-21 after 2 and 7 days from 0.3% to 4% and 11% total miRNA content, respectively (Figure 5C).
Interferon treatment was shown to induce hundreds of interferon-regulated genes (Stark et al., 1998; van Boxel-Dezaire et al., 2006). Treatment of human A549 and HeLa S3 cell lines with interferon α, β or γ, however, did not cause any substantial changes in miRNA profiles, while we confirmed by qRT/PCR the strong upregulation of interferon-induced OAS1 (50-fold) and IRF1 (200-fold) mRNAs (Figure S13).
Discussion
The estimated number of miRNA genes in mammalian genomes has been steadily increasing, reaching currently tens of thousands (Berezikov et al., 2005; Xie et al., 2005; Rigoutsos et al., 2006). These numbers appear to be supported, at least at first glance, by studies using deep sequencing technologies (Berezikov et al., 2006a; Berezikov et al., 2006b; Cummins et al., 2006; Kloosterman et al., 2006a) or specialized small RNA isolation and cloning procedures (Bentwich et al., 2005; Takada et al., 2006). Furthermore, the cell-type specific expression of some miRNAs suggested that many more miRNAs could surface if only a sufficiently large number of cell types or tissues were studied. In order to derive a miRNA gene expression atlas that sets into perspective the number and functional relevance of miRNA genes, we have taken the approach of cloning and sequencing over 250 small RNA libraries from different cell types and tissues, with ~1,000 miRNA clones per library. We provide the data as well as analysis tools on a web-accessible interface. We believe that these will be instrumental in guiding the rapidly developing field of miRNA biology.
We characterized the profiles of complex tissues, sorted cells, and individual cell lines and found on average ~70 miRNA genes expressed per sample. Only a small fraction of these appeared to be regulated in cell differentiation experiments. We did not find evidence for substantial induction of many of the new candidate miRNAs identified as rare cloning events in deep sequencing surveys, despite the analysis of over 330,000 small RNA clones obtained from 256 small RNA libraries from different cell types and tissues.
The large overall number of miRNA clones obtained from human, mouse and rat samples allowed us to analyze the processing patterns for each miRNA. We found that at least 40% of miRNA sequences deposited in miRBase (v8.2) do not represent the predominantly cloned sequence either because of differences in 3′ or 5′ end processing, or because of miRNA/miRNA* strand selection. In addition, we analyzed a large number of features of miRNA genes, including sequence conservation and their relationship to genomic repeats. This analysis led us to conclude that more than 97% of all miRNA clones originated from less than 300 miRNA genes per species, genes that satisfied a series of general properties expected to be met by prototypical miRNAs (Ambros et al., 2003; Bartel, 2004). Surprisingly, only 17% of all miRNA sequences identified by a recent deep-sequencing study (Cummins et al., 2006) were prototypical. The remaining miRNAs, though clearly originating from hairpin precursors, showed unusual maturation or sequence conservation patterns. With rare exceptions, these miRNAs were also found at low levels and/or did not exhibit cell-type specific expression. For many more recently reported miRNA candidates, we do not have cloning evidence. We speculate that these small RNAs originate from dsRNA structures that only accidentally enter the RNAi pathway, such as fold-back elements controlled by dsRNA deaminases or the binding sites of RNAi-unrelated dsRNA-binding proteins.
Several different methods have been used in the past years for miRNA expression profiling (reviewed in Aravin and Tuschl, 2005). Unfortunately, there was no universal or synthetic miRNA reference standard available to assess the correlation of signal intensities with the absolute miRNA concentration of the sample. We therefore compared some miRNA profiles from small RNA cloning to those obtained from our newly developed miRNA array platform as well as to absolute amounts determined by quantitative Northern blotting of a small subset of miRNAs. For the new miRNA array platform, we synthesized a universal miRNA standard, from which we also prepared small RNA libraries that we cloned and sequenced. Relative clone frequencies for a given miRNA correlate significantly with quantitative Northern blotting results for these miRNAs across samples, but we also detect a systematic bias in our library preparation method. When we compared our brain and liver cloning profiles with those obtained by various other methods in the literature, we generally observed a good correlation, suggesting that the discrepancies between actual miRNA concentrations and those measured by various high-throughput methods are intrinsic to the miRNA sequence and their specific secondary self-structure.
From our miRNA expression atlas we computed the tissue and cell type specificity of miRNA expression. Our data are in good agreement with previously published in situ analysis in mouse (Kloosterman et al., 2006b). A more detailed comparison of miRNA profiles originating from similar tissues showed differences not only in cell-type specific miRNAs, but also in ubiquitously expressed miRNAs. The relative expression of these ubiquitous miRNAs in different cell types is difficult to quantify by in situ analysis.
We included in our study a large number of cell lines and primary patient material from many cancers to define starting points for higher-throughput approaches like miRNA microarrays. Understanding deregulation of miRNA expression, and making decisions on the selection of candidates to examine in a certain disease context, is much more informative in the context of a large expression database. We have provided a thorough overview of miRNA profiles in diverse leukemia, lymphoma, and brain tumors, and defined a conservative set of relevant miRNA cistrons mediating miRNA regulation in these diseases.
Although we explicitly searched in all of our libraries for possible mutations in miRNAs that might impair or change function, we found only low abundant variations that could be explained by RNA editing events such as deamination or 3′ end processing of stable intermediates or miRNAs by transferases and poly(A) and poly(U) polymerases. The frequency of these modifications was low for the vast majority of miRNAs, suggesting that these modifications play a limited role in miRNA function.
In summary, we established a first mammalian miRNA gene atlas based on sequencing from small RNA libraries. We included miRNA profiles from a large collection of mammalian cell and tissue samples and we developed tools for miRNA profile analysis and presentation, which will assist future research on miRNA function and its implication in disease. This atlas will continue to grow in utility as additional libraries are cloned and sequenced.
Experimental Procedures
Details about the procedures and protocols used in our studies are given in the Supplemental Experimental Procedures. Here we summarize briefly the main features of our methods.
Cell lines and tissues
Cell lines were cultured according to guidelines provided by ATCC (www.atcc.org) and DSMZ (www.dsmz.de) and incubated at 37°C in 5% CO2. Cells were differentiated as published elsewhere with details and references given in the Supplemental Experimental Procedures.
Human tissue samples or RNA isolated thereof was obtained following the written consent of the person and/or legal guardian (where applicable) and the identity was obscured for reasons of privacy. Normal tissue was dissected postmortem. Human pancreatic islet tissue was obtained from pancreatic islet donors for transplantation. Sorted cells were obtained using magnetic DynaBeads (Dynal) according to the manufacturer’s instructions from peripheral blood of healthy donors that were stimulated with G-CSF. Brain tumor samples hsa_FC020688, hsa_WL210995, and hsa_DD040800 were obtained at diagnosis at the University Hospital of Giessen, Germany, and flash frozen in liquid nitrogen. The diagnosis of those was confirmed histologically. ALL, acute myeloblastic leukemia (AML) and Burkitt lymphoma patient samples were obtained from bone marrow aspirate at indicated time points while CLL patient samples were isolated from peripheral blood. Marginal zone lymphoma, follicular lymphoma and mantle cell lymphoma samples were obtained at diagnosis by tumor biopsy.
Total RNA isolation, small RNA library preparation, sequencing, and sequence annotation
Total RNA isolation and small RNA cloning was performed as described previously (Pfeffer et al., 2003). Most samples were cloned using pre-adenylated 3′ adapter oligonucleotides and the T4 RNA ligase Rnl2(1–249) as described (Pfeffer et al., 2005). The annotation procedure is outlined in Figure S1 in the Supplemental data.
Construction of miRNA precursor clusters
Previous studies indicate that miRNAs that are closely spaced in the genome are co-expressed presumably due to being part of the same transcription unit (Lee et al., 2004; Baskerville and Bartel, 2005). We have therefore defined clusters of potentially co-expressed precursors based on the relative genomic distance of the precursors or their sharing of a mature form.
Construction of miRNA sequence groups
Since miRNAs that are highly similar in sequence are likely to have similar functions, we grouped miRNAs based on their similarity over their entire length, and not only based on the sequence identity of the 5′ end of the miRNA (“seed” or “nucleus”).
Construction of the orthology table
We identified orthologous miRNA genes by mapping all human/mouse/rat precursors to the three genomes, and using the pairwise genome alignments provided by the UCSC Genome Informatics group (http://genome.cse.ucsc.edu) and the synteny information from the published synteny maps (Murphy et al., 2005).
Comparing miRNA expression between samples
miRNA samples were compared in a Bayesian framework. The distance between two samples was computed as a function of the relative ratio of likelihoods of two models, one assuming that the relative cloning frequency of each of the miRNAs was the same, the other that they are different between the two samples.
Hierarchical clustering of samples
We clustered samples hierarchically, using the distance measure defined above. At every step during the clustering procedure, we computed the number of miRNAs of a given type in the cluster as the sum of the counts for that miRNA type in all the samples in the cluster. We then grouped the two clusters whose distance is minimal among all pairs of clusters.
Comparative analysis of expression of miRNAs from the same and from different precursor clusters
All mature miRNAs for which we had at least 5 copies in at least 10 samples were used to investigate the correlation in expression of miRNAs that are and those that are not closely spaced in the genome. As a measure of correlation we used the Pearson correlation coefficient between the frequencies of any two miRNAs across tissues.
Analysis of miRNA tissue specificity
To quantify the specificity of the expression of mature miRNAs or miRNA precursor clusters in the various tissues types and samples, we first used the hierarchical tissue classification scheme to determine the clone count of each miRNA or miRNA precursor cluster for each tissue class. We then normalized the miRNA counts within each sample, and calculated a “normalized tissue enrichment” for each miRNA as the ratio of the normalized clone count for one tissue type relative to the total clone count. Finally, we calculated the information content of the “normalized tissue enrichment” distribution across tissues classes.
Sequence modification analysis
To identify sequence changes of mature miRNA sequences relative to their genomic sequence we first built a position-independent background model of all sequence changes (Figure S14) and then reported sequence changes that were significantly above the background in a chi-squared analysis. Those sequence changes with at least two observations at the same position in at least one library were collected for each individual sequence change (Tables S24, S25) and summarized by position relative to the mature form for all sequence changes (Table S26).
The tissue specificity or enrichment of the observed modifications was calculated in a manner analogous to the tissue specificity of miRNA expression described above. The computed information scores for each modification are given in Tables S27, S28 and modifications that occurred highly tissue-specifically are indicated in the modification Table (Tables S24, S25).
miRNA microarray design and hybridization
miRNA microarrays were designed as deoxyoligonucleotide arrays (Miltenyi Biotech) and hybridized using standards condition. Samples and a synthetic pool of 816 oligoribonucleotides were separately fluorescently labeled using miRCURY LNA microRNA Array labeling kit (Exiqon). The obtained data was submitted to GEO (accession number: GPL4867).
Supplementary Material
Acknowledgments
We thank R. Darnell, A. Tarakhovsky, S. A. Muljo, K. Rajewsky, L. van Dyk, F. Grässer, C. Rice, D. Ho, N. Rosen, W. Gerald, M. Ladanyi, C. Münz, M. Stoffel, M. Poy, P. Greengard, and Q. Zhao for providing cell lines and tissues (for details see Table S1), S. Tomiuk for advice on array design and J. Pena for critical review of the manuscript. U. Bissels and A. Bosio are employees of Miltenyi Biotec. T. Tuschl is a cofounder of Alnylam Therapeuticals and serves on its Scientific Advisory Board. P. Landgraf is supported by the Dr. Mildred Scheel Stiftung für Krebsforschung of the Deutsche Krebshilfe (German Cancer Aid) and A. Aravin by a FRAXA Research Foundation Fellowship. The project was supported by NIH grant P01 GM073047-01 and Howard Hughes Medical Institute to T. Tuschl, by the SNF grant 205321-105945 to M. Zavolan and by the Associazione Italiana per la Ricerca sul Cancro (AIRC) to R. Di Lauro.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ambros V, Bartel B, Bartel DP, Burge CB, Carrington JC, Chen X, Dreyfuss G, Eddy SR, Griffiths-Jones S, Marshall M, et al. A uniform system for microRNA annotation. RNA. 2003;9:277–279. doi: 10.1261/rna.2183803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravin A, Tuschl T. Identification and characterization of small RNAs involved in RNA silencing. FEBS Lett. 2005;579:5830–5840. doi: 10.1016/j.febslet.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Baskerville S, Bartel DP. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA. 2005;11:241–247. doi: 10.1261/rna.7240905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentwich I, Avniel A, Karov Y, Aharonov R, Gilad S, Barad O, Barzilai A, Einat P, Einav U, Meiri E, et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet. 2005;37:766–770. doi: 10.1038/ng1590. [DOI] [PubMed] [Google Scholar]
- Berezikov E, Guryev V, van de Belt J, Wienholds E, Plasterk RH, Cuppen E. Phylogenetic shadowing and computational identification of human microRNA genes. Cell. 2005;120:21–24. doi: 10.1016/j.cell.2004.12.031. [DOI] [PubMed] [Google Scholar]
- Berezikov E, Plasterk RH. Camels and zebrafish, viruses and cancer: a microRNA update. Hum Mol Genet. 2005;14(Suppl 2):R183–190. doi: 10.1093/hmg/ddi271. [DOI] [PubMed] [Google Scholar]
- Berezikov E, Thuemmler F, van Laake LW, Kondova I, Bontrop R, Cuppen E, Plasterk RH. Diversity of microRNAs in human and chimpanzee brain. Nat Genet. 2006a;38:1375–1377. doi: 10.1038/ng1914. [DOI] [PubMed] [Google Scholar]
- Berezikov E, van Tetering G, Verheul M, van de Belt J, van Laake L, Vos J, Verloop R, van de Wetering M, Guryev V, Takada S, et al. Many novel mammalian microRNA candidates identified by extensive cloning and RAKE analysis. Genome Res. 2006b;16:1289–1298. doi: 10.1101/gr.5159906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blow MJ, Grocock RJ, van Dongen S, Enright AJ, Dicks E, Futreal PA, Wooster R, Stratton MR. RNA editing of human microRNAs. Genome Biol. 2006;7:R27. doi: 10.1186/gb-2006-7-4-r27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Yeo G, Muotri AR, Kuwabara T, Gage FH. Noncoding RNAs in the mammalian mentral nervous system. Annu Rev Neurosci. 2006;29:77–103. doi: 10.1146/annurev.neuro.29.051605.112839. [DOI] [PubMed] [Google Scholar]
- Caudy AA, Myers M, Hannon GJ, Hammond SM. Fragile X-related protein and VIG associate with the RNA interference machinery. Genes & Dev. 2002;16:2491–2496. doi: 10.1101/gad.1025202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 Is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005;65:6029–6033. doi: 10.1158/0008-5472.CAN-05-0137. [DOI] [PubMed] [Google Scholar]
- Cullen BR. Viruses and microRNAs. Nat Genet. 2006;38(Suppl 1):S25–30. doi: 10.1038/ng1793. [DOI] [PubMed] [Google Scholar]
- Cummins JM, He Y, Leary RJ, Pagliarini R, Diaz LA, Jr, Sjoblom T, Barad O, Bentwich Z, Szafranska AE, Labourier E, et al. The colorectal microRNAome. Proc Natl Acad Sci USA. 2006;103:3687–3692. doi: 10.1073/pnas.0511155103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vita G, Bauer L, da Costa VM, De Felice M, Baratta MG, De Menna M, Di Lauro R. Dose-dependent inhibition of thyroid differentiation by RAS oncogenes. Mol Endocrinol. 2005;19:76–89. doi: 10.1210/me.2004-0172. [DOI] [PubMed] [Google Scholar]
- Diederichs S, Haber DA. Sequence variations of microRNAs in human cancer: alterations in predicted secondary structure do not affect processing. Cancer Res. 2006;66:6097–6104. doi: 10.1158/0008-5472.CAN-06-0537. [DOI] [PubMed] [Google Scholar]
- Eisenberg E, Nemzer S, Kinar Y, Sorek R, Rechavi G, Levanon EY. Is abundant A-to-I RNA editing primate-specific? Trends Genet. 2005;21:77–81. doi: 10.1016/j.tig.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Farh KK, Grimson A, Jan C, Lewis BP, Johnston WK, Lim LP, Burge CB, Bartel DP. The widespread impact of mammalian microRNAs on mRNA repression and evolution. Science. 2005;310:1817–1821. doi: 10.1126/science.1121158. [DOI] [PubMed] [Google Scholar]
- Gottwein E, Cai X, Cullen BR. A novel assay for viral microRNA function identifies a single nucleotide polymorphism that affects Drosha processing. J Virol. 2006;80:5321–5326. doi: 10.1128/JVI.02734-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai N, Naraba H. Polymorphisms in human pre-miRNAs. Biochem Biophys Res Commun. 2005;331:1439–1444. doi: 10.1016/j.bbrc.2005.04.051. [DOI] [PubMed] [Google Scholar]
- Jin P, Zarnescu DC, Ceman S, Nakamoto M, Mowrey J, Jongens TA, Nelson DL, Moses K, Warren ST. Biochemical and genetic interaction between the fragile X mental retardation protein and the microRNA pathway. Nat Neurosci. 2004;7:113–117. doi: 10.1038/nn1174. [DOI] [PubMed] [Google Scholar]
- Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science. 2007;315:1137–1140. doi: 10.1126/science.1138050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DD, Kim TT, Walsh T, Kobayashi Y, Matise TC, Buyske S, Gabriel A. Widespread RNA editing of embedded alu elements in the human transcriptome. Genome Res. 2004;14:1719–1725. doi: 10.1101/gr.2855504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloosterman WP, Steiner FA, Berezikov E, de Bruijn E, van de Belt J, Verheul M, Cuppen E, Plasterk RH. Cloning and expression of new microRNAs from zebrafish. Nucleic Acids Res. 2006a;34:2558–2569. doi: 10.1093/nar/gkl278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloosterman WP, Wienholds E, de Bruijn E, Kauppinen S, Plasterk RH. In situ detection of miRNAs in animal embryos using LNA-modified oligonucleotide probes. Nat Methods. 2006b;3:27–29. doi: 10.1038/nmeth843. [DOI] [PubMed] [Google Scholar]
- Kluiver J, Kroesen BJ, Poppema S, van den Berg A. The role of microRNAs in normal hematopoiesis and hematopoietic malignancies. Leukemia. 2006;20:1931–1936. doi: 10.1038/sj.leu.2404387. [DOI] [PubMed] [Google Scholar]
- Koegler G, Sensken S, Airey JA, Trapp T, Muschen M, Feldhahn N, Liedtke S, Sorg RV, Fischer J, Rosenbaum C, et al. A new human somatic stem cell from placental cord blood with intrinsic pluripotent differentiation potential. J Exp Med. 2004;200:123–135. doi: 10.1084/jem.20040440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krützfeldt J, Poy MN, Stoffel M. Strategies to determine the biological function of microRNAs. Nat Genet. 2006;38(Suppl 1):S14–19. doi: 10.1038/ng1799. [DOI] [PubMed] [Google Scholar]
- Krützfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Meyer J, Borkhardt A, Tuschl T. New microRNAs from mouse and human. RNA. 2003;9:175–179. doi: 10.1261/rna.2146903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002;12:735–739. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294:858–862. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science. 2001;294:862–864. doi: 10.1126/science.1065329. [DOI] [PubMed] [Google Scholar]
- Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23:4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Shih I, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- Linsley PS, Schelter J, Burchard J, Kibukawa M, Martin MM, Bartz SR, Johnson JM, Cummins JM, Raymond CK, Dai H, et al. Transcripts targeted by the microRNA-16 family cooperatively regulate cell cycle progression. Mol Cell Biol. 2007;27:2240–2245. doi: 10.1128/MCB.02005-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciano DJ, Mirsky H, Vendetti NJ, Maas S. RNA editing of a miRNA precursor. RNA. 2004;10:1174–1177. doi: 10.1261/rna.7350304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallory AC, Vaucheret H. Functions of microRNAs and related small RNAs in plants. Nat Genet. 2006;38(Suppl 1):S31–36. doi: 10.1038/ng1791. [DOI] [PubMed] [Google Scholar]
- Moss EG, Lee RC, Ambros V. The cold shock domain protein LIN-28 controls developmental timing in C. elegans and is regulated by the lin-4 RNA. Cell. 1997;88:637–646. doi: 10.1016/s0092-8674(00)81906-6. [DOI] [PubMed] [Google Scholar]
- Murphy WJ, Larkin DM, Everts-van der Wind A, Bourque G, Tesler G, Auvil L, Beever JE, Chowdhary BP, Galibert F, Gatzke L, et al. Dynamics of mammalian chromosome evolution inferred from multispecies comparative maps. Science. 2005;309:613–617. doi: 10.1126/science.1111387. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Baldwin DA, Kloosterman WP, Kauppinen S, Plasterk RH, Mourelatos Z. RAKE and LNA-ISH reveal microRNA expression and localization in archival human brain. RNA. 2006;12:187–191. doi: 10.1261/rna.2258506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obernosterer G, Leuschner PJ, Alenius M, Martinez J. Post-transcriptional regulation of microRNA expression. RNA. 2006;12:1161–1167. doi: 10.1261/rna.2322506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquinelli AE, Reinhart BJ, Slack F, Martindale MQ, Kuroda MI, Maller B, Hayward DC, Ball ED, Degnan B, Müller P, et al. Conservation of the sequence of let-7 heterochronic regulatory RNA. Nature. 2000;408:86–89. doi: 10.1038/35040556. [DOI] [PubMed] [Google Scholar]
- Pfeffer S, Lagos-Quintana M, Tuschl T. Cloning of small RNA molecules. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidmann JG, Smith JA, Struhl K, editors. Current Protocols in Molecular Biology. New York: John Wiley and Sons; 2003. pp. 26.24.21–26.24.18. [Google Scholar]
- Pfeffer S, Sewer A, Lagos-Quintana M, Sheridan R, Sander C, Zavolan M, Grässer FA, van Dyk LF, Ho CK, Shuman S, et al. Identification of microRNAs of the herpesvirus family. Nature Meth. 2005;2:269–276. doi: 10.1038/nmeth746. [DOI] [PubMed] [Google Scholar]
- Plante I, Davidovic L, Ouellet DL, Gobeil LA, Tremblay S, Khandjian EW, Provost P. Dicer-derived microRNAs are utilized by the Fragile X Mental Retardation Protein for assembly on target RNAs. J Biomed Biotechnol. 2006;2006:64347. doi: 10.1155/JBB/2006/64347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plasterk RH. Micro RNAs in animal development. Cell. 2006;124:877–881. doi: 10.1016/j.cell.2006.02.030. [DOI] [PubMed] [Google Scholar]
- Poy MN, Eliasson L, Krutzfeldt J, Kuwajima S, Ma X, Macdonald PE, Pfeffer S, Tuschl T, Rajewsky N, Rorsman P, Stoffel M. A pancreatic islet-specific microRNA regulates insulin secretion. Nature. 2004;432:226–230. doi: 10.1038/nature03076. [DOI] [PubMed] [Google Scholar]
- Rajewsky N. microRNA target predictions in animals. Nat Genet. 2006;38(Suppl 1):S8–S13. doi: 10.1038/ng1798. [DOI] [PubMed] [Google Scholar]
- Rigoutsos I, Huynh T, Miranda K, Tsirigos A, McHardy A, Platt D. Short blocks from the noncoding parts of the human genome have instances within nearly all known genes and relate to biological processes. Proc Natl Acad Sci USA. 2006;103:6605–6610. doi: 10.1073/pnas.0601688103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivdasani RA. MicroRNAs: regulators of gene expression and cell differentiation. Blood. 2006;108:3646–3653. doi: 10.1182/blood-2006-01-030015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalheiser NR, Torvik VI. Mammalian microRNAs derived from genomic repeats. Trends Genet. 2005;21:322–326. doi: 10.1016/j.tig.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Sood P, Krek A, Zavolan M, Macino G, Rajewsky N. Cell-type-specific signatures of microRNAs on target mRNA expression. Proc Natl Acad Sci USA. 2006;103:2746–2751. doi: 10.1073/pnas.0511045103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark A, Brennecke J, Bushati N, Russell RB, Cohen SM. Animal MicroRNAs confer robustness to gene expression and have a significant impact on 3′UTR evolution. Cell. 2005;123:1133–1146. doi: 10.1016/j.cell.2005.11.023. [DOI] [PubMed] [Google Scholar]
- Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- Takada S, Berezikov E, Yamashita Y, Lagos-Quintana M, Kloosterman WP, Enomoto M, Hatanaka H, Fujiwara SI, Watanabe H, Soda M, et al. Mouse microRNA profiles determined with a new and sensitive cloning method. Nucleic Acids Res. 2006;34:e115. doi: 10.1093/nar/gkl653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson JM, Newman M, Parker JS, Morin-Kensicki EM, Wright T, Hammond SM. Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes & Dev. 2006;20:2202–2207. doi: 10.1101/gad.1444406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Boxel-Dezaire AH, Rani MR, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity. 2006;25:361–372. doi: 10.1016/j.immuni.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Wienholds E, Kloosterman WP, Miska E, Alvarez-Saavedra E, Berezikov E, de Bruijn E, Horvitz RH, Kauppinen S, Plasterk RH. MicroRNA expression in zebrafish embryonic development. Science. 2005;309:310–311. doi: 10.1126/science.1114519. [DOI] [PubMed] [Google Scholar]
- Wienholds E, Plasterk RH. MicroRNA function in animal development. FEBS Lett. 2005;579:5911–5922. doi: 10.1016/j.febslet.2005.07.070. [DOI] [PubMed] [Google Scholar]
- Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75:855–862. doi: 10.1016/0092-8674(93)90530-4. [DOI] [PubMed] [Google Scholar]
- Xie X, Lu J, Kulbokas EJ, Golub TR, Mootha V, Lindblad-Toh K, Lander ES, Kellis M. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature. 2005;434:338–345. doi: 10.1038/nature03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Chendrimada TP, Wang Q, Higuchi M, Seeburg PH, Shiekhattar R, Nishikura K. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat Struct Mol Biol. 2006;13:13–21. doi: 10.1038/nsmb1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.