

Abstract
The initiation of viral RNA replication by the transfection of viral RNA is an integral tool in dissecting the life cycles, susceptibility, and pathogenesis of numerous RNA viruses. Many different transfection methods deliver viral RNA into mammalian cells, including DEAE-dextran and lipid-based reagents, but electroporation is one of the most popular methods. Unfortunately, electroporation suffers from many limitations, including high cell death, serum-free transfection conditions, and requires many cells and relatively large amounts of RNA. To optimize and facilitate the introduction of viral RNAs into mammalian cells, different commercially available RNA transfection reagents were compared for their ability to deliver yellow fever virus (YFV) and hepatitis C virus (HCV) RNA replicons into Huh7 cells. The performance of the commercial transfection reagents was also compared directly to electroporation. When properly optimized, certain reagents were superior to electroporation, with much less cell death, less RNA required and increased transfection efficiency. The factors associated with high efficiency transfection, and the advantage of being able to deliver RNA in the presence of serum are discussed.
Keywords: viral, RNA, transfection, electroporation, lipid, HCV
1. Introduction
RNA viruses are important causes of many different human and animal diseases. Researchers are continually investigating how viruses enter and exit cells, replicate and cause disease. Molecular clones are critical to understanding these processes as well as developing effective therapies and vaccines. While for some viruses infectious RNA can be generated in situ by delivery of cDNA and a DNA-dependent RNA polymerase, for other viruses a more physiologically relevant delivery method is preferred, or even required. One of the great breakthroughs in the study of positive-strand RNA viruses was the discovery that transfection of genomic RNA synthesized by in vitro transcription of a cDNA copy of the RNA genome could initiate the viral life cycle and produce infectious virus (Kaplan et al., 1985). This technique allowed researchers to create defined mutations within a viral cDNA clone and then produce viruses with specific mutations. This advance greatly facilitated the study of positive-strand RNA viruses and the roles of specific viral genes and RNA regulatory sequences in viral life cycles. The production of virus via transfection of genomic RNA has been applied to the study of many different positive strand RNA viruses, including but not limited to, poliovirus (Kaplan et al., 1985), yellow fever virus (YFV) (Rice et al., 1989), dengue virus (Lai et al., 1991), and hepatitis C virus (HCV) (Lindenbach et al., 2005).
Many different methods are used to deliver viral RNA to mammalian cells, including DEAE-dextran, commercial transfection reagents (lipid and/or polymer based) and electroporation. Researchers often use electroporation to deliver viral RNAs to mammalian cells, and we have routinely used this method to deliver HCV and YFV mini-replicon RNAs to Huh7 cells. Unfortunately, there are many disadvantages to using electroporation over other transfection methods, including the need for large cell numbers, large amounts of RNA, serum-free transfection conditions, and the high level of cell death. To facilitate our HCV and YFV studies, we investigated the efficacy of several commercially available RNA transfection reagents. Our hypothesis was that the commercially available transfection reagents would not be subject to the limitations of electroporation, and we could identify a reagent that would facilitate high transfection efficiency with low cellular toxicity. To test our hypothesis, we evaluated three commercial RNA transfection reagents: DMRIE-C Reagent (Invitrogen, Carlsbad, CA), TransMessenger™ Reagent (Qiagen Inc.-USA, Valencia, CA), and the TransIT®-mRNA Transfection Kit (Mirus Bio Corporation, Madison, WI) in viral RNA transfection assays. While all transfection reagents required fewer cells and less RNA than electroporation in both transient and stable transfection assays, the TransIT®-mRNA Transfection Kit had the broadest applicability in both assay conditions. In direct comparisons, two commercially available transfection reagents had higher signal to noise ratio with much less cell death than electroporation.
2. Materials and methods
2.1 Cell culture
The human hepatoma cell line, Huh7, (kindly provided by Stanley Lemon) was grown in Dulbecco's Modification of Eagle's Medium (DMEM) (Mediatech, Inc., Herndon, VA) supplemented 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA) and 100 units of penicillin and 10 μg/ml streptomycin (Invitrogen) in a humidified incubator at 37°C and 5% CO2.
2.2 Viral replicons and RNA synthesis
The yellow fever virus replicons YF-R.Luc2A-RP, YF-IRES-Luc (ΔDD), and YF-GFP2A-RP (Jones et al., 2005) (kindly provided by Richard Kuhn) were derived from pACNR/FLYF, a full length cDNA clone of YFV 17D (genus Flavivirus, family Flaviviridae) (Bredenbeek et al., 2003). These plasmids were linearized with XhoI and then purified using QIAEX II Kit (Qiagen Inc.-USA) before serving as templates for the in vitro transcription reactions. The hepatitis C virus replicon Ntat2ANeo(SI) (kindly provided by Stanley Lemon) (Yi et al., 2002) was derived from the HCV 1bN cDNA clone (genus Hepaciviruses, family Flaviviridae) (Ikeda et al., 2002). The Ntat2ANeo(SI)(GND) replication defective mutant was created by site-directed mutagenesis of the Ntat2ANeo(SI) plasmid. Amino acid 318 of the NS5B gene was mutated from aspartic acid to asparagine using oligos 5′-CACGATGCTCGTGAACGGGAACGACCTTGTCGTTATCTG-3′ and 5′-CGACAAGGTCGTTCCCGTTCACGAGCATCGTGCAGT-3′. The Ntat2ANeo plasmids were linearized with XbaI and purified with the QIAEX II kit before serving as templates for in vitro transcription. The YFV RNA replicons were synthesized by in vitro transcription of the linearized YFV replicon plasmids using the mMESSAGE mMACHINE SP6 Kit (Ambion, Austin, TX) according to the manufacturer's recommendations. The HCV RNA replicons were in vitro transcribed using the linearized HCV replicon plasmids and the mMESSAGE mMACHINE T7 Kit (Ambion). After RNA synthesis was complete, the in vitro transcription reactions were treated with 1 μl of RNase-free DNase (Ambion) at 37°C for 15 min to degrade the DNA templates, and the RNA was then purified from each reaction by LiCl precipitation.
2.3 Viral RNA transfections using commercial transfection reagents
2.3.1 Initial optimization transfections in 12-well plates
Huh7 cells were plated in 12-well plates (2.5 × 105 cells/well) 24 hours prior to transfection and were approximately 80% confluent at the time of transfection. This level of confluency is in agreement with the recommended transfection conditions of all three reagent protocols. Transfection of cells at confluencies below the manufacturers' recommended range correlated with lower efficiency than the results presented here (data not shown). The viral RNA replicons were transfected into the Huh7 cells using three different RNA transfection reagents, DMRIE-C, TransMessenger™, and the TransIT-mRNA Kit. All transfections were performed in parallel on the same day. Transfections were performed according to the manufacturer's recommended protocols. Varying the amount of RNA within the manufacturer's suggested range had little or no effect on luciferase yield. For each transfection condition tested, two separate complex tubes were set up. Each tube had a 3X master mix that was aliquoted into triplicate wells; there were a total of 6 samples per condition. Briefly, the DMRIE-C transfections were performed by first washing the cells with 1 ml of Opti-MEM Reduced Serum Media (Invitrogen). Opti-MEM (0.5 ml) was then added to a sterile polystyrene tube, followed by 1, 2, 3, or 4 μl of DMRIE-C, and mixed gently. The viral RNA (1 μg) was added to each tube, mixed and immediately added drop wise to the appropriate well. The cells were incubated for 6 hours at 37°C and 5% CO2, and then the medium containing the transfection complexes was removed from each well and replaced with 1 ml of complete growth medium (DMEM with 10% serum). The TransMessenger™ Reagent transfections were performed by first diluting 2 μl of Enhancer R in 46 μl of Buffer EC-R in a sterile polystyrene tube. One microgram of viral RNA was added to the tube and mixed. The mixture was incubated at room temperature for 5 min. Next, 2, 4, 6, or 8 μl of TransMessenger™ Reagent was added to each tube, mixed, and incubated for 10 min at room temperature. While the transfection complexes were forming, the cells were washed with 2 ml/well of sterile phosphate buffered saline (PBS). After removal of the PBS, 450 μl of Opti-MEM was added to each well, and the transfection complexes were added drop wise to each well. The cells were then incubated for 6 hours at 37°C and 5% CO2. The medium was removed from each well, the cells were gently washed with PBS, and then 1 ml of complete growth medium (DMEM with 10% serum) was added to each well. The TransIT-mRNA Kit transfections were performed by first adding 1 μg of viral RNA to 100 μl of Opti-MEM in a sterile polystyrene tube. Immediately the mRNA Boost Reagent was added to each tube followed by the TransIT-mRNA Reagent. The following amounts of each reagent were used based on the protocol; 0.5/0.5, 0.5/1, 0.5/1.5, 1/1, 1/2, 1/3 (μl of mRNA Boost Reagent/μl of TransIT-mRNA Reagent). The tubes were incubated at room temperature for 3 min, and the complexes were added drop wise to the cells in complete growth media with no media change. Following transfection, all the transfected cells were incubated at 37°C and 5% CO2 for 24 hours and then harvested for luciferase assays. Based on these assays, transfections were optimized for 6-well plates by doubling all reagents, including media components, cells, RNA and transfection reagents.
2.4 Colony forming assay
Huh7 cells were plated in 6-well plates (5 × 105 cells/well) 24 hours prior to transfection and were approximately 90% confluent at the time of transfection. The HCV Ntat2ANeo(SI) and Ntat2ANeo(SI)(GND) RNA replicons were transfected into duplicate wells of Huh7 cells using optimal DMRIE-C, TransMessenger™ Reagent, and the TransIT-mRNA Kit conditions found in section 2.3.1. Twenty-four hours post-transfection, each well of transfected cells was trypsinized and resuspended in fresh complete growth media containing 0.5 mg/ml G418 (Invitrogen). Each well of trypsinized cells was plated in two 10 cm dishes in the presence of G418. One dish received 1/10 of the cells and the other received 9/10 of the cells. The cells were grown for one week in 0.5 mg/ml G418 and then shifted to 1.0 mg/ml G418 containing media for 2 additional weeks, during which time the media was changed every 72 hours. The G418 resistant colonies that developed were visualized by staining with Crystal Violet (Sigma Aldrich, St. Louis, MO) as previously described (Kato et al., 2005), and the number of colonies on the 1/10 plates were determined and averaged for each set of duplicates.
2.5 Viral RNA electroporation
Subconfluent Huh7 cells in a T-75 flask (4 × 106 cells) were trypsinized, washed twice with Hank's Buffered Salt Solution (Cellgro Mediatech, Herndon, VA), and resuspended in 500 μl of PBS. The cells were transferred to a 4 mm gap electroporation cuvette (Molecular Bio Products, San Diego, CA). Five micrograms of either the YF-GFP2A-RP or the YF-R.Luc2A-RP RNAs was added to a cuvette and electroporated with two pulses at 100 V, 125Ω, and 2300 μF using an ECM630 Electroporator (BTX Molecular Delivery Systems, Holliston, MA). After electroporation, the cells were allowed to recover at room temperature for 5 min, resuspended in 2 ml of complete growth media, and plated in 6-well plates. Cells were incubated at 37°C and 5% CO2 for 24 hours, and then the media was replaced with fresh complete growth media. The plates were incubated another 24 hours at 37°C at which time they were approximately 30% confluent. Cells were harvested and subjected to luciferase assays or flow cytometry analyses.
2.6 Luciferase assays
YF-R.Luc2A-RP, or YF-IRES-Luc (ΔDD) transfected Huh7 cells were washed with PBS (1 ml per 12-well and 2 ml per 6-well). The cells were lysed by the addition of Cell Lysis Buffer (100 μl per 12-well and 250 μl per 6-well) (Promega Corp., Madison, WI) to each well followed by a 15-minute incubation at room temperature. Renilla luciferase assays were performed using the Renilla Luciferase Assay System (Promega). Briefly, 5 μl of each cell lysate was added to 100 μl of Luciferase Assay Reagent I (LARI) and measured in a DLReady TD-20/20 Turner Designs Luminometer (Turner Biosystems, Sunnyvale, CA). In the comparison of the electroporation to the TransIT-mRNA Transfection Kit, the concentration of protein in each sample was determined using the BCA™ Protein Assay Kit (Pierce, Rockford, IL) according to the manufacturer's recommendations. The amount of luciferase activity in each sample was then normalized to the amount of protein in the sample to take into account the low cellular density of the electroporated cells at harvest compared to the TransIT-mRNA Kit transfected cells.
2.7 Fluorescence microscopy and flow cytometry analysis
2.7.1 Fluorescence microscopy
The YF-GFP2A-RP and YF-IRES-Luc (ΔDD) RNA transfected cells were washed once with 1 ml of PBS, followed by addition of 2 ml of PBS. Both phase contrast and GFP fluorescent images of the transfected or electroporated cells were acquired using an inverted fluorescence microscope with a 10X lens and digital images were collected using a Zeiss Axiocam MRC digital camera (Axio Cam MRm, Zeiss, Germany) and Axio-Vision software (Axio Cam MRm, Zeiss, Germany).
2.7.2 Flow cytometry analysis
Huh7 cells electroporated or transfected with the YF-GFP2A-RP or YF-IRES-Luc (ΔDD) RNA replicons were harvested by trypsinization and washed twice with PBS. The cells were resuspended in 1 ml of PBS, and subjected to flow cytometry on a LSR Flow Cytometer (Becton Dickinson, Franklin Lakes, NJ). The YF-IRES-Luc (ΔDD) transfected cells served as the negative GFP control and were used to set the gates to identify the GFP positive cells.
3. Results
3.1 Comparison of three different commercial RNA transfection reagents
3.1.1 Transfection of the YF-R.luc2A-RP RNA replicon
To identify a commercial transfection reagent that might outperform electroporation, three commercially available transfection reagents capable of delivering large RNA molecules to mammalian cells were tested. DMRIE-C, TransMessenger™ Reagent, and the TransIT-mRNA Transfection Kit were tested in parallel for their ability to transfect the yellow fever virus YF-R.Luc2A-RP RNA replicon. A range of reagent concentrations were tested based on manufacturer's recommendations to test the performance of each reagent and identify the optimal conditions for transfecting the YF-R.Luc2A-RP RNA into Huh7 cells. As expected, all three transfection reagents were capable of delivering the YFV RNA replicon to Huh7 cells (Fig. 1). The TransIT-mRNA Kit was the most efficient at transfecting the replicon RNA, producing levels of Renilla luciferase that were approximately 250% and 84% greater than the DMRIE-C and TransMessenger™ Reagent transfected cells, respectively. The optimal TransIT-mRNA Kit transfection conditions per well of a 12-well plate were 2 μl TransIT-mRNA Reagent, 1 μl mRNA Boost Reagent, and 1 μg of replicon RNA.
Fig. 1.
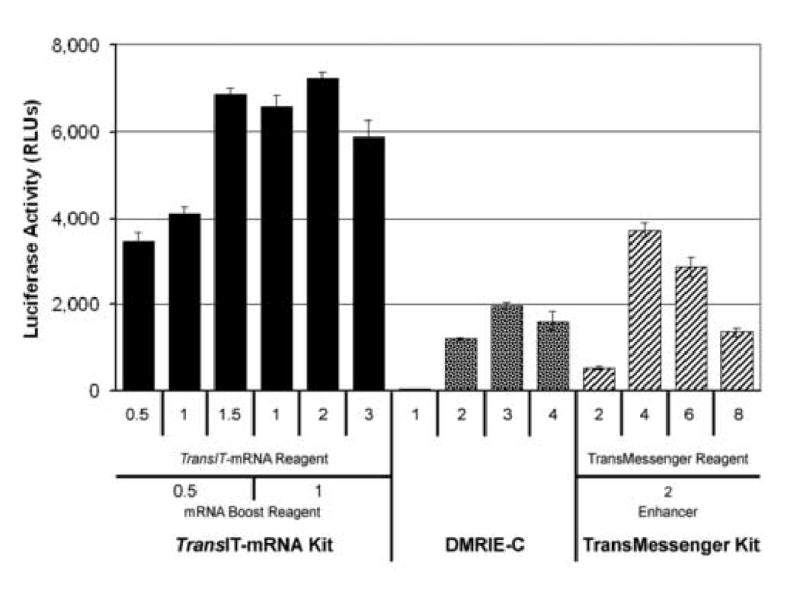
Comparison of three commercially available RNA transfection reagents. Huh7 cells in 12-well plates were transfected with 1 μg of YF-R.Luc2A-RP RNA per well in parallel using either the TransIT®-mRNA Transfection Kit, DMRIE-C, or TransMessenger™ Reagent. The amounts of each reagent tested are indicated. Each transfection condition was tested in duplicate, and each complex formation tube was set up as a 3X master mix and transfected into three wells of a 12-well plate for a total of six wells per condition. Cells were harvested 24 hours post-transfection and assayed for Renilla luciferase activity. The results presented are the average of the 6 wells transfected per condition.
3.1.2 Colony forming assay using the HCV Ntat2ANeo(SI) RNA replicon
Because the optimal delivery method for transient transfections may differ from the optimal method for stable transfections, a colony forming assay was performed in which the HCV Ntat2ANeo(SI) and Ntat2ANeo(SI)(GND) RNA replicons were transfected into Huh7 cells using the optimal transfection conditions for each transfection reagent determined previously (section 3.1.1) (Fig. 1). The Ntat2ANeo(SI) replicon replicates in transfected cells and produces G418 resistant cells, while the polymerase mutant Ntat2ANeo(SI)(GND) replicon does not replicate and served as a negative control. After transfecting Huh7 cells with each RNA replicon, the cells were split and replated in media containing G418 and allowed to grow for three weeks. The cultures were then stained with Crystal Violet to visualize the G418 resistant colonies formed (Fig. 2a). Very few colonies formed after transfection of the Ntat2ANeo(SI)(GND) negative control replicon, indicating that the G418 selection was effective (Fig. 2a). Many colonies formed after transfection of the Ntat2ANeo(SI) RNA with the three different transfection reagents. In this case, the TransIT-mRNA Kit produced a larger number of colonies than the DMRIE-C and TransMessenger™ Reagents by 20% and 53%, respectively (Fig 2b).
Fig. 2.
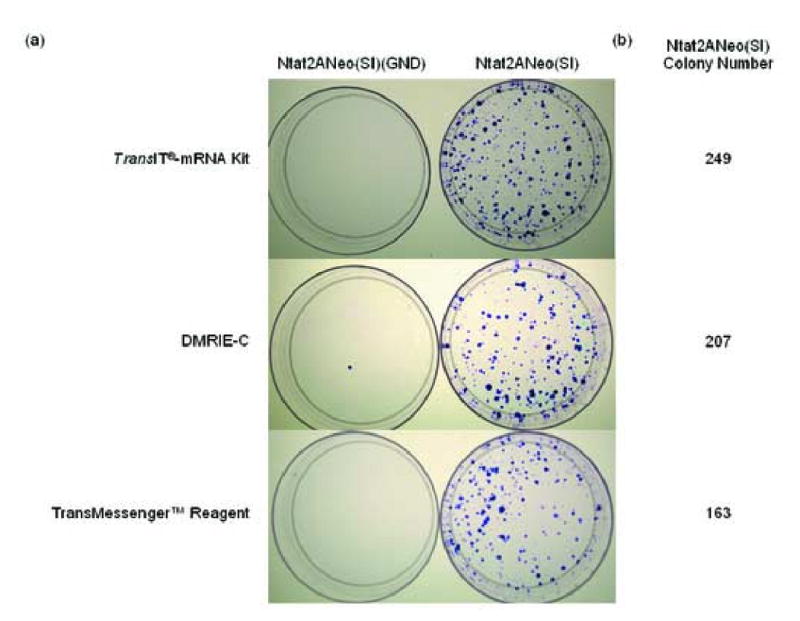
Performance of the RNA transfection reagents in a colony formation assay. Huh7 cells in 6-well plates were transfected in duplicate with 2 μg of the Ntat2ANeo(SI) RNA or the negative control Ntat2ANeo(SI)(GND) RNA using the TransIT®-mRNA Transfection Kit, DMRIE-C, or TransMessenger™ Reagent. Twenty-four hours post-transfection, each well of transfected cells was trypsinized and 1/10 of the trypsinized cells were plated in 10 cm dishes in the presence of G418 to select for cells stably replicating the Ntat2ANeo(SI) replicon. Three weeks post-transfection, the 10 cm dishes were stained with crystal violet blue to visualize the G418 resistant colonies. (a) Colony formation after transfection using each of the three transfection reagents. (b) Average number of colonies formed from the duplicate transfections using the three transfection reagents.
In summary, the three commercial transfection reagents were effective in both transient and stable RNA transfections, but the efficiency was both assay and reagent dependent. The TransIT-mRNA Transfection Kit outperformed the DMRIE-C and the TransMessenger™ Reagents in both sets of tests, although the difference in the colony-forming assay was marginal.
3.2 Comparison of TransIT-mRNA Kit transfections to electroporation
3.2.1 YFV luciferase replicon delivery comparison
Electroporation has commonly been used successfully to deliver viral RNA, however since many HCV replicons are not robust replicators in vitro, these experiments require optimal viral RNA delivery, particularly for mutants with decreased replication capacity (data not shown). Because of this requirement and the high performance of the transfection reagents in Figure 1, the TransIT-mRNA Kit was directly compared to electroporation. Huh7 cells with 5 μg of either the YF-R.Luc2A-RP RNA or the YF-IRES-Luc (ΔDD) were electroporated using optimal conditions identified previously (data not shown) (Fig. 3). The YF-IRES-Luc (ΔDD) RNA mutant is incapable of replicating in transfected cells due to a polymerase mutation. This replicon is a negative control and produced minimal luciferase activity after delivery using either method (data not shown). In parallel, Huh7 cells were transfected with the same two viral replicons using the TransIT-mRNA Kit, but in these transfections only 2 μg of viral RNA per well was transfected, based on the optimal results obtained in section 3.1.1. Due to the high cell death associated with electroporation, the luciferase activity in each sample was normalized to the amount of protein in the sample, and those normalized luciferase activities were presented. We found that the TransIT-mRNA Kit was much more effective at delivering the YFV replicon than electroporation. Normalized luciferase activities were approximately 10-fold higher in the TransIT-mRNA Kit transfected samples than the electroporated samples (Fig. 3).
Fig. 3.
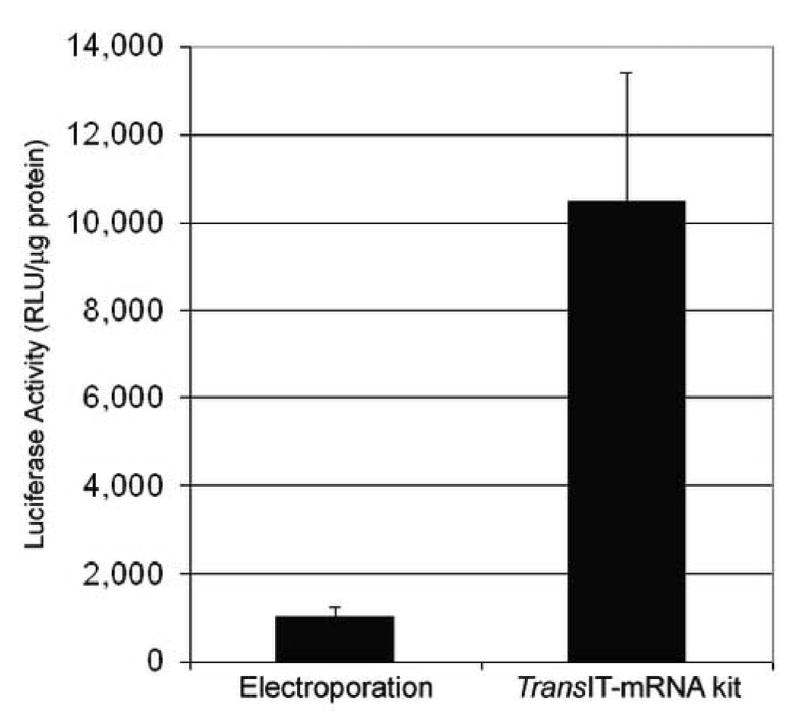
Performance comparison of electroporation to TransIT®-mRNA Kit transfected cells using a luciferase expressing replicon. Huh7 cells were either electrorporated with 5 μg or transfected with 2 μg of the YF-R.luc2A-RP RNA using the TransIT-mRNA Transfection Kit. Twenty-four hours post-delivery, the cells were harvested, lysed and assayed for both Renilla luciferase activity and total protein concentration. The luciferase activities were then normalized to total protein to account for differences in sample cell number.
3.2.2 YFV GFP replicon delivery comparison
The YFV luciferase replicon transfections (Section 3.2.1) demonstrated the highly effective transfection of viral RNA using the commercial transfection reagents, however there was no direct assessment of transfection efficiency. To extend the results of these experiments, we transfected or electroporated Huh7 cells in parallel as described in section 3.2.1 with either the YFV YF-GFP2A-RP or the YF-IRES-Luc (ΔDD) RNAs. The YF-GFP2A-RP replicon was used to determine transfection efficiency (percent GFP expressing cells in the population) of each delivery method. Forty-eight hours post-transfection the cells were examined by fluorescence microscopy to visualize the GFP expressing cells (Fig 4a). In parallel, duplicate transfected cells were harvested and analyzed by flow cytometry to determine the percentage of GFP expressing cells in the transfected or electroporated samples. As expected, the cells transfected or electroporated with the YF-IRES-Luc (ΔDD) negative control RNA did not produce any GFP expressing cells (Fig. 4b). In contrast, GFP expressing cells were detected in the Huh7 cells transfected or electroporated with the YF-GFP2A-RP RNA replicon. Quantitation by flow cytometry demonstrated that the cells transfected with only 2 μg of the YF-GFP2A-RP RNA using the TransIT-mRNA Kit produced approximately 50% GFP positive cells, while the cells electroporated with five times more viral RNA yielded approximately 1% GFP positive cells (Fig. 4b). Transfection with the other commercial transfection reagents (using 2 ug of RNA, the same as TransIT-mRNA Kit, but less than electroporation) with DMRIE-C yielding results similar to electroporation (∼ 1%) and TransMessenger™ yielding ∼10% transfection efficiency.
Fig. 4.
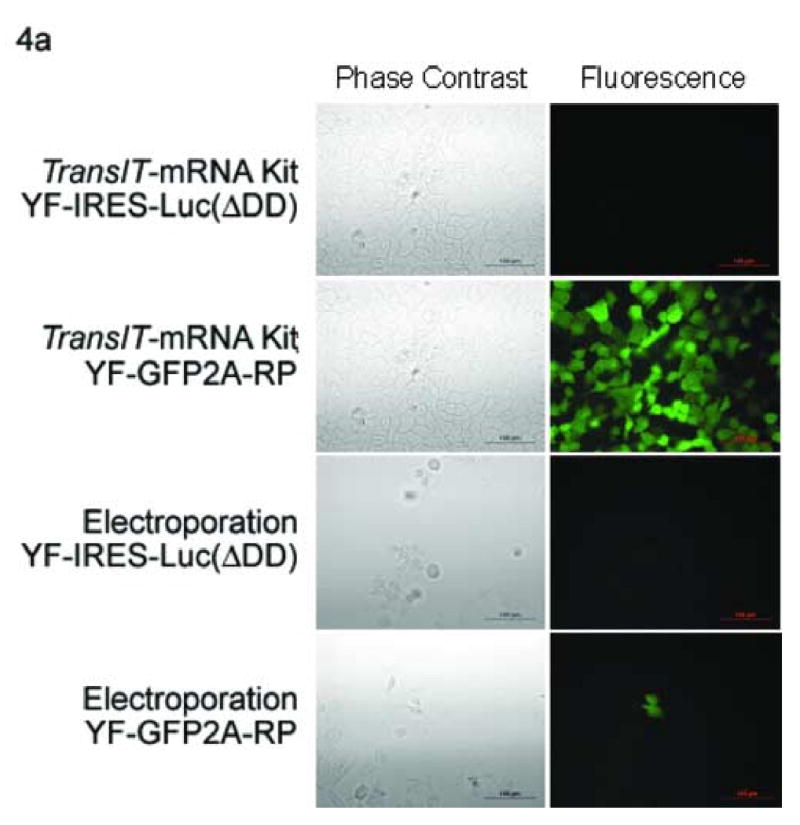
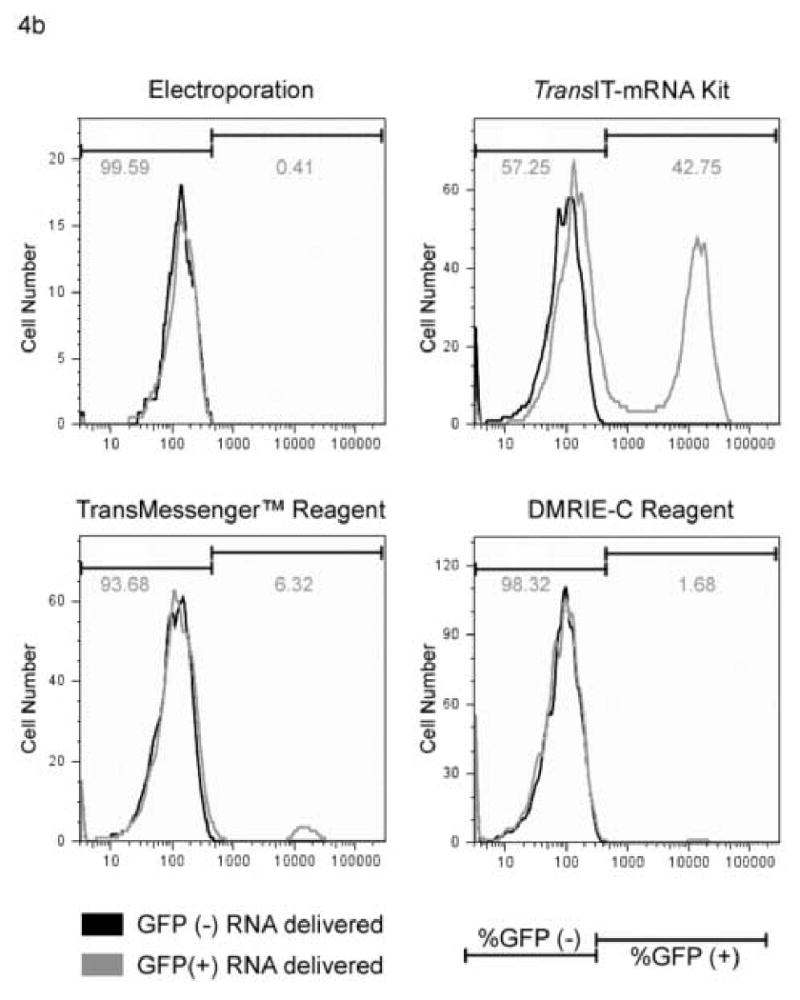
Efficiency comparison of electroporation to transfection using a GFP expressing replicon. Huh7 cells were electroporated with 5 μg or transfected using the TransIT-mRNA Transfection Kit, TransMessenger™ or DMRIE-C with 2 μg of the yellow fever virus YF-GFP2A-RP or the YF-IRES-Luc (ΔDD) (negative control) RNA using optimal conditioned determined previously. Forty-eight hours post-delivery, the cells were examined by fluorescence microscopy to visualize the GFP transfected cells (a), or quantitated by flow cytometry to determine the number of GFP positive cells in the population (b). The YF-IRES-Luc (ΔDD) RNA transfected cells served as the negative control to set the gates on the flow cytometer. Note the y axis differs between RNA delivery methods and denotes cell survival, the x axis denotes fluorescence intensity.
4. Discussion
The delivery of in vitro transcribed RNA to cells is a crucial and common technique in RNA virology. This process has been the only reliable way to study HCV replication in cell culture and typically has been performed by electroporation. Since electroporation of HCV replicons was a particularly low efficiency process, it became clear that selection at both the viral and cellular levels occurred (Bartenschlager et al., 2003; Blight et al., 2002). For example, multiple labs have shown only subpopulations of Huh7 cells are highly transfectable (Blight et al., 2002). Since electroporation results in a large amount of cell death, cells that receive and propagate the replicons have passed through a significant genetic bottleneck. Examining the aberrant cell signaling in the resultant highly permissive subpopulations such as the Huh7.5 cells has been fruitful (Sumpter et al., 2005), but this amount of cellular selection greatly increases the complexity of experimental interpretation. The HCV field is particularly preoccupied with drug susceptibility testing of replicons, and therefore often resorts to transient assays with less host cell selective pressure (Murray et al., 2003).
Although electroporation is capable of delivering viral RNA to mammalian cells with reproducible and interpretable results (Bredenbeek et al., 2003; Jones et al., 2005; Lohmann et al., 1999), it has several undesirable features. First, electroporation of mammalian cells in standard sized cuvettes generally requires ∼5 × 106 cells per electroporation, and the number of cells required for a series of electroporations quickly becomes unmanageable. Second, due to the constraints on cuvette size and electroporation equipment available, electroporations are not easily scaled up, although 96-well, high-throughput electroporation plates are becoming more common. Third, electroporation generally results in significant cell death (hence, the need for large numbers of cells), adding to the complexity of the experiments and a lower yield of cells in the electroporated samples. Fourth, large amounts of RNA (2-10 μg) are generally required for each electroporation, and the time and expense involved in preparation of large quantities of RNA can be prohibitive. Fifth and finally, electroporations are performed in the absence of serum, which increases the complexity of the procedure and decreases cell survival.
As described here, the TransIT-mRNA Transfection Kitefficiently transfects YFV and HCV replicons into Huh7 cells, a liver hepatoma cell line, yet it is highly likely that this transfection kit will also be a useful tool for the study of other viruses and other assays. For example, packaging assays frequently require serial electroporations to deliver both genomic RNA, and structural protein mRNAs separately, and serial delivery using a transfection reagent would significantly decrease cell death compared to serial electroporations (Jones et al., 2005). The TransIT-mRNA Transfection Kit has also been used to transfect RNA into other cell lines such as Vero, A549, and BHK-21 (data not shown). These cell lines are popular hosts cells for other viruses and will likely make this reagent a useful tool for those viruses as well. One of the more interesting characteristics of this transfection reagent is functional in the presence of serum. It has been demonstrated previously that this reagent protects RNA from RNase degradation in vitro (data not shown), and this protection is likely critical to facilitating RNA transfection in the presence of serum. It is possible that this trait could be utilized to allow the rescue of negative strand RNA viruses. For example, the rescue of respiratory syncytial virus (RSV) currently involves the infection of Hep-2 cells with vaccinia virus that express T7 RNA polymerase. The infected cells are then transfected with T7 promoter driven expression plasmids for the RSV replication/transcription proteins N, P, L, and M2-1 as well as the (+) strand anti-genomic plasmid (Bridgen and Elliott, 1996). Packaging the replication/transcription protein encoding mRNAs with TransIT-mRNA Transfection Reagent in one tube and the (-) genomic RNA in a separate tube would theoretically prevent hybridization, decrease toxicity of the current rescue procedure, obviate the need for vaccinia virus in the rescue of negative strand RNA viruses, and simplify the entire rescue procedure.
The goal of this research was to determine whether a commercially available transfection reagent could overcome the inherent drawbacks in electroporation and replace it. As described here, the three commercially available transfection reagents were all effective in both transient and stable assays, but the TransIT-mRNA Kit outperformed the other two reagents in all three assays. We used bothenzymatic and flow cytometry assays to compare the transfection reagents to electroporation, and independent of the assay used, the transfection reagents compared favorably to electroporation although the magnitude of the differences varied. There are many possible reasons for these differences, but the two most likely reasons are differences in the assays and the viral RNAs transfected. The luciferase assays detect luciferase activity in all the luciferase expressing cells independent of the level of luciferase present in any one cell, while the GFP flow cytometry assay only detects GFP positive cells once a threshold of GFP protein level in the cell is achieved. Second there were also slight sequence variations between the viral luciferase reporters and the viral GFP reporters, which could alter slightly the experimental outcomes.
When compared directly to electroporation, the transfection reagents, particularly the TransIT-mRNA Kit, were more versatile, and overcame most of the problems associated with electroporation. There are many advantages to using the transfection reagents. First, transfections can be performed on adherent cells in any size culture vessel, and the number of cells required is dependent on the size of the culture vessel and not the transfection reagent. As a result, optimal transfection conditions in one cell culture plate can be adjusted to different culture vessels simply by scaling the transfection conditions based on surface area of the culture vessel. Second, transfection reagents generally required less RNA for each transfection compared to electroporation, yet produced better transfection results. Finally, because the cells can be transfected in the presence of serum (not true for DMRIE-C and TransMessenger™), the transfection procedure is greatly simplified and likely leads to increased viability of the transfected cells.
Acknowledgments
TransIT®-mRNA Transfection Kit used in this study was provided as a gift by Mirus Bio Corporation. G.G. was supported in part by a grant to R.S. by The Partnership Fund for a Healthy Future and by NIH KO8 AZ0557750 to RS and the NIH/NIAID Regional Center of Excellence for Biodefense and Emerging Infectious Diseases Research (RC) Program. The authors wish to acknowledge membership within and support from the Region V ‘Great Lakes’ RCE (NIH award 1-U54-AI-057153). The authors also wish to thank Bryan Hughes for his help with the flow cytometry analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bartenschlager R, Kaul A, Sparacio S. Replication of the hepatitis C virus in cell culture. Antiviral Res. 2003;60:91–102. doi: 10.1016/j.antiviral.2003.08.016. [DOI] [PubMed] [Google Scholar]
- Blight KJ, McKeating JA, Rice CM. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol. 2002;76:13001–14. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredenbeek PJ, Kooi EA, Lindenbach B, Huijkman N, Rice CM, Spaan WJ. A stable full-length yellow fever virus cDNA clone and the role of conserved RNA elements in flavivirus replication. J Gen Virol. 2003;84:1261–8. doi: 10.1099/vir.0.18860-0. [DOI] [PubMed] [Google Scholar]
- Bridgen A, Elliott RM. Rescue of a segmented negative-strand RNA virus entirely from cloned complementary DNAs. Proc Natl Acad Sci U S A. 1996;93:15400–4. doi: 10.1073/pnas.93.26.15400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda M, Yi M, Li K, Lemon SM. Selectable subgenomic and genome-length dicistronic RNAs derived from an infectious molecular clone of the HCV-N strain of hepatitis C virus replicate efficiently in cultured Huh7 cells. J Virol. 2002;76:2997–3006. doi: 10.1128/JVI.76.6.2997-3006.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CT, Patkar CG, Kuhn RJ. Construction and applications of yellow fever virus replicons. Virology. 2005;331:247–59. doi: 10.1016/j.virol.2004.10.034. [DOI] [PubMed] [Google Scholar]
- Kaplan G, Lubinski J, Dasgupta A, Racaniello VR. In vitro synthesis of infectious poliovirus RNA. Proc Natl Acad Sci U S A. 1985;82:8424–8. doi: 10.1073/pnas.82.24.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato N, Nakamura T, Dansako H, Namba K, Abe K, Nozaki A, Naka K, Ikeda M, Shimotohno K. Genetic variation and dynamics of hepatitis C virus replicons in long-term cell culture. J Gen Virol. 2005;86:645–56. doi: 10.1099/vir.0.80479-0. [DOI] [PubMed] [Google Scholar]
- Lai CJ, Zhao BT, Hori H, Bray M. Infectious RNA transcribed from stably cloned full-length cDNA of dengue type 4 virus. Proc Natl Acad Sci U S A. 1991;88:5139–43. doi: 10.1073/pnas.88.12.5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623–6. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110–3. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- Murray EM, Grobler JA, Markel EJ, Pagnoni MF, Paonessa G, Simon AJ, Flores OA. Persistent replication of hepatitis C virus replicons expressing the beta-lactamase reporter in subpopulations of highly permissive Huh7 cells. J Virol. 2003;77:2928–35. doi: 10.1128/JVI.77.5.2928-2935.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice CM, Grakoui A, Galler R, Chambers TJ. Transcription of infectious yellow fever RNA from full-length cDNA templates produced by in vitro ligation. New Biol. 1989;1:285–96. [PubMed] [Google Scholar]
- Sumpter R, Jr, Loo YM, Foy E, Li K, Yoneyama M, Fujita T, Lemon SM, Gale M., Jr Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J Virol. 2005;79:2689–99. doi: 10.1128/JVI.79.5.2689-2699.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi M, Bodola F, Lemon SM. Subgenomic hepatitis C virus replicons inducing expression of a secreted enzymatic reporter protein. Virology. 2002;304:197–210. doi: 10.1006/viro.2002.1652. [DOI] [PubMed] [Google Scholar]