

Abstract
Eicosanoids have been implicated in a vast number of devastating inflammatory conditions, including arthritis, atherosclerosis, pain, and cancer. Currently, over a hundred different eicosanoids have been identified, with many having potent bioactive signaling capacity. These lipid metabolites are synthesized de novo by at least 50 unique enzymes, many of which have been cloned and characterized. Due to the extensive characterization of eicosanoid biosynthetic pathways, this field provides a unique framework for integrating genomics, proteomics, and metabolomics toward the investigation of disease pathology. To facilitate a concerted systems biology approach, this review outlines the proteins implicated in eicosanoid biosynthesis and signaling in human, mouse, and rat. Applications of the extensive genomic and lipidomic research to date illustrate the questions in eicosanoid signaling that could be uniquely addressed by a thorough analysis of the entire eicosanoid proteome.
Keywords: genomics, proteomics, lipidomics, cyclooxygenase, lipoxygenase, cytochrome P450, prostaglandin, leukotriene, eicosanoid
SYSTEMS BIOLOGY AND EICOSANOID “OMICS”
Biological processes are comprised of numerous converging signals that concertedly create a coherent effect. Any individual signaling element may elicit a physiological response, yet its loss is not fatal to the organism. In fact, a wide range of genetic variation can be observed within individual members of a given species without leading to a dramatic loss of function. As these signals have redundant and emergent properties, it can be difficult to explain how a biological process works using a limited number of molecular indicators. For this reason, systems biology has emerged to address the question of how molecular biology works as an integrated process (1).
Systems biology has advanced exponentially during the past two decades, with transcriptomics, proteomics, and metabolomics each playing an integral role. Each of these platforms brings its own unique advantages and limitations in facilitating the investigation of disease pathology. A transcriptomic approach can detect the upregulation and downregulation of important biosynthetic and signaling genes; however, gene changes often don't directly correlate with changes in protein levels (2). Proteomic analyses can identify enzymes and posttranslational protein changes in a cell or tissue, but fall short of determining which particular enzymes actively produce metabolites under disease conditions; likewise, metabolic approaches successfully identify bioactive signaling molecules, but therapeutic intervention generally requires definitive protein targets.
The eicosanoid class of signaling molecules highlights many of the issues facing comprehensive systems biology analyses. Eicosanoids comprise a class of bioactive lipid mediators derived from the metabolism of polyunsaturated fatty acids by cyclooxygenases (3–5), lipoxygenases (3, 6), cytochrome P450s, or nonenzymatic pathways (Fig. 1). Technically, “eicosanoids” refers to fatty acids containing 20 carbons (eicosa), but the field generally uses the term eicosanoids more broadly to also include similar metabolites of other polyunsaturated fatty acids. This lipid class has been intensely studied over the past 30 years because of its contribution to the inflammatory response in diseases such as arthritis and asthma, yet the majority of this work has focused on a select few genes, proteins, or metabolites within it. In many instances, the regulation of one arm has important regulatory implications for another arm of the eicosanoid biosynthetic pathway. Many of these enzymes produce multiple lipid products; likewise, many lipid products can be formed by a number of different enzymes acting in parallel or in concert (Fig. 2). For these reasons, a comprehensive systems biology approach would be invaluable for understanding and treating diseases implicating eicosanoid signaling.
Fig. 1.

Overview of eicosanoid biosynthesis through COX, LOX, CYP P450, and nonenzymatic pathways acting on arachidonic acid.
Fig. 2.

Major eicosanoid biosynthetic pathways. The metabolites of the major pathways are indicated in color: COX (purple), 5-LOX (orange), 15-LOX (green), 12-LOX (yellow), CYP epoxygenase (red), CYP ω-hydroxylase (cyan), and nonenzymatic oxidation (gray). The products of arachidonic acid metabolism are illustrated, but similar products can be formed from other fatty acids (e.g., linoleic acid, eicosapentenoic acid, and docosahexaenoic acid).
Transcriptomic and lipidomic analyses of eicosanoid biosynthesis can be performed using existing methodologies. With the completion of the major mammalian genome projects, commercial gene array technology has rapidly progressed and allowed the generation of whole-genome data sets of mRNA changes to become a commonplace tool at many research institutions. This proliferation allowed academic research groups to focus on the more challenging task of interpreting this data and made transcriptomic analysis the gold standard in systems biology research. Due to its immense scope, metabolomics has not yet achieved this level of standardization in scientific practice. However, as a part of their goal to create the infrastructure to identify and quantify all lipid molecular species, the Lipid Metabolites and Pathways Strategies (LIPID MAPS) consortium has developed liquid chromatography tandem mass spectrometry methodology that comprehensively covers nearly all the known metabolites of this class (7, 8). Specifically, a complete lipidomic analysis of eicosanoids is now available (9), along with corresponding gene microarray data (www.lipidmaps.org). Thus, is it timely to consider a proteomic analysis that could complete an integrated picture of the entire eicosanoid signaling network.
A quantitative proteomic analysis of the eicosanoid pathway has thus far lagged behind concomitant transcriptomic and lipidomic studies. These tandem mass spectrometry techniques can be divided into two philosophical approaches (10). The classically practiced MS1 and MS2 proteomic scanning techniques have demonstrated the potential to rapidly identify thousands of proteins from a single sample, yet these often represent only a fraction of the total protein content of a cell or tissue, and the investigator has little control over which proteins make the cut (11). On the other hand, by measuring the levels of a selected population of proteins using multiple reaction monitoring, significantly lower limits of detection for can be achieved (12). However, to create a method to investigate a selected proteome, one must first have a fundamental understanding of what proteins could be involved and how these proteins fit into the larger scheme.
In the book Functional Lipidomics, Bowers-Gentry et al. (7) give a broad outline for using a lipidomic approach to classify and measure eicosanoids. To facilitate a concerted systems biology approach, this review provides a systematic overview of the proteins involved in eicosanoid biosynthesis and signaling. The genes and proteins responsible for these activities are listed in supplementary Tables I–VII online, provided one of the following pieces of evidence: 1) the gene has been expressed and characterized in vitro; 2) the purified protein has been sequenced and characterized in vitro; 3) the gene has been overexpressed (or for transcription factors, expressed with a reporter gene) and characterized in a cellular system; 4) loss of activity has been demonstrated in cells or tissue from gene knockout animals. Furthermore, as studies of human biology and disease commonly employ mouse or rat models as a surrogate, important differences between these species are highlighted.
PHOSPHOLIPASE A2
Arachidonic acid and other polyunsaturated fatty acids serve as the metabolic precursors for eicosanoid synthesis (13, 14). Biologically, these molecules are generally not available in large quantities in the free acid form, but are stored at the sn-2 position on the glycerol backbone of membrane phospholipids. To be used for biosynthesis, phospholipase A2 (PLA2) liberates sn-2 fatty acids from phospholipids at the membrane interface.
Phospholipase A2 represents a superfamily of at least 15 groups that have wide-ranging roles in biological processes. These enzymes can be considered as five types: cytosolic PLA2 (cPLA2), secreted PLA2 (sPLA2), calcium-independent PLA2, platelet-activating factor acetylhydrolase, and lysosomal PLA2; their classification and biological functions have been extensively reviewed (13–15).
Current literature strongly implicates the Group IVA cPLA2 as the chief enzyme involved in polyunsaturated fatty acid release for eicosanoid biosynthesis (16, 17), with sPLA2s potentially playing a supplementary role (see supplementary Table I). Bonventre et al. (18) and Uozumi et al. (19) independently demonstrated that peritoneal macrophages from Group IVA PLA2 knockout mice were incapable of producing eicosanoids. More recently, Adler et al. (20) identified a patient with inherited Group IVA deficiency and determined that nearly all arachidonic acid used for eicosanoid production by platelets and circulating leukocytes was attributable to this enzyme. The Group IVA cPLA2 uses a catalytic Ser/Asp dyad to hydrolyze fatty acids and contains a C2 domain that facilitates calcium-dependent translocation from the cytosol to the membrane surface where it encounters phospholipid substrate. Overall, while cPLA2s have a broad range of homology between human, mouse, and rat, the Group IVA cPLA2 has 95% identity between these species. Other cPLA2 show between 54 and 82% identity in these species and have been less well-characterized to date (13), yet may still play a supplementary role in eicosanoid biosynthesis.
While Group IVA cytosolic PLA2 is the primary enzyme involved in eicosanoid biosynthesis, sPLA2s have been demonstrated to play a supplementary role, reviewed extensively by Lambeau and Gelb (21). In particular, the Group IIA, Group V, and Group X PLA2s have all been shown to modulate eicosanoid levels in mammalian cellular systems. However, the majority of these studies have been performed by either overexpression or exogenous supplementation of sPLA2, which complicates the investigation of its role in pathology. Future work using sPLA2 knockout mice may help further elucidate the role these enzymes play in eicosanoid biosynthesis during an inflammatory response.
CYCLOOXYGENASE METABOLITES
Cyclooxygenases
Prostaglandins (PGs) are bioactive signaling molecules derived from cyclooxygenase (COX) and subsequent PG synthase activity on arachidonic acid (4, 5, 22) (see supplementary Table II). COXs contain two distinct active sites, a COX and peroxidase site, both of which use the same tyrosyl radical and heme-iron for catalysis. The COX site incorporates molecular O2 at the 11- and 15-carbon on arachidonic acid to form PGG2, which contains the following moieties: a five-member ring linked at C-8 and C-12, an endoperoxide bridge across C-9 and C-11, and a peroxide at C-15. The peroxidase site reduces the peroxide to a hydroxyl to form PGH2, the substrate for the various PG synthases. In addition to forming PGH2, arachidonic acid can situate in the active site pocket in a limited number of suboptimal conformations that incorporate a single molecular O2, forming trace amounts of either 11(R)-hydroxyeicosatetraenoic acid (HETE) or 15(S)-HETE. COX activity does not exhibit long-term stability, as the catalytic tyrosyl radical can be transferred to a nearby tyrosyl residue and cause “suicide inactivation” after ∼300 turnovers. Because the enzyme can perform only a limited number of reactions, it must be constantly reexpressed to generate metabolites. The details of these mechanisms have been reviewed extensively (5).
COXs constitute two distinct genes, COX-1 and COX-2. These two isoforms exhibit ∼60% identity and have nearly identical active site residues (4). The most significant structural difference between these enzymes is an isoleucine to valine substitution, which results in a larger COX-2 active site pocket. This allows COX-2 to be more permissive in selecting substrates, and unlike COX-1, it can metabolize dihomo-γ-linolenic and eicosapentaenoic acid in addition to arachidonic acid. Pharmaceutical companies have also taken advantage of the larger COX-2 active site, creating isoform selective inhibitors such as Celecoxib (Celebrex™). While COX-1 and COX-2 have similar structural and catalytic features, these isozymes exhibit different expression patterns. COX-1 is constitutively expressed by most cell types and has been implicated in a number of homeostatic processes, including stomach acidity control, endometrial cycling, and renal function. In contrast, COX-2 expression is controlled by the pro-inflammatory transcription factor NF-κB and highly upregulated in response to infection, atherosclerosis, and a number of cancers.
Prostaglandins
PGH2 can form a number of different bioactive products through the action of PG synthases. This includes a number of important signaling molecules, including PGI2 (also known as prostacyclin), thromboxane A2, PGE2 (also known as dinoprostone), PGD2, and PGF2α. Following the PG abbreviation (Fig. 3), the nomenclature highlights the two important structural features: the components of the five-member prostane ring, as denoted by a letter A–K, and the number of double bonds, denoted by a subscript number (23). The lone exception is thromboxane, which has a six-member oxane ring, and is abbreviated TX.
Fig. 3.

PG nomenclature and structure. Arachidonic acid carbons are numbered 1–20, starting from the carboxylate. The prostaglandin letter indicates composition of the prostane ring, with PGH2 as an example.
Prostaglandin I2 and thromboxane A2
PGI2 was first identified in 1976 (24) and is formed by the prostacyclin synthase, a member of the cytochrome P450 monooxygenase superfamily (25). Structural elucidation of the human prostacyclin synthase was not obtained until 2006 (26), confirming the mechanism of PGH2 endoperoxide bridge rearrangement proposed by Hecker and Ullrich in 1989 (27). The active site heme-iron interacts with the C-11 oxygen, promoting the hemolytic cleavage of the endoperoxide bond and the formation of an ether linkage between C-9 and C-5. The PGI2 ring is highly labile and rapidly hydrolyzed to form the stable but biologically inactive 6-keto PGF1α, and because of this, PGI2 and 6-keto PGF1α are often used interchangeably in the literature. PGI2 binds the G-coupled protein receptor IP (28, 29) as well as the transcription factors peroxisome proliferator-activated receptor (PPAR) α, PPARδ, and PPARγ (30, 31).
Identified in 1975 (32), thromboxane A2 (TXA2) is the physiological counterbalancing signal to PGI2. Similar to PGI2, its biosynthesis is catalyzed by a member of the cytochrome P450 superfamily member, thromboxane A synthase (27, 33). This enzyme facilitates rearrangement of the PGH2 endoperoxide bridge by a complimentary mechanism to prostacyclin synthase, interacting with the C-9 oxygen to promote endoperoxide bond cleavage. The C-11 oxygen radical initiates intramolecular rearrangement, resulting in either the formation of TXA2 or 12-hydroxyheptadecatrienoic acid. TXA2 contains an unstable ether linkage that is rapidly hydrolyzed in vivo under aqueous conditions to form the biologically inert TXB2. It also binds a specific G protein-coupled receptor (GPCR), termed the TP receptor, which culminates in increasing intracellular calcium concentrations (33).
Prostaglandin E2
PGE2 may be the best characterized signaling molecule within the eicosanoid class. A PubMed search for “PGE2” reveals that, since the identification and isolation of PGE2 from human seminal plasma in 1963 (34), well over 27,000 articles have been published studying this molecule. The effects of PGE2 have been implicated in innumerable biological processes and disease pathologies, including parturition, bronchodilation, pain signaling, innate and adaptive immune responses, cancer, arthritis, and atherosclerosis (4, 35). The wide spectrum of PGE2 signaling can be partially attributed to differences in expression and function of its four known receptors, EP1-4. The EP receptors are all GPCRs, and their most potent natural ligand is PGE2. Functionally, EP1 and EP3 mediate their responses through calcium signaling, whereas EP2 and EP4 do so by increasing cAMP levels. Likewise, PGE2 synthesis occurs through three unique enzymes, named the cytosolic PGE synthase (cPGES), microsomal PGE synthase-1 (mPGES-1) and mPGES-2 (35).
Cytosolic PGE2 synthase was identified by Tanioka et al. (36) in 2000 from rat brain and determined it to be identical to human p23. Previously, this protein was identified as a chaperone protein that interacts with heat shock protein-90, which upregulates cPGES activity in vitro. The catalytic activity of cPGES requires glutathione and the conserved Tyr-9 residue for activity, similar to other cytosolic glutathione S-transferases, including hematopoietic PGD synthase (35, 37). In this case, the glutathione thiol is expected to interact with the C-9 oxygen of PGH2, facilitating cleavage of the endoperoxide bridge and the formation of PGE2.
The cloning and characterization of human mPGES-1 was reported by Jakobsson et al. in 1999 (38). As a member of the membrane-associated proteins in eicosanoid and glutathione (MAPEG) superfamily of transmembrane proteins, this enzyme requires glutathione for activity. While mPGES-1 has not yet been crystallized, Huang et al. (39) examined it using site-directed mutagenesis and computational modeling onto the microsomal glutathione S-transferase (mGST) structure, its nearest subfamily member. This method identified Arg-110 and Tyr-114 interactions with the C terminus of PGH2 and determined that glutathione thiol was positioned to catalyze PGE2 biosynthesis. The mechanism of PGE2 formation likely proceeds by a similar mechanism to cPGES and h-PGDS; however, the structural element activating glutathione remains undetermined. mPGES-1 expression is coregulated with COX-2, and both proteins colocalize to the endoplasmic reticulum (40); however, COX-2 resides in the lumen of the endoplasmic reticulum, whereas the mPGES-1 active site appears to face the cytosol. A thorough examination of mPGES-1 lature has been reviewed by Samuelsson, Morgenstern, and Jakobsson (40).
mPGES-2 activity was first reported by Wantanabe et al. (41) as a distinct PGES from cPGES and m-PGES-1 in rats. While it has no specific requirement for glutathione, its activity was enhanced by the presence of free thiols. The human isoform was cloned in 2002 by Tanikawa et al. (42) and contains a putative C-x-x-C sequence found in glutaredoxin and thioredoxin, but not cPGES or m-PGES-1. Subsequent crystallization by Yamada et al. in 2005 (43) confirmed it contained significant structural differences from other PGES. Using molecular modeling, they proposed a mechanism for mPGES-2 catalysis. A hydrogen bonding network between Phe-112, Cys-110, Cys-113, and Tyr-107 lowers the Pka of Cys-110 such that it can donate its thiol hydrogen to the C-11 oxygen. This leads to cleavage of the endoperoxide bridge and the creation of a ketone by the C-9 oxygen, forming PGE2. The final step is facilitated by free thiols, activated by Tyr-107; however, this does not appear to be necessary for productive catalysis.
Prostaglandin D2
PGD2 is a structural isomer of PGE2, and early studies of D-series PGs regarded them as side-products of E-series biosynthesis (44). Whereas the prostane ring on PGE2 has a 9-keto and 11-hydroxy moiety, the positions of these substituents are reversed on PGD2. In the late 1970s, PGD2 was identified as a major product in rat brain homogenates (45) and activated mast cells (46). PGD2 is formed by two evolutionarily distinct, but functionally convergent, prostaglandin D synthases (PGDS): the lipocalin-type (l-PGDS) and hematopoietic-type (h-PGDS). One critical difference between the PGDSs is the requirement for glutathione; l-PGDS can function without this cofactor, while h-PGDS uses it for catalysis. Urade et al. (47) proposed a mechanism for l-PGDS, whereby the thiol from Cys-65 forms a transient bond with the C-11 oxygen of PGH2, opening the endoperoxide ring. Exogenous sulfhydryl compounds then remove the C-11 hydrogen, culminating in PGD2 production and release. NMR structural studies and molecular modeling estimated a 5 Å distance between the Cys-65 thiol and the C-11 oxygen, consistent with this model (48). Following crystallization in 1997, Kanaoka et al. (49) proposed that h-PGDS contains a pocket that activates glutathione via Tyr-8, such that the glutathione thiol functions analogous to Cys-65 of l-PGDS.
PGD2 has two known receptors, DP1 and chemoattractant receptor-homologous, molecule expressed on Th2 cells (CRTH2) (50). DP1 belongs to the family of GPCR that includes IP, TP, EP1-4, and FP, and ligand binding leads to intracellular increases of cAMP. While exhibiting similar PGD2 binding, CRTH2 shares little homology with DP1 and appears more closely related to the leukotriene B4 receptors BLT1 and BLT2. CRTH2 activation leads to elevation of intracellular calcium and decreased cAMP levels (51).
Prostaglandin F2α
PGF2α was isolated and structurally characterized in 1963 by Samuelsson (34) from human seminal fluid and demonstrably stimulated smooth muscles from rabbit duodenum. Since then, it has been implicated in a number of physiological processes and disease states (52), including endometrial cycling, embryo development parturition, vasoconstriction, as well as acute inflammation, oxygen-depravation injury, and atherosclerosis. Only one PGF2α-specific receptor has been cloned (53), a GPCR termed FP, which upon binding ligand results in an elevation of intracellular calcium. To date, three PGF2α biosynthetic enzymes have been cloned and characterized (54): prostaglandin F synthase (PGFS) (55), prostamide/PGFS (56), and 9-keto prostaglandin reductase (9K-PGR).
The human PGFS was cloned in 1999 by Suzuki-Yamamoto et al. (55) and determined to be identical to aldo-keto reductase 1C3 (AKR1C3). Similar to mPGES-2 and h-PGDS, PGFS evolved from the thioredoxin protein family and contains the putative C-x-x-C motif. Crystal structures of PGFS were published in 2004 (57) and 2006 (58) by Komoto et al. and used to propose a mechanism for PGF2α catalysis. In this model, no specific amino acids coordinate with the PGH2 endoperoxide; however, the reactive hydrogen of NADPH interacts with the C-9 oxygen of PGH2. This leads to a hydride shift and cleavage of the endoperoxide bond, whereby the C-11 oxygen can be protonated by H2O to complete the reaction. Interestingly, PGFS can also take PGD2 as a substrate. They suggest that His-117 and Tyr-55 form hydrogen bonds with the carbonyl at C-9, promoting an analogous hydride shift from NADPH onto C-9. This forms a stable hydroxyl moiety and the creation of 11β-PGF2α. Structural comparisons show that PGFS is nearly identical to AKR1C1 and AKR1C2, with only seven amino acid differences, and their structures superimposable (57); however, these substitutions all incorporate smaller amino acids to form a larger active site cavity and mitigate potential steric hindrance with PG substrates predicted in AKR1C1 and AKR1C2. An in vitro analysis of purified AKR-1C1, AKR-1C2, and AKR-1C4 confirms they have no significant activity toward PG substrate (59). To date, neither the mouse or rat homologs of PGFS have been conclusively cloned and identified.
Prostamide/PGFS was identified in mouse and swine brain in 2008 (56), and the recombinant murine protein was expressed and characterized. Like PGFS, prostamide/PGFS is classified as a thioredoxin protein, and its C-x-x-C motif is required for productive catalysis. This protein facilitates the reduction of PGH2 to PGF2α by a similar mechanism to PGFS, involving a hydride transfer from NADPH; in contrast to PGFS, it does not appear to productively reduce either PGE2 or PGD2 but instead takes PGH2-ethanolamide as a substrate. A comparison between the swine and murine protein demonstrated similar catalytic parameters, and based on sequence homology, it is predicted that its function is well conserved in mammalian species, including human and rat.
In addition to using PGH2 and PGD2 as substrates for the formation of PGF2α, it has been shown that PGE2 can be reduced by 9K-PGR activity to form this molecule (60). In 2000, Asselin and Fortier (61) identified and characterized bovine AKR1C5 as the putative 9K-PGR but have yet to definitively characterize a functional homolog in human, mouse, or rat. Since previous work has shown that only a few substitutions can radically alter the substrate specificity of enzymes in the AKR family, homology modeling in absence of in vitro data must be viewed with caution.
Carbonyl reductase-1 (CBR-1) has also been demonstrated to reduce a number of exogenous and endogenous metabolic substrates, including 9-keto reductase activity on PGE2 to form PGF2α (62, 63). Its high Km value compared with other substrates indicates that PGE2 may not be an endogenous substrate; however, a number of studies have implicated murine Carbonyl reductase-1 in vivo with lower PGE2 levels (64) and alterations in the PGE2/PGF2α ratio (65), warranting more thorough investigation into its role in eicosanoid metabolism.
Cyclopentenone prostaglandins
Cyclopentenone PGs comprise a family of molecules that are formed by dehydration of hydroxyl moieties PGE2 and PGD2 (66). Dehydration of PGE2 leads to PGA2, which has been shown to isomerize and form PGC2. This highly unstable molecule rapidly undergoes a secondary isomerization, culminating in PGB2. Analogous to PGE2, PGD2 dehydration of the prostane ring forms PGJ2. When PGJ2 isomerizes to form Δ12-PGJ2, it promotes a secondary dehydration of the C-15 hydroxyl culminating in 15d-PGJ2. Cyclopentenone PGs contain highly electrophilic α,β-unsaturated carbonyls that have been shown to react with the thiol moiety on cysteinyl residues, either on glutathione or cellular proteins. PGA2 and PGJ2 contain a single α,β-unsaturated carbonyl, while both Δ12-PGJ2 and 15d-PGJ2 contain two of these electrophilic centers.
Cyclopentenones can exert their effects through both receptor-mediated signaling and covalent protein interaction. Dehydration dramatically lowers the affinity of a cyclopentenone for the PG receptor of its precursor PG molecule. However, 15d-PGJ2 has been identified as a high affinity ligand for the transcription factor PPARγ as well as a less potent activator of PPARα and PPARδ. It is unclear whether the levels of 15d-PGJ2 necessary to activate PPARs are high enough to elicit this response in vivo (67). Evidence suggests that nonreceptor-mediated effects of cyclopentenones can be attributed to their electrophilic α,β-unsaturated carbonyl centers. Many of these biological actions can be recapitulated by cyclopentenone, while structurally related molecules lacking a reactive center, such as PGB2, are unable to elicit these effects.
5-LIPOXYGENASE METABOLITES
5-Lipoxygenase and 5-lipoxygenase activating protein
Metabolites from the 5-lipoxygenase (5-LOX) pathway were first structurally characterized in 1979, when Murphy, Hammarstrom, and Samuelsson (68) identified leukotriene C4 as a component of the slow-reacting substance of anaphylaxis. Leukotrienes comprise a family of bioactive signaling molecules formed by the activity of 5-LOX on arachidonic acid (see supplementary Table III). 5-LOX contains a nonheme iron bound by three histidine residues, which it uses to catalyze the addition of molecular oxygen into arachidonic acid. 5-LOX was cloned in 1988 (69), but a three-dimensional structure has not yet been determined. However, attempts have been made to model the human 5-LOX sequence onto the rabbit 15-LOX crystal structure, which has 57% sequence identity. Similar to cPLA2, 5-LOX has a catalytic domain linked to a C2 domain that facilitates translocation to the membrane surface. Phosphorylation of Ser271 and Ser663 increase activity, whereas Ser523 phosphorylation inactivates this enzyme. 5-LOX also appears to have an ATP binding site that does not require hydrolysis, but instead stabilizes its structure.
Unlike other lipoxygenases, 5-LOX requires the presence of 5-LOX activating protein (FLAP) for productive leukotriene synthesis in vivo. FLAP is a member of the MAPEG superfamily, localizing to the nuclear envelope. Recently, a group at Merck Research Laboratories led by Joseph W. Becker succeeded in crystallizing FLAP bound either both MK-886 and MK-591, inhibitors of 5-LOX metabolism (70). They suggest FLAP exists as a trimer, creating a 3200 Å binding pocket that allows arachidonic acid to laterally diffuse into the protein complex from the membrane. The cytosolic loops of FLAP interact with the 5-LOX catalytic domain and transfer arachidonic acid into the 5-LOX active site. The rat 5-LOX and FLAP have not been expressed for specific characterization by in vitro systems; however, both the 5-LOX and FLAP have been studied using chemical inhibitors, their sequences are highly conserved (>90% identity with their human and mouse homologs) and are generally accepted to play a role in leukotriene biosynthesis.
5-LOX performs the initial enzymatic step in leukotriene synthesis (71), creating 5-hydroperoxy-eicosatetraenoic acid (5-HpETE) by incorporating one molecular oxygen at the C-5 position of arachidonic acid. Depending on cellular conditions, 5-HpETE has a number of potential metabolic fates. It can be secreted in its peroxide form, reduced to 5-HETE, or undergo a catalytic rearrangement in the 5-LOX active site to form leukotriene (LT) A4 (Fig. 4). When produced in vivo, LTA4 can be acted on by LTA4 hydrolase (LTAH) to generate the stereospecific LTB4 or used as a substrate by LTC4 synthase (LTCS) to catalyze the conjugation of the glutathione to form LTC4. Additionally, LTA4 serves as a precursor for lipoxin biosynthesis, described in greater detail in subsequent sections. Nonenzymatic hydrolysis of the LTA4 epoxide creates all four potential steroisomers of LTB4, including Δ6-trans LTB4, 12-epi LTB4, and Δ6-trans, 12-epi LTB4. The enzymes involved in leukotriene metabolism are described here, and a thorough review of the subject has been published by Murphy and Gijon (71).
Fig. 4.

Structures of 5-lipoxygenase metabolites. 5-Lipoxygenase creates the labile epoxide LTA4, which can be enzymatically converted into LTB4, LTC4, and LXA4.
5-HETE and 5-oxoETE
Current literature suggests that 5-HETE itself does not appear to play a significant role in biological signaling. However, it can be further reduced by 5-hydroxy-eicosatetraenoic acid dehydrogenase (5-HEDH) to form the bioactive 5-oxo-eicosatetraenoic acid (5-oxoETE). While the gene has not yet been cloned, the biophysical properties of the human enzyme have been well characterized by Powell and Rokach (72). It localizes to the microsomal fraction of a number of cell types, including neutrophils, monocytes, eosinophils, and platelets. It is highly selective for 5-hydroxyl lipids, as HETEs with hydroxyl groups at the 8 through 15 position are not significantly metabolized. It requires NADP+ as a cofactor, which regulates activity levels in vivo. When neutrophils activate NADPH oxidase, responsible for superoxide formation during the respiratory burst, it generates NADP+ and leads to full activity of 5-hydroxy-eicosatetraenoic acid dehydrogenase in vivo. Activation of the human 5-oxo-eicosatetraenoic acid receptor (OXE, previously known as orphan G-coupled protein receptor R527) leads to increased intracellular calcium concentrations and inhibition of cAMP production (73). This receptor is highly expressed in neutrophils, eosinophils, and alveolar macrophages and appears to plays a role in chemotaxis. The mouse and rat homologs have not yet been identified.
Leukotriene B4
Leukotriene B4 was characterized as a 5-LOX metabolite produced by polymorphonuclear leukocytes (74) and demonstrates potent autocrine activation of chemotaxis and aggregation (75). Leukotriene B4 signals through two GPCR receptors, BLT1 and BLT2, which lead to increased intracellular calcium levels (76). BLT1 has ∼20-fold higher affinity for LTB4 compared with BLT2 and is highly expressed in leukocytes. Studies using BLT1 knockout mice have primarily attributed a number of LTB4-dependant signaling responses to this receptor, including neutrophil chemotaxis (77), neutrophil recruitment in vivo (78), and LTB4-induced leukocyte rolling to firm adhesion (77). BLT2 has broader substrate specificity (76), with 12-HETE, 12-HpETE, and 15-HETE demonstrating dose-dependant binding properties; furthermore, it displays a more ubiquitous expression pattern in humans, where in addition to leukocytes it has been detected in the spleen, liver, ovary, pancreas, heart, prostate, lung, and colon. In 2008, Okuno et al. (79) demonstrated that 12-hydroxyheptadecatrienoic acid showed 10-fold higher affinity for human BLT2 compared with LTB4, leading to reconsideration of LTB4 binding as this receptor's primary function.
LTAH is zinc-dependant metalloprotein that leads to the stereospecific hydrolysis of the LTA4 to form the dihydroxy LTB4 (80, 81). Its activity was characterized from plasma samples in a wide range of mammalian species (82) prior to being sequenced from human lung cDNA libraries in 1987 (83). Homology studies revealed that, in addition to other zinc hydrolases, this enzyme shares sequence similarity with a number of zinc-dependant aminopeptidases (71). LTAH exhibits aminopeptidase activity, and LTB4 biosynthesis can be blocked by peptidase inhibitors (81), suggesting a potential evolutionary origin. The first high-resolution crystal structure of LTAH was published in 2001 (84) and used to propose its catalytic mechanism. The active-site zinc ion and Glu-271 coordinate with a free water molecule and position it for deprotonation by the LTA4 epoxide oxygen. The delocalized carbocation created between C-6 and C-12 is attacked by a second water molecule, directed to C-12 by Asp-375 in a stereospecific manner forming LTB4.
Cysteinyl leukotrienes
Cysteinyl leukotrienes LTC4, LTD4, and LTE4 together make up the slow-acting substance of anaphylaxis and play an important signaling role in inflammation (85). They can be synthesized by a number of different cell types, including mast cells, eosinophils, neutrophils, basophils, and macrophages. Two receptors have been implicated in cysteinyl leukotriene signaling, cysLT1 and cysLT2. The expression of cysLT1 appears to be highest in cell types that produced these metabolites, whereas cysLT2 is more broadly expressed and can be found in the heart, brain, and vasculature (86). These receptors are both GPCRs that culminates in intracellular calcium mobilization and have similar binding affinities; cysLT1 has a rank order of LTD4 > LTC4 >> LTE4, whereas cysLT2 binds LTC4 and LTD4 equipotently.
Leukotriene C4
In 1979, it was demonstrated that LTA4 could be conjugated to glutathione to form leukotriene C4, the first identified component of the slow-reacting substance of anaphylaxis (68). The enzyme mainly responsible for LTC4 production is LTCS, identified as a transmembrane protein from the MAPAG superfamily that includes FLAP and mPGES-1 (87, 88). Other MAPEG enzymes, mGST-2 and mGST-3, also exhibit LTC4 synthase activity and appear to play a role in LTC4 biosynthesis in epithelial cells and the testis (89, 90). However, LTCS knockout mice demonstrate its role as the primary contributor to LTC4 biosynthesis in vivo.
It wasn't until 2007 that the LTCS structure was solved using X-ray crystallography (91, 92), suggesting the following mechanism of catalysis. Leukotriene C4 synthase forms a homotrimer, with each monomer containing four transmembrane α-helices. The LTCS active site forms at the interface between two adjacent monomers. One monomer binds glutathione in a U-shaped conformation that positions the thiol for deprotonation by Arg-104 and subsequent nucleophilic attack of C-6. The Arg-31 of the second monomer donates a proton to form a hydroxyl group at C-5. The ω-terminus of LTA4 binds a hydrophobic cavity in the interface that is capped by Trp-116, serving as a molecular “ruler” to facilitate LTCS selectivity for 20-carbon polyunsaturated fatty acids.
Leukotriene D4
Soon after its discovery, it was recognized that the glutathione amino acids of LTC4 could be cleaved by endogenous peptidases. The first enzyme identified to work on LTC4 was γ-glutamyl transpeptidase (GGT) (93), which removes glutamic acid to form LTD4. In addition to LTC4, it can hydrolyze a broad range of glutathione-containing metabolites proceeding through a modified ping-pong mechanism thought to involve Arg-107 and Asp-423 in the human isoform (94, 95). GGT resides on the extracellular membrane surface, and its activity can be detected in a number of tissues and fluids, including blood plasma (93, 94).
Interestingly, separate studies using GGT knockout mice and human subjects deficient in GGT both demonstrated residual activity, suggesting GGT was not solely responsible for LTD4 production. Using a human cDNA library, Heisterkamp et al. (96) identified related GGT gene, originally termed GGT-rel, with 40% amino acid similarity. This enzyme effectively hydrolyzed LTC4 while having low activity toward other glutathione substrates for GGT, suggesting it could act specifically on cysteinyl leukotrienes. In the GGT-knockout mouse study, the residual peptidase activity toward LTC4 was purified from intestinal tissue homogenates (95). This activity was originally termed γ-glutamyl leukotrienase (GGL) due to its selectivity for LTC4 as a substrate. Subsequent cloning and sequencing confirmed that murine GGL was a homolog to human GGT-rel gene (97). Both GGT and GGL appear to play a functional role in LTC4 metabolism, as demonstrated by metabolism studies using single and double knockout mice (98). Neither of the rat homologs have been characterized.
Leukotriene E4
Leukotriene D4 can be further metabolized by dipeptidases. The first such enzyme characterized was the human membrane-bound dipeptidase-1 (MBD-1) (99), which facilitates the hydrolysis of a broad range of cysteinyl-glycine adducts. MBD-1 is a metalloproteinase that uses an active-site zinc ion to facilitate the hydrolysis of the amide between dipeptides; in leukotriene biochemistry, it catalyzes the metabolism of LTD4 to LTE4. Using an MBD-knockout mouse, overall LTE4 levels decreases, but significant levels of LTE4 were detectable; furthermore, the changes in activity varied between tissues, with some losing >50% of their activity with others remaining unaffected (100), suggesting the presence of additional genes in LTD4 metabolism. Two additional MBD genes, MBD-2 and MBD-3, were identified from a BLAST search of the mouse genome (101); expression of MBD-2 determined it was capable of hydrolyzing LTD4 into LTE4, whereas MBD-3 did not productively metabolize LTD4. The rat homologs have not been studied in vitro to date.
OTHER LIPOXYGENASE METABOLITES
Lipoxygenases
Lipoxygenases are nonheme iron metalloproteins that catalyze the stereoselective insertion of molecular oxygen into arachidonic acid (71, 102). While the three-dimensional structures of most mammalian lipoxygenases have not been solved, the structure of soybean LOX (103, 104) and rabbit leukocyte-type 12/15-LOX (105) have been reported and used as the basis for understanding other lipoxygenases through homology modeling. The LOX reaction mechanism involves an iron-catalyzed hydrogen abstraction from arachidonic acid at C-7, C-10, or C-13, forming a conjugated radical reaching two carbons in either direction (106). The structure of a given LOX enzyme generally conforms tightly around the fatty acid, and small channels within the protein direct molecular oxygen toward selected carbons, facilitating the formation of the following HpETEs and corresponding HETEs: 8-HETE, 12-HETE, and 15-HETE (see supplementary Table III). With the exception of 12R-LOX, mammalian LOXs typically direct the oxygen to attack the pro-S face of arachidonic acid. The positional specificity by which a LOX incorporates oxygen onto arachidonic acid is determined by how deeply it penetrates the cavity, as well as the orientation (either C terminus or ω terminus) it enters the active site. Work by Jisaka et al. suggests that two amino acid positions opposite of the catalytic iron ion determine the entry orientation. When a histidine and a bulky aromatic amino acid fill this position, arachidonic acid C terminus enters the active side and promotes 5-LOX and 8-LOX activity. Substitution of these amino acids with either a glutamine or aspartate alongside an aliphatic residue promotes the entry of the ω terminus and facilitates 12-LOX and 15-LOX activity.
Hydroxyeicosatetraenoic acids
In mammalian systems, lipoxygenase activities have been implicated in the biosynthesis of 5-HETE (see above), 8-HETE, 12-HETE, and 15-HETE (71, 106). The HETEs have been implicated as potential ligands for PPARα (107) and PPARγ (108). They exert numerous biological effects, including, but not limited to, mitogen-activated protein kinase signaling (109), monocyte chemoattractant protein-1 expression (110), angiogenesis (111), cancer growth and metastasis (112, 113), and neuronal apoptosis (114); however, the precise molecular mechanisms behind these effects remain unclear. Interestingly, 12- and 15-HpETE, but not the corresponding HETEs, have been shown to directly activate the capsaicin-sensitive vanilloid receptor (VR1) (115), which is involved in inflammatorypain signaling.
Hepoxilins
Hepoxilins are biologically relevant signaling molecules produced by certain 12-LOXs (116) (Fig. 5). Two hepoxilins have been identified, HXA3 and HXB3, both of which incorporate an epoxide across the C-11 and C-12 double bond, as well as an additional hydroxyl moiety (117); HXA3 has a C-8 hydroxyl, whereas the HXB3 hydroxyl occurs at C-10. The epoxy moiety is labile and can be hydrolyzed either by a hepoxilin specific epoxide hydrolase (HXEH) (118) or in acidic aqueous solution to form the corresponding diol metabolites trioxilin A3 and B3 (117). Most of the known bioactive effects can be attributed to HXA3, which include intracellular calcium release (119), neutrophil migration (120), insulin secretion (121), ichthyosis (122), and modulation of neuronal signaling (123). Current evidence suggests the existence of a HXA3 receptor (124), but to date one has not been identified.
Fig. 5.

Structures of 12-lipoxygenase metabolites. 12-Lipoxygenase creates 12-HpETE, which can further isomerize to form HXA3.
Lipoxins
Early studies of arachidonic acid metabolism demonstrated that multiple lipoxygenases can act on the same substrate to generate a new class of lipid mediators termed lipoxins (LX) (125). Lipoxins A4 and B4, structurally characterized from human neutrophils incubated with 15-HpETE, each contain three hydroxyl moieties and a conjugated tetraene. The third hydroxyl of LXA4 is positioned at C-6, and of LXB4 at C-14.
The action of 5-LOX, in concert with a 12-LOX or 15-LOX activity, has been shown to produce lipoxins by three distinct pathways (126), which often occur between heterogeneous transcellular interactions in vivo. Neutrophil 5-LOX can produce and secrete LTA4 that is taken up by platelets, where it is acted upon by 12-LOX-p to form lipoxins (127). Likewise, 15-LOXs can generate either 15-HpETE or 15-HETE that can be taken up by monocytes and neutrophils, where highly expressed 5-LOX uses it to generate lipoxins (125). Finally, aspirin acetylated COX-2, rendered unable to synthesize PGs, can act as a 15-LOX. This leads to the formation of 15R-HETE and culminates in creation of epi-lipoxins (128), which have altered stereochemistry at the C-15 hydroxyl but similar biological potency.
Lipoxins are the first lipid mediators discovered that demonstrated anti-inflammatory activity as well as the capacity to promote the resolution of inflammation and return to tissue homeostasis (6, 126, 129). Their biological activity has been shown to be mediated through the lipoxin A4 receptor (ALX), cysLT1, and the aryl hydrocarbon receptor, a nuclear transcription factor (129). The ALX receptor is a GPCR that upon binding ligand can lead to intracellular calcium increases, PLD activation, and decreased NF-κB activity. Additionally, ALX cross-talks with vascular endothelial growth factor receptor, connective tissue growth factor receptor, and platelet-derived growth factor receptor to downregulate their activity. This receptor can also bind a number of small peptides; some dock the LXA4 binding site, while other others appear to function at a different site and lead to distinct downstream signaling events. LXA4 binds to recombinant cysLT1 with approximately equal affinity as LTD4 (130) and has been suggested to function as a competitive inhibitor. Lipoxin binding to the aryl hydrocarbon receptor transcription factor leads to the expression of suppressor of cytokine signaling-2 in dendritic cells (131, 132). On a pathological level, the lipoxin signaling promotes inhibition of neutrophil migration, the recruitment of nonphlogistic macrophages, and increased phagocytosis among the countless documented effects (6, 126). The complex nature of lipoxin receptor signaling has been reviewed by Chiang et al. (129) in greater detail.
Eoxins
Analogous to 5-LOX biosynthesis of LTA4, 12/15-LOX-l can form an epoxide across C-14 and C-15 to form 14,15-LTA4 (133). Feltenmark et al. (134) demonstrated that murine eosinophils and mast cells can use 14,15-LTA4 to generate structural analogs of the cysteinyl leukotrienes, which they termed eoxins (EX). The enzymatic mechanism of eoxin biosynthesis has not been definitively elucidated, but likely occurs by a similar pathway as the cysteinyl leukotrienes (Fig. 6). Eoxins had weaker contractile activity on guinea pig pulmonary parenchyma or ileum (135, 136); however, in a cell culture model of epithelial barrier function EXC4, EXD4, and EXE4 demonstrably effected epithelial permeability at a lower dose than histamine (134).
Fig. 6.

Structure of eoxin C4. Eoxins are the 15-LOX analogs of the cysteinyl leukotrienes, where the thiol attachment occurs at C-14.
Phylogenetic classification of LOX enzymes
Historically, lipoxygenases have been classified according to their activity (102). This system works well for the 5-LOX, where the human isoform catalyzes 5-HETE formation in a similar manner to the mouse and rat homologs. However, it becomes more confusing when applied to other lipoxygenases, whose positional specificities are not as clearly defined or tightly conserved between mammalian homologs. For example, the leukocyte 12/15-LOX (12/15-LOX-l) from mouse produces significant amounts of both 12-HETE and 15-HETE (137). The human isoform of 15-LOX-2 produces 15-HETE, whereas the mouse homolog forms 8-HETE (138). For these reasons, an alternative classification scheme based on phylogeny has been proposed (102), describing the following 8-LOXs, 12-LOXs, and 15-LOXs: platelet-type 12-LOX (12-LOX-p), epidermis-type 12-LOXs (12-LOX-e, 12R-LOX, and e-LOX-3), leukocyte-type 12/15-LOX (12/15-LOX-l), and 8/15-lipoxygenase (8/15-LOX-2).
Platelet-type 12-LOX
In 1974, Hamberg and Samuelsson (139) described the activity of a lipoxygenase in platelets that converted arachidonic acid into 12-HETE. The human enzyme was concurrently cloned by Funk, Furci, and FitzGerald (140) and Isumi et al. (141) in 1990, and due to its high level of expression in these cells has been termed platelet-type 12-lipoxygenase (12-LOX-p). However, it's also abundant in epithelial tissue, and has been demonstrated to provide human skin melanomas with survival signals and promote tumor metastasis (142–144). Examination of the human 12-LOX-p by in vitro and in vivo analysis confirmed the native gene's positional specificity for producing 12-HETE (140, 145). While mutations designed to knock 12-lipoxygenase activity into the rabbit leukocyte-type 12/15-LOX (primarily a 15-LOX) succeeded (146, 147), the reverse mutagenesis experiments failed to convert 12-LOX-p into an effective 15-lipoxygenase (145). Other studies indicate that 12-LOX-p can function as a hepoxilin synthase on 12-HpETE (148), as well as a lipoxin synthase on LTA4 (149).
Epidermal-type 12-LOXs (12-LOX-e, 12R-LOX, and e-LOX-3)
The epidermal type 12-lipoxygenase (12-LOX-e) was separately cloned from mice by Funk et al. in 1996 (150) and Kinzig et al. in 1997 (151). The gene was the same size as the platelet-type and leukocyte-type 12-LOX and had 60% identity to these enzymes. Expression of the murine 12-LOX-e gene confirmed its lipoxygenase activity, specifically producing 12(S)-HETE (150). By analogy, 12-LOX-e likely functions by a similar catalytic mechanism as other 12-LOX. Using in situ hybridization, 12-LOX-e expression was demonstrated in keratinocytes and targeted areas of the hair follicle. The human isoform of 12-LOX-e was identified as a nonfunctional pseudogene (152), while in rat its activity has not been investigated.
As early as 1975, scaly lesions caused by psoriasis, an inflammatory disorder that affects the skin and joints, were known to contain increased levels of stereochemically pure 12-HETE (153). Distinct from other lipoxygenase metabolites, the hydroxyl moiety of 12-HETE found in these lesions was in the R-configuration (154). In 1998, Boeglin, Kim, and Brash (155) discovered the source of the 12R-HETE originated from 12R-lipoxygenase (12R-LOX), the first known mammalian R-lipoxygenase. Expression of 12R-LOX demonstrated that it produced 12R-HETE to the exclusion of significant amounts of 12S-HETE, as well as not making 5-HETE, 8-HETE, 9-HETE, 11-HETE, and 15-HETE. Using mutants of two R-lipoxygenases (human 12R-LOX and coral 8R-LOX) and two S-lipoxygenases (human and mouse 8/15-LOX-2 isoforms), Coffa and Brash (156) identified a conserved amino acid, corresponding with 12R-LOX Gly-441, that confers a substantial amount of stereochemical control to these enzymes. At this position, an alanine-to-glycine mutation in S-LOXs reversed the chirality of lipid metabolites. However, while mutating the glycyl residue of coral 8R-LOX fully converted it to a 12S-LOX, the 12R-LOX mutant had mixed activity. Meruvu et al. (157) identified Val631 as another important residue in facilitating the positional and stereochemical specificity of this enzyme.
Another epidermal-type LOX, e-LOX-3, has been cloned from both mouse (158) and human (159). While it contained the putative lipoxygenases' structural features and had >50% homology with 12R-LOX, gene expression in a number of cell lines indicated it had no activity toward arachidonic acid (158). However, mutations in both 12R-LOX and e-LOX-3 were identified in patients afflicted with psoriasis (160), and in 2003, Yu et al. (161) identified e-LOX-3 as a hepoxilin synthase that specifically used 12R-HpETE as a substrate. This suggests that impairment of hepoxilin production by a 12R-LOX/e-LOX-3 pathway leads to psoriasis in human patients.
Leukocyte-type 12/15-LOX (12/15-LOX-l)
The human 12/15-LOX-l was originally cloned as a 15-LOX from a cDNA library in 1988 (162), followed by rabbit (163), rat (164), and mouse (165). The first structural elucidation of a mammalian lipoxygenase was reported using X-ray crystallography on the rabbit 12/15-LOX-l homolog (105). The substrate binding pocket is lined by hydrophobic residues, and four residues comprise its base: Phe-353, Ile-418, Met-419, and Ile-593 (147). These residues are conserved in the human isoform and are considered critical for promoting 15-LOX activity. By comparison, the mouse and rat 12/15-LOX-l have amino acid substitutions at each of these positions that could explain their predilection toward 12-LOX activity. The rat 12/15-LOX-l also has intrinsic hepoxilin synthase activity (148), whereas human homolog does not. Based on the evidence that human 12-LOX-p can synthesize hepoxilins, Nigam et al. (116) surmised that lipoxygenase positional specificity, and not genetic homology, confer an enzyme with hepoxilin synthase activity. This would implicate the mouse 12/15-LOX-l as a hepoxilin synthase, but to date it has not been experimentally examined. The human 12/15-LOX-l exhibits eoxygenase activity that generates 14,15-LTs (134); the rabbit (166), pig (133), and sheep (167) homologs also demonstrate this activity, suggesting that mouse and rat enzymes retain this capacity.
From even preliminary investigations (168), 12/15-LOX-l has demonstrated positional promiscuity when oxidizing arachidonic acid, forming 15-HETE, 12-HETE, and 8,15-diHETE. The production of 12-HETE by 12/15-LOX-l has been shown to increase expression of monocyte chemoattractant protein-1, which increases vascular adhesion and the recruitment of macrophages (168). In addition to action on free arachidonic acid, 12/15-LOX-l can function at the membrane surface and on LDL to oxidize phospholipid and cholesterol ester fatty acids, leading to the development of atherosclerosis (169). In 2008, Harkewicz et al. (170) identified 12/15-LOX-l cholesterol ester peroxides as the active components of minimally oxidized LDL. In contrast to producing pro-inflammatory molecules, 12/15-LOX-l can take LTA4 as a substrate and produce the anti-inflammatory lipoxins (6, 129). Using both knockout and overexpression transgenic mouse models, Merched et al. (171) propose that 12/15-LOX-l helps suppress atherosclerosis. Undoubtedly, the capacity of 12/15-LOX-l to make pro-inflammatory as well anti-inflammatory metabolites complicates the study of this enzyme at the whole-animal level, and future research will help elucidate its role in signaling.
8/15-lipoxygenase-2
In 1997, Brash, Boeglin, and Chang (172) reported the discovery of a second 15-LOX gene in humans. The murine homolog has primarily 8-LOX activity (173), while the rat homolog has not been characterized to date. Mutational analyses of these enzymes determined that substitution of Tyr-603 and His-604 to the corresponding residues in the human isoform (aspartate and valine) confers 15-LOX activity (174). Likewise, mutation of the human enzyme to the murine residues confers 8-LOX activity. Though these enzymes appear to have different metabolic output, overexpression of either isoform leads to similar biological consequences in murine keratinocytes (138). In this model, 8/15-LOX-2 expression, as well as exogenous addition of 15-HETE and 8-HETE, blocked premalignant cell growth in a manner attributed to the inhibition of DNA synthesis. Interestingly, both human and murine 8/15-LOX-2 have been shown to produce 8,15-diHETE in vitro (175), a more potent agonist of PPARα than 8-HETE. In these studies, the murine enzyme was capable of both reactions and sequentially oxygenates the C-8 and C-15 position. The human homolog could only perform the oxygenation of C-15 but was able to use 8-HETE as a substrate. Future investigations may yet elucidate other novel metabolites produced by 8/15-LOX-2 that have important bioactivities.
CYTOCHROME P450 METABOLITES
Cytochrome P450
Cytochrome P450 (CYP) enzymes comprise a diverse superfamily of enzymes present in bacteria, fungi, prostists, plants, and animals (176, 177). The name was coined in 1962 to describe the first characterized cytochrome P450 based on the unusual absorbance peak of 450 nm by its carbon monoxide-bound form (178). CYP facilitate the addition of oxygen on a wide range of substrates. To date, 7232 genes in 781 families of have been categorized, and the list is constantly growing as more complete genome sequences become available; the generally accepted nomenclature system and current list of known CYPs is curated by Dr. David R. Nelson (http://drnelson.utmem.edu/CytochromeP450.html). They play an important role in the metabolism of toxins in the liver as well as signaling throughout the body. In eicosanoid biosynthesis, CYP activities are classified by hydrolase and epoxygenase activities.
Similar to LOXs, CYPs catalyze the hydroxylation and epoxygenation of arachidonic acid (Fig. 7). However, whereas LOX uses an active nonheme iron to abstract hydrogen directly from arachidonic acid, CYPs contain a heme-iron active site that oxidizes its substrate by a different mechanism (179, 180). Using NADPH as a cofactor, CYP performs a stepwise reduction of the catalytic iron and molecular oxygen to form a highly reactive oxo-iron species. Depending on the relative position of arachidonic acid, the reactive oxygen can perform an abstraction of an sp3 hybridized hydrogen or removal of an electron from an sp2 hybridized carbon. In both cases, the transfer of oxygen to the unstable arachidonic acid intermediate terminates the reaction by forming HETE or epoxy-eicosatrienoic acid, respectively.
Fig. 7.

Structures of cytochrome P450 metabolites. Cytochrome P450 enzymes can catalyze ω-oxidation (example: 20-HETE) and epoxidation (11,12-EET) reactions.
Each species has its own unique set of CYPs (181). In 2004, Nelson et al. (182) performed an extensive analysis of the human and mouse genomes, identifying 57 human and 102 mouse functional CYPs. They identified the same CYP families in human and mouse; however, four subfamilies have been evolutionarily discarded in humans, and a significant level of gene cluster size and organization has occurred. These gene expansions and mutations have significant impact on function, with subtle changes in the size and shape of the binding pocket capable of imparting dramatic changes in substrate selectivity and alignment, as well as product formation. In many cases, a single amino acid mutation can drastically alter the function of a CYP. For example, an S473V substitution on CYP2C2 allows a lauric acid hydoxylase to accept the steroid progesterone as a substrate (183). Thus, CYP homology modeling in the absence of experimental data may inaccurately represent in vivo function. For these reasons, we did not attempt to match CYP homologs between species (see supplementary Tables IV and V).
ω-Hydrolase HETEs
In addition to hydroxylating arachidonic acid between C-5 and C-15 to produce LOX-like HETEs, the CYP enzyme can add a hydroxyl moiety to the sp3-hybridized ω-carbons to form a unique class of HETEs (177). Historically, ω-hydrolase activity has been attributed to CYP4A and 4F in mammalian systems, though CYP from other families have demonstrable activity as well (see supplementary Table IV). The most well characterized of these is 20-HETE, hypothesized to play an important role in hypertension (184). 20-HETE has been shown promote systemic vasoconstriction by inhibiting KCa channel activity (185, 186), and in the kidney it blocks sodium reabsorbtion by inhibiting Na+-K+-ATPase activity (187). Other ω-HETEs demonstrate considerable bioactivity that often act in opposition to 20-HETE. For example, 18-HETE and 19-HETE dose-dependently induce vasodilatation by inhibiting the effects of 20-HETE (188). They also, in addition to 16-HETE and 17-HETE, have been shown to induce sodium reuptake in the kidney (188). More recently, 16-HETE has demonstrated the unique capacity among ω-HETEs to inhibit neutrophil adhesion, suggesting an important role in inflammation (189). Though it has been suggested that ω-HETEs signal through a putative receptor, neither the receptor nor its active second-messenger component has be identified (177).
Epoxyeicosatrienoic acids
The epoxidation of arachidonic acid by CYPs results in the formation of unique bioactive lipid mediators termed epoxyeicosatrienoic acids (EETs) (190). Each double bond has been shown to be susceptible to oxidation, resulting in 5,6-EET, 8,9-EET, 11,12-EET, and 14,15-EET. In mammals, the most well documented CYP epoxygenases include the 2C and 2J families, although currently numerous others have been implicated in EET biosynthesis (see supplementary Table V). The CYP epoxidases display significant promiscuity, with most enzymes making more than one EET, as well as retaining a significant level of ω-hydrolase activity.
EETs have been implicated in a number of important biological processes, including vascular tone, renal function, leukocyte adhesion, neuronal signaling, and angiogenesis. However, the effect of specific EET isomers often remains uncertain. In 1999, Node et al. (191) determined that 11,12-EET specifically block the expression of vascular cell adhesion molecule-1 by inhibiting NF-κB, an important mediator of leukocyte recruitment during inflammation. Using a model of vascular laminar flow, Liu et al. (192) suggest NF-κB inhibition can be achieved by any of the four EET stereoisomers.
Current evidence suggests that EET signaling occurs through a putative GPCR, though to date it has not been identified. Membranes from human U937 monocyte cell line were discovered by Wong et al. (193) to contain a high affinity binding site for 11,12-EET and 14,15-EET, which has recently been characterized as a GPCR that induces PKA activation and increases in cAMP (194). The EET-induced GPCR activity leads to the opening of BKCa channels through the Gsα g-protein (195, 196). Falck et al. (197) synthesized a library of 11,12-EET analogs and, using vascular cell adhesion molecule-1 expression in human endothelial cells, identified a number of structural elements required for functional recognition by the putative GPCR binding site. A corresponding study measuring BKCa activity induced by a series of 14,15-EET analogs identified similar structural elements (198), suggesting these two EETs may bind the same receptor in vivo.
In addition to GPCR signaling, EETs have been identified as bioactive ligands for cation channels and PPARs. Watanabe et al. (199) identified 5,6-EET, and to a lesser degree 8,9-EET, as an endogenous ligand for the vanilloid type 4 receptor (TRPV4). When activated, TRPV4 acts as a cation channel to allow the influx of extracellular calcium, and its activity has been implicated in rodent models of inflammatory hyperalgesia (200–202). Downstream activation of BKCa channels also result from TRPV4 (203). While these studies implicate EETs in the induction of hyperalgesia, Inceoglu et al. (204) demonstrate that inhibiting soluble epoxide hydrolase, which raises endogenous levels of EETs, blocks inflammatory hyperalgesia in the rat model. Receptor binding assays identified micrometer EET affinity for cannabinoid CB2, neurokinin NK1, and dopamine D3 receptors. In rat cardiomyocytes, EETs activate KATP channels by reducing their sensitivity to endogenous ATP inhibition (205). Studies have also implicated EETs in the direct activation of transcription factors PPARα and PPARγ (192, 206). However, the selectivity of these receptors for specific EET regioisomers as well as the downstream biological consequences have remained unclear, and undoubtedly further work will characterize the role that EETs play in biological signaling (207, 208).
Soluble epoxide hydrolase and dihydroxyeicosatrienoic acids
The majority of the EET biological activities are diminished by the hydrolysis to the corresponding dihydroxyeicosatrienoic acids (DHET). In mammals, this process is primarily catalyzed by the enzyme soluble epoxide hydrolase (sEH). The murine and human isoforms have been cloned and structurally characterized by X-ray crystallography (209–211), allowing for a detailed mechanistic description of its catalytic function using the human sequence (212). The sEH active site forms a deep, L-shaped hydrophobic pocket with a catalytic aspartyl at the L-turn accessible to both openings. Upon entering the active site, the EET epoxide moiety interacts with Tyr-382 and Tyr-465. His-523 positions and activates Asp-334 for a nucleophilic attack on an epoxide carbon, opening the ring and facilitating water hydrolysis to the corresponding diol. Unlike other eicosanoid biosynthetic enzymes, sEH does not require any metal cofactor for catalysis. However, its activity can be inhibited by the presence of zinc, suggesting a straightforward mechanism for regulating EET metabolism during inflammation (213). Due to the loss of EET bioactivity, sEH formation of DHETs has been considered the metabolic inactivation step of EET signaling. However, recent work has demonstrated that DHETs can retain certain EET signaling capacities, effectively activating BKCa channels in smooth muscle cells (214), as well as PPARα and PPARγ (192, 215).
Nonenzymatic lipid metabolites
Free radicals generated by oxidative stress have long been believed to participate in the development of a number of neurodegenerative and inflammatory diseases by causing damage to DNA, proteins, and lipids (216). In 1990, Morrow et al. (217) described a class of PG-like compounds formed in vitro by free radical catalyzed peroxidation. Abstraction of hydrogen from C-13 forms a five carbon delocalized radical that can form a prostane ring when followed by the addition of molecular oxygen at the activated C-11. Due to the lack of enzymatic control in the reaction, these PG-like compounds comprise a series of stereoisomers termed 15-series due to the location of the nonprostane hydroxyl moiety. Furthermore, the reaction can also be initiated at C-7, leading to the 5-series, as well as at C-10, leading to both the 8- and 12-series of isoprostanes. In total, 64 possible isoprostane isomers can be generated by radical initiated peroxidation (216). To date, no specific isoprostane receptors have been identified. However, isoprostanes have demonstrated binding affinity for PG receptors, including the FP and TP receptor (218, 219).
Lipid peroxidation can also proceed forward in a lipoxygenase-like activity. Following the abstraction of hydrogen, the peroxide formed by oxygenation of the radical can simply be reduced to a corresponding HETE. By this pathway, free radical reactions can produce all the HETEs generated by LOX and CYP. Additionally, nonenzymatic peroxidation can generate 9-HETE, which has no identifiable biological activity and has been used as a marker of nonenzymatic lipoxygenase-like activity (220). In practice, both 9-HETE and isoprostanes have been shown to serve as useful biomarkers of oxidative stress in vivo. In a recent study of major coronary heart disease (221), human plasma levels of isoprostanes and 9-HETE highly correlated with the pathogenesis and may serve as an indicator in clinical assessments of heart disease.
EICOSANOID CATABOLISM
Eicosanoid catabolism plays an important role in the control of bioactive lipid signaling (see supplementary Table VI). Not all molecules from the same class break down by the same pathway, which helps further differentiate the functional signaling role of each eicosanoid in vivo. In particular, the dysfunction of PG catabolizing enzymes, reviewed in detail by Tai et al. (222), has been implicated in susceptibility to disease. Generally, the catabolism of eicosanoids leads to diminished bioactive signaling and urinary excretion from the body by facilitating changes that increase their water solubility.
11-Hydroxythromboxane B2 reductase
Studies investigating thromboxane clearance indicate that a significant portion undergoes dehydrogenation at C-11 to form 11dh-TXB2. This degradation metabolite is readily detected in both human blood plasma and urine (223, 224). The enzyme responsible for catalysis in vivo has been termed 11-dehydroxythromboxane B2 dehydrogenase (11-TXDH) and has two distinct isoforms. Using amino acid composition analysis, the liver-type isoform from swine has been classified as a cytosolic aldehyde dehydrogenase family member (225). The second enzyme, the kidney-type 11-TXDH, can catalyze the dehydrogenation but not the reverse reductions, distinguishing it from the liver type. While the human, mouse, and rat 11-TXDH isoforms have not been identified, 11dh-TXB2 has been detected in urine samples from these species. Historically, TXB2 and 11dh-TXB2 have been considered biologically inert due to their inability to activate the thromboxane TP receptor (226), but work by Böhm et al. (227) demonstrates that 11dh-TXB2 is an agonist of the CRTH2 receptor.
15-Prostaglandin dehydrogenase
The catabolism of selected PGs, leukotrienes, and HETEs is initiated by oxidation by 15-prostaglandin dehydrogenase (15-PGDH). Two distinct enzymes have been identified with this activity, 15-PGDH-I and 15-PGDH-II; however, the high Km values of 15-PGDH-II for most PGs suggest that the majority of the in vivo activity can be attributed to 15-PGDH-I (222). The 15-PGDH-I is NAD+ dependant and metabolizes E-series PGs, lipoxins, 15-HETE, 5,15-diHETE, and 8,15-diHETE, while other PGs appear to be less susceptible substrates (228). In all likelihood, 15-PGDH can metabolize a larger array of eicosanoids than those which have been tested.
The 15-PGDH-I genes have been cloned in human, mouse, and rat, but a crystal structure of the enzyme has not yet been determined. In 2006, Cho et al. (229) used the molecular modeling to develop a three-dimensional structure of the human protein and propose a catalytic mechanism. Following the sequential binding of NAD+ and PGE2, the 15-hydroxyl is coordinated with Tyr-151, Ser-138, and Gln-148. Tyr-151 deprotonates the 15-hydroxyl, facilitating a hydride transfer from C-15 and the release of 15k-PGE2. The oxidation to a 15-ketone reduces the biological activity of most eicosanoid substrates, and loss of function has been implicated in the development of certain cancers (230). However, in 2007, Chou et al. (231) identified 15k-PGE2 as a ligand for PPARγ and an active component of murine 3T3-L1 fibroblast differentiation into adipocytes.
13-Prostaglandin reductase
The 13-prostaglandin reductases (13-PGR) metabolize eicosanoids by catalyzing NADH/NADPH-dependant double bond reduction. The gene was originally cloned from human and pig sources as leukotriene B4 12-hydroxydehydrogenase (LTB4DH) (232), but later discovered to have dual functionality as a PG reductase (13-PGR-1) (233). In addition to reducing PGs, 13-PGR has been shown to use lipoxins as substrates (234). The resulting metabolites from 13-PGR activity show reduced biological activity, and PGE2 metabolites from this pathway are readily detected in urinary excretion (235). The rat protein has LTB4DH and 13-PGR activities, suggesting that these functions are retained in the murine homolog (see supplementary Table VI).
Structural determinations of human 13-PGR-1 in complex with NADP+ and 15k-PGE2 were solved in 2004 and used to propose the following mechanism of catalysis (236). 13-PGR-1 forms a homodimer, with an active site pocket formed at the dimer interface. The 2'-hydroxyl group of NADPH forms a hydrogen bond with the C-15 oxygen on PGE2, promoting the stabilization of an enolate intermediate. The NADPH hydride attacks the carbocation at C-13, followed by ketone formation and the release of the reduced metabolite 13,14-dihydro,15-keto-PGE2 (dhk-PGE2). LTB4 oxidation likely proceeds by a similar mechanism as 15-PGDH, with a conserved tyrosine serving as the catalytic residue. In 2007, Chou et al. (231) reported the discovery of a second PG reductase gene (13-PGR-2) in mice that they demonstrate works by a similar reductive mechanism on 15k-PGE2 but have not exhaustively studied its substrate selectivity or potential LTB4DH activity.
Further metabolism for urinary excretion
As part of a Phase I clinical trial for LTB4 administration, Berry et al. (237) performed an exhaustive study of its catabolism. In this study, urine samples from subjects injected with LTB4 were analyzed, and 11 LTB4 metabolites were identified. Based on the qualitative and quantitative analysis of these metabolites, they proposed a pathway for LTB4 metabolism that, in addition to LTB4-specific enzymes, proceeds through three main pathways of catabolism: β-oxidation, CYP ω-hydrolases, and glucuronidation (Fig. 8).
Fig. 8.
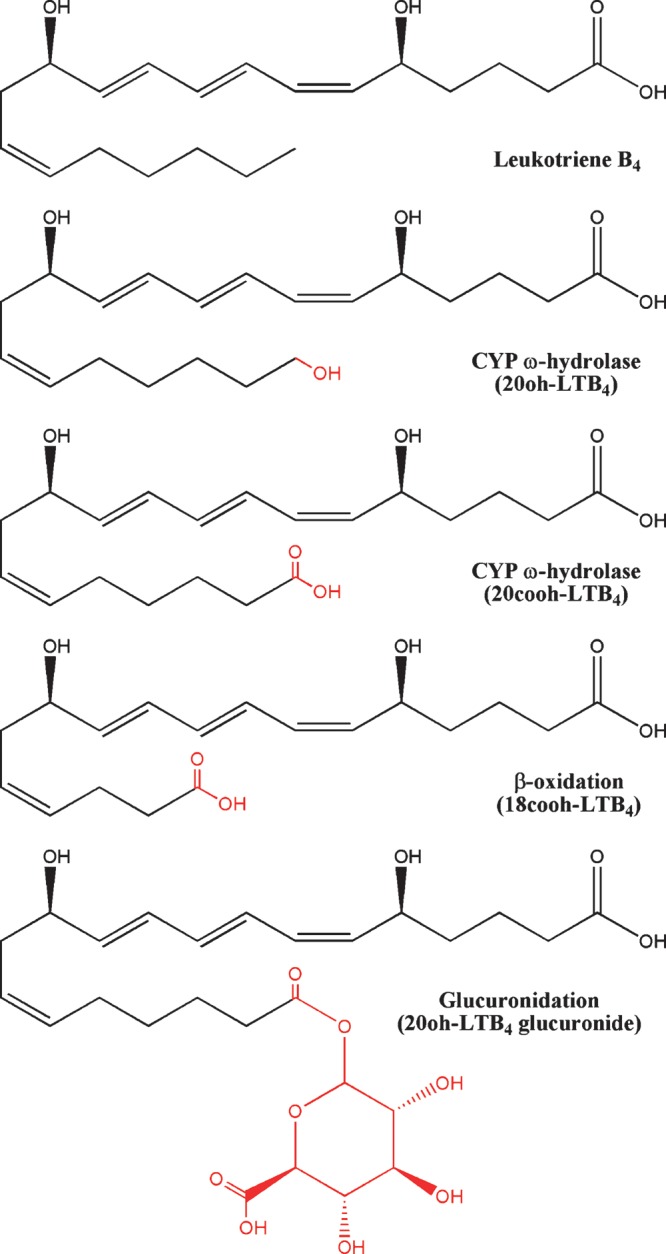
Examples of LTB4 metabolism by β-oxidation, CYP ω-hydrolases, and glucuronidation.
β-Oxidation
In addition to working on fatty acids, the β-oxidation metabolic pathway can capably act on the carboxyl moiety of PGs, leukotrienes, and HETEs, reviewed in detail by Diczfalusy (238). Studies in patients with Zellweger syndrome, a congenital disorder characterized by the absence of intact peroxisomes, determined that the majority of eicosanoid chain shortening occurs though peroxisomal β-oxidation pathways. Peroxisomal β-oxidation requires acyl-CoA oxidase, multifunctional enzyme, and acetyl-CoA acyltransferase activities (239); however, the specific isoforms implicated in eicosanoid degradation have not been conclusively identified. The subsequent losses of two (dinor) or four (tetranor) carbons significantly increases the hydrophilicity of these molecules for urinary excretion. With thromboxane as an exception, the majority of PG metabolites detected in urine have undergone β-oxidation of the C terminus. Pharmacokinetic analysis of first generation lipoxin analogs suggests that this class also undergoes β-oxidation (240). The HETEs also undergo β-oxidation from the C terminus, with the exception of 5-HETE.
Likewise, leukotrienes also appear to only be β-oxidized from the ω-terminus following CYP ω-hydroxylation and subsequent carboxylate formation by further oxidation, suggesting that C-5 hydroxyl moieties modulate the efficacy of this degradation pathway. Consistent with this, LTB4 injections in human patients lead to β-oxidation from the ω terminus (237).
CYP ω-hydrolases
The CYP ω-hydroxylated metabolites have a number of important bioactivities, as previously described. In addition, CYP hydrolases facilitate the catabolism of PGs, leukotrienes, and DHETs for urinary excretion (177). Hydroxylation of eicosanoids both reduces their bioactivity and increases their water solubility. Some ω-hydroxyl eicosanoids can be further metabolized by CYPs to form carboxylic acids (241, 242), allowing for β-oxidation of this end of the molecule. Following LTB4 administration, LTB4 metabolites were found to be hydroxylated at C-17 though C-20; however, the majority of these compounds had undergone further catabolism to form glucuronides at these sites (237).
Glucuronidation
In addition to action by CYPs, lipophilic molecules are further catabolized into water-soluble metabolites through the addition of the sugar glucuronide (243). In mammals, this reaction is catalyzed by members of the UDP-glucuronosyltransferase enzymes. To date, 15 UDP enzymes have been found in humans, and a study by Turgeon et al. (244) in 2003 using a HEK293 expression system found that most of them were capable of metabolizing LTB4 and LOX-derived HETEs. Glucuronidation also plays a significant role in the catabolism of DHETs in humans, but rats excrete the diol form directly (245). In the human trials of LTB4 metabolism, nearly all metabolites with a hydroxyl moiety were found to be glucuronidated, suggesting nonspecific catalysis of eicosanoids by UDPs plays a significant role in their removal from the body (237).
APPLICATIONS OF A SYSTEMS APPROACH TO EICOSANOID BIOLOGY
The LIPID MAPS consortium has produced preliminary studies comparing comprehensive lipidomic and transcriptomic changes in macrophages (7–9). In response to the lipopolysaccharide Kdo2-Lipid A, RAW264.7 murine macrophage cells were analyzed for eicosanoids by LC/MS/MS, as well as transcriptomic changes using Affemetrix gene arrays. Using publicly available data (www.lipidmaps.org), these changes can be mapped together to investigate the correlation between genes and metabolites (Fig. 9). This stimulatory pathway causes upregulation of the numerous COX genes and downregulation of the 5-LOX pathway, causing primarily increases in COX metabolites (9); PGDH, which metabolically inactivates PGE2, was downregulated, suggesting that it plays an important role in eicosanoid degradation. It remains to be addressed if other LOX or CYP enzymes are basally expressed and play an important role in this cell model. Work by Buczynski et al. (9) highlights the issue of basal protein activity, demonstrating that calcium agonists can activate the constitutively expressed 5-LOX pathway in RAW264.7 cells to make cysteinyl leukotrienes. Clearly, a proteomic analysis could address a number of such remaining questions in this cellular system. Thus, it is timely to carry out a proteomic analysis of the eicosanoid proteins to give a fully integrated biological picture.
Fig. 9.

Temporal genomic and lipidomic changes in eicosanoid biosynthesis in RAW264.7 macrophages in response to Kdo2-Lipid A stimulation. Greater intensity of red indicates increasing levels, greater intensity of green indicates decreasing levels, and gray represents no change in levels relative to unstimulated cells. Changes are represented as a function of time (left to right), where rectangles indicate mRNA data and circles indicate lipid data. When enzyme activities can result from multiple genes, each is represented as a separate line. The transcriptomic data will be reported by S. Subramaniam and S. Gupta (unpublished observations), and the lipidomic data will be reported by Gupta et al. (unpublished observations); both data sets are also publicly available at www.lipidmaps.org.
In a similar murine macrophage model, Shui et al. (246) compared the efficacy of two state of the art quantitative proteomic approaches. Using either the isobaric tag for relative and absolute quantitation chemical tagging or the stable isotope labeling with amino acids in cell culture labeling technique, they identified 1286 total unique proteins from the entire activated macrophage proteome, with 512 of those being reliably detectable for quantitation. However, probing these data sets for specific changes in the eicosanoid proteome only identified two proteins, FLAP and 13-PGR. None of the PG biosynthetic proteins were identified using these approaches, including the highly upregulated COX-2. In contrast, Jenkins et al. (247) have developed a selected population procedure for quantifying changes in the entire CYP protein family. Combining the advantages of isobaric tag for relative and absolute quantitation with the selectivity of multiple-reaction monitoring mass spectrometry, they could measure 16 different CYPs at femtomolar levels from murine liver microsome samples. Provided purified protein standards are available, this methodology could be readily expanded to investigate the entire eicosanoid proteome.
The information contained herein provides the basis necessary to decipher the eicosanoid proteome and complete the integrated pathway map. Microarray analysis of gene regulation and mass spectrometry methodology for lipid analysis capable of comprehensively analyzing these pathways now exist. This review provides the tools necessary to create a complimentary proteomic methodology. The sequential flow of information from gene to protein to metabolite remains a central focus in biological research. However, the integration of eicosanoid transcriptomic, proteomic, and lipidomic platforms, if carried out, would represent the first time such an analysis was performed on any biological system. This would undoubtedly have enormous implications for our understanding of biology and disease pathology and serve as a model for future systems biology endeavors.
Supplementary Material
Acknowledgments
The authors thank Rebecca C. Bowers-Gentry for her invaluable advice and input in drafting this manuscript. Sequence alignments found in the supplementary tables were done using Jalview 2.3 (248). The authors are also grateful to the entire LIPID MAPS Consortium for the use of their data, available at www.lipidmaps.org for public use.
Abbreviations
AKR, aldo-keto reductase
ALX, lipoxin A4 receptor
COX, cyclooxygenase
CRTH2, chemoattractant receptor-homologous, molecule expressed on Th2 cells
CYP, cytochrome P450
cysLT, cysteinyl leukotrienes
DHET, dihydroxyeicosatrienoic acid
EET, epoxyeicosatrienoic acid
EX, eoxin
FLAP, 5-lipoxygenase activating protein
GGL, γ-glutamyl leukotrienase
GGT, γ-glutamyl transpeptidase
GPCR, G protein-coupled receptor
HETE, hydroxyeicosatetraenoic acid
HpETE, hydroperoxy-eicosatetraenoic acid
HX, hepoxilin
9K-PGR, 9-keto prostaglandin reductase
LT, leukotriene
LTAH, leukotriene A4 hydrolase
LTB4DH, leukotriene B4 12-hydroxydehydrogenase
LTCS, leukotriene C4 synthase
LIPID MAPS, Lipid Metabolites and Pathways Strategies
LOX, lipoxygenase
LX, lipoxin
MAPEG, membrane-associated protein in eicosanoid and glutathione
MBD, membrane-bound dipeptidase
mGST, microsomal glutathione S-transferase
PG, prostaglandin
PGDH, prostaglandin dehydrogenase
PGDS, prostaglandin D synthase
PGES, PGE synthase
PGFS, prostaglandin F synthase
PGR, prostaglandin reductase
PLA2, phospholipase A2
PPAR, peroxisome proliferator-activated receptor
sEH, soluble epoxide hydrolase
TRPV4, vanilloid type 4 receptor
TXA2, thromboxane A2
11-TXDH, 11-dehydroxythromboxane B2 dehydrogenase
This work was supported by the National Institutes of Health LIPID MAPS Large Scale Collaborative Grant GM069338 and R01 GM64611 and GM20501. M.W.B. was supported by Gastroenterology Pre-doctoral Training Grant 2T32DK007202-32 from the National Institutes of Health.
Published, JLR Papers in Press, February 24, 2009.
Footnotes
Guest editor for this article was Jay W. Heinecke, JLR Associate Editor, University of Washington.
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of seven tables.
References
- 1.Aderem A. 2005. Systems biology: its practice and challenges. Cell. 121 511–513. [DOI] [PubMed] [Google Scholar]
- 2.de Godoy L. M., J. V. Olsen, J. Cox, M. L. Nielsen, N. C. Hubner, F. Frohlich, T. C. Walther, and M. Mann. 2008. Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature. 455 1251–1254. [DOI] [PubMed] [Google Scholar]
- 3.Funk C. D. 2001. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 294 1871–1875. [DOI] [PubMed] [Google Scholar]
- 4.Simmons D. L., R. M. Botting, and T. Hla. 2004. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol. Rev. 56 387–437. [DOI] [PubMed] [Google Scholar]
- 5.Smith W. L., D. L. DeWitt, and R. M. Garavito. 2000. Cyclooxygenases: structural, cellular, and molecular biology. Annu. Rev. Biochem. 69 145–182. [DOI] [PubMed] [Google Scholar]
- 6.Serhan C. N., N. Chiang, and T. E. Van Dyke. 2008. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 8 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feng, L., and G. D. Prestwich. 2006. Functional Lipidomics. Taylor & Francis, Boca Raton, FL.
- 8.Schmelzer K., E. Fahy, S. Subramaniam, and E. A. Dennis. 2007. The lipid maps initiative in lipidomics. Methods Enzymol. 432 171–183. [DOI] [PubMed] [Google Scholar]
- 9.Buczynski M. W., D. L. Stephens, R. C. Bowers-Gentry, A. Grkovich, R. A. Deems, and E. A. Dennis. 2007. TLR-4 and sustained calcium agonists synergistically produce eicosanoids independent of protein synthesis in RAW264.7 cells. J. Biol. Chem. 282 22834–22847. [DOI] [PubMed] [Google Scholar]
- 10.Domon B., and R. Aebersold. 2006. Mass spectrometry and protein analysis. Science. 312 212–217. [DOI] [PubMed] [Google Scholar]
- 11.Bantscheff M., M. Schirle, G. Sweetman, J. Rick, and B. Kuster. 2007. Quantitative mass spectrometry in proteomics: a critical review. Anal. Bioanal. Chem. 389 1017–1031. [DOI] [PubMed] [Google Scholar]
- 12.Keshishian H., T. Addona, M. Burgess, E. Kuhn, and S. A. Carr. 2007. Quantitative, multiplexed assays for low abundance proteins in plasma by targeted mass spectrometry and stable isotope dilution. Mol. Cell. Proteomics. 6 2212–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schaloske R. H., and E. A. Dennis. 2006. The phospholipase A2 superfamily and its group numbering system. Biochim. Biophys. Acta. 1761 1246–1259. [DOI] [PubMed] [Google Scholar]
- 14.Six D. A., and E. A. Dennis. 2000. The expanding superfamily of phospholipase A(2) enzymes: classification and characterization. Biochim. Biophys. Acta. 1488 1–19. [DOI] [PubMed] [Google Scholar]
- 15.Burke, J. E., and E. A. Dennis. Phospholipase A2 structure/function, mechanism and signaling. J. Lipid Res. Epub ahead of print. November, 14, 2008; doi: 10.1194/jlr.R800033-JLR200. [DOI] [PMC free article] [PubMed]
- 16.Uozumi N., and T. Shimizu. 2002. Roles for cytosolic phospholipase A2alpha as revealed by gene-targeted mice. Prostaglandins Other Lipid Mediat. 68–69 59–69. [DOI] [PubMed] [Google Scholar]
- 17.Ghosh M., D. E. Tucker, S. A. Burchett, and C. C. Leslie. 2006. Properties of the Group IV phospholipase A2 family. Prog. Lipid Res. 45 487–510. [DOI] [PubMed] [Google Scholar]
- 18.Bonventre J. V., Z. Huang, M. R. Taheri, E. O'Leary, E. Li, M. A. Moskowitz, and A. Sapirstein. 1997. Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature. 390 622–625. [DOI] [PubMed] [Google Scholar]
- 19.Uozumi N., K. Kume, T. Nagase, N. Nakatani, S. Ishii, F. Tashiro, Y. Komagata, K. Maki, K. Ikuta, Y. Ouchi, et al. 1997. Role of cytosolic phospholipase A2 in allergic response and parturition. Nature. 390 618–622. [DOI] [PubMed] [Google Scholar]
- 20.Adler D. H., J. D. Cogan, J. A. Phillips, N. Schnetz-Boutaud, G. L. Milne, T. Iverson, J. A. Stein, D. A. Brenner, J. D. Morrow, O. Boutaud, et al. 2008. Inherited human cPLA(2alpha) deficiency is associated with impaired eicosanoid biosynthesis, small intestinal ulceration, and platelet dysfunction. J. Clin. Invest. 118 2121–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lambeau G., and M. H. Gelb. 2008. Biochemistry and physiology of mammalian secreted phospholipases A2. Annu. Rev. Biochem. 77 495–520. [DOI] [PubMed] [Google Scholar]
- 22.Rouzer, C. A., and L. J. Marnett. Cyclooxygenases: structural and functional insights. J. Lipid Res. Epub ahead of print. October 23, 2008; doi10.1194/jlr.R800042-JLR200.. [DOI] [PMC free article] [PubMed]
- 23.Nelson N. A. 1974. Prostaglandin nomenclature. J. Med. Chem. 17 911–918. [DOI] [PubMed] [Google Scholar]
- 24.Whittaker N., S. Bunting, J. Salmon, S. Moncada, J. R. Vane, R. A. Johnson, D. R. Morton, J. H. Kinner, R. R. Gorman, J. C. McGuire, et al. 1976. The chemical structure of prostaglandin X (prostacyclin). Prostaglandins. 12 915–928. [DOI] [PubMed] [Google Scholar]
- 25.Wu K. K., and J. Y. Liou. 2005. Cellular and molecular biology of prostacyclin synthase. Biochem. Biophys. Res. Commun. 338 45–52. [DOI] [PubMed] [Google Scholar]
- 26.Chiang C. W., H. C. Yeh, L. H. Wang, and N. L. Chan. 2006. Crystal structure of the human prostacyclin synthase. J. Mol. Biol. 364 266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hecker M., and V. Ullrich. 1989. On the mechanism of prostacyclin and thromboxane A2 biosynthesis. J. Biol. Chem. 264 141–150. [PubMed] [Google Scholar]
- 28.Katsuyama M., Y. Sugimoto, T. Namba, A. Irie, M. Negishi, S. Narumiya, and A. Ichikawa. 1994. Cloning and expression of a cDNA for the human prostacyclin receptor. FEBS Lett. 344 74–78. [DOI] [PubMed] [Google Scholar]
- 29.Boie Y., T. H. Rushmore, A. Darmon-Goodwin, R. Grygorczyk, D. M. Slipetz, K. M. Metters, and M. Abramovitz. 1994. Cloning and expression of a cDNA for the human prostanoid IP receptor. J. Biol. Chem. 269 12173–12178. [PubMed] [Google Scholar]
- 30.Aubert J., G. Ailhaud, and R. Negrel. 1996. Evidence for a novel regulatory pathway activated by (carba)prostacyclin in preadipose and adipose cells. FEBS Lett. 397 117–121. [DOI] [PubMed] [Google Scholar]
- 31.Kojo H., M. Fukagawa, K. Tajima, A. Suzuki, T. Fujimura, I. Aramori, K. Hayashi, and S. Nishimura. 2003. Evaluation of human peroxisome proliferator-activated receptor (PPAR) subtype selectivity of a variety of anti-inflammatory drugs based on a novel assay for PPAR delta(beta). J. Pharmacol. Sci. 93 347–355. [DOI] [PubMed] [Google Scholar]
- 32.Hamberg M., J. Svensson, and B. Samuelsson. 1975. Thromboxanes: a new group of biologically active compounds derived from prostaglandin endoperoxides. Proc. Natl. Acad. Sci. USA. 72 2994–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kinsella B. T., D. J. O'Mahony, and G. A. Fitzgerald. 1997. The human thromboxane A2 receptor alpha isoform (TP alpha) functionally couples to the G proteins Gq and G11 in vivo and is activated by the isoprostane 8-epi prostaglandin F2 alpha. J. Pharmacol. Exp. Ther. 281 957–964. [PubMed] [Google Scholar]
- 34.Samuelsson B. 1963. Isolation and identification of prostaglandins from human seminal plasma. 18. Prostaglandins and related factors. J. Biol. Chem. 238 3229–3234. [PubMed] [Google Scholar]
- 35.Park J. Y., M. H. Pillinger, and S. B. Abramson. 2006. Prostaglandin E2 synthesis and secretion: the role of PGE2 synthases. Clin. Immunol. 119 229–240. [DOI] [PubMed] [Google Scholar]
- 36.Tanioka T., Y. Nakatani, N. Semmyo, M. Murakami, and I. Kudo. 2000. Molecular identification of cytosolic prostaglandin E2 synthase that is functionally coupled with cyclooxygenase-1 in immediate prostaglandin E2 biosynthesis. J. Biol. Chem. 275 32775–32782. [DOI] [PubMed] [Google Scholar]
- 37.Murakami M., Y. Nakatani, T. Tanioka, and I. Kudo. 2002. Prostaglandin E synthase. Prostaglandins Other Lipid Mediat. 68–69 383–399. [DOI] [PubMed] [Google Scholar]
- 38.Jakobsson P. J., S. Thoren, R. Morgenstern, and B. Samuelsson. 1999. Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc. Natl. Acad. Sci. USA. 96 7220–7225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang X., W. Yan, D. Gao, M. Tong, H. H. Tai, and C. G. Zhan. 2006. Structural and functional characterization of human microsomal prostaglandin E synthase-1 by computational modeling and site-directed mutagenesis. Bioorg. Med. Chem. 14 3553–3562. [DOI] [PubMed] [Google Scholar]
- 40.Samuelsson B., R. Morgenstern, and P. J. Jakobsson. 2007. Membrane prostaglandin E synthase-1: a novel therapeutic target. Pharmacol. Rev. 59 207–224. [DOI] [PubMed] [Google Scholar]
- 41.Watanabe K., K. Kurihara, Y. Tokunaga, and O. Hayaishi. 1997. Two types of microsomal prostaglandin E synthase: glutathione-dependent and -independent prostaglandin E synthases. Biochem. Biophys. Res. Commun. 235 148–152. [DOI] [PubMed] [Google Scholar]
- 42.Tanikawa N., Y. Ohmiya, H. Ohkubo, K. Hashimoto, K. Kangawa, M. Kojima, S. Ito, and K. Watanabe. 2002. Identification and characterization of a novel type of membrane-associated prostaglandin E synthase. Biochem. Biophys. Res. Commun. 291 884–889. [DOI] [PubMed] [Google Scholar]
- 43.Yamada T., J. Komoto, K. Watanabe, Y. Ohmiya, and F. Takusagawa. 2005. Crystal structure and possible catalytic mechanism of microsomal prostaglandin E synthase type 2 (mPGES-2). J. Mol. Biol. 348 1163–1176. [DOI] [PubMed] [Google Scholar]
- 44.Ito S., S. Narumiya, and O. Hayaishi. 1989. Prostaglandin D2: a biochemical perspective. Prostaglandins Leukot. Essent. Fatty Acids. 37 219–234. [DOI] [PubMed] [Google Scholar]
- 45.Abdel-Halim M. S., M. Hamberg, B. Sjoquist, and E. Anggard. 1977. Identification of prostaglandin D2 as a major prostaglandin in homogenates of rat brain. Prostaglandins. 14 633–643. [DOI] [PubMed] [Google Scholar]
- 46.Roberts 2nd L. J., R. A. Lewis, J. A. Oates, and K. F. Austen. 1979. Prostaglandin thromboxane, and 12-hydroxy-5,8,10,14-eicosatetraenoic acid production by ionophore-stimulated rat serosal mast cells. Biochim. Biophys. Acta. 575 185–192. [DOI] [PubMed] [Google Scholar]
- 47.Urade Y., T. Tanaka, N. Eguchi, M. Kikuchi, H. Kimura, H. Toh, and O. Hayaishi. 1995. Structural and functional significance of cysteine residues of glutathione-independent prostaglandin D synthase. Identification of Cys65 as an essential thiol. J. Biol. Chem. 270 1422–1428. [DOI] [PubMed] [Google Scholar]
- 48.Shimamoto S., T. Yoshida, T. Inui, K. Gohda, Y. Kobayashi, K. Fujimori, T. Tsurumura, K. Aritake, Y. Urade, and T. Ohkubo. 2007. NMR solution structure of lipocalin-type prostaglandin D synthase: evidence for partial overlapping of catalytic pocket and retinoic acid-binding pocket within the central cavity. J. Biol. Chem. 282 31373–31379. [DOI] [PubMed] [Google Scholar]
- 49.Kanaoka Y., H. Ago, E. Inagaki, T. Nanayama, M. Miyano, R. Kikuno, Y. Fujii, N. Eguchi, H. Toh, Y. Urade, et al. 1997. Cloning and crystal structure of hematopoietic prostaglandin D synthase. Cell. 90 1085–1095. [DOI] [PubMed] [Google Scholar]
- 50.Pettipher R. 2008. The roles of the prostaglandin D(2) receptors DP(1) and CRTH2 in promoting allergic responses. Br. J. Pharmacol. 153 (Suppl 1.): S191–S199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sawyer N., E. Cauchon, A. Chateauneuf, R. P. Cruz, D. W. Nicholson, K. M. Metters, G. P. O'Neill, and F. G. Gervais. 2002. Molecular pharmacology of the human prostaglandin D2 receptor, CRTH2. Br. J. Pharmacol. 137 1163–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Basu S. 2007. Novel cyclooxygenase-catalyzed bioactive prostaglandin F2alpha from physiology to new principles in inflammation. Med. Res. Rev. 27 435–468. [DOI] [PubMed] [Google Scholar]
- 53.Abramovitz M., Y. Boie, T. Nguyen, T. H. Rushmore, M. A. Bayne, K. M. Metters, D. M. Slipetz, and R. Grygorczyk. 1994. Cloning and expression of a cDNA for the human prostanoid FP receptor. J. Biol. Chem. 269 2632–2636. [PubMed] [Google Scholar]
- 54.Daiyasu H., K. Watanabe, and H. Toh. 2008. Recruitment of thioredoxin-like domains into prostaglandin synthases. Biochem. Biophys. Res. Commun. 369 281–286. [DOI] [PubMed] [Google Scholar]
- 55.Suzuki-Yamamoto T., M. Nishizawa, M. Fukui, E. Okuda-Ashitaka, T. Nakajima, S. Ito, and K. Watanabe. 1999. cDNA cloning, expression and characterization of human prostaglandin F synthase. FEBS Lett. 462 335–340. [DOI] [PubMed] [Google Scholar]
- 56.Moriuchi H., N. Koda, E. Okuda-Ashitaka, H. Daiyasu, K. Ogasawara, H. Toh, S. Ito, D. F. Woodward, and K. Watanabe. 2008. Molecular characterization of a novel type of prostamide/prostaglandin F synthase, belonging to the thioredoxin-like superfamily. J. Biol. Chem. 283 792–801. [DOI] [PubMed] [Google Scholar]
- 57.Komoto J., T. Yamada, K. Watanabe, and F. Takusagawa. 2004. Crystal structure of human prostaglandin F synthase (AKR1C3). Biochemistry. 43 2188–2198. [DOI] [PubMed] [Google Scholar]
- 58.Komoto J., T. Yamada, K. Watanabe, D. F. Woodward, and F. Takusagawa. 2006. Prostaglandin F2alpha formation from prostaglandin H2 by prostaglandin F synthase (PGFS): crystal structure of PGFS containing bimatoprost. Biochemistry. 45 1987–1996. [DOI] [PubMed] [Google Scholar]
- 59.Penning T. M., S. Steckelbroeck, D. R. Bauman, M. W. Miller, Y. Jin, D. M. Peehl, K. M. Fung, and H. K. Lin. 2006. Aldo-keto reductase (AKR) 1C3: role in prostate disease and the development of specific inhibitors. Mol. Cell. Endocrinol. 248 182–191. [DOI] [PubMed] [Google Scholar]
- 60.Levine L., K. Y. Wu, and S. S. Pong. 1975. Stereospecificity of enzymatic reduction of prostaglandin E2 to F2alpha. Prostaglandins. 9 531–544. [DOI] [PubMed] [Google Scholar]
- 61.Asselin E., and M. A. Fortier. 2000. Detection and regulation of the messenger for a putative bovine endometrial 9-keto-prostaglandin E(2) reductase: effect of oxytocin and interferon-tau. Biol. Reprod. 62 125–131. [DOI] [PubMed] [Google Scholar]
- 62.Wermuth B. 1981. Purification and properties of an NADPH-dependent carbonyl reductase from human brain. Relationship to prostaglandin 9-ketoreductase and xenobiotic ketone reductase. J. Biol. Chem. 256 1206–1213. [PubMed] [Google Scholar]
- 63.Miura T., T. Nishinaka, and T. Terada. 2008. Different functions between human monomeric carbonyl reductase 3 and carbonyl reductase 1. Mol. Cell Biochem. 315 113–121. [DOI] [PubMed] [Google Scholar]
- 64.Yokoyama Y., B. Xin, T. Shigeto, M. Umemoto, A. Kasai-Sakamoto, M. Futagami, S. Tsuchida, F. Al-Mulla, and H. Mizunuma. 2007. Clofibric acid, a peroxisome proliferator-activated receptor alpha ligand, inhibits growth of human ovarian cancer. Mol. Cancer Ther. 6 1379–1386. [DOI] [PubMed] [Google Scholar]
- 65.Waclawik A., and A. J. Ziecik. 2007. Differential expression of prostaglandin (PG) synthesis enzymes in conceptus during peri-implantation period and endometrial expression of carbonyl reductase/PG 9-ketoreductase in the pig. J. Endocrinol. 194 499–510. [DOI] [PubMed] [Google Scholar]
- 66.Straus D. S., and C. K. Glass. 2001. Cyclopentenone prostaglandins: new insights on biological activities and cellular targets. Med. Res. Rev. 21 185–210. [DOI] [PubMed] [Google Scholar]
- 67.Powell W. S. 2003. 15-Deoxy-delta12,14–PGJ2: endogenous PPARgamma ligand or minor eicosanoid degradation product? J. Clin. Invest. 112 828–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Murphy R. C., S. Hammarstrom, and B. Samuelsson. 1979. Leukotriene C: a slow-reacting substance from murine mastocytoma cells. Proc. Natl. Acad. Sci. USA. 76 4275–4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Matsumoto T., C. D. Funk, O. Radmark, J. O. Hoog, H. Jornvall, and B. Samuelsson. 1988. Molecular cloning and amino acid sequence of human 5-lipoxygenase. Proc. Natl. Acad. Sci. USA. 85 26–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ferguson A. D., B. M. McKeever, S. Xu, D. Wisniewski, D. K. Miller, T. T. Yamin, R. H. Spencer, L. Chu, F. Ujjainwalla, B. R. Cunningham, et al. 2007. Crystal structure of inhibitor-bound human 5-lipoxygenase-activating protein. Science. 317 510–512. [DOI] [PubMed] [Google Scholar]
- 71.Murphy R. C., and M. A. Gijon. 2007. Biosynthesis and metabolism of leukotrienes. Biochem. J. 405 379–395. [DOI] [PubMed] [Google Scholar]
- 72.Powell W. S., and J. Rokach. 2005. Biochemistry, biology and chemistry of the 5-lipoxygenase product 5-oxo-ETE. Prog. Lipid Res. 44 154–183. [DOI] [PubMed] [Google Scholar]
- 73.Jones C. E., S. Holden, L. Tenaillon, U. Bhatia, K. Seuwen, P. Tranter, J. Turner, R. Kettle, R. Bouhelal, S. Charlton, et al. 2003. Expression and characterization of a 5-oxo-6E,8Z,11Z,14Z-eicosatetraenoic acid receptor highly expressed on human eosinophils and neutrophils. Mol. Pharmacol. 63 471–477. [DOI] [PubMed] [Google Scholar]
- 74.Borgeat P., and B. Samuelsson. 1979. Metabolism of arachidonic acid in polymorphonuclear leukocytes. Structural analysis of novel hydroxylated compounds. J. Biol. Chem. 254 7865–7869. [PubMed] [Google Scholar]
- 75.Ford-Hutchinson A. W., M. A. Bray, M. V. Doig, M. E. Shipley, and M. J. Smith. 1980. Leukotriene B, a potent chemokinetic and aggregating substance released from polymorphonuclear leukocytes. Nature. 286 264–265. [DOI] [PubMed] [Google Scholar]
- 76.Tager A. M., and A. D. Luster. 2003. BLT1 and BLT2: the leukotriene B(4) receptors. Prostaglandins Leukot. Essent. Fatty Acids. 69 123–134. [DOI] [PubMed] [Google Scholar]
- 77.Tager A. M., J. H. Dufour, K. Goodarzi, S. D. Bercury, U. H. von Andrian, and A. D. Luster. 2000. BLTR mediates leukotriene B(4)-induced chemotaxis and adhesion and plays a dominant role in eosinophil accumulation in a murine model of peritonitis. J. Exp. Med. 192 439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Haribabu B., M. W. Verghese, D. A. Steeber, D. D. Sellars, C. B. Bock, and R. Snyderman. 2000. Targeted disruption of the leukotriene B(4) receptor in mice reveals its role in inflammation and platelet-activating factor-induced anaphylaxis. J. Exp. Med. 192 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Okuno T., Y. Iizuka, H. Okazaki, T. Yokomizo, R. Taguchi, and T. Shimizu. 2008. 12(S)-Hydroxyheptadeca-5Z, 8E, 10E-trienoic acid is a natural ligand for leukotriene B4 receptor 2. J. Exp. Med. 205 759–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Haeggstrom J. Z. 2004. Leukotriene A4 hydrolase/aminopeptidase, the gatekeeper of chemotactic leukotriene B4 biosynthesis. J. Biol. Chem. 279 50639–50642. [DOI] [PubMed] [Google Scholar]
- 81.Tholander F., F. Kull, E. Ohlson, J. Shafqat, M. M. Thunnissen, and J. Z. Haeggstrom. 2005. Leukotriene A4 hydrolase, insights into the molecular evolution by homology modeling and mutational analysis of enzyme from Saccharomyces cerevisiae. J. Biol. Chem. 280 33477–33486. [DOI] [PubMed] [Google Scholar]
- 82.Fitzpatrick F., J. Haeggstrom, E. Granstrom, and B. Samuelsson. 1983. Metabolism of leukotriene A4 by an enzyme in blood plasma: a possible leukotactic mechanism. Proc. Natl. Acad. Sci. USA. 80 5425–5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Funk C. D., O. Radmark, J. Y. Fu, T. Matsumoto, H. Jornvall, T. Shimizu, and B. Samuelsson. 1987. Molecular cloning and amino acid sequence of leukotriene A4 hydrolase. Proc. Natl. Acad. Sci. USA. 84 6677–6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Thunnissen M. M., P. Nordlund, and J. Z. Haeggstrom. 2001. Crystal structure of human leukotriene A(4) hydrolase, a bifunctional enzyme in inflammation. Nat. Struct. Biol. 8 131–135. [DOI] [PubMed] [Google Scholar]
- 85.Peters-Golden M., M. M. Gleason, and A. Togias. 2006. Cysteinyl leukotrienes: multi-functional mediators in allergic rhinitis. Clin. Exp. Allergy. 36 689–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rovati G. E., and V. Capra. 2007. Cysteinyl-leukotriene receptors and cellular signals. ScientificWorldJournal. 7 1375–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lam B. K., J. F. Penrose, G. J. Freeman, and K. F. Austen. 1994. Expression cloning of a cDNA for human leukotriene C4 synthase, an integral membrane protein conjugating reduced glutathione to leukotriene A4. Proc. Natl. Acad. Sci. USA. 91 7663–7667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Welsch D. J., D. P. Creely, S. D. Hauser, K. J. Mathis, G. G. Krivi, and P. C. Isakson. 1994. Molecular cloning and expression of human leukotriene-C4 synthase. Proc. Natl. Acad. Sci. USA. 91 9745–9749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Scoggan K. A., P. J. Jakobsson, and A. W. Ford-Hutchinson. 1997. Production of leukotriene C4 in different human tissues is attributable to distinct membrane bound biosynthetic enzymes. J. Biol. Chem. 272 10182–10187. [DOI] [PubMed] [Google Scholar]
- 90.Kanaoka Y., A. Maekawa, J. F. Penrose, K. F. Austen, and B. K. Lam. 2001. Attenuated zymosan-induced peritoneal vascular permeability and IgE-dependent passive cutaneous anaphylaxis in mice lacking leukotriene C4 synthase. J. Biol. Chem. 276 22608–22613. [DOI] [PubMed] [Google Scholar]
- 91.Ago H., Y. Kanaoka, D. Irikura, B. K. Lam, T. Shimamura, K. F. Austen, and M. Miyano. 2007. Crystal structure of a human membrane protein involved in cysteinyl leukotriene biosynthesis. Nature. 448 609–612. [DOI] [PubMed] [Google Scholar]
- 92.Martinez Molina D., A. Wetterholm, A. Kohl, A. A. McCarthy, D. Niegowski, E. Ohlson, T. Hammarberg, S. Eshaghi, J. Z. Haeggstrom, and P. Nordlund. 2007. Structural basis for synthesis of inflammatory mediators by human leukotriene C4 synthase. Nature. 448 613–616. [DOI] [PubMed] [Google Scholar]
- 93.Anderson M. E., R. D. Allison, and A. Meister. 1982. Interconversion of leukotrienes catalyzed by purified gamma-glutamyl transpeptidase: concomitant formation of leukotriene D4 and gamma-glutamyl amino acids. Proc. Natl. Acad. Sci. USA. 79 1088–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Keillor J. W., R. Castonguay, and C. Lherbet. 2005. Gamma-glutamyl transpeptidase substrate specificity and catalytic mechanism. Methods Enzymol. 401 449–467. [DOI] [PubMed] [Google Scholar]
- 95.Carter B. Z., A. L. Wiseman, R. Orkiszewski, K. D. Ballard, C. N. Ou, and M. W. Lieberman. 1997. Metabolism of leukotriene C4 in gamma-glutamyl transpeptidase-deficient mice. J. Biol. Chem. 272 12305–12310. [DOI] [PubMed] [Google Scholar]
- 96.Heisterkamp N., E. Rajpert-De Meyts, L. Uribe, H. J. Forman, and J. Groffen. 1991. Identification of a human gamma-glutamyl cleaving enzyme related to, but distinct from, gamma-glutamyl transpeptidase. Proc. Natl. Acad. Sci. USA. 88 6303–6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Carter B. Z., Z. Z. Shi, R. Barrios, and M. W. Lieberman. 1998. Gamma-glutamyl leukotrienase, a gamma-glutamyl transpeptidase gene family member, is expressed primarily in spleen. J. Biol. Chem. 273 28277–28285. [DOI] [PubMed] [Google Scholar]
- 98.Shi Z. Z., B. Han, G. M. Habib, M. M. Matzuk, and M. W. Lieberman. 2001. Disruption of gamma-glutamyl leukotrienase results in disruption of leukotriene D(4) synthesis in vivo and attenuation of the acute inflammatory response. Mol. Cell. Biol. 21 5389–5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Adachi H., I. Kubota, N. Okamura, H. Iwata, M. Tsujimoto, H. Nakazato, T. Nishihara, and T. Noguchi. 1989. Purification and characterization of human microsomal dipeptidase. J. Biochem. 105 957–961. [DOI] [PubMed] [Google Scholar]
- 100.Habib G. M., Z. Z. Shi, A. A. Cuevas, Q. Guo, M. M. Matzuk, and M. W. Lieberman. 1998. Leukotriene D4 and cystinyl-bis-glycine metabolism in membrane-bound dipeptidase-deficient mice. Proc. Natl. Acad. Sci. USA. 95 4859–4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Habib G. M., Z. Z. Shi, A. A. Cuevas, and M. W. Lieberman. 2003. Identification of two additional members of the membrane-bound dipeptidase family. FASEB J. 17 1313–1315. [DOI] [PubMed] [Google Scholar]
- 102.Kuhn H., and V. B. O'Donnell. 2006. Inflammation and immune regulation by 12/15-lipoxygenases. Prog. Lipid Res. 45 334–356. [DOI] [PubMed] [Google Scholar]
- 103.Minor W., J. Steczko, B. Stec, Z. Otwinowski, J. T. Bolin, R. Walter, and B. Axelrod. 1996. Crystal structure of soybean lipoxygenase L-1 at 1.4 A resolution. Biochemistry. 35 10687–10701. [DOI] [PubMed] [Google Scholar]
- 104.Skrzypczak-Jankun E., L. M. Amzel, B. A. Kroa, and M. O. Funk, Jr. 1997. Structure of soybean lipoxygenase L3 and a comparison with its L1 isoenzyme. Proteins. 29 15–31. [PubMed] [Google Scholar]
- 105.Gillmor S. A., A. Villasenor, R. Fletterick, E. Sigal, and M. F. Browner. 1997. The structure of mammalian 15-lipoxygenase reveals similarity to the lipases and the determinants of substrate specificity. Nat. Struct. Biol. 4 1003–1009. [DOI] [PubMed] [Google Scholar]
- 106.Coffa G., C. Schneider, and A. R. Brash. 2005. A comprehensive model of positional and stereo control in lipoxygenases. Biochem. Biophys. Res. Commun. 338 87–92. [DOI] [PubMed] [Google Scholar]
- 107.Murakami K., T. Ide, M. Suzuki, T. Mochizuki, and T. Kadowaki. 1999. Evidence for direct binding of fatty acids and eicosanoids to human peroxisome proliferators-activated receptor alpha. Biochem. Biophys. Res. Commun. 260 609–613. [DOI] [PubMed] [Google Scholar]
- 108.Huang J. T., J. S. Welch, M. Ricote, C. J. Binder, T. M. Willson, C. Kelly, J. L. Witztum, C. D. Funk, D. Conrad, and C. K. Glass. 1999. Interleukin-4-dependent production of PPAR-gamma ligands in macrophages by 12/15-lipoxygenase. Nature. 400 378–382. [DOI] [PubMed] [Google Scholar]
- 109.Hsi L. C., L. C. Wilson, and T. E. Eling. 2002. Opposing effects of 15-lipoxygenase-1 and -2 metabolites on MAPK signaling in prostate. Alteration in peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 277 40549–40556. [DOI] [PubMed] [Google Scholar]
- 110.Wen Y., J. Gu, G. E. Vandenhoff, X. Liu, and J. L. Nadler. 2008. Role of 12/15-lipoxygenase in the expression of MCP-1 in mouse macrophages. Am. J. Physiol. Heart Circ. Physiol. 294 H1933–H1938. [DOI] [PubMed] [Google Scholar]
- 111.Zhang B., H. Cao, and G. N. Rao. 2005. 15(S)-hydroxyeicosatetraenoic acid induces angiogenesis via activation of PI3K-Akt-mTOR-S6K1 signaling. Cancer Res. 65 7283–7291. [DOI] [PubMed] [Google Scholar]
- 112.Nie D., S. Krishnamoorthy, R. Jin, K. Tang, Y. Chen, Y. Qiao, A. Zacharek, Y. Guo, J. Milanini, G. Pages, et al. 2006. Mechanisms regulating tumor angiogenesis by 12-lipoxygenase in prostate cancer cells. J. Biol. Chem. 281 18601–18609. [DOI] [PubMed] [Google Scholar]
- 113.Kumar K. A., K. M. Arunasree, K. R. Roy, N. P. Reddy, A. Aparna, G. V. Reddy, and P. Reddanna. 2009. Effects of (15S)-hydroperoxyeicosatetraenoic acid and (15S)-hydroxyeicosatetraenoic acid on the acute- lymphoblastic-leukaemia cell line Jurkat: activation of the Fas-mediated death pathway. Biotechnol. Appl. Biochem. 52 121–133. [DOI] [PubMed] [Google Scholar]
- 114.Kwon K. J., Y. S. Jung, S. H. Lee, C. H. Moon, and E. J. Baik. 2005. Arachidonic acid induces neuronal death through lipoxygenase and cytochrome P450 rather than cyclooxygenase. J. Neurosci. Res. 81 73–84. [DOI] [PubMed] [Google Scholar]
- 115.Hwang S. W., H. Cho, J. Kwak, S. Y. Lee, C. J. Kang, J. Jung, S. Cho, K. H. Min, Y. G. Suh, D. Kim, et al. 2000. Direct activation of capsaicin receptors by products of lipoxygenases: endogenous capsaicin-like substances. Proc. Natl. Acad. Sci. USA. 97 6155–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nigam S., M. P. Zafiriou, R. Deva, R. Ciccoli, and R. Roux-Van der Merwe. 2007. Structure, biochemistry and biology of hepoxilins: an update. FEBS J. 274 3503–3512. [DOI] [PubMed] [Google Scholar]
- 117.Pace-Asciak C. R., E. Granstrom, and B. Samuelsson. 1983. Arachidonic acid epoxides. Isolation and structure of two hydroxy epoxide intermediates in the formation of 8,11,12- and 10,11,12-trihydroxyeicosatrienoic acids. J. Biol. Chem. 258 6835–6840. [PubMed] [Google Scholar]
- 118.Pace-Asciak C. R., and W. S. Lee. 1989. Purification of hepoxilin epoxide hydrolase from rat liver. J. Biol. Chem. 264 9310–9313. [PubMed] [Google Scholar]
- 119.Dho S., S. Grinstein, E. J. Corey, W. G. Su, and C. R. Pace-Asciak. 1990. Hepoxilin A3 induces changes in cytosolic calcium, intracellular pH and membrane potential in human neutrophils. Biochem. J. 266 63–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mrsny R. J., A. T. Gewirtz, D. Siccardi, T. Savidge, B. P. Hurley, J. L. Madara, and B. A. McCormick. 2004. Identification of hepoxilin A3 in inflammatory events: a required role in neutrophil migration across intestinal epithelia. Proc. Natl. Acad. Sci. USA. 101 7421–7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pace-Asciak C. R., J. M. Martin, and E. J. Corey. 1986. Hepoxilins, potential endogenous mediators of insulin release. Prog. Lipid Res. 25 625–628. [DOI] [PubMed] [Google Scholar]
- 122.Nigam S., M. P. Zafiriou, R. Deva, N. Kerstin, C. Geilen, R. Ciccoli, M. Sczepanski, and M. Lohse. 2008. Hepoxilin A3 (HXA3) synthase deficiency is causative of a novel ichthyosis form. FEBS Lett. 582 279–285. [DOI] [PubMed] [Google Scholar]
- 123.Carlen P. L., N. Gurevich, L. Zhang, P. H. Wu, D. Reynaud, and C. R. Pace-Asciak. 1994. Formation and electrophysiological actions of the arachidonic acid metabolites, hepoxilins, at nanomolar concentrations in rat hippocampal slices. Neuroscience. 58 493–502. [DOI] [PubMed] [Google Scholar]
- 124.Reynaud D., P. Demin, and C. R. Pace-Asciak. 1996. Hepoxilin A3-specific binding in human neutrophils. Biochem. J. 313 537–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Serhan C. N., M. Hamberg, and B. Samuelsson. 1984. Lipoxins: novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc. Natl. Acad. Sci. USA. 81 5335–5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Serhan C. N. 2005. Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostaglandins Leukot. Essent. Fatty Acids. 73 141–162. [DOI] [PubMed] [Google Scholar]
- 127.Serhan C. N., and K. A. Sheppard. 1990. Lipoxin formation during human neutrophil-platelet interactions. Evidence for the transformation of leukotriene A4 by platelet 12-lipoxygenase in vitro. J. Clin. Invest. 85 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Claria J., and C. N. Serhan. 1995. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc. Natl. Acad. Sci. USA. 92 9475–9479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chiang N., C. N. Serhan, S. E. Dahlen, J. M. Drazen, D. W. Hay, G. E. Rovati, T. Shimizu, T. Yokomizo, and C. Brink. 2006. The lipoxin receptor ALX: potent ligand-specific and stereoselective actions in vivo. Pharmacol. Rev. 58 463–487. [DOI] [PubMed] [Google Scholar]
- 130.Gronert K., T. Martinsson-Niskanen, S. Ravasi, N. Chiang, and C. N. Serhan. 2001. Selectivity of recombinant human leukotriene D(4), leukotriene B(4), and lipoxin A(4) receptors with aspirin-triggered 15-epi-LXA(4) and regulation of vascular and inflammatory responses. Am. J. Pathol. 158 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Schaldach C. M., J. Riby, and L. F. Bjeldanes. 1999. Lipoxin A4: a new class of ligand for the Ah receptor. Biochemistry. 38 7594–7600. [DOI] [PubMed] [Google Scholar]
- 132.Machado F. S., J. E. Johndrow, L. Esper, A. Dias, A. Bafica, C. N. Serhan, and J. Aliberti. 2006. Anti-inflammatory actions of lipoxin A4 and aspirin-triggered lipoxin are SOCS-2 dependent. Nat. Med. 12 330–334. [DOI] [PubMed] [Google Scholar]
- 133.Yokoyama C., F. Shinjo, T. Yoshimoto, S. Yamamoto, J. A. Oates, and A. R. Brash. 1986. Arachidonate 12-lipoxygenase purified from porcine leukocytes by immunoaffinity chromatography and its reactivity with hydroperoxyeicosatetraenoic acids. J. Biol. Chem. 261 16714–16721. [PubMed] [Google Scholar]
- 134.Feltenmark S., N. Gautam, A. Brunnstrom, W. Griffiths, L. Backman, C. Edenius, L. Lindbom, M. Bjorkholm, and H. E. Claesson. 2008. Eoxins are proinflammatory arachidonic acid metabolites produced via the 15-lipoxygenase-1 pathway in human eosinophils and mast cells. Proc. Natl. Acad. Sci. USA. 105 680–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Drazen J. M., R. A. Lewis, K. F. Austen, M. Toda, F. Brion, A. Marfat, and E. J. Corey. 1981. Contractile activities of structural analogs of leukotrienes C and D: necessity of a hydrophobic region. Proc. Natl. Acad. Sci. USA. 78 3195–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sala A., M. Civelli, D. Oliva, B. Spur, A. E. Crea, G. C. Folco, and S. Nicosia. 1990. Contractile and binding activities of structural analogues of LTC4 in the longitudinal muscle of guinea-pig ileum. Eicosanoids. 3 105–110. [PubMed] [Google Scholar]
- 137.Burger F., P. Krieg, F. Marks, and G. Furstenberger. 2000. Positional- and stereo-selectivity of fatty acid oxygenation catalysed by mouse (12S)-lipoxygenase isoenzymes. Biochem. J. 348 329–335. [PMC free article] [PubMed] [Google Scholar]
- 138.Schweiger D., G. Furstenberger, and P. Krieg. 2007. Inducible expression of 15-lipoxygenase-2 and 8-lipoxygenase inhibits cell growth via common signaling pathways. J. Lipid Res. 48 553–564. [DOI] [PubMed] [Google Scholar]
- 139.Hamberg M., and B. Samuelsson. 1974. Prostaglandin endoperoxides. Novel transformations of arachidonic acid in human platelets. Proc. Natl. Acad. Sci. USA. 71 3400–3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Funk C. D., L. Furci, and G. A. FitzGerald. 1990. Molecular cloning, primary structure, and expression of the human platelet/erythroleukemia cell 12-lipoxygenase. Proc. Natl. Acad. Sci. USA. 87 5638–5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Izumi T., S. Hoshiko, O. Radmark, and B. Samuelsson. 1990. Cloning of the cDNA for human 12-lipoxygenase. Proc. Natl. Acad. Sci. USA. 87 7477–7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Virmani J., E. N. Johnson, A. J. Klein-Szanto, and C. D. Funk. 2001. Role of ‘platelet-type’ 12-lipoxygenase in skin carcinogenesis. Cancer Lett. 162 161–165. [DOI] [PubMed] [Google Scholar]
- 143.Pidgeon G. P., K. Tang, Y. L. Cai, E. Piasentin, and K. V. Honn. 2003. Overexpression of platelet-type 12-lipoxygenase promotes tumor cell survival by enhancing alpha(v)beta(3) and alpha(v)beta(5) integrin expression. Cancer Res. 63 4258–4267. [PubMed] [Google Scholar]
- 144.Raso E., B. Dome, B. Somlai, A. Zacharek, W. Hagmann, K. V. Honn, and J. Timar. 2004. Molecular identification, localization and function of platelet-type 12-lipoxygenase in human melanoma progression, under experimental and clinical conditions. Melanoma Res. 14 245–250. [DOI] [PubMed] [Google Scholar]
- 145.Chen X. S., and C. D. Funk. 1993. Structure-function properties of human platelet 12-lipoxygenase: chimeric enzyme and in vitro mutagenesis studies. FASEB J. 7 694–701. [DOI] [PubMed] [Google Scholar]
- 146.Sloane D. L., R. Leung, J. Barnett, C. S. Craik, and E. Sigal. 1995. Conversion of human 15-lipoxygenase to an efficient 12-lipoxygenase: the side-chain geometry of amino acids 417 and 418 determine positional specificity. Protein Eng. 8 275–282. [DOI] [PubMed] [Google Scholar]
- 147.Borngraber S., M. Browner, S. Gillmor, C. Gerth, M. Anton, R. Fletterick, and H. Kuhn. 1999. Shape and specificity in mammalian 15-lipoxygenase active site. The functional interplay of sequence determinants for the reaction specificity. J. Biol. Chem. 274 37345–37350. [DOI] [PubMed] [Google Scholar]
- 148.Nigam S., S. Patabhiraman, R. Ciccoli, G. Ishdorj, K. Schwarz, B. Petrucev, H. Kuhn, and J. Z. Haeggstrom. 2004. The rat leukocyte-type 12-lipoxygenase exhibits an intrinsic hepoxilin A3 synthase activity. J. Biol. Chem. 279 29023–29030. [DOI] [PubMed] [Google Scholar]
- 149.Romano M., X. S. Chen, Y. Takahashi, S. Yamamoto, C. D. Funk, and C. N. Serhan. 1993. Lipoxin synthase activity of human platelet 12-lipoxygenase. Biochem. J. 296 127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Funk C. D., D. S. Keeney, E. H. Oliw, W. E. Boeglin, and A. R. Brash. 1996. Functional expression and cellular localization of a mouse epidermal lipoxygenase. J. Biol. Chem. 271 23338–23344. [DOI] [PubMed] [Google Scholar]
- 151.Kinzig A., G. Furstenberger, F. Burger, S. Vogel, K. Muller-Decker, A. Mincheva, P. Lichter, F. Marks, and P. Krieg. 1997. Murine epidermal lipoxygenase (Aloxe) encodes a 12-lipoxygenase isoform. FEBS Lett. 402 162–166. [DOI] [PubMed] [Google Scholar]
- 152.Sun D., S. H. Elsea, P. I. Patel, and C. D. Funk. 1998. Cloning of a human “epidermal-type” 12-lipoxygenase-related gene and chromosomal localization to 17p13. Cytogenet. Cell Genet. 81 79–82. [DOI] [PubMed] [Google Scholar]
- 153.Hammarstrom S., M. Hamberg, B. Samuelsson, E. A. Duell, M. Stawiski, and J. J. Voorhees. 1975. Increased concentrations of nonesterified arachidonic acid, 12L-hydroxy-5,8,10,14-eicosatetraenoic acid, prostaglandin E2, and prostaglandin F2alpha in epidermis of psoriasis. Proc. Natl. Acad. Sci. USA. 72 5130–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Woollard P. M. 1986. Stereochemical difference between 12-hydroxy-5,8,10,14-eicosatetraenoic acid in platelets and psoriatic lesions. Biochem. Biophys. Res. Commun. 136 169–176. [DOI] [PubMed] [Google Scholar]
- 155.Boeglin W. E., R. B. Kim, and A. R. Brash. 1998. A 12R-lipoxygenase in human skin: mechanistic evidence, molecular cloning, and expression. Proc. Natl. Acad. Sci. USA. 95 6744–6749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Coffa G., and A. R. Brash. 2004. A single active site residue directs oxygenation stereospecificity in lipoxygenases: stereocontrol is linked to the position of oxygenation. Proc. Natl. Acad. Sci. USA. 101 15579–15584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Meruvu S., M. Walther, I. Ivanov, S. Hammarstrom, G. Furstenberger, P. Krieg, P. Reddanna, and H. Kuhn. 2005. Sequence determinants for the reaction specificity of murine (12R)-lipoxygenase: targeted substrate modification and site-directed mutagenesis. J. Biol. Chem. 280 36633–36641. [DOI] [PubMed] [Google Scholar]
- 158.Kinzig A., M. Heidt, G. Furstenberger, F. Marks, and P. Krieg. 1999. cDNA cloning, genomic structure, and chromosomal localization of a novel murine epidermis-type lipoxygenase. Genomics. 58 158–164. [DOI] [PubMed] [Google Scholar]
- 159.Krieg P., F. Marks, and G. Furstenberger. 2001. A gene cluster encoding human epidermis-type lipoxygenases at chromosome 17p13.1: cloning, physical mapping, and expression. Genomics. 73 323–330. [DOI] [PubMed] [Google Scholar]
- 160.Jobard F., C. Lefevre, A. Karaduman, C. Blanchet-Bardon, S. Emre, J. Weissenbach, M. Ozguc, M. Lathrop, J. F. Prud'homme, and J. Fischer. 2002. Lipoxygenase-3 (ALOXE3) and 12(R)-lipoxygenase (ALOX12B) are mutated in non-bullous congenital ichthyosiform erythroderma (NCIE) linked to chromosome 17p13.1. Hum. Mol. Genet. 11 107–113. [DOI] [PubMed] [Google Scholar]
- 161.Yu Z., C. Schneider, W. E. Boeglin, L. J. Marnett, and A. R. Brash. 2003. The lipoxygenase gene ALOXE3 implicated in skin differentiation encodes a hydroperoxide isomerase. Proc. Natl. Acad. Sci. USA. 100 9162–9167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sigal E., C. S. Craik, E. Highland, D. Grunberger, L. L. Costello, R. A. Dixon, and J. A. Nadel. 1988. Molecular cloning and primary structure of human 15-lipoxygenase. Biochem. Biophys. Res. Commun. 157 457–464. [DOI] [PubMed] [Google Scholar]
- 163.Fleming J., B. J. Thiele, J. Chester, J. O'Prey, S. Janetzki, A. Aitken, I. A. Anton, S. M. Rapoport, and P. R. Harrison. 1989. The complete sequence of the rabbit erythroid cell-specific 15-lipoxygenase mRNA: comparison of the predicted amino acid sequence of the erythrocyte lipoxygenase with other lipoxygenases. Gene. 79 181–188. [DOI] [PubMed] [Google Scholar]
- 164.Watanabe T., J. F. Medina, J. Z. Haeggstrom, O. Radmark, and B. Samuelsson. 1993. Molecular cloning of a 12-lipoxygenase cDNA from rat brain. Eur. J. Biochem. 212 605–612. [DOI] [PubMed] [Google Scholar]
- 165.Chen X. S., U. Kurre, N. A. Jenkins, N. G. Copeland, and C. D. Funk. 1994. cDNA cloning, expression, mutagenesis of C-terminal isoleucine, genomic structure, and chromosomal localizations of murine 12-lipoxygenases. J. Biol. Chem. 269 13979–13987. [PubMed] [Google Scholar]
- 166.Bryant R. W., T. Schewe, S. M. Rapoport, and J. M. Bailey. 1985. Leukotriene formation by a purified reticulocyte lipoxygenase enzyme. Conversion of arachidonic acid and 15-hydroperoxyeicosatetraenoic acid to 14, 15-leukotriene A4. J. Biol. Chem. 260 3548–3555. [PubMed] [Google Scholar]
- 167.Sailesh S., Y. V. Kumar, M. Prasad, and P. Reddanna. 1994. Sheep uterus dual lipoxygenase in the synthesis of 14,15-leukotrienes. Arch. Biochem. Biophys. 315 362–368. [DOI] [PubMed] [Google Scholar]
- 168.Sigal E., D. Grunberger, E. Highland, C. Gross, R. A. Dixon, and C. S. Craik. 1990. Expression of cloned human reticulocyte 15-lipoxygenase and immunological evidence that 15-lipoxygenases of different cell types are related. J. Biol. Chem. 265 5113–5120. [PubMed] [Google Scholar]
- 169.Glass C. K., and J. L. Witztum. 2001. Atherosclerosis. the road ahead. Cell. 104 503–516. [DOI] [PubMed] [Google Scholar]
- 170.Harkewicz R., K. Hartvigsen, F. Almazan, E. A. Dennis, J. L. Witztum, and Y. I. Miller. 2008. Cholesteryl ester hydroperoxides are biologically active components of minimally oxidized low density lipoprotein. J. Biol. Chem. 283 10241–10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Merched A. J., K. Ko, K. H. Gotlinger, C. N. Serhan, and L. Chan. 2008. Atherosclerosis: evidence for impairment of resolution of vascular inflammation governed by specific lipid mediators. FASEB J. 22 3595–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Brash A. R., W. E. Boeglin, and M. S. Chang. 1997. Discovery of a second 15S-lipoxygenase in humans. Proc. Natl. Acad. Sci. USA. 94 6148–6152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Jisaka M., R. B. Kim, W. E. Boeglin, L. B. Nanney, and A. R. Brash. 1997. Molecular cloning and functional expression of a phorbol ester-inducible 8S-lipoxygenase from mouse skin. J. Biol. Chem. 272 24410–24416. [DOI] [PubMed] [Google Scholar]
- 174.Jisaka M., R. B. Kim, W. E. Boeglin, and A. R. Brash. 2000. Identification of amino acid determinants of the positional specificity of mouse 8S-lipoxygenase and human 15S-lipoxygenase-2. J. Biol. Chem. 275 1287–1293. [DOI] [PubMed] [Google Scholar]
- 175.Jisaka M., C. Iwanaga, N. Takahashi, T. Goto, T. Kawada, T. Yamamoto, I. Ikeda, K. Nishimura, T. Nagaya, T. Fushiki, et al. 2005. Double dioxygenation by mouse 8S-lipoxygenase: specific formation of a potent peroxisome proliferator-activated receptor alpha agonist. Biochem. Biophys. Res. Commun. 338 136–143. [DOI] [PubMed] [Google Scholar]
- 176.Nelson D. R. 2006. Cytochrome P450 nomenclature, 2004. Methods Mol. Biol. 320 1–10. [DOI] [PubMed] [Google Scholar]
- 177.Roman R. J. 2002. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol. Rev. 82 131–185. [DOI] [PubMed] [Google Scholar]
- 178.Omura T. 1999. Forty years of cytochrome P450. Biochem. Biophys. Res. Commun. 266 690–698. [DOI] [PubMed] [Google Scholar]
- 179.Schlichting I., J. Berendzen, K. Chu, A. M. Stock, S. A. Maves, D. E. Benson, R. M. Sweet, D. Ringe, G. A. Petsko, and S. G. Sligar. 2000. The catalytic pathway of cytochrome p450cam at atomic resolution. Science. 287 1615–1622. [DOI] [PubMed] [Google Scholar]
- 180.Hirao H., D. Kumar, W. Thiel, and S. Shaik. 2005. Two states and two more in the mechanisms of hydroxylation and epoxidation by cytochrome P450. J. Am. Chem. Soc. 127 13007–13018. [DOI] [PubMed] [Google Scholar]
- 181.Nelson D. R. 1999. Cytochrome P450 and the individuality of species. Arch. Biochem. Biophys. 369 1–10. [DOI] [PubMed] [Google Scholar]
- 182.Nelson D. R., D. C. Zeldin, S. M. Hoffman, L. J. Maltais, H. M. Wain, and D. W. Nebert. 2004. Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics. 14 1–18. [DOI] [PubMed] [Google Scholar]
- 183.Ramarao M., and B. Kemper. 1995. Substitution at residue 473 confers progesterone 21-hydroxylase activity to cytochrome P450 2C2. Mol. Pharmacol. 48 417–424. [PubMed] [Google Scholar]
- 184.Fleming I. 2005. Cytochrome P-450 under pressure: more evidence for a link between 20-hydroxyeicosatetraenoic acid and hypertension. Circulation. 111 5–7. [DOI] [PubMed] [Google Scholar]
- 185.Ma Y. H., D. Gebremedhin, M. L. Schwartzman, J. R. Falck, J. E. Clark, B. S. Masters, D. R. Harder, and R. J. Roman. 1993. 20-Hydroxyeicosatetraenoic acid is an endogenous vasoconstrictor of canine renal arcuate arteries. Circ. Res. 72 126–136. [DOI] [PubMed] [Google Scholar]
- 186.Croft K. D., J. C. McGiff, A. Sanchez-Mendoza, and M. A. Carroll. 2000. Angiotensin II releases 20-HETE from rat renal microvessels. Am. J. Physiol. Renal Physiol. 279 F544–F551. [DOI] [PubMed] [Google Scholar]
- 187.Schwartzman M., N. R. Ferreri, M. A. Carroll, E. Songu-Mize, and J. C. McGiff. 1985. Renal cytochrome P450-related arachidonate metabolite inhibits (Na+ + K+)ATPase. Nature. 314 620–622. [DOI] [PubMed] [Google Scholar]
- 188.Carroll M. A., M. Balazy, P. Margiotta, D. D. Huang, J. R. Falck, and J. C. McGiff. 1996. Cytochrome P-450-dependent HETEs: profile of biological activity and stimulation by vasoactive peptides. Am. J. Physiol. 271 R863–R869. [DOI] [PubMed] [Google Scholar]
- 189.Bednar M. M., C. E. Gross, S. R. Russell, S. P. Fuller, T. P. Ahern, D. B. Howard, J. R. Falck, K. M. Reddy, and M. Balazy. 2000. 16(R)-hydroxyeicosatetraenoic acid, a novel cytochrome P450 product of arachidonic acid, suppresses activation of human polymorphonuclear leukocyte and reduces intracranial pressure in a rabbit model of thromboembolic stroke. Neurosurgery. 47 1410–1418 (discussion 1418–1419). [PubMed] [Google Scholar]
- 190.Spector, A. A. Arachidonic acid cytochrome P450 epoxygenase pathway. J. Lipid Res. Epub ahead of print. October 23, 2008; doi: 10.1194/jlr.R800038-JLR200. [DOI] [PMC free article] [PubMed]
- 191.Node K., Y. Huo, X. Ruan, B. Yang, M. Spiecker, K. Ley, D. C. Zeldin, and J. K. Liao. 1999. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 285 1276–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Liu Y., Y. Zhang, K. Schmelzer, T. S. Lee, X. Fang, Y. Zhu, A. A. Spector, S. Gill, C. Morisseau, B. D. Hammock, et al. 2005. The antiinflammatory effect of laminar flow: the role of PPARgamma, epoxyeicosatrienoic acids, and soluble epoxide hydrolase. Proc. Natl. Acad. Sci. USA. 102 16747–16752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Wong P. Y., P. S. Lai, S. Y. Shen, Y. Y. Belosludtsev, and J. R. Falck. 1997. Post-receptor signal transduction and regulation of 14(R),15(S)-epoxyeicosatrienoic acid (14,15-EET) binding in U-937 cells. J. Lipid Mediat. Cell Signal. 16 155–169. [DOI] [PubMed] [Google Scholar]
- 194.Yang W., V. R. Tuniki, S. Anjaiah, J. R. Falck, C. J. Hillard, and W. B. Campbell. 2008. Characterization of epoxyeicosatrienoic acid binding site in U937 membranes using a novel radiolabeled agonist, 20–125i-14,15-epoxyeicosa-8(Z)-enoic acid. J. Pharmacol. Exp. Ther. 324 1019–1027. [DOI] [PubMed] [Google Scholar]
- 195.Node K., X. L. Ruan, J. Dai, S. X. Yang, L. Graham, D. C. Zeldin, and J. K. Liao. 2001. Activation of Galpha s mediates induction of tissue-type plasminogen activator gene transcription by epoxyeicosatrienoic acids. J. Biol. Chem. 276 15983–15989. [DOI] [PubMed] [Google Scholar]
- 196.Li P. L., and W. B. Campbell. 1997. Epoxyeicosatrienoic acids activate K+ channels in coronary smooth muscle through a guanine nucleotide binding protein. Circ. Res. 80 877–884. [DOI] [PubMed] [Google Scholar]
- 197.Falck J. R., L. M. Reddy, Y. K. Reddy, M. Bondlela, U. M. Krishna, Y. Ji, J. Sun, and J. K. Liao. 2003. 11,12-epoxyeicosatrienoic acid (11,12-EET): structural determinants for inhibition of TNF-alpha-induced VCAM-1 expression. Bioorg. Med. Chem. Lett. 13 4011–4014. [DOI] [PubMed] [Google Scholar]
- 198.Gauthier K. M., J. R. Falck, L. M. Reddy, and W. B. Campbell. 2004. 14,15-EET analogs: characterization of structural requirements for agonist and antagonist activity in bovine coronary arteries. Pharmacol. Res. 49 515–524. [DOI] [PubMed] [Google Scholar]
- 199.Watanabe H., J. Vriens, J. Prenen, G. Droogmans, T. Voets, and B. Nilius. 2003. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature. 424 434–438. [DOI] [PubMed] [Google Scholar]
- 200.Alessandri-Haber N., O. A. Dina, E. K. Joseph, D. Reichling, and J. D. Levine. 2006. A transient receptor potential vanilloid 4-dependent mechanism of hyperalgesia is engaged by concerted action of inflammatory mediators. J. Neurosci. 26 3864–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Alessandri-Haber N., O. A. Dina, E. K. Joseph, D. B. Reichling, and J. D. Levine. 2008. Interaction of transient receptor potential vanilloid 4, integrin, and SRC tyrosine kinase in mechanical hyperalgesia. J. Neurosci. 28 1046–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Sipe W. E., S. M. Brierley, C. M. Martin, B. D. Phillis, F. B. Cruz, E. F. Grady, W. Liedtke, D. M. Cohen, S. Vanner, L. A. Blackshaw, et al. 2008. Transient receptor potential vanilloid 4 mediates protease activated receptor 2-induced sensitization of colonic afferent nerves and visceral hyperalgesia. Am. J. Physiol. Gastrointest. Liver Physiol. 294 G1288–G1298. [DOI] [PubMed] [Google Scholar]
- 203.Earley S., T. J. Heppner, M. T. Nelson, and J. E. Brayden. 2005. TRPV4 forms a novel Ca2+ signaling complex with ryanodine receptors and BKCa channels. Circ. Res. 97 1270–1279. [DOI] [PubMed] [Google Scholar]
- 204.Inceoglu B., K. R. Schmelzer, C. Morisseau, S. L. Jinks, and B. D. Hammock. 2007. Soluble epoxide hydrolase inhibition reveals novel biological functions of epoxyeicosatrienoic acids (EETs). Prostaglandins Other Lipid Mediat. 82 42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Lu T., T. Hoshi, N. L. Weintraub, A. A. Spector, and H. C. Lee. 2001. Activation of ATP-sensitive K(+) channels by epoxyeicosatrienoic acids in rat cardiac ventricular myocytes. J. Physiol. 537 811–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Ng V. Y., Y. Huang, L. M. Reddy, J. R. Falck, E. T. Lin, and D. L. Kroetz. 2007. Cytochrome P450 eicosanoids are activators of peroxisome proliferator-activated receptor alpha. Drug Metab. Dispos. 35 1126–1134. [DOI] [PubMed] [Google Scholar]
- 207.Kotlikoff M. I. 2005. EDHF redux: EETs, TRPV4, and Ca2+ sparks. Circ. Res. 97 1209–1210. [DOI] [PubMed] [Google Scholar]
- 208.Spector A. A., and A. W. Norris. 2007. Action of epoxyeicosatrienoic acids on cellular function. Am. J. Physiol. Cell Physiol. 292 C996–C1012. [DOI] [PubMed] [Google Scholar]
- 209.Beetham J. K., T. Tian, and B. D. Hammock. 1993. cDNA cloning and expression of a soluble epoxide hydrolase from human liver. Arch. Biochem. Biophys. 305 197–201. [DOI] [PubMed] [Google Scholar]
- 210.Gomez G. A., C. Morisseau, B. D. Hammock, and D. W. Christianson. 2004. Structure of human epoxide hydrolase reveals mechanistic inferences on bifunctional catalysis in epoxide and phosphate ester hydrolysis. Biochemistry. 43 4716–4723. [DOI] [PubMed] [Google Scholar]
- 211.Argiriadi M. A., C. Morisseau, B. D. Hammock, and D. W. Christianson. 1999. Detoxification of environmental mutagens and carcinogens: structure, mechanism, and evolution of liver epoxide hydrolase. Proc. Natl. Acad. Sci. USA. 96 10637–10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Morisseau C., and B. D. Hammock. 2005. Epoxide hydrolases: mechanisms, inhibitor designs, and biological roles. Annu. Rev. Pharmacol. Toxicol. 45 311–333. [DOI] [PubMed] [Google Scholar]
- 213.Draper A. J., and B. D. Hammock. 1999. Inhibition of soluble and microsomal epoxide hydrolase by zinc and other metals. Toxicol. Sci. 52 26–32. [DOI] [PubMed] [Google Scholar]
- 214.Lu T., P. V. Katakam, M. VanRollins, N. L. Weintraub, A. A. Spector, and H. C. Lee. 2001. Dihydroxyeicosatrienoic acids are potent activators of Ca(2+)-activated K(+) channels in isolated rat coronary arterial myocytes. J. Physiol. 534 651–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Fang X., S. Hu, B. Xu, G. D. Snyder, S. Harmon, J. Yao, Y. Liu, B. Sangras, J. R. Falck, N. L. Weintraub, et al. 2006. 14,15-Dihydroxyeicosatrienoic acid activates peroxisome proliferator-activated receptor-alpha. Am. J. Physiol. Heart Circ. Physiol. 290 H55–H63. [DOI] [PubMed] [Google Scholar]
- 216.Milne G. L., H. Yin, and J. D. Morrow. 2008. Human biochemistry of the isoprostane pathway. J. Biol. Chem. 283 15533–15537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Morrow J. D., K. E. Hill, R. F. Burk, T. M. Nammour, K. F. Badr, and L. J. Roberts 2nd. 1990. A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc. Natl. Acad. Sci. USA. 87 9383–9387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Kunapuli P., J. A. Lawson, J. Rokach, and G. A. FitzGerald. 1997. Functional characterization of the ocular prostaglandin f2alpha (PGF2alpha) receptor. Activation by the isoprostane, 12-iso-PGF2alpha. J. Biol. Chem. 272 27147–27154. [DOI] [PubMed] [Google Scholar]
- 219.Takahashi K., T. M. Nammour, M. Fukunaga, J. Ebert, J. D. Morrow, L. J. Roberts 2nd, R. L. Hoover, and K. F. Badr. 1992. Glomerular actions of a free radical-generated novel prostaglandin, 8-epi-prostaglandin F2 alpha, in the rat. Evidence for interaction with thromboxane A2 receptors. J. Clin. Invest. 90 136–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Guido D. M., R. McKenna, and W. R. Mathews. 1993. Quantitation of hydroperoxy-eicosatetraenoic acids and hydroxy-eicosatetraenoic acids as indicators of lipid peroxidation using gas chromatography-mass spectrometry. Anal. Biochem. 209 123–129. [DOI] [PubMed] [Google Scholar]
- 221.Shishehbor M. H., R. Zhang, H. Medina, M. L. Brennan, D. M. Brennan, S. G. Ellis, E. J. Topol, and S. L. Hazen. 2006. Systemic elevations of free radical oxidation products of arachidonic acid are associated with angiographic evidence of coronary artery disease. Free Radic. Biol. Med. 41 1678–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Tai H. H., C. M. Ensor, M. Tong, H. Zhou, and F. Yan. 2002. Prostaglandin catabolizing enzymes. Prostaglandins Other Lipid Mediat. 68–69 483–493. [DOI] [PubMed] [Google Scholar]
- 223.Kumlin M., and E. Granstrom. 1986. Radioimmunoassay for 11-dehydro-TXB2: a method for monitoring thromboxane production in vivo. Prostaglandins. 32 741–767. [DOI] [PubMed] [Google Scholar]
- 224.Catella F., D. Healy, J. A. Lawson, and G. A. FitzGerald. 1986. 11-Dehydrothromboxane B2: a quantitative index of thromboxane A2 formation in the human circulation. Proc. Natl. Acad. Sci. USA. 83 5861–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Westlund P., A. C. Fylling, E. Cederlund, and H. Jornvall. 1994. 11-Hydroxythromboxane B2 dehydrogenase is identical to cytosolic aldehyde dehydrogenase. FEBS Lett. 345 99–103. [DOI] [PubMed] [Google Scholar]
- 226.Roberts 2nd L. J., A. R. Brash, and J. A. Oates. 1982. Metabolic fate of thromboxane A2 and prostacyclin. Adv. Prostaglandin Thromboxane Leukot. Res. 10 211–225. [PubMed] [Google Scholar]
- 227.Böhm E., G. J. Sturm, I. Weiglhofer, H. Sandig, M. Shichijo, A. McNamee, J. E. Pease, M. Kollroser, B. A. Peskar, and A. Heinemann. 2004. 11-Dehydro-thromboxane B2, a stable thromboxane metabolite, is a full agonist of chemoattractant receptor-homologous molecule expressed on TH2 cells (CRTH2) in human eosinophils and basophils. J. Biol. Chem. 279 7663–7670. [DOI] [PubMed] [Google Scholar]
- 228.Ensor C. M., and H. H. Tai. 1995. 15-Hydroxyprostaglandin dehydrogenase. J. Lipid Mediat. Cell Signal. 12 313–319. [DOI] [PubMed] [Google Scholar]
- 229.Cho H., L. Huang, A. Hamza, D. Gao, C. G. Zhan, and H. H. Tai. 2006. Role of glutamine 148 of human 15-hydroxyprostaglandin dehydrogenase in catalytic oxidation of prostaglandin E2. Bioorg. Med. Chem. 14 6486–6491. [DOI] [PubMed] [Google Scholar]
- 230.Myung S. J., R. M. Rerko, M. Yan, P. Platzer, K. Guda, A. Dotson, E. Lawrence, A. J. Dannenberg, A. K. Lovgren, G. Luo, et al. 2006. 15-Hydroxyprostaglandin dehydrogenase is an in vivo suppressor of colon tumorigenesis. Proc. Natl. Acad. Sci. USA. 103 12098–12102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Chou W. L., L. M. Chuang, C. C. Chou, A. H. Wang, J. A. Lawson, G. A. FitzGerald, and Z. F. Chang. 2007. Identification of a novel prostaglandin reductase reveals the involvement of prostaglandin E2 catabolism in regulation of peroxisome proliferator-activated receptor gamma activation. J. Biol. Chem. 282 18162–18172. [DOI] [PubMed] [Google Scholar]
- 232.Yokomizo T., Y. Ogawa, N. Uozumi, K. Kume, T. Izumi, and T. Shimizu. 1996. cDNA cloning, expression, and mutagenesis study of leukotriene B4 12-hydroxydehydrogenase. J. Biol. Chem. 271 2844–2850. [DOI] [PubMed] [Google Scholar]
- 233.Ensor C. M., H. Zhang, and H. H. Tai. 1998. Purification, cDNA cloning and expression of 15-oxoprostaglandin 13-reductase from pig lung. Biochem. J. 330 103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Clish C. B., B. D. Levy, N. Chiang, H. H. Tai, and C. N. Serhan. 2000. Oxidoreductases in lipoxin A4 metabolic inactivation: a novel role for 15-onoprostaglandin 13-reductase/leukotriene B4 12-hydroxydehydrogenase in inflammation. J. Biol. Chem. 275 25372–25380. [DOI] [PubMed] [Google Scholar]
- 235.Gross N. D., J. O. Boyle, J. D. Morrow, M. K. Williams, C. S. Moskowitz, K. Subbaramaiah, A. J. Dannenberg, and A. J. Duffield-Lillico. 2005. Levels of prostaglandin E metabolite, the major urinary metabolite of prostaglandin E2, are increased in smokers. Clin. Cancer Res. 11 6087–6093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Hori T., T. Yokomizo, H. Ago, M. Sugahara, G. Ueno, M. Yamamoto, T. Kumasaka, T. Shimizu, and M. Miyano. 2004. Structural basis of leukotriene B4 12-hydroxydehydrogenase/15-Oxo-prostaglandin 13-reductase catalytic mechanism and a possible Src homology 3 domain binding loop. J. Biol. Chem. 279 22615–22623. [DOI] [PubMed] [Google Scholar]
- 237.Berry K. A., P. Borgeat, J. Gosselin, L. Flamand, and R. C. Murphy. 2003. Urinary metabolites of leukotriene B4 in the human subject. J. Biol. Chem. 278 24449–24460. [DOI] [PubMed] [Google Scholar]
- 238.Diczfalusy U. 1994. Beta-oxidation of eicosanoids. Prog. Lipid Res. 33 403–428. [DOI] [PubMed] [Google Scholar]
- 239.Wanders R. J., and H. R. Waterham. 2006. Biochemistry of mammalian peroxisomes revisited. Annu. Rev. Biochem. 75 295–332. [DOI] [PubMed] [Google Scholar]
- 240.Guilford W. J., and J. F. Parkinson. 2005. Second-generation beta-oxidation resistant 3-oxa-lipoxin A4 analogs. Prostaglandins Leukot. Essent. Fatty Acids. 73 245–250. [DOI] [PubMed] [Google Scholar]
- 241.Soberman R. J., J. P. Sutyak, R. T. Okita, D. F. Wendelborn, L. J. Roberts 2nd, and K. F. Austen. 1988. The identification and formation of 20-aldehyde leukotriene B4. J. Biol. Chem. 263 7996–8002. [PubMed] [Google Scholar]
- 242.Sutyak J., K. F. Austen, and R. J. Soberman. 1989. Identification of an aldehyde dehydrogenase in the microsomes of human polymorphonuclear leukocytes that metabolizes 20-aldehyde leukotriene B4. J. Biol. Chem. 264 14818–14823. [PubMed] [Google Scholar]
- 243.Tukey R. H., and C. P. Strassburg. 2000. Human UDP-glucuronosyltransferases: metabolism, expression, and disease. Annu. Rev. Pharmacol. Toxicol. 40 581–616. [DOI] [PubMed] [Google Scholar]
- 244.Turgeon D., S. Chouinard, P. Belanger, S. Picard, J. F. Labbe, P. Borgeat, and A. Belanger. 2003. Glucuronidation of arachidonic and linoleic acid metabolites by human UDP-glucuronosyltransferases. J. Lipid Res. 44 1182–1191. [DOI] [PubMed] [Google Scholar]
- 245.Newman J. W., T. Watanabe, and B. D. Hammock. 2002. The simultaneous quantification of cytochrome P450 dependent linoleate and arachidonate metabolites in urine by HPLC-MS/MS. J. Lipid Res. 43 1563–1578. [DOI] [PubMed] [Google Scholar]
- 246.Shui W., S. A. Gilmore, L. Sheu, J. Liu, J. D. Keasling, and C. R. Bertozzi. 2008. Quantitative proteomic profiling of host-pathogen interactions: the macrophage response to mycobacterium tuberculosis lipids. J. Proteome Res. 8 282–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Jenkins R. E., N. R. Kitteringham, C. L. Hunter, S. Webb, T. J. Hunt, R. Elsby, R. B. Watson, D. Williams, S. R. Pennington, and B. K. Park. 2006. Relative and absolute quantitative expression profiling of cytochromes P450 using isotope-coded affinity tags. Proteomics. 6 1934–1947. [DOI] [PubMed] [Google Scholar]
- 248.Clamp M., J. Cuff, S. M. Searle, and G. J. Barton. 2004. The Jalview Java alignment editor. Bioinformatics. 20 426–427. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.