

Abstract
The fetus has a high requirement for cholesterol and synthesizes cholesterol at elevated rates. Recent studies suggest that fetal cholesterol also can be obtained from exogenous sources. The purpose of the current study was to examine the transport of maternal cholesterol to the fetus and determine the mechanism responsible for any cholesterol-driven changes in transport. Studies were completed in pregnant hamsters with normal and elevated plasma cholesterol concentrations. Cholesterol feeding resulted in a 3.1-fold increase in the amount of LDL-cholesterol taken up by the fetus and a 2.4-fold increase in the amount of HDL-cholesterol taken up. LDL-cholesterol was transported to the fetus primarily by the placenta, and HDL-cholesterol was transported by the yolk sac and placenta. Several proteins associated with sterol transport and efflux, including those induced by activated liver X receptor, were expressed in hamster and human placentas: NPC1, NPC1L1, ABCA2, SCP-x, and ABCG1, but not ABCG8. NPC1L1 was the only protein increased in hypercholesterolemic placentas. Thus, increasing maternal lipoprotein-cholesterol concentrations can enhance transport of maternal cholesterol to the fetus, leading to 1) increased movement of cholesterol down a concentration gradient in the placenta, 2) increased lipoprotein secretion from the yolk sac (shown previously), and possibly 3) increased placental NPC1L1 expression.
Keywords: placenta, yolk sac, Smith-Lemli-Opitz Syndrome, low density lipoprotein, high density lipoprotein, liver X receptor, NPC1 Like 1
Cholesterol is essential for normal fetal development. Possibly the most noted role of cholesterol is as a structural component of membranes. Sterols are also the precursor for bile acids, steroid hormones, and oxysterols, which are all synthesized by the fetus and regulate various cellular processes. Cholesterol is also required for activation of sonic hedgehog (Shh), a protein involved in brain development, and for propagation of the Shh signal (1, 2). Cholesterol is obtained endogenously by de novo synthesis and exogenously by transfer of maternal cholesterol to the fetus. Interestingly, maternal plasma triglyceride and cholesterol concentrations increase during pregnancy in humans (3, 4), possibly an adaptation to maternal and fetal needs. However, whether these changes in maternal lipoprotein concentrations result in increased transport of maternal cholesterol to the fetus is unclear.
Since the fetus does not come in direct contact with the maternal circulation, maternal cholesterol destined for the fetus must initially be taken up by the placenta and yolk sac prior to transport and delivery to the fetus. Indeed, the placenta and yolk sac take up maternal LDL- and HDL-cholesterol at relatively elevated rates compared with other peripheral tissues (5). LDL is taken up by the LDL receptor (LDLR), which is expressed abundantly in the placenta but expressed at low levels, if at all, in the yolk sac (5, 6). LDL-derived sterol is transported to the lysosome/endosome pathway where the ester bond is hydrolyzed (7). The unesterified cholesterol is then transported by Niemann-Pick C1 (NPC1) to a metabolically active pool of cholesterol, to the plasma membrane, or to be esterified [reviewed in (8, 9)]. HDL-cholesterol is taken up by SR-BI and cubilin, both of which are expressed highly in the yolk sac and in the placenta at somewhat lower levels (5, 10–13). The ester bonds are hydrolyzed, possibly through one of the cytosolic esterases. Once internalized, sterols from either source are transported by way of a number of putative transport proteins, including NPC1 Like 1 (NPC1L1), sterol carrier protein (SCP)-x/2, and ABCA2 (14, 15) [reviewed in (16, 17)]. The sterol is secreted from cells as lipoprotein particles or apoE:lipid complexes or effluxed by way of ABCA1, ABCG1, or ABCG5/8 [reviewed in (18)].
Several lines of evidence suggest that maternally derived cholesterol taken up by the placenta and yolk sac is indeed transported to the fetus. First, evidence is found in human fetuses with the Smith-Lemli-Opitz syndrome (SLOS) that are unable to synthesize cholesterol at normal rates due to null/null mutations in 3β-hydroxysteroid Δ7-reductase, the enzyme that converts 7-dehydrocholesterol to cholesterol (19, 20). SLOS fetuses and newborns, even infants with the null/null genotype, contain cholesterol (21, 22). Importantly, the severity of the SLOS phenotype is affected by the apoE isoform expressed in the mothers but not the fathers (23). Second, though the reported rates of transport vary, some of the early human studies did indeed demonstrate a transfer of maternal cholesterol to the fetal circulation (24–26). Third, more recent studies in the mouse determined that ∼20% of murine fetal cholesterol is derived from the maternal circulation (27, 28). Fourth, fetal cholesterol concentrations are positively correlated with circulating cholesterol concentrations in pregnant hamsters (29), and, fifth, the amount of maternally derived cholesterol secreted from yolk sacs can be manipulated with plasma cholesterol concentrations (30). Finally, lipoprotein-derived cholesterol is transported from the apical (maternal) side of a confluent monolayer of cultured trophoblasts to the basolateral (fetal) side (31).
Why might one be interested in the ability to transport maternal cholesterol to the fetus? It has been shown that an increase in dietary sterol can improve the quality of life of persons with SLOS (32, 33), as one might expect since the severity of the syndrome is correlated with circulating cholesterol concentrations (34–36). Since the congenital defects found in individuals with SLOS are due to metabolic changes in utero, an increase in exogenous cholesterol in fetuses with SLOS could have a significant impact on the improvement of congenital defects. More globally, epidemiological studies have shown that fetal metabolism is altered when maternal cholesterol concentrations vary in fetuses unaffected by SLOS, including fetal growth rates (37–40). This is not specific for humans in that murine fetuses of dams with low HDL-cholesterol levels have reduced growth rates as well (41).
Thus, the goal of the current studies was to determine whether the amount of maternal cholesterol transported to the fetus could be enhanced by increasing circulating levels of cholesterol in the pregnant dam and to determine which lipoprotein is the primary source of cholesterol. Based on studies in cultured trophoblasts and yolk sacs (30, 31), we hypothesize that more LDL- and HDL-cholesterol would be transported to the fetus when maternal plasma cholesterol concentrations are elevated. We further hypothesize that enhanced movement across the placenta in vivo would be mediated through cholesterol-induced activation of the iver X receptor (LXR)-inducible proteins ABCG1 and apoE. To test our hypotheses, female hamsters were fed 0 or 2% added cholesterol to render them normo- or hypercholesterolemic (29), respectively. The fractional catabolic rates (FCRs) and cholesterol uptake rates of maternal LDL- and HDL-cholesterol by the placenta, yolk sac, and fetus were determined. Elevating maternal plasma cholesterol concentrations does indeed result in an increase in the uptake of maternal lipoprotein-cholesterol by the placenta and yolk sac. Importantly, an increase in uptake by the extraembryonic fetal tissues was parlayed into an increase in the transport of maternally derived cholesterol to the fetus. The increases in uptake by extraembryonic tissues are not due to a change in expression of lipoprotein receptors. Increased sterol movement across and out of the placenta is most likely due to movement down a concentration gradient and possibly increased NPC1L1, but not a sterol-induced change in LXR-inducible protein expression (apoE and ABCG1).
MATERIALS AND METHODS
Animals and diets
Male and female hamsters (Charles River Laboratories, Kingston, NJ) weighing between 110 and 120 g were maintained in a temperature- and humidity-controlled room and fed a pelleted chow (diet #7012; Harlan Teklad, Madison, WI). Once acclimated, females were fed diets containing ground chow (#7012; Harlan Teklad) or ground chow plus 2% cholesterol (wt/wt; MP Biomedicals, Eschwege, Germany). After 3 weeks, females were mated as described (29), continued to be fed the same diets, and studied at 11.5 d postconception (dpc); hamsters have a gestational period of 15.5 d. All protocols were approved by the Institutional Animal Care and Use Committee of the University of Cincinnati.
Plasma, lipoprotein, and tissue cholesterol concentrations
Blood was collected from the descending aorta and plasma isolated. Total cholesterol concentrations were determined enzymatically (Thermo Scientific, Waltham, MA). Plasma LDL- and HDL-cholesterol concentrations were determined by simultaneously separating plasma at densities of 1.020 and 1.063 g/ml and measuring the content of cholesterol in the top and bottom of each tube by gas-liquid chromatography using stigmastanol as an internal standard (42). Tissues were saponified and cholesterol content measured by gas-liquid chromatography.
Lipoprotein clearance studies
Lipoproteins were collected from older donor hamsters by sequential ultracentrifugation: LDL was defined as particles with densities of 1.020<d<1.055 g/ml, and HDL was defined as particles with densities of 1.070<d<1.21 g/ml; tighter densities were used to ensure no labeling of large LDL or small HDL. Lipoproteins were radiolabeled with [1α, 2α(n)-3H]cholesteryl oleate (GE Healthcare, Giles, UK) or [1α, 2α(n)-3H]cholesteryl oleyl ether (GE Healthcare) as described (31, 43) using hamster plasma as a source of cholesteryl ester transfer protein.
Three different types of studies were completed. To determine the FCR and uptake rates of maternal lipoproteins by the placenta and yolk sac, a bolus of radiolabeled lipoprotein-[3H]cholesteryl ether was injected into the femoral vein of females at 11.5 dpc. Cholesteryl ether was used in these studies because the ether cannot be hydrolyzed in the lysosomes and thus radiolabel is trapped in tissues (43, 44). Females were awake within minutes and were allowed to roam free within their cages. Blood was collected from the jugular vein of each lightly anesthetized hamster at 10 and 35 min postinjection. At 1.5 h postinjection, tissues and blood were collected. Tissues were saponified, and sterol was extracted. Amount of radiolabel in plasma and tissues was measured.
To determine the length of time required to measure uptake, transport (when the fetus took up ∼5 μg of cholesterol for either lipoprotein) and efflux/secretion of cholesterol from the maternal circulation to the fetus, boluses of LDL- and HDL-[3H]cholesteryl ester, a metabolizable marker, were injected to the femoral vein of another set of females. Blood was collected from the jugular vein of the hamsters at different times postinjection, including 0.75, 1.5, 3, and 6 h. Animals were anesthetized at 6, 12, 18, and 24 h postinjection, and tissues collected and assayed for radiolabel; studies were completed at 11.5 dpc.
Based on our timed studies with cholesteryl ester, the amount of maternally derived LDL- and HDL-cholesterol that was transported to fetuses was determined at 22 and 6 h postinjection, respectively. Boluses of LDL- or HDL-[3H]cholesteryl ester were injected into the femoral vein of a third set of females. Plasma was collected at 0.75, 1.5, 3, 6, and 22 h postinjection. Tissues were collected 22 and 6 h postinjection, and radiolabeled sterol concentrations in plasma and fetuses were measured; studies were completed at 11.5 dpc.
Kinetic analysis
The analysis of the maternal plasma radioactivity was completed using the SAAM II program (SAAM Institute, Seattle, WA). The plasma die-away curves were biphasic, and the plasma kinetic data were modeled using a two-pool model, with a single plasma compartment that exchanges with an extravascular compartment. For the analyses of placenta, yolk sac, and fetus uptake data, the model was modified to include rate constants from the plasma pool to the placenta, yolk sac, and fetus to account for the accumulation of radioactivity in these organs at the end of the kinetic studies. The model-predicted rate constants from plasma to the placenta, yolk sac, and fetus were used to calculate the transfer of maternal cholesterol to the placenta, yolk sac, and fetus.
mRNA expression levels
Hamster placentas were collected. Tissue RNA was isolated using TRIzol (Invitrogen, Carlsbad, CA) and stored in FORMAzol® (Molecular Research Center, Cincinnati, OH) at −80°C. RNA was treated with RNase-free DNase I and reverse transcribed to cDNA by SuperScript II reverse transcriptase using random hexamers. PCR assays were performed on a Bio-Rad iCycler iQ real-time PCR Detection System using SYBR green as our fluorophore. A serial dilution of a randomly picked sample was used to generate a standard curve for each gene examined. This standard curve was used to calculate the relative levels of mRNA for the gene of interest and the reference/housekeeping gene (cyclophilin). Primers for ABCG1 and ABCA2 were previously described (45), as were primers for ABCG8 (46). Remaining primer sequences were 5′-ACTTTGGGCCTTTCTTTCGATGG-3′ (forward) and 5′-ATCTCTTTGTTCAGTGGAGGCCCA-3′ (reverse) for NPC1, 5′-TGAACCGCTTCTGGGATTACCT-3′ (forward) and 5′-AGTGCCGTCAGTTCTTGTGTGACT-3′ (reverse) for apoE, and 5′-TGGTTTCCAATAGGAGCAGGAGCA-3′ (forward) and 5′-TAAAGGACAGCGCCAAGACAGTGA-3′ (reverse) for NPC1L1.
Protein expression levels
Placentas and yolk sacs were collected from hamsters, and microsomes were isolated as described (47). The sample from each dam contained multiple (8–12) placentas or yolk sacs. In addition, placentas were collected from women at term. Random sampling of whole placentas was performed from beneath the chorionic plate. Placental samples were stored at −80°C until used. Tissues were homogenized and unbroken cells removed. Protocols were approved by the Institutional Review Board of the University of Cincinnati.
Protein concentrations were determined by the Lowry assay, equal amounts of protein pooled from three to four samples, and proteins separated on a Bio-Rad 4-15% Tris-HCl Ready gel (Bio-Rad Laboratories, Hercules, CA) under denaturing conditions. Proteins were transferred to a polyvinylidene difluoride membrane that was blocked in Tris-buffered saline containing 0.1% Tween and 5% dry milk for 1 h. After blocking, blots were incubated with chicken anti-LDL receptor IgG (Novus Biologicals, Littleton, CO) or rabbit anti-SR-BI receptor IgG (Novus Biologicals). After 1 h at 37°C, the membranes were washed and then incubated with the appropriate secondary antibody conjugated with peroxidase (rabbit anti-chicken IgG and donkey anti-rabbit; GE Healthcare). Chemiluminescence from ECL Plus (GE Healthcare) was detected by a Storm 450 PhosphorImager. Samples were tested similarly for presence of ABCA2 (Santa Cruz Biotechnologies, Santa Cruz, CA), NPC1 (Novus Biologicals), NPC1L1 (Cayman Chemicals, Ann Arbor, MI), ABCG1 (Novus Biologicals), ABCG8 (48), and SCP-2/x (49) in human placentas with the exception that some proteins were separated on 7.5% gels poured in the laboratory or as described.
Statistics
All data are presented as means ± SEM. Comparisons between groups were deemed significant by Student's t-test (P < 0.05).
RESULTS
The fetus is a unique tissue in that it does not come in direct contact with the maternal circulation (50). Thus, any maternally derived lipoprotein-cholesterol that is in the fetus would have been taken up first by the placenta and/or yolk sac, transported across the trophoblasts and visceral endoderm cells of the placenta and yolk sac, respectively, and effluxed and/or secreted into the fetal circulation.
For these studies, FCR was defined as pools of LDL- or HDL-cholesterol cleared from plasma/time per gram of tissue or whole tissue by the placenta, yolk sac, and fetus of normo- and hypercholesterolemic dams. FCR parallel clearance rates, the microliter of plasma cleared of its lipoproteins per hour per gram (51). FCR were determined for two reasons. First, plasma cholesterol concentrations are different in dams fed 0 or 2% dietary cholesterol (29). Since clearance rates and FCR can change with plasma cholesterol concentrations (52, 53), measuring the amount of dpm per tissue could be misleading. Second, one can convert FCR to the actual micrograms of lipoprotein-cholesterol taken up and/or transported by multiplying FCR by the maternal pool size (52).
Dams were fed different levels of dietary cholesterol that are known to lead to an increase in cholesterol in embryonic and extraembryonic fetal tissues (29, 54). In dams fed a normocholesterolemic diet, FCR was 0.050 ± 0.001 maternal pools/1.5 h per g placenta (Fig. 1A). Each gram of yolk sac cleared ∼20% of that cleared per gram of placenta (Fig. 1B). There was no difference in FCR per gram of placenta or yolk sac when tissues of dams fed no or 2% cholesterol were compared. A unique aspect of these studies is that one can calculate the mass of maternal LDL-cholesterol taken up by the entire placenta or yolk sac, which will account for differences in maternal LDL-cholesterol concentrations and differences in the sizes of the tissue. The placentas of dams fed control diets took up 1.740 ± 0.356 μg of maternal LDL-cholesterol during the time of the study (1.5 h) (Fig. 1C). Due to the several-fold increase in LDL-cholesterol concentration in dams fed cholesterol, uptake of maternal LDL-cholesterol was 3.4-fold greater in placentas of dams fed cholesterol (P < 0.001). A similar effect was found in the yolk sac; the yolk sac of dams fed control diets took up 0.052 ± 0.007 μg in 1.5 h, whereas the yolk sac in dams fed cholesterol took up 0.200 ± 0.026 μg (Fig. 1D) (P = 0.005).
Fig. 1.

FCR and uptake of maternal LDL-cholesterol by the placenta and yolk sac at 11.5 dpc. Female hamsters were fed 0 or 2% added cholesterol prior to and during mating. Hamster LDL was radiolabeled with [3H]cholesteryl oleyl ether. A bolus of radiolabel was injected into animals at 11.5 dpc. Using the SAAM II program, we calculated FCR (maternal pools/1.5 h per gram tissue; A and B) and calculated the uptake (or transfer) rates of maternal cholesterol to the entire placenta and yolk sac (μg/1.5 h per tissue; C and D). Data are presented as means (n = 3) ± SEM. The asterisk depicts significant differences between animals fed different diets. LDL-cholesterol concentrations were 4.1 ± 0.5 and 17.5 ± 2.2 mg/dl for dams fed 0 or 2% cholesterol, respectively, at 11.5 dpc.
For HDL-cholesterol, the FCR was 0.045 ± 0.004 maternal pools of HDL-cholesterol/1.5 h per gram of placenta in control-fed dams (Fig. 2A). There was no effect of dietary cholesterol on FCR per gram of placentas of dams fed cholesterol. Similar to our previously reported results (5), the yolk sac took up significantly more HDL-cholesterol as compared with the placenta. In the yolk sac, FCR per gram of yolk sac of HDL-cholesterol was almost 15-fold greater than rates for the placenta (0.612 ± 0.006 pools/1.5 h per g yolk sac) (Fig. 2B). As with the placenta, FCR per gram of yolk sac of dams fed either diet remained relatively similar. As with LDL, the uptake rates per whole tissue were measured next. The calculated mass of HDL-cholesterol taken up by the placentas of control dams was 2.622 ± 0.469 μg maternal cholesterol/1.5 h per placenta (Fig. 2C). The uptake of maternal cholesterol from HDL was 3.5-fold greater in dams fed cholesterol (P = 0.017). Even though the tissue is much smaller, the yolk sac actually took up more HDL-cholesterol compared with the placenta (5.81 ± 1.13 μg/1.5 h per yolk sac) (Fig. 2D) (29). As with the placenta, the yolk sac of cholesterol-fed dams tended to take up more cholesterol when compared with the yolk sac of control dams (P = 0.059).
Fig. 2.

FCR and uptake of maternal HDL-cholesterol by the placenta and yolk sac at 11.5 dpc. Female hamsters were fed 0 or 2% added cholesterol prior to and during mating. Hamster HDL was radiolabeled with [3H]cholesteryl oleyl ether. A bolus of radiolabel was injected into animals at 11.5 dpc. FCR (pools/1.5 h per gram tissue) was calculated initially. Using the SAAM II program, we calculated FCR (maternal pools/1.5 h per gram tissue; A and B) and calculated the uptake rates of maternal cholesterol to the entire placenta and yolk sac (μg/1.5 h per gram tissue; C and D). Data are presented as means (n = 3–4) ± SEM. The asterisk depicts significant differences between animals fed different diets. HDL-cholesterol concentrations were 7.6 ± 1.3 and 25.2 ± 2.5 mg/dl for dams fed 0 or 2% cholesterol, respectively, at 11.5 dpc.
Since we demonstrated a significant increase in the amount of maternally derived cholesterol taken up by the placenta and yolk sac, the question remains: Can the amount of cholesterol that is transported to the fetus be changed in parallel with an increase in the amount of cholesterol being taken up by the placenta and yolk sac? Since the fetus does not come in direct contact with the maternal circulation, fetal uptake of maternal cholesterol is in effect a summation of all the processes involved in transport, including uptake of lipoprotein-cholesterol by the placenta and/or yolk sac, transport of lipoprotein-cholesterol across the cells to the basolateral side, and secretion or efflux of lipoprotein-cholesterol into the fetal circulation either directly or by way of endothelial cells. To examine this question, we initially determined when the radiolabeled sterol appeared in the fetus and when the rate of uptake was linear. Dams were injected with a bolus of radiolabel, and fetuses were collected at 6, 12, 18, and 24 h postinjection (Fig. 3). LDL clearance rates were measured first. Within 6 h of the injection, the fetus had taken up 0.27 ± 0.06 μg of LDL-cholesterol. The rate of uptake or transport of maternal cholesterol appeared linear through 24 h postinjection. In the HDL studies, the fetus took up as much maternal HDL-cholesterol within 6 h of the bolus injection as the fetus did of maternal LDL-cholesterol in 24 h (4.01 ± 0.33 μg). We chose to complete the studies at times when similar amounts of LDL- and HDL-cholesterol were taken up, which was 22 h for LDL-cholesterol and 6 h for HDL-cholesterol.
Fig. 3.

Uptake of maternal lipoprotein-cholesterol by the fetus. Female hamsters were mated. Hamster LDL and HDL were radiolabeled with [3H]cholesteryl oleate. At various times, radiolabeled LDL and HDL were injected into the femoral vein between 10.5 and 11.5 dpc. At 6, 12, 18, and 24 h postinjection, tissues were collected and the uptake of maternal cholesterol by the fetus was calculated. Data are presented as means (n = 3–4) ± SEM.
For LDL-cholesterol, the FCR per gram fetus was 0.088 ± 0.11 maternal pools/d per gram fetus (Fig. 4A). Unlike the placenta and yolk sac, the FCR was 36% lower per gram fetus of dams fed cholesterol compared with fetuses of control dams. The FCR per gram fetus for HDL-cholesterol was ∼3-fold greater than FCR per gram fetus for LDL-cholesterol (Fig. 4B). Though the FCR for HDL-cholesterol was 47% lower per gram fetus of dams fed cholesterol compared with FCR in control dams, the differences were not significant. However, the calculated mass of maternal LDL- and HDL-cholesterol taken up by the fetus was 3.0-fold (P < 0.001) and 2.0-fold (P = 0.037) greater, respectively, in the fetuses of cholesterol-fed dams due to the elevated lipoprotein-cholesterol concentrations in these dams (Fig. 4C, D). After 24 h, there was still significant maternal cholesterol in the placenta and yolk sac that was not yet transferred to the fetus.
Fig. 4.

FCR and uptake rates of maternal lipoprotein-cholesterol by the fetus. Female hamsters were fed 0 or 2% added cholesterol prior to and during mating. Hamster LDL and HDL were radiolabeled with [3H]cholesteryl oleate. A bolus of radiolabel was injected into animals at 10.5 (LDL) or 11.25 (HDL) dpc. At 22 (LDL) and 6 (HDL) h postinjection, tissues were collected and amount of radiolabel measured. FCR (pools of maternal LDL- and HDL-cholesterol/d per g fetus; A and B) and uptake or transport rates (μg maternal LDL- and HDL-cholesterol/d per fetus; C and D) were calculated. Uptake rates include the uptake by the placenta and yolk sac, transport across cells, and efflux/secretion into the fetal circulation. Data are presented as means (n = 14 for LDL and = 3 for HDL studies) ± SEM. The asterisk depicts significant differences between animals fed different diets. LDL-cholesterol concentrations were 5.4 ± 0.7 and 30.7 ± 2.7 mg/dl for dams fed 0 or 2% cholesterol, respectively, at 11.5 dpc. HDL-cholesterol concentrations were 9.9 ± 0.9 and 38.0 ± 3.9 mg/dl for dams fed 0 or 2% cholesterol, respectively, at 11.5 dpc.
The expression levels of the lipoprotein receptors responsible for the uptake of LDL and HDL in the placenta and yolk sac, respectively (5), were measured initially. There was no difference in the expression of the LDLR or SR-BI in the placentas or yolk sacs of dams fed different amounts of cholesterol (Fig. 5).
Fig. 5.
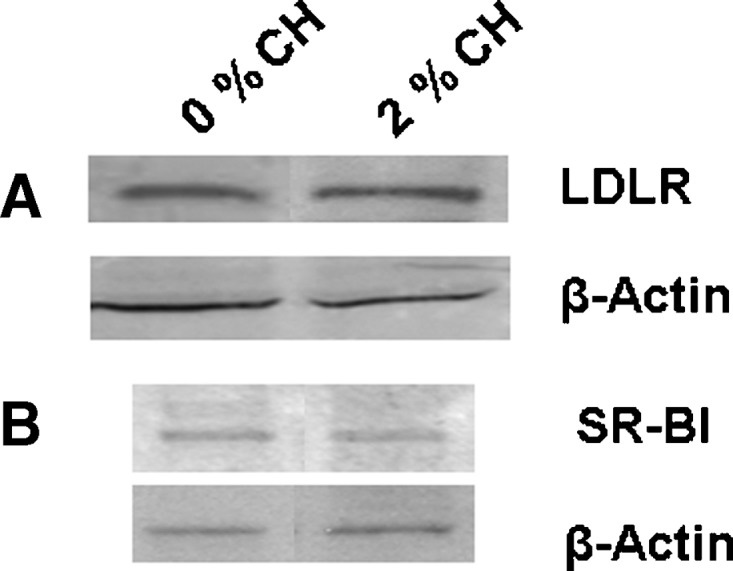
Expression of lipoprotein receptors in extraembryonic fetal tissues in the hamster. Tissues were collected from dams treated as in Fig. 1. Tissues were pooled (n = 3–4) and the relative amount of the LDLR in the placenta (A) and SR-BI in the yolk sac (B) were determined by immunoblotting; 150 μg protein was loaded in each lane, and β-Actin was used as a control for loading.
The remainder of the studies were focused on the placenta because 1) LDL, the primary lipoprotein particle in humans, is taken up primarily by the rodent placenta, and 2) we already know that the yolk sac of cholesterol-fed hamsters secrete more lipoprotein-cholesterol (30), as occurs in cholesterol-enriched livers (55). To determine if a difference in sterol movement across cells occurred as a result of changes in intracellular proteins, mRNA levels of various proteins involved in sterol transport or export out of cells also were measured (Fig. 6); regulation of posttranscriptional/posttranslational mechanisms was not measured. There was no effect of tissue cholesterol concentration in the amount of mRNA for ABCG1, ABCA2, or apoE. The amount of NPC1 mRNA decreased (P = 0.016) and NPC1L1 mRNA increased (P = 0.003) in placentas of dams fed cholesterol compared with placentas of control dams. Additionally, there was little to no message for ABCG8 in the hamster placenta versus the hamster liver, which had abundant mRNA levels (1.25 relative mRNA ABCG8/cyclophilin). SCP-2/x protein levels but not mRNA levels were determined in the hamster. Expression levels were very low compared with the liver, and the amounts of SCP-x plus SCP-2 did not vary much between hamsters fed different levels of cholesterol (data not shown). Expression of proteins not yet demonstrated to be expressed by the human placenta but known to be involved in LDL transport were then tested by Western blotting; ABCG1, NPC1, ABCA2, NPC1L1, and SCP-x were all expressed in the human placenta (Fig. 7). ABCG8 and SCP-2 were not detectable by Western blotting (data not shown).
Fig. 6.

Relative mRNA levels in hamster placentas. Tissues were collected from dams treated as in Fig. 1. The relative amount of mRNA in the placenta compared with cyclophilin is presented. Data are presented as means (n = 5) ± SEM. The asterisk depicts significant differences between animals fed different diets.
Fig. 7.
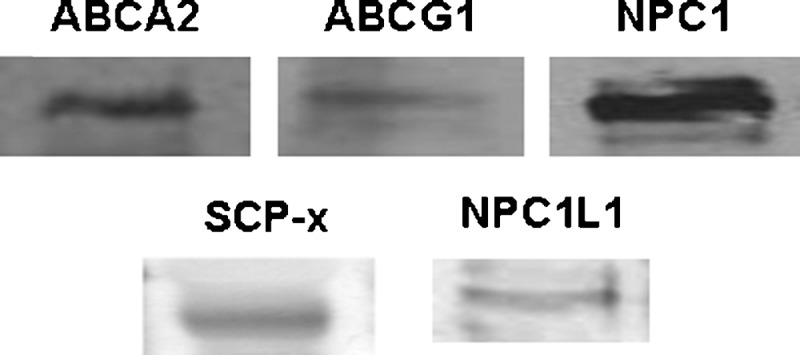
Expression of proteins in the human placenta. Placentas were collected at term from women with uncomplicated pregnancies, homogenized, and proteins separated. The presence of proteins not yet shown in the human placenta was determined by immunoblotting using 150 μg of protein in each for all proteins, except SCP-x when 20 μg protein was used.
DISCUSSION
The fetus requires cholesterol for development. Too little cholesterol due to a lack of synthesis leads to a spectrum of congenital defects as seen in infants with SLOS (19, 20, 56). Too little cholesterol due to a lack of maternal cholesterol or reduced expression of placental lipoprotein receptors is correlated with smaller fetuses and a trend for microcephaly (37–39). Thus, the ability to manipulate the amount of cholesterol that is transported to the fetus could have a significant impact upon fetal development.
The in vivo data presented here support our hypothesis that increased amounts of cholesterol are transported from the maternal to fetal circulation when maternal cholesterol concentrations are elevated. There was a 2.4-fold increase in the amount of maternal HDL-cholesterol taken up by fetuses and a 3.1-fold increase in the amount of maternal LDL-cholesterol taken up by fetuses of hypercholesterolemic dams. Though most of the maternal cholesterol transferred to the fetus was derived from maternal HDL, a significant increase in the amount of both LDL- and HDL-cholesterol transported to the fetus occurred when maternal cholesterol concentrations were increased. When the amount of cholesterol taken from LDL and HDL was presented as a percentage of total body cholesterol, ∼12% of fetal cholesterol was derived from maternal circulation in dams fed no added cholesterol and ∼25% of fetal cholesterol was derived from maternal circulation in dams fed 2% added cholesterol. Increases in uptake of the placenta and yolk sac were also measured and were greater than those in the fetus since not all maternally derived cholesterol is transported to the fetus. LDL delivered markedly more maternal cholesterol to the placenta than to the yolk sac. HDL delivered more maternal cholesterol to the yolk sac than to the placenta, though HDL-cholesterol was delivered to both tissues. Thus, transport of LDL would represent transport across the placenta since LDL is not taken up by the yolk sac in appreciable amounts (Fig. 1C, D). The transport of HDL would represent transport across the yolk sac as well as the placenta (Fig. 2).
Yolk sac
The yolk sac is patent throughout gestation in the rodent and in the first trimester in human [reviewed in (50)]. The marked increase in uptake of maternal HDL in hypercholesterolemic hamsters measured here was not due to increased expression of SR-BI (see below) and was responsible for the increase in yolk sac cholesterol concentration and secretion (30); yolk sac sterol synthesis rates were reduced in dams fed 2% cholesterol (54). Because we had already known that hypercholesterolemic sacs secrete more cholesterol (30) and we knew that rodent and human yolk sacs secrete lipoproteins (30, 57–59), we focused the remainder of this manuscript on delineating the cholesterol-induced metabolic changes in the placenta.
Placenta
The placenta is important in both the rodent and human throughout mid to late gestation. Increases in the uptake of maternal lipoprotein-cholesterol lead to a modest suppression of sterol synthesis rates and moderately elevated sterol concentrations, at least in the rodent in early/mid gestation and in cultured human choriocarcinoma cells (29, 31, 41). Since the placenta expresses LXR and LXR-inducible proteins (60), our initial hypothesis was that at least part of any increase in movement of maternally derived cholesterol to the fetal circulation in hypercholesterolemic dams was due to an LXR-induced increase in placental ABCG1 and apoE (61). As in other cell types, cholesterol can be effluxed (via ABCG1) or secreted (via apoE:lipid complexes) from trophoblasts (31). Though data in our laboratory have demonstrated that LDL-cholesterol is effluxed to phospholipid vesicles and HDL from the basolateral (fetal) membranes of BeWo cells most likely by way of ABCG1 (62) and not ABCA1 or SR-BI (31), a recent study in mice suggests that ABCA1 may have a role in transport of maternally derived cholesterol to the fetus (63). Part of the difference in the studies could be that a significant amount of radiolabeled plasma cholesterol in the study by Lindegaard et al. (63) was as HDL and as such was likely taken up and transported via the yolk sac and not the placenta (30, 58, 59). It was somewhat surprising that the levels of ABCG1 and apoE in the placentas of dams fed cholesterol were not elevated compared with control placentas, as a 2-fold increase in liver cholesterol concentration will result in activation of LXR (64), and placental cholesterol concentrations were ∼2-fold greater in cholesterol-fed dams (29); it should be noted that the placenta consists of several cell types, though transport proteins should be expressed highly in trophoblasts. Consequently, our hypothesis now states that even though placental cholesterol concentrations are elevated in dams fed cholesterol, the enzymes required to form the specific oxysterols required to activate LXR and increase transcription of various proteins are absent; which oxysterols activate LXR in the placenta is currently unknown. It should be noted that activation of LXR through other means, such as with an LXR agonist, could affect apoE and ABCG1 levels and thereby enhance sterol transport to the fetus (63).
Even if LXR-inducible proteins were not affected, other sterol-affected processes involved with sterol uptake and transport may have been affected. We initially examined the LDLR because this receptor is responsible for uptake of LDL and LDL is the primary lipoprotein in human circulation. There was no change in the expression of the LDLR. This was not to be unexpected in the hamsters because recent studies have demonstrated that fetal tissues at times of rapid growth (mid/early gestation), including the placenta, have a blunted metabolic response to cellular cholesterol concentrations, possibly due to constitutive processing of sterol regulatory element binding protein-2 (SREBP-2) (54). Thus, the increase in uptake of LDL-cholesterol (and HDL-cholesterol) would be dependent on maternal lipoprotein-cholesterol concentrations. A series of elegant studies by Dietschy and coworkers showed that in the face of acutely elevated plasma LDL-cholesterol concentrations and no change in lipoprotein receptor levels, more LDL-cholesterol is taken up (65, 66). Since LDLR levels were not changed in the placenta and plasma cholesterol levels were elevated in hypercholesterolemic dams, more sterol would have to be taken up; the same is likely true for HDL uptake by SR-BI in the yolk sac. Consequently, a concentration gradient across trophoblasts would occur and cholesterol could move down the gradient and ultimately to the fetal circulation. Even in trophoblasts that are not replete with cholesterol, basolateral membranes have less cholesterol than apical membranes (67), leading to primarily unidirectional transport of cholesterol (31).
Interestingly, a recent study in humans demonstrated that placental LDLR levels were reduced in females with hypercholesterolemia (68). The decrease was not due to changes in tissue cholesterol concentrations or SREBP-2 levels (69). While this might initially suggest that the rodent does not regulate placental proteins as does the human, the human studies were performed at term, whereas these studies were preformed in early to mid gestation when growth rates were more dramatic. As in the hamster, cholesterol concentrations in BeWo cells, human choriocarcinoma-derived trophoblasts of which subclone 30 becomes polarized on transwells (31), increased when cells were exposed to exogenous sterol. It is likely that the regulation of proteins in the placenta in early and late gestation differs, possibly due to different rates of proliferation and sterol requirements for membrane substrates, and is an area of research still underdeveloped.
Potential cholesterol-induced changes in expression levels of key proteins involved with transport were also measured, some of which have not been previously shown in the placenta. The expression level of NPC1 was reduced in cholesterol-replete placentas, as demonstrated in other tissues (70), and as such would not enhance transport from lysosomes. Though the mechanism of regulation is thought to be via SREBP-2, a different regulatory mechanism exists in the rapidly growing placenta since SREBP-2 levels are not affected significantly in the cholesterol-replete placenta (54). Interestingly, though not surprising since NPC1L1 is thought to be involved with sterol transport across the intestine [reviewed in (16, 17)], NPC1L1 was also expressed in the placenta. In contrast to NPC1, expression levels were increased when cholesterol concentrations were elevated. Even though the role of NPC1L1 is still debatable, one can theorize that an increase in NPC1L1 could aide in enhanced sterol movement (from LDL or HDL) across trophoblasts (71). SCP-2/x and ABCA2 (14, 15) were also expressed by the placenta, and as in other tissues, expression appeared unaffected by placental cholesterol concentrations (72, 73).
Summary
How do these data relate to the human? In both humans and rodents, plasma LDL and HDL are taken up by the placenta, both of which are hemochorial [reviewed in (50)]. Rodent and human placentas both synthesize sterol as well (74–77), though the amount of newly synthesized sterol transported to the fetus, if any, is unknown. Additionally, the machinery needed to transport sterol across the placenta and efflux/secreted sterol from cells is also present in both the human and rodent as demonstrated here. Likewise, yolk sacs of both species secrete lipoprotein particles (30, 57–59). One difference between transport of nutrients from the maternal to fetal circulations in the rodent and human is that the human has a viable, active yolk sac in the first trimester and a viable, active placenta in the second and third trimesters, whereas the rodent has a viable, active yolk sac and placenta throughout most of gestation [reviewed in (50)]. A second difference between the rodent and human is that the rodent is primarily an HDL-carrying animal and the yolk sac and placenta take up significant amounts of HDL. However, the human carries most cholesterol in LDL, but little LDL is taken up by the yolk sac. Thus, transport of LDL-cholesterol from the dam to fetus in the rodent would model transport across the human placenta. Transport of HDL-cholesterol in the rodent model would include transport across the placenta and yolk sac. Importantly, there was an increase in transport of both HDL- and LDL-derived cholesterol to the fetus in hypercholesterolemic hamsters, indicating that more cholesterol could be transported to the human fetus in early (via yolk sac via HDL) as well as middle and late (via placenta via LDL/HDL) gestation.
The ability to manipulate cholesterol transport to the fetus should have a dramatic impact on in utero development, especially of fetuses affected with SLOS and other defects in sterol biosynthesis. Since membrane integrity and function is altered in SLOS (78–80) and since a number of signaling proteins are located in the membrane, the benefit of enhanced sterol concentrations could be as simple as a change in membrane composition and a normalization of various processes in utero. Shh signaling is likely also improved (2, 81). In support of this, the outcome of pregnancy for fetuses with SLOS, as defined by a standardized scoring process, was modified by apoE isoform of the mother, but not the father (23). The ability to transport more cholesterol across the placenta could play a role in fetal metabolism of unaffected fetuses as well. Recent data demonstrate that women with low serum cholesterol concentrations or abnormal lipoprotein receptor levels birth smaller infants (37–40), and mice with low plasma HDL-cholesterol levels have fetuses with reduced growth rates (41). While these human data do not represent a cause and effect situation, they raise the possibility that an adequate amount of cholesterol is required for growth, directly or indirectly, and growth rates are reduced and metabolism affected if less cholesterol is available. The ability to affect growth rates would be of major significance to the health industry because infants with altered growth rates have an increased risk to develop a number of diseases later in life (82–84).
Acknowledgments
The authors thank M. Hayden Lichtenberg for her excellent technical assistance.
Abbreviations
dpc, days postconception
FCR, fractional catabolic rate
LDLR, LDL receptor
NPC1, Niemann-Pick C1
NPC1L1, NPC1 Like Protein
SCP, sterol carrier protein
Shh, sonic hedgehog
SLOS, Smith-Lemli-Opitz Syndrome
SREBP-2, sterol regulatory element binding protein-2
These studies were supported by grants HD34089 (LAW) and GM31651 (FS) from the NIH and 053025N from the AHA (GAG).
Published, JLR Papers in Press, January 3, 2009.
References
- 1.Porter J. A., K. E. Young, and P. A. Beachy. 1996. Cholesterol modification of hedgehog signaling proteins in animal development. Science. 274 255–259. [DOI] [PubMed] [Google Scholar]
- 2.Cooper M. K., C. A. Wassif, P. A. Krabowiak, J. Taipale, R. Gong, R. I. Kelley, F. D. Porter, and P. A. Beachy. 2003. A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis. Nat. Genet. 33 508–513. [DOI] [PubMed] [Google Scholar]
- 3.Salameh W. A., and D. S. Matrogiannis. 1994. Maternal hyperlipidemia in pregnancy. Clin. Obstet. Gynecol. 37 66–77. [DOI] [PubMed] [Google Scholar]
- 4.Potter J. M., and P. J. Nestel. 1979. The hyperlipidemia of pregnancy in normal and complicated pregnancies. Am. J. Obstet. Gynecol. 133 165–170. [DOI] [PubMed] [Google Scholar]
- 5.Wyne K. L., and L. A. Woollett. 1998. Transport of maternal LDL and HDL to the fetal membranes and placenta of the Golden Syrian hamster is mediated by receptor-dependent and receptor-independent processes. J. Lipid Res. 39 518–530. [PubMed] [Google Scholar]
- 6.Shi W., K. F. Swan, S. R. Lear, J. S. O'Neil, S. K. Erickson, and M. C. Henson. 1999. Regulation of pathways determining cholesterol availability in the baboon placenta with advancing gestation. Biol. Reprod. 61 1499–1505. [DOI] [PubMed] [Google Scholar]
- 7.Brown M. S., and J. L. Goldstein. 1986. A receptor-mediated pathway for cholesterol homeostasis. Science. 232 34–47. [DOI] [PubMed] [Google Scholar]
- 8.Ory D. S. 2000. Niemann-Pick type C: a disorder of cellular cholesterol trafficking. Biochim. Biophys. Acta. 1529 331–339. [DOI] [PubMed] [Google Scholar]
- 9.Wojtanik K. M., and L. Liscum. 2003. The transport of low density lipoprotein-derived cholesterol to the plasma membrane is defective in NPC1 cells. J. Biol. Chem. 278 14850–14856. [DOI] [PubMed] [Google Scholar]
- 10.Hatzopoulos A. K., A. Rigotti, R. D. Rosenberg, and M. Krieger. 1998. Temporal and spatial pattern of expression of the HDL receptor SR-BI during murine embryogenesis. J. Lipid Res. 39 495–508. [PubMed] [Google Scholar]
- 11.Hammad S. M., S. Stefansson, W. O. Twal, C. J. Drake, P. Fleming, A. Remaley, H. B. Brewer, Jr., and W. S. Argraves. 1999. Cubilin, the endocytic receptor for intrinsic factor-vitamin B12 complex, mediates high-density lipoprotein holoparticle endocytosis. Proc. Natl. Acad. Sci. USA. 96 10158–10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kozyraki R., J. Fyfe, M. Kristiansen, C. Gerdes, C. Jacobsen, S. Cui, E. I. Christensen, M. Aminoff, A. de la Chapelle, R. Krahe, et al. 1999. The intrinsic factor-vitamin B12 receptor, cubilin, is a high-affinity apolipoprotein A-I receptor facilitating endocytosis of high-density lipoprotein. Nat. Med. 5 656–661. [DOI] [PubMed] [Google Scholar]
- 13.Smith B. T., J. C. Mussell, P. A. Fleming, J. L. Barth, D. D. Spyropoulos, M. A. Cooley, C. J. Drake, and W. S. Argraves. 2006. Targeted disruption of cubilin reveals essential developmental roles in the structure and function of endoderm and in somite formation. BMC Dev. Biol. 6 30–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zanlungo S., A. Rigotti, and F. Nervi. 2004. Hepatic cholesterol transport from plasma into bile: implications for gallstone disease. Curr. Opin. Lipidol. 15 279–286. [DOI] [PubMed] [Google Scholar]
- 15.Mack J. T., D. M. Townsend, V. Beljanski, and K. D. Tew. 2007. The ABCA2 transporter: intracellular roles in trafficking and metabolism of LDL-derived cholesterol and sterol-related compounds. Curr. Drug Metab. 8 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu L. 2008. The structure and function of Niemann-Pick C1-like 1 protein. Curr. Opin. Lipidol. 19 263–269. [DOI] [PubMed] [Google Scholar]
- 17.Hui D. Y., E. D. Labonte, and P. N. Howles. 2008. Development and physiological regulation of intestinal lipid absorption. III. Intestinal transporters and cholesterol absorption. Am. J. Physiol. Gastrointest. Liver Physiol. 294 G839–G843. [DOI] [PubMed] [Google Scholar]
- 18.Oram J. F., and A. M. Vaughan. 2006. ATP-Binding cassette cholesterol transporters and cardiovascular disease. Circ. Res. 99 1031–1043. [DOI] [PubMed] [Google Scholar]
- 19.Irons M., E. R. Elias, G. Salen, G. S. Tint, and A. K. Batta. 1993. Defective cholesterol biosynthesis in Smith-Lemli-Opitz syndrome. Lancet. 341 1414. [DOI] [PubMed] [Google Scholar]
- 20.Tint G. S., M. Irons, E. R. Elias, A. K. Batta, R. Frieden, T. S. Chen, and G. Salen. 1994. Defective cholesterol biosynthesis associated with the Smith-Lemli-Opitz syndrome. N. Engl. J. Med. 330 107–113. [DOI] [PubMed] [Google Scholar]
- 21.Linck L. M., S. J. Hayflick, D. S. Lin, K. P. Battalie, S. Ginat, T. Burlingame, K. M. Gibson, M. Honda, A. Honda, G. Salen, et al. 2000. Fetal demise with Smith-Lemli-Opitz syndrome confirmed by tissue sterol analysis and the absence of measurable 7-dehydrocholesterol Δ7-reductase activity in chorionic villi. Prenat. Diagn. 20 238–240. [PubMed] [Google Scholar]
- 22.Nowaczyk M. J. M., S. A. Farrell, W. L. Sirkin, L. Velsher, P. A. Krakowiak, J. S. Waye, and F. D. Porter. 2001. Smith-Lemli-Opitz (RHS) syndrome: holoprosencephaly and homozygous IVS8–1G to C genotype. Am. J. Med. Genet. 103 75–80. [DOI] [PubMed] [Google Scholar]
- 23.Witsch-Baumgartner M., M. Gruber, H. G. Kraft, M. Rossi, P. Clayton, M. Giros, D. Haas, R. I. Kelley, M. Krajewska-Walasek, and G. Utermann. 2004. Maternal apo E genotype is a modifier of the Smith-Lemli-Opitz syndrome. J. Med. Genet. 41 577–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin D. S., R. M. Pitkin, and W. E. Connor. 1977. Placental transfer of cholesterol into the human fetus. Am. J. Obstet. Gynecol. 128 735–739. [DOI] [PubMed] [Google Scholar]
- 25.Pitkin R. M., W. E. Connor, and D. S. Lin. 1972. Cholesterol metabolism and placental transfer in the pregnant rhesus monkey. J. Clin. Invest. 51 2584–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Plotz E. J., J. J. Kabara, M. E. Davis, G. V. LeRoy, and R. G. Gould. 1968. Studies on the synthesis of cholesterol in the brain of the human fetus. Am. J. Obstet. Gynecol. 101 534–538. [DOI] [PubMed] [Google Scholar]
- 27.Tint G. S., H. Yu, Q. Shang, G. Xu, and S. B. Patel. 2006. The use of the Dhcr7 knockout mouse to accurately determine the origin of fetal sterols. J. Lipid Res. 47 1535–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshida S., and Y. Wada. 2005. Transfer of maternal cholesterol to embryo and fetus in pregnant mice. J. Lipid Res. 46 2168–2174. [DOI] [PubMed] [Google Scholar]
- 29.McConihay J. A., P. S. Horn, and L. A. Woollett. 2001. The effect of maternal hypercholesterolemia on fetal sterol metabolism in the Golden Syrian hamster. J. Lipid Res. 42 1111–1119. [PubMed] [Google Scholar]
- 30.Hayden Lichtenberg M., C. S. Wilke, J. A. McConihay, N. A. Granholm, and L. A. Woollett. 2005. Yolk sac cholesteryl ester secretion rates can be manipulated in the Golden Syrian hamster: effect of yolk sac cholesterol concentrations. Biochim. Biophys. Acta. 1735 214–221. [DOI] [PubMed] [Google Scholar]
- 31.Schmid K. E., W. S. Davidson, L. Myatt, and L. A. Woollett. 2003. The transport of cholesterol across a BeWo cell monolayer: implications for net transport of sterol from the maternal to fetal circulation. J. Lipid Res. 44 1909–1918. [DOI] [PubMed] [Google Scholar]
- 32.Irons M., E. R. Elias, D. Abuelo, M. J. Bull, C. L. Greene, V. P. Johnson, L. Keppen, C. Schanen, G. S. Tint, and G. Salen. 1997. Treatment of Smith-Lemli-Opitz syndrome: results of a multi-center trial. Am. J. Med. Genet. 68 311–314. [PubMed] [Google Scholar]
- 33.Elias E. R., M. B. Irons, A. D. Hurley, G. X. Tint, and G. Salen. 1997. Clinical effects of cholesterol supplementation in six patients with the Smith-Lemli-Opitz syndrome (SLOS). Am. J. Med. Genet. 68 305–310. [DOI] [PubMed] [Google Scholar]
- 34.Tint G. S., G. Salen, A. K. Batta, S. Shefer, M. Irons, E. R. Elias, D. N. Abuelo, V. P. Johnson, M. Lambert, and R. Lutz. 1995. Correlation of severity and outcome with plasma sterol levels in variants of the Smith-Lemli-Opitz syndrome. J. Pediatr. 127 82–87. [DOI] [PubMed] [Google Scholar]
- 35.Witsch-Baumgartner M., B. U. Fitzky, M. Ogorelkova, H. G. Kraft, F. F. Moebius, H. Glossmann, U. Seedorf, G. Gillessen-Kaesbach, G. F. Hoffmann, P. Clayton, et al. 2000. Mutational spectrum in the Δ7-sterol reductase gene and genotype-phenotype correlation in 84 patients with Smith-Lemli-Opitz syndrome. Am. J. Hum. Genet. 66 402–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cunniff, C. M. 2004. Smith-Lemli-Opitz Syndrome. Gene Reviews. www.genetests.org.
- 37.Clausen T., T. K. Burski, N. Oyen, K. Godang, J. Bollerslev, and T. Henriksen. 2005. Maternal anthropometric and metabolic factors in the first half of pregnancy and risk of neonatal macrosomia in term pregnancies. A prospective study. Eur. J. Endocrinol. 153 887–894. [DOI] [PubMed] [Google Scholar]
- 38.Wadsack C., S. Tabano, A. Maier, U. Hiden, G. Alvino, V. Cozzi, M. Huttinger, W. J. Schneider, U. Lang, I. Cetin, et al. 2006. Intrauterine growth restriction (IUGR) is associated with alterations in placental lipoprotein receptors and maternal lipoprotein composition. Am. J. Physiol. Endocrinol. Metab. 292 E476–E484. [DOI] [PubMed] [Google Scholar]
- 39.Edison R. J., K. Berg, A. Remaley, R. Kelley, C. Rotimi, R. E. Stevenson, and M. Muenke. 2007. Adverse birth outcome among mothers with low serum cholesterol. Pediatrics. 120 723–733. [DOI] [PubMed] [Google Scholar]
- 40.Napoli C., F. P. D'Armiento, F. P. Mancini, A. Postiglione, J. L. Wiztum, G. Palumbo, and W. Palinski. 1997. Fatty streak formation occurs in human fetal aortas and is greatly enhanced by maternal hypercholesterolemia. Intimal accumulation of LDL and its oxidation precede monocyte recruitment into early atherosclerotic lesions. J. Clin. Invest. 100 2680–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McConihay J. A., A. M. Honkomp, N. A. Granholm, and L. A. Woollett. 2000. Maternal high density lipoproteins affect fetal mass and extra-embryonic fetal tissue sterol metabolism in the mouse. J. Lipid Res. 41 424–432. [PubMed] [Google Scholar]
- 42.Woollett L. A., D. K. Spady, and J. M. Dietschy. 1989. Mechanisms by which saturated triacylglycerols elevate the plasma low density lipoprotein-cholesterol concentration in hamsters. Differential effects of fatty acid chain length. J. Clin. Invest. 84 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Woollett L. A., D. Kearney, and D. K. Spady. 1997. Diet modification alters plasma HDL cholesterol concentrations but not the transport of HDL cholesteryl esters to the liver in the hamster. J. Lipid Res. 38 2289–2302. [PubMed] [Google Scholar]
- 44.Halperin G., O. Stein, and Y. Stein. 1986. Synthesis of ether analogs of lipoprotein lipids and their biological applications. Methods Enzymol. 129 816–848. [DOI] [PubMed] [Google Scholar]
- 45.Su Y. R., M. F. Linton, and S. Fazio. 2002. Rapid quantification of murine ABC mRNAs by real time reverse transcriptase-polymerase chain reaction. J. Lipid Res. 43 2180–2187. [DOI] [PubMed] [Google Scholar]
- 46.Jia X., N. Ebine, I. Demonty, Y. Wang, R. Beech, V. Muise, M. G. Fortin, and P. J. H. Jones. 2007. Hypocholesterolaemic effects of plant sterol analogues are independent of ABCG5 and ABCG8 transporter expressions in hamsters. Br. J. Nutr. 98 550–555. [DOI] [PubMed] [Google Scholar]
- 47.Jelinek D. F., S. Andersson, C. A. Slaughter, and D. W. Russell. 1990. Cloning and regulation of cholesterol 7α-hydroxylase, the rate-limiting enzyme in bile acid biosynthesis. J. Biol. Chem. 265 8190–8197. [PMC free article] [PubMed] [Google Scholar]
- 48.Graf G. A., L. Yu, W. P. Li, R. Gerard, P. L. Tuma, J. C. Cohen, and H. H. Hobbs. 2003. ABCG5 and ABCG8 are obligate heterodimers for protein trafficking and biliary cholesterol excretion. J. Biol. Chem. 278 48275–48282. [DOI] [PubMed] [Google Scholar]
- 49.Atshaves B. P., A. Petrescu, O. Starodub, J. Roths, A. B. Kier, and F. Schroeder. 1999. Expression and intracellular processing of the 58 kDa Sterol Carrier Protein 2/3-Oxoacyl-CoA Thiolase in transfected mouse L-cell fibroblasts. J. Lipid Res. 40 610–622. [PubMed] [Google Scholar]
- 50.Woollett L. A. 2008. Where does fetal and embryonic cholesterol originate and what does it do? Annu. Rev. Nutr. 28 97–114. [DOI] [PubMed] [Google Scholar]
- 51.Dietschy J. M., S. D. Turley, and D. K. Spady. 1993. Role of liver in the maintenance of cholesterol and low density lipoprotein homeostasis in different animal species, including humans. J. Lipid Res. 34 1637–1659. [PubMed] [Google Scholar]
- 52.Spady D. K., D. W. Bilheimer, and J. M. Dietschy. 1983. Rates of receptor-dependent and -independent low density lipoprotein uptake in the hamster. Proc. Natl. Acad. Sci. USA. 80 3499–3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Spady D. K., and J. M. Dietschy. 1985. Dietary saturated triacylglycerols suppress hepatic low density lipoprotein receptor activity in the hamster. Proc. Natl. Acad. Sci. USA. 82 4526–4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yao L., K. Jenkins, P. S. Horn, M. H. Lichtenberg, and L. A. Woollett. 2007. Inability to fully suppress sterol synthesis rates with exogenous sterol in embryonic and extraembyronic fetal tissues. Biochim. Biophys. Acta. 1171 1372–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xie C., L. A. Woollett, S. D. Turley, and J. M. Dietschy. 2002. Fatty acids differentially regulate hepatic cholesteryl ester formation and incorporation into lipoproteins in the liver of the mouse. J. Lipid Res. 43 1508–1519. [DOI] [PubMed] [Google Scholar]
- 56.Porter F. D. 2000. RSH/Smith-Lemli-Opitz Syndrome: a multiple congenital anomaly/mental retardation syndrome due to an inborn error of cholesterol biosynthesis. Mol. Genet. Metab. 71 163–174. [DOI] [PubMed] [Google Scholar]
- 57.Lanford R. E., D. L. Bronson, L. E. Estlack, and F. H. Wians, Jr. 1991. Plasma protein and apolipoprotein synthesis by human yolk sac carcinoma cells in vitro. In Vitro Cell. Dev. Biol. 27A 205–210. [DOI] [PubMed] [Google Scholar]
- 58.Farese R. V., Jr., S. Cases, S. L. Ruland, H. J. Kayden, J. S. Wong, S. G. Young, and R. L. Hamilton. 1996. A novel function for apolipoprotein B: lipoprotein synthesis in the yolk sac is critical for maternal-fetal lipid transport in mice. J. Lipid Res. 37 347–360. [PubMed] [Google Scholar]
- 59.Plonné D., L. Winkler, H. Franke, and R. Dargel. 1992. The visceral yolk sac - an important site of synthesis and secretion of apolipoprotein B containing lipoproteins in the feto-placental unit of the rat. Biochim. Biophys. Acta. 1127 174–185. [DOI] [PubMed] [Google Scholar]
- 60.Plosch T., E. M. E. van Straten, and S. F. Kuipers. 2007. Cholesterol transport by the placenta: placental liver X receptor activity as a modulator of fetal cholesterol metabolism? Placenta. 28 604–610. [DOI] [PubMed] [Google Scholar]
- 61.Laffitte B. A., J. J. Repa, S. B. Joseph, D. C. Wilpitz, H. R. Kast, D. J. Mangelsdorf, and P. Tontonoz. 2001. LXRs control lipid-inducible expression of the apolipoprotein E gene in macrophages and adipocytes. Proc. Natl. Acad. Sci. USA. 98 507–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang N., D. Lan, W. Chen, F. Matsuruar, and A. R. Tall. 2004. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc. Natl. Acad. Sci. USA. 101 9774–9779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lindegaard M. L., C. A. Wassif, B. Varisman, M. Amar, E. V. Wasmuth, R. Shamburek, L. B. Nielsen, A. T. Remaley, and F. D. Porter. 2008. Characterization of placental cholesterol transport: ABCA1 is a potential target for in utero therapy of Smith-Lemli-Opitz syndrome. Hum. Mol. Genet. 17 3806–3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Repa J. J., K. K. Buhman, R. V. J. Farese, J. M. Dietschy, and S. D. Turley. 2004. ACAT2 deficiency limits cholesterol absorption in the cholesterol-fed mouse: impact on hepatic cholesterol homeostasis. Hepatology. 40 1088–1097. [DOI] [PubMed] [Google Scholar]
- 65.Spady D. K., J. B. Meddings, and J. M. Dietschy. 1986. Kinetic constants for receptor-dependent and receptor-independent low density lipoprotein transport in the tissues of the rat and hamster. J. Clin. Invest. 77 1474–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Spady D. K., L. A. Woollett, and J. M. Dietschy. 1993. Regulation of plasma LDL-cholesterol levels by dietary cholesterol and fatty acids. Annu. Rev. Nutr. 13 355–381. [DOI] [PubMed] [Google Scholar]
- 67.Powell T. L., T. Jansson, N. P. Illsley, M. Wennergren, M. Korotkova, and B. Strandvik. 1999. Composition and permeability of syncytiotrophoblast plasma membranes in pregnancies complicated by intrauterine growth restriction. Biochim. Biophys. Acta. 1420 86–94. [DOI] [PubMed] [Google Scholar]
- 68.Ethier-Chiasson M., A. Duchesne, J-C. Forest, Y. Giguère, A. Masse, C. Mounier, and J. Lafond. 2007. Influence of maternal lipid profile on placental protein expression of LDLr and SR-BI. Biochem. Biophys. Res. Commun. 359 8–14. [DOI] [PubMed] [Google Scholar]
- 69.Marseille-Tremblay C., M. Ethier-Chiasson, J-C. Forest, Y. Giguère, A. Masse, C. Mounier, and J. Lafond. 2008. Impact of maternal circulating cholesterol and gestational diabetes mellitus on lipid metabolism in human term placenta. Mol. Reprod. Dev. 75 1054–1062. [DOI] [PubMed] [Google Scholar]
- 70.Gèvry N., K. Schoonjams, F. Guay, and B. D. Murphy. 2008. Cholesterol supply and SREBPs modulate transcription of the Niemann-Pick C-1 gene in steroidogenic tissues. J. Lipid Res. 49 1024–1033. [DOI] [PubMed] [Google Scholar]
- 71.Ge L., J. Wang, W. Qi, H. H. Hiao, J. Cao, Y. X. Qu, B. L. Li, and B. L. Song. 2008. The cholesterol absorption inhibitor ezetimibe acts by blocking the sterol-induced internalization of NPC1L1. Cell Metab. 7 508–519. [DOI] [PubMed] [Google Scholar]
- 72.Baum C. L., S. Kansal, and N. O. Davidson. 1993. Regulation of sterol carrier protein-2 gene expression in rat liver and small intestine. J. Lipid Res. 34 729–739. [PubMed] [Google Scholar]
- 73.Kaminski W. E., A. Piehler, K. Püllmann, M. Porsch-Ozcürümez, C. Duong, G. M. Bared, C. Buchler, and G. Schmitz. 2001. Complete coding sequence, promoter region, and genomic structure of the human ABCA2 gene and evidence for sterol-dependent regulation in macrophages. Biochem. Biophys. Res. Commun. 281 249–258. [DOI] [PubMed] [Google Scholar]
- 74.Boguslawski W., and W. Sokolowski. 1984. HMG-CoA reductase activity in the microsomal fraction from human placenta in early and term pregnancy. Int. J. Biochem. 16 1023–1026. [DOI] [PubMed] [Google Scholar]
- 75.Simpson E. R., J. C. Porter, L. Milewich, D. W. Bilheimer, and P. C. MacDonald. 1978. Regulation by plasma lipoproteins of progesterone biosynthesis and 3-hydroxy-3-methyl glutaryl coenzyme A reductase activity in cultured human choriocarcinoma cells. J. Clin. Endocrinol. Metab. 47 1099–1105. [DOI] [PubMed] [Google Scholar]
- 76.Woollett L. A. 1996. Origin of cholesterol in the fetal Golden Syrian hamster: contribution of de novo sterol synthesis and maternal-derived lipoprotein cholesterol. J. Lipid Res. 37 1246–1257. [PubMed] [Google Scholar]
- 77.Belknap W. M., and J. M. Dietschy. 1988. Sterol synthesis and low density lipoprotein clearance in vivo in the pregnant rat, placenta, and fetus. Sources for tissue cholesterol during fetal development. J. Clin. Invest. 82 2077–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tulenko T. N., K. Boeze-Battaglia, R. P. Mason, G. S. Tint, R. D. Steiner, W. E. Connor, and E. F. Labelle. 2006. A membrane defect in the pathogenesis of the Smith-Lemli-Opitz syndrome. J. Lipid Res. 47 134–143. [DOI] [PubMed] [Google Scholar]
- 79.Kovarova M., C. A. Wassif, S. Odom, K. Liao, F. D. Porter, and J. Rivera. 2006. Cholesterol deficiency in a mouse model of Smith-Lemli-Opitz syndrome reveals increased mast cell responsiveness. J. Exp. Med. 203 1161–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Keller R. K., T. P. Arnold, and S. J. Fliesler. 2004. Formation of 7-dehydrocholesterol-containing membrane rafts in vitro and in vivo, with relevance to the Smith-Lemli-Opitz syndrome. J. Lipid Res. 45 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cooper M. K., J. A. Porter, K. E. Young, and P. A. Beachy. 1998. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science. 280 1603–1607. [DOI] [PubMed] [Google Scholar]
- 82.Barker D. J. P. 2004. The developmental origins of adult disease. J. Am. Coll. Nutr. 23 588S–595S. [DOI] [PubMed] [Google Scholar]
- 83.Boney C. M., A. Verma, R. Tucker, and B. R. Vohr. 2005. Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics. 115 e290–e296. [DOI] [PubMed] [Google Scholar]
- 84.Samaras T. T., H. Elrick, and L. H. Storms. 2003. Birthweight, rapid growth, cancer, and longevity: a review. J. Natl. Med. Assoc. 95 1170–1183. [PMC free article] [PubMed] [Google Scholar]