

Abstract
Kaposi's sarcoma-associated herpesvirus (KSHV) is the causative agent of three human proliferative disorders, namely, Kaposi's sarcoma, primary effusion lymphomas (PEL), and multicentric Castleman's disease. Lytic DNA replication of KSHV, which is essential for viral propagation, requires the binding of at least two KSHV proteins, replication and transactivation activator (RTA) and K-bZIP, on the lytic origin of replication. Moreover, K-bZIP physically interacts with RTA and represses its transactivation activity on several viral promoters in transient transfection assays. To evaluate the physiological roles of K-bZIP in the context of PEL, we generated BCBL-1 cells with a tetracycline (Tet)-inducible small hairpin RNA (shRNA) directed against the K8 mRNA to knock down K-bZIP expression at different points during KSHV's life cycle. Using this model, we demonstrate that in the absence of K-bZIP expression, dramatic decreases in orf50, orf57, and orf26 transcript expression are observed. Similar effects were seen at the protein level for RTA (immediate-early protein) and K8.1 (late protein) expression. Interestingly, a direct correlation between K-bZIP levels and viral lytic mRNAs was noticed. As a consequence of K-bZIP knockdown, viral DNA replication and virion production were severely impaired. The same effects were observed following knockdown of K-bZIP in another PEL cell line, BC3. Finally, using shRNA-K8-inducible 293 cells, we report that de novo synthesis of K-bZIP is not necessary for initiation of infection and latency establishment. These data support the concept that K-bZIP is essential for lytic viral gene expression, viral DNA replication, and virus propagation in PEL cells.
Kaposi's sarcoma-associated herpesvirus (KSHV), also known as human herpesvirus 8 (HHV-8), is a gammaherpesvirus that closely resembles Epstein-Barr virus (EBV). KSHV infection is associated with the development of all types of Kaposi's sarcoma (KS) and with B-cell lymphoproliferative diseases, such as primary effusion lymphoma (PEL) and multicentric Castleman's disease (4, 6, 7, 29). The KSHV life cycle includes both latent and lytic replication phases (21, 27). During latency, a limited number of viral genes are expressed, and no infectious virions are produced. In latently infected cells, multiple copies of the viral genome are maintained as extrachromosomal episomes and are replicated in synchrony with cell division (4). During a productive infection, a complex of viral enzymes including the viral DNA polymerase ensures viral DNA replication. The lytic KSHV life cycle is characterized by the production of progeny viruses from infected cells and can be induced using chemicals such as 12-O-tetradecanoylphorbol 13-acetate (TPA) and butyric acid (BAc) (22, 23). Chemical induction of PEL-derived B cells initiates the expression of the replication and transcription factor activator (RTA), encoded by open reading frame 50 (orf50), which is necessary and sufficient for the lytic infectious cycle and reactivation from latency (20, 24, 37). RTA modulates and activates the expression of several viral genes involved in the lytic replication process (9, 30, 31), and in particular, RTA binds directly to a 12-bp palindromic sequence shared by the orf57 and orfk8 promoters (19).
KSHV orfk8 encodes an early viral protein that is activated during the lytic replication cycle of KSHV (18). K-bZIP, the major product of orfk8, is a nuclear homodimerizing protein of 237 amino acids (11, 26) that colocalizes with PML oncogenic domains (36). K-bZIP is a transcriptional repressor which affects the transcription of a subset of viral and cellular genes (12, 15, 16, 32). In particular, K-bZIP directly binds RTA and represses RTA-mediated transactivation of the K-bZIP promoter, leading to a negative autoregulation system during lytic infection (16). K-bZIP was also reported to associate with the CREB-binding protein (CBP) and with p53 and to inhibit the transactivation potential of these molecules (10, 25).
K-bZIP is the positional and structural analogue of EBV Zta, which is a strong transcriptional factor capable of triggering EBV reactivation and lytic replication and transactivating a number of viral and cellular genes (8, 17, 28). K-bZIP has also been found to be important for KSHV replication. Indeed, K-bZIP binds to ori-Lyt (origin of lytic DNA replication), through the transcription factor CAAT enhancer binding protein α (C/EBPα), and to RTA, which leads to interaction with the core replication complex (35). Thus, RTA and K-bZIP play key roles in initiation of KSHV ori-Lyt-dependent DNA replication. The generation of an orfk8-deleted recombinant HHV-8, studied in the context of monkey Vero cells, showed that K-bZIP is important for virus replication, as this mutant failed to reactivate and produce infectious viruses (13). On the other hand, Izumiya et al. suggested that overexpression of K-bZIP in B cells results in less viral DNA replication (12). However, considering that overexpression systems can be misleading and that the recombinant study was limited to monkey Vero cells, which are not physiological targets of KSHV infection, we decided to design a B-cell model with a K-bZIP defect, using small interfering RNA technology. In this study, we generated BCBL-1 cells, designated TREx-BCBL-1-shRNAK8 cells, expressing a Tet-inducible small hairpin RNA (shRNA) against orfk8. Using this system, we observed decreased expression of K-bZIP in response to doxycycline (a Tet analog), and as a consequence, we saw a dramatic reduction in virus production and viral genomic copy levels following cell activation with TPA. Moreover, our data indicated marked inhibitions of orf50, orf57, and orf26 mRNA levels, without effects on viral latent mRNA levels. These observations were confirmed with the BC3 PEL cell line stably expressing shRNA-K8. Hence, our results provide evidence for the importance of K-bZIP in the reactivation process leading to the lytic viral cycle in the context of PEL cell lines. Finally, the use of an analogous TREx-shRNA system in 293 cells led us to conclude that de novo K-bZIP synthesis is not essential for the initial phase of infection and the establishment of KSHV latency.
MATERIALS AND METHODS
Reagents and antibodies.
TPA was purchased from Invivogen (San Diego, CA). BAc was purchased from Acros Organics (Geel, Belgium). Anti-K-bZIP was generously given by Henri Gruffat (Lyon, France) (26). Anti-LANA and Anti-K8.1 were obtained from Advanced Biotechnologies (Columbia, MD), anti-cyclin was obtained from ABCAM (Baltimore, MD), and rabbit polyclonal antibody against KSHV RTA (5) was a kind gift from Georges Miller (Yale University). Anti-actin antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Cell culture.
HEK 293T and HEK 293 cells (ATCC, Manassas, VA) were cultured in Dulbecco's modified Eagle's medium (Sigma) containing 10% heat-inactivated fetal bovine serum. BCBL-1 cells (27) were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH. BC3 (1) cells were purchased from ATCC. PEL cells were grown in RPMI 1640 medium (Sigma-Aldrich Canada Ltd., Oakville, Ontario, Canada) supplemented with 10% fetal bovine serum.
Plasmids and constructs.
A plasmid expressing wild-type K8 was described previously (15). A plasmid expressing K8 resistant to shRNA-K8 was made by mutagenesis with the following primers: 5′-TCGCAACAGCTTCCAACTCGCAGATCCAAAAGGAGATTGCACCGTAAGTTTGAAGAGGAACGCTTATGCACTAAG-3′ and 5′-CTTAGTGCATAAGCGTTCCTCTTCAAACTTACGGTGCAATCTCCTTTTGGATCTGGCGAGTTGGAAGCTGTTGCGA-3′. Underlined bases represent the silent mutations introduced. The shRNA-K8 construct was made by cloning annealed shRNA primers into HindIII and BglII doubly digested pTER vector (33), provided by Hans Clevers. shRNA-K8 primer sequences were as follows: 5′-GATCCAGAGGCGACTACATAGAAATTGATATCCGTTTCTATGTAGTCGCCTCT-3′ and 5′-AGCTTTTGGAAAAAAAGAGGCGACTACATAGAAACGGATATCAATTTCTATGTAGTCGCCTCTGG-3′. In the same way, an irrelevant control, shRNA-ctl, was designed from the HHV-6 IE1 mRNA, using the following primers: 5′-GATCCCAGAGGCTGGTATTAGAACATTGATATCCGTGTTCTAATACCAGCCTCTTTTTTTCCAAA-3′ and 5′-AGCTTTTGGAAAAAAAGAGGCTGGTATTAGAACACGGATATCAATGTTCTAATACCAGCCTCTGG-3′.
Generation of shRNA-inducible system.
To establish cell lines constitutively expressing the Tet repressor, pcDNA6/TR plasmid (Invitrogen) was electroporated (960 μF, 200 V) into BCBL-1 cells. Forty-eight hours after transfection, BCBL-1 cells were selected with 20 μg of blasticidin S HCl (Invitrogen)/ml in six-well plates for 4 weeks. This TREx BCBL-1 cell line was next transfected with pcDNA/FRT/TO-βGal to test the repressor's efficacy. Next, to establish the Tet-inducible shRNA-K8 or shRNA-ctl cell line (TREx BCBL-1-shRNA-K8 or TREx BCBL-1-shRNA-ctl, respectively), pTER-shRNA-K8 or pTER-shRNA-ctl was transfected into TREx BCBL-1 cells by electroporation. Forty-eight hours after electroporation, the cells were selected with 80 μg of Zeocin (Invitrogen)/ml for 4 weeks. pTER-shRNA-K8 and pTER-shRNA-ctl were also transfected into BC3 cells, and Zeocin selection was performed for 4 weeks. 293 cells expressing inducible shRNA-K8 and shRNA-ctl were generated in a similar way. The experiments were done with a pool of cells selected during stable transfection.
Production of rKSHV.219 and infection of 293 cells.
Vero cells containing latent rKSHV.219 and a baculovirus containing RTA (BacK50) were a kind gift of Jeffrey Vieira (Seattle, WA) (34). Briefly, for the generation of rKSHV.219 stocks, Vero cells containing latent rKSHV.219 (80 to 90% confluent) were infected with BacK50 for 2 h in a minimum volume, the inoculum was removed, the cells were washed once with phosphate-buffered saline (PBS), and fresh medium with 1.25 mM BAc was added. Twenty-four hours later, the medium with BAc was removed and fresh medium was added. At 50 to 70 h post-baculovirus infection, the medium was collected, the cells were removed by centrifugation (300 × g, 5 min), and the supernatant was passed through a 0.45-μm filter and then used as a KSHV inoculum for 293 cell infection. Centrifugation enhancement was used for infection by centrifuging the culture plates at 450 × g for 20 min, with replacement of the medium 2 h after centrifugation. This increased the infection levels approximately two- to threefold.
Transfection.
Transfections of HEK 293T and HEK 293 cells were performed using the calcium phosphate precipitation procedure. Cells were plated at 100,000 cells/well (12-well plate) the day prior to transfection. Cells were transfected with up to 2 μg of expression vector per well and brought to a total of 2.5 μg of DNA per well for each condition, using the pCMV3T control plasmid. Cells were lysed 48 h after transfection, and proteins from whole-cell extract samples were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis.
RT-QPCR.
Total RNA was extracted from transfected 293T cells, transfected 293 cells, or B-cell-derived cell lines by use of Trizol reagent (Invitrogen, Ontario, Canada). All RNA samples were treated with DNase I to eliminate residual genomic DNA prior to amplification. cDNA was synthesized, and real-time quantitative PCR (RT-QPCR) analysis was performed on a Rotorgene instrument (Corbett Research) with SyBRGreener technology (Invitrogen). The specificity of amplification was assessed for each sample by melting curve analysis. All transcripts were normalized according to gapdh housekeeping gene expression. The following primers were used: gapdh forward, 5′-CGAGATCCCTCCAAAATCAA-3′; gapdh reverse, 5′-TTCACACCCATGACGAACAT-3′; orfk8 forward, 5′-CAAGAGGCGACTACATAGAAA-3′; orfk8 reverse, 5′-GATCACATACTTCGGCCTTAAC-3′; orf26 forward, 5′-GCTCGAATCCAACGGATTTG-3′; orf26 reverse, 5′-AATAGCGTGCCCCAGTTGC-3′; orf57 forward, 5′-CATCCTAGAGGACTCTGT-3′; orf57 reverse, 5′-TTGCTCGTCTTCCAGTGT-3′; orf50 forward, 5′-CGCAATGCGTTACGTTGTTG-3′; and orf50 reverse, 5′-GCCCGGACTGTTGAATCG-3′.
Detection of viral genome copies in supernatants and cells.
To test virus production from TPA/BAc-induced B cells, 1 ml of supernatant was harvested at different time points and processed as described above. To measure the number of genome equivalents, an aliquot of 200 μl of supernatant was treated for 30 min at 37°C with DNase I (Roche) to eliminate unencapsidated DNA. After DNase inactivation, samples were spiked with 106 copies of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) plasmid to normalize the extraction procedure. Genomic DNA was isolated with a QIAamp DNA blood mini kit (Qiagen, Mississauga, Ontario, Canada) according to the manufacturer's instructions. The DNA was then subjected to QPCR using ORF26 and GAPDH primers. To assess the presence of HHV-8 DNA in B cells exposed to reactivation, cellular DNA was extracted and analyzed by QPCR with ORF26 and GAPDH primers.
Immunoblot analysis.
Proteins from whole-cell extracts were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane (Bio-Rad, Mississauga, Ontario, Canada). Nonspecific sites were blocked by incubating the membranes in PBS containing 0.05% Tween 20 (PBS-T) and 5% nonfat dry milk for 1 h, followed by a 1-h incubation with primary antibodies (anti-K-bZIP, anti-RTA, anti-LANA, and anti-cyclin). After three 5-minute washes in PBS-T, the membranes were incubated for 1 h with horseradish peroxidase-conjugated goat anti-rabbit or goat anti-mouse immunoglobulin G (IgG; KPL Laboratories, Gaithersburg, MD) in blocking solution. Immunoreactive proteins were visualized by chemiluminescence using Supersignal Pico West substrate (Pierce, Rockford, IL).
Immunofluorescence assay.
BCBL-1-shRNA-K8 and BCBL-1-shRNA-ctl cells were fixed in cold acetone for 10 min and air dried. Cells were first incubated with rabbit anti-K-bZIP or mouse anti-K8.1 for 1 h at room temperature. Slides were washed three times for 5 min in PBS and then incubated with Alexa 588-labeled anti-mouse IgG or Alexa 588-labeled anti-rabbit IgG (Invitrogen) for 1 h at room temperature. Slides were washed three times and mounted with SlowFade Gold antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen). Slides were observed by fluorescence microscopy (BX51 microscope; Olympus, Canada).
Flow cytometry.
293 cells (2 × 105) were washed three times in PBS and fixed with 2% paraformaldehyde, and fluorescence-activated cell sorting (FACS) analysis was performed with a FACSCalibur flow cytometer (Becton Dickinson, Mountainview, CA) to detect green fluorescent protein (GFP) and dsRed.
Statistical analysis.
Statistical analysis was performed using Graph Pad In Stat software, using unpaired Student's t test or analysis of variance. Statistical significance was achieved when the P value was <0.05.
RESULTS
K-bZIP is associated with the viral DNA replication complex, which is composed of at least six other viral proteins (e.g., SSB, POL, PAF, HEL, PRI, and PPF) and is known to act as a KSHV origin of replication binding protein (36). K-bZIP is also described as a transcriptional repressor of RTA activity. To further confirm these roles in a PEL cell line model, we generated an inducible shRNA against orfk8. The shRNA was designed using Dharmacon's software and then cloned into the pTER vector. To test the efficiency of shRNA-K8, we first transfected 293T cells with a K8 expression vector in the absence or in the presence of increasing doses of pTER expressing shRNA-K8 or shRNA-ctl. As shown in Fig. 1A, transfection of the K8 expression vector led to abundant K-bZIP expression in the presence of pTER-ctl. On the other hand, we typically observed a >60% decrease in K-bZIP expression in the presence of shRNA against orfk8. To explore the impact of this shRNA-K8 on KSHV's life cycle, we generated stable TREx-BCBL-1-shRNA-K8 or TREx-BCBL-1-shRNA-ctl cells, in which the expression of the shRNA can be induced by adding doxycycline/Tet to the culture medium. PEL-derived BCBL-1 cells are latently infected with KSHV and can be induced to undergo lytic replication upon treatment with TPA-BAc, which activates the expression of KSHV RTA. TREx BCBL-1-shRNA-ctl and TREx BCBL-1-sh-RNA-K8 cells were simultaneously incubated with TPA-BAc and doxycycline for 48 h, and then whole-cell lysates were used for immunoblotting using anti-K-bZIP antibodies. In TREx BCBL-1-shRNA-ctl cells, a time-dependent increase of K-bZIP expression was observed following TPA-BAc stimulation, with no impact from shRNA-ctl (Fig. 1B). In contrast, in TREx BCBL-1-shRNA-K8 cells, a time-dependent decrease in K-bZIP expression was observed, with a nearly complete shutdown of K-bZIP expression observed at 48 h. The expression of both isoforms of K-bZIP (the three-exon and two-exon forms) was reduced by the shRNA-K8. Similar results were obtained with BC3 cells expressing a stable shRNA-K8 in comparison to BC3-shRNA-ctl cells (data not shown). These results confirmed that the shRNA-K8 could effectively inhibit TPA-BAc-induced K-bZIP expression in two PEL cell lines, BCBL-1 and BC3.
FIG. 1.

Inhibition of K-bZIP by shRNA at the protein level. (A) 293T cells were transfected with K8 expression vector and increasing amounts of pTER-shRNA-K8, pTER-shRNA-ctl, or empty vector. Thirty-six hours later, the cultures were harvested and total cell extracts were made and analyzed by Western blotting, using specific anti-K-bZIP and anti-actin antibodies. (B) TREx BCBL-1shRNA-ctl and TREx BCBL-1-shRNA-K8 cells were stimulated with or without 1 μg/ml doxycycline for 48 h in the presence of TPA (20 ng/ml) and BAc (0.3 mM). Whole-cell lysates were used for immunoblotting with anti-K-bZIP and anti-actin antibodies.
Given that the K-bZIP protein is associated with DNA replication due to its ori-Lyt binding properties, we were interested in determining the impact of K-bZIP knockdown on viral replication. TREx BCBL-1-sh-RNA-K8 cells were stimulated with TPA-BAc and with or without 1 μg/ml doxycycline for 72 h. Cells were harvested at different time points, DNA extracts were processed, and genome copies were measured by QPCR using orf26-specific primers. As shown in Fig. 2A, in the absence of doxycycline, we observed a gradual increase in viral genome copies, with a 10-fold increase at 72 h post-TPA/BAc stimulation. On the other hand, when doxycycline was present, resulting in K-bZIP suppression, no significant increases in KSHV episomal copies were detected for any of the time points studied (Fig. 2A). Similarly, in BC3-shRNA-ctl cells, a 30-fold increase in viral genome copies was detected at 72 h post-TPA stimulation (Fig. 2B). In comparison, we noticed an 80% reduction in the number of viral genome copies in BC3 cells expressing shRNA-K8. Our data suggest that K-bZIP is necessary for proper DNA replication to occur.
FIG. 2.
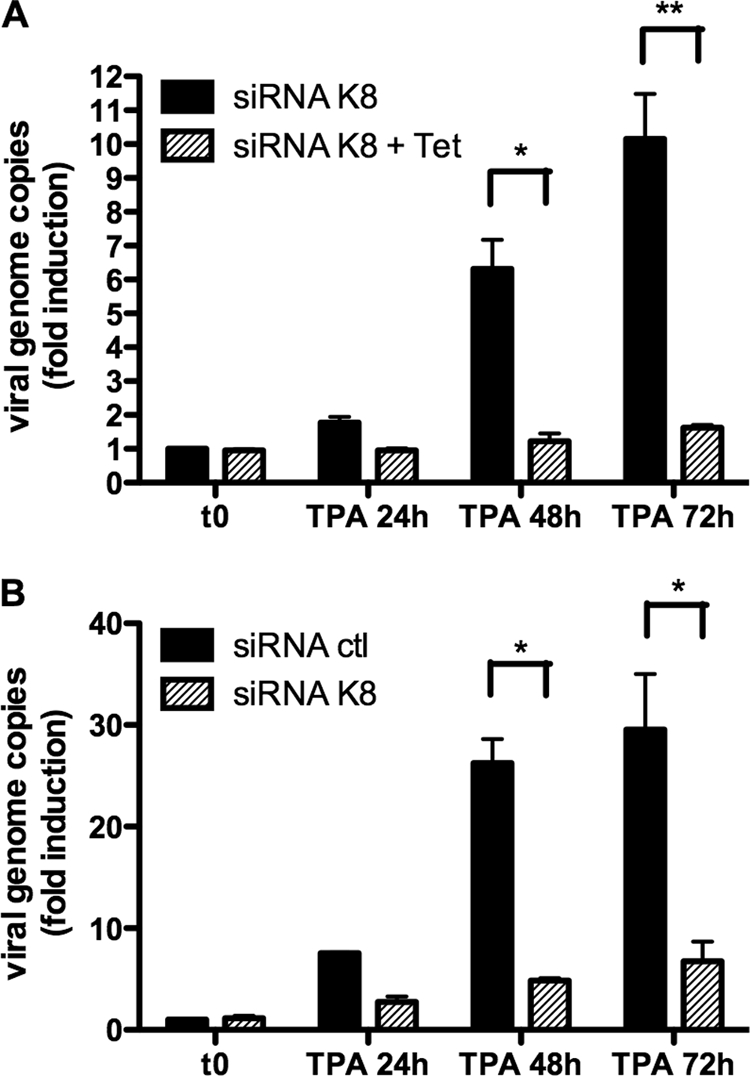
K-bZIP expression is required for viral replication in BC3 and BCBL-1 cells. (A) TREx BCBL-1-shRNA-K8 cells were stimulated with or without 1 μg/ml doxycycline for 72 h in the presence of TPA (20 ng/ml) and BAc (0.3 mM). Cells were harvested at 0, 24, 48, and 72 h, followed by DNA extraction, and the number of viral genome copies was determined by QPCR using orf26-specific primers. (B) BC3-shRNA-K8 or BC3-shRNA-ctl cells were stimulated with TPA (20 ng/ml) and BAc (0.3 mM). Cells were harvested at 0, 24, 48, and 72 h, followed by DNA extraction, and the number of viral genome copies was determined by QPCR using orf26-specific primers. Results are expressed as mean (triplicate) inductions (n-fold) ± standard deviations (SD) relative to nonstimulated cells after normalization of samples with GAPDH expression. Results are representative of three independent experiments. *, P < 0.05; **, P < 0.01.
We next tested the impact of K-bZIP knockdown on virion production. TREx BCBL-1-sh-RNA-K8 cells were stimulated with TPA-BAc and with or without 1 μg/ml doxycycline for 72 h. Supernatants were harvested at different time points and treated with DNase to eliminate naked (unencapsidated) viral DNA. After DNase inactivation, supernatants were processed for DNA extraction and genome copies were measured by QPCR. In the absence of doxycycline, DNase-resistant viral DNA was detected in supernatants, with maximum detection observed at 48 h post-TPA/BAc treatment, indicating proper reactivation of the lytic cycle (Fig. 3A). This pattern was completely different in the absence of K-bZIP expression (with doxycycline), where a 90% inhibition in viral genome copy number was noted at 48 h post-TPA/BAc stimulation (Fig. 3A). To confirm this decrease in virion production, we repeated this experiment in BC3-containing shRNA cells. BC3-shRNA-K8 or BC3-shRNA-ctl cells were stimulated with TPA-BAc for 96 h, and viral output was quantified in supernatants. The result showed that for each time point studied, a >65% reduction in the number of viral genome copies was observed in BC3-shRNA-K8 cells compared to BC3-shRNA-ctl cells (Fig. 3B). Thus, our results indicated that K-bZIP is important for the DNA replication process and for virion production in BCBL-1 and BC3 cells.
FIG. 3.
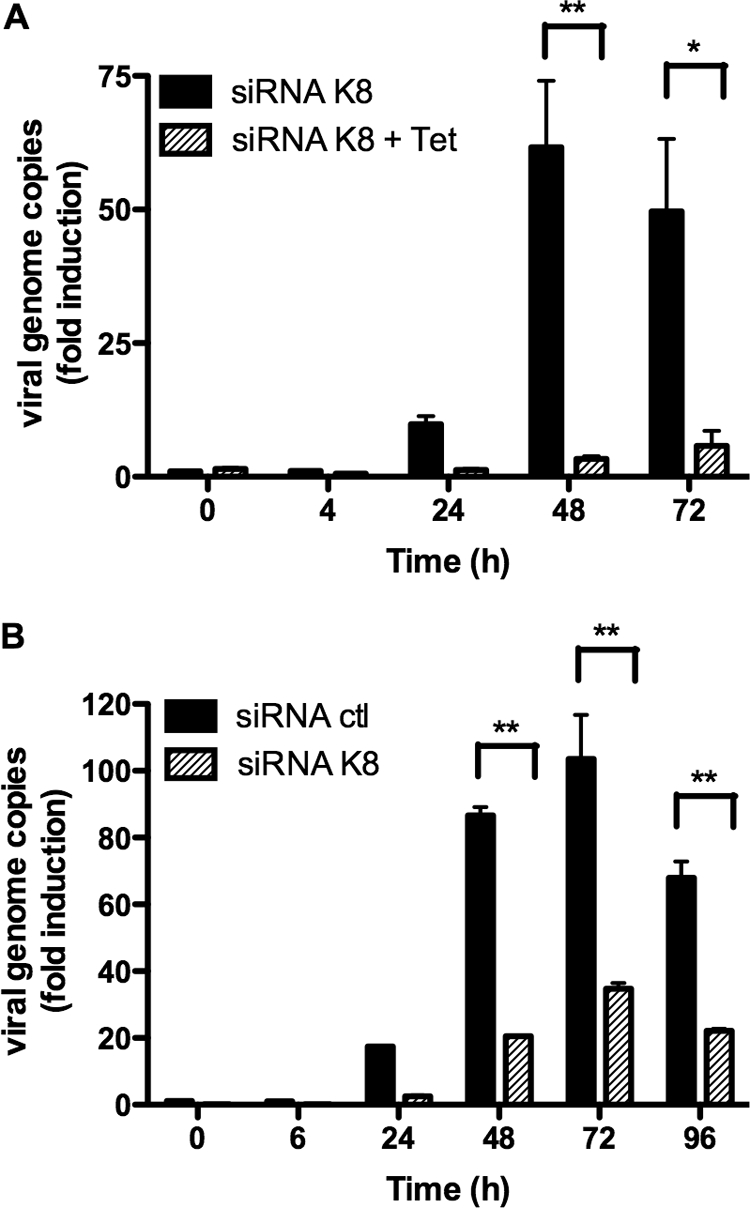
K-bZIP expression is required for virus production in BC3 and BCBL-1 cells. (A) TREx BCBL-1-shRNA-K8 cells were stimulated with or without 1 μg/ml doxycycline for 72 h in the presence of TPA (20 ng/ml) and BAc (0.3 mM). Cells were harvested at 0, 4, 24, 48, and 72 h, followed by DNA extraction from virions in the supernatants after DNase treatment. Viral genome copies were measured by QPCR using orf26-specific primers. (B) BC3-shRNA-K8 or BC3-shRNA-ctl cells were stimulated with TPA (20 ng/ml) and BAc (0.3 mM). Cells were harvested at 0, 6, 24, 48, 72, and 96 h, followed by DNA extraction from virions in the supernatants after DNase treatment. Viral genome copies were measured by QPCR using orf26-specific primers. Results are expressed as mean (triplicate) inductions (n-fold) ± SD relative to nonstimulated cells after normalization of samples with GAPDH expression. Results are representative of three independent experiments. *, P < 0.05; **, P < 0.01.
Previous studies using transient assays have shown that K-bZIP is a transcriptional repressor that affects RTA-induced transcription of a subset of viral genes, such as orf57 and orfk8 itself (12, 16). To determine whether this role of K-bZIP could be observed under more physiological conditions, we tested the effects of K-bZIP knockdown on the expression of various viral mRNAs in PEL cell lines. TREx BCBL-1-shRNA-K8 cells were stimulated with TPA-BAc, with or without 1 μg/ml doxycycline, for 72 h. In the same way, BC3-shRNA-K8 or BC3-shRNA-ctl cells were stimulated with TPA-BAc. For each time point, total RNA was isolated and processed for orfk8, orf50, orf57, and orf26 RT-QPCR. As shown in Fig. 4A, we first confirmed the efficiency of our shRNA-K8 at inhibiting orfk8 mRNA in BC3 cells (left panel) and TREx BCBL-1 cells (right panel) for each time point. Second, considering that we observed less viral DNA replication and virion production in B cells containing shRNA-K8, we measured the expression of the orf50 mRNA, encoding RTA. A time-dependent increase in orf50 mRNA was observed in TREx BCBL-1-shRNA-ctl cells and BC3-shRNA-ctl cells, confirming the efficiency of TPA stimulation. For all time points tested, we typically observed 90 to 95% reductions in orf50 mRNA expression in BC3 (Fig. 4B, left panel) and TREx BCBL-1 (Fig. 4B, right panel) cells. We also evaluated the mRNA levels of orf57 in TPA-BAc-stimulated B cells containing shRNA-ctl or shRNA-K8. In BC3-shRNA-ctl cells, orf57 expression increased up to 70-fold at 6 h poststimulation and stayed at high levels until 72 h post-TPA/BAc induction (Fig. 4C, left panel). In TREx BCBL-1-shRNA-ctl cells, the accumulation of orf57 mRNA reached a maximum at 48 h, with 1,000-fold induction (Fig. 4C, right panel). In both BC3-shRNA-K8 and BCBL-1-shRNA-K8 cells, we observed an almost complete shutdown of orf57 mRNA expression (Fig. 4C). The mRNA levels of the late gene orf26 were also determined. In BC3 and TREx BCBL-1 cells, orf26 mRNA expression was maximal at 24 h post-TPA stimulation (Fig. 4D). The induction of orf26 mRNA was much less in B cells containing shRNA-K8, with greatest effects observed in TREx BCBL-1-shRNA-K8 cells (Fig. 4D, right panel). Significant inhibition was also observed for other lytic transcripts, such as vIL6 and vGPCR mRNAs, but not for latent transcripts, including LANA and vCyclin mRNAs (data not shown). Finally, we investigated the expression of orfk8, orf50, and orf57 mRNAs at earlier time points post-TPA/BAc stimulation. As shown in Fig. 4E, we observed an induction of orfk8 (Fig. 4E) and orf50 (Fig. 4F), as early as 2 h poststimulation and for up to 8 h, in TREx BCBL-1-shRNA-K8 cells. Similar patterns for orf57 mRNA were observed, with increases starting at 4 h poststimulation (Fig. 4G). In the absence of K-bZIP, no orf57 mRNA induction was noted, and a significant reduction in orf50 mRNA was observed at 8 h poststimulation (Fig. 4F). Overall, these data suggest that the expression of immediate-early, early, and late lytic genes, but not latent genes, is greatly affected when K-bZIP is absent.
FIG. 4.

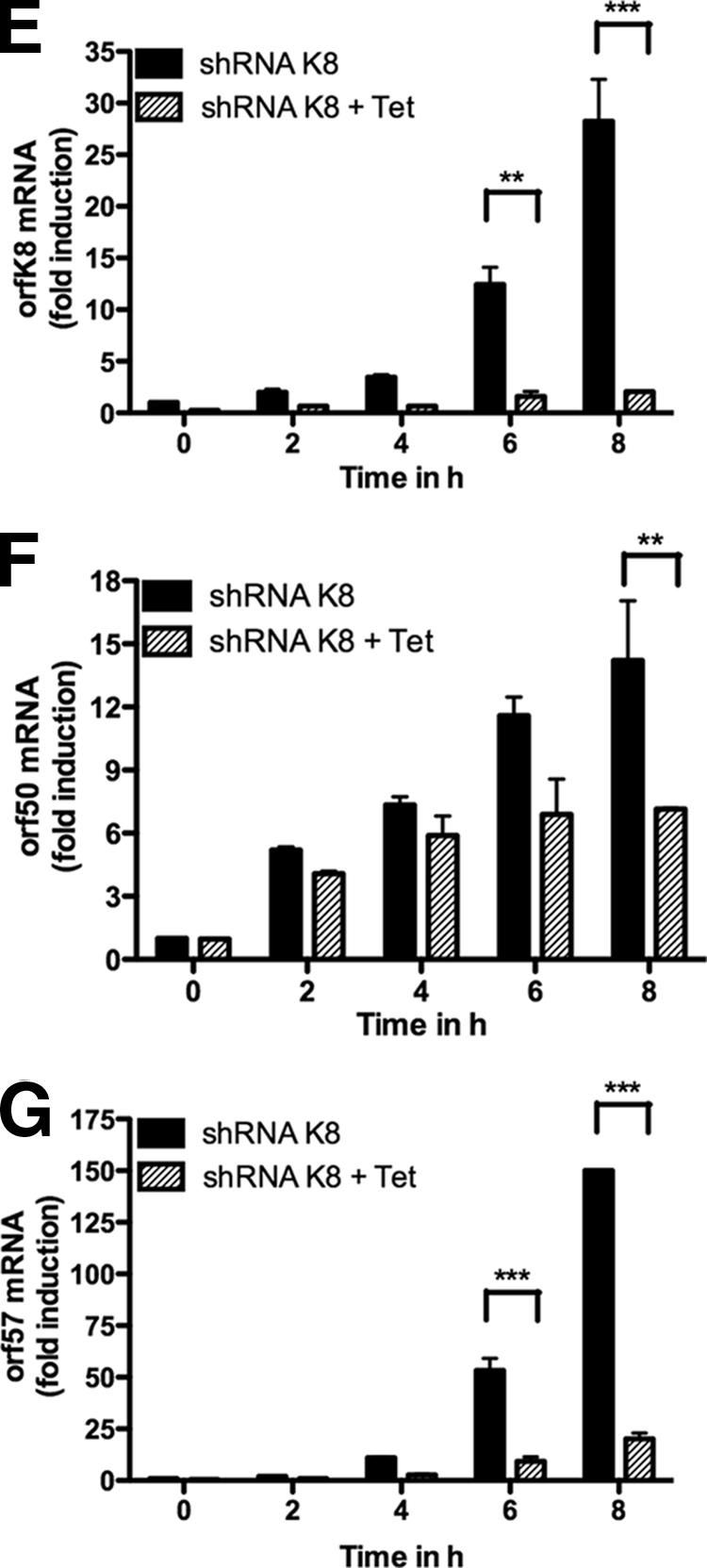
BC3 and BCBL-1 cell transactivation of mRNAs is inhibited in the absence of K-bZIP. TREx BCBL-1-shRNA-K8 cells were stimulated with or without 1 μg/ml doxycycline for 72 h in the presence of TPA-BAc. Cells were harvested at 0, 4, 24, 48, and 72 h. BC3-shRNA-K8 or BC3-shRNA-ctl cells were stimulated with TPA-BAc. Cells were harvested at 0, 6, 24, 48, and 72 h. For each time point, total RNA was isolated and processed for ORFK8 (A), ORF50 (B), ORF57 (C), and ORF26 (D) by RT-QPCR. (E to G) TREx BCBL-1-shRNA-K8 cells were stimulated with or without 1 μg/ml doxycycline for 24 h in the presence of TPA (20 ng/ml) and BAc (0.3 mM). Cells were harvested at 0, 2, 4, 6, and 8 h. For each time point, total RNA was isolated and processed for ORFK8 (E), ORF50 (F), and ORF57 (G) RT-QPCR. Results are expressed as mean (triplicate) inductions (n-fold) ± SD relative to nonstimulated cells after normalization of samples with GAPDH expression. Results are representative of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To confirm that the overall decrease in viral mRNAs is reflected at the protein level, TREx BCBL-1-shRNA-K8 cells were stimulated with TPA-BAc and with or without doxycycline for 48 h, followed by Western blot analyses. As presented in Fig. 5A, in the absence of doxycycline, we observed an increase of K-bZIP expression, with highest expression detected at 24 h, with no significant effect of shRNA-ctl (Fig. 5A). TREx BCBL-1-shRNA-K8 cells stimulated overnight with doxycycline before TPA/BAc induction showed dramatic reductions in K-bZIP expression. As a consequence of K-bZIP's knockdown, RTA was not detected at 8, 24, or 48 h poststimulation in BCBL-1-shRNA-K8 cells (Fig. 5A). No differences were noted in LANA expression. No detectable vCyclin was observed in TREx BCBL-1 cells (not shown). To further confirm these results, we repeated the same experiment with BC3-shRNA-ctl or BC3-shRNA-K8 cells stimulated with TPA-BAc for 48 h. As we observed a time-dependent increase of K-bZIP in BC3-shRNA-ctl cells, a concomitant augmentation of RTA was noticed (Fig. 5B). This was not the case in BC3-shRNA-K8 cells, where RTA levels remained very low to undetectable. No significant differences were seen for LANA and vCyclin expression in BC3-shRNA-ctl and BC3-shRNA-K8 cells. Following these results, we performed an immunofluorescence assay on TREx BCBL-1-shRNA-K8 or TREx BCBL-1-shRNA-ctl cells stimulated with TPA-BAc and treated with or without doxycycline for 24 h. First, the absence of nuclear K-bZIP upon doxycycline induction was confirmed at 2 and 24 h in BCBL-1-shRNA-K8 cells (Fig. 5C). Next, in TREx BCBL-1-shRNA-ctl cells, the presence of the cytoplasmic late protein K8.1 was observed at 24 h post-TPA stimulation. On the other hand, in TREx BCBL-1-shRNA-K8 cells stimulated with doxycycline, we could not detect K8.1 at 24 h poststimulation (Fig. 5D). In light of these results, it appears that K-bZIP is required for the proper expression of early and late proteins, but not latent proteins, in PEL cell lines. To our knowledge, K-bZIP has not been shown as an activator of viral transcription yet. To study this, 293T cells were cotransfected with luciferase reporters driven by the orf50 or orf57 promoter, together with increasing doses of pCMV3T-K8 vector. Dose-dependent increases in orf50 and orf57 promoter activation were observed following K-bZIP expression (see Fig. S1 in the supplemental material). These results argue that K-bZIP can indeed behave as a transactivator of gene expression.
FIG. 5.

K-bZIP expression is necessary for lytic immediate-early and late protein expression in BC3 and BCBL-1 cells. (A) TREx BCBL-1-shRNA-K8 cells were stimulated with or without 1 μg/ml doxycycline for 48 h in the presence of TPA (20 ng/ml) and BAc (0.3 mM). Cells were harvested at 0, 8, 24, and 48 h. (B) BC3-shRNA-K8 or BC3-shRNA-ctl cells were stimulated with TPA (20 ng/ml) and BAc (0.3 mM). Cells were harvested at 0, 8, 24, and 48 h. (A and B) For each time point, the cultures were harvested and total cell extracts were made and analyzed by Western blotting, using specific anti-K-bZIP, anti-RTA, anti-LANA, anti-cyclin, and anti-actin antibodies. (C and D) Immunofluorescence assay on TREx BCBL-1-shRNA-K8 or TREx BCBL-1-shRNA-ctl cells, stimulated with or without 1 μg/ml doxycycline for 24 h in the presence of TPA (20 ng/ml) and BAc (0.3 mM) for 2 or 24 h. Acetone-fixed cells were incubated with rabbit anti-K-bZIP (C) or mouse anti-K8.1 (D), followed by incubation with Alexa 588-conjugated secondary antibodies (green). Nuclei were stained with DAPI (blue). The localization of the proteins relative to the nucleus is presented in the merged pictures.
Given that in the absence of K-bZIP neither viral mRNA transcripts nor viral lytic proteins were expressed at their normal levels, we next determined whether the kinetics of K-bZIP knockdown relative to TPA stimulation is important. To answer this question, the expression of the K8 shRNA was turned on at different time points (−16 h, 0 h, 24 h, and 48 h) relative to TPA/BAc stimulation. Little or no K8 mRNA induction was noted when doxycycline was added before (−16 h) or simultaneously with (0 h) TPA-BAc (Fig. 6A). Similar effects were detected with orf26 mRNA (Fig. 6B), suggesting that K-bZIP is essential during the early reactivation phase. When cells were first treated with TPA-BAc and allowed to express K-bZIP at its normal level for 24 h and 48 h before its expression was knocked down, half of the orf26 mRNA level was recovered, suggesting that once the cycle has been initiated properly, the requirement for K-bZIP is minimal. Next, to determine whether the amount of K-bZIP was important for proper viral reactivation, TREx BCBL-1-shRNA-K8 cells were stimulated with TPA-BAc together with different doses of doxycycline to induce partial suppression of K-bZIP expression (Fig. 6C). Our results indicate that the lower the level of K8 mRNA, the lower the level of orf26 mRNA (Fig. 6D). These results suggest that a direct correlation exists between the levels of K-bZIP and the efficiency at which the lytic cycle is initiated.
FIG. 6.

Dose-dependent transactivation of viral mRNAs in the presence of K-bZIP. (A and B). TREx BCBL-1-shRNA-K8 cells were stimulated with TPA (20 ng/ml) and BAc (0.3 mM), with or without 1 μg/ml doxycycline, 16 h before, simultaneously with, or 24 h or 48 h after TPA-BAc stimulation. Cells were harvested at 72 h, and for each time point, total RNA was isolated and processed for ORFK8 (A) and ORF26 (B) by RT-QPCR. (C and D) TREx BCBL-1-shRNA-K8 cells were stimulated with TPA-BAc, with or without 5, 50, or 500 pg/ml or 2 ng/ml of doxycycline. Cells were harvested at 24 h, and for each time point, total RNA was isolated and processed for ORFK8 (C) and ORF26 (D) by RT-QPCR. Results are expressed as mean (triplicate) inductions (n-fold) ± SD relative to nonstimulated cells after normalization of samples with GAPDH expression. Results are representative of three independent experiments. *, P < 0.05; **, P < 0.01.
To confirm that the effects we observed are specific to the inhibition of K-bZIP expression, we generated a K8 expression vector that is resistant to inhibition by shRNA-K8 with hopes of rescuing the KSHV lytic cycle. First, 293T cells were transfected with a K8 expression vector or a K8 expression vector resistant to shRNA-K8 in the presence of increasing doses of pTER expressing shRNA-K8. As shown in Fig. 7A, transfection of shRNA-K8 led to the inhibition of K-bZIP expression. On the other hand, we did not observe any differences in K-bZIP expression in the presence of the K8 expression vector resistant to shRNA against orfk8. We then electroporated TREx BCBL-1-sh-RNA-K8 cells with the K8 expression vector resistant to shRNA-K8 or with pCMV3T, as the control plasmid. After 6 h, cells were stimulated with TPA-BAc and with or without 1 μg/ml doxycycline for 48 h, and total RNA was isolated and processed for orf50 and orf26 mRNAs by RT-QPCR (Fig. 7B). We typically observed 80% and 90% inhibition of orf50 and orf26 mRNAs, respectively, in TREx BCBL-1-sh-RNA-K8 cells transfected with empty pCMV3T vector in the presence of doxycycline (Fig. 7B). However, when TREx BCBL-1-sh-RNA-K8 cells were transfected with the K8 expression vector resistant to shRNA-K8, a twofold increase in orf50 and orf26 mRNAs was observed. Considering that the electroporation efficiency of BCBL-1 cells was 15 to 20% (as determined using a GFP expression vector and FACS analysis), this increase is significant. Our ability to rescue, at least partly, the KSHV replicative cycle through expression of a shRNA-resistant K8 indicates that K-bZIP is essential for viral growth and argues against an off-target effect of shRNA-K8.
FIG. 7.

Overexpression of a K8 expression plasmid resistant to shRNA-K8 is able to reverse the inhibition of viral orf50 and orf26 mRNAs. (A) 293T cells were transfected with K8 or shRNA-K8-resistant expression vector and increasing amounts of pTER-shRNA-K8 or empty vector. Thirty-six hours later, the cultures were harvested and total cell extracts were analyzed by Western blotting, using specific anti-K-bZIP and anti-actin antibodies. (B) TREx BCBL-1-shRNA-K8 cells were electroporated with shRNA-K8-resistant expression vector (K8shRNA Mut) or control vector and then stimulated with or without 1 μg/ml doxycycline for 48 h in the presence of TPA-BAc. Total RNA was isolated and processed for ORF50 and ORF26 by RT-QPCR. Results are representative of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Considering the importance of K-bZIP for viral gene expression, DNA replication, and virion production in PEL cells, we next asked the importance of K-bZIP in the establishment of the infection. In order to evaluate the impact of K-bZIP on the infection, we generated a TREx 293-shRNA model inducible for the expression of shRNA-K8. We first transfected TREx 293-shRNA-ctl or TREx 293-shRNA-K8 cells with a K8 expression vector, with or without doxycycline. Induction of shRNA with doxycycline led to a significant inhibition of the K-bZIP protein (see Fig. S2a in the supplemental material). Activation of shRNA-ctl had no effect on K-bZIP expression. We next carried out the infection of TREx 293-shRNA-ctl or TREx 293-shRNA-K8 cells with rKSHV.219 virus in the presence or absence of doxycycline. As shown in Fig. 8A and B, a dose-dependent infection of both 293-shRNA cell lines was observed, as determined by GFP expression. The addition of doxycycline had no impact on the infection levels, reflecting the fact that K-bZIP expression is dispensable for the initial phase of infection and establishment of latency. As reported previously, the rKSHV.219 virus can be reactivated with BAc (34). Tet-treated TREx 293-shRNA-ctl and TREx 293-shRNA-K8 cells infected with rKSHV.219 were stimulated with BAc for 4 days to induce reactivation. The number of cells displaying reactivated KSHV, as determined by dsRed expression (under the control of the PAN promoter) (Fig. 8B), and expression of orfk8, orf26, and orf57 mRNAs were determined (Fig. 8C). FACS analysis revealed no differences in the number of dsRed-positive cells between TREx 293-shRNA-ctl and TREx 293-shRNA-K8 cells (Fig. 8B). In the presence of BAc and the absence of doxycycline, orfk8 mRNA was increased up to fourfold in both TREx 293-shRNA-ctl and TREx 293-shRNA-K8 cells (Fig. 8C). Upon addition of doxycycline and activation of shRNA-K8, orfk8 mRNA levels dropped significantly, to near basal levels. In these cells, however, no significant differences were noticed for the expression of orf57 and orf26 mRNAs in both cell lines. A potential effect of K-bZIP on PAN promoter expression was excluded considering the report of Izumiya et al. (12) indicating that K-bZIP does not have any effect on this promoter. Overall, these results indicated that K-bZIP does not affect the initial phases of infection and establishment of latency. Furthermore, unlike what we observed in PEL cell lines, K-bZIP knockdown in 293 cells had marginal effects, at best, on the transcription of viral mRNAs associated with the KSHV lytic cycle, highlighting important differences between both models.
FIG. 8.

KSHV infection of 293 cells is independent of the presence of K-bZIP. (A) TREx 293-shRNA-ctl and TREx 293-shRNA-K8 cells were infected with different doses of rKSHV.219 virus, with or without 1 μg/ml doxycycline, for 96 h. Infected 293 cells were analyzed for GFP expression by observation under a fluorescence microscope. (B) TREx 293-shRNA-ctl and TREx 293-shRNA-K8 cells were infected with rKSHV.219 virus, with or without 1 μg/ml doxycycline, for 96 h in the presence or absence of 5 mM BAc. FACS analysis was then performed to measure GFP- and dsRed-positive cells. (C) TREx 293-shRNA-ctl and TREx 293-shRNA-K8 cells were infected with rKSHV.219 virus, with or without 1 μg/ml doxycycline, for 96 h in the presence or absence of 5 mM BAc. Cells were harvested, and total RNA was isolated and processed for orfk8 (top), orf26 (middle), and orf57 (bottom) mRNAs by RT-QPCR.
DISCUSSION
K-bZIP, the KSHV K8 gene product, is the positional and structural analogue of EBV Zta (18). Like Zta, K-bZIP is required for ori-Lyt-dependent DNA replication (2). Indeed, the prereplication complexes are recruited to ori-Lyt DNA through RTA, which interacts with RTA-responsive elements, as well as K-bZIP, which binds to a cluster of C/EBP binding motifs with the aid of C/EBPα (35). Izumiya et al. showed that BCBL-1 cells overexpressing K-bZIP harbored less viral DNA than control cells did under conditions where viral reactivation was favored (12). On the other hand, a recent study using a bacmid deletant for orfk8 indicated that virus replication in the absence of K-bZIP could be rescued when cells were overexpressing very large amounts of RTA (13). This study characterized the effects of orfk8 deletion in monkey Vero cells. In order to determine the roles of K-bZIP for KSHV in PEL cells, we generated a doxycycline-inducible shRNA targeting orfk8 in BCBL-1 cells. Since K-bZIP is known to play a role in DNA replication, we first studied the kinetics of episomal copy accumulation following TPA/BAc treatment. In TREx BCBL-1-shRNA-K8 cells, no significant increase in genome copies was detectable when K-bZIP was knocked down, in contrast to a 10-fold increase in copy number in control cells. Similar results were obtained using BC3 PEL cells expressing stable shRNA-K8. Hence, K-bZIP plays an essential role in virus DNA replication and virus production in PEL cells. Similar data were obtained by Kato-Noah et al., who showed that a recombinant KSHV, BAC36ΔK8, failed to replicate and produce infectious virions following TPA/BAc stimulation (13). However, using a combination of RTA overexpression plus TPA stimulation, K-bZIP was found to be dispensable for the replication process in this bacmid system (13). In PEL cells, TPA/BAc stimulation is generally effective at inducing levels of RTA expression that are sufficient for the launching of the KSHV lytic cycle (27). This is supported by our data using BCBL-1 and BC3 cells, where RTA induction and virion production were obtained following TPA/BAc treatment. In this system, in the absence of K-bZIP expression, TPA/BAc stimulation proved insufficient to allow the KSHV cycle to proceed in PEL cells. One hypothesis that could reconcile our results and those of Kato-Noah et al. (13) is that the adenoviral vector used produces more RTA than our TPA/BAc stimulation, which can override the need for K-bZIP. Further work is needed to determine whether a threshold level of RTA is required to bypass the need for K-bZIP or whether these differences are attributable to the various experimental models.
A role as a repressor of viral and cellular genes has been attributed to K-bZIP. For example, K-bZIP has been shown to inhibit transforming growth factor beta signaling pathway and beta interferon gene transcription (15, 32). Moreover, K-bZIP represses RTA-mediated transactivation of RTA and MTA, but not the PAN promoter, in transient assays (12, 16). In our TREx BCBL-1-shRNA model, the absence of K-bZIP is associated with much reduced expression of lytic (orf50, orf57, and orf26) genes. Interestingly, at 2 h and 4 h post-TPA/BAc stimulation, the induction levels of orf50 were similar in control and K-bZIP knockdown cells, suggesting that initial gene activation occurred normally in the absence of K-bZIP. At all other time points tested, reduced transcriptional activities were noted. Under conditions where lytic replication was induced using TPA/BAc treatment, the study using BACΔK8 showed identical inhibitions of early orf57 and late K8.1 mRNAs (13). These mRNA inhibitions were confirmed at the protein level. In contrast, K-bZIP does not seem to have any effects on the expression of latent mRNAs or proteins. Hence, K-bZIP's effects might occur predominantly on the expression of lytic cycle genes. Moreover, we observed that BC3 shRNA-K8 cells cultured for long periods of times (>4 weeks) had similar proliferation kinetics and viral episome copy numbers to control cells, indicating that K-bZIP does not play a role in viral episomal maintenance. Further analysis revealed that the amount of K-bZIP is very important for its impact on mRNA transactivation. Indeed, using various doses of doxycycline to obtain various levels of K-bZIP suppression, we observed a direct correlation between the amount of K-bZIP present and the activation of orf57 and orf26 mRNAs. At the highest dose of doxycycline used, K-bZIP shutdown was nearly complete, and as a consequence, 90% of orf26 mRNA induction was inhibited. On the other hand, with limited suppression of K-bZIP expression (50%), expression of lytic transcripts was reduced only 50%. In addition, we found that overexpression of a K8 expression vector resistant to shRNA-K8 was able to partially restore (due to the transfection efficiency) the inhibition seen in our model. In our PEL cell model, K-bZIP behaves as a positive regulator of the viral cycle rather than as a repressor. In fact, under no circumstances have we observed a negative (repressor) effect of K-bZIP on the KSHV life cycle. Furthermore, in transient transfection assays with a luciferase reporter under the control of various promoters, including the orf50 or orf57 promoter or a simple TATA box, K-bZIP behaved as a weak transactivator. Our results suggest that K-bZIP behaves more like an amplifier that facilitates/coordinates gene expression. In its absence, limited RTA transcription takes place but cannot be sustained over time, resulting in impaired progression to the later phases of the lytic cycle.
Bechtel et al. showed that the K-bZIP protein is encapsidated in the virions, suggesting that K-bZIP could be important for initial and late stages of KSHV infection (3). Primary infection is also known to start with a lytic burst before the establishment of latency (14). In order to study the impact of K-bZIP on primary infection, we used a TREx 293-shRNA-K8 model where shRNA-K8 expression is inducible. Infection experiments with rKSHV.219 have shown no significant difference between TREx 293-shRNA-K8 and TREx 293-shRNA-ctl cells for the establishment of latency. Similarly, the presence of K-bZIP seems not to be essential for reactivation in these TREx 293-shRNA cells. One possible explanation accounting for the difference between 293 cells and PEL cells is that the K-bZIP role in mRNA transactivation seen in B cells is cell type specific. Alternatively, the fact that infecting virions bring along K-bZIP, and thus bypass the inhibitory action of the shRNA, which acts at the level of mRNA, might result in sufficient levels of K-bZIP inside the cells to allow the cycle to proceed. One other important difference we noted between PEL cells and KSHV-infected 293 cells is the efficiency at which TPA-BAc induces reactivation. We detected much higher inductions (10- to 100-fold) in our TREx B-cell model than in the TREx 293 cell model. Unfortunately, we were not able to show greater reactivation with different BAc stimulations or with the use of BacK50 (data not shown).
To date, our report is the first to describe the roles of K-bZIP in PEL cells in the context of the entire viral genome and without viral protein overexpression. Our data confirmed the roles of K-bZIP in the DNA replication machinery and in virus production. In addition, and in contrast to previous studies, our results indicate that K-bZIP behaves as a facilitator of viral mRNA transcription, as no viral lytic genes are expressed when K-bZIP is knocked down in PEL cells. Moreover, de novo synthesis of K-bZIP is dispensable for the establishment of infection in 293 cells. Overall, this work provides a better understanding and highlights new functions and roles of K-bZIP in PEL cells, as well as pinpointing important differences depending on the experimental model used.
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Cancer Institute of Canada and by a senior scholarship from the Fonds de la Recherche en Santé du Québec, awarded to Louis Flamand.
Footnotes
Published ahead of print on 25 March 2009.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Arvanitakis, L., E. A. Mesri, R. G. Nador, J. W. Said, A. S. Asch, D. M. Knowles, and E. Cesarman. 1996. Establishment and characterization of a primary effusion (body cavity-based) lymphoma cell line (BC3) harboring Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) in the absence of Epstein-Barr virus. Blood 882648-2654. [PubMed] [Google Scholar]
- 2.AuCoin, D. P., K. S. Colletti, S. A. Cei, I. Papouskova, M. Tarrant, and G. S. Pari. 2004. Amplification of the Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 lytic origin of DNA replication is dependent upon a cis-acting AT-rich region and an ORF50 response element and the trans-acting factors ORF50 (K-Rta) and K8 (K-bZIP). Virology 318542-555. [DOI] [PubMed] [Google Scholar]
- 3.Bechtel, J. T., R. C. Winant, and D. Ganem. 2005. Host and viral proteins in the virion of Kaposi's sarcoma-associated herpesvirus. J. Virol. 794952-4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cesarman, E., Y. Chang, P. S. Moore, J. W. Said, and D. M. Knowles. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 3321186-1191. [DOI] [PubMed] [Google Scholar]
- 5.Chang, P. J., and G. Miller. 2004. Autoregulation of DNA binding and protein stability of Kaposi's sarcoma-associated herpesvirus ORF50 protein. J. Virol. 7810657-10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang, Y., E. Cesarman, M. S. Pessin, F. Lee, J. Culpepper, D. M. Knowles, and P. S. Moore. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 2661865-1869. [DOI] [PubMed] [Google Scholar]
- 7.Dupin, N., C. Fisher, P. Kellam, S. Ariad, M. Tulliez, N. Franck, E. van Marck, D. Salmon, I. Gorin, J. P. Escande, R. A. Weiss, K. Alitalo, and C. Boshoff. 1999. Distribution of human herpesvirus-8 latently infected cells in Kaposi's sarcoma, multicentric Castleman's disease, and primary effusion lymphoma. Proc. Natl. Acad. Sci. USA 964546-4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fixman, E. D., G. S. Hayward, and S. D. Hayward. 1995. Replication of Epstein-Barr virus oriLyt: lack of a dedicated virally encoded origin-binding protein and dependence on Zta in cotransfection assays. J. Virol. 692998-3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gradoville, L., J. Gerlach, E. Grogan, D. Shedd, S. Nikiforow, C. Metroka, and G. Miller. 2000. Kaposi's sarcoma-associated herpesvirus open reading frame 50/Rta protein activates the entire viral lytic cycle in the HH-B2 primary effusion lymphoma cell line. J. Virol. 746207-6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hwang, S., Y. Gwack, H. Byun, C. Lim, and J. Choe. 2001. The Kaposi's sarcoma-associated herpesvirus K8 protein interacts with CREB-binding protein (CBP) and represses CBP-mediated transcription. J. Virol. 759509-9516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Izumiya, Y., T. J. Ellison, E. T. Yeh, J. U. Jung, P. A. Luciw, and H. J. Kung. 2005. Kaposi's sarcoma-associated herpesvirus K-bZIP represses gene transcription via SUMO modification. J. Virol. 799912-9925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Izumiya, Y., S. F. Lin, T. Ellison, L. Y. Chen, C. Izumiya, P. Luciw, and H. J. Kung. 2003. Kaposi's sarcoma-associated herpesvirus K-bZIP is a coregulator of K-Rta: physical association and promoter-dependent transcriptional repression. J. Virol. 771441-1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kato-Noah, T., Y. Xu, C. C. Rossetto, K. Colletti, I. Papouskova, and G. S. Pari. 2007. Overexpression of the Kaposi's sarcoma-associated herpesvirus transactivator K-Rta can complement a K-bZIP deletion bacmid and yields an enhanced growth phenotype. J. Virol. 8113519-13532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krishnan, H. H., P. P. Naranatt, M. S. Smith, L. Zeng, C. Bloomer, and B. Chandran. 2004. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi's sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J. Virol. 783601-3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lefort, S., A. Soucy-Faulkner, N. Grandvaux, and L. Flamand. 2007. Binding of Kaposi's sarcoma-associated herpesvirus K-bZIP to interferon-responsive factor 3 elements modulates antiviral gene expression. J. Virol. 8110950-10960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liao, W., Y. Tang, S. F. Lin, H. J. Kung, and C. Z. Giam. 2003. K-bZIP of Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 (KSHV/HHV-8) binds KSHV/HHV-8 Rta and represses Rta-mediated transactivation. J. Virol. 773809-3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lieberman, P. M., J. M. Hardwick, J. Sample, G. S. Hayward, and S. D. Hayward. 1990. The Zta transactivator involved in induction of lytic cycle gene expression in Epstein-Barr virus-infected lymphocytes binds to both AP-1 and ZRE sites in target promoter and enhancer regions. J. Virol. 641143-1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin, S. F., D. R. Robinson, G. Miller, and H. J. Kung. 1999. Kaposi's sarcoma-associated herpesvirus encodes a bZIP protein with homology to BZLF1 of Epstein-Barr virus. J. Virol. 731909-1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lukac, D. M., L. Garibyan, J. R. Kirshner, D. Palmeri, and D. Ganem. 2001. DNA binding by Kaposi's sarcoma-associated herpesvirus lytic switch protein is necessary for transcriptional activation of two viral delayed early promoters. J. Virol. 756786-6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lukac, D. M., J. R. Kirshner, and D. Ganem. 1999. Transcriptional activation by the product of open reading frame 50 of Kaposi's sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J. Virol. 739348-9361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller, G., L. Heston, E. Grogan, L. Gradoville, M. Rigsby, R. Sun, D. Shedd, V. M. Kushnaryov, S. Grossberg, and Y. Chang. 1997. Selective switch between latency and lytic replication of Kaposi's sarcoma herpesvirus and Epstein-Barr virus in dually infected body cavity lymphoma cells. J. Virol. 71314-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller, G., M. O. Rigsby, L. Heston, E. Grogan, R. Sun, C. Metroka, J. A. Levy, S. J. Gao, Y. Chang, and P. Moore. 1996. Antibodies to butyrate-inducible antigens of Kaposi's sarcoma-associated herpesvirus in patients with HIV-1 infection. N. Engl. J. Med. 3341292-1297. [DOI] [PubMed] [Google Scholar]
- 23.Moore, P. S., S. J. Gao, G. Dominguez, E. Cesarman, O. Lungu, D. M. Knowles, R. Garber, P. E. Pellett, D. J. McGeoch, and Y. Chang. 1996. Primary characterization of a herpesvirus agent associated with Kaposi's sarcomae. J. Virol. 70549-558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakamura, H., M. Lu, Y. Gwack, J. Souvlis, S. L. Zeichner, and J. U. Jung. 2003. Global changes in Kaposi's sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J. Virol. 774205-4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park, J., T. Seo, S. Hwang, D. Lee, Y. Gwack, and J. Choe. 2000. The K-bZIP protein from Kaposi's sarcoma-associated herpesvirus interacts with p53 and represses its transcriptional activity. J. Virol. 7411977-11982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Portes-Sentis, S., E. Manet, G. Gourru, A. Sergeant, and H. Gruffat. 2001. Identification of a short amino acid sequence essential for efficient nuclear targeting of the Kaposi's sarcoma-associated herpesvirus/human herpesvirus-8 K8 protein. J. Gen. Virol. 82507-512. [DOI] [PubMed] [Google Scholar]
- 27.Renne, R., W. Zhong, B. Herndier, M. McGrath, N. Abbey, D. Kedes, and D. Ganem. 1996. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 2342-346. [DOI] [PubMed] [Google Scholar]
- 28.Sarisky, R. T., Z. Gao, P. M. Lieberman, E. D. Fixman, G. S. Hayward, and S. D. Hayward. 1996. A replication function associated with the activation domain of the Epstein-Barr virus Zta transactivator. J. Virol. 708340-8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Soulier, J., L. Grollet, E. Oksenhendler, P. Cacoub, D. Cazals-Hatem, P. Babinet, M. F. d'Agay, J. P. Clauvel, M. Raphael, L. Degos, et al. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 861276-1280. [PubMed] [Google Scholar]
- 30.Sun, R., S. F. Lin, L. Gradoville, Y. Yuan, F. Zhu, and G. Miller. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 9510866-10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun, R., S. F. Lin, K. Staskus, L. Gradoville, E. Grogan, A. Haase, and G. Miller. 1999. Kinetics of Kaposi's sarcoma-associated herpesvirus gene expression. J. Virol. 732232-2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tomita, M., J. Choe, T. Tsukazaki, and N. Mori. 2004. The Kaposi's sarcoma-associated herpesvirus K-bZIP protein represses transforming growth factor beta signaling through interaction with CREB-binding protein. Oncogene 238272-8281. [DOI] [PubMed] [Google Scholar]
- 33.van de Wetering, M., I. Oving, V. Muncan, M. T. Pon Fong, H. Brantjes, D. van Leenen, F. C. Holstege, T. R. Brummelkamp, R. Agami, and H. Clevers. 2003. Specific inhibition of gene expression using a stably integrated, inducible small-interfering-RNA vector. EMBO Rep. 4609-615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vieira, J., and P. M. O'Hearn. 2004. Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology 325225-240. [DOI] [PubMed] [Google Scholar]
- 35.Wang, Y., Q. Tang, G. G. Maul, and Y. Yuan. 2006. Kaposi's sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: dual role of replication and transcription activator. J. Virol. 8012171-12186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu, F. Y., J. H. Ahn, D. J. Alcendor, W. J. Jang, J. Xiao, S. D. Hayward, and G. S. Hayward. 2001. Origin-independent assembly of Kaposi's sarcoma-associated herpesvirus DNA replication compartments in transient cotransfection assays and association with the ORF-K8 protein and cellular PML. J. Virol. 751487-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu, Y., D. P. AuCoin, A. R. Huete, S. A. Cei, L. J. Hanson, and G. S. Pari. 2005. A Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 ORF50 deletion mutant is defective for reactivation of latent virus and DNA replication. J. Virol. 793479-3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.