

Abstract
Insulin and insulin-like growth factor I (IGF-I) are ubiquitous hormones that regulate growth and metabolism of most mammalian cells, including pancreatic β-cells. In addition to being an insulin secretagogue, glucose regulates proliferation and survival of β-cells. However, it is unclear whether the latter effects of glucose occur secondary to autocrine activation of insulin signaling proteins by secreted insulin. To examine this possibility we studied the effects of exogenous glucose or insulin in β-cell lines completely lacking either insulin receptors (βIRKO) or insulin receptor substrate 2 (βIRS2KO). Exogenous addition of either insulin or glucose activated proteins in the insulin signaling pathway in control β-cell lines with the effects of insulin peaking earlier than glucose. Insulin stimulation of βIRKO and βIRS2KO cells led to blunted activation of phosphatidylinositol 3-kinase and Akt kinase, while surprisingly, glucose failed to activate either kinase but phosphorylated extracellular signal-regulated kinase. Control β-cells exhibited low expression of IGF-1 receptors compared to compensatory upregulation in βIRKO cells. The signaling data support the slow growth and reduced DNA and protein synthesis in βIRKO and βIRS2KO cells in response to glucose stimulation. Together, these studies provide compelling evidence that the growth and survival effects of glucose on β-cells require activation of proteins in the insulin signaling pathway.
Pancreatic islet β-cell regeneration and function are regulated by multiple stimuli, including nutrients, hormones, and growth factors acting via diverse intracellular signaling pathways (4, 43). Glucose is the primary regulator of insulin secretion and insulin biosynthesis, and its effects on growth and survival have been suggested to occur by activation of insulin receptor substrate 2 (IRS-2), a protein in the insulin/insulin-like growth-factor I (IGF-I) signaling pathway (40, 45). Indeed, over the last decade most components in the insulin signaling pathway have been identified in murine and human pancreatic β-cells (1, 4, 17, 36), and their cross talk with other signaling pathways in β-cells is being systematically unraveled using genetic approaches in mice (reviewed in references 4 and 31). For example, insulin signaling has been reported to regulate many effects in β-cells that are also promoted by glucose, such as enhancing insulin gene expression, insulin secretion, proinsulin biosynthesis, and cell cycle progression (25, 28-30, 38). Considering the similar effects of insulin and glucose in β-cells that occur by activation of largely similar proteins in the insulin/IGF-I signaling pathway, it is unclear whether the effects of glucose require activation of insulin receptors via secreted insulin.
Examination of the independent effects of glucose versus insulin on β-cell function in vivo is limited by a lack of suitable mouse models. Further, the difficulty in separating the downstream effects of exogenous glucose from those of exogenous insulin in cultured β-cells and isolated islets is confounded by the continuous secretion of insulin via the regulated and constitutive pathways (19, 22). This has prompted investigators to study β-cell lines that stably or transiently express small interfering RNA (siRNA) against insulin receptors (9, 38) in an attempt to render them unresponsive to insulin or whole islets treated with siRNA against IRS-2 (41). However, in the former approaches a complete reduction in insulin receptor expression has not been achieved, with the knockdowns ranging from 80 to 90%, which would allow residual receptor activity and potential activation of downstream pathways. In the latter approach, the use of siRNA in whole islets would target all islet cell types, thereby making it difficult to identify the effects in only β-cells. To circumvent these limitations and to directly examine the signaling effects of exogenous glucose in the complete absence of insulin action, we have derived β-cell lines from βIRKO or IRS2KO mice in which the insulin receptor or IRS-2 protein is undetectable and compared them with β-cells expressing all proteins in the insulin/IGF-1 signaling cascade. We report that the growth and survival effects of glucose require the activation of insulin receptors and IRS-2 and implicate a dominant role for insulin in the regulation of β-cell regeneration and function.
MATERIALS AND METHODS
Cells and cell culture.
Insulin-secreting cell lines were established from three independent mice each from groups of βIRKO, IRS-1KO, and IRS-2-KO mice as described previously (29). Clonal and polyclonal cell lines exhibited qualitatively similar signaling properties; however, considering their reported advantages (21) we have used polyclonal cells for all experiments reported in the manuscript. Cells were routinely maintained at 37°C and 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) containing 25 mM glucose, 10% fetal bovine serum, and penicillin and streptomycin. Experiments were performed on 80% confluent cells. Cells were initially starved overnight in DMEM containing 0.1% bovine serum albumin (BSA), 8.3 mM glucose, and penicillin-streptomycin. Subsequently, cells were cultured for 2 h in glucose-free modified KRB buffer (KRBH; 125 mM NaCl, 4.74 mM KCl, 1 mM CaCl2, 1.2 mM KH2PO4, 1.2 mM, MgSO4, 5 mM NaHCO3, 25 mM HEPES [pH 7.4]) with 0.1% BSA to minimize endogenous insulin secretion. The stimulation was performed in the same buffer with glucose and insulin concentrations as indicated. Insulin stimulation was performed in the absence of glucose.
Immunoprecipitation and Western blot analyses.
For immunoprecipitations, cells were lysed (50 mM HEPES, 150 mM NaCl, 10 mM EDTA, 10 mM Na4P2O7, 100 mM NaF, 1% Triton, pH 7.5, 1× protease inhibitor cocktail [Roche], and 2 mM vanadate), and 500 μg of total protein lysate was subjected to immunoprecipitation. Briefly, a total of 30 μl of protein A-agarose beads (Upstate) per sample was washed twice in protein lysis buffer in the presence of protease and phosphatase inhibitors. The beads were then prebound with the specific antibody for 1 h at room temperature. For 500 μg of total protein we used the following amounts of antibodies: 1.5 μg anti IRβ (C-19; Santa Cruz Biotechnology), 1.5 μg anti IGF-1Rβ (C-20; Santa Cruz Biotechnology), 4 μg anti-IRS-1 (Upstate), and 2.5 μg anti-IRS-2 (Upstate). Unbound antibody was then removed by centrifugation and beads were washed to reduce background signal. A total of 500 to 1,000 μg of protein was added to the beads and binding was allowed overnight at 4°C. Total unbound protein was removed by centrifugation and beads were washed three times in the lysis buffer before proteins were released from the beads by boiling the samples for 15 min in 2× Laemmli buffer. Total eluate was subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting.
For immunoblotting, cells were lysed in radioimmunoprecipitation assay buffer. Protein lysates were precleared by centrifugation at 13,000 rpm and 4°C and total protein content was determined using the bicinchoninic acid assay (Pierce, Rockford, IL). Samples were resuspended in reducing SDS-PAGE sample buffer at a concentration of 1 μg/μl and boiled for 5 min, and 20 to 50 μg of total protein was resolved by SDS-polyacrylamide gel electrophoresis in a discontinuous buffer system. Then, proteins were transferred onto nitrocellulose membranes (Schleicher & Schuell, Dassel, Germany), blocked in 5% BSA in Tris-buffered saline-Tween, and incubated with primary antibodies diluted (1:1,000) in 5% BSA in Tris-buffered saline-Tween overnight at 4°C followed by a 1-h incubation at room temperature with secondary antibodies at a 1:5,000 dilution. Antibodies against Akt, phospho-Akt (Ser473), FoxO1, phospho-FoxO1 (Ser256), p42/44 mitogen-activated protein kinase (MAPK; extracellular signal-regulated kinase [ERK]), phospho-p42/p44 MAPK (p-ERK; Thr202/Tyr204), and cleaved caspase-3 (Asp175) were from Cell Signaling Technology.
Anti-phosphatidylinositol 3-kinase (PI3K; p85α; n-SH2 domain) was obtained from Upstate. Anti-α-tubulin antibody was from Abcam. Horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G and goat anti-mouse immunoglobulin G were from Santa Cruz Biotechnology. The phosphotyrosine antibody used (mouse monoclonal) was obtained from an in-house source. Adenovirus for Δp85α was obtained from M. Kasuga (Kobe University, Japan) (44), and the adenovirus for lacZ was prepared as described previously (50). The adenoviruses were used at a multiplicity of infection of 100, and equal protein expression in the cell lines was confirmed by Western blotting.
Measurement of PI3K activity.
Following incubation the cells were solubilized in lysis buffer (50 mM HEPES, 150 mM NaCl, 10 mM EDTA, 10 mM Na4P2O7, 100 mM NaF, pH 7.5, 1% Nonidet P-40 substitute, protease inhibitor cocktail [Roche], and 2 mM vanadate) for 15 min at 4°C and then sonicated and precleared at 13,000 rpm for 15 min. A 750-μg aliquot of protein lysate was incubated with 10 μl of antiphosphotyrosine antibody (in-house source) overnight at 4°C. Protein complexes were then coupled to 30 μl of protein A/G-Sepharose beads (Upstate) for 90 min. Immunopellets then were washed twice in lysis buffer, twice in 0.5 M LiCl -0.1 M Tris, and twice in PI3K assay buffer (20 mM HEPES, pH 7.4, 5 mM MgCl2). After the final wash the pellet was resuspended in 30 μl PI3K assay buffer.
For lipid kinase assays, anti-PI was obtained from Avanti Polar Lipids, Inc. (Alabaster, AL), and thin-layer chromatography (TLC) silica plates were obtained from Merck (Darmstadt, Germany). Phosphorylated lipids were detected by autoradiography on Bioexpress Blue Basic Autorad films.
Phosphatidylinositol (10 μg/condition), which had been dried under air and dispersed at a concentration of 1 mg/ml in PI3K assay buffer by sonication at 4°C, was added to each sample. The reaction was started by adding 10 μl of a solution containing 50 mM MgCl2, 250 μM ATP, and 0.5 μl (5 μCi) of [33P]ATP. After 15 min of incubation at room temperature the reaction was terminated by adding 15 μl of 4 N HCl. Phospholipids were immediately extracted with 130 μl CHCl3-MeOH (1:1) in an Eppendorf tube. The organic phase was spotted onto Silica Gel 60 TLC plates. The TLC plates were developed in CHCl3-MeOH-H2O-NH4OH (45:35:8.5:1.5) for 1 h and then exposed to autoradiography films for 3 days.
Semiquantitative RT-PCR.
Cells were infected with the indicated adenovirus followed by starvation and glucose stimulation for 12 h or, alternatively, after starvation the cells were treated with 10 mM compound LY294002 for 30 min followed by glucose stimulation. Total RNA was isolated from cells and 500 ng of RNA was applied to a reverse transcription (RT-PCR) One-Step system (Invitrogen). The PCR was carried out as described previously (39).
Apoptosis and [3H]thymidine assays.
Cells were starved overnight in serum-free DMEM containing 1.1 mM glucose. Subsequently, cells were incubated in DMEM containing 1.1 mM glucose and the indicated concentration of insulin for 5 h, and the lysates were applied to an enzyme-linked immunosorbent assay kit (Boehringer Mannheim/Roche) to determine the amount of nucleosomes as a marker of apoptosis as described previously (51). For caspase-3 experiments, the cells were incubated for the designated periods in the presence of the indicated glucose concentrations for 24 h. For the [3H]thymidine assays, cells were plated at a density of 3 × 105 cells/well in 24-well dishes. After 24 h, the medium was changed to DMEM and incubated with 0.1% insulin-free BSA. After a further 48 to 72 h of incubation the cells were pulsed with 2 mCi of [methyl-3H]thymidine (NEN) per well for 1 h at 37°C followed by washing with ice-cold phosphate-buffered saline and then lysed in 0.1% SDS. Trichloroacetate-precipitable DNA incorporated radioactivity was determined using a scintillation counter. All assays were performed in triplicate.
Statistical methods.
All experiments were performed at least three times unless otherwise indicated and quantified for changes as described in Results. Statistical methods included Student's t test, analysis of variance, or analysis of variance with post hoc comparisons as appropriate. A P value of <0.05 was considered statistically significant.
RESULTS
Insulin activates the insulin signaling pathway in cultured β-cells.
To evaluate the effects of exogenous insulin, we stimulated cultured control or βIRKO β-cells with insulin for different periods and harvested the cells for analyses of protein expression. As expected, insulin phosphorylated the insulin receptor in control but not in βIRKO cells (Fig. 1a, left panel). In contrast, the phosphorylation of IGF-1R was observed in control cells and hardly detectable in the βIRKO cells upon insulin stimulation (Fig. 1a, right panel). In controls, IRS-1 was maximally tyrosine phosphorylated at 1 minute upon insulin stimulation, and virtually no effects were observed in βIRKO cells (Fig. 1b, left upper panel). IRS-2 was also maximally tyrosine phosphorylated 1 minute after insulin stimulation in controls. In contrast, in βIRKO cells, a higher basal phosphorylation of IRS-2 was evident that was not enhanced further upon insulin stimulation (Fig. 1b, right upper panel). This correlated with an increase in total IRS-2 protein in βIRKO cells (data not shown). The expression of the IRS-1- or IRS-2-associated p85 regulatory subunit of PI3K was comparable to the phosphorylation states of IRS proteins (Fig. 1b, lower panels). The amount of p85 protein bound to the total tyrosine-phosphorylated complex rapidly increased at 1 minute upon insulin stimulation in controls, whereas in βIRKO cells, it was low under basal conditions and remained low even upon insulin stimulation (Fig. 1b, left lower panel). PI3K activity associated with the phosphotyrosine complex paralleled the levels of p85 bound to the complex in both cell types (Fig. 1c). Consequently, Akt phosphorylation was stimulated by insulin in controls but not in βIRKO cells (Fig. 1d).
FIG. 1.
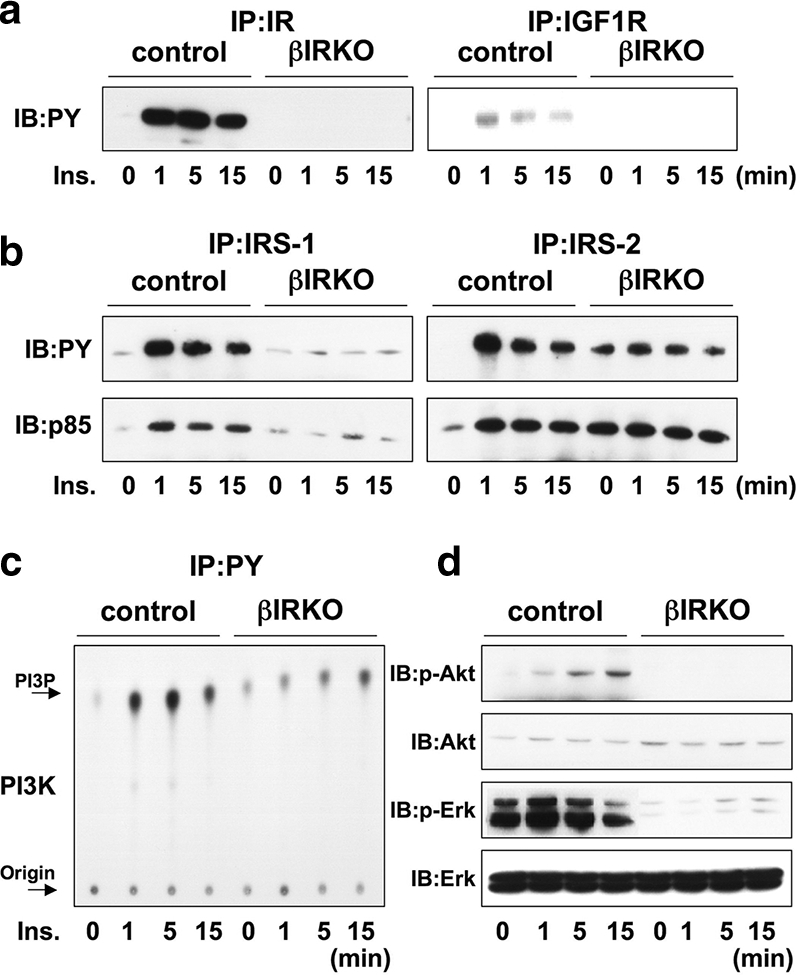
Insulin activates proteins in the insulin/IGF-1 signaling pathway in control β-cells, but not in βIRKO cells. Cells were stimulated with 100 nM insulin for 0, 1, 5, or 15 min. (a) Tyrosine phosphorylation of the insulin receptor and IGF-1 receptor. (b) Tyrosine phosphorylation of IRS-1 and IRS-2. The immunoprecipitates (IP) with anti-IR, anti-IRS-1, or anti-IRS-2 from the indicated cell lysates with or without insulin stimulation were immunoblotted (IB) with anti-PY (top panels) or the p85 antibody (bottom panels). (c) PI3K activity associated with the phosphotyrosine complex. PI3K activity was measured after immunoprecipitation with anti-PY from the indicated cell lysates. (d) Phosphorylation of Akt and ERK. The cell lysates were immunoblotted with the indicated antibodies. Representative blots are shown from three independent experiments.
ERK was mildly activated by insulin in control cells, while in βIRKO cells very low levels of phosphorylation were detected in the basal state and remained low upon insulin stimulation (Fig. 1d).
Glucose activates proteins in the insulin signaling pathway in cultured β-cells.
To explore whether the absence of the insulin receptor impacted the signaling effects of glucose, we stimulated βIRKO or control cells with glucose for similar periods as for insulin. In controls, glucose stimulated tyrosine phosphorylation of the insulin receptor, but not IGF-1R, albeit more slowly and weakly compared with insulin stimulation (Fig. 2a, left panel). The stimulatory effect of glucose was virtually undetectable in βIRKO cells (Fig. 2a, right panel). Similarly, the phosphorylation of IRS-1 and IRS-2 also occurred more slowly and weakly in control cells in response to glucose compared to the rapid effects observed with insulin stimulation (Fig. 2b, upper left and right panels). The protein levels of p85 bound to IRS-1 or IRS-2 paralleled the levels of phosphorylation of each IRS protein (Fig. 2b, lower panels). By contrast, in βIRKO cells, glucose failed to stimulate phosphorylation of IRS-1 or IRS-2, despite upregulation in the basal phosphorylation of IRS-2 (Fig. 2b). PI3K associated with the phosphotyrosine complex also paralleled the changes in p85 protein bound to IRS proteins in controls, while in βIRKO cells the elevated high basal activity was not altered further in response to glucose stimulation (Fig. 2c). Akt phosphorylation was enhanced in control cells after glucose stimulation with maximum phosphorylation occurring 15 min after stimulation (Fig. 2d), in comparison to the rapid activation of Akt by insulin within 5 minutes (Fig. 1d). Interestingly, glucose failed to activate Akt in βIRKO cells (Fig. 2d), suggesting that the effect of glucose on Akt signaling requires functional insulin receptors. One interpretation of these data is that the effects of glucose on PI3K and Akt kinase occur more slowly than the effects of exogenous insulin because either glucose has to be metabolized to generate additional signals to enhance signaling and/or glucose has to first secrete insulin for activation of the insulin receptor in an autocrine/paracrine manner. In contrast and consistent with previous reports (23), ERK was activated by glucose in both control and βIRKO cells (Fig. 1d), confirming that the effects of glucose on ERK activity occur independently of insulin signaling.
FIG. 2.
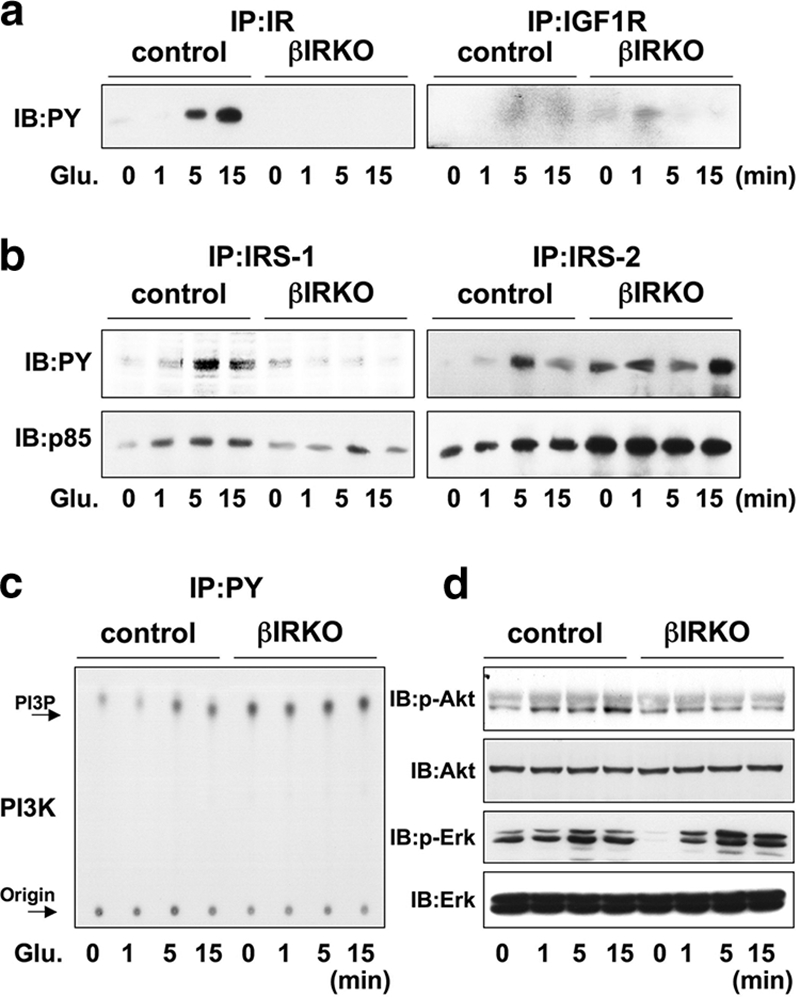
Glucose activates proteins in the insulin/IGF-1 signaling pathway in control β-cells, but not in βIRKO cells. Cells were treated with glucose (25 mM) for 0, 1, 5, or 15 min. (a) Tyrosine phosphorylation of the insulin receptor and IGF-1 receptor. (b) Tyrosine phosphorylation of IRS-1 and IRS-2. The immunoprecipitates (IP) with anti-IR, anti-IRS-1, or anti-IRS-2 from the indicated cell lysates with or without glucose stimulation were immunoblotted (IB) with anti-PY (top panels) or the p85 antibody (bottom panels). (c) PI3K activity associated with the phosphotyrosine complex. PI3K activity was measured in the immunoprecipitation with anti-PY from the indicated cell lysates. (d) Phosphorylation of Akt and ERK. The cell lysates were immunoblotted with the indicated antibodies. Representative blots are shown from three independent experiments.
βIRKO cells exhibit a compensatory increase in IGF-1 receptor expression.
It is evident from previous reports including our own that pancreatic β-cells express both insulin and IGF-I receptors, with the former being expressed at a higher density (Fig. 3a) (16, 54). Since the two receptors exhibit a high degree of homology (7) it is possible that disruption of one receptor type leads to enhanced expression of the related receptor to compensate in signaling. We therefore undertook experiments to examine the effects of exogenous IGF-I on control and βIRKO cells. IGF-I treatment did not phosphorylate the insulin receptor in control or βIRKO cells (Fig. 3b, left panel). While exogenous IGF-I did not phosphorylate its own receptor in controls (Fig. 3b, right panel), interestingly, we observed a rapid and striking phosphorylation of the IGF-1 receptor in βIRKO cells (Fig. 3b, right panel), suggesting compensatory upregulation of the IGF-1 receptor in the absence of insulin receptor expression. Examination of downstream proteins revealed a lack of effect of IGF-I on phosphorylation of IRS-1 or IRS-2 in controls (Fig. 3c, left and right upper panels). In βIRKO cells we observed a mild effect of IGF-I on phosphorylation of IRS-1 that peaked 15 min after stimulation (Fig. 3c, upper left panel), compared to a rapid, strong, and sustained phosphorylation of IRS-2 (Fig. 3c, upper right panel). Similar effects were observed in protein levels of p85 associated with IRS-1 and IRS-2 (Fig. 3c, lower panels).
FIG. 3.
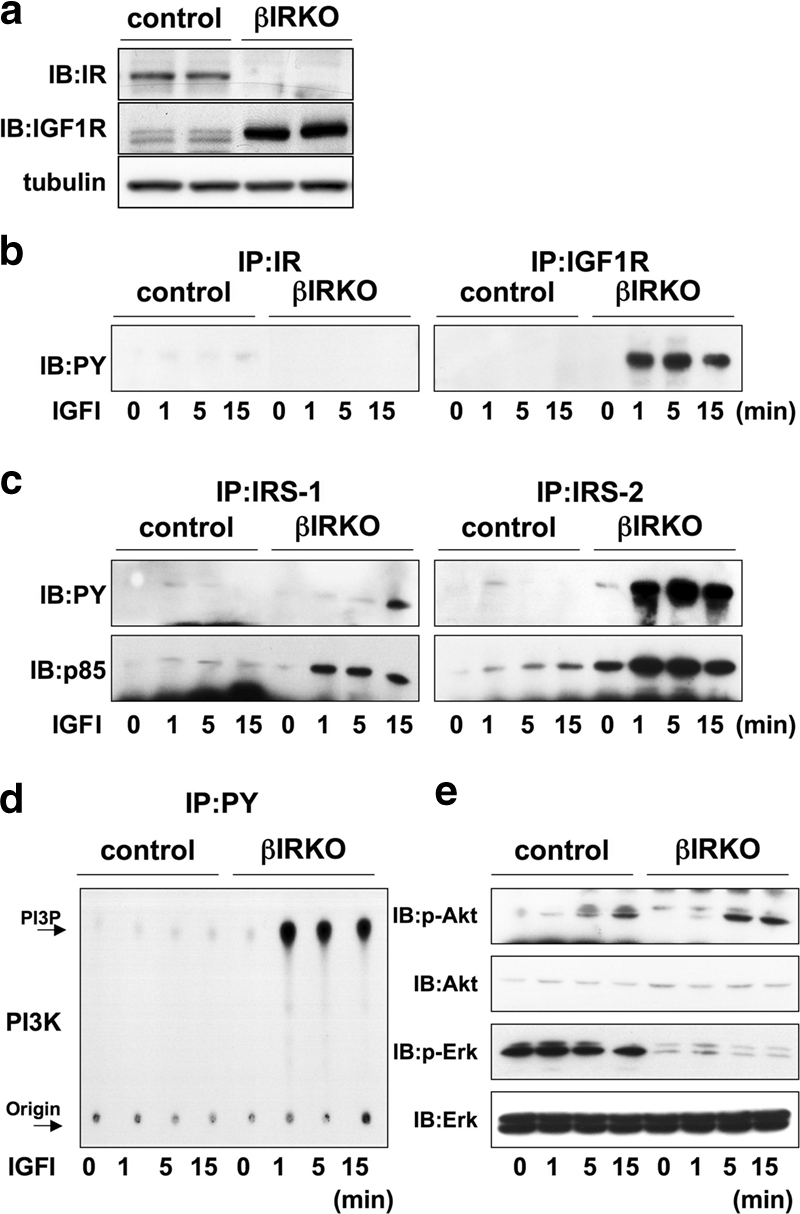
βIRKO cells exhibit a compensatory increase in expression of IGF-1 receptors. (a) To determine the presence and cross-reactivity of insulin and IGF-1 receptors we treated control β-cells with IGF-I (100 nM) and performed immunoprecipitation and blotting for either the insulin receptor or the IGF-1 receptor. For experiments in panels b to d, cells were treated with IGF-I (100 nM) for 0, 1, 5 or 15 min. (b) Tyrosine phosphorylation of insulin receptor (right panel) and IGF-1 receptor (left panel). (c) Tyrosine phosphorylation of IRS-1 and IRS-2. The immunoprecipitates (IP) with anti-IRS-1 or anti-IRS-2 from the indicated cell lysates with or without IGF-1 stimulation were immunoblotted (IB) with anti-PY (top panels) or the p85 antibody (bottom panels). (d) PI3K activity associated with the phosphotyrosine complex. PI3K activity was measured in the immunoprecipitation reaction with anti-PY from the indicated cell lysates. (e) Phosphorylation of Akt and ERK. The cell lysates were immunoblotted with the indicated antibodies. Representative blots are shown from three independent experiments.
IGF-I treatment failed to activate PI3K in control cells while rapidly activating the kinase in βIRKO cells (Fig. 3d), which in turn led to activation of p-Akt but not p-ERK (Fig. 3e). We also observed mild activation of Akt kinase in control cells that peaked between 5 and 15 min and an increase in p-ERK (Fig. 3e). These data suggest that the lack of insulin receptors leads to upregulation of IGF-1 receptor expression and a consequent activation of IRS-2, PI3K, and Akt. Whether the effects of IGF-I become significant in vivo in states of β-cell insulin resistance requires further investigation.
We would like to emphasize that the relative concentrations of glucose, insulin, and/or IGF-I in the treatment medium play a critical role in determining the signaling effects in the β-cell lines. It is possible that a minor change in the concentration of one stimulus, which would alter the overall ratio with the other stimuli, significantly impacts both the rapidity and magnitude of activation of the proteins in the insulin/IGF-I signaling cascade with consequent differential end points.
Lack of insulin signaling promotes apoptosis and poor cell growth.
Glucose has been reported to act as a mitogen, suggesting a role for the nutrient in the modulation of β-cell mass (6, 56). On the other hand, mice lacking insulin receptors in β-cells (25, 52) exhibit reduced β-cell mass and develop age-dependent diabetes; this implies an antiapoptotic role for insulin itself in the regulation of β-cell growth and survival. Furthermore, insulin signaling through PI3K/Akt has been reported to activate survival pathways in different cells, including β-cells (10, 49). To assess the effects of glucose on antiapoptosis/proliferation in β-cells lacking insulin receptors, we treated control or βIRKO cells with a range of concentrations of either glucose or exogenous insulin in the presence of a constant concentration of glucose. Serum depletion-induced apoptosis was detectable in control and βIRKO cells. As expected, addition of increasing amounts of glucose to the medium significantly decreased apoptosis in control cells. However, in βIRKO cells, the rescue from apoptosis was significantly lower at all tested glucose concentrations (Fig. 4a, upper and lower panels), suggesting that signaling via the insulin receptor is necessary to activate one or more downstream proteins to mediate the protective effects of exogenous glucose.
FIG. 4.

Insulin signaling protects β-cells from apoptosis and promotes growth. (a) Glucose protects β-cells from serum deprivation-induced apoptosis. Cells were cultured in the indicated glucose concentrations without serum for 24 h. The levels of apoptosis were assessed by determining the levels of cleaved caspase-3 by Western blotting (IB). Shown is a representative result from three independent experiments. Data (lower graph) for the changes are expressed as the ratio of cleaved caspase-3 to total caspase. *, P < 0.05 versus the respective control (FCS plus glucose). (b) Insulin protects β-cells from serum deprivation-induced apoptosis. Cells were cultured in the indicated concentration of insulin without serum for 24 h. The levels of apoptosis were assessed using an enzyme-linked immunosorbent assay for nucleosomal DNA as described in Materials and Methods. Data are expressed as the percentage of cells rescued from apoptosis. Data are from three independent experiments. (c) Glucose promotes activation of cleaved caspase-3 in βIRKO but not in control β-cells. Data are representative of three independent experiments. (d) Blocking the insulin receptor with anti-insulin receptor antibody enhances serum depletion-induced cleaved caspase-3 in control β-cells. Data are representative of three independent experiments. (e) Insulin or glucose stimulates p4E-BP1 in control but not βIRKO β-cells. Cells were stimulated with insulin (100 nM) or glucose (20 mM) for 30 min, and the cell lysates were subjected to immunoblotting (IB) with anti-p-4E-BP1. Data are representative of three independent experiments. (f) βIRKO β-cells exhibit significantly reduced [3H]thymidine incorporation compared to control β-cells over a 96-h incubation period. *, P < 0.05 versus the respective control (day 0); #, P < 0.05 for βIRKO versus control.
We next performed studies to directly examine the antiapoptotic effects of insulin, and we observed that in control cells serum depletion-induced apoptosis was not only prevented by the presence of exogenous insulin but significantly increased their survival in a dose-dependent manner (Fig. 4b). In contrast, βIRKO cells showed poor survival in the presence of exogenous insulin, suggesting that lack of insulin action promotes apoptosis (Fig. 4b).
Consistent with reduced β-cell survival, βIRKO cells exhibited increased cleaved caspase-3 even in low glucose concentrations, and this was enhanced further by high glucose levels. In contrast, in control cells, cleaved caspase-3 was virtually undetectable when the cells were exposed to high glucose (Fig. 4c). Indeed, high glucose activated Akt kinase and promoted phosphorylation of its downstream target, FoxO1, in controls (but not in βIRKO cells), presumably through activation of insulin receptors by autocrine effects of secreted insulin (Fig. 4c). Similarly, blocking insulin signaling, using an antibody against the α-subunit of the insulin receptor, increased expression of cleaved caspase-3 in both the presence and absence of glucose (Fig. 4d). Together, these data indicate that high glucose protects against cell death by activating Akt and downstream antiapoptotic signaling through activation of the insulin receptor.
To examine the effects of insulin on DNA synthesis we used a thymidine incorporation assay. While a dose-dependent response was evident in control β-cells, a blunted effect was observed in the βIRKO group (Fig. 4e). These data suggest that a lack of insulin signaling impairs β-cell growth by directly decreasing proliferation. Next, we compared the effects of glucose versus insulin on protein synthesis by assessing the phosphorylation of 4E-BP1, an inhibitory protein involved in translation initiation (15). While insulin and glucose equally promoted phosphorylation of 4E-BP1 in controls, a poor response to either stimulus in βIRKO cells suggested that glucose stimulation of mTOR, a key regulator of protein synthesis and cell cycle progression, requires intact insulin receptors in β-cells (Fig. 4f).
Insulin and glucose promote expression of GLUT2 in a class IA PI3K-dependent manner.
Ablation of insulin receptors in β-cells leads to decreased glucose-stimulated insulin secretion that is associated with reduced expression of GLUT2 (39). Consistent with a role in glucose sensing, expression of GLUT2 was increased by both glucose and insulin stimulation, while IGF-1 minimally enhanced GLUT2 expression in controls and showed little effect in βIRKO cells (Fig. 5a, left and right panels). The glucose-stimulated increase in GLUT2 expression was abolished by treatment with LY294002, a specific inhibitor of PI3K (55), while GLUT2 expression was downregulated under basal conditions in βIRKO cells and not increased further by glucose (Fig. 5b, left and right panels). Moreover, expression of a dominant negative form of the PI3K subunit p85 (Δp85) in control cells downregulated both basal and glucose-stimulated expression of GLUT2, while the low levels of GLUT2 in βIRKO cells were further reduced by the expression of Δp85 (Fig. 5c, upper and lower panels). These data suggest that glucose regulation of GLUT2 expression by class IA PI3K requires activation of the insulin receptor.
FIG. 5.

Glucose and insulin promote expression of GLUT2. (a) Insulin and glucose enhance expression of GLUT2. Cells were starved overnight and stimulated with insulin (100 nM), IGF-I (100 nM), or glucose (20 mM) for 12 h. The right panel shows the the data normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) levels. *, P < 0.05 for untreated versus insulin or IGF-I or glucose. Data are representative of three independent experiments. (b) Inhibition of PI3K blocks the effect of glucose on the expression of GLUT2. Cells were preincubated with 10 μM LY294002 for 30 min and stimulated with glucose (20 mM) in the presence of the inhibitor for 12 h. The right panel shows data normalized to GAPDH levels. *, P < 0.05 for untreated versus glucose; #, P < 0.05 for glucose versus glucose plus LY. Data are representative of three independent experiments. (c) Expression of the dominant negative p85 in control β-cells downregulates the expression of GLUT2. Cells were infected with the indicated adenovirus for 48 h and stimulated with glucose (20 mM) for 12 h. Total RNA was isolated and subjected to semiquantitative RT-PCR for detecting GLUT2. The lower panel shows quantification of the data. Comparisons with controls: *, P < 0.05 for LacZ glucose treated versus LacZ glucose untreated or for p85 glucose untreated versus LacZ glucose untreated; #, P < 0.05 for p85 glucose treated versus LacZ glucose treated. Comparisons with βIRKOs: #, P < 0.05 for p85 glucose untreated or glucose treated versus LacZ glucose untreated versus glucose treated. For comparisons of LacZ controls versus LacZ βIRKOs: *, P < 0.05 for glucose treated or untreated. Representative data from three independent experiments are shown.
IRS-2 is a key substrate for insulin signaling in β-cells.
The phenotype of the IRS-2KO mouse is similar to the βIRKO mouse, especially with regard to β-cell mass, with both mutants exhibiting hypoplastic islets and a susceptibility to develop overt diabetes (24, 25, 59). In contrast, IRS-1 knockout mice are insulin resistant but do not develop diabetes due to compensatory β-cell growth and enhanced survival, in part due to an upregulation in IRS-2 (20, 46, 58). Surprisingly, βIGF1RKO mice do not exhibit major defects in development of β-cells or maintenance of β-cell mass in adults (26, 52, 61). Considering the similar phenotypes in the βIRKO and IRS2KO mice, we addressed the hypothesis that the effects of glucose in β-cells are blunted due to the absence of IRS-2 by performing signaling experiments in βIRS2KO β-cells.
In control β-cells exogenous application of insulin phosphorylated the insulin receptor rapidly within 1 minute, compared to a weak phosphorylation in βIRS2KO cells. Exogenous insulin also phosphorylated the IGF-1R in both control and KO cells to a similar extent, but the effect was generally weaker compared to the robust effects on the insulin receptor. These data suggest that the ablation of IRS-2 does not directly modulate the functionality of IGF-1 receptors by feedback effects (Fig. 6a, right panel). Insulin stimulated phosphorylation of IRS-1 in controls, with maximal effects between 1 and 5 minutes (Fig. 6b, left upper panel). In βIRS2KO cells we observed hyperphosphorylation of IRS-1 in the basal state that did not increase further upon addition of exogenous insulin, suggesting an attempt to compensate for lack of IRS-2 (20). As expected, we did not detect phosphorylation of IRS-2 in βIRS2KO cells, whereas in control cells IRS-2 phosphorylation was evident with maximal effects between 1 and 5 minutes after insulin stimulation (Fig. 6b, right upper panel). The levels of phosphorylation of the p85 subunit of PI3K corresponded with the phosphorylation status of the insulin receptor substrates located upstream (Fig. 6b, lower panels).
FIG. 6.
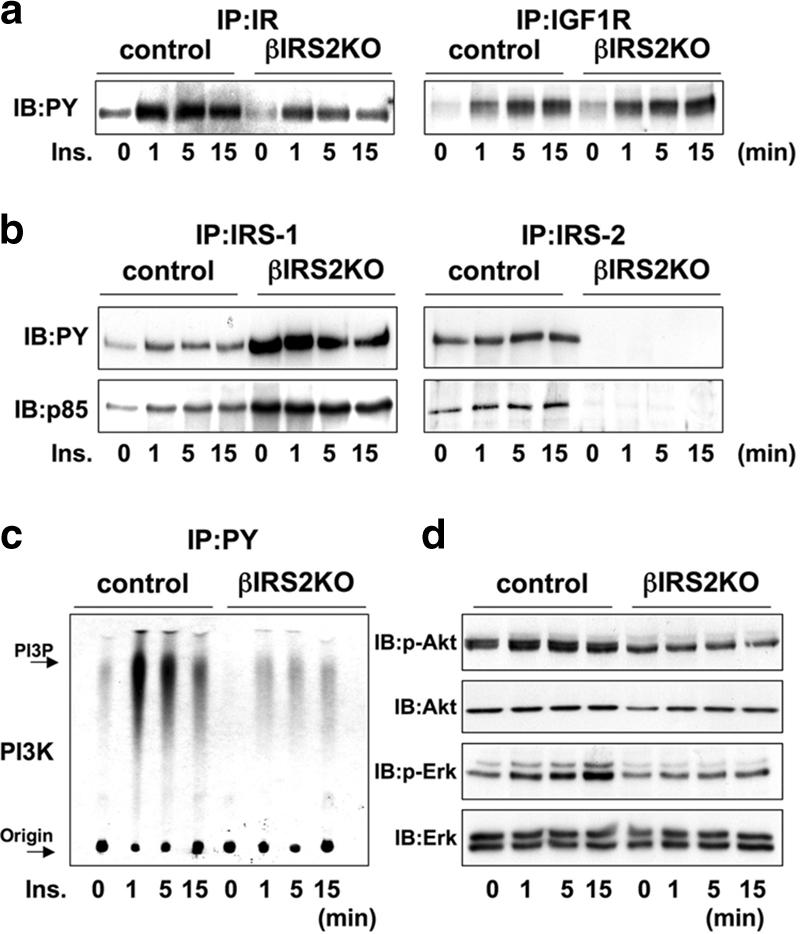
Insulin activates proteins in the insulin/IGF-I signaling pathway in control but not in βIRS2KO β-cells. Cells were stimulated with 100 nM insulin for 0, 1, 5, or 15 min. (a) Tyrosine phosphorylation of insulin receptor and IGF-1 receptor. (b) Tyrosine phosphorylation of IRS-1 and IRS-2. The immunoprecipitates (IP) with anti-IR, anti-IRS-1, or anti-IRS-2 antibodies from the indicated cell lysates with or without insulin stimulation were immunoblotted (IB) with anti-PY (top panels) or the p85 antibody (bottom panels). (c) PI3K activity associated with the phosphotyrosine complex. PI3K activity was measured in the immunoprecipitate with anti-PY from the indicated cell lysates. (d) Phosphorylation of Akt kinase and ERK. The cell lysates were immunoblotted with the indicated antibodies. Representative blots are shown from three independent experiments.
PI3K and Akt kinase activity were enhanced by insulin stimulation of control cells and peak effects were detectable 1 to 5 minutes after addition of insulin. On the other hand, in βIRS2KO cells both PI3 kinase and Akt kinase were activated but the effects were weaker compared to those observed in control cells (Fig. 6c). ERK, a key downstream kinase in the insulin/IGF-I signaling pathway, displayed weak and delayed phosphorylation in βIRS2KO cells compared to controls (Fig. 6d). Together these data demonstrate the similarities in the defects observed in insulin action between the βIRKO and βIRS2KO cells.
Glucose effects on insulin signaling require activation of IRS-2.
The effects of glucose stimulation of βIRS2KO cells were largely similar to those observed in βIRKO β-cells and occurred much slower compared to the rapid effects of insulin stimulation (Fig. 7a and b). We focused on key kinases in the insulin/IGF-1 signaling pathway and observed that glucose stimulation of control cells enhanced PI3 kinase and Akt kinase activities, with maximal effects occurring 15 min after addition (Fig. 7c). In contrast, in βIRS2KO cells a blunted response was evident even 15 min after glucose addition. However, ERK was activated by glucose to a similar extent in both control and βIRS2KO cells, with maximal effects occurring after 15 min, confirming that the effects of glucose on p-ERK are part of an insulin-independent pathway (Fig. 7d).
FIG. 7.
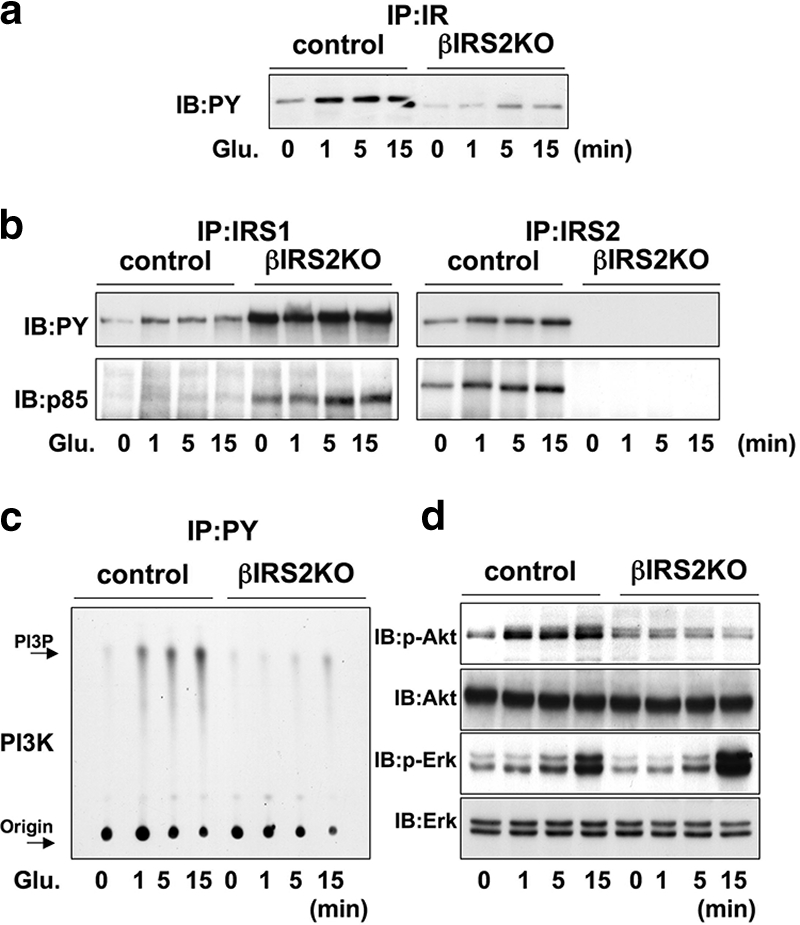
Glucose activates proteins in the insulin/IGF-I signaling pathway in control but not in βIRS2KO β-cells. Cells were treated with glucose (25 mM) for 0, 1, 5, or 15 min. (a) Tyrosine phosphorylation of insulin receptor and IGF-1 receptor. (b) Tyrosine phosphorylation of IRS-1 and IRS-2. The immunoprecipitates (IP) with anti-IR, anti-IRS-1, or anti-IRS-2 from the indicated cell lysates with or without glucose stimulation were immunoblotted (IB) with anti-PY (top panels) or the p85 antibody (bottom panels). (c) PI3K activity associated with the phosphotyrosine complex. PI3K activity was measured in the immunoprecipite with anti-PY from the indicated cell lysates. (d) Phosphorylation of Akt kinase and ERK. The cell lysates were immunoblotted with the indicated antibodies. Representative blots are shown from three independent experiments.
Activation of ERK requires glucose metabolism.
The metabolism of glucose is a critical requirement for insulin secretion (37, 43). To further examine whether the effects of glucose metabolism are dependent on IRS-2 for subsequent phosphorylation of ERK we incubated cells with 2-deoxyglucose (2-DOG), which cannot undergo glycolysis (60). Incubation of control or βIRS2KO cells with 2-DOG uniformly failed to phosphorylate ERK, suggesting that the products of glucose metabolism that trigger ERK phosphorylation act independently of activation of IRS-2 protein (Fig. 8), consistent with previous reports (23).
FIG. 8.

Glucose effects on ERK occur independently of insulin signaling. Cells were exposed to glucose (25 mM) (a) or 2-DOG (25 mM) (b) for 0, 1, 5, or 15 min. The cell lysates were immunoblotted (IB) with the indicated antibody. Representative blots from three experiments are shown.
DISCUSSION
Using β-cells that completely lack either the insulin receptor or IRS-2, we found that the signaling effects of glucose require the presence of an intact insulin/IGF-I signaling pathway. Glucose stimulation of the mutant β-cells failed to activate key kinases that mediate important downstream effects, including proliferation, survival, and protein synthesis, indicating the significance of insulin signaling for the maintenance of β-cell mass.
Although previous studies, using β-cell lines with a partial knockdown of insulin receptors, have reported that the signaling effects of glucose in β-cells include the effects of secreted insulin, the presence of residual insulin signaling in these cells raised the confounding possibility that the effects of glucose were diluted by the ambient insulin that is continuously secreted by β-cells via the constitutive pathway. For example, in independent experiments Da Silva Xavier et al. (9) and Ohsugi et al. (38) achieved transient knockdown of insulin receptor expression levels in MIN6 cells that ranged between 80 and 90%. In these approaches the residual insulin signaling activity can potentially confound the interpretation of the effects of glucose in β-cells. Further, previous reports indicate potential nonspecific effects of siRNA (2). Similarly, experiments designed to dissociate the effects of glucose from insulin secretion using somatostatin (57) are unlikely to completely block regulated and constitutive insulin secretion that occurs in primary β-cells (19, 22). The use of β-cell lines from βIRKO and IRS-2-KO mice that lack expression of the insulin receptor or IRS-2, respectively, circumvents these limitations and allows for direct interpretation of the signaling effects of glucose.
Consistent with the signaling effects of insulin reported in the classical insulin target tissues (7), insulin rapidly activated proteins in the insulin/IGF-1 signaling pathway in control β-cells, with peak effects occurring within a minute, in contrast to the delayed effects of glucose. While the delayed effects of glucose in control β-cells may be explained, in part, by the time required for the sugar to be transported into the cell and metabolized (43), the failure of glucose to activate PI3 kinase or Akt kinase in the β-cells only when either the insulin receptor or IRS-2 is lacking clearly indicates the dependence of glucose on the two proteins for its signaling effects (Fig. 9).
FIG. 9.

Schematic of a link between glucose and insulin signaling in β-cells. (A) Potential direct effects of glucose and/or its metabolites on proteins in the insulin/IGF-1 signaling pathway. (B) Potential indirect effects of glucose and direct effects of insulin following exocytosis of insulin. Akt, v-akt murine thymoma viral oncogene homolog; FoxO-1, forkhead box O1; GRB2, growth factor receptor-bound protein 2; PIP3, phosphatidylinositol (3,4,5)-trisphosphate; mTOR, mammalian target of rapamycin; 4EBP1, translation initiation factor 4e binding protein 1.
The defects in glucose-stimulated signaling also provide insights into the phenotypes of the βIRKO mice. For example, the activation of cleaved capsase-3, either by glucose stimulation of β-cells lacking insulin receptors (βIRKO β-cells) or by blocking insulin action in control β-cells by using an anti-insulin receptor antibody, indicates that functional insulin receptors activate antiapoptotic signaling in control β-cells. Indeed, βIRKO mice exhibit an age-dependent decrease in β-cell mass and a susceptibility to develop type 2 diabetes (25, 27, 34). Thus, it is conceivable that ambient hyperglycemia promotes apoptosis of β-cells (12, 14, 33) in insulin-resistant states due to poor insulin-stimulated activation of Akt (5, 11, 49). Furthermore, the reduced stimulation of mTOR by glucose in βIRKO cells suggests that blunted protein synthesis also contributes to a reduced islet cell mass in βIRKO mice (35).
We and others have previously reported that loss of insulin signaling in β-cells impairs acute-phase insulin secretion in response to glucose (9, 25). In agreement with these reports, expression of GLUT2 was stimulated by glucose and insulin, but not by IGF-1. This effect is diminished in βIRKO cells, suggesting that functional insulin receptors play a role in glucose-stimulated expression of GLUT2. It is possible that a reduced expression of GLUT2 in βIRKO cells plays some role in the ability of glucose to directly activate insulin signaling proteins. Our studies using the PI3 kinase blocker (LY294002) and the dominant negative p85 provide further evidence that the effect of glucose occurs in a PI3K-dependent manner (62). In contrast, the effects of glucose on p-ERK and its potential downstream effects on cell proliferation and differentiation occur independently of insulin signaling (23). While our studies provide a systematic characterization of alterations in the signaling proteins in the insulin/IGF-1 pathway in βIRKO versus βIRS2KO cell lines, our findings in the βIRKO cells are largely in agreement with those of Da Silva Xavier et al. (9) and Ohsugi et al. (38).
Insulin and glucose minimally activated IGF-1 receptors in controls, while phosphorylation of the IGF-1 receptor and activation of downstream proteins in the insulin/IGF-1 signaling cascade were clearly evident upon IGF-I stimulation of βIRKO cells. The upregulation of IGF-1 receptors in βIRKO cells indicates a potential long-term compensatory effect in the absence of functional insulin receptors. Thus, the significance of IGF-1 receptor activation in vivo for the maintenance of β-cell proliferation and survival, especially during states of β-cell insulin resistance, requires careful investigation (32).
The similarities in signaling defects between βIRKO and IRS-2KO β-cells are consistent with the largely similar phenotypes of the mutant mouse models (24, 25, 59). In contrast, IRS-1KO mice exhibit a compensatory increase in β-cell mass (3, 24, 29) that is, in part, due to upregulation of IRS-2 (20). The contrasting signaling effects due to loss of IRS-1 versus loss of IRS-2 indicate that these two substrates serve different downstream pathways in β-cells, with IRS-1 having a dominant effect in insulin secretion (28, 29) and IRS-2 being relevant for growth and/or apoptosis (8, 20). Cell-specific roles for the two substrates have also been reported in other cell types. For example, brown preadipocytes that lack IRS-1 fail to differentiate into mature adipocytes due to altered UCP-1 expression, compared to a mild defect in cells lacking IRS-2 (48, 53). In hepatocytes, IRS-1 and IRS-2 have been suggested to play complementary roles in regulating lipid and carbohydrate metabolism (13, 18, 42, 47). The availability of stable knockout β-cell lines created for the studies reported in this paper provides us with an opportunity to further dissect the differential signaling responses between IRS-1 and IRS-2 in β-cells.
In summary, our studies point to an important role for insulin/IGF-I signaling in the signaling responses activated by glucose stimulation with implications for a direct effect of insulin in the maintenance of β-cell mass and function (Fig. 9).
ADDENDUM IN PROOF
The use of insulin receptor-deficient β-cells has been reported in a previous study (C. Guillen et al., Endocrinology 147:1959-1968, 2006). An important distinction, however, is that Guillen et al. used “fetal” islets, in contrast to β-cell lines isolated from adult mice in our experiments. Considering that fetal β-cells are immature, unresponsive to glucose, and differ in nutrient sensing and potentially other properties compared to glucose-responsive adult β-cells, it is likely that the β-cell lines reported in our study differ in their signaling properties from β-cell lines derived from fetal islets.
Acknowledgments
We thank Lindsay Huse for excellent assistance with preparation of the manuscript.
This work was supported by NIH RO1 DK 67536 (R.N.K.) and RO1 DK 68721 (R.N.K.) and the DERC Specialized Assay Core (P30 DK36836).
Footnotes
Published ahead of print on 9 March 2009.
REFERENCES
- 1.Accili, D. 2004. Lilly lecture 2003. The struggle for mastery in insulin action: from triumvirate to republic. Diabetes 531633-1642. [DOI] [PubMed] [Google Scholar]
- 2.Anonymous. 2003. Whither RNAi? Nat. Cell Biol. 5489-490. [DOI] [PubMed] [Google Scholar]
- 3.Araki, E., M. A. Lipes, M. E. Patti, J. C. Bruning, B. Haag III, R. S. Johnson, and C. R. Kahn. 1994. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature 372186-190. [DOI] [PubMed] [Google Scholar]
- 4.Assmann, A., C. Hinault, and R. N. Kulkarni. 2009. Growth factor control of pancreatic islet regeneration and function. Pediatr. Diabetes 1014-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernal-Mizrachi, E., S. Fatrai, J. D. Johnson, M. Ohsugi, K. Otani, Z. Han, K. S. Polonsky, and M. A. Permutt. 2004. Defective insulin secretion and increased susceptibility to experimental diabetes are induced by reduced Akt activity in pancreatic islet beta cells. J. Clin. Investig. 114928-936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonner-Weir, S., and F. E. Smith. 1994. Islet cell growth and the growth factors involved. Trends Endocrinol. Metab. 560-64. [DOI] [PubMed] [Google Scholar]
- 7.Cheatham, B., and C. R. Kahn. 1995. Insulin action and the insulin signaling network. Endocr Rev. 16117-142. [DOI] [PubMed] [Google Scholar]
- 8.Choudhury, A. I., H. Heffron, M. A. Smith, H. Al-Qassab, A. W. Xu, C. Selman, M. Simmgen, M. Clements, M. Claret, G. Maccoll, D. C. Bedford, K. Hisadome, I. Diakonov, V. Moosajee, J. D. Bell, J. R. Speakman, R. L. Batterham, G. S. Barsh, M. L. Ashford, and D. J. Withers. 2005. The role of insulin receptor substrate 2 in hypothalamic and beta cell function. J. Clin. Investig. 115940-950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Da Silva Xavier, G., Q. Qian, P. J. Cullen, and G. A. Rutter. 2004. Distinct roles for insulin and insulin-like growth factor-1 receptors in pancreatic beta-cell glucose sensing revealed by RNA silencing. Biochem. J. 377149-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Datta, S. R., A. Brunet, and M. E. Greenberg. 1999. Cellular survival: a play in three Akts. Genes Dev. 132905-2927. [DOI] [PubMed] [Google Scholar]
- 11.Dickson, L. M., and C. J. Rhodes. 2004. Pancreatic beta-cell growth and survival in the onset of type 2 diabetes: a role for protein kinase B in the Akt? Am. J. Physiol. Endocrinol. Metab. 287E192-E198. [DOI] [PubMed] [Google Scholar]
- 12.Donath, M. Y., D. J. Gross, E. Cerasi, and N. Kaiser. 1999. Hyperglycemia-induced beta-cell apoptosis in pancreatic islets of Psammomys obesus during development of diabetes. Diabetes 48738-744. [DOI] [PubMed] [Google Scholar]
- 13.Dong, X., S. Park, X. Lin, K. Copps, X. Yi, and M. F. White. 2006. Irs1 and Irs2 signaling is essential for hepatic glucose homeostasis and systemic growth. J. Clin. Investig. 116101-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Efanova, I. B., S. V. Zaitsev, B. Zhivotovsky, M. Kohler, S. Efendic, S. Orrenius, and P. O. Berggren. 1998. Glucose and tolbutamide induce apoptosis in pancreatic beta-cells. A process dependent on intracellular Ca2+ concentration. J. Biol. Chem. 27333501-33507. [DOI] [PubMed] [Google Scholar]
- 15.Gingras, A. C., S. G. Kennedy, M. A. O'Leary, N. Sonenberg, and N. Hay. 1998. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev. 12502-513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goren, H. J., R. N. Kulkarni, and C. R. Kahn. 2004. Glucose homeostasis and tissue transcript content of insulin signaling intermediates in four inbred strains of mice: C57BL/6, C57BLKS/6, DBA/2, and 129X1. Endocrinology 1453307-3323. [DOI] [PubMed] [Google Scholar]
- 17.Gunton, J. E., R. N. Kulkarni, S. Yim, T. Okada, W. J. Hawthorne, Y. H. Tseng, R. S. Roberson, C. Ricordi, P. J. O'Connell, F. J. Gonzalez, and C. R. Kahn. 2005. Loss of ARNT/HIF1β mediates altered gene expression and pancreatic-islet dysfunction in human type 2 diabetes. Cell 122337-349. [DOI] [PubMed] [Google Scholar]
- 18.Haeusler, R. A., and D. Accili. 2008. The double life of Irs. Cell. Metab. 87-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Halban, P. A. 1994. Proinsulin processing in the regulated and the constitutive secretory pathway. Diabetologia 37(Suppl. 2)S65-S72. [DOI] [PubMed] [Google Scholar]
- 20.Hennige, A. M., U. Ozcan, T. Okada, U. S. Jhala, M. Schubert, M. F. White, and R. N. Kulkarni. 2005. Alterations in growth and apoptosis of insulin receptor substrate-1-deficient beta-cells. Am. J. Physiol. Endocrinol. Metab. 289E337-E346. [DOI] [PubMed] [Google Scholar]
- 21.Hohmeier, H. E., H. Mulder, G. Chen, R. Henkel-Rieger, M. Prentki, and C. B. Newgard. 2000. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 49424-430. [DOI] [PubMed] [Google Scholar]
- 22.Irminger, J. C., F. M. Vollenweider, M. Neerman-Arbez, and P. A. Halban. 1994. Human proinsulin conversion in the regulated and the constitutive pathways of transfected AtT20 cells. J. Biol. Chem. 2691756-1762. [PubMed] [Google Scholar]
- 23.Khoo, S., and M. H. Cobb. 1997. Activation of mitogen-activating protein kinase by glucose is not required for insulin secretion. Proc. Natl. Acad. Sci. USA 945599-5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kubota, N., K. Tobe, Y. Terauchi, K. Eto, T. Yamauchi, R. Suzuki, Y. Tsubamoto, K. Komeda, R. Nakano, H. Miki, S. Satoh, H. Sekihara, S. Sciacchitano, M. Lesniak, S. Aizawa, R. Nagai, S. Kimura, Y. Akanuma, S. I. Taylor, and T. Kadowaki. 2000. Disruption of insulin receptor substrate 2 causes type 2 diabetes because of liver insulin resistance and lack of compensatory beta-cell hyperplasia. Diabetes 491880-1889. [DOI] [PubMed] [Google Scholar]
- 25.Kulkarni, R. N., J. C. Bruning, J. N. Winnay, C. Postic, M. A. Magnuson, and C. R. Kahn. 1999. Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell 96329-339. [DOI] [PubMed] [Google Scholar]
- 26.Kulkarni, R. N., M. Holzenberger, D. Q. Shih, U. Ozcan, M. Stoffel, M. A. Magnuson, and C. R. Kahn. 2002. β-Cell-specific deletion of the Igf1 receptor leads to hyperinsulinemia and glucose intolerance but does not alter β-cell mass. Nat. Genet. 31111-115. [DOI] [PubMed] [Google Scholar]
- 27.Kulkarni, R. N., and C. R. Kahn. 2001. Genetic mouse models of insulin resistance, p. 299-323. In J. F. Habener and M. A. Hussain (ed.), Molecular basis of pancreas development and function. Kluwer Academic Publishers, New York, NY.
- 28.Kulkarni, R. N., M. G. Roper, G. Dahlgren, D. Q. Shih, L. M. Kauri, J. L. Peters, M. Stoffel, and R. T. Kennedy. 2004. Islet secretory defect in insulin receptor substrate 1 null mice is linked with reduced calcium signaling and expression of sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA)-2b and -3. Diabetes 531517-1525. [DOI] [PubMed] [Google Scholar]
- 29.Kulkarni, R. N., J. N. Winnay, M. Daniels, J. C. Bruning, S. N. Flier, D. Hanahan, and C. R. Kahn. 1999. Altered function of insulin receptor substrate-1-deficient mouse islets and cultured beta-cell lines. J. Clin. Investig. 104R69-R75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leibiger, B., I. B. Leibiger, T. Moede, S. Kemper, R. N. Kulkarni, C. R. Kahn, L. M. de Vargas, and P. O. Berggren. 2001. Selective insulin signaling through A and B insulin receptors regulates transcription of insulin and glucokinase genes in pancreatic beta cells. Mol. Cell 7559-570. [DOI] [PubMed] [Google Scholar]
- 31.Leroith, D., and D. Accili. 2008. Mechanisms of disease: using genetically altered mice to study concepts of type 2 diabetes. Nat. Clin. Pract. Endocrinol. Metab. 4164-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu, J. L. 2007. Does IGF-I stimulate pancreatic islet cell growth? Cell. Biochem. Biophys. 48115-125. [DOI] [PubMed] [Google Scholar]
- 33.Maedler, K., G. A. Spinas, R. Lehmann, P. Sergeev, M. Weber, A. Fontana, N. Kaiser, and M. Y. Donath. 2001. Glucose induces beta-cell apoptosis via upregulation of the Fas receptor in human islets. Diabetes 501683-1690. [DOI] [PubMed] [Google Scholar]
- 34.Mauvais-Jarvis, F., A. Virkamaki, M. D. Michael, J. N. Winnay, A. Zisman, R. N. Kulkarni, and C. R. Kahn. 2000. A model to explore the interaction between muscle insulin resistance and beta-cell dysfunction in the development of type 2 diabetes. Diabetes 492126-2134. [DOI] [PubMed] [Google Scholar]
- 35.McDaniel, M. L., C. A. Marshall, K. L. Pappan, and G. Kwon. 2002. Metabolic and autocrine regulation of the mammalian target of rapamycin by pancreatic beta-cells. Diabetes 512877-2885. [DOI] [PubMed] [Google Scholar]
- 36.Muller, D., G. C. Huang, S. Amiel, P. M. Jones, and S. J. Persaud. 2006. Identification of insulin signaling elements in human beta-cells: autocrine regulation of insulin gene expression. Diabetes 552835-2842. [DOI] [PubMed] [Google Scholar]
- 37.Newgard, C. B., and J. D. McGarry. 1995. Metabolic coupling factors in pancreatic beta-cell signal transduction. Annu. Rev. Biochem. 64689-719. [DOI] [PubMed] [Google Scholar]
- 38.Ohsugi, M., C. Cras-Meneur, Y. Zhou, E. Bernal-Mizrachi, J. D. Johnson, D. S. Luciani, K. S. Polonsky, and M. A. Permutt. 2005. Reduced expression of the insulin receptor in mouse insulinoma (MIN6) cells reveals multiple roles of insulin signaling in gene expression, proliferation, insulin content, and secretion. J. Biol. Chem. 2804992-5003. [DOI] [PubMed] [Google Scholar]
- 39.Otani, K., R. N. Kulkarni, A. C. Baldwin, J. Krutzfeldt, K. Ueki, M. Stoffel, C. R. Kahn, and K. S. Polonsky. 2004. Reduced beta-cell mass and altered glucose sensing impair insulin-secretory function in betaIRKO mice. Am. J. Physiol. Endocrinol. Metab. 286E41-E49. [DOI] [PubMed] [Google Scholar]
- 40.Patti, M. E., X. J. Sun, J. C. Bruening, E. Araki, M. A. Lipes, M. F. White, and C. R. Kahn. 1995. 4PS/insulin receptor substrate (IRS)-2 is the alternative substrate of the insulin receptor in IRS-1-deficient mice. J. Biol. Chem. 27024670-24673. [DOI] [PubMed] [Google Scholar]
- 41.Persaud, S. J., D. Muller, and P. M. Jones. 2008. Insulin signalling in islets. Biochem. Soc. Trans. 36290-293. [DOI] [PubMed] [Google Scholar]
- 42.Previs, S. F., D. J. Withers, J. M. Ren, M. F. White, and G. I. Shulman. 2000. Contrasting effects of IRS-1 versus IRS-2 gene disruption on carbohydrate and lipid metabolism in vivo. J. Biol. Chem. 27538990-38994. [DOI] [PubMed] [Google Scholar]
- 43.Rutter, G. A. 2001. Nutrient-secretion coupling in the pancreatic islet beta-cell: recent advances. Mol. Aspects Med. 22247-284. [DOI] [PubMed] [Google Scholar]
- 44.Sakaue, H., W. Ogawa, M. Matsumoto, S. Kuroda, M. Takata, T. Sugimoto, B. M. Spiegelman, and M. Kasuga. 1998. Posttranscriptional control of adipocyte differentiation through activation of phosphoinositide 3-kinase. J. Biol. Chem. 27328945-28952. [DOI] [PubMed] [Google Scholar]
- 45.Sun, X. J., L. M. Wang, Y. Zhang, L. Yenush, M. G. Myers, Jr., E. Glasheen, W. S. Lane, J. H. Pierce, and M. F. White. 1995. Role of IRS-2 in insulin and cytokine signalling. Nature 377173-177. [DOI] [PubMed] [Google Scholar]
- 46.Tamemoto, H., T. Kadowaki, K. Tobe, T. Yagi, H. Sakura, T. Hayakawa, Y. Terauchi, K. Ueki, Y. Kaburagi, S. Satoh, et al. 1994. Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature 372182-186. [DOI] [PubMed] [Google Scholar]
- 47.Taniguchi, C. M., K. Ueki, and R. Kahn. 2005. Complementary roles of IRS-1 and IRS-2 in the hepatic regulation of metabolism. J. Clin. Investig. 115718-727. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48.Tseng, Y. H., K. M. Kriauciunas, E. Kokkotou, and C. R. Kahn. 2004. Differential roles of insulin receptor substrates in brown adipocyte differentiation. Mol. Cell. Biol. 241918-1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tuttle, R. L., N. S. Gill, W. Pugh, J. P. Lee, B. Koeberlein, E. E. Furth, K. S. Polonsky, A. Naji, and M. J. Birnbaum. 2001. Regulation of pancreatic beta-cell growth and survival by the serine/threonine protein kinase Akt1/PKBα. Nat. Med. 71133-1137. [DOI] [PubMed] [Google Scholar]
- 50.Ueki, K., P. Algenstaedt, F. Mauvais-Jarvis, and C. R. Kahn. 2000. Positive and negative regulation of phosphoinositide 3-kinase-dependent signaling pathways by three different gene products of the p85α regulatory subunit. Mol. Cell. Biol. 208035-8046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ueki, K., D. A. Fruman, S. M. Brachmann, Y. H. Tseng, L. C. Cantley, and C. R. Kahn. 2002. Molecular balance between the regulatory and catalytic subunits of phosphoinositide 3-kinase regulates cell signaling and survival. Mol. Cell. Biol. 22965-977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ueki, K., T. Okada, J. Hu, C. W. Liew, A. Assmann, G. M. Dahlgren, J. L. Peters, J. G. Shackman, M. Zhang, I. Artner, L. S. Satin, R. Stein, M. Holzenberger, R. T. Kennedy, C. R. Kahn, and R. N. Kulkarni. 2006. Total insulin and IGF-I resistance in pancreatic beta cells causes overt diabetes. Nat. Genet. 38583-588. [DOI] [PubMed] [Google Scholar]
- 53.Valverde, A. M., M. Arribas, C. Mur, P. Navarro, S. Pons, A. M. Cassard-Doulcier, C. R. Kahn, and M. Benito. 2003. Insulin-induced up-regulated uncoupling protein-1 expression is mediated by insulin receptor substrate 1 through the phosphatidylinositol 3-kinase/Akt signaling pathway in fetal brown adipocytes. J. Biol. Chem. 27810221-10231. [DOI] [PubMed] [Google Scholar]
- 54.Van Schravendijk, C. F., A. Foriers, J. L. Van den Brande, and D. G. Pipeleers. 1987. Evidence for the presence of type I insulin-like growth factor receptors on rat pancreatic A and B cells. Endocrinology 1211784-1788. [DOI] [PubMed] [Google Scholar]
- 55.Vlahos, C. J., W. F. Matter, K. Y. Hui, and R. F. Brown. 1994. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-1 (LY294002). J. Biol. Chem. 2695241-5248. [PubMed] [Google Scholar]
- 56.Weir, G. C., and S. Bonner-Weir. 2007. A dominant role for glucose in beta cell compensation of insulin resistance. J. Clin. Investig. 11781-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wicksteed, B., C. Alarcon, I. Briaud, M. K. Lingohr, and C. J. Rhodes. 2003. Glucose-induced translational control of proinsulin biosynthesis is proportional to preproinsulin mRNA levels in islet beta-cells but not regulated via a positive feedback of secreted insulin. J. Biol. Chem. 27842080-42090. [DOI] [PubMed] [Google Scholar]
- 58.Withers, D. J., D. J. Burks, H. H. Towery, S. L. Altamuro, C. L. Flint, and M. F. White. 1999. Irs-2 coordinates Igf-1 receptor-mediated beta-cell development and peripheral insulin signalling. Nat. Genet. 2332-40. [DOI] [PubMed] [Google Scholar]
- 59.Withers, D. J., J. S. Gutierrez, H. Towery, D. J. Burks, J. M. Ren, S. Previs, Y. Zhang, D. Bernal, S. Pons, G. I. Shulman, S. Bonner-Weir, and M. F. White. 1998. Disruption of IRS-2 causes type 2 diabetes in mice. Nature 391900-904. [DOI] [PubMed] [Google Scholar]
- 60.Woodward, G. E., and M. T. Hudson. 1954. The effect of 2-desoxy-d-glucose on glycolysis and respiration of tumor and normal tissues. Cancer Res. 14599-605. [PubMed] [Google Scholar]
- 61.Xuan, S., T. Kitamura, J. Nakae, K. Politi, Y. Kido, P. E. Fisher, M. Morroni, S. Cinti, M. F. White, P. L. Herrera, D. Accili, and A. Efstratiadis. 2002. Defective insulin secretion in pancreatic beta cells lacking type 1 IGF receptor. J. Clin. Investig. 1101011-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zawalich, W. S., G. J. Tesz, and K. C. Zawalich. 2002. Inhibitors of phosphatidylinositol 3-kinase amplify insulin release from islets of lean but not obese mice. J. Endocrinol. 174247-258. [DOI] [PubMed] [Google Scholar]