

Abstract
NF-κB signaling is critical to the survival and transformation of cells infected by the human gammaherpesviruses Epstein-Barr virus and Kaposi's sarcoma-associated herpesvirus. Here we have examined how elimination of the NF-κB transcription factor p50 from mice affects the life cycle of murine gammaherpesvirus 68 (MHV68). Notably, mice lacking p50 in every cell type were unable to establish a sufficiently robust immune response to control MHV68 infection, leading to high levels of latently infected B cells detected in the spleen and persistent virus replication in the lungs. The latter correlated with very low levels of virus-specific immunoglobulin G (IgG) in the infected p50−/− mice at day 48 postinfection. Because the confounding impact of the loss of p50 on the host response to MHV68 infection prevented a direct analysis of the role of this NF-κB family member on MHV68 latency in B cells, we generated and infected mixed p50+/+/p50−/− bone marrow chimeric mice. We show that the chimeric mice were able to control acute virus replication and exhibited normal levels of virus-specific IgG at 3 months postinfection, indicating the induction of a normal host immune response to MHV68 infection. However, in p50+/+/p50−/− chimeric mice the p50−/− B cells exhibited a significant defect compared to p50+/+ B cells in supporting MHV68 latency. In addition to identifying a role for p50 in the establishment of latency, we determined that the absence of p50 in a subset of the hematopoietic compartment led to persistent virus replication in the lungs of the chimeric mice, providing evidence that p50 is required for controlling virus reactivation. Taken together, these data demonstrate that p50 is required for immune control by the host and has distinct tissue-dependent roles in the regulation of murine gammaherpesvirus latency during chronic infection.
Murine gammaherpesvirus 68 (MHV68) is a natural pathogen of rodents and shares many biologic properties with the human pathogens Epstein-Barr virus (EBV) and Kaposi's sarcoma-associated herpesvirus (KSHV). As a gammaherpesvirus, MHV68 is lymphotropic and is associated with pathologies and neoplasia that increase upon loss of host immune control (21, 56, 75). Chronic infection with MHV68 initiates with an acute productive phase of infection and dissemination to secondary lymphoid tissues, leading to the establishment of a quiescent latent virus program in B cells and ultimately memory B cells, with intermittent reactivation events that are controlled by the immune system (27, 56, 87). The genetic tractability of MHV68 and the murine host permit a molecular dissection of key factors and signaling pathways required for chronic infection by the virus, in addition to those processes utilized for immune control by the host (29). NF-κB signaling is one host pathway that is utilized by gammaherpesviruses to promote latency and cell survival (8, 18, 40, 65).
Gammaherpesviruses encode numerous gene products that hijack NF-κB signaling by distinct mechanisms to regulate virus and host gene expression, which in turn promote virus latency and the survival, proliferation, and transformation of infected cells (18, 40, 65). Latent membrane protein 1 of EBV is thought to mimic the function of a constitutively active CD40 receptor, activating NF-κB signaling pathways to promote both survival and proliferation of newly infected B cells (35, 62, 77). The KSHV viral-flice inhibitory protein (vFLIP) induces NF-κB-mediated upregulation of antiapoptotic genes and interleukin-6 (IL-6), driving cell survival and proliferation of lymphoma cells (1, 14, 39). The viral G protein-coupled receptor (vGPCR) of KSHV constitutively activates NF-κB to upregulate angiogenic and proinflammatory factors (3, 51, 69, 80). KSHV vFLIP and vGPCR expression in endothelial cells induces the hallmark spindle-cell morphology of KS tumors (37, 41, 52). Although not constitutively active, the MHV68 vGPCR activates NF-κB, AKT, and p44/p42 mitogen-activated protein kinase in response to the chemokines GROα, IL-8, KC, and MIP-2 (81). The MHV68 vGPCR is required for KC-mediated inhibition of MHV68 lytic replication in culture and influences virus reactivation from latency (47, 53, 81). Notably, pharmacologic inhibitors of NF-κB activation induce apoptosis of KSHV-positive (KSHV+) and EBV+ lymphomas in culture and delay tumor progression of EBV+ and KSHV+ lymphomas in mice, suggesting that the NF-κB pathway may be a good therapeutic target for the elimination of gammaherpesvirus-associated lymphomas (43, 44, 92). However, the roles of NF-κB signaling and the impact of NF-κB inhibition during chronic gammaherpesvirus infection in a natural host setting have not been clearly defined.
MHV68 is detected in proliferating, germinal center B cells at the peak of splenic expansion and is found in isotype class-switched memory B cells at late times postinfection (27, 50, 55, 87). We previously reported the generation of a recombinant MHV68 (MHV68-IκBαM) that expresses a mutant form of IκBα (IκBαM) that potently blocks activation of NF-κB mediated through the canonical activation pathway (46). Notably, we observed that IκBαM expression did not affect acute replication in the lungs upon intranasal infection. However, in vivo inhibition of NF-κB activation substantially reduced the establishment of latency in B cells (46). This is consistent with analyses of other upstream signaling molecules that feed into the canonical and alternative NF-κB activation pathways; B cells that lack the Toll-like receptor-responsive MyD88 and the B-cell costimulatory membrane protein CD40 are also defective reservoirs for latency in vivo (33, 45, 88). Taking the findings together, NF-κB signaling appears to be a key host determinant in the establishment and maintenance of MHV68 latency, yet the roles of individual NF-κB subunits and their impact on viral and cellular gene expression in the context of virus infection have not been determined.
The NF-κB family of transcription factors is comprised of five subunits, NF-κB1 (p105/p50), NF-κB2 (p105/p52), p65/RelA, cRel, and RelB, that form dimers and bind recognition sequences in the promoter regions of genes critical to many aspects of development, inflammation, cell growth, and survival (58). B cells, the primary reservoir for gammaherpesvirus latency, utilize two pathways of NF-κB activation to drive transcriptional events that mediate maturation, response to infection, upregulation of gene products essential for proliferation, protection from apoptosis, and isotype class switching during germinal center reactions (5, 25, 38). In the canonical pathway, engagement of the B-cell receptor, as well as Toll-like receptor ligands and proinflammatory cytokines (e.g., tumor necrosis factor alpha [TNF-α] and IL-1β), leads to the activation of the IKK kinase complex IKK1/IKK2/NEMO, degradation of IκBα, and nuclear translocation of NF-κB dimers (most commonly p50-p65). The alternative pathway of NF-κB activation in B cells initiates with surface receptor engagement of BAFF or CD40 to activate NIK and IKK1-mediated proteolysis of p100 to p52 and subsequent nuclear translocation of p52-containing dimers (6, 16, 17, 42).
NF-κB1 encodes the inactive precursor molecule p105, which is constitutively processed to p50 by the proteasome (54). NF-κB p50 lacks a transactivation domain, and thus its function as an activator or repressor of transcription depends largely on heterodimerization with other NF-κB subunits and interactions with cofactors (59, 91). NF-κB1 p50−/− mice generate typical numbers of lymphocytes and form germinal centers, but they have functional defects in the B-cell compartment (60, 70). B cells lacking p50 proliferate normally in response to surface immunoglobulin cross-linking and membrane-bound CD40L, but they fail to respond to the mitogens lipopolysaccharide and soluble CD40L (72). NF-κB1 p50−/− mice have a substantial reduction in the generation of isotype class-switched immunoglobulin responses to T-dependent antigens (60, 70, 72). The infection of NF-κB p50−/− mice with pathogens can result in heightened or decreased pathology, investigations of which have led to the identification of organ- and cell type-specific roles for NF-κB in the protection from apoptosis and the generation of innate and adaptive host immune responses (5, 58, 59, 70).
Here we report a further examination of the role of NF-κB signaling during chronic gammaherpesvirus infection, initially through the identification of NF-κB subunits that are activated during productive versus latent MHV68 infection. These analyses led to examining the role of p50 in vivo, which determined that this NF-κB family member plays distinct roles in the host response, as well as in regulating the establishment and maintenance of gammaherpesvirus latency.
MATERIALS AND METHODS
Viruses and tissue culture.
γHV68/MHV68 WUMS (ATCC VR1465) was the wild-type (WT) virus. Virus passage and titer determination were performed as previously described (84). In addition, a recombinant MHV68, MHV68-eYFP, which contains a cytomegalovirus (CMV)-driven enhanced yellow fluorescent protein (eYFP) expression construct flanked by a chromatin insulator element isolated from the major histocompatibility complex class II locus (49) that was cloned into the PmlI site of the 27-29B intergenic region of MHV68 to generate MHV68-eYFP, was used in some studies (C. Collins, J. M. Boss, and S. H. Speck, unpublished results). YFP expression could be detected in splenocytes of infected mice between 14 and 18 days postinfection (dpi) at frequencies similar to those determined by limiting-dilution nested PCR (Collins et al., unpublished results). NIH 3T12 cells and mouse embryonic fibroblast (MEF) cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10 U of penicillin per ml, 10 μg of streptomycin per ml, 10% fetal calf serum (FCS), and 2 mM l-glutamate (cMEM). S11 MHV68+ lymphoma cells were maintained in RPMI supplemented with 100 U of penicillin per ml, 100 mg of streptomycin per ml, 10% FCS, and 50 μM beta-mercaptoethanol. Cells were cultured at 37°C in a 5% CO2 environment. MEF cells were prepared as previously described (61) from NF-κB1 p50−/− mice and the control mice on both 129 × C57BL/6 and C57BL/6 backgrounds (strain information is described below).
Mice, infections, and organ harvests.
NF-κB1−/− mice on two genetic backgrounds were acquired from The Jackson Laboratory (Bar Harbor, ME): NF-κB1−/− mice on a 129 × B6 genetic background (B6;129P-Nfkb1tm1Bal/J, catalog no. 00284) and control B6129PF2/J mice (catalog no. 100903); and NF-κB1−/− mice on a BL6 background (B6.Cg-Nfkb1tm1Bal/J, catalog no. 006097) and control C57BL/6J mice (catalog no. 000664). Mice were housed in the Whitehead Vivarium of Emory University in accordance with federal and university guidelines. The p50−/− mice and the control mice were bred and housed under identical conditions in static, microisolator cages with autoclaved water and food. The bone marrow (BM) chimeras were also maintained in sterile conditions throughout the experiments. All protocols for animal studies were approved by the Institutional Animal Care and Use Committee of Emory University.
Mice were placed under isofluorane anesthesia prior to intranasal inoculation with 100 PFU or 1,000 PFU of virus in 20 μl of cMEM. For the global p50−/− experiments, mice were used at between 2 and 4 months of age. For the BM chimera experiments, mice were generally 4 months of age at the time of infection. Mice were sacrificed by use of isofluorane or carbon dioxide. Spleens were harvested into cMEM, homogenized, and filtered through a 100-μm-pore-size nylon cell strainer (Becton Dickinson, Franklin Lakes, NJ). Erythrocytes were removed with red blood cell lysis buffer (Sigma, St. Louis, MO). Except where indicated, pooled splenocytes from three to five mice were used in all experiments. Whole lungs or specific lobes of lung tissue were harvested and immediately frozen in cMEM at −80°C. Blood was acquired by a retro-orbital bleed, and the serum was isolated and stored at −80°C.
Generation of NF-κB1 mixed BM chimeric mice.
B6.SJL-PtprcaPepb/BoyJ WT (Ly5.1) mice (catalog no. 002014; The Jackson Laboratory) were lethally irradiated (950 rads) with two doses of 475 rads at a 20-h interval and reconstituted with BM from both p50+/+ (Ly5.1+) and p50−/− (Ly5.2+, B6.Cg-Nfkb1tm1Bal/J) mice. BM cells were flushed from femurs, depleted of red blood cells, and mixed. Recipient mice (Ly5.1) were anesthetized with 2,2,2-tribromoethanol, and a total of 1.4 × 107 to 2 × 107 cells in 100 μl phosphate-buffered saline (PBS) were injected intraocularly. Mice were rested for 8 weeks to allow for reconstitution, which was confirmed by flow cytometry analysis of peripheral blood lymphocytes, before MHV68 infection. Chimeric mice received neomycin and polymyxin B (Sigma-Aldrich) in acidified drinking water until reconstitution was confirmed.
Plaque assays.
Plaque assays were performed as previously described (15), with minor modifications. NIH 3T12 cells were plated in six-well pates 1 day prior to infection at 2 × 105 cells per well. Organs were subjected to four rounds of mechanical disruption of 1 min each using 1.0-mm zirconia/silica beads (Biospec Products, Bartsville, OK) in a Mini-Beadbeater-8 (Biospec Products). Serial 10-fold dilutions of organ homogenate were plated on NIH 3T12 monolayers in a 200-μl volume. Infections were performed for 1 h at 37°C with rocking every 15 min. Immediately after infection, plates were overlaid with 1 to 2% methylcellulose in cMEM. After 6 to 8 days, the methylcellulose was aspirated and washed with PBS, the monolayer was fixed with methanol and stained with 0.1% crystal violet in 20% methanol, and plaques were scored. The limit of detection for this assay is 50 PFU per organ.
Nuclear extract preparation and electrophoretic mobility shift analysis.
S11 cells were harvested, washed once in PBS, and resuspended in sucrose buffer (320 mM sucrose, 10 mM Tris HCl [pH 8.0], 3 mM CaCl2, 2 mM magnesium acetate, 0.1 mM EDTA, 0.5% Nonidet P-40 [NP-40], 1 mM dithiothreitol [DTT], 0.5 mM phenylmethylsulfonyl fluoride [PMSF]). Nuclei were spun down, washed, and resuspended in a low-salt buffer (20 mM HEPES [pH 7.9], 1.5 mM MgCl2, 20 mM KCl, 0.2 mM EDTA, 25% glycerol, 0.5 mM DTT, 0.5 mM PMSF) prior to the gradual addition of an equal volume of high-salt buffer (20 mM HEPES [pH 7.9], 1.5 mM MgCl2, 800 mM KCl, 0.2 mM EDTA, 25% glycerol, 1% NP-40, 0.5 mM DTT, 0.5 mM PMSF, protease inhibitor cocktail [Roche, Mannheim, Germany]) followed by vigorous shaking at 4°C. The supernatant collected after centrifugation at 14,000 × g for 15 min at 4°C was the nuclear extract.
Adherent cells were harvested, washed once in PBS, and resuspended in hypotonic lysis buffer (10 mM HEPES [pH 7.9], 10 mM KCl, 1.5 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 0.5 mM PMSF, protease inhibitor cocktail [Roche]) for 15 min prior to the addition of 1/20 volume of 10% NP-40. Nuclei were spun down, washed in hypotonic lysis buffer, and then resuspended in high-salt buffer (25% glycerol, 20 mM HEPES [pH 7.9], 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 0.5 mM PMSF, protease inhibitor cocktail [Roche]) with vigorous shaking at 4°C. The supernatant collected after centrifugation at 10,000 × g for 10 min at 4°C was the nuclear extract.
Nuclear extracts were assayed for NF-κB activation by electrophoretic mobility shift assay (EMSA). A 2.5- to 7.5-μg amount of nuclear extract was incubated with a 32P-labeled oligonucleotide containing the NF-κB consensus site, AGTTGAGGGGACTTTCCCAGGC, in a binding reaction mixture containing 2 mM HEPES (pH 7.9), 1 mM EDTA, 5 mM DTT, 0.05% Triton X-100, 5% glycerol, and 2 μg poly(dI-dC) (Roche) for 30 min at room temperature. Competition experiments were performed with 20- and 100-fold molar excesses of unlabeled oligonucleotides containing the WT or mutated NF-κB consensus site (AGTTGAGGCGACTTTCCCAGGC). Supershift experiments were performed by overnight incubation of nuclear extracts with 0.2 to 4.0 μg of antibodies against NF-κB subunits (Santa Cruz Biotechnology, Santa Cruz, CA) at 4°C. Nucleoprotein complexes were subjected to electrophoresis in 5% native polyacrylamide gels at 180 V, dried under vacuum, and analyzed by phosphorimager analysis (Typhoon 9410; Amersham Biosciences, Piscataway, NJ).
Magnetic cell separation.
Murine B cells were isolated by depletion of non-B cells using a B-cell isolation kit (Miltenyi Biotec, Cologne, Germany) per the manufacturer's recommendations. Briefly, cells were resuspended at 2 × 108 cells/ml in 1× PBS containing 0.5% FCS, followed by staining with Fc block (0.125 μg/106 cells) on ice for 15 min. Cells were labeled with biotin-antibody cocktail (biotin-conjugated antibodies against CD43, CD4, and Ter-119) at 10 μl per 1 × 107 cells for 15 min on ice, followed by staining with antibiotin microbeads at 20 μl per 1 × 107 cells for 15 min on ice. Cells were washed twice with PBS containing 0.5% FCS and subjected to magnetic separation using the autoMACS (Miltenyi Biotec, Cologne, Germany). Following separation, stained cell populations were analyzed by flow cytometry as described below.
Flow cytometry.
Cells resuspended in PBS supplemented with 2% FCS were stained for fluorescence-activated cell sorting (FACS) using a combination of the following antibodies: R-phycoerythrin (PE)-conjugated antibodies to CD19, CD95, CD44, and immunoglobulin D (IgD); fluorescein isothiocyanate (FITC)-conjugated antibodies to GL7, Vβ4, Ly5.1/CD45.1, IgG1, IgG2a/2b, and IgG3; allophycocyanin (APC)-conjugated antibodies to CD19, CD62L, Ly5.2/CD45.2, and Ly5.1; PE-Cy7-conjugated antibodies to CD95 and CD69; Pacific Blue-conjugated antibody to CD8; and peridinin chlorophyll protein-Cy5.5-conjugated antibody to Ly5.2/CD45.2. Rat anti-mouse CD16/CD32 (Fc block) was used to block Fc receptors prior to staining. All reagents were obtained from BD Biosciences (San Jose, CA) and EBioscience (San Diego, CA).
For FACS, cells were resuspended at 2 × 107cells per ml and incubated for 30 min on ice in PBS containing 1% FCS and Fc block. Cells were stained with PE- and FITC-conjugated antibodies at 5 μl per 1 × 107 cells by incubation for 30 min on ice in the dark. Cells were washed twice with PBS containing 1% FCS and resuspended at 1 × 108 cells/ml. Stained cell populations were acquired using a FACSvantage instrument (BD Biosciences). Sorted and unsorted cell populations were resuspended in cMEM supplemented with 10% dimethyl sulfoxide and stored at −80°C for limiting-dilution PCR analyses or in cMEM at 4°C for limiting-dilution ex vivo reactivation analyses as described below.
For flow cytometry analysis, cells were resuspended at 1 × 106 cells per ml in PBS containing 2% FCS for 20 min on ice in the dark and stained with a 1:100 to 1:300 dilution of the conjugated antibody. Data was collected on an LSRII instrument (BD Biosciences) and analyzed using FlowJo software (Treestar, Ashland, Oregon).
Limiting-dilution nested-PCR detection of MHV68 genome-positive cells.
Limiting-dilution analysis to determine the frequency of cells harboring the viral genome was performed using a single-copy-sensitive nested PCR assay amplifying a region of ORF50, as previously described (85). Briefly, frozen samples were thawed, counted, resuspended in isotonic buffer, and plated in serial threefold dilutions in a background of 104 uninfected NIH 3T12 cells in 96-well plates (Eppendorf Scientific, Westbury, NY). Plates were covered with PCR foil (Eppendorf), and cells were lysed with proteinase K for 6 h at 56°C. Ten microliters of round 1 PCR mix was added to each well by foil puncture. Following first-round PCR, 10 μl of round 2 PCR mix was added to each well by foil puncture, and samples were subjected to round 2 PCR. All cell lysis and PCR were performed on a PrimusHT thermal cycler (MWG Biotech, High Point, NC). Products were resolved by ethidium bromide staining on 2% agarose gels. Twelve PCRs were performed for each sample dilution, and a total of six dilutions were performed per sample. Every PCR plate contained control reactions (uninfected cells and 10 copies, 1 copy, and 0.1 copy of plasmid DNA in a background of 104 cells). All of the assays demonstrated approximately single-copy sensitivity with no false positives.
Limiting-dilution ex vivo reactivation analyses.
Limiting-dilution analysis to determine the frequency of cells containing virus capable of reactivating from latency was performed as previously described (85, 86). Briefly, bulk splenocytes or sorted cell populations were resuspended in cMEM and plated in serial twofold dilutions (starting with 105 cells) onto MEF monolayers in 96-well tissue culture plates. Twelve dilutions were plated per sample, and 24 wells were plated per dilution. Wells were scored for cytopathic effect (CPE) at 21 to 28 days postplating. To detect preformed infectious virus, parallel samples of mechanically disrupted cells were plated onto MEF monolayers. This process kills >99% of live cells, which allows preformed infectious virus to be discerned from virus reactivating from latently infected cells. Unless indicated otherwise, significant levels of preformed virus were not detected in these assays.
Limiting-dilution analysis of persistent viral replication in the lungs.
Preformed infectious virus in the lungs was measured by a modified form of the limiting-dilution ex vivo reactivation assay described above. Briefly, the lower left lobe of the lung from each animal was subjected to four rounds of mechanical disruption of 1 min each using 1.0-mm zirconia/silica beads followed by an additional four rounds of disruption using 0.5-mm zirconia/silica beads (BioSpec Products, Bartlesville, OK). Disruption of cells was verified by trypan blue exclusion (Sigma). The homogenate was brought up to 3.4 ml with cMEM and plated in twofold serial dilutions onto MEFs in 96-well tissue culture plates. Twelve dilutions of 16 replicate wells were plated. CPE was monitored microscopically at 14 to 21 days postplating.
Antibody ELISAs.
For MHV68-specific IgG assay, plates were first coated with a 1% final concentration of paraformaldehyde-fixed viral antigen for sample wells, with standard wells coated with a 2-μg/ml concentration of donkey anti-mouse IgG (Jackson ImmunoResearch) in coating buffer (0.1 M Na2CO3, 0.2% NaN3, pH 9.6), and incubated either at 37°C for 2 h or at 4°C overnight, and then plates were blocked with 3% bovine serum albumin in PBS and incubated for 2 h at 37°C. After washing of the plates with enzyme-linked immunosorbent assay (ELISA) wash buffer (200 ml 10× PBS, 1 ml Tween 20, H2O added to 2000 ml), serum samples or mouse IgG at a concentration of 1 μg/ml (Jackson ImmunoResearch) was added to each well at a threefold dilution in ELISA diluent (BD Bioscience) and then incubated for 3 h at 37°C. Plates were then washed, and horseradish peroxidase-conjugated donkey anti-mouse IgG (Jackson ImmunoResearch) was added at a 1:5,000 dilution and incubated for 2 h at 37°C. Plates were washed, and 100 μl of 1:1 developer was added (BD Bioscience). The reaction was stopped after 10 min with 50 μl stop solution (BD Bioscience), and plates were read on a microplate reader (Synergy HT; BioTek). The concentration of antibody in serum samples was determined by using a standard curve generated by threefold serial dilutions of standard in the same assay.
Statistical analyses.
All data were analyzed by using GraphPad Prism software (GraphPad Software, San Diego, CA). Titer data were statistically analyzed with the Mann-Whitney nonparametric, unpaired two-tailed t test. Based on the Poisson distribution, the frequencies of reactivation and viral genome-positive cells were obtained from the nonlinear regression fit of the data where the regression line intersected 63.2%. The frequencies of reactivation and genome-positive cells were statistically analyzed by unpaired two-tailed t test of the log 63.2% effective concentration.
RESULTS
Characterization of NF-κB activation during infection.
The S11 cell line harbors MHV68 in a predominantly latent state (78) and was observed to have constitutive NF-κB activation as assessed using an NF-κB luciferase reporter assay (data not shown). To identify the NF-κB subunits that are activated in this latently infected B-cell line, EMSAs were performed with nuclear extracts prepared from S11 cells cultured under normal growth conditions. Two distinct mobility shifts were evident upon incubation with a labeled oligonucleotide containing a consensus NF-κB recognition sequence (Fig. 1A). The addition of antibody against the NF-κB subunit p50 largely resulted in an additional shift in the mobility of the lower complex. Antibody against c-Rel ablated detection of the upper complex, whether added alone or in combination with antibodies against p50 or p65. Taken together, these findings indicate that p50 and cRel are constitutively present in the nuclei of the latently MHV68-infected S11 B lymphoma cells.
FIG. 1.

NF-κB p50 is active during latent and lytic infection. EMSA analysis of NF-κB binding in MHV68-infected cells is shown. (A) Nuclear extracts were prepared from S11 cells, MHV68+ B-cell lymphoma cells, cultured under normal growth conditions. The molar fold excess of WT or mutant (mut) competitor and antibodies against NF-κB subunits for supershift analysis are indicated. (B) Nuclear extracts were prepared from primary MEFs infected at an MOI of 5.0 from 30 min to 24 h. PAA, phosphonoacetic acid treatment. (C) Identification of NF-κB subunits by supershift analysis of nuclear extracts from MEFs treated for 15 min with TNF-α or infected for 24 h. Antibodies against NF-κB subunits are indicated. CTRL and p50−/− reflect the nuclear extracts from MEFs of control and p50−/− mice on a C57BL/6 background, which were prepared and infected in parallel. Arrows point to specific complexes, brackets indicate supershifted complexes, and dots indicate nonspecific complexes.
We previously observed that MHV68 activates NF-κB during virus replication in permissive murine fibroblasts (46). To further examine the activation profile upon de novo infection of permissive cells, nuclear extracts were prepared from primary MEFs during a 24-h time course following infection at a high multiplicity of infection (MOI) of 5 PFU per cell and analyzed for NF-κB binding by EMSA. Low levels of NF-κB activation were evident by 6 h postinfection, but robust activation was not observed until 12 to 24 h postinfection in MEF cultures (Fig. 1B). Activation of NF-κB was largely repressed upon the inhibition of viral DNA synthesis by phosphonoacetic acid, indicative of late lytic cycle activation of NF-κB signaling in fibroblast cells by MHV68. Supershift analyses revealed that the NF-κB complexes from the nuclear extracts of infected fibroblasts contained the p50 and p65/RelA and subunits, as seen upon treatment with TNF-α (Fig. 1C). The NF-κB-specific complex was also present in the nuclear extracts of an independent preparation of MHV68-infected control MEFs but was absent in the nuclear extracts of infected MEFs from p50−/− mice on the C57BL/6 genetic background that were prepared in parallel with control MEFs (Fig. 1C). The p65/p50 heterodimer is consistent with a classical-pathway-mediated activation of NF-κB during a post-DNA replication stage of MHV68 infection. Taken together, the gel shift analyses identified distinct NF-κB complexes activated during latent infection and virus replication, with the p50 subunit being common to both.
Altered kinetics of establishment of MHV68 latency in the absence of NF-κB p50.
Based on the identification of p50 as a component of NF-κB activation during MHV68 latency, we further characterized the role of p50 during MHV68 infection of mice. We first examined the ability of p50−/− mice to control and clear acute MHV68 replication. Upon intranasal inoculation, the acute phase of MHV68 infection is characterized by lytic replication in the lung that is cleared from normal immunocompetent mice within 2 weeks. However, following intranasal infection of p50−/− mice with 100 PFU of WT MHV68, ca. 10- and 100-fold-higher levels of virus replication in the lungs were observed compared to that in control mice at 9 and 12 dpi, respectively (Fig. 2A). However, by 17 dpi, both control and p50−/− mice had cleared MHV68 from the lungs as assessed using a standard plaque assay (Fig. 2A). Notably, our previous investigation of the role of NF-κB signaling during MHV68 infection, utilizing a recombinant MHV68 that expresses the NF-κB superrepressor IκBαM under the control of the CMV immediate-early promoter (MHV68-IκBαM), determined that NF-κB activation was dispensable for virus replication in fibroblasts and in the lungs of mice (46). In addition, the recombinant MHV68-IκBαM did not exhibit a replication defect in SVEC 4-10 murine endothelial cells or RLE-6TN rat lung epithelial cells (data not shown). However, this approach did not address NF-κB-dependent roles during the earliest stages of infection prior to CMV immediate-early promoter-driven IκBαM expression.
FIG. 2.

Infection of NF-κB1 p50−/− mice with MHV68 results in elevated levels of acute replication in the lungs and a delay in the peak of splenic latency. (A) Acute replication in the lung. Control (CTRL, B6x129PF2/J) mice or NF-κB1 p50−/− (B6;129P) mice were infected with 100 PFU. On the indicated dpi, lungs were harvested and disrupted, and titers were determined on NIH 3T12 fibroblasts. The data were compiled from one or two experiments with three to five mice analyzed per experiment. Data are shown as log10 titer, and the bar indicates the geometric mean titer. The dashed line indicates the limit of detection of this assay as log101.7 or 50 PFU/ml of sample homogenate. Significant differences between CTRL and p50−/− mice at 9 dpi (P = 0.0060) and 12 dpi (P < 0.0001) were identified. (B) Multistep growth curve in MEFs isolated from CTRL or p50−/− mice infected with an MOI of 0.001 PFU per cell. Samples were harvested at the indicated time points, and titers were determined on NIH 3T12 cells. (C and D) Frequency of splenocytes harboring viral genomes. Splenocytes were harvested from control mice (CTRL, B6) or NF-κB1 p50−/− mice (B6) at the indicated dpi with 100 PFU of WT MHV68. Limiting-dilution viral genome PCR analysis was utilized to determine the frequency of latency for CTRL mice and p50−/− mice at 18 dpi (1/425 and 1/5,066, respectively; P = 0.0378) and 28 to 29 dpi (1/1,847 and 1/71, respectively). (E and F) Frequency of splenocytes reactivating virus. Splenocytes were harvested from control mice (CTRL, B6) or NF-κB1 p50−/− mice (p50−/−, B6) at the indicated dpi with 100 PFU of WT MHV68. Limiting-dilution reactivation analysis was utilized to determine the frequency of ex vivo reactivation for CTRL mice and p50−/− mice at 18 dpi (1/6,161 and 1/82,514, respectively; P < 0.0001) and 28 to 29 dpi (1/18,784 and <1/100,000, respectively). In parallel, as indicated by the open symbols, mechanically disrupted cells were plated to detect the presence of preformed infectious virus. For both limiting-dilution assays, curve fit lines were derived from nonlinear regression analysis, and symbols represent the mean percentage of wells positive for virus (viral DNA or CPE) ± the standard error of the mean. The dotted line represents 63.2%, from which the frequency of viral genome-positive cells or the frequency of cells reactivating virus was calculated based on the Poisson distribution. The data shown represent at least two independent experiments with spleen cells pooled from three to five mice per experimental group.
To determine whether the complete absence of NF-κB p50 affects the kinetics of MHV68 replication, multistep growth curves in primary WT and p50−/− MEFs were performed. Following a low-MOI (0.001) infection, the kinetics of MHV68 replication in p50−/− MEFs were very similar to that observed in control MEFs (Fig. 2B). Therefore, based on the in vivo results previously obtained with the MHV68-IκBαM recombinant virus, coupled with the absence of an obvious replication defect in p50−/− MEFs, it seems unlikely that the observed heightened levels of acute virus replication in the p50−/− mice are the consequence of p50 regulating MHV68 replication. Rather, these results suggest that the impact of the loss of p50 on the host immune response leads to an inability to efficiently control MHV68 replication.
We extended our analyses of MHV68 infection in p50−/− mice by examining latency in the spleen. At 18 dpi, the peak period of splenic latency in normal mice infected with 100 PFU of MHV68, there was a substantial, 12-fold defect in the establishment of latency in p50−/− mice compared to control mice (1/5,066 versus 1/425), despite increased levels of acute virus replication in the lungs of p50−/− mice at 9 and 12 dpi (Fig. 2C). Using an ex vivo limiting-dilution reactivation assay, we observed a 13-fold defect in reactivation from latency at 18 dpi in the p50−/− mice (1/82,514) compared to control C57BL/6 mice (1/6,161) (Fig. 2D), which closely correlates with the defect in establishment of latency and suggests that p50 has no additional role in MHV68 reactivation once latency is established in the spleen.
To assess whether there might be a delay in dissemination and/or expansion of splenic latency in p50−/− mice, we examined a later time point. In striking contrast to the results obtained at 16 to 18 dpi, at 28 to 29 dpi we observed a complete reversal of the phenotype. In p50−/− mice, the levels of latency were increased 26-fold (1/225) over that observed in the control C57BL/6 (1/1,847) (Fig. 2E). The observed delay in seeding splenic latency in the p50−/− mice was further substantiated by the increased levels of ex vivo reactivation of p50−/− (1/18,784) compared to those in their normal C57BL/6 counterparts at 28 to 29 dpi (<1/100,000) (Fig. 2F). Thus, it appears that MHV68-infected p50−/− mice exhibit a pronounced delay in the peak expansion of splenic latency in spite of the heightened levels of acute virus replication in the lungs.
MHV68 gains access to germinal center B cells in p50−/− mice.
The dramatic alteration in the kinetics of splenic latency in p50−/− mice raised the possibility that MHV68 infection in these mice might be skewed toward different B-cell subsets than observed in p50-sufficient B cells, in which the majority of infected cells at the peak of latency exhibit a germinal center phenotype (27, 50, 55, 87). To characterize B-cell infection, we infected p50−/− and control mice with a recombinant MHV68 (MHV68-eYFP) that expresses YFP at detectable levels in splenocytes at periods of peak splenic latency (Fig. 3). As expected, the mean percentage of CD19+/YFP+ cells was consistently higher in control mice (0.6% ± 0.6%) than p50−/− mice (0.11% ± 0.03%) at 18 dpi. By 31 dpi the percentage of CD19+/YFP+ cells (0.02% ± 0.01%) in control mice (p50 sufficient) was dramatically reduced, in agreement with the characteristic decay of latency in the spleen as determined by limiting-dilution PCR analyses for the frequency of cells harboring the viral genome (Fig. 2). In addition, our characterization of YFP marking in WT C57BL/6 mice has revealed that transgene expression is extinguished at later times postinfection (C. M. Collins and S. H. Speck, unpublished data). However, as determined by limiting-dilution analysis of the frequency of viral genome-positive cells (Fig. 2), p50−/− mice had a greater percentage of YFP+ cells (1.4% ± 1.9%) at 31 dpi (Fig. 3). It is important to note that following low-dose intranasal inoculation (100 PFU), we have observed, upon analyzing latency in individual mice, significant mouse-to-mouse variation in the levels of viral latency (unpublished data), which underscores the need to pool latently infected cells from groups of four or five mice per experiment. Regardless, the kinetics and frequency of YFP+ B cells in both control and p50−/− mice closely correlated with the frequency of infected cells determined by the limiting-dilution PCR assay, indicating efficient marking of the infected cells with the YFP-expressing recombinant MHV68.
FIG. 3.

MHV68-eYFP gains access to similar B-cell reservoirs in p50−/− mice and control C57/BL6 mice. Cells were prepared from spleens harvested from uninfected age-matched (naïve) mice or from three to five mice that were infected 18 or 31 days prior to harvest with 100 PFU of MHV68-eYFP. Subsets were derived from initial FACS gating on live lymphocyte populations, CD19+, and then eYFP+. Activated cells were gated on CD19+/YFP+/CD69hi. Germinal center cells were gated on CD19+/YFP+/GL7hi/CD95hi. The data in the gated populations are the percentage of YFP+ cells that fall within the gate and are shown for a representative mouse. The means and standard deviations for these subsets are shown on each flow plot in brackets and were determined from the analysis of three infected mice.
To assess the distribution of MHV68 in specific p50-deficient B-cell populations, we analyzed the expression at 18 and 31 dpi of B-cell surface markers indicative of activated (CD69+) and germinal center (GL7+ CD95+) (Fig. 3). Remarkably, the distribution of MHV68-eYFP among these B-cell populations appeared to be very similar in control versus p50−/− mice. We observed that a slightly higher percentage of infected YFP+ p50−/− B cells had upregulated expression of the activation marker CD69 at 18 dpi, while a lower percentage of infected YFP+ p50−/− B cells upregulated CD69 at 31 dpi compared to control infected YFP+ B cells (Fig. 3). However, the frequency of activation at the peak of splenic latency for the control mice at 18 dpi (58.7 ± 2.0) closely approximated the frequency for p50−/− mice at 31 dpi (64.0 ± 5.4) (Fig. 3). As previously noted (27, 50, 55, 87), in both control and p50−/− mice, virus infection was found predominately in B cells exhibiting surface markers characteristic of germinal center B cells. Overall, while the absence of p50 led to significant alterations in the kinetics of establishing splenic latency, ultimately the virus appeared to reach the same B-cell reservoir independent of p50 expression.
Increased splenic latency at late times postinfection correlates with both a decreased humoral immune response against MHV68 and increased persistent virus replication in the lungs of p50−/− mice.
In normal immunocompetent mice the peak of MHV68 latency at 16 to 18 dpi is followed by a significant decline in the frequency of splenocytes harboring virus, due to either egress of the latently infected cells from the spleen and/or clearance of these cells. The latter likely reflects a combination of normal turnover of specific B-cell populations, virus-induced cell death, and immune-mediated clearance of MHV68-infected B cells. Thus, we assessed whether the expansion of latency observed at 28 to 29 dpi in p50−/− was transient or persisted in these animals. Notably, at 48 to 49 dpi (Fig. 4A), the frequency of viral genome-positive splenocytes (1/135) was comparable to the levels observed at 28 to 29 dpi (1/71), in contrast to the expected decline in the frequency of viral genome-positive splenocytes observed in control mice at 48 to 49 dpi (1/5,685). Importantly, by 48 to 49 dpi, there was little or no preformed infectious virus detected in the spleens of p50−/− mice, and the frequency of splenocytes that reactivate virus upon explant was below the limit of accurate quantitation of the limiting-dilution reactivation assay (data not shown). By 3 months postinfection (105 to 106 dpi), the frequency of viral genome-positive splenocytes was ca. 100-fold higher in the p50−/− mice than in the control mice, with p50−/− mice maintaining latency at approximately the same level as observed at 28 to 29 and 48 to 49 dpi (Fig. 4B). We further purified splenic B cells and determined that, as expected, B cells remain the primary latency reservoir for MHV68, even in the absence of p50; the frequency of viral genome-positive B cells (1/118) was nearly identical to the frequency of bulk splenocytes harboring MHV68 (1/142) (Fig. 4B).
FIG. 4.

Hyperestablishment of splenic latency and persistence in the lungs is associated with a lack of serum IgG against MHV68 in NF-κB1 p50−/− mice. (A) Frequency of splenocytes harboring viral genomes at 45 to 49 dpi. Bulk splenocytes were prepared from two sets of control mice (CTRL, B6 and B6x129PF2/J) or NF-κB1 p50−/− mice (p50−/−, B6 and B6;129P) infected with 100 PFU of WT MHV68. Limiting-dilution viral genome PCR analysis was utilized to determine the frequency of latency for bulk splenocytes from CTRL mice and p50−/− mice at 45 to 49 dpi (1/5,685 and 1/135, respectively). (B) Frequency of splenocytes harboring viral genomes at 105 to 106 dpi. Bulk splenocytes and CD19+ B cells were prepared from control mice (CTRL, B6) or NF-κB1 p50−/− mice (p50−/−, B6) with 100 PFU of WT MHV68. Limiting-dilution viral genome PCR analysis was utilized to determine the frequency of latency for bulk splenocytes from CTRL mice and p50−/− mice at 105 to 106 dpi (1/35,950 and 1/118, respectively) and for B cells (1/32,500 and 1/142, respectively). Postseparation FACS analysis indicated that the mean purities for CD19+ cells were 93% ± 2.1% for CTRL and 91.4% ± 0.1% for p50−/−. Curve fit lines were derived from nonlinear regression analysis, and symbols represent the mean percentage of wells positive for viral DNA ± the standard error of the mean. The dashed line represents 63.2%, from which the frequency of viral genome-positive cells was calculated based on the Poisson distribution. The data shown represent at least two independent experiments with spleen cells pooled from three to five mice per experimental group. (C) Persistence in the lungs. Lung tissue was isolated from three or four infected control mice (CTRL, BL6) or NF-κB1 p50−/− mice (p50−/−, BL6) at the indicated dpi with 100 PFU of WT MHV68. Bars for each sample represent the mean percentage for 16 wells positive for CPE upon plating fourfold dilutions (1:10, 1:40, and 1:160) of mechanically disrupted lung tissue from an individual mouse on an indicator MEF monolayer. (D) Determination of total IgG production in sera of individual naïve versus infected CTRL and p50−/− mice. The bar represents the geometric mean titer. The mean total IgG was significantly different for comparisons of naïve or infected sera within or between CTRL and p50−/− mice (P < 0.006). (E) Determination of MHV68-specific IgG production in sera of individual infected mice. The bar represents the geometric mean titer. Naïve sera from both strains of mice had undetectable levels of MHV68-specific IgG (data not shown). The mean anti-MHV68 IgG titer was significantly different (P < 0.0001).
How are the high levels of MHV68 latency maintained in the p50−/− mice? We hypothesized that this might be the consequence of virus reactivation fueling persistent virus replication, likely at a distal site, since there was no evidence of ongoing virus replication in the spleens of p50−/− mice at late times postinfection. The lungs are one site that has previously been shown to support persistent virus replication in some settings (11, 73, 88). To assess the presence of low-level virus replication in the lungs of p50−/− and control mice at late times postinfection, lungs of infected mice were disrupted and plated in a limiting-dilution assay on permissive MEF monolayers. This approach substantially increases the sensitivity for detecting preformed infectious virus compared to a standard plaque assay (79), enabling the detection of low levels of virus replication at 18 dpi (Fig. 4C) that were not detected in the lungs at 17 dpi by plaque assay (Fig. 2A and 4C and data not shown). Notably, intermittent levels of virus were detected in lung homogenates at early and intermediate time points in control mice, consistent with sporadic reactivation or low-level persistent virus replication in the lungs (11, 73, 88). However, by 105 to 106 dpi, preformed infectious virus was barely detectable in one of eight control mice examined (Fig. 4C). Unlike the case in the control mice, increasing levels of virus replication were detected in the p50−/− mice with time, and an end point dilution was not reached in the lung homogenates from p50−/− mice at 105 to 106 dpi (Fig. 4C). This indicates that the p50−/− mice are unable to adequately control MHV68 infection in the lungs, leading to persistent low-level virus replication that was not observed in the control mice 3 months after infection.
The high levels of splenic latency, coupled with the detection of increased persistent virus replication in the lungs at late times after infection of p50−/− mice, suggest that these animals mount a suboptimal immune response to chronic MHV68 infection. It is known that B cells in NF-κB−/− mice fail to proliferate in response to some mitogens, have reduced levels of serum immunoglobulin and isotype class-switched B cells, and fail to generate high-affinity antibodies in response to T-dependent antigens (38, 60, 70). Thus, we assessed antibody responses in the p50−/− mice. As previously observed for responses to T-dependent antigens, there was a log reduction in total IgG in naïve and infected p50−/− mice compared to control mice (Fig. 4D). Strikingly, there was a substantially more severe defect in the IgG response generated against MHV68 antigens in the p50−/− mice compared to control mice (Fig. 4E). Notably, failure to generate a robust humoral immune response against MHV68 has been associated with higher viral loads in latency reservoirs, as well as recrudescence and persistent virus replication in the lungs of immunodeficient mice (32, 48, 88).
The number and size of germinal centers in p50−/− mice are smaller than those in control mice upon immunization with nitrophenol-conjugated keyhole limpet hemocyanin (60). This led us to further examine the response of p50−/− B cells to MHV68 infection. Initially, we characterized B- and T-cell populations in control and p50−/− mice and did not detect any gross alterations over the course of MHV68 infection in the frequencies of B cells, CD4+ T, cells or CD8+ T cells, with the exception that the relative percentage of CD4+ T cells was elevated in p50−/− compared to control mice at 105 dpi (Table 1). We then extended these analyses to further examine B-cell responses. Germinal center participation in infected p50−/− mice and control mice was assessed by flow cytometry, with staining for the surface markers GL7 and CD95. Germinal center expansion of CD19+ B cells was observed at 28 dpi in p50−/− mice (Table 2). However, at all time points examined, the infected p50−/− mice had significantly fewer germinal center B cells than MHV68-infected control mice.
TABLE 1.
Analysis of B- and T-cell populations in control C57BL/6 versus p50−/− micea
| dpi | Mean (SD) %b
|
|||||
|---|---|---|---|---|---|---|
| B cells
|
CD4+ T cells
|
CD8+ T cells
|
||||
| Control | p50−/− | Control | p50−/− | Control | p50−/− | |
| 0 (naïve mice) | 58.9 | 62.0 | 19.9 (3.2) | 18.8 (2.8) | 13.8 (1.8) | 11.9 (3.3) |
| 18 | 56.9 (3.4) | 52.8 (4.6) | 16.4 (3.1) | 20.0 (1.7) | 11.3 (1.8) | 12.1 (3.2) |
| 28 | 53.0 (3.3) | 47.5 (5.0) | 18.3 (2.5) | 15.4 (2.5) | 14.0 (2.8) | 7.9 (0.7) |
| 45-56 | 52.7 (6.6) | 49.6 (6.0) | 26.4 (2.8) | 26.3 (1.8) | 18.8 (3.6) | 10.5 (1.9) |
| 105 | NA | NA | 15.7 (3.1) | 25.1 (4.2)* | 15.7 (4.0) | 14.1 (6.4) |
Mice were infected with 100 PFU of MHV68 by intranasal inoculation. Subsets were derived from initial FACS gating on live lymphocytes.
The data shown are the percentages for each subset derived from FACS analysis of 3 to 5 individual infected mice or 13 naïve mice pooled from the individual harvest days. *, significant difference (P < 0.05) between infected control and p50−/− mice. B cells were determined by gating on CD19+ live lymphocytes, CD4+ T cells were gated on CD4+/CD8− live lymphocytes, and CD8+ T cells were gated on CD4−/CD8+ live lymphocytes. NA, not analyzed.
TABLE 2.
Analysis of B-cell and T-cell responses to MHV68 in control C57BL/6 and p50−/− micea
| dpi | Mean (SD) %b
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Germinal center B cells
|
Effector CD4+ T cells
|
Effector CD8+ T cells
|
Vβ4+ CD8+ T cells
|
|||||
| Control | p50−/− | Control | p50−/− | Control | p50−/− | Control | p50−/− | |
| 0 (naïve mice) | 0.5 | 0.6 | 15.2 (3.2) | 21.7 (5.2)* | 2.8 (0.4) | 3.9 (1.7) | 3.9 (0.2) | 3.8 (0.3) |
| 18 | 5.3 (3.6) | 0.7 (0.3)* | 32.2 (3.9) | 17.1 (2.3)* | 27.2 (5.0) | 6.4 (1.4)* | 9.0 (3.3) | 4.0 (0.2) |
| 28 | 8.9 (1.9) | 2.5 (1.4)* | 38.3 (1.8) | 28.6 (2.0)* | 36.2 (20.2) | 19.8 (9.5) | 19.5 (14.7) | 10.9 (6.5)* |
| 45-56 | 3.6 (0.8) | 0.7 (0.8)* | 19.3 (4.4) | 23.6 (2.2) | 28.9 (7.3) | 12.2 (3.4)* | 23.3 (5.3) | 8.8 (1.4)* |
| 105 | NA | NA | 23.2 (6.3) | 22.2 (6.0) | 32.7 (9.5) | 5.1 (1.4)* | 31.0 (9.8) | 5.7 (0.9)* |
Mice were infected with 100 PFU of MHV68 by intranasal inoculation. Specific cell subsets were derived from initial FACS gating on live lymphocytes.
The data shown are the percentage for each subset derived from FACS analysis of three to five individual infected mice or naïve mice pooled from the individual harvest days. *, significant difference (P < 0.05) between infected control and p50−/− mice. B cells were analyzed at 18, 28, and 56 dpi. T cells were analyzed at 18, 28, 45, and 105 dpi. NA, not analyzed. Germinal center CD19+ B cells were gated as percent CD95+/GL7+ of CD19+ B cells, effector CD4+ T cells were gated as percent CD62Llo/CD44hi of CD4+ T cells, effector CD8+ T cells were gated as percent CD62Llo/CD44hi of CD8+ T cells, and Vβ4+ CD8+ T cells were gated as percent Vβ4+ of CD8+ T cells.
Given the defect in B-cell participation in germinal center reactions, coupled with a severe defect in the humoral response against MHV68 in p50−/− mice, global T-cell responses were investigated. While the percentages of CD4+ and CD8+ T cells did not differ significantly between p50−/− mice and control mice at between 18 and 45 dpi (data not shown), dramatic differences were observed in the percentages of effector T cells as characterized by the downregulation of CD62L and the upregulation of CD44 (Table 2). The population of effector CD4+ T cells in p50−/− mice did not increase until 28 dpi and remained lower than the population of effector CD4+ T cells in control mice, which doubled to 32.2% by 18 dpi (Table 2). p50−/− mice responded to MHV68 infection with a peak of 20% effector CD8+ T cells at 28 dpi, which fell to nearly naïve levels by 105 dpi (Table 2). This is in sharp contrast to the effector CD8+ T-cell response in control mice, where approximately 30% of CD8+ T cells were CD62Llo/CD44hi from 18 dpi through the duration of the infection (largely owing to the robust and sustained expansion of Vβ4+ CD8+ T cells, as discussed below).
A hallmark of MHV68 infection is the expansion of Vβ4+ CD8+ T cells in response to the MHV68 superantigen-like secreted M1 protein (23). Here we observed that upon WT MHV68 infection of control C57BL/6 mice, ca. 20% of CD8+ T cells were Vβ4+ by 28 dpi, increasing to 30% of CD8+ T cells by 105 dpi (Table 2). A slight expansion of the Vβ4+ CD8+ T cells was evident at 28 dpi in the p50−/− mice (11%), but, as observed with the effector CD8+ T-cell population, this T-cell subset failed to further expand as infection progressed in the p50−/− mice (Table 2). Our previous studies have provided evidence that Vβ4+ CD8+ T cells are an important source of gamma interferon (IFN-γ) during MHV68 infection, which serves to suppress virus reactivation from some latently infected reservoirs (7, 23). Thus, the absence of a robust Vβ4+ CD8+ T-cell response may also contribute to the observed persistent virus replication in the lungs of MHV68-infected p50−/− mice. Taken together, these data indicate that p50 plays an important role in T-cell responses to MHV68 infection, and thus, in addition to a significant defect in the humoral immune response to MHV68, compromised T-cell responses likely also contribute to the phenotype of MHV68 in NF-κB p50−/− mice.
Evaluation of immune control in MHV68-infected mixed BM chimeric mice.
The infection of p50−/− mice revealed substantial defects in host immune control, which undoubtedly significantly affect control of chronic MHV68 infection. Such alterations in virus-host interactions prevent a clear determination of a role for p50 in the infected cell versus a requirement for p50 in the hematopoietic or nonhematopoietic, stromal cell compartments of the host in response to the virus. Therefore, mixed p50+/+/p50−/− BM chimeric mice were generated to restore normal host responses and enable investigation of the intrinsic requirements for p50 in MHV68 B cells in the context of a p50-competent host background. Normal C57BL/6 mice congenic for Ly5.1 were lethally irradiated to deplete their BM and then reconstituted with an 8:2 ratio (based on cell number) of BM isolated from normal C57BL/6 Ly5.1+ mice and p50−/− C57BL/6 mice congenic for Ly5.2. This input ratio was used to ensure that the reconstituted mice had a normal, p50-sufficient immune system. Eight weeks after reconstitution, the ratio of Ly5.1+ (p50+/+) to Ly5.2 (p50−/−) splenocytes was 7:3, demonstrating no intrinsic defect in p50−/− B-cell maturation in the presence of p50+/+ B cells.
The p50+/+/p50−/− BM chimeras were examined for their ability to control MHV68 infection compared to their parental counterparts. At 12 dpi, MHV68 replication in the lungs of the BM chimeras was restored to levels seen in the control p50+/+ mice, unlike the higher levels observed in p50−/− mice (Fig. 5A). In addition, both total IgG and anti-MHV68 IgG levels were restored to the levels observed in control mice at 3 months postinfection (Fig. 5B and C). These analyses provide strong evidence that the BM chimeric approach ameliorates the defects in the host response observed in the p50−/− knockout mice.
FIG. 5.

Mixed BM p50+/+/p50−/− chimeric mice have restored control of acute replication and normal IgG responses. (A) Acute replication in the lung. Control mice on a C57BL/6 or Ly5.1 background, NF-κB1 p50−/− mice (BL6), and mixed BM p50+/+/p50−/− mice were infected with 100 PFU. At 12 dpi, lungs were harvested and disrupted, and titers were determined on NIH 3T12 fibroblasts. Data from individual mice are shown as log10 titer, and the bar indicates the geometric mean titer. The dashed line indicates the limit of detection of this assay as log101.7 or 50 PFU/ml of sample homogenate. Significant differences were identified between CTRL (B6 and Ly5.1) and p50−/− (P < 0.05) mice. (B) Determination of total IgG production in sera of individual naïve versus infected mice. The bar represents the geometric mean titer. Infected CTRL mice and BM chimeras differed significantly from naïve CTRL mice (P < 0.008). (C) Determination of MHV68-specific IgG production in sera of individual infected mice. The bar represents the geometric mean titer. Naïve sera had undetectable levels of MHV68-specific IgG (data not shown). Infected CTRL mice and BM chimeras differed significantly from p50−/− mice (P < 0.0008 and P = 0.0306, respectively).
Identification of a role for p50 in latently MHV68-infected B cells.
To determine whether there is a direct role for p50 in chronic MHV68 infection, aside from its role in the host immune response to MHV68, splenocytes from MHV68-infected p50+/+/p50−/− mixed BM chimeric mice were separated by flow cytometry into p50+/+ B cells and p50−/− B cells based on the expression of Ly5.1 or Ly5.2, respectively. As shown in Fig. 6A, post-FACS analyses demonstrate that each population was >99% pure. Analysis of MHV68 latency at 18 dpi revealed a nearly sixfold decrease in the frequency of viral genome-positive Ly5.2 (p50−/−) B cells compared to Ly5.1 (p50+/+) B cells recovered from the same mouse (Fig. 6B). MHV68 reactivation from p50−/− B cells was also reduced at 18 dpi (Fig. 6C), but at levels consistent with the defect in the establishment of latency at this time point, thus discounting a role for p50 in virus reactivation from latently infected B cells.
FIG. 6.

NF-κB1 p50 null B cells are poor reservoirs for MHV68 latency in mixed BM chimeras. (A) Representative flow cytometric analysis of cell populations of p50+/+/p50−/− mixed BM chimeras before and after sorting. Eight weeks after reconstitution of wild-type Ly5.1+ mice with a 7:3 ratio of Ly5.1+ WT to Ly5.2+ p50−/− hematopoietic cells, BM chimeric mice were infected with 100 PFU of MHV68. At the indicated times below, splenocytes were harvested and then stained with anti-CD19-PE, anti-Ly5.1-FITC, and anti-Ly5.2-APC and sorted into p50+/+, Ly5.1+, and p50−/− Ly5.2+ B-cell populations. Postsort FACS analysis indicated that the mean purities were 99.8% ± 0.2% for p50+/+ Ly5.1+ CD19+ cells and 99.3% ± 0.6% for p50−/− Ly5.2+ cells. (B, D, F, and G) Frequency of sorted p50+/+ and p50−/− CD19+ B cells harboring viral genomes. Limiting-dilution viral genome PCR analysis was utilized to determine the frequency of latency in p50+/+ and p50−/− B cells at 18 dpi (1/46 and 1/255), 26 to 29 dpi (1/330 and 1/3,727), 46 to 48 dpi (1/679 and 1/16,238), and 89 to 92 dpi (1/3,418 and 1/30,634). (C and E) Frequency of sorted p50+/+ and p50−/− CD19+ B cells reactivating virus. Limiting-dilution reactivation analysis was utilized to determine the frequency of ex vivo reactivation in p50+/+ and p50−/− B cells at 18 dpi (1/7,876 and 1/28,065) and 26 to 29 dpi (below the limit of detection). For both limiting-dilution assays, curve fit lines were derived from nonlinear regression analysis, and symbols represent the mean percentage of wells positive for virus (viral DNA or CPE) ± the standard error of the mean. The dashed line represents 63.2%, from which the frequency of viral genome-positive cells or the frequency of cells reactivating virus was calculated based on the Poisson distribution. The data shown represent two independent experiments with spleen cells pooled from three to five mice per experimental group.
In contrast to the enhanced latency observed in p50−/− mice at 28 to 29 dpi, the frequency of viral genome-positive Ly5.2 (p50−/−) B cells was 11-fold lower than the frequency of viral genome-positive Ly5.1 (p50+/+) B cells in the context of the mixed BM chimeric mouse at 26 to 29 dpi (Fig. 6D). As expected, by 26 to 29 dpi there was little or no detectable virus reactivation from either the Ly5.1+ or Ly5.2+ B-cell population (Fig. 6E). A similar defect in MHV68 latency in p50−/− B cells was also observed at 46 to 48 dpi (Fig. 6F) and at 89 to 92 dpi (Fig. 6G). Therefore, in the context of a p50-sufficient host, p50−/− B cells exhibit a significant latency defect at all time points examined. However, latently infected p50−/− B cells reactivated from latency as efficiently as p50-sufficient B cells, demonstrating that p50 is dispensable for MHV68 reactivation. Taken together, the analyses of mixed BM chimeric mice clearly demonstrated a B-cell intrinsic role for NF-κB p50 in the establishment of MHV68 latency.
Given the failure to support the efficient establishment of latency in the absence of p50, p50+/+ (Ly5.1+) B cells and p50−/− (Ly5.2+) B cells isolated from mixed BM chimeric mice were analyzed for phenotypic alterations in response to MHV68 in two independently generated sets of BM chimeric mice at 18 and 19 dpi (Fig. 7). B-cell participation in a germinal center reaction is a critical step in B-cell differentiation into a memory B cell and is a property of latently MHV68-infected cells in normal mice (27, 56, 87). The p50+/+ and p50−/− B-cell subsets present in the mixed BM chimeric mice were analyzed for dual surface expression of GL7 and CD95/Fas, a property of germinal center B cells (Fig. 7A). B cells that lacked p50 were twofold less likely than their p50+/+ counterparts (2.40% ± 1.07% versus 4.91% ± 3.02%; P = 0.0052) to participate in germinal center reactions (Fig. 7B). Polyclonal activation of B cells, as demonstrated by the upregulation of the activation marker CD69, is a common response to MHV68 infection and has been previously reported to correspond to splenomegaly and viral load (19, 46). Although there was an overall higher level of CD69 on naïve splenocytes from p50−/− mice (24.5% ± 1.27%) versus p50+/+ mice (9.68% ± 1.29%), a smaller proportion of p50−/− B cells became activated upon infection (1.3-fold increase to 32.8% ± 4.45%) compared to the p50+/+ B cells (2.2-fold increase to 21.7% ± 5.77%) (data not shown). Thus, the phenotypic analyses indicate that p50 plays an intrinsic role in normal B-cell responses to MHV68 infection, independent of the presence of p50-sufficient cells. Overall, the data obtained from the mixed BM chimeric mice indicate that the failure of p50−/− B cells to efficiently compete with the p50+/+ B cells as a reservoir for MHV68 latency is linked to the loss of p50-dependent B-cell functions.
FIG. 7.

B cells lacking NF-κB1 p50 in the BM chimeras have defective responses to MHV68 infection. Cells were prepared from spleens harvested from uninfected age-matched (naïve) mice or from two independent sets of infected p50+/+/p50−/− mixed BM chimeric mice at 18 to 19 dpi with 100 PFU of WT MHV68. Splenocytes from individual mice were stained with anti-Ly5.1-APC, anti-Ly5.2-peridinin chlorophyll protein-Cy5.5, anti-GL7-FITC, and anti-CD95-PE and then analyzed by flow cytometry. (A) Representative flow cytometry plots of the germinal center B-cell population (percent GL7+/CD95+ of CD19+) in naïve and infected p50+/+ versus p50−/− subsets. (B) Scatter plot of germinal center cells in each population, compiled from two independent sets of infected BM chimeras. Each symbol represents data from an individual mouse, and the bar depicts the mean percentage. One of two representative experiments is shown. All comparisons except for the percent differences of naïve p50−/− and p50+/+ germinal center B cells are statistically significant (P < 0.01); *, P = 0.0052.
Role for p50-mediated control of virus replication in the lungs.
The MHV68-infected mixed BM p50+/+/p50−/− chimeric mice were nearly indistinguishable from control C57BL/6 mice in levels of acute virus replication, frequency of latently MHV68-infected B cells, and antibody production. One additional phenotype uncovered in the p50−/− mice was the substantially elevated levels of preformed infectious virus in the lungs of infected mice at late times postinfection. Loss of control of persistent virus replication in the lungs has been previously reported upon MHV68 infection of immune-deficient mice (11, 32, 48, 88) and is consistent with the immune dysfunction uncovered in the infected p50−/− mice. However, NF-κB has also been implicated in controlling virus reactivation from latently infected B cells (8). Thus, to assess whether persistent virus replication is controlled in the mixed BM chimeric mice, virus replication in the lungs at late times postinfection was examined. Unexpectedly, appreciable levels of persistent virus replication were detected in the lungs of MHV68-infected mixed BM chimeras, albeit at reduced levels compared to p50−/− mice (Fig. 8). This is in striking contrast to the nearly complete absence of ongoing replication in the lungs of C57BL6 mice at 3 and 7 months after infection with a 10-fold-higher dose of WT virus (Fig. 8 and data not shown). This indicates that in the mixed p50+/+/p50−/− BM chimeras, the persistence of MHV68 replication in the lungs can be, at least in part, attributed to an intrinsic defect in MHV68-infected p50−/− hematopoietic cells (likely p50−/− B cells). This reveals a new role for p50 in controlling virus reactivation from latency and suggests that resident p50−/− B cells are likely contributing to viral persistence in the lungs.
FIG. 8.
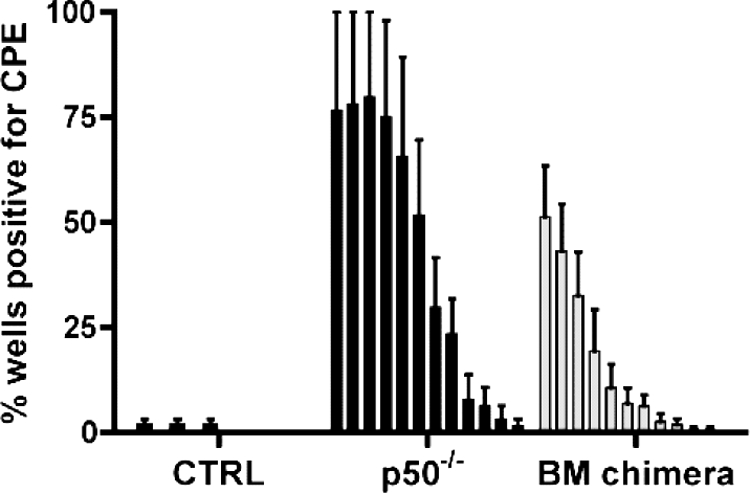
MHV68 replication persists in the lungs of mice harboring a subset of p50−/− hematopoietic cells at 3 months postinfection. Lung tissue was isolated from infected control mice (CTRL, BL6), NF-κB1 p50−/− mice (p50−/−, BL6), or p50+/+/p50−/− BM chimeric mice at 3 months postinfection with 100 PFU of WT MHV68. Bars for each sample represent the percentage for 16 wells positive for CPE upon plating twofold serial dilutions (starting at 1:10) of mechanically disrupted lung tissue on an indicator MEF monolayer.
DISCUSSION
The analyses reported here have uncovered several novel findings regarding the role of NF-κB signaling during MHV68 infection. First, the NF-κB activation profile differs in productive and latent MHV68 infections, with the activation of the NF-κB subunit p50 common to both. Second, a striking increase in WT MHV68 replication was observed in the lungs of p50−/− mice but not in p50−/− MEFs in culture, suggesting a role for p50 in host control during acute infection. Third, the kinetics of latency establishment in the splenocytes of p50−/− mice were grossly distorted such that the expansion of latency in the spleens of p50−/− mice occurred 10 days later than in control mice and was maintained at high levels as late as 3 months postinfection, leading to the identification of persistent MHV68 replication in the lungs of p50−/− at late times postinfection and identification of defects in the host adaptive immune response. Fourth, the generation and infection of p50+/+/p50−/− mixed BM chimeric mice restored virus control and humoral immunity such that the intrinsic role of p50 in latently infected B cells could be examined. Defects in typical B-cell responses such as activation and germinal center participation demonstrated a B-cell intrinsic requirement for p50 in response to infection. Of particular significance, the p50−/− B cells failed to effectively compete with their p50+/+ counterparts as reservoirs for MHV68 latency. Finally, we demonstrated persistent virus replication in the lungs of the p50+/+/p50−/− BM chimeras, indicating a role for NF-κB p50 in maintaining MHV68 latency. Taken together, these data identify NF-κB p50 as an important regulatory factor that plays a role in the host immune response to MHV68 infection, as well as having a direct impact on MHV68 latency in B cells.
The NF-κB activation profile varies in lytic and latent infections.
As in lymphoma cell lines latently infected with KSHV or EBV (8, 39), we observed that NF-κB was constitutively activated in the MHV68+ S11 B lymphoma cell line (Fig. 1A). As is typical of mature B cells, c-Rel and p50 were detected in the nuclei of S11 cells. In KSHV and EBV, latency-associated gene products drive NF-κB activation and cell survival. NF-κB protects against apoptosis in MHV68+ S11 lymphoma cells (data not shown), but the viral gene products that potentially mediate this activation have not been identified. Ideally, NF-κB activation should be examined upon de novo MHV68 infection of primary murine B cells. However, the requirement for exogenous stimulation by cytokines or mitogens to maintain murine B-cell survival ex vivo leads to NF-κB activation and prevents analyses of virus-mediated NF-κB activation.
An EMSA analysis revealed activation of NF-κB late in the MHV68 replication cycle following high-MOI productive infection in both primary (Fig. 1B) and transformed (data not shown) fibroblast cells. The observed activation was reduced by inhibiting viral DNA synthesis, suggesting that NF-κB is activated following early viral gene expression. The NF-κB subunits p65 and p50 were identified by antibody supershift analyses, similar to responses to TNF-α and indicative of classical NF-κB pathway activation (Fig. 1C). There was no significant increase in NF-κB activation during the earliest stages of productive MHV68 infection, consistent with our previous studies with the MHV68-IκBαM virus, which failed to identify a role for NF-κB activation during virus replication in permissive fibroblasts (46). Experiments with UV-inactivated virus or infection at a high MOI (MOI = 50) did not result in the detection of NF-κB activation, arguing against an early phase of activation during MHV68 infection of fibroblast cells (data not shown).
The absence of NF-κB activation early during the productive infection of fibroblasts by MHV68 is consistent with data indicating that NF-κB inhibits MHV68 replication. Overexpression of the NF-κB subunit p65 inhibits the viral lytic transactivator RTA and blocks MHV68 replication in cell culture (8). In addition, inhibition of NF-κB leads to reactivation of KSHV and EBV from B cells in culture (8, 9). Infection of endothelial cells in culture with KSHV, which appears to biased toward establishment of a latent infection, results in a rapid and sustained period of NF-κB activation that is associated with an upregulation of NF-κB-dependent proinflammatory and prosurvival genes (36, 51, 63). Furthermore, inhibition of NF-κB activation in KSHV-infected endothelial cells can lead to an increase in lytic gene expression (36).
The role of NF-κB activation during the late phase of MHV68 infection in permissive cells has not been defined. The CMV-driven IκBαM construct is expressed within 4 h of NIH 3T3 infection with the MHV68-IκBαM recombinant virus (L. T. Krug, J. C. Forrest, and S. H. Speck, data not shown) and blocks NF-κB activation during virus infection in permissive cells, but it does not affect replication in fibroblasts (46) or in transformed endothelial and epithelial cell lines (data not shown). Furthermore, comparison of MHV68 replication in WT and p50−/− MEFs demonstrated that p50 is dispensable for virus replication following infection at a low MOI (Fig. 2B). Taken together, these data indicate that MHV68 does not require NF-κB activation for productive infection of primary or immortalized fibroblasts. However, we cannot rule out a role for NF-κB activation in some permissive MHV68-infected cell population in vivo during the acute phase of MHV68 replication. NF-κB-dependent cues during the acute phase of infection might protect the cell from apoptosis or affect the recruitment and activation of B cells that are necessary for seeding splenic latency (78, 85). As discussed below, a delay in inflammation might explain higher lytic replication in the lungs of p50−/− mice (Fig. 2A). Both the KSHV vGPCR and vFLIP appear to play dual roles in promoting cell survival and induction of paracrine factors with inflammatory properties (37, 51). Therefore, NF-κB activation might play role in the inflammatory and effector responses of other cell types infected in vivo (e.g., epithelial and endothelial cells, dendritic cells, and macrophages) (28, 73, 74).
MHV68 pathogenesis in the absence of NF-κB p50.
MHV68 replicated to higher levels in the lungs of p50−/− mice, yet there was no indication of a delay in clearance. Alterations in the kinetics or levels of virus were not recapitulated upon a low-multiplicity infection of p50−/− MEFs in culture, suggesting a requirement for p50 by the host. Heighted levels of acute-phase replication have been reported for IFN-α/βR−/−, IFN-γR−/−, and STAT1−/− mice (4, 22, 83). However, the delayed kinetics of establishing splenic latency in p50−/− mice observed here differs from the rampant dissemination of MHV68 in the absence of type I IFN signaling (4, 22).
Intranasal inoculation of MHV68 results in the infiltration of immune cells and production of cytokines and chemokines in the lungs of mice, many of which are typically regulated by NF-κB (66, 68). The critical role for NF-κB-dependent innate immune responses is also apparent upon the infection of NF-κB p50−/− mice with bacterial pathogens. Streptococcus pneumoniae infection leads to rapid death and sepsis in p50−/− mice (70). The failure to clear the enteric pathogen Citrobacter rodentium from the guts of p50−/− mice correlated with a decrease in inflammatory cell infiltration into colonic tissue, not a lack of anti-C. rodentium antibodies (20). Extracellular Listeria monocytogenes was cleared normally in the peritoneums of p50−/− mice, but the defect in clearance of intracellular Listeria monocytogenes was attributed to a defect in cytokine-induced activation of p50−/− macrophages (70). The activation or inhibition of NF-κB activity can lead to an increase or decrease, respectively, in models of acute lung inflammation and associated injury (13, 24, 57). For instance, mice receiving adenovirus-delivered NF-κB p65/RelA had greater clearance of Pseudomonas aeruginosa, while mice administered Ad-IκBDN, a dominant inhibitor of NF-κB, exhibited decreased clearance (34). Inhibition of NF-κB was associated with a decrease in TNF-α, the proinflammatory chemokine MIP-2, and neutrophil infiltration (64). Further examination of the inflammatory response in p50−/− mice might identify inflammatory events that mediate immune control of acute MHV68 replication in the lungs.
The altered kinetics of establishment of MHV68 latency in the p50−/− mice led us to investigate whether the absence of p50 alters the latency reservoir(s) for MHV68. Since B cells are the predominant latency reservoir for MHV68 in the spleen, we assessed the distributions of MHV68 in specific p50-deficient B-cell populations at 18 and 31 dpi, examining expression of surface markers indicative of activated (CD69+) and germinal center (GL7+ CD95+) B cells (Fig. 3). Strikingly, the analysis of surface marker expression revealed that even though there was a lower frequency of MHV68-infected B cells at 18 dpi in p50−/− mice, the infected B cells become activated, participated in germinal center reactions, and were detected in IgG+/IgD− class-switched B cells (Fig. 3 and data not shown). The ability of MHV68 to gain access to the same subset of B cells in the p50−/− mice as found in the control mice suggests that p50 is not absolutely required for MHV68 latency once the virus gains access to the spleen. For instance, the absence of a severe alteration in the phenotype of MHV68-infected p50−/− B cells might be due to compensation by other NF-κB subunits. Alternatively, the accumulation and higher levels of MHV68-infected cells at 31 dpi in p50−/− mice may be a result of a delay in trafficking to the spleen and/or a delay in germinal center expansion in the absence of NF-κB p50. We were unable to determine the differential distribution of MHV68-eYFP-infected B cells in the newly formed (CD21lo CD23lo), marginal zone (CD21hi CD23lo), or follicular (CD21+ CD23+) compartments in the p50−/− and control mice, since MHV68 infection alters CD21 and CD23 expression, as reported for KSHV (12, 26) (C. Collins and S. H. Speck, unpublished observations.
In the p50−/− mice there was a high frequency of splenic latency that is likely due to reseeding B-cell infection via ongoing persistent virus replication in these animals. Notably, the level of virus replication in the lungs was below the limit of detection using a traditional plaque assay, instead requiring a limiting-dilution overlay of MEFs and monitoring for the appearance of CPE. There was intermittent virus replication observed in control mice during early stages of chronic infection. This indicated that there is a low level of sporadic reactivation in the lungs that is held in check by the host immune response in normal mice. However, this reactivation/persistent replication was not controlled in the p50−/− mice, leading to persistence in the lungs. The increased persistence of MHV68 in the p50−/− mice suggests that there is a dysfunction of the immune system that is influencing B-cell biology and the virus life cycle.
Immune defects of the NF-κB1 p50−/− mice prevent control of MHV68.
The phenotypes observed in p50−/− mice infected with MHV68 (heightened levels of acute replication in the lungs and delayed seeding of splenic latency, followed by increased viral load in the spleen and ongoing persistent virus replication in the lungs at late times postinfection) bear a striking similarity to the phenotypes observed in mice lacking CD4+ T cells (11). CD4+ T cells are critical for the generation of splenomegaly, nonspecific B-cell activation and increased nonspecific antibody production, the establishment of B-cell latency, the expansion of Vβ4 CD8+ T cells, and the generation of an effective CD8+ T-cell response (21). The absence of the costimulatory molecules CD40, CD40L, and CD80/CD86 (but, unexpectedly, not CD28), which are critical for the interactions of B cells and CD4+ T cells before and during germinal center reactions, also results in virus recrudescence (21, 31, 48, 66). Evaluation of several markers of immune function in p50−/− mice uncovered immune defects in both B- and T-lymphocyte compartments.
NF-κB p50 knockout mice generate normal B-cell numbers and follicles but have defects in generating high-affinity T-dependent antibodies (30, 70). NF-κB p50 is critical for the germ line CHγ3 transcription in response to stimulation and likely mediates chromatin remodeling at the Sγ3 and Sγ1 region through direct binding (72, 82). Administration of the T-dependent nitrophenol-conjugated keyhole limpet hemocyanin immunogen to p50−/− mice leads to smaller and decreased numbers of germinal centers (59, 60). Thus, the failure to generate an anti-MHV68 antibody response in p50−/− mice is not altogether unexpected given the role of p50 in isotype class switching. The combined lack of antiviral antibody and failure to control latency in the lungs of the p50−/− mice seemingly underscores the importance of antibodies in the control of chronic MHV68 infection (32, 76). However, the loss of humoral immunity is compounded by defects in T-cell function in the p50−/− mice.
The absence of p50 in mice infected with Leishmania major leads to a failure to generate an antigen-specific CD4+ T-cell response and decreased production of IFN-γ, which was attributed to a lack of CD4+ T-cell proliferation (2). The maintenance of a high level of activated, effector CD8+ T cells is a hallmark of MHV68 infection that is dependent on CD4+ T cells (11). In our investigation of CD4+ T-cell responses, there was a defect in the generation of effector CD4+ T cells in the p50−/− mice at 18 and 28 dpi (Table 2). In turn, there was a profound deficiency in effector CD8+ T cells (Table 2). The added failure to expand the M1-dependent Vβ4+ CD8+ T-cell population in the p50−/− mice would also be expected to reduce the production of antiviral cytokines such as IFN-γ and TNF-α, which likely hold reactivation in check (7, 23, 71).
Taken together, our results indicate that while there were no gross defects in the numbers of B, CD4+, or CD8+ T cells (Table 1), the impairment of the effector functions of these cells in the absence of NF-κB1 p50 led to a failure of humoral and cell-mediated immunity. This lack of adaptive immune function and the concomitant loss of virus control parallel the increase in gammaherpesvirus pathologies associated with immune dysfunction in AIDS and transplant patients. Further characterization of differences in the inflammatory response in the lungs, coupled with analyses of abnormal immune cell functions in lymphoid tissues, in p50−/− versus control mice may provide insights into MHV68 dissemination and establishment of latency.
B-cell intrinsic role for NF-κB p50 to support MHV68 latency.
NF-κB-mediated immune dysfunction in the global p50 knockout mice led to many disruptions in the typical life cycle of MHV68 in the host. Characterizing the role for p50 in MHV68-infected B cells could be realized only in the context of a p50-sufficient host. Therefore, mixed p50+/+/p50−/− BM chimeras were generated in a WT host such that p50−/− B cells were the minority population, thereby minimizing the impact on the overall immune response of the animal. As expected, the infection and generation of p50+/+/p50−/− BM chimeras resulted in a restoration of virus control during acute replication and the production of antiviral antibodies. During both early and late phases of chronic infection, an examination of the frequency of viral genome-positive cells revealed a ca. 10-fold-lower frequency of latency in the p50−/− cells at all time points. This demonstrated a defect in the ability of the p50−/− B compartment to compete with p50+/+ B cells as a latency reservoir for MHV68. The finding of a role for p50 in the establishment of MHV68 latency substantiates and extends our previous identification of the NF-κB signaling pathway as a critical element of latency establishment using the IκBαM-transgenic virus approach (46).
MHV68 latency is intimately tied to B-cell differentiation. MHV68 is found predominantly in the spleen in proliferating B cells undergoing a germinal center reaction, ultimately establishing a latent program in isotype class-switched memory B cells (27, 50, 55, 87). In the mixed BM chimeric mice, p50−/− B cells exhibited intrinsic defects in activation and germinal center participation. B cells lacking p50 have a higher turnover rate in vivo and have decreased survival in culture (38, 60). Thus, NF-κB signaling might be critical for responding to activation cues, such as CD40 or engagement by surface Ig/B-cell receptor, that mediate proliferative expansion and survival during a germinal center reaction, consistent with the requirement for CD40 to maintain MHV68 latency (45).
The cues and signals that lead to p50 activation, and the transcriptional and functional consequences of that activation in the infected B cell, have not been defined. Interestingly, p50−/− B cells in mixed BM chimeras have similar defects in B-cell responses that correspond to a defect in MHV68 latency, as was also observed in MyD88−/− and CD40−/− cell populations in mixed BM chimeric mice (33, 45). This identifies two upstream pathways leading to NF-κB activation that have critical roles in MHV68 latency. Interestingly, the inefficient establishment of latency in p50−/− B cells of the BM chimeras at the onset of splenic latency differs from the latency phenotype of CD40−/− B cells in CD40+/+/CD40−/− mixed BM chimeric mice. CD40−/− B cells were found to support latency at an early time point but diminished in frequency over time (45). This suggests that NF-κB p50 plays a critical role that precedes CD40-driven NF-κB activation and germinal center functions. Furthermore, while we uncovered a substantial defect in latency in B cells lacking p50, there remains a population of MHV68-infected p50−/− B cells at late times postinfection. The failure to completely abrogate latency in the absence of single virus or host factors suggests that there are numerous pathways and/or multiple reservoirs in the host. A p50−/− cell that is able to support latency might be complemented by other NF-κB subunits activated via the canonical and/or alternative pathways. Alternatively, the p50−/− cell population might result from the direct infection of a fully differentiated, quiescent cell, or it might merely represent a snapshot of ongoing de novo infections stemming from reactivation or persistence in a distal latency reservoir. Future studies will more closely examine phenotypic differences of infected p50−/− cells versus their infected p50+/+ counterparts with the aim of discerning differences in the viral latency program and the status of the infected cell.
An additional novel finding in the p50+/+/p50−/− mixed BM chimeras was the presence of persistent MHV68 replication in the lungs at late times postinfection, albeit at levels slightly lower than those observed in p50−/− mice. Infectious virus was found intermittently in immunocompetent control mice but was consistently undetectable in the lungs of these mice at ≥3 months of infection. Notably, we did not observe an increase in ex vivo reactivation of p50−/− B cells isolated from the spleen; however, we also did not detect any persistent virus replication in the spleens of these animals, indicating that the anatomical site plays a major role in this phenomenon (e.g., the microenvironment of the lungs is likely quite distinct from that of the spleen). As such, lung tissue might lack factors such as CD40/CD40L interactions that mediate repression from reactivation, thus creating an environment more conducive to gammaherpesvirus reactivation (67). In addition, the transcriptional consequence of p50 activation in a latently infected B cell likely varies with the stimuli encountered. Interestingly, vFLIP inhibits viral gene expression and RTA transactivation during reactivation from 293 cells indirectly through NF-κB-mediated repression of AP1 (89, 90). Also, the KSHV vGPCR inhibits lytic viral gene expression from lymphoma cells, providing additional support for a role for NF-κB in promoting the maintenance of latency (10). The p50−/− hematopoietically derived cell reservoir that is the source for reactivation and/or persistent virus replication is most likely B lymphocytes, but other latent reservoirs such as macrophages and dendritic cells may be involved (28). For instance, infected MyD88−/− mice exhibit hyperreactivation from the peritoneal exudate cells, which consist primarily of macrophages (33).
Understanding how gammaherpesviruses utilize and manipulate host responses continues to provide fundamental insights into pathways that are involved in disease progression and has the potential to identify novel targets for antiviral therapies. NF-κB signaling is critical to the survival and transformation of cells infected by the human gammaherpesviruses EBV and KSHV. Here, we have identified NF-κB p50 as a critical component of the NF-κB signaling pathway that controls the establishment and maintenance of MHV68 latency in a tissue-dependent manner. The analyses provide an impetus for further studies to identify virus and host genes regulated by NF-κB p50 in different tissues during chronic MHV68 infection.
Acknowledgments
We thank Robert Karaffa for expertise and assistance with flow cytometry and members of the Speck laboratory for helpful discussions and critical reading of the manuscript. We thank Clinton R. Paden and Soojung Park for technical assistance.
This research was supported by NIH grant R01 CA95318 to S.H.S. S.H.S. was also supported by NIH grants R01 CA43143, CA52004, CA58524, and AI58057. L.T.K. was supported by NIH Ruth L. Kirschstein National Research Service Award F32 AI066818.
Footnotes
Published ahead of print on 4 March 2009.
REFERENCES
- 1.An, J., Y. Sun, R. Sun, and M. B. Rettig. 2003. Kaposi's sarcoma-associated herpesvirus encoded vFLIP induces cellular IL-6 expression: the role of the NF-kappaB and JNK/AP1 pathways. Oncogene 223371-3385. [DOI] [PubMed] [Google Scholar]
- 2.Artis, D., K. Speirs, K. Joyce, M. Goldschmidt, J. Caamaño, C. A. Hunter, and P. Scott. 2003. NF-kappa B1 is required for optimal CD4+ Th1 cell development and resistance to Leishmania major. J. Immunol. 1701995-2003. [DOI] [PubMed] [Google Scholar]
- 3.Arvanitakis, L., E. Geras-Raaka, A. Varma, M. C. Gershengorn, and E. Cesarman. 1997. Human herpesvirus KSHV encodes a constitutively active G-protein-coupled receptor linked to cell proliferation. Nature 385347-350. [DOI] [PubMed] [Google Scholar]
- 4.Barton, E. S., M. L. Lutzke, R. Rochford, and H. W. T. Virgin. 2005. Alpha/beta interferons regulate murine gammaherpesvirus latent gene expression and reactivation from latency. J. Virol. 7914149-14160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beinke, S., and S. C. Ley. 2004. Functions of NF-kappaB1 and NF-kappaB2 in immune cell biology. Biochem. J. 382393-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bossen, C., and P. Schneider. 2006. BAFF, APRIL and their receptors: structure, function and signaling. Semin. Immunol. 18263-275. [DOI] [PubMed] [Google Scholar]
- 7.Braaten, D. C., J. S. McClellan, I. Messaoudi, S. A. Tibbetts, K. B. McClellan, J. Nikolich-Zugich, and H. W. Virgin. 2006. Effective control of chronic gamma-herpesvirus infection by unconventional MHC class Ia-independent CD8 T cells. PLoS Pathog. 2e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown, H. J., M. J. Song, H. Deng, T. T. Wu, G. Cheng, and R. Sun. 2003. NF-κB inhibits gammaherpesvirus lytic replication. J. Virol. 778532-8540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cahir-McFarland, E. D., D. M. Davidson, S. L. Schauer, J. Duong, and E. Kieff. 2000. NF-kappa B inhibition causes spontaneous apoptosis in Epstein-Barr virus-transformed lymphoblastoid cells. Proc. Natl. Acad. Sci. USA 976055-6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cannon, M., E. Cesarman, and C. Boshoff. 2006. KSHV G protein-coupled receptor inhibits lytic gene transcription in primary-effusion lymphoma cells via p21-mediated inhibition of Cdk2. Blood 107277-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cardin, R. D., J. W. Brooks, S. R. Sarawar, and P. C. Doherty. 1996. Progressive loss of CD8+ T cell-mediated control of a gamma-herpesvirus in the absence of CD4+ T cells. J. Exp. Med. 184863-871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang, H., Y. Gwack, D. Kingston, J. Souvlis, X. Liang, R. E. Means, E. Cesarman, L. Hutt-Fletcher, and J. U. Jung. 2005. Activation of CD21 and CD23 gene expression by Kaposi's sarcoma-associated herpesvirus RTA. J. Virol. 794651-4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng, D. S., W. Han, S. M. Chen, T. P. Sherrill, M. Chont, G. Y. Park, J. R. Sheller, V. V. Polosukhin, J. W. Christman, F. E. Yull, and T. S. Blackwell. 2007. Airway epithelium controls lung inflammation and injury through the NF-kappa B pathway. J. Immunol. 1786504-6513. [DOI] [PubMed] [Google Scholar]
- 14.Chugh, P., H. Matta, S. Schamus, S. Zachariah, A. Kumar, J. A. Richardson, A. L. Smith, and P. M. Chaudhary. 2005. Constitutive NF-kappaB activation, normal Fas-induced apoptosis, and increased incidence of lymphoma in human herpes virus 8 K13 transgenic mice. Proc. Natl. Acad. Sci. USA 10212885-12890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clambey, E. T., H. W. T. Virgin, and S. H. Speck. 2002. Characterization of a spontaneous 9.5-kilobase-deletion mutant of murine gammaherpesvirus 68 reveals tissue-specific genetic requirements for latency. J. Virol. 766532-6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Claudio, E., K. Brown, S. Park, H. Wang, and U. Siebenlist. 2002. BAFF-induced NEMO-independent processing of NF-kappa B2 in maturing B cells. Nat. Immunol. 3958-965. [DOI] [PubMed] [Google Scholar]
- 17.Coope, H. J., P. G. Atkinson, B. Huhse, M. Belich, J. Janzen, M. J. Holman, G. G. Klaus, L. H. Johnston, and S. C. Ley. 2002. CD40 regulates the processing of NF-kappaB2 p100 to p52. EMBO J. 215375-5385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Damania, B. 2004. Modulation of cell signaling pathways by Kaposi's sarcoma-associated herpesvirus (KSHVHHV-8). Cell Biochem. Biophys. 40305-322. [DOI] [PubMed] [Google Scholar]
- 19.de Lima, B. D., J. S. May, S. Marques, J. P. Simas, and P. G. Stevenson. 2005. Murine gammaherpesvirus 68 bcl-2 homologue contributes to latency establishment in vivo. J. Gen. Virol. 8631-40. [DOI] [PubMed] [Google Scholar]
- 20.Dennis, A., T. Kudo, L. Kruidenier, F. Girard, V. F. Crepin, T. T. Macdonald, G. Frankel, and S. Wiles. 2008. The p50 subunit of NF-κB is critical for in vivo clearance of the noninvasive enteric pathogen Citrobacter rodentium. Infect. Immun. 764978-4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doherty, P. C., J. P. Christensen, G. T. Belz, P. G. Stevenson, and M. Y. Sangster. 2001. Dissecting the host response to a gamma-herpesvirus. Philos. Trans. R. Soc. London B 356581-593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dutia, B. M., D. J. Allen, H. Dyson, and A. A. Nash. 1999. Type I interferons and IRF-1 play a critical role in the control of a gammaherpesvirus infection. Virology 261173-179. [DOI] [PubMed] [Google Scholar]
- 23.Evans, A. G., J. M. Moser, L. T. Krug, V. Pozharskaya, A. L. Mora, and S. H. Speck. 2008. A gammaherpesvirus-secreted activator of Vbeta4+ CD8+ T cells regulates chronic infection and immunopathology. J. Exp. Med. 205669-684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Everhart, M. B., W. Han, T. P. Sherrill, M. Arutiunov, V. V. Polosukhin, J. R. Burke, R. T. Sadikot, J. W. Christman, F. E. Yull, and T. S. Blackwell. 2006. Duration and intensity of NF-kappaB activity determine the severity of endotoxin-induced acute lung injury. J. Immunol. 1764995-5005. [DOI] [PubMed] [Google Scholar]
- 25.Feng, B., S. Cheng, W. S. Pear, and H. C. Liou. 2004. NF-kB inhibitor blocks B cell development at two checkpoints. Med. Immunol. 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferguson, A. R., and R. B. Corley. 2005. Accumulation of marginal zone B cells and accelerated loss of follicular dendritic cells in NF-kappaB p50-deficient mice. BMC Immunol. 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flano, E., I. J. Kim, D. L. Woodland, and M. A. Blackman. 2002. Gamma-herpesvirus latency is preferentially maintained in splenic germinal center and memory B cells. J. Exp. Med. 1961363-1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flano, E., I. J. Kim, J. Moore, D. L. Woodland, and M. A. Blackman. 2003. Differential gamma-herpesvirus distribution in distinct anatomical locations and cell subsets during persistent infection in mice. J. Immunol. 1703828-3834. [DOI] [PubMed] [Google Scholar]
- 29.Forrest, J. C., L. T. Krug, and S. H. Speck. 2008. Murine gammaherpesvirus 68 infection of mice: a small animal model for characterizing basic aspects of gamma-herpesvirus pathogenesis, p. 735-775. In B. Damania and J. Pipas (ed.), DNA tumor viruses. Springer, New York, NY.
- 30.Franzoso, G., L. Carlson, L. Poljak, E. W. Shores, S. Epstein, A. Leonardi, A. Grinberg, T. Tran, T. Scharton-Kersten, M. Anver, P. Love, K. Brown, and U. Siebenlist. 1998. Mice deficient in nuclear factor (NF)-kappa B/p52 present with defects in humoral responses, germinal center reactions, and splenic microarchitecture. J. Exp. Med. 187147-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fuse, S., J. J. Obar, S. Bellfy, E. K. Leung, W. Zhang, and E. J. Usherwood. 2006. CD80 and CD86 control antiviral CD8+ T-cell function and immune surveillance of murine gammaherpesvirus 68. J. Virol. 809159-9170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gangappa, S., S. B. Kapadia, S. H. Speck, and H. W. T. Virgin. 2002. Antibody to a lytic cycle viral protein decreases gammaherpesvirus latency in B-cell-deficient mice. J. Virol. 7611460-11468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gargano, L. M., J. M. Moser, and S. H. Speck. 2008. Role for MyD88 signaling in murine gammaherpesvirus 68 latency. J. Virol. 823853-3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giannoni, F., A. Shea, C. Inglis, L. N. Lee, and S. R. Sarawar. 2008. CD40 engagement on dendritic cells, but not on B or T cells, is required for long-term control of murine gammaherpesvirus 68. J. Virol. 8211016-11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grimm, T., S. Schneider, E. Naschberger, J. Huber, E. Guenzi, A. Kieser, P. Reitmeir, T. F. Schulz, C. A. Morris, and M. Stürzl. 2005. EBV latent membrane protein-1 protects B cells from apoptosis by inhibition of BAX. Blood 1053263-3269. [DOI] [PubMed] [Google Scholar]
- 36.Grossmann, C., and D. Ganem. 2008. Effects of NFkappaB activation on KSHV latency and lytic reactivation are complex and context-dependent. Virology 37594-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grossmann, C., S. Podgrabinska, M. Skobe, and D. Ganem. 2006. Activation of NF-κB by the latent vFLIP gene of Kaposi's sarcoma-associated herpesvirus is required for the spindle shape of virus-infected endothelial cells and contributes to their proinflammatory phenotype. J. Virol. 807179-7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grumont, R. J., I. J. Rourke, L. A. O'Reilly, A. Strasser, K. Miyake, W. Sha, and S. Gerondakis. 1998. B lymphocytes differentially use the Rel and nuclear factor kappaB1 (NF-kappaB1) transcription factors to regulate cell cycle progression and apoptosis in quiescent and mitogen-activated cells. J. Exp. Med. 187663-674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guasparri, I., S. A. Keller, and E. Cesarman. 2004. KSHV vFLIP is essential for the survival of infected lymphoma cells. J. Exp. Med. 199993-1003. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 40.Hiscott, J., H. Kwon, and P. Génin. 2001. Hostile takeovers: viral appropriation of the NF-kappaB pathway. J. Clin. Investig. 107143-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jensen, K. K., D. J. Manfra, M. G. Grisotto, A. P. Martin, G. Vassileva, K. Kelley, T. W. Schwartz, and S. A. Lira. 2005. The human herpes virus 8-encoded chemokine receptor is required for angioproliferation in a murine model of Kaposi's sarcoma. J. Immunol. 1743686-3694. [DOI] [PubMed] [Google Scholar]
- 42.Kalled, S. L. 2006. Impact of the BAFF/BR3 axis on B cell survival, germinal center maintenance and antibody production. Semin. Immunol. 18290-296. [DOI] [PubMed] [Google Scholar]
- 43.Keller, S. A., D. Hernandez-Hopkins, J. Vider, V. Ponomarev, E. Hyjek, E. J. Schattner, and E. Cesarman. 2006. NF-kappaB is essential for the progression of KSHV- and EBV-infected lymphomas in vivo. Blood 1073295-3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keller, S. A., E. J. Schattner, and E. Cesarman. 2000. Inhibition of NF-kappaB induces apoptosis of KSHV-infected primary effusion lymphoma cells. Blood 962537-2542. [PubMed] [Google Scholar]
- 45.Kim, I. J., E. Flano, D. L. Woodland, F. E. Lund, T. D. Randall, and M. A. Blackman. 2003. Maintenance of long term gamma-herpesvirus B cell latency is dependent on CD40-mediated development of memory B cells. J. Immunol. 171886-892. [DOI] [PubMed] [Google Scholar]
- 46.Krug, L. T., J. M. Moser, S. M. Dickerson, and S. H. Speck. 2007. Inhibition of NF-kappaB activation in vivo impairs establishment of gammaherpesvirus latency. PLoS Pathog. 3e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee, B. J., U. H. Koszinowski, S. R. Sarawar, and H. Adler. 2003. A gammaherpesvirus G protein-coupled receptor homologue is required for increased viral replication in response to chemokines and efficient reactivation from latency. J. Immunol. 170243-251. [DOI] [PubMed] [Google Scholar]
- 48.Lyon, A. B., and S. R. Sarawar. 2006. Differential requirement for CD28 and CD80/86 pathways of costimulation in the long-term control of murine gammaherpesvirus-68. Virology 35650-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Majumder, P., J. A. Gomez, and J. M. Boss. 2006. The human major histocompatibility complex class II HLA-DRB1 and HLA-DQA1 genes are separated by a CTCF-binding enhancer-blocking element. J. Biol. Chem. 28118435-18443. [DOI] [PubMed] [Google Scholar]
- 50.Marques, S., S. Efstathiou, K. G. Smith, M. Haury, and J. P. Simas. 2003. Selective gene expression of latent murine gammaherpesvirus 68 in B lymphocytes. J. Virol. 777308-7318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martin, D., R. Galisteo, Y. Ji, S. Montaner, and J. S. Gutkind. 2008. An NF-kappaB gene expression signature contributes to Kaposi's sarcoma virus vGPCR-induced direct and paracrine neoplasia. Oncogene 271844-1852. [DOI] [PubMed] [Google Scholar]
- 52.Montaner, S., A. Sodhi, A. Molinolo, T. H. Bugge, E. T. Sawai, Y. He, Y. Li, P. E. Ray, and J. S. Gutkind. 2003. Endothelial infection with KSHV genes in vivo reveals that vGPCR initiates Kaposi's sarcomagenesis and can promote the tumorigenic potential of viral latent genes. Cancer Cell 323-36. [DOI] [PubMed] [Google Scholar]
- 53.Moorman, N. J., H. W. T. Virgin, and S. H. Speck. 2003. Disruption of the gene encoding the gammaHV68 v-GPCR leads to decreased efficiency of reactivation from latency. Virology 307179-190. [DOI] [PubMed] [Google Scholar]
- 54.Moorthy, A. K., O. V. Savinova, J. Q. Ho, V. Y. Wang, D. Vu, and G. Ghosh. 2006. The 20S proteasome processes NF-kappaB1 p105 into p50 in a translation-independent manner. EMBO J. 251945-1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moser, J. M., J. W. Upton, R. D. Allen III, C. B. Wilson, and S. H. Speck. 2005. Role of B-cell proliferation in the establishment of gammaherpesvirus latency. J. Virol. 799480-9491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nash, A. A., B. M. Dutia, J. P. Stewart, and A. J. Davison. 2001. Natural history of murine gamma-herpesvirus infection. Philos. Trans. R. Soc. London B 356569-579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pantano, C., J. L. Ather, J. F. Alcorn, M. E. Poynter, A. L. Brown, A. S. Guala, S. L. Beuschel, G. B. Allen III, L. A. Whittaker, M. Bevelander, and others. 2008. Nuclear factor kappa B activation in airway epithelium induces inflammation and hyperresponsiveness. Am. J. Respir. Crit. Care Med. 177959-969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pasparakis, M., T. Luedde, and M. Schmidt-Supprian. 2006. Dissection of the NF-kappaB signalling cascade in transgenic and knockout mice. Cell Death Differ. 13861-872. [DOI] [PubMed] [Google Scholar]
- 59.Pereira, S. G., and F. Oakley. 2008. Nuclear factor-kappaB1: regulation and function. Int. J. Biochem. Cell Biol. 401425-1430. [DOI] [PubMed] [Google Scholar]
- 60.Pohl, T., R. Gugasyan, R. J. Grumont, A. Strasser, D. Metcalf, D. Tarlinton, W. Sha, D. Baltimore, and S. Gerondakis. 2002. The combined absence of NF-kappa B1 and c-Rel reveals that overlapping roles for these transcription factors in the B cell lineage are restricted to the activation and function of mature cells. Proc. Natl. Acad. Sci. USA 994514-4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pollock, J. L., and H. W. T. Virgin. 1995. Latency, without persistence, of murine cytomegalovirus in the spleen and kidney. J. Virol. 691762-1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rastelli, J., C. Hömig-Hölzel, J. Seagal, W. Müller, A. C. Hermann, K. Rajewsky, and U. Zimber-Strobl. 2008. LMP1 signaling can replace CD40 signaling in B cells in vivo and has unique features of inducing class-switch recombination to IgG1. Blood 1111448-1455. [DOI] [PubMed] [Google Scholar]
- 63.Sadagopan, S., N. Sharma-Walia, M. V. Veettil, H. Raghu, R. Sivakumar, V. Bottero, and B. Chandran. 2007. Kaposi's sarcoma-associated herpesvirus induces sustained NF-κB activation during de novo infection of primary human dermal microvascular endothelial cells that is essential for viral gene expression. J. Virol. 813949-3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sadikot, R. T., H. Zeng, M. Joo, M. B. Everhart, T. P. Sherrill, B. Li, D. S. Cheng, F. E. Yull, J. W. Christman, and T. S. Blackwell. 2006. Targeted immunomodulation of the NF-kappaB pathway in airway epithelium impacts host defense against Pseudomonas aeruginosa. J. Immunol. 1764923-4930. [DOI] [PubMed] [Google Scholar]
- 65.Santoro, M. G., A. Rossi, and C. Amici. 2003. NF-kappaB and virus infection: who controls whom. EMBO J. 222552-2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sarawar, S. R., B. J. Lee, and F. Giannoni. 2004. Cytokines and costimulatory molecules in the immune response to murine gammaherpesvirus-68. Viral Immunol. 173-11. [DOI] [PubMed] [Google Scholar]
- 67.Sarawar, S. R., B. J. Lee, S. K. Reiter, and S. P. Schoenberger. 2001. Stimulation via CD40 can substitute for CD4 T cell function in preventing reactivation of a latent herpesvirus. Proc. Natl. Acad. Sci. USA 986325-6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sarawar, S. R., R. D. Cardin, J. W. Brooks, M. Mehrpooya, R. A. Tripp, and P. C. Doherty. 1996. Cytokine production in the immune response to murine gammaherpesvirus 68. J. Virol. 703264-3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schwarz, M., and P. M. Murphy. 2001. Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor constitutively activates NF-kappa B and induces proinflammatory cytokine and chemokine production via a C-terminal signaling determinant. J. Immunol. 167505-513. [DOI] [PubMed] [Google Scholar]
- 70.Sha, W. C., H. C. Liou, E. I. Tuomanen, and D. Baltimore. 1995. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell 80321-330. [DOI] [PubMed] [Google Scholar]
- 71.Simas, J. P., S. Marques, A. Bridgeman, S. Efstathiou, and H. Adler. 2004. The M2 gene product of murine gammaherpesvirus 68 is required for efficient colonization of splenic follicles but is not necessary for expansion of latently infected germinal centre B cells. J. Gen. Virol. 852789-2797. [DOI] [PubMed] [Google Scholar]
- 72.Snapper, C. M., P. Zelazowski, F. R. Rosas, M. R. Kehry, M. Tian, D. Baltimore, and W. C. Sha. 1996. B cells from p50/NF-kappa B knockout mice have selective defects in proliferation, differentiation, germ-line CH transcription, and Ig class switching. J. Immunol. 156183-191. [PubMed] [Google Scholar]
- 73.Stewart, J. P., E. J. Usherwood, A. Ross, H. Dyson, and T. Nash. 1998. Lung epithelial cells are a major site of murine gammaherpesvirus persistence. J. Exp. Med. 1871941-1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Suárez, A. L., and L. F. van Dyk. 2008. Endothelial cells support persistent gammaherpesvirus 68 infection. PLoS Pathog 4e1000152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tarakanova, V. L., F. Suarez, S. A. Tibbetts, M. A. Jacoby, K. E. Weck, J. L. Hess, S. H. Speck, and H. W. Virgin. 2005. Murine gammaherpesvirus 68 infection is associated with lymphoproliferative disease and lymphoma in BALB β2 microglobulin-deficient mice. J. Virol. 7914668-14679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tibbetts, S. A., J. S. McClellan, S. Gangappa, S. H. Speck, and H. W. T. Virgin. 2003. Effective vaccination against long-term gammaherpesvirus latency. J. Virol. 772522-2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Uchida, J., T. Yasui, Y. Takaoka-Shichijo, M. Muraoka, W. Kulwichit, N. Raab-Traub, and H. Kikutani. 1999. Mimicry of CD40 signals by Epstein-Barr virus LMP1 in B lymphocyte responses. Science 286300-303. [DOI] [PubMed] [Google Scholar]
- 78.Usherwood, E. J., J. P. Stewart, and A. A. Nash. 1996. Characterization of tumor cell lines derived from murine gammaherpesvirus-68-infected mice. J. Virol. 706516-6518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.van Dyk, L. F., H. W. T. Virgin, and S. H. Speck. 2003. Maintenance of gammaherpesvirus latency requires viral cyclin in the absence of B lymphocytes. J. Virol. 775118-5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vart, R. J., L. L. Nikitenko, D. Lagos, M. W. Trotter, M. Cannon, D. Bourboulia, F. Gratrix, Y. Takeuchi, and C. Boshoff. 2007. Kaposi's sarcoma-associated herpesvirus-encoded interleukin-6 and G-protein-coupled receptor regulate angiopoietin-2 expression in lymphatic endothelial cells. Cancer Res. 674042-4051. [DOI] [PubMed] [Google Scholar]
- 81.Verzijl, D., C. P. Fitzsimons, M. van Dijk, J. P. Stewart, H. Timmerman, M. J. Smit, and R. Leurs. 2004. Differential activation of murine herpesvirus 68-and Kaposi's sarcoma-associated herpesvirus-encoded ORF74 G protein-coupled receptors by human and murine chemokines. J. Virol. 783343-3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang, L., R. Wuerffel, and A. L. Kenter. 2006. NF-kappa B binds to the immunoglobulin S gamma 3 region in vivo during class switch recombination. Eur. J. Immunol. 363315-3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Weck, K. E., A. J. Dal Canto, J. D. Gould, A. K. O'Guin, K. A. Roth, J. E. Saffitz, S. H. Speck, and H. W. Virgin. 1997. Murine gamma-herpesvirus 68 causes severe large-vessel arteritis in mice lacking interferon-gamma responsiveness: a new model for virus-induced vascular disease. Nat. Med. 31346-1353. [DOI] [PubMed] [Google Scholar]
- 84.Weck, K. E., M. L. Barkon, L. I. Yoo, S. H. Speck, and H. I. Virgin. 1996. Mature B cells are required for acute splenic infection, but not for establishment of latency, by murine gammaherpesvirus 68. J. Virol. 706775-6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Weck, K. E., S. S. Kim, H. I. Virgin, and S. H. Speck. 1999. B cells regulate murine gammaherpesvirus 68 latency. J. Virol. 734651-4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Weck, K. E., S. S. Kim, H. I. Virgin, and S. H. Speck. 1999. Macrophages are the major reservoir of latent murine gammaherpesvirus 68 in peritoneal cells. J. Virol. 733273-3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Willer, D. O., and S. H. Speck. 2003. Long-term latent murine gammaherpesvirus 68 infection is preferentially found within the surface immunoglobulin D-negative subset of splenic B cells in vivo. J. Virol. 778310-8321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Willer, D. O., and S. H. Speck. 2005. Establishment and maintenance of long-term murine gammaherpesvirus 68 latency in B cells in the absence of CD40. J. Virol. 792891-2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ye, F. C., F. C. Zhou, J. P. Xie, T. Kang, W. Greene, K. Kuhne, X. F. Lei, Q. H. Li, and S. J. Gao. 2008. Kaposi's sarcoma-associated herpesvirus latent gene vFLIP inhibits viral lytic replication through NF-κB-mediated suppression of the AP-1 pathway: a novel mechanism of virus control of latency. J. Virol. 824235-4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhao, J., V. Punj, H. Matta, L. Mazzacurati, S. Schamus, Y. Yang, T. Yang, Y. Hong, and P. M. Chaudhary. 2007. K13 blocks KSHV lytic replication and deregulates vIL6 and hIL6 expression: a model of lytic replication induced clonal selection in viral oncogenesis. PLoS One 2e1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhong, H., M. J. May, E. Jimi, and S. Ghosh. 2002. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol. Cell 9625-636. [DOI] [PubMed] [Google Scholar]
- 92.Zou, P., J. Kawada, L. Pesnicak, and J. I. Cohen. 2007. Bortezomib induces apoptosis of Epstein-Barr virus (EBV)-transformed B cells and prolongs survival of mice inoculated with EBV-transformed B cells. J. Virol. 8110029-10036. [DOI] [PMC free article] [PubMed] [Google Scholar]