

1. Introduction
The effects of low-molecular-weight (<1000 g/mol) chemicals on biosystems ranging from membranes to organisms are of interest in medicinal chemistry, chemical biology, pharmacokinetics, pesticide science, environmental toxicology, and several other areas of science and technology. Most effects are difficult to predict, because of their heavy dependence on the chemical structures. The effects in homologous series in biosystems containing at least one membrane, however, are often (6174 cases documented)1,2 a smooth function of the physicochemical properties of chemicals, most frequently represented by the reference partition coefficients (4223 cases),1,2 but also by electronic parameters and steric characteristics. The structure-nonspecific effects described by nonlinear dependencies on the reference partition coefficients (553 cases)1,2 are mostly related to the passive distribution of chemicals in the aqueous and less-polar phases of biosystems rather than to the structure-specific interactions with the receptors, enzymes, and transporters. The disposition-related homologous series data provide initial outlines of the relationships between structure and disposition, equivalent to the cross sections along one variable through complex dependencies. Can the relationships be extended to cover broader classes of chemicals or eventually the entire chemical universe?
The research area that is striving to understand the principles of distribution of chemicals at the subcellular level of biosystems, in terms of the structures and properties of chemicals, can be appropriately called structure-based subcellular pharmacokinetics (SBSP). Theoretical fundamentals, status, applications to experimental data, and probable future development trends of SBSP are reviewed here. Numerous reviews are available on different aspects of distribution and how it is related to the properties of the chemicals,3–25 and the subject is covered, to some extent, in several monographs on quantitative structure−activity relationships (QSARs),26–32 drug design,33–41 pesticide design,42,43 and medicinal chemistry.44–50 This treatise introduces several new concepts and presents a systematic, critical, integrative, and chemically oriented view. To foster readability, a unified nomenclature is used throughout the text, and the details of the model development are deferred to section . These details can be skipped without a major impact on the understanding of the remaining content.
For a quick, semi-quantitative overview of the disposition of chemicals in biosystems and its relation to structure of chemicals, the reader is referred to the following psrts of the paper. In section , relevant physiology is concisely summarized and prevailing transport routes in individual organs are identified. Section relates the trans-bilayer transport rates to the interactions of chemicals in the bilayer regions, which can be characterized using surrogate solvent systems, as described in section . These results are used in section to obtain coarse estimates of equilibration times for crossing a single bilayer on a gross time scale, in regard to its dependence on the structure of the chemicals.
1.1. Subcellular Pharmacokinetics
The main goal of structure-based subcellular pharmacokinetics (SBSP) is a model-based description of the kinetics of the distribution of chemicals, in terms of the properties of both chemicals and biosystems. For this purpose, the conceptual kinetic models are constructed, comprising physically distinct subcellular compartments such as membranes or their regions, and the extracellular and intracellular aqueous phases. The considered processes include transport and accumulation in a set of the aqueous phases and membranes, as well as protein binding, metabolism, hydrolysis, and other reactions of chemicals with body constituents. The resulting differential equations are solved either numerically (see section ) or explicitly (see sections and ), depending on the complexity of the model. To simplify the solutions, the complexity of mathematical description of the subcellular models is often reduced in the process of solving the pertinent differential equations, using the experimentally verified time hierarchy of the processes that determine the disposition of chemicals. Individual rate and equilibrium parameters are related to the computed characteristics or experimentally determined physicochemical properties by the extra-thermodynamic linear free-energy relationships (LFERs).51,52 The properties of chemicals characterize their behavior in water (hydration, ionization, spontaneous hydrolysis rate) or in macroscopic surrogate systems imitating parts or processes in the biosystem, which are either difficult to analyze directly, or for which generalizations are sought (reference partitioning systems (section ), protein binding (section ), in vitro enzymatic reactions (section ), and surrogate chemical reactions (section )).
Like most other models of biological processes, the SBSP models belong to the category of a posteriori semi-empirical models,53,54 because the modeled system is not known in sufficient detail to allow for the formulation of a priori theoretical models. The unknown properties, which do not change under given experimental conditions and are difficult to measure, are usually collected in adjustable coefficients, which are then optimized by regression analysis to provide the best agreement between the model and experiment. To formulate tractable models, decisive features governing the behavior of the system must be identified and captured in the description. For a meaningful optimization of the regression coefficients in the SBSP models, a proper balance between the number of adjustable coefficients and the information content of available experimental data is required. In the following, every attempt will be made to follow these principles in the descriptions of chemicals’ disposition. Individual steps in the SBSP model construction are analyzed in section .
The functional form, in which the time is incorporated in the SBSP models, has been repeatedly proven by classical pharmacokinetics. Therefore, a proper application of the SBSP concepts to the chemicals’ disposition in organisms should result in expressions that provide similar time dependencies as the equations of classical pharmacokinetics (sections and ).
As the ultimate outcome of the SBSP modeling, the kinetics of disposition is expressed as a nonlinear disposition function of properties, with adjustable coefficients containing the biological and chemical attributes, which do not vary under given experimental conditions. The properties are represented by the reference partition coefficients, acidity, reactivity, and other parameters, which may also include three-dimensional (3D) structures of chemicals and structure-related characteristics. The disposition function is calibrated using experimental data of, typically, several dozens of compounds, and then used for the prediction of distribution of untested chemicals under identical experimental conditions. The SBSP models have a better predictive ability than empirical models, especially outside the ranges of tested properties(55) (section ). Finally yet importantly, the conceptual nature of the SBSP models facilitates their flexible extensions accommodating some changes in experimental protocols (e.g., section ).
SBSP has its roots in the distribution-based quantitative structure−activity relationships (QSAR or QSTAR, if the exposure time, T, is included), as defined by the pioneering works of Hansch and Fujita,(56) Stehle and Higuchi,(57) Penniston,(58) McFarland,(59) Yalkowsky and Flynn,60,61 Leo,(62) Martin,(63) Kubinyi,(64) Dearden,(65) van de Waterbeemd,(66) Cooper,(67) Seydel,(68) Schaper,(69) Franke,(70) Berner,(71) Amidon,(72) Anderson and Xiang,(73) Benet,(74) and others. SBSP is also related to classical pharmacokinetics,(75) with which it shares the representation of biosystems as a set of compartments. However, the compartment size has been reduced to subcellular dimensions, with the SBSP compartments representing the membranes or their regions, and extracellular and intracellular aqueous phases. The nanoscale dimensions ensure fast diffusion of chemicals inside the less-structured subcellular compartments (section ). Fast diffusion makes the SBSP models suitable for modeling the effects of chemicals by facilitating a coherent description of their concentrations in the immediate surroundings of the receptors, as known in chemical kinetics. The rate/equilibrium constants, characterizing elementary steps in the subcellular distribution, are related to physicochemical properties of chemicals via the extra-thermodynamic LFERs, as known in the QSAR research and physical organic chemistry.51,52
SBSP relies on other disciplines for methodological support: principles of dynamic modeling(53) are used to simplify the models; computational chemistry provides simulation techniques and molecular characteristics; chemical kinetics supplies some modeling schemes and approximations; physical organic chemistry lends LFER; biophysical methods are used to study transport; and biochemical approaches deliver the binding data for the interactions of chemicals with body constituents. The interplay between SBSP and other disciplines is outlined in Figure 1. A special effort has been made to compare and integrate, where feasible, the approaches to the analysis of the same phenomenon in different areas.
Figure 1.

Structure-based subcellular pharmacokinetics and related sciences. Two-sided arrows indicate mutual influence; one-sided arrows indicate supportive roles.
Experimental techniques providing data for the calibration of the SBSP models can be classified as indirect and direct, depending on whether the phase, where the concentration of chemicals is determined, has been separated in the course of the experiment from the rest of the biosystem or not, respectively. The indirect techniques include measurement of the chemical’s uptake into membranes,(76) vesicles,(77) subcellular organelles,(78) cells,(79) tissues,(80) and organs.(81) The direct approaches, suitable for simpler biosystems, are represented mainly by spectroscopic techniques: ultraviolet and visible (UV Vis) difference spectroscopy,82–85 circular dichroism,84–86 fluorescence lifetime measurements,(87) fluorescence quenching techniques,88–95 fluorescence anisotropy,(96) Fourier-transform infrared (FT-IR) spectroscopy(84) combined with attenuated total reflection (ATR)(97) and high pressure,(98) electron paramagnetic resonance (EPR) with phospholipids containing spin labels in different positions,99–104 and various nuclear magnetic resonance (NMR) techniques,84,87,90,99,105–129 including the measurement of nuclear Overhauser enhancement spectroscopy (NOESY)130,131 cross-relaxation rates.(127) Other direct techniques include neutron diffraction,132–138 small-angle X-ray diffraction (XRD),(139) autoradiography,(140) and fluorescence microscopy.141,142
The time courses of the concentrations of chemicals in individual cells and their organelles are difficult to measure precisely, and the intracellular data for the calibration of the SBSP models are quite scarce. For this purpose, the uptake and release kinetics of chemicals, monitored in the extracellular media, are often used. This is a rigorous approach, because the extracellular and intracellular concentrations are variables of the same model and any of them can be, in principle, used for the model calibration. Although this statement is not valid for all types of compartment models, it does hold for the SBSP models. Another option is to use a biological effect, which has been assumed or shown to be (i) an immediate consequence of the chemical’s presence in a biological phase,(143) and (ii) proportional to the chemical’s concentration,(144) as a measure of the time course of the chemical’s concentration in the relevant biological phase.
According to the dependence on the detailed molecular structures of chemicals, the processes in which chemicals participate in biosystems can be classified in two groups: (i) disposition of chemicals in biosystems, except protein-mediated transport and enzymatic biotransformation; and (ii) interactions of chemicals with specific macromolecules, which are responsible for biological effects, mediated transport, and biotransformations of the compounds. The processes in the first group can be fully characterized by properties of chemicals in aqueous solutions (ionization, hydrolysis) and surrogate systems (e.g., partitioning, reactivity, redox potential). The processes in the second group depend predominantly on the molecular structure of compounds, given as the 3D constitution of atoms, and a spatial distribution of the structure-related properties. These processes can be examined in vitro, using the relevant macromolecule(s). The first-group processes governing disposition of chemicals, except enzymatic biotransformation and protein-mediated transport, are considered property-related or conformation-averaged processes, and the second-group interactions with the fate- and effect-determining macromolecules are deemed structure-specific or conformation-dependent processes. The property-related processes are easier to model and the models can be generalized for all biosystems. The conformation-dependent processes convey structural specificity to the disposition of chemicals and must be modeled individually for each biosystem. The overall description of disposition for diverse chemicals requires that the results for conformation-dependent processes are embedded in the models for property-dependent processes.
With the improvement of experimental techniques, multiple binding modes are detected with increased frequencies in some binding processes.(145) The processes still retain a significant level of structural specificity and require specific treatment in QSAR modeling.(146) The methods for descriptions of both process types have been developed separately. SBSP conceptually combines modeling of both the property-related processes and the structure-specific processes, as illustrated in eq 7 in section . Therefore, SBSP represents a suitable framework for integrated description of the fates of chemicals in biosystems. One of the outcomes is the cell-QSAR approach, which extends, by incorporating the disposition function, conceptual 3D-QSAR techniques for the application to cell-level assay data.
1.2. Disposition in Organisms
To elicit an effect in a biosystem, a chemical must reach the site of action, frequently located far away from the points of entry, which, for higher organisms, are typically the gastrointestinal and pulmonary tracts, skin, mucosa, and cornea. We will first focus on the physiologic aspects, and later, in section , cover the mechanisms of the transport processes.
In higher organisms, the first step is absorption, which brings the molecules from the point of entry or the site of administration into the bloodstream or the lymphatic system. Although the lymph circulation is much slower than that of the blood,(147) very lipophilic chemicals may use the former route for distribution, piggy-backing on the transport systems of lipophilic nutrients.148–150 These compounds may also exhibit slow absorption into the blood and lymph capillaries in the villi, and enter the lymph via endocytosis in the Peyer’s patches in ileum, especially in lipid-based formulations.151–160
Absorption mostly proceeds by the transcellular route, with the molecules crossing epithelia and other tissues by passive and active transport through the bilayers and cell interiors. An exception is the absorption through the skin, where the only cells in the external stratum corneum layer are keratinized corneoytes.(161) Corneocytes are embedded in a lamellar phase of lipids,(162) with the composition differing from that of other membranes. Absorption through stratum corneum is assumed to proceed mainly by passive lateral diffusion(163) through the lipid phase.(164) Could a similar passage through a cell layer by lateral diffusion in the bilayers,(165) without entering cell interiors, be accomplished by amphiphilic molecules in other tissues? This does not seem to be a widespread mechanism. Tight junctions would need to be bypassed by the flip-flop to the cytoplasmic side of the bilayer, followed by the return to the outside leaflet before the release into medium on the opposite site of the cell layer. Moreover, for many amphiphilic compounds, the flip-flop is a slow process (section ).
Once in the blood or lymph, chemicals quickly reach distant body parts by convection and diffusion, although circulation of the lymph is slower. The details of vasculature have been long known in great detail, starting with the description of capillaries by Malpighi in 1661, the capillary walls by Schwann in 1839, and the cellular nature of the capillary walls by von Recklinghausen in 1862, as well as the introduction of the term “endothelium” by His in 1865.(166) The distribution of chemicals from the biological fluids into tissues involves (i) paracellular transport into interstitial fluid directly mixing with the content of blood and lymph capillaries and (ii) transcellular transport bringing the molecules of chemicals into the cells. In the hierarchy of the vasculature, we shall focus on the capillaries, because (i) their walls are composed of a single layer of endothelial cells and have openings in some organs, in contrast to larger blood vessels, which have continuous and more-complex structures, and are more difficult to permeate; and (ii) they represent the majority of ∼300 m2 of the vascular endothelium surface and of ∼19 000 km of overall length of blood vessels in a standard 70-kg man.(167)
The walls of blood capillaries in individual organs differ in porosity (Table 1). Continuous capillary walls with endothelial cells connected by tight junctions and supported by a complete basement membrane are encountered in places requiring a barrier protection, as in brain (but not choroid plexus), lungs, intestine, placenta, and testis. Continuous endothelia are still capable of paracellular transport of small molecules, with diameters of <3 nm, such as urea and ions.(168) The endothelial junctions are regulated by various signaling mechanisms and can operatively respond to physiological conditions.(168) The pores in capillary walls are associated with specific functions. Renal glomerulus, serving for plasma filtration in the kidney, exhibits 30−60 nm openings between the endothelial cells, which are supported by a complete basement membrane. The most porous capillaries allow the transcellular passage of macromolecules and even some cells, thanks to the broken basement membrane and the openings with the diameters reaching 100−300 nm in the bone marrow, spleen, and sinusoids of the liver,169–171 and up to 1.2 μm in tumors.(172) For a point of reference, the average capillary diameter is only slightly larger than the average diameter of erythrocytes (7.5 μm),(173) and the minimum capillary diameter is ∼4 μm.(167) Lymph capillaries are also porous.(167) The exchange of water between the blood and interstitium through the leaky blood capillaries attains 70% of the volume per minute(174) and is somewhat slower for the lymph capillaries, because of the difference in hydrostatic pressures. Because all pores are substantially larger than small organic molecules, the transport through the leaky capillary walls into the interstitial space proceeds mainly through the paracellular route on the time scale of minutes. This type of transport has the specificity determined only by the interactions of chemicals with cells that affect the free concentrations and, thus, the overall transport rates. In contrast, the transcellular route that the chemicals must use to cross the nonporous capillaries exhibits widely differing transport rates and mechanisms, which depend on the properties of the chemicals (section ).
Table 1. Perfusion Characteristics of Some Organs in a Standard 70-kg Human167,175,176.
| Blood Flow |
Capillary Density |
|||||
|---|---|---|---|---|---|---|
| organ | relative volume (%) | fraction of cardiac output (%) | (mL g−1 min−1) | capillary type a | served cellsb | per mm3 |
| lungs | 1.6 | 100 | 5 | C | 1 | 5000 |
| kidneys | 0.5 | 22 | 4 | C, F c | 2 | 2500 |
| intestines | 1.7 | 21 | 0.9 | C | 5 | 500 |
| liver | 2.3 | 27 | 0.8 | S | 5 | 500 |
| heart | 0.4 | 4 | 0.7 | C | 5 | 500 |
| brain | 2.0 | 14 | 0.5 | C, F d | 5 | 500 |
| spleen | 0.3 | 1.5 | 0.4 | S | 5 | 500 |
| skin | 11 | 6 | 0.04 | C | 15 | 50 |
| fat | 20 | 4 | 0.03 | C | 15 | 50 |
| muscles | 43 | 15 | 0.02 | C | 15 | 50 |
| bones | 16 | 5 | 0.02 | S | 15 | 50 |
C = continuous, complete endothelial layer and basement membrane; F = fenestrated, complete basement membrane, endothelial layer has openings 30-60 nm in diameter; S = sinusoids, broken basement membrane, endothelial layer has openings 100−300 nm in diameter.
The average number of cell layers served.
C in proximal and distal tubule and the loop of Henle, F in glomerulus.
F only in choroid plexus.
Chemicals can enter the tissues cells directly from the capillary walls, from the interstitial fluid or from other tissue cells. Individual organs differ in density of vasculature and, consequently, in the number of cells, which do not maintain a direct contact with the capillaries or primary interstitial fluid directly mixing with the blood.175,176 According to the decreasing capillary densities, the tissues can be classified into four categories: (i) lungs; (ii) kidney; (iii) brain, liver, intestines, and heart; and (iv) muscles, connective tissues, fat, and bones (Table 1). The average thickness of the cell layer served by a capillary increases in this order. The basal blood flows for individual organs are also summarized in Table 1. The basal blood flow can be increased 5−10 times in most organs during physical exercise and other conditions. The exceptions are brain and kidneys, where the blood flow is buffered, and the basal flow cannot increase more than twice.(167) These data determine relative equilibration times for individual tissues, which are primarily given by the number of crossed bilayers, including, in each cell, cellular, and organelle membranes, as well as endoplasmic reticulum. For a single bilayer, the equilibration times vary between less than a second and several days, depending on the structure of the transported chemical (section ). The growing number of crossed bilayers increasingly magnifies these differences.
Chemicals can be eliminated by processes that do or do not change their molecular structure, i.e., by metabolism or excretion, respectively. Metabolism and excretion processes collectively represent elimination.
The prevailing portion of metabolism in higher organisms happens in the liver, where the majority of metabolizing enzymes, predominantly cytochromes P450 (CYPs, section ), are located. However, CYPs and other metabolizing enzymes are also expressed in the intestine and some tissues. Many chemicals can also participate in spontaneous reactions with body constituents (section ).
The excretion of chemicals and their metabolites proceeds mainly in the kidneys, although excretion into feces, bile, saliva, and sweat, as well as transpiration into air, may contribute. From more than a liter of the blood passing through the kidneys every minute, ∼100 mL of plasma are filtered at the glomerulus of nephrons, through the 30−60 nm pores, which are normally too narrow to allow the passage of proteins and protein-bound chemicals. Active, protein-mediated secretion and reabsorption of chemicals proceed in the proximal tubule (sections and ), and passive reabsorption happens along the entire tubule. Water is also reabsorbed, reducing the volume of the filtrate by a factor of ∼100.
The time courses of concentrations of chemicals in individual parts of higher organisms are determined by absorption, distribution, metabolism, and excretion (ADME). We will refer to these processes collectively as disposition, although, in classical pharmacokinetics, the term disposition denotes only the last three processes.(75) In vitro cellular systems (e.g., microbial populations) do not possess specialized structures participating in absorption and excretion, and the processes that the chemicals undergo can be classified as distribution (or transport) and metabolism.
During absorption, distribution, and elimination, the majority of chemicals seem to enter and leave the majority of the cells through phospholipid bilayers of the membranes by passive diffusion, without the help of protein carriers, unless the chemicals are analogs of physiologic molecules with active or passive carrier-mediated transport. The baseline diffusional trans-bilayer transport rates of many chemicals may be increased by influx transporters in the intestine, liver, and kidney(177) (section ) or decreased by the efflux pumps and transporters (section ), which are expressed in organs with excretory functions such as the kidney and liver; in sheets or tissues with barrier functions such as the intestine and capillaries of brain, testis, and placenta; and also in tumors.(178) The list of chemicals undergoing facilitated or active transport is steadily growing, because this area is a subject of extensive research, because of the obvious importance in drug delivery.(179) It is assumed that practically each drug is a substrate of a transporter somewhere in the body but the phenomenon may not always be clinically relevant.(180) If the trans-bilayer transport is fast, its further enhancement has no significant impact on the overall process. Understanding of both passive and mediated transport mechanisms is necessary for prediction of distribution of chemicals. The generalizing conclusions about the key roles of transporters for the entry of all drugs into all cell types(181) are not based on sound arguments. The transport mechanisms utilizing protein carriers can be described in terms of structure in a similar way as other structure-specific processes, using receptor-based or ligand-based approaches (section ).
After crossing the cell membrane, intracellular distribution of chemicals proceeds chiefly by passive diffusion through the bilayers of organelles and of endoplasmic reticulum, although several transporter proteins are expressed in intracellular membranes and may affect the transport rates. Some hydrophobic chemicals, planar aromatic hydrocarbons, and similar compounds(182) enter the nucleus in a complex with the aryl hydrocarbon receptor,183,184 but the passive diffusion across the nuclear envelope cannot be excluded.
In summary, passive trans-bilayer diffusion is a ubiquitous and most important process affecting the distribution of chemicals in biosystems. Because of frequent references to this process, we will use the term “transport” to denote the flux and the term “accumulation” to characterize the pseudo-equilibrium retention of chemicals in bilayers. The SBSP models are essentially common mass-action based models for the interactions of chemicals with body constituents, which are placed within the framework of intracellular and extracellular compartments separated by membranes. The term “baseline disposition” can be used to denote the disposition that is not affected by protein-mediated processes, such as active or passive mediated transport or enzyme-catalyzed metabolism. The baseline disposition is always occurring in any biosystem and is implicitly assumed if no specific knowledge about the concentration and kinetics of transporters or metabolizing enzymes is available. The carrier-mediated influx or efflux become important if they significantly change the baseline disposition. Understanding the transport and its interplay with other property-related processes (ionization, hydrolysis) is a crucial requirement for the development of the relationships between the baseline disposition kinetics and structure or properties of chemicals. The interactions of chemicals with biosystems will be analyzed at the elementary level of individual processes in section , and for the biosystems of increasing complexity in section .
2. Individual Steps vs. Properties
To describe chemical disposition via explicit or numerical solutions of appropriate differential equations, the rate and equilibrium parameters for individual processes must be expressed as functions of physicochemical properties of chemicals and biosystems. In sections −, we will focus on the structural dependencies of the interactions with lipids, protein carriers, inert proteins, and enzymes, which are involved in disposition of chemicals.
2.1. Interactions with Lipids
Lipids of mammalian cells can be classified as bilayer-forming lipids and other lipids. The first category is represented by phospholipids, cholesterol, sphingomyelins, and other species,(185) whereas the second category consists mainly of triglycerides and their derivatives. Triglycerides are contained in lipoproteins and lipid droplets, which are present in practically all cells, and accumulate to a large extent in adipocytes (section ).(186)
From the viewpoint of chemicals’ disposition in biosystems, transport through phospholipid bilayers and accumulation in phospholipid bilayers are among the most important processes. Chemicals usually interact with lipid aggregates, not with individual lipid molecules. Therefore, the interactions are mostly of structure-nonspecific nature, resemble interactions with phases or solvents, and can be analyzed using the data obtained in macroscopic surrogate systems (section ). These facts permit generalization of the rules for the interactions of chemicals with phospholipids to the level of predictions of the kinetics and equilibria from the structure of chemicals.
2.1.1. Bilayer Structure and Properties
History of the research on bilayer structure and properties is tightly interwoven with structural studies of biological membranes. The timeline showing the major events in the history of the bilayer structural studies is given in Figure 2, with the details and references summarized in the next section. The key findings about the protein interactions with the bilayer, if not related to the bilayer structure, are only mentioned in the text. The bilayer structure is analyzed in more detail in section .
Figure 2.

Historical milestones in the elucidation of bilayer structure.
2.1.1.1. History of Membrane Structure
The concept of the cell membrane originated in the second half of the 19th century from the work of botanists, who recognized that the protoplasm of plant cells was separated from the cell walls by an osmotic barrier.187–189 The determination of surface tension was established as the measurement of the force needed to pull a metal plate out of the interface.(190) The predecessor of the Langmuir trough was invented and used to measure the dynamic surface tension.(191) Numerous observations of lipid-soluble compounds partitioning into the membrane led to the hypothesis about the lipid character of the membrane.(192) The measurements of the area to which oils were able to spread at the air/water interface resulted in the proposal for the monolayer structure of fatty acids having the headgroups immersed in water and chains protruding into air.(193) The notion of a double lipid layer came from the observations showing that the lipid content of erythrocytes from several animals was just sufficient to cover the cell surface by two monolayers.(194) The resulting structural model of the membrane as a bilayer sandwiched between two protein layers holding the lipids in place(195) was later modified, on the basis of electron-microscopic observations, to a self-standing bilayer with adsorbed polypeptide and possibly polysaccharide layers,(196) and finally to the fluid mosaic model with the bilayer representing a support for peripheral and integral proteins.(197) The geometry of phospholipid molecules was considered a factor in formation of the maximal bilayer curvature.(198) The X-ray(199) and calorimetric(200) observations indicated the presence of the gel and liquid-ordered lipid phases in the membranes. The X-ray structural studies clarified the orientation of hydrocarbon chains.(201) The hydrophobic effect, attributed to entropy-driven changes in water structure,(202) which was initially studied in connection with colloid behavior,203–205 waterproofing of textiles,(206) and protein folding,207,208 was recognized as the driving force in the formation of the bilayer.(209) The structure of the phospholipid bilayer in the gel phase was studied by neutron diffraction.210–212 The headgroup dipoles change orientations in response to the overall charge of the bilayer.(213) Integral proteins were shown to interact with the bilayer core via hydrophobic residues.(214) In a bilayer composed of a lipid mixture, the lipids with the lengths of the fatty acid chains matching the dimensions of the hydrophobic exteriors of integral proteins concentrate around the proteins, to minimize hydrophobic mismatch.(215) Combined X-ray and neutron diffraction analyses provided a detailed picture of distribution of individual phospholipid fragments along the normal of a liquid-ordered phosphatidylcholine (PC) bilayer.216–220 High-resolution X-ray diffraction was used to determine the key structural parameters of the fully hydrated PC bilayer.(221) Coexistence of the gel and liquid-ordered phases was directly observed by atomic force microscopy.(222) These observations contributed to the current view of biological membranes as dynamic structures, where peripheral and integral proteins interact with the bilayer containing different phospholipids and other lipid species in liquid-ordered and gel states.
2.1.1.2. Membrane Composition
Phospholipid bilayers form the basis of endomembranes, including cell membrane, endoplasmic reticulum, Golgi bodies, vesicles, and nuclear envelope, which participate in several vital processes in eukaryotic cells, such as attachment, intracellular compartmentalization, energy transduction, signaling, protein sorting, and other secretory and endocytic processes. The protein and lipid contents are roughly equal on the weight basis, with significant deviations for specialized membranes. For instance, myelin, the multilayered insulating sheath of neural axons contains 80% lipid, and mitochondrial membranes, the locale of respiration, oxidative phosphorylation, and intense metabolism, contain 80% protein. In the simplest case of inert proteins, the significant protein content decreases the area available for passive trans-bilayer transport and modifies the structural arrangement of surrounding bilayer phospholipids. The irregularities may increase the transport rates, as described for the perturbed bilayer structures at the transition temperature (sections and ).
Mammalian bilayers are composed of phospholipids, sphingomyelin, glycolipids, cholesterol and other sterols, and many other non-bilayer-forming lipid species.(185) Phospholipids, mainly PC, less phosphatidylserine, phosphatidylethanolamine, and phosphatidylinositides, account for 40%−80% of the bilayer weight. Cholesterol, the preponderant mammalian sterol, is equimolar with phospholipids in hepatocytes, erythrocytes, and myelin; it represents 20% w/w of stratum corneum(223) and is less abundant in other cells and tissues. The lipid composition of individual membranes varies widely, even between the membranes of the same cell,(224) and affects the permeation rates. The tubular cells of barrier epithelia in the kidney nephrons, manifesting low membrane permeability, contain the outer bilayer leaflets composed of a specific mixture of PC, sphingomyelin, glycosphingolipids, and cholesterol. The omission of any component dramatically increases permeability.(225) Caveolae, invaginated membrane structures with high levels of cholesterol, sphingomyelin, and caveolin (section ), exhibit high permeabilities, thanks to a high degree of unsaturation of fatty acid chains.(226)
2.1.1.3. Phospholipid Distribution in Membranes
Phospholipids in membrane bilayers do not form homogeneous mixtures. Lipid distribution may differ, under physiological conditions, for different cells or different membranes of the same cell, for the two monolayers forming a bilayer, and for a monolayer. For different cell membranes, sphingolipids and sterols form gradients along the secretory pathways, with the highest abundance in the plasma membranes and the lowest level in endoplasmic reticulum.
In a bilayer, lipids display asymmetric trans-bilayer arrangements maintained by energy-dependent transporters: sphingomyelin, glycolipids, and PC concentrate in the outer leaflet of the normal cell membrane, whereas phosphatidylserine and other phospholipids prefer the inner monolayer.227,228 These differences are particularly pronounced in renal and intestinal epithelial cells, which exhibit low permeabilities to carbon dioxide and ammonia.(229) Perturbation of this asymmetry, especially the appearance of phosphatidylserine at the cellular surface, is a hallmark of apoptosis, which enables an orderly removal of the affected cell by phagocytes.(230)
In any of the two monolayers of the bilayer, some mixtures of lipids are immiscible to various degrees and may form lateral domains, occasionally termed lipid rafts, especially in the surroundings of proteins.215,231–237 Formation of a domain in a monolayer affects the phospholipid distribution in the opposite monolayer.238–242 The domains participate in many of the membrane-related vital processes mentioned in the introductory paragraph of section ,243–252 as well as in the transport253,254 and accumulation255–258 of chemicals. The perturbed bilayer structures at the raft perimeters may affect the transport rates, because increased permeabilities were observed for coexisting phases at transition temperatures (see the next section).
2.1.1.4. Phase Behavior of Bilayers
Phospholipid molecules in aggregates exhibit collective behavior. The main determinant of this behavior, for the given composition and normal pressure, is temperature. At lower temperatures, the bilayer is in the gel phase, with the fatty acid chains tightly packed and ordered, and mostly assuming extended trans conformations.(259) When the temperature increases above the main transition temperature, the bilayer enters the liquid-ordered phase, with less-regular conformations of fatty acid chains. Among additional chain-ordered phases, the ripple phase of PC and phosphatidylglycerols, exhibiting periodic ripples several hundred angstroms apart,(260) is most frequently studied. The profile of individual ripples may be symmetrical or asymmetrical, with the two shoulders differing in the thickness: one side has the same thickness as the gel phase and the rest is thinner, possibly because of a different tilt or the melted state of the chains.(261) The formation of ripples, consisting of gel and liquid-ordered regions in individual monolayers, which were periodically arranged for geometrical and topological reasons, was modeled by Monte Carlo (MC) simulations(260) and other computational methods (section ). In PC, the ripple phase occurs just below the gel-to-liquid transition chain-melting temperature that marks the coexistence of the gel and fluid phases.(261)
In biological membranes, the main chain-melting transition occurs within the range of 20−60 °C. The heat capacity, as well as the volume and area compressibility, reach their peak values during this transition.(262) The heat-capacity changes seem to be proportional to the volume changes,(263) as well as to the relaxation times, which range from seconds to approximately a minute.(264) Usually, the transition temperature is affected less by the headgroup composition than by that of the tails: it increases with the length and saturation of the fatty acid chains. The transition temperature exhibits a decreasing sigmoidal dependence on hydration: the upper limit is observed for dehydrated bilayers, whereas the lower limit is achieved at the saturation of water-binding capacity of the lipids. The gel-liquid transition occurs in a narrow temperature interval in pure phospholipid bilayers, especially for multi-bilayer liposomes.(264) Additives usually broaden the transition temperature interval. In the gel phase, cholesterol decreases the fatty acid chain ordering and reduces the main transition temperature.(265)
The gel−liquid phase change results in an abrupt increase in bilayer volume (∼4%),(266) area (∼25%),(267) enthalpy (20−40 kJ/mol),(268) and the headgroup hydration(269) (approximately three-fold in PC).(270) The thickness of the bilayer decreases upon the chain-melting transition,(271) as reflected in the disproportionate changes of the volume and area. Above the transition temperature, further temperature elevation results in an increased abundance of folded conformations, leading to a continuous reduction of phospholipid surface density and bilayer thickness(272) and a concomitant increase in headgroup hydration.(270) Within a range of ∼20°, these changes are dependent on temperature in an approximately linear way.(271) The relationship between the phospholipid surface area and the average number of water molecules intercalated in the headgroup region is almost linear.(273) Cholesterol increases the fatty acid chain order in the liquid-ordered phase, as opposed to its effect in the gel phase.(265) The increased order, thanks to cholesterol or diminished unsaturation, is associated with a decrease in the water concentration in the core.(274) Hydration of the headgroups is not closely correlated with the chain order.(274)
2.1.1.5. Bilayer Structure at Atom Level
The dynamic bilayer structure is characterized by average attributes. The most important structural characteristic is the surface area per lipid molecule that is related to the bilayer thickness via the specific volume. To describe the thickness, one can use (i) the steric thickness, characterizing interbilayer distances in multilayer preparations; (ii) the electron density thickness, obtained from electron density profiles as the head-head separation; (iii) the hydrophobic thickness, which is most important for hydrophobic matching of protein structures to bilayers; and (iv) the Luzzati’s thickness, obtained by the gravimetric X-ray method,(275) as amended by inclusion of the compression to eliminate hydration defects.(276)
XRD determines the electron density profiles along the bilayer normal.199,201,275 Neutron diffraction provides the mean positions of the deuterium atoms in bilayers containing selectively deuterated lipids.219,277,278 The experimental data have been available earlier for the more-ordered gel phase210–212 than for the liquid phase. A combination of the XRD and neutron diffraction data provided a detailed picture of distribution of individual phospholipid atoms along the normal of a liquid-ordered PC bilayer with comparatively low hydration.216–220 High-resolution XRD and a computational reduction of the fluctuations enabled determination of the key structural parameters of the fully hydrated PC bilayer, including the area and volume per lipid.(221)
The status of the fatty acid chains can be monitored in the bilayers containing selectively deuterated lipids. 2H NMR results allow for the calculation of the order parameters, isomerization times, and trans/gauche populations of the fatty acid chains.279,280 The latter attribute can also be studied by Raman spectroscopy(281) and IR spectroscopy: the CD2 bands differ for trans and gauche orientations of the adjacent C−C bonds.259,282
2H-NMR spectroscopy was used to study hydration of the headgroups,(283) and, in combination with 31P-NMR, to characterize conformations and orientations of the headgroups.(284) Monitoring of the phosphate and carbonyl bands by FT-IR allowed quantification of hydration and interactions with cations.(285) For oriented bilayers, 2H-NMR was used to determine the overall tilt of the phospholipid molecules.(286)
The bilayers formed by the most abundant mammalian phospholipids, PCs, are studied most frequently. Individual parts of the PC molecules exhibit the following approximate time-averaged orientations, with respect to the bilayer plane. The dipoles formed by the zwitterionic phosphocholine groups align at angles starting at ∼0° and increasing(287) up to ∼50°, when the surface charge becomes more positive.(213) A similar movement of the dipoles, from the in-plane orientation to an orientation more parallel to the bilayer plane normal, was observed in the PC monolayer at the air/water interface upon compression from the liquid-ordered phase to the gel phase.(269) The glycerol backbones are almost perpendicular or slightly slanted, to improve the access of sn-1 carbonyl to water molecules,(288) although the sn-2 carbonyl remains better hydrated.(289) The fatty acid chains are tilted to compensate for the larger surface area of the headgroup, compared to the relevant cross section of the extended acyl chains. The charged phosphocholine groups form a regular network of electrostatic interactions.(290) The thickness of the bilayer increases(291) and the area per phospholipid decreases with prolongation of the acyl chains.272,273
Despite intense thermal motion,220,221 the subregions of (i) low headgroup density, (ii) high headgroup density, (iii) high tail density, and (iv) low tail density were discerned in the structure of a hydrated PC bilayer patch that was simulated by molecular dynamics (MD).(292) The borders of the subregions cannot be precisely expressed in terms of the phospholipid structural elements because of their thermal fluctuations. The ranges of thermal motions and the subregion borders of a fluid PC bilayer are outlined in Figure 3.
Figure 3.

Thermal motion and subregions of the liquid-ordered dioleoylphosphatidylcholine bilayer. The arrows indicate the range of 95% probability of occurrence of (from left) terminal methyls (red), double bonds (blue), carbonyls (red), glycerol (green), phosphate (light blue), choline (black), and hydration water (blue).(220) The subregions are approximated according to the MD simulation results.(292)
Subregion 1 (perturbed water) is represented by a water layer, whose structure is affected by interactions with trimethylammonium and phosphate groups, which temporarily protrude into this subregion. Subregion 1 starts where the orientational ordering of water by the headgroups can be observed and ends where the densities of water and lipid become comparable. In this direction, the overall mass density is gradually increasing along the normal to the bilayer plane. Significant lipid movement in the direction of the bilayer normal creates a rough bilayer/water interface that averages in time to a smooth density profile. Just above the gel−liquid transition temperature, the primary hydration shell consists of 12−16 water molecules per the PC headgroup.293,294 The thickness of this subregion is ∼1 nm in PC and is affected by the surface charge density.(213) Sometimes, this subregion is considered to be part of the bulk water phase.(295)
Subregion 2 (interphase) extends over 0.8 nm and includes, most of the time, the hydrated headgroup atoms up to the ester groups and the first two or three tail methylenes. The distance between the phosphate groups and the headgroups/core interface is 0.41−0.54 nm.(273) This subregion has the highest density and the lowest free volume fraction. Just above the gel−fluid transition temperature, practically all water molecules are engaged in hydration shells of the headgroup atoms. The extent of hydration and the phospholipid surface area increase with temperature as described previously.270–273
Subregion 3 (soft polymer) starts close to the carbonyl groups, where the water density drops to 1% of bulk water density. It contains the first 6−8 methylene segments of the fatty acid chains, with the thickness equal to ∼0.7 nm. The chains are tightly packed, with restricted atom movement, so that the subregion behaves like a viscous liquid. The chain order parameters do not vary much for individual methylene segments. The exception is the second sn-2 methylene, which seems to be less ordered than the other methylenes due to two prevalent conformations.(265) The mass density in subregion 3 is lower than that in subregion 2.
The decrease in density continues in subregion 4 (alkane),(296) which starts where the mass density becomes equal to the density of hexadecane. Subregion 4 represents the center of the hydrocarbon core and is formed by the remaining portions of the fatty acid chains. This subregion resembles a liquid alkane with low viscosity.(292) Thanks to the diffusive hopping transport,292,297,298 some water is present in the bilayer core formed by subregions 3 and 4;299,300 however, the actual concentrations may be too low to measure by some techniques.218,301
Phosphatidylethanolamines (PEs), the most abundant bacterial phospholipids, differ from PC mainly in the hydration of the headgroups. The amine hydrogens participate in hydrogen bonds with water molecules and phosphates of other PE molecules. These interactions, along with the smaller headgroup size, result in a significant decrease in the surface area per lipid molecule.(302)
The surface charge of the bilayer is dependent on the ion composition of the aqueous phase, and on adsorption of charged chemicals, in addition to the headgroup composition. A difference in the monovalent salt concentrations on the two sides of the bilayer can induce significant asymmetry in structure, dynamics, and electrostatic properties of the bilayer.(303) Surface charge density affects the conformation of the zwitterionic phosphate−nitrogen dipoles(213) and the partitioning of charged solutes.304–306 The surface charge at the shear plane of the bilayer, ∼2 Å from the surface,(307) is characterized by the zeta potential,(308) which is related to electrophoretic mobility. This attribute was used to determine the liposome/water partition coefficients of charged and neutral solutes by capillary electrophoresis with liposomes (section ) acting as a pseudo-stationary phase.(309) The electrostatics of the surface are also affected by the dipole potential310,311 which originates from the alignment of electric fields of the charged groups, carbonyls of the ester groups,(213) and the first layer of hydrating water molecules.(312) The influence of the dipole potential on electrostatic properties of the headgroup region is smaller than that of surface charge but not negligible.
2.1.2. Phospholipid Systems
Interactions of chemicals with phospholipids represent critical factors for the kinetics of both disposition and effects of chemicals. Accumulation in biological membranes is important primarily for understanding pseudo-equilibrium pharmacokinetic parameters such as the volume of distribution and the tissue/blood partition coefficients, as well as for elucidation of the biological effects of chemicals. The trans-bilayer transport is a key process in absorption and distribution. Direct measurements on biological membranes are obstructed by interactions of chemicals with proteins and other membrane components, complex membrane architecture, and variability of membrane composition. To avoid these complications, self-aggregating phospholipid systems are frequently used instead of biological membranes to study the interactions of chemicals with phospholipids.
The bilayer/water partition coefficients are expected to be good descriptors of equilibrium partitioning in tissues and organisms, because the bilayers are the parts of biological membranes where many compounds accumulate. Published studies indicate successful descriptions for a limited series of compounds.313–315 A broader application to more-diverse compounds may be limited by the variable phospholipids composition of biological membranes.
An important goal of the SBSP modeling is the prediction of the bilayer interactions from structure of chemicals. The bilayer partitioning is a composite process that generally includes interactions with the headgroups, interface, and core. Consequently, the overall bilayer/water partition coefficients are more difficult to scale in the extrathermodynamic relationships (e.g. eq 1 in section ) than the elementary core/water, interface/water, and headgroups/water partition coefficients. Deconvolution of the liposome partitioning into core/water and interface/water contributions was attempted for limited datasets.316,317
An inherent drawback common to all bilayer systems is the potentially long incubation time needed to achieve the equilibrium for amphiphilic compounds: the flip-flop between the two interfaces of the bilayer can significantly slow the partitioning process,(20) as well as lead to experimental errors, because of the termination of the experiment before reaching the equilibrium. Transport is also slow for compounds with extreme lipophilicities (section );(20) however, the errors caused by a premature experiment termination are less frequent and less significant. For very hydrophilic chemicals, there is no fast step providing the illusion of reaching the equilibrium. Very lipophilic chemicals may have biphasic kinetics but the slow second phase is the partitioning into the small volume of internal water, which is less extensive and causes comparatively small errors. For the monolayer systems, the risk of a slow kinetics is limited to hydrophilic chemicals, which may exhibit a slow entry into the core.
Depending on the preparation, the support used, the lipid composition, the lipid concentration, and the temperature, the phospholipid aggregates can have nanoscale dimensions and spherical, discoid, or oblate ellipsoid shapes (see Figure 4), or macroscopic spherical or planar geometries. The aggregates include free and gel-trapped liposomes and other bilayer-based corpuscles, supported bilayers, and monolayers at solid, liquid, and gas interfaces with water.
Figure 4.

Cross sections of spherical or discoid phospholipid aggregates: (A) liposomes, (B) supported bilayers on microspheres, (C) monolayers on alkylated microspheres, (D) nanodisks, (E) immobilized artificial membranes, and (F) bicelles. The schematic structures are not drawn to proportion. More details are given in the text.
2.1.2.1. Liposomes and Other Bilayer Microsystems
Liposomes are spherical corpuscles consisting of the aqueous central phase surrounded by one (Figure 4A) or more phospholipid bilayers, which are separated by thin layers of water. They were first described by Bangham et al. in 1965,(318) although the suspensions of phospholipids, stained by histological dyes, have been microscopically observed much earlier.(192) Free (Figure 4A), immobilized, and supported liposomes (Figure 4B) have a long history of being used as surrogate systems for membranes. In these studies, unilamellar vesicles are preferred to multilamellar liposomes because of better-defined and simpler structures. Based on the size, liposomes are classified into three categories: the minimum-size unilamellar vesicles (diameters of 15−30 nm), large unilamellar vesicles (up to several hundred nanometers), and giant unilamellar vesicles (on the order of micrometers). The enthalpy and entropy contributions to binding may differ in small and large unilamellar vesicles,(319) although the area per lipid, chain ordering, and dynamics seem to be similar.(320) The enthalpy−-entropy compensation may lower the differences in the overall free energies.(319)
Giant unilamellar vesicles are generated from specific lipid mixtures by various techniques including detergent dialysis from aqueous mixtures of organic solvents321,322 and chaotropic solutions,(323) freezing-thawing,(324) gentle hydration,325,326 rapid solvent evaporation,(327) and electroformation on electrodes.(328) The vesicles are sufficiently large to be observed by optical microscopes and manipulated by micropipettes.(329) They are well-suited for the fluorescence monitoring of lateral inhomogeneities also called lipid rafts or domains,224,330,331 examination of mechanical(332) and electrical(333) properties, and patch-clamp recording of incorporated ion channels.(334)
Limitations such as a complicated kinetics with the difficult-to-predict duration of transport for some compounds,(20) a tedious separation,335,336 a laborious spectroscopic data analysis that is due to the light scattering if the separation step is omitted,337–339 and low stability of some preparations(340) restrict the use of liposomes in routine experiments with increased throughput. The direct approaches, avoiding the separation of liposomes, are limited to fluorescent and UV-Vis-absorbing compounds(82) or ionized compounds.341,342
Immobilization of liposomes by embedding in agarose gel beads,(343) adsorption on alkylated surfaces,(344) or by biotin−avidin interactions alleviate the problem of liposome separation but introduce the risk of the matrix effects.(345) Similar issues may arise with the liposomes that were noncovalently attached to a sensor chip and had the kinetics and equilibria of interactions with drugs analyzed using the surface plasmon resonance technology that measures the changes in liposome mass caused by drug binding.346–349 The examination of matrix effects significantly increase the workload, especially if it is rigorously performed for each studied compound.
Planar bilayers of nanoscale dimensions and circular shape, reminiscent of some plasma lipoproteins (section ), can be stabilized if their edges are covered by either membrane scaffold proteins350,351 (nanodisks; see Figure 4D) or by phospholipids with shorter fatty acids and detergents352–354 (bicelles; see Figure 4F). They are mostly used in imaging,(355) NMR studies,(356) and stabilization(357) of membrane proteins and less in the studies of chemicals’ accumulation, because the stabilizing proteins, lipids, or detergents could interfere with the solute binding. The lipid−protein interactions in nanodisks were analyzed using MD simulations(358) and mesoscale modeling.(359) The self-assembly of lipoprotein nanodisks and their disassembly caused by cholate addition were studied by mesoscale modeling and small-angle X-ray scattering.360,361
To improve stability and separation properties of liposomes, microspherical supported phospholipid bilayer systems (Figure 4B) have been developed. The most commonly used solid supports are silica362,363 (Transil beads(364)), mica,(365) glass366,367 (supported spherical vesicles(368)), and polydimethylacrylamide hydrogel (Lipobeads).(369) The attachment of phospholipids is generally based on either chemisorption on the contact monolayer366,370,371 or adhesion of the bilayer due to preferred planar shape of the bilayer formed by certain type of phospholipids.(365)
Polymerized liposomes containing rafts of nonpolymerized phospholipids exhibit concentration-dependent blue−red color shift upon interaction with chemicals.(372) The effective concentration ranges seem to be specific for three elementary mechanisms: interaction with the surface, bilayer penetration, and no interaction. Polymerized liposomes undergo color transitions in response to external stimuli such as changes of pH or temperature, or binding of chemicals even if no phospholipids are incorporated.(373) If the importance of phospholipids in the response to binding is confirmed, the system could be used for a fast screening of chemicals for interactions with phospholipids.
2.1.2.2. Planar Bilayer Systems
The use of planar membranes for elucidation of some aspects of membrane transport dates back to the 1920s. The first membranes’ only attribute shared with the biological membrane was the presence of pores. The gelatine membranes that had been hardened in formaldehyde(374) and contained copper ferrocyanide,375,376 as well as collodion membranes(377) exhibited selective permeability, based on the size of the permeants.
The membranes with the structure of phospholipid bilayers were produced four decades later. A macroscopic planar bilayer can be generated at an aperture in a thin Teflon barrier separating the donor and acceptor aqueous compartments.378−380 A drop of the solution of phospholipids in an organic solvent is transferred on the aperture that is ∼1 mm in diameter and is immersed in an aqueous phase. The bilayer forms spontaneously, creating a black spot in the microscope view of the rainbow-colored droplet. Based on this phenomenon, the system is called the black lipid membrane (BLM). Asymmetrical bilayers were created using two different air/water monolayers separated by a barrier with an aperture, upon lowering the barrier across the interface.(381) A similar principle was used for the formation of the bilayer in a microfluidic chip.(382) Polycarbonate filters with the apertures in the micrometer range were used as a support for the formation of the PC/cholesterol bilayers and provided enhanced mechanical stability.383–385 Accessibility of the apparatus to analytical, electrical,(386) optical, and mechanical measurements makes BLM an excellent tool for detailed studies of transport,387–390 bilayer electroporation,(391) reconstitution of membrane proteins(392) or channels,393–395 and other bilayer-related phenomena.
The bilayer stacks of skin lipids—cholesterol, ceramides, and free fatty acids—can be reconstituted in vitro.(396) The multilamellar sheets mimic the real situation better than liposomes397,398 and can be used for permeability measurements.(399)
Macroscopic solvent-free planar bilayers were formed from the corresponding monolayers or by bilayer adsorption between two porous cellulose sheets,(400) on patch-clamp pipette opening,(401) quartz,(402) glass, mica,(362) silicon,(403) agar gel,(404) organosulfate- and organophosphate-modified gold surfaces,(405) metal (stainless steel,(406) silver,(407) platinum(408)) and metal-coated (gold,(370) platinum(408)) or polymer-coated(409) glass plates, and porous supports.410–412
The throughput of the experiments with planar bilayers was significantly increased by the introduction of the Parallel Artificial Membrane Permeation Assay (PAMPA).413–415 A multiwell microtiter plate is sandwiched with the multiwell filter plate of the same format. The porous hydrophobic filter is impregnated with a mixture of PC (2%−10%) in alkane solvents. The opposite well pairs, separated by the filter, represent the aqueous donor and acceptor compartments. Individual wells can be stirred to regulate the thickness of the stagnant water layer so that it imitates that in the intestine, and to accelerate the procedure for lipophilic compounds.(416) Other support materials were examined,417,418 and the composition of the impregnating mixture was optimized to reproduce permeation through the Caco-2 cell monolayer,(419) intestine,420,421 blood-brain barrier,(422) and skin.(423) The corrections for the effects of the filter porosity(424) and the unstirred water layers were introduced to provide more-realistic permeabilities.(425) Later, the PAMPA approach was modified to measure the permeabilities and the partition coefficients of chemicals in hexadecane(424) and 1-octanol.(426)
The PAMPA permeabilities of a limited set of compounds depend in a bilinear way on the reference partition coefficients, and linearly on the indicator variables for some ionizable groups.(427) The permeability parameters were modeled in terms of hydrogen-bond donor and acceptor abilities, refraction, and polarizability using the solvatochromic LFER approach.(428)
In view of the extensive industrial use, it is interesting that the structure of the impregnated PAMPA filters has not been investigated in detail, although the formation of multiple bilayers in the filter pores was anticipated.429,430 Some assurance about the bilayer formation comes from the following facts. The phospholipid-alkane mixtures used for the impregnation of the filters have similar composition as those used to prepare BLM.378−380 The phospholipids in the filters are capable of fusion with phospholipid vesicles.431–433 A multiple-bilayer structure was inferred from electrical resistance measurements on a hydrophobic filter impregnated with a synthetic lipid analog.434–436 The formation of a single PC/cholesterol bilayer in the pores of a polycarbonate filter was inferred from the current−voltage relationships and the response to the polyene antibiotics amphotericin-B(383) that forms a trans-bilayer channel in association with sterols. A better defined bilayer structure in the filter pores could definitely improve the reproducibility and interlaboratory variation of the PAMPA measurements, as well as interpretation of the results. A nonsolvent version of the impregnation, with liposomes filling the pores and forming a layer on the filter, was recently reported.437–439
2.1.2.3. Monolayers at the Air/Water Interface
Lipid monolayers have been widely used to study the behavior of lipid bilayers, with the underlying assumption that two weakly coupled monolayers, having appropriate surface pressure,440,441 imitate the bilayer’s adsorption and partitioning behavior. From the viewpoint of the interactions with chemicals, monolayer systems differ from the bilayer, inter alia, by the absence of the opposite headgroup region that leads to a faster achievement of the equilibrium due to the removal of the potentially slowest step, the flip-flop of amphiphilic molecules between individual bilayer leaflets (section ). Phospholipid monolayers have been deposited on the interfaces between water and other phases, which can exist in the gas, liquid, or solid state. Each system has unique advantages and disadvantages. However, the monolayers with liquid support or solid support formed by hydrocarbon chains have the cores, which have the appropriate structure and sufficient size to solvate the permeants in a similar way as the bilayer core.
The Langmuir trough with a movable barrier that can regulate the surface pressure in the monolayers191,193,442 at the air/water or argon/water(441) interfaces is a classical tool of surface chemistry. Its invention(191) and use greatly contributed to the elucidation of monolayer and bilayer structures. Adsorbed phospholipids have the headgroups solvated in the aqueous phase. Their fatty acid chains are facing the air,(193) where they may exhibit a higher ordering of the end methylenes than that in bilayers.(443) The typical trough experiments monitor surface pressure, i.e., the tendesurface pressurency to keep the current volume, surface area, and shape, in the relation to the overall surface area. The dependence of surface pressure on the overall surface area leads to a straightforward determination of the phospholipid surface area and its change upon binding of the chemicals.(444) The entire dose-response curve can be measured for one monolayer using the continuous exchange of the chemical solution in the subphase for the stock chemical solution under the conditions of rapid mixing.(441) The surface pressure versus area measurement, combined with other techniques, provides a basis for the inference about the approximate positions of interacting chemicals in the monolayer.445,446 Surfactants, amphiphilic compounds with clearly separated polar and nonpolar parts, reduce the surface pressure at the air/water and lipid/water interfaces.
The lateral dimensions of the monolayer allow the use of a broad palette of techniques, ranging from electrochemical methods such as the surface potential measurements,(445) to analytical methods that examine the composition of the aqueous phase, and surface imaging. The last category includes fluorescence microscopy examining surface domains in phospholipid monolayers doped with fluorescent phospholipid analogs447–449 or lateral diffusion of phospholipids after photobleaching,(450) as well as specular neutron reflection,(451) ellipsometry, FT-IR reflectance,(452) X-ray reflectance,(453) and grazing incidence XRD(454) determining some geometry aspects of the molecules of phospholipids or interacting chemicals.(455) Binding of radioactive compounds to phospholipid monolayers can be easily tracked using a detector positioned closely above the interface. The method was widely applied to monitoring the adsorption of Ca2+ ions(456) and proteins.457,458
There are two concerns with the phospholipid monolayer at the air/water interface as a mimic for phospholipid bilayers and membranes. First, only the aqueous subphase can be sampled for the change in the concentrations of the studied chemical so the experiments do not clearly locate the chemical in the headgroup or core regions of the monolayer. Second, the hydrocarbon core is too thin to effectively solvate larger hydrophobic molecules. The hydrocarbon layer can be made thicker by adding hydrocarbons that float on top of the fatty acid chains. This phenomenon, however, was only observed for hexane. Longer hydrocarbons (12−16 carbons), which would be preferred because of lower volatility, intercalate between the fatty acid chains and reduce their tilt angle so that the chains become practically perpendicular to the interface.453,454
2.1.2.4. Monolayers at the Solvent/Water Interface
The hydrocarbon-doped monolayers described in the previous paragraph represent a transition between air/water and alkane/water interfaces. While a microscopic alkane layer at the air/water interface in the former setup is a prerequisite for surface imaging, thicker layers in the latter arrangement enable one to use conventional analytical and electrochemical techniques to monitor concentration changes in the nonpolar phase.(459) The performed experiments focused on the behavior of phospholipids at the interfaces between water solutions and 2,2,4-trimethylpentane,(460) heptane,(461) and other alkanes.(462) If the alkanes are of comparable lengths with the fatty acyl chains, they intercalate into the monolayer, which then assumes a different state than that of the bilayer.(462)
The first systems of this nature were created at the beginning of the last century. The globules, formed by mixing the solutions of PC in various water-immiscible solvents with water,192,463 were ∼0.1 mm in diameter and could be observed microscopically, after staining with histological dyes. The structure of the monolayer was not known at that time. If sufficiently stable and uniform, the globules could become a valuable tool for studying the interactions of chemicals with the phospholipids, thanks to the absence of the second interface, which eliminates the potentially slow flip-flop step (section ).
2.1.2.5. Monolayers on Solid Supports
The phospholipid monolayers on solid supports, which can be easily adapted for routine experiments with an increased throughput, are more relevant to pre-clinical drug candidate screening and other high-throughput applications than other monolayer setups. The monolayers can be bound to the support by covalent or noncovalent forces.
Phospholipids or their derivatives, which are covalently immobilized to the surfaces of porous silica microspheres, served as a column material in high-pressure liquid chromatography. They are called immobilized artificial membranes (IAMs) and the structure is sketched in Figure 4E (the monolayer can also form inside the pores).(464) The retention factors were used as descriptors for the prediction of trans-bilayer transport of chemicals.465–469 The liposome and IAM binding were correlated for some compounds(470) and not correlated for other compounds.471,472 A systematically higher partitioning to IAMs than to liposomes was measured for several other series of compounds.343,473 The IAMs were not recommended for the characterization of ionized chemicals, because the matrix charges seemed to be insufficiently shielded.(474) In the molecular dynamics simulations,(475) no structural differences were observed between the PC headgroup in IAMs and the liquid-ordered bilayer. However, the 31P-NMR studies(476) indicated that the surface density of IAM headgroups is significantly lower than that in liquid-ordered membranes. The decrease is ∼30% for ether-linked PC and ∼40% for ester-linked PC.(477) The hydrocarbon core of IAMs is characterized, in comparison with the bilayer core, by smaller thickness,(470) higher packing density,(475) and, consequently, lower rotational freedom of the fatty acid chains. Moreover, the ends of the phospholipid fatty acid chains in IAMs are covalently bound to porous silica beads, which may exhibit extreme surface curvatures, especially in the pores.(478) At these spots, disturbances in the continuous bilayer may exist, which are not seen in the averaging NMR experiments. Diffusion of chemicals in the pores may complicate the measurement of the transport rates, if attempted.
Monolayers adsorbed on alkylated surfaces are expected to resemble the bilayer more closely than IAMs. Adsorbed monolayers can be prepared by several techniques. A multistep immersion process has been used to create layers of dimyristoylphosphatidylcholine (DMPC) on the surface of silica plates with covalently attached octadecyl chains.(479) The uneven coverage of the hydrophobic surface and the formation of multilamellar islands make this technique unsuitable for the exact bilayer partitioning studies where a well-defined system is needed. The Langmuir−Blodgett technique(480) produced a phospholipid monolayer on the surface of gold-coated plates with covalently attached octadecyl chains.(481) The obtained monolayer was uniform and stable in both dry and rehydrated forms. Alkylated glass plates were used to obtain a phospholipid monolayer by vesicle fusion.(482) The solid lipid nanoparticles with the triglyceride core covered with adsorbed phospholipids and other surfactants have been obtained by industry-scale processes, such as emulsification of melted triglycerides and homogenizaton.483–491 The slow diffusion of compounds in the solid core that makes the suspension a useful drug delivery vehicle, is a drawback for mechanistic studies of interactions of chemicals with phospholipids, along with the presence of surfactants, irregular coating, and nonspherical shapes.(488)
We have developed a monolayer system that overcomes most of the aforementioned problems. The system consists of uniform nonporous octadecylated silica (ODS) particles (1.5 μm in diameter) that are coated with phospholipids (Figure 4C).(492) The coating is based on a spontaneous self-assembly of phospholipid molecules that is expected to lead to formation of a monolayer with the packing density comparable to that in liposomes. Formation of a continuous monolayer in a similar setting has also been demonstrated for planar surfaces.(481)
The presence of a single interface eliminates the possibility of the time-consuming flip-flop process and leads to a significantly faster binding and simpler kinetics, compared to that observed in free or immobilized liposomes. The smooth surface of the nonporous particles is a prerequisite for formation of a defect-free monolayer. The hydrocarbon core, formed by phospholipid acyls and the octadecyl chains attached to the microspheres, is expected to be more similar to the bilayer core in thickness and solvation properties than the core in IAMs. The used ODS particles provide a significantly larger specific surface area than that of planar systems. Easy separation of the DMPC-coated ODS-particles from the medium is a prerequisite for a plate-friendly assay that is suitable for routine testing with higher throughput than using liposomes or IAMs. If an estimate of the overall bilayer partitioning is of interest, the monolayer partition coefficients are first deconvoluted into the core and headgroup contributions, and the headgroup contribution is counted twice.(492)
2.1.3. Surrogate Solvent Systems
Individual regions of phospholipid bilayers are difficult to access experimentally, especially in biological membranes. Various organic solvents, imitating the bilayers differing in lipid composition, packing, and hydration levels, have been examined as membrane or bilayer surrogates, with the objective of obtaining thermodynamic or kinetic information on the partitioning process.52,56,73,272,389,493–508 Curiously, air was also used as the surrogate phase imitating the nonpolar hydrocarbon core of the bilayer. The measurements of the surface pressure of chemicals at the air/water interface have been miniaturized and adapted for a high throughput.(509) A parameter approximating the bilayer/water partition coefficient was derived using the experimental data for the air/water partition coefficient and the surface area.
Partitioning equilibria of solutes in bilayer regions result from the interplay of several factors, which include the interactions of solutes with phospholipids and water molecules, the energy for creation of the cavities for solute molecules,510–512 electrostatic interactions with the membrane and dipole potentials,(513) and entropic consequences of partitioning in individual regions.514–516 Estimates for the first factor have traditionally been obtained using organic solvents as surrogates of the bilayer or one of its regions.
2.1.3.1. Collander Equation
The partition coefficients (Px) of a set of compounds in a solvent system that is capable of similar interactions with the studied compounds as the reference system, are related to the reference partition coefficient P via the Collander equation52,517 as
where Ax (A represents accumulation) and βx are empirical adjustable coefficients, which are specific for system x. The coefficients Ax and βx are obtained by the fit to experimental data. The approach has been widely used in the design of bioactive compounds,(518) computational chemistry,(519) protein folding,(520) and other areas.(31) At the biological levels, from cells to organisms, the empirical coefficients also account for the variability in the composition of bilayers.
2.1.3.2. 1−Octanol as the Reference Solvent
After early experiments with loosely defined oils192,493–497,521 and other solvents,52,498,517,522,523 the studies of Hansch and Fujita,(56) as well as those of Leo et al.,(524) have established 1-octanol as a widely used bilayer-mimicking solvent. The authors, and many followers in the areas of QSAR and the design of bioactive compounds, intuitively considered hydrated 1-octanol to be a hydrocarbon core surrogate, despite its comparatively high equilibrium water content at room temperature (2.18 mol/L).(525) There is some water in the bilayer core, because of trans-bilayer water transport.292,297,298 The equilibrium water concentrations in the core increase as the fatty acyl chains become more disordered,(274) but they are generally low218,301 (section ).
Contrary to the core-surrogate hypothesis, studies in peptide binding to liposomes(526) (section ) and in transport through BLM(507) (section ) assumed that 1-octanol mimics the interfacial region of the bilayer. Interestingly, both views can be correct for different compound sets: XRD analyses,(527) spectroscopic experiments,(528) and MD simulations529,530 of the structure of liquid, water-saturated 1-octanol revealed fluctuating inverted micellar aggregates encompassing polar and nonpolar regions, which could imitate hydrated headgroup regions and cores, respectively. Inverted micelles consist of the centers comprising hydrated hydroxyl groups,(531) which are surrounded by outward-extending alkyl chains.527,529 The centers are thin and prolonged in neat 1-octanol and become longer and more spherical in hydrated 1-octanol.(530)
As the criteria for selection of a proper bilayer surrogate, early studies used the uptake of chemicals into cells and transport through biological sheets, which were affected by protein binding and other factors. These problems are avoided when using phospholipid liposomes, which are more suitable for evaluation of the surrogate phases.
Liposome partitioning is affected by several factors,(185) including surface charge density, especially for ionized solutes,304–306 and phospholipid surface density, which, in turn, is dependent on temperature,272,504 fatty acid composition,(532) and cholesterol content272,504 (section ). Ideally, all data used for comparisons should be measured under identical conditions that imitate the studied biosystem, so that the variation can be taken into account by the empirical regression coefficients in eq 1. Simple chemicals affect various bilayer properties at higher concentrations: surface charge,304,533 thickness,(534) fluidity in individual regions,397,398 and transition temperatures, to name just a few. Chemicals in biosystems act at low concentrations, except for the first bilayers coming into contact with the dose (section ). To mimic the reality for the majority of chemical−phospholipid interactions, the liposome partitioning data should be measured at low concentrations of chemicals or, if feasible, extrapolated to zero concentrations.
The correlations of the partition coefficients of chemicals in liposomes and in the 1-octanol/water system become nonlinear for large molecules, presumably because of the energy needed to create a cavity(535) in the structured bilayer subregions 2 and 3 (section ). The correlations displayed separate trends for neutral, negatively charged, and positively charged molecules, with the last category exhibiting the largest scatter.(536) The relationships broke down even for a homologous series exhibiting different preferred bilayer locales for individual members.(537) If more-diverse chemicals were considered, the relationship seemed to hold, with a broader scatter and within certain size and lipophilicity limits,535,538 for neutral molecules(539) but not for charged molecules.342,540 The differences are more significant for acids, as opposed to bases, because the former show more-pronounced pH dependences of the bilayer/water partition coefficients (PB/W) than the latter. This phenomenon is caused by the interactions with the headgroups of neutral and acidic phospholipids, which lead to a rearrangement in the headgroups.541,542 The former profiles are still rather shallow in comparison with those for the 1-octanol/water partition coefficients (PO/W). The differences of the PB/W values for different pH of the aqueous phases are dependent on the ionization character of the chemical: they are comparatively small for bases and can reach a magnitude100-fold larger for acids.(543) The PO/W values of both acids and bases can differ by 3−4 orders of magnitudes in the systems with differing pH values of the aqueous phase. A careful comparison of the data measured above the gel-to-liquid transition temperature with the experimental or calculated PO/W values(544) showed that, even for neutral chemicals, different lines can be discerned for polar compounds with or without hydrogen-bond donor ability(538) and nonpolar compounds,(532) as shown in Figure 5. On the bilogarithmic scale, the lines have the slopes of 0.63 for nonpolar compounds, 0.82 for polar compounds without hydrogen-bond donor groups, and 1.27 for the group consisting of hydrogen-bond donors and acceptors (phenols and alcohols). However, the entire dataset exhibits a reasonable linear relationship with the squared correlation coefficient r2 = 0.950, as noticed previously.(539)
Figure 5.

Dimyristoylphosphatidylcholine bilayer/water532,538 versus 1-octanol/water partition coefficients(544)P for (●) nonpolar compounds, (▲) phenols, (△) alcohols, (▼) anilines, and (★) polar compounds with no hydrogen-bond donor group. Lines represent linear fits to the data for (—) nonpolar compounds, (- - -) phenols and alcohols, and (· · ·) polar compounds without hydrogen-bond donor ability. More details are given in the text.
In summary, the PO/W describes partitioning in the phospholipid bilayer very well for a homologous set of compounds,(537) and acceptably for diverse neutral molecules (Figure 5), within certain size limits. In both cases, the relative affinities of the chemicals for the headgroups and core must not significantly vary among the studied chemicals. PO/W is a perfect reference coefficient in the cases when overall partitioning is targeted, e.g., for the description of pseudo-equilibrium disposition (section ). Other surrogate solvent systems are needed if the distribution between bilayer regions is of interest: the hydroxyl group of 1-octanol can hardly emulate the interaction capabilities of phosphate, ammonium, and carbonyl functionalities of the phospholipid headgroups. In addition to the main advantage that 1-octanol resembles some membranes as assessed through the application of eq 1, other desirable properties of this solvent include chemical stability, easy availability and purification, UV-Vis transparency (which is important for the spectrophotometric determination of many chemicals), and a low volatility.(518)
2.1.3.3. Other Solvent Systems
The studies with BLM (section ) showed that the selectivity of the bilayer to the partitioning of the hydrogen-bonding groups lies somewhere between those of 1-octanol and pure hydrocarbons,(389) but it is significantly closer to the latter.545,546 Additional support for the use of hydrocarbons as core surrogates500,501 comes from the experimental observation of similar molecular packing of the fatty acid methylene and methyl groups in subregion 4 of the bilayer and in bulk liquid alkanes(219) (section ). The hydrocarbon cores seem to have similar solvation properties in all bilayers, although fine differences were noted.507,508 Most likely, a single reference phase, e.g. n-hexadecane, can be used, with the differences being taken into account by applying the Collander equation (eq 1).
Whereas alkanes approximate solvation properties of the hydrocarbon core, is there a phase that emulates most interaction capabilities of the headgroups? Four solvents were used to model the amphiprotic, hydrogen-bond acceptor and donor, and hydrophobic properties of membranes: 1-octanol, propylene glycol dipelargonate (PGDP), chloroform, and alkanes, respectively.(503) The structure of PGDP contains some fragments found in phospholipids; however, no substructure imitates the charged functional groups, and an equivalent of the hydroxy group of PGDP is not found in PC. Other tested solvents include isopropylmyristate(547) and N-butyl-acetate.(548) The ethylene glycol−heptane system549–551 represents a rare attempt to imitate separately the headgroup and core regions of the bilayer. None of the solvents used so far emulates the charged functional groups of phospholipids.
A straightforward approach to inclusion of all functional groups of a phospholipid into the surrogate system is represented by the direct use of acylated headgroups. We have found(552) that the PC headgroup with truncated fatty acid chains, diacetylphosphatidylcholine (DAcPC), when dissolved in water in a molar ratio of 1:14, resembling the conditions in the hydration shell of the PC headgroups in the bilayer slightly above the transition temperature,265,553 forms a homogeneous and only slightly viscous solution. Albeit isotropic, the hydrated DAcPC imitates the type of solvation that solutes experience in the PC headgroup region, including the lack of free water molecules. The methyl groups in acetyls mimic the first two or three methylenes of the sn-2 fatty acid chain, which thermally fluctuate at the level of sn-1 carbonyls in a PC bilayer and can be considered a part of the headgroup region.
The DAcPC molecules do not measurably partition from the concentrated aqueous solution (1.96 mol/L) into n-hexadecane (C16) in the two-phase system. For a set of 16 compounds, the C16/DAcPC partition coefficients were measured and used to estimate the prevalent location of the compounds in the bilayer. The difference logPC16/W − logPO/W (formally equal to logPC16/O) is frequently considered to be an indicator of the hydrogen-bonding ability,549,550,554,555 a correlate for permeability,(556) and a parameter characterizing the preferential location of solutes in the bilayer. The fraction of compounds present in the core, as estimated from the C16/DAcPC partition coefficients, is plotted against log PC16/O in Figure 6. To connect the compounds with known locale in the headgroups or in the core, the data were fitted(557) with the Boltzmann sigmoidal function:(552)
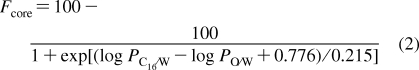 |
Figure 6.

Fractions of the chemicals in the core region, as predicted from the C16/DAcPC partitioning, versus the difference between C16/W and C16/O partition coefficients. The compounds with experimentally determined locale are shown as full points. The sigmoidal curve connects the compounds with known locales in the headgroups or the core and corresponds to eq 2.(552)
Standard deviations of the two optimized coefficients are <20% of the coefficient values, and the squared correlation coefficient is r2 = 0.992. The locales estimated using the C16/DAcPC partitioning (Figure 6) are in perfect agreement with the known locales for all compounds where this information is available. However, eq 2 can be used to obtain approximate estimates in the absence of C16/DAcPC data.
The solvation energies of a solute in C16 and in hydrated DAcPC phase can be rationalized in terms of two contributions: the energies needed to form cavities in both phases, and the interactions of the solute molecules with the solvent molecules lining the cavities.(548) The cavity formation energy is maximal in bulk water and decreases in the headgroup region, to a minimum in the core.513,558 The cavities in the isotropic DAcPC solution can probably be formed easier than those in the headgroups, because the headgroup anchoring effect of the high-density tail subregion 3 (section ) is missing in DAcPC. Based on these facts, we expect that the main contributions to the solvation energies in DAcPC and C16 will come from the solute−solvent interactions. Thus partitioning in the C16/DAcPC system is expected to provide only information about one of the factors contributing to the location in the bilayer, namely, the energy of interactions with the phospholipids. The cavity formation energy, the energy of electrostatic interactions of the permeants with the membrane and dipole potentials, and entropic factors are not included. The C16/DAcPC data can help factorize the contributions to solvation energies in individual bilayer regions.
Lipophilic chemicals also partition into plasma lipoproteins and the adipose tissue (section ). Triacylglycerols (TAGs), representing ∼90% of the adipocyte’s mass(176) and forming the lipoprotein cores, lack structural similarity to common surrogate molecules: alkanes miss the hydrogen-bond acceptor abilities of the TAG ester carbonyls, 1-octanol has a surplus hydrogen-bond donor function, and DAcPC features additional charged groups. Olive oil has been advocated as the best surrogate;(559) however, a chemically defined phase would provide more-reproducible results. Long-chain TAGs with saturated chains are solid at room temperature. The melting temperatures decrease with shortening the chains, e.g., trioctanoylglycerol melts at 6 °C. This clear liquid boils at 233 °C and can be distilled easily. Trioctanoylglycerol seems to be suitable for partitioning studies at room temperature, although no experiments have been performed yet. Triolein (trioleoylglycerol)192,560–563 represents another option.
Surrogate phases were also used to mimic the binding sites of proteins, using solvents that represent the fragments of amino acid side chains: benzene, toluene, pyrrole, 1-propane-thiol, and methyl ethyl sulfide.(564) The applicability of the approach to protein interactions is limited, because the protein binding sites are much more heterogeneous than the bilayer regions. The protein phases, which could represent homogeneous environments, are not large enough to completely solvate the interacting drug molecules, as observed in the macroscopic surrogate phases.
2.1.3.4. Partitioning and Interfacial Transfer
Experimental techniques for monitoring transport of chemicals through the planar387,389,390,506,565–567 or spherical bilayers73,397,507,546,568–585 focus on overall permeability. The elementary steps have not been studied, with the sole exception of the flip-flop rates for amphiphilic compounds.586,587 Computational simulations of chemical distribution in hydrated bilayer patches(588) (section ) do not analyze this phenomenon either, because no experimental data are available for comparison. Despite this lack of interest, the rate of interfacial transfer is an important characteristic for all chemicals, which accumulate in the bilayer regions. Some information about the process can be obtained from the data on interfacial transfer between water-immiscible organic solvents and water (see below).
Structure and dynamics of liquid−liquid interfaces is a matter of intense experimental research relying mainly on voltammetry,(589) vibrational sum frequency spectroscopy,590–596 and X-ray reflectance,597–603 as well as on computational simulations.558,604–607 The interface structures are rather different, even for similar organic solvents. For lower alkanes(597) and carbon tetrachloride systems, where the nonpolar phase has no hydrated groups interacting with water, the interface is only a few angstroms wide and exhibits molecular sharpness, with a slight mutual permeation of both solvents due to thermally excited capillary waves. Interfaces of higher alkane systems are wider than predicted from the capillary-wave theory, and the deviations increase with the chain length.608,609 Dichloromethane also exhibits a wide, more diffuse interface in contact with water.(610) The interface is broader for the solvents with well-hydrated functional groups, including alcohols602,611,612 and ketones,(600) as well as for other solvents containing amphiphilic compounds, such as surfactants590,613 and phospholipids,(614) which adsorb at the interface. Physical resemblance of the interfaces between bilayer/water and organic solvent/water systems is closer for the solvents with functionalized molecules than for alkanes.(558)
Computational simulation of partitioning at the interfaces suggested interesting behavior of several polar solutes, such as alcohols, anesthetics,(615) monofluoromethane, difluoromethane, and trifluoromethane (but not methane and tetrafluoromethane),(513) as well as trifluoroethane (but not hexafluoroethane)(616) at the hexane/water and glycerol-1-monooleate/water interfaces. The solutes exhibited accumulation at the interface, although molecular structures of at least some of them did not show a clear separation of polar and nonpolar fragments as known for surfactants. This fact indicates that interactions of functional groups and substructures with interfaces may be difficult to extrapolate from hydration energies.
Despite the dissimilarities of interfaces, solvent/water systems have been used to monitor the solute transfer, in the hope that relative trends can be discerned, which would also hold for the bilayer/water interface. The experimental arrangement for the measurement of the transfer rate parameters from an organic phase to water and backwards is simple.(498) Both immiscible phases sit one above the other in a temperature-controlled jar and are stirred at an optimal rate that secures sufficiently rapid equilibration in the bulks. Under the stirring conditions, the interface is usually not planar and adopts a slightly conical shape, but the interfacial area can be approximately calculated. The studied chemical is added to the phase in which it has a higher solubility, and the kinetics of its partitioning is monitored, preferably in both phases. From the time courses of the concentrations, the rate parameters of transfer, li and lo (for the direction from water to the organic phase and backwards, respectively), are determined. This so called slow-stirring method precisely determines the partition coefficient P = li/lo, because the achievement of the equilibrium is verified in the course of the experiment, and the risk of the mutual droplet cross-contamination of the phases is completely eliminated,(617) as opposed to the more frequently used shake-flask approach. The slow-stirring method was also used to measure the P values of unstable compounds with the rate of decomposition comparable to the rate of transfer.(618) Arrangements of multiple phases have been used in the water−1-octanol−water system619,620 and in the system consisting of a series of 11 alternating aqueous and 1-octanol phases.(621) A moving-drop technique was developed and used to measure the transfer rates of short-chain alcohols and tritiated water across the 1-octanol/water interface.(622) Electrochemical techniques are useful for the monitoring of the interfacial transfer in solvent/water systems.589,623–630
For the QSARs of the compounds, which do not interact with the headgroups, the relationships between P and the rate parameters li and lo are of special interest. Experimental observations(620) were quantified as(631)
where At and Bt (the subscript “t” denotes the transfer) are empirical coefficients, which are dependent on the organic phase, the geometry of the apparatus, and the stirring rate, but are independent of the molecular structure of the tested chemicals. The rate parameter li for the transfer from water to the organic phase initially increases with the partition coefficient (P = li/lo) and levels off for high P values. The rate parameter for the reverse direction (lo) is constant for lower P values and drops for high P values. The observation, summarized in eqs 3, was later extended to chemicals that do not belong to a homologous series, may ionize or form ion pairs.(66) The plateaus in the dependencies were assumed to originate as a consequence of the diffusional control of partitioning in the diffusion layers.
In a steady-state treatment of the interfacial transport process,(66) the empirical coefficients in eqs 3 were expressed using the diffusion coefficient D and the effective thickness h of the corresponding diffusion layer as At = Dorg/horg and Bt = Dorghaq/(Daqhorg). The apparent independence on the molecular structure of the coefficient At is rather puzzling as the diffusion coefficients in the apolar solvents are known to vary with the molar volume.(632) A small dependence of the permeability coefficient on the molar volume was observed(633) in experiments with rotating diffusion chamber,(634) which, however, monitor transport through planar systems (section ) that is more complex than the simple interfacial transfer. Another view of the empirical coefficients At and Bt was provided by considering convective diffusion caused by the laminar flow in the vicinity of the interface.(635) Practical impact of these observations on the distribution of chemicals in biosystems remains to be assessed.
A further matter of dispute is the interfacial resistance to transfer of chemicals through the phase interface, which is sometimes found to be an effective barrier to the chemical transfer634,636 and, in other cases, turns out to be negligible.637,638 This phenomenon probably is dependent on the ability of transported chemicals to bind to individual bilayer regions (see sections and ), as shown in MD simulations of trans-bilayer transport.(588)
2.1.4. Accumulation and Transport
Historically, transport and accumulation were first measured using cells (section ) and biological sheets (section ). Most organic molecules with molecular weights up to 1000 g/mol are expected to pass the bilayer through the bilayers. The measurements on spontaneously aggregating phospholipid systems, void of the effects of other membrane components, started after liposomes (section ) and BLM (section ) were discovered.
Typically, the measurements in phospholipid systems use high chemical-to-lipid ratios, corresponding to molar fractions of 0.1 and more. Such high concentrations of chemicals are relevant only for the bilayers, which come into a direct contact with certain types of local doses of chemicals. Upon absorption from the gastrointestinal and pulmonary tracts or through the skin, and subsequent distribution, however, the chemicals become significantly diluted. A typical ballpark value for the drug dosing is 1 mg/kg, because only ∼10% of the drug candidates show some potency in the 0.1 mg/kg range.(639) For the lipid weight fractions in lean human tissues,(176) ranging from 0.01 to 0.1, there are 104−105 times more lipids than drug molecules under pseudo-equilibrium conditions (section ). Even if all molecules would accumulate in the lipid phases, the chemical-to-lipid ratios are much lower than in the experiments with phospholipid aggregates. The measured data usually must be extrapolated to zero concentrations, to become relevant for the majority of observed chemical-bilayer interactions in human body. From this viewpoint, the experimental methods that “see” the molecules of chemicals reflect the real conditions better than the methods that examine lipids.
While the used biological systems such as cells and biological sheets contain several bilayers, the data were frequently processed by the models consisting of a single bilayer. In several studies of transport mechanisms, the results from different test systems were used interchangeably, although the number of crossed membranes varied significantly. The outcomes of these studies will be sorted out and presented separately for different test systems: bilayers in sections −, and membrane vesicles, subcellular organelles, cells, cell monolayers, biological sheets, tissues, organs, and organisms in sections −, respectively.
2.1.4.1. Binding in Bilayer Regions
The headgroup and core regions of the phospholipid bilayer differ in the types of interactions with the transported molecules of chemicals, and in the ability to shield electrostatic charges. Both regions are sufficiently voluminous to fully solvate the molecules of chemicals.(220) The headgroup region can be subdivided into perturbed water and interphase (subregions 1 and 2), and the core region consists of soft polymer and alkane (subregions 3 and 4), as described in section . Chemicals can accumulate in the headgroup subregions 1 and 2 (cephalophiles), subregions 2−3 (amphiphiles), and core subregions 3−4 (lipophiles). Taking into account that the solvation properties of the headgroup region differ from those of bulk water, the existence of the headgroups/water interface is justified. Published examples of individual interaction modes are summarized in Charts 1−3, with Tables 2−4 providing more details. The headgroup region is well-hydrated but the water molecules are tightly bound to the phospholipids headgroups. Consequently, the solvation properties of the headgroup region differ from those of bulk water. Therefore, we prefer to use the term “cephalophilicity” to denote the tendency to accumulate in the headgroup region, instead of “hydrophilicity”, as occasionally seen.640–643
Chart 1. Cephalophiles Accumulating in the Headgroup Region of the Phosphatidylcholine Bilayer.

a The headgroup/core interface is assumed to pass through the deeper ester group of phosphatidylcholine.
Chart 3. Lipophiles Accumulating in the Hydrocarbon Core of the Phosphatidylcholine Bilayer.

Table 2. Cephalophiles: Compounds Experimentally Shown To Accumulate in the Headgroup Region of the Phosphatidylcholine Bilayer.
| number | namea | method(s) | remark(s) |
|---|---|---|---|
| 1 | xenon | 129Xe−1H NOE(131) | |
| 2 | alcohols | fluorescence lifetime,(87)2H and 31P NMR,114,1271H and 13C NMR,126,647 XRD and 1H NMR(646) | R = CH3,(87) C2H5, C3H7;87,114,126,127 R= benzyl,(647) R = 1-[2-(methylamino)-1-phenyl]-propyl (ephedrine);(646) however, R = C4H9−C8H17 are amphiphilic(87) |
| 3 | halothane | 19F NMR,(106) FT-IR ATR,(97)2H and NOE NMR(119) | amphiphilic or lipophilic(119) |
| 4 | methoxyfluorane | NOE and NOESY NMR(130) | |
| 5 | isofluorane | 19F NMR,(120)2H and NOE NMR(119) | amphiphilic or lipophilic(119) |
| 6 | enfluorane | FT IR ATR,(97)2H and NOE NMR(119) | amphiphilic or lipophilic(119) |
| 7 | 1-chloro-1,2,2-trifluorocyclobutane | 2H 121 and 19F NMR120,121 | |
| 8 | pyridine | MASb13C NMR(115) | |
| 9 | procaine | 1H and 13C NMR,(126)2H NMR,107,109,110 EPR102,107 | charged species accumulate in subregion 1,(126) neutral species are lipophilic102,107 |
| 10 | indoles | 2-D NOESY 1H MASb NMR,, solid state 2H NMR,123,124 fluorescence quenching(92) | X = NH, R = H; X = NH, R = CH3; X = N(CH3), R = H; X = CH2; R = H;123,124 X = NH; R = (CH2)3COOH89,92 |
| 11 | ciprofloxacin | fluorescence quenching(93) | |
| 12 | 2-[4-N(CH3)2-phenyl]-3-OH-flavone | fluorescence quenching(95) | |
| 13 | 1-anilinonaphthalene-8-sulfonic acid | fluorescence quenching(91) | carbons 4 and 5, and associated hydrogens may protrude into subregion 3 |
| 14 | 9-anthracenes | fluorescence quenching88,92 | R = (CH2)2N+(CH3)3, N+H2CH3, and OH88,92 |
| 15 | carbazoles | fluorescence quenching89,92 | R=H92 and CH389 |
| 16 | 1-pyrenesulfonic acid | fluorescence quenching(91) | some carbons and associated hydrogens may protrude into subregion 3 |
Structures in Chart 1.
Magic-angle spinning.
Table 4. Lipophiles: Compounds Experimentally Shown To Accumulate in the Core of Phosphatidylcholine Bilayer.
| number | namea | method(s) | remark(s) |
|---|---|---|---|
| 37 | n-hexane112,132,134,135 and n-decane(137) | neutron diffraction,112,132,134,1352H NMR(112) | |
| 38 | poly-isoprenes (carotenoid pigments) | neutron diffraction,(138)13C and 31P NMR,(117) EPR,(101) XRD, 1H NMR, UV Vis, CD83–86 | R = CH2CH(CH3)2 and all bonds saturated (squalaneb);(138) R = 2,6,6-tri-CH3-cyclohexen-1-yl (β-carotenec);85,101,117 R = 2,6,6-tri-CH3-4-hydroxy-cyclohexen-1-yl (zeaxanthinc);(85) R = 2,6,6-tri-CH3-4-hydroxy-cyclohexen-2-yl (luteinc,d);83,85,86 R = 2,6,6-tri-CH3-1,2-epoxy-cyclohexyl (violaxanthinc);(85) R = 2,6,6-tri-CH3-cyclohexen-1-yl-4-one (canthaxanthinc,d)(84) |
| 39 | 1,2-dichlorohexafluorocyclobutane | 19F NMR120,121 | |
| 40 | 2,3-dichlorooctofluorobutane | 19F NMR(120) | |
| 41 | benzocaine | EPR103 | may interact with the more dense subregion 3 of the core |
| 42 | benzenes | 1H126,646 and 13C NMR,(126) XRD and 1H NMR,(646) small-angle XRD;646,648 | R = H (benzene);644,645 CH3;(645) 1,4-diCH3; (646) R = 1-OH, 2,6-di-C(CH3)3, 4-CH3 or 4-Br; (648) R = (CH2)2CH3;(647) R = Pr > Et > Me > H, moving more to headgroups in this order(126) |
| 43 | amiodarone | small-angle XRD(139) | N is charged in water (pKa = 9.1−9.4) |
| 44 | O-CH3-Δ8 -tetrahydrocannabinol | 31P and 13C solid-state NMR122,125 | |
| 45 | 1,6-diphenyl-1,3,5-hexatriene | fluorescence quenching(92) |
Structures in Chart 3.
The molecule lies parallel to the membrane surface in the core center.
The molecules span both monolayers and form dimers and higher aggregates.
The molecules form additional pool in the headgroup region.
Structural determinants of binding in individual bilayer regions are quite subtle, and the overall outcome is dependent on a fine balance of individual structural elements of the molecule, including their spatial arrangement. Benzene derivatives, for instance, accumulate in the core (42 in Table 4 and Chart 3),644–646 unless a hydrogen-bonding substituent is available to anchor them in the headgroup region as seen for benzyl alcohol (2 in Table 2 and Chart 1).(647) The partitioning in the hydrocarbon core prevails, however, if the hydrogen-bonding ability of the substituent is obstructed by other groups as in 2,6-di-C(CH3)3-phenols (42 in Table 4 and Chart 3).(648)
Several experimental techniques, summarized in Tables 2−4, have been used to characterize—often only qualitatively or semi-quantitatively—preferred locations of compounds in the phospholipid bilayer. This information is important for quantitative assessment of the transport rates, because analyses of published trans-bilayer transport data (section ) indicate that the transport rates are dependent on the tendencies of chemicals to interact with the hydrocarbon core and the headgroup region. The two tendencies of chemicals are lipophilicity and cephalophilicity, respectively. Amphiphilicity denotes a mixed scenario, when a molecule interacts with both headgroup and hydrocarbon regions. Smaller amphiphilic molecules bind in subregions 2 and 3; larger molecules reach into subregions 1 and 4. Our preliminary data indicate that some chemicals could also accumulate at the interface between the bulk water and the headgroups, pointing to the possibility that there may be two types of amphiphilicity.
Lipophilicity is usually parametrized by a reference partition coefficient (section ), which is defined as the ratio of activities (or concentrations, if they are sufficiently low) of the solute in a surrogate phase and the aqueous phase under equilibrium conditions. Because lipophilicity is given by the relative levels of accumulation in the hydrocarbon core of the bilayer and in bulk water, the core/water partition coefficient is the most suitable parameter for characterization of lipophilicity in the context of bilayer transport and accumulation.
Lipophilicity generally is defined vaguely, because there are different nonpolar phases in the body: bilayer cores, lipid droplets, and hydrophobic lipoprotein interiors. To avoid having different lipophilicities reflecting composition variability of biological nonpolar phases, we prefer to define lipophilicity as the core/water partition coefficient for a saturated PC, which should be properly parametrized by the hexadecane (C16)/water partition coefficient (section ), according to eq 1 in section . Fatty acid chains are hydrocarbons, which only differ in the length, number, and positions of double bonds, and sometimes in branching. This variability should not lead to pronounced differences in the interactions of the chains with chemicals. Therefore, the Collander equation (eq 1) can be used to correlate the core/water partition coefficients for the cores of varying composition with the C16/water partition coefficient. Other reference partition coefficients may be more suitable for lipid droplets and lipoprotein cores (section ). All partition coefficients are additive-constitutive properties, which can be, in principle, partitioned among individual fragments of the molecule.501,518,649–652 This procedure allows for a fast structure-based prediction of all partition coefficients.
Cephalophilicity denotes the tendency of a chemical to bind in the headgroup region, forming the interactions in subregion 1 and, predominantly, in subregion 2. Experimentally, cephalophilicity can be quantified as the headgroups/core or headgroups/water partition coefficients, which are quite tedious to measure directly in the bilayers. In the absence of fast and convenient microtechniques, which would be capable of a routine monitoring of the concentrations in the bilayer regions, solvation energies in the systems imitating the bilayer environments have been used to provide initial estimates. As mentioned in section , we have found that diacetylphosphatidylcholine (DAcPC) forms a homogeneous solution with acceptable viscosity, when mixed with water in a similar ratio as in the fully hydrated bilayer headgroup region. From this solution, the DAcPC molecules practically do not partition into nonpolar solvents. The two-phase system n-hexadecane/DAcPC aqueous solution was used to precisely determine the differences in solvation energies of organic chemicals, which can be used to estimate the preferred bilayer location (section )(552) and the transport rates (section ).
Amphiphilicity corresponds to the ability of a molecule to have, in at least one conformation, lipophilic and cephalophilic parts placed in two subspaces separated by a plane, so that they are able to interact with the headgroup region and the hydrocarbon core across the headgroups/core interface. Amphiphilicity has been roughly expressed as asymmetry of the spatial distribution of lipophilicity in the molecule. For a given conformation, amphiphilicity was characterized by the lipophilic moment(653) and similar parameters,316,654–656 or by an empirical function describing position-dependent solvation energies.(640) All these approaches utilize the hydration characteristics of chemicals in bulk water instead of true cephalophilic characteristics. These characteristics may not be adequate because the water molecules, although abundant in the headgroup region, are mostly confined forming strong hydrogen bonds in the hydration shells and behave differently than those in bulk water. Cephalophilic fragment contributions must be developed, in a similar way as known for lipophilic contributions.(518)
2.1.4.2. Interactions with Plasma Lipoproteins and Lipid Droplets
A common feature of plasma lipoproteins and intracellular lipid droplets is the presence of a core that is composed mainly of triacylglycerols (TAGs). TAGs have different solvation properties than phospholipids. Specific surrogate phases are needed for a precise estimation of the partitioning of chemicals into TAGs.
Plasma lipoproteins are spherical or discoidal particles, which are composed of apolipoproteins, a phospholipid−cholesterol monolayer coat, and the core that mainly contains TAGs and esterified cholesterol. They are classified into four groups, based on the particle density, decreasing from 1.21 g/mL to <0.95 g/mL, as follows: high-density lipoproteins, low-density lipoproteins, very-low-density lipoproteins, and chylomicrons. In this order, the diameter of the particles rises from 35 Å to 6 μm, the TAG content increases from 2 % to 88 % d.w., and the protein, cholesterol, and phospholipid contents decrease.(657) The self-assembly of lipoprotein nanodisks and their disassembly caused by cholate addition were analyzed by mesoscale modeling and small-angle X-ray scattering.360,361 The physiological function of plasma lipoproteins is the transport of, respectively, cholesterol from tissues to the liver, cholesterol to tissues, endogenous TAGs, and exogenous TAGs and cholesterol. Interactions with lipoproteins can significantly contribute to intestinal absorption of lipophilic chemicals via lymph.148–150 The potential to be absorbed by this pathway was assessed using the binding to chylomicrons(658) and predicted using the partial least squares (PLS) regression analysis of numerous VolSurf descriptors.(659)
Lipid droplets are ubiquitous cellular structures, composed mainly of TAGs. They are highly concentrated in adipocytes, which contain ∼90% (w/w) TAGs. Partitioning into adipose tissues heavily affects the distribution of lipophilic compounds.
Synthetic TAGs with saturated fatty acids melt in the range of 60−80 °C, and the melting temperatures rise with chain prolongation. Solid TAGs mixed with emulsifiers form platelets with discrete molecular layers.660,661 Liquid TAGs under physiological conditions are not completely isotropic,(662) and loose remnants of the layered organization from the solid state can be recognized.(663) The aggregates of the ester groups are more diffuse than the headgroup regions observed for phospholipid bilayers (section ), bind much less water than the phospholipid headgroups, and do not form distinct phases. Consequently, the solvation behavior of TAGs can probably be emulated by one solvent, although the solubilization process can be complex in some cases.(664) Vegetable oil has been proposed as a TAG surrogate,(665) but the dependence of the composition on the source may be a problem. The partition coefficients of chemicals have been measured in the triolein (trioleoylglycerol)-water system,192,562,563 and used for structure-based predictions(561) and in correlations with bioconcentration in fish.(666)
2.1.4.3. Transport versus Bilayer Fluidity
The bilayers exhibit generally higher permeabilities in the liquid-ordered phase than in the gel phase.(667) Several barrier epithelia such as gastric mucosa, and those in the renal collecting duct and urinary bladder, maintain high solute gradients between body compartments and the blood. Low fluidity of their apical membranes has been postulated as the mechanism underlying the low permeabilities.225,668–670 Regulation by the lipid composition of the bilayer fluidity at the body temperature has been considered a major mechanism for controlling bilayer permeability.225,226,670,671 Expulsion from the bilayer into bulk water of di-tert-butyl nitroxide,(672) perylene,(673) propranolol (34 in Table 3 and Chart 2), timolol,(674) benzene, hexane,(272) and Bay K 8644, which is a 1,4-dihydropyridine derivative (27 in Table 3 and Chart 2),(675) was observed after cooling liquid-ordered bilayers to the gel state. Increasing the surface density of the liquid-ordered phospholipids by addition of cholesterol, cooling in the temperature range above the transition temperature, or extending the fatty acid chains resulted in decreased partitioning of benzene,(272) hexane,(504) non-ionized acetic acid,(505) and polychlorinated biphenyls.(532) Increased surface density also reduced permeability of acetic acid through the bilayer composed of DMPC and cholesterol.(505)
Table 3. Amphiphiles: Compounds Experimentally Shown To Accumulate at the Interface between the Headgroup and Core Regions of the Phosphatidylcholine Bilayer.
| number | namea | method(s) | remark(s) |
|---|---|---|---|
| 17 | 4-tert-octyl phenol | fluorescence quenching(89) | |
| 18 | 9-anthracenes | fluorescence quenching | R = CH2NH3+,88,92 COOH,(92) COOCH3;(89) however, R = H(92) and R = CH3,88,89,92 are lipophilic |
| 19 | 11-(9-carbazole)-undecanoic acid | fluorescence quenching89,92 | |
| 20 | ibuprofen | PFGb MAS 1H NMR(129) | phenyl ring interacts with glycerol and fatty-acid chains |
| 21 | lidocaine | 1H and 13C NMR(126) | charged species interacting with the first two methylenes of fatty-acid chains; neutral lipophilic |
| 22 | tetracaine | 2H and 31P NMR,(107)1H118,126 and 13C NMR,105,126 fluorescence quenching(94) | neutral species protruding deeper into core90,107,108 or lipophilic(107) |
| 23 | dibucaine | 1H and 2H NMR,(116)1H and 13C NMR shifts(126) | charged species interact with the first two methylenes of fatty acids only; neutral species are lipophilic |
| 24 | acemetacin | fluorescence quenching(94) | |
| 25 | indomethacin | fluorescence quenching(94) | |
| 26 | 4-C(CH3)3-[3-(CH2)2Cl-ureido] benzene | FT-IR and high pressure IR(98) | |
| 27 | 4-phenyl-dihydropyridines | small-angle XRD,(675) neutron diffraction,(136)2H and 31P NMR(113) | Bay K 8644: R1= 2-CF3; R2=CH3; R3=NO2; R4=CH3;675 amlodipine: R1 = 2-Cl; R2 = CH2CH3; R3 = CH3; R4 = CH2O(CH2)2N+H3;113,675 Bay P 8857: R1 = 3-NO2; R2 = (CH2)2I; R3 = COOCH(CH3)2; R4 = CH3;675 nimodipine (Bay E 9736): R1 = 3-NO2, R2 = (CH2)2OCH3; R3 = COOCH(CH3)2; R4 = CH3 is amphiphilic136,675 or lipophilic(113) |
| 28 | Δ8-tetrahydrocannabinol | 31P and 13C solid-state NMR122,125 | |
| 29 | 2-CF3-10-(4-MSAB)-phenothiazinec | fluorescence, calorimetry, EPR(104) | |
| 30 | (+)-totarol | MAS 13C NMR(128) | |
| 31 | bisphenol A | 1H and 13C NMR(647) | interacts with the first two methylenes of fatty acids |
| 32 | 2-anilino-naphthalene-6-sulfonic acids | fluorescence quenching(91) | R1 = H; R2 = CH3; R1 = CH3; R2 = H |
| 33 | 1,6-diphenyl-1,3,5-hexatrienes | fluorescence quenching,(92) fluorescence anisotropy,(96)2H NMR(111) | R = (CH2)2COOH, N+(CH3)3, (CH2)3N+(CH3)392 R= H and R = CH3 are lipophilic92,96,111 |
| 34 | propranolol | 13C NMR and EPR,(99) neutron diffraction(674) | |
| 35 | 2-[4-N(R1)2-phenyl]-3-OH-6-N+(CH3)2R2-flavones | fluorescence quenching(95) | R1 = C2H5; R2 = C8H17 (lower interface in Chart 2); R1 = C4H9; R2 = CH3 (upper interface in Chart 2) |
| 36 | 4-[4-[4′-(3-OH-flavonyl)]piperazino]-1(3-sulfopropyl)pyridinium | fluorescence quenching(95) |
Chart 2. Amphiphiles Accumulating at the Interface between the Headgroups and Core of the Phosphatidylcholine Bilayer.

a The headgroup/core interface is assumed to pass through the deeper ester group of phosphatidylcholine. The position of the headgroup/core interface is shown, where available. For 35, the upper interface is valid for R1 = C4H9, R2 = CH3, and the lower interface holds for R1 = C2H5, R2 = C8H17 (the molecule is tilted to allow a part of the n-octyl chain to interact with the core).
The temperature dependence of the bilayer transport is not monotonously sigmoidal. At the gel-to-liquid transition temperature, the inhomogeneous bilayer structure results in the peak values of the transport rates, as observed for water,(676) Na+ cations,673,677,678 glucose, methylphosphoethanolamine,(580) and trimeprazine,(572) as well as in a faster insertion of an amphiphilic hydroxycoumarin alkylammonium fluorophore.(256) These phenomena have been observed in the bilayers composed of pure phospholipids, where the transition occurs in a narrow temperature interval, as opposed to the bilayers consisting of mixed phospholipids and membranes. In membranes, the interactions of phospholipids with integral proteins may cause distortions in the bilayer structure. These irregularities could have a similar enhancing effect on the transport rates as observed during gel-to-liquid transitions.
2.1.4.4. Steady-State Transport
Most experimental data on the steady-state trans-bilayer transport were obtained using the BLM technique, utilizing a small-area bilayer separating two aqueous compartments (section ). Many models used to describe the BLM data were previously derived for the cellular and sheet systems (sections and , respectively), represented by the oversimplified one-membrane or one-compartment systems.
Before looking at detailed descriptions, let us make a few general remarks on the steady-state disposition of chemicals in biosystems. A flow-through transport system reaches the steady state, when a constant flux passes through each cross section of the system, and the concentrations of the transported chemical in all parts of the system become time-invariant. For this situation to happen, the concentration in the entry compartment—and, consequently, the entry rate—must be constant for a sufficiently long time. Although membrane transport studies are mostly performed in closed systems, the data are often processed using the assumption of the steady-state conditions in the membrane, because the concentrations of the permeant in the bilayer regions are assumed to be much smaller than those in the donor aqueous compartment. Consequently, the respective gradients are close to zero and considered to be approximately constant.
The constant flux through the bilayer is described by the permeability coefficient, which is quantified as the slope of the approximately linear increasing part of the concentration−time curve in the acceptor compartment. The shape of this curve documents the adherence of the situation to the steady-state conditions. The presence or absence of an initial lag phase indicates that the permeant at least temporarily does or does not accumulate in one or more bilayer regions. The leveling off at the end of the transport experiment reflects the achievement of the equilibrium, when the chemical potentials in both aqueous compartments become equal. For the non-ionized solute not interacting with the bilayer, the equilibrium concentrations are easy to calculate using the initial concentration in the donor compartment and the volumes of the two compartments. For the opposite case, membrane accumulation can be determined from the difference between the experimental equilibrium concentration and the hypothetical concentration for the situation without membrane accumulation. In summary, for chemicals interacting with the bilayer, the permeability coefficient alone does not suffice to describe and predict the overall distribution of chemicals, and additional parameters are needed to account for the possible lag phase and the final phase of the distribution kinetics. Despite this shortcoming, the permeability coefficient is frequently used as a characteristic of the interactions of chemicals with bilayers in pharmaceutics and in membrane biology.
As shown several decades ago for the cellular transport,521,679,680 the permeability coefficient for the steady-state flux through the bilayer was linearly related, on the bilogarithmic scale, to the surrogate partition coefficient (section ) of the transported chemicals forming a homologous series.297,668,681–685 In terms of elementary steps, this dependence was explained by the solubility-diffusion mechanism, which assumes a fast dissolution of the permeant into the entry region of the bilayer, slow diffusion through the interior, and another fast dissolution step at the distal bilayer/water interface. Within the framework of this mechanism, the permeability coefficient is proportional to the product of the partition coefficient and the diffusion coefficient, with both quantities mostly measured in surrogate systems. The linear log-log relationships between trans-bilayer permeability and the partition coefficients for more-diverse chemicals were augmented by molecular weight as another linear term, because the diffusion coefficient is inversely related to molecular weight. This relation was obtained based on a structural model of the bilayer as a homogeneous, hydrophobic slab. The model has been made more realistic by the addition of two resistances at the interfaces,(499) and significantly improved by inclusion of scaling factors accounting for the effect of chain ordering(506) and the permeant size and shape.(73)
2.1.4.5. Specific Mechanism for Small Molecules
Water,(298) urea, formic acid,(387) formamide,(297) oxygen,(686) and nitric oxide(687) are frequently assumed to permeate the bilayer using a mechanism that is specific for small molecules.292,688 Details of this mechanism may include mobile chain kinks taking the permeants along,(689) hopping of the permeant molecules between the kinks(690) or between free volumes created by thermal fluctuations in the headgroup region and in the core,677,691 and collective density fluctuations (phonons).(692) Longer, transient, and water-filled pores(693) were implicated in trans-bilayer permeation of monovalent cations including protons and potassium.(684) Molecular simulations indicated that the mechanisms involves active participation of the ions such as protons, Na+ cations, and hydroxyls.(694) The main support for the involvement of transient pores comes from the fact that the measured transport rates are higher than predicted from the corresponding permeation models. Imperfections in these models, demonstrated in sections −, indicate that this criterion may not sufficiently support the claim that the special mechanisms are the main transport route, at least for some small molecules and in some membranes.
A rare experimental opportunity to examine the trans-bilayer transport mechanism is based on the fact that the pores produced by thermal fluctuations occur less frequently for longer phospholipid molecules,(695) resulting in an exponentially decreasing dependence of the permeability coefficient on the bilayer thickness.(696) Experiments with the bilayers made of PC with unsaturated fatty acids of varying lengths showed that water, urea, glycerol,(684) and halide anions(685) do not use the short-lived pores as the dominant transport mechanism. Protons and Na+ cations do utilize this mechanism as the main route, but only for thinner membranes.
Apparently, more experiments are needed to reconcile the contradictory views. The thermal fluctuations occur with differing frequencies in individual bilayer regions. Therefore, it is possible that some molecules will cross the bilayer using a combined mechanism consisting of classical diffusion in the more-dense subregions 2 and 3 and a pore-hopping mechanism in subregion 4. The question of the transport mechanism is fundamental for the phenomenological description of transport kinetics. For a pore-hopping mechanism, the maximum concentration of a chemical in a bilayer region would be limited by the pore density, whereas no such limits are expected for diffusing chemicals.
2.1.4.6. Kinetics of Transport
The overall kinetics of transport and accumulation is described completely and exhaustively by mechanistic models with several rate constants, characterizing the transfers between individual bilayer regions. The use of transient kinetics prevails in the areas of drug design and membrane biophysics. The observed trans-bilayer transport rates of diverse chemicals range from seconds to days, which are needed to achieve the partitioning equilibrium across a single liquid-ordered bilayer. The transient kinetics of transport provides a more-complete picture of the overall process, including accumulation in individual regions of the bilayer that is important for the description of the interactions of chemicals with the transport proteins, metabolizing enzymes, and the receptors. The first kinetic model of transport in a multicellular system assumed slow transfers through both bilayer interfaces, which can include subregions 1−3 (section ), and fast diffusion in the core, where the chemical could accumulate.(58) This mechanism is in accordance with the bilayer density profile (section ), and accurately describes the trans-bilayer transport of some chemicals, as summarized below. The resulting peak-shaped dependencies of the transport rates on the partition coefficients were frequently observed at the cellular level.(1) The model was extended by considering the different solvation abilities of the headgroup regions and the core in some cases.667,697 The prediction of trans-bilayer transport rates remains elusive, despite tremendous efforts to develop descriptors of compounds that would correlate with this property.(469) Here, we present an analysis of the transport data and properties of the permeants that could help in resolving the prediction problem.
Tendency to associate with the headgroups, interface, and core, as reflected in cephalophilicity, amphiphilicity, and lipophilicity of chemicals (section ), leads to the compartmentalization of the bilayer for transport models. The observed situations are summarized in Figure 7. Cephalophilicity is not included in this scheme because, at this point, we do not completely understand structural determinants of this property and cannot tell whether a permeant accumulates in the headgroups, as compared to the bulk water or not. Lipophilicity is precisely characterized by the reference partition coefficient and, therefore, three levels of lipophilicity are distinguished in Figure 7. Amphiphilicity is considered in a semi-quantitative way, with high magnitudes expected for compounds with separated polar and nonpolar parts, and low magnitudes otherwise. The interface regions, where the amphiphiles partition, are drawn in Figure 7 as the volumes comprising parts of subregions 2 and 3, i.e., the high-density headgroup and core regions (section ), which are sometimes called the diffusion layers.(698)
Figure 7.

Compartmentalization of the bilayer for the description of passive transport of chemicals as dependent on lipophilicity and amphiphilicity of chemicals. The headgroups and the core are represented by the low-density bilayer subregions 1 and 4, respectively. The core/headgroups interface is depicted as a volume, where amphiphilic chemicals accumulate and includes (portions of) the high-density subregions 2 and 3. The headgroups/water interface is not shown, because of insufficient information about the accumulation of chemicals there. The schematic phospholipid structures in the bottom right corner indicate these correspondences. The overall transfer time is the time needed for a complete equilibration of a chemical across a liquid-ordered bilayer, based on the experimental data referenced in the text. The transfer rate parameters are listed in the order of individual steps, from the donor aqueous phase (left) to the acceptor aqueous phase (right). For the backward processes, the order of individual steps is in the right-to-left direction.
The compartmentalization rules are simple. The aqueous phases are always regarded as compartments. The bilayer can be represented by maximally five compartments: two headgroup regions, two interfaces, and the core. The actual number of compartments is given by the permeant properties. The compartments, in which the permeant exhibits measurable accumulation, are included in the scheme. Although the headgroups are shown in Figure 7, we do not yet know which types of chemicals prefer to partition there. Therefore, in the following paragraphs, we will only deal with the amphiphilicity and lipophilicity of chemicals.
Let us first consider non-amphiphilic chemicals. If hydrophilic, they do not interact significantly with any part of the bilayer, which then becomes just an inert barrier between the two aqueous compartments (row 1). If lipophilic, they accumulate in the hydrocarbon core of the membrane, which then represents a compartment (rows 2 and 3). Amphiphilic chemicals tend to interact with the headgroup/core interface, forming one additional compartment on each side of the bilayer. For hydrophilic amphiphiles, the interfaces are the only two compartments of the bilayer. The third bilayer compartment, corresponding to the hydrocarbon core, is added for chemicals that are both amphiphilic and lipophilic.
The experimentally observed rates of trans-bilayer transport of chemicals vary in a wide range. The results of published transport studies are summarized in a common framework in Figure 7. Diffusion in bulk water, as well as in the low-density headgroup and core subregions 1 and 4, is much faster than the steps, which involve crossing of the more-structuralized interface subregions 2 and 3 (sections and ).(699) The four schemes represent known mechanisms that have been used to describe the trans-bilayer transport.
The first row, which is valid for non-amphiphilic hydrophilic chemicals, represents the inert barrier concept.(700) A single parameter, the permeability coefficient PC is sufficient for a complete description of the process. A typical duration of the transport process is minutes to hours,569,701–703 because molecules of the bilayer-inert chemicals enter the bilayer with difficulty. Because the membrane accumulation is negligible and the intracellular aqueous phases are usually much smaller than their extracellular counterparts, the transport of these chemicals is ideally measured as their release from preloaded vesicles. The scheme in the first row is also suitable for description of kink-based and pore-based mechanisms for the permeants with a low bilayer accumulation.
The second and third rows describe the situation of the non-amphiphilic chemicals with a higher lipophilicity, which can accumulate in the bilayer core(56) but do not interact with the interfaces. Two rate parameters, li and lo—for entering and leaving the core, respectively—are needed for a complete description in this case. Both rate parameters and, consequently, the overall transport rate are dependent on lipophilicity(631) (see eq 3 in section ). The fastest transport, with the equilibrium reached in the time interval ranging from milliseconds to several seconds,82,577,704 is observed for chemicals with intermediate lipophilicity. Very hydrophilic chemicals (row 1) and very lipophilic chemicals (row 3) cross the bilayer at much slower rates.56,143,705 This mechanism has been most extensively used in simulations of transport in biosystems.16,58,706 The agreement between phenomenological simulations and the experiment (sections and ) supports the adequacy of the used description, at least for this mechanism.
The fourth row applies to chemicals, which are both hydrophilic and amphiphilic. The adsorption and desorption rates, characterized by the parameters la and ld, respectively, are usually high (on the order of milliseconds(707) or seconds(708)). The flip-flop between the two interfaces of the bilayer, characterized by the rate parameter lf, can take minutes,578,584 hours,(709) or days,710–712 depending on the strength of the interaction between the chemical and the headgroup region. This point is illustrated by the fact that the flip-flop rates are much higher for the non-ionized species of amphiphilic molecules compared with the ionized species.(713)
The fifth and sixth rows in Figure 7 describe the interactions with the interfaces, combined with the accumulation in the core, which are typical for amphiphilic and lipophilic chemicals. Two rate parameters (ldc and lac) for the desorption/adsorption between the interface and the core were added. The desorption from the interface into water was accomplished within a fraction of a second.(714) This mechanism has been described for ionizable chemicals, with the desorption from the interface into the core caused by charge neutralization.107–110,429
The conclusions summarized in Figure 7 can be extended by including cephalophilicity. The four laminae, two phases (the headgroups and the core) and two interfaces (between the bulk water and the headgroups and between the headgroups and core) play important roles in the overall bilayer permeability. Our hypothesis is that an intermediate strength of interactions of a chemical with all four laminae is necessary for its fast trans-bilayer transport. Any deviation of the interaction strengths from the intermediate values, even for a single lamina, leads to a slowdown in the overall transport. The hypothesis is based on simple and straightforward logic: weak interactions with a lamina prevent a compound from entering it, and strong interactions keep the compound inside the lamina. This behavior has been previously postulated for the bilayer core by Hansch and Fujita,(56) and used as a theoretical basis for an interpretation of the concave activity−lipophilicity profiles.
For the frequent case of non-amphiphilic chemicals, a cell or a higher biosystem can be represented by a catenary set of alternating aqueous and membrane compartments, as it has been proposed for pure transport of non-amphiphilic compounds.(58) All presented models reviewed below in sections and apply to chemicals, which do not differ in cephalophilicity and amphiphilicity. The assumption of the instantaneous diffusion in bulk water and the core allows the use of the conventional mass balance description of the kinetics and equilibria of all processes affecting the fates of chemicals within the compartments (part 10.1.1).
2.1.5. Computational Approaches
Simulation of different types of the bilayer behavior is complicated by widely differing length and time scales of the studied system: microscopic thickness contrasts with almost macroscopic surface area, and the rates of the bilayer processes including the transport (section ) vary in a broad range. Another complicating factor is the rich phase behavior of phospholipid aggregates, which may adopt the form of micellar, lamellar, hexagonal, and cubic phases.
The time scales of individual motions in the bilayer(715) increase in the following order: fluctuations of bonds, angles, and dihedrals within the same conformation (femtoseconds for covalent bonds to non-hydrogen-bonded hydrogens, picoseconds for other covalent bonds); trans−gauche isomerizations of dihedrals (tens of picoseconds in the core to nanoseconds in the headgroups); lipid rotation along the long axis and “wobbling” (nanoseconds);(716) lipid position changes in the monolayer due to lateral diffusion (nanoseconds to microseconds);(265) collective undulatory motions of lipids (on the order of 0.1−10 ms, and sped up, with a smaller amplitude, by protein binding),(717) relaxation during transitions,264,718 trans-bilayer transport of drugs and other small molecules lacking strong interactions with bilayer regions as summarized in section (seconds to minutes); and, finally, the spontaneous flip-flop of some lipids and lipid derivatives between monolayers of the same bilayer,711,719–721 and transport of chemicals with extreme lipophilicity and amphiphilicity, as described in section (minutes to days). The major events and their time scales are summarized in Figure 8.
Figure 8.

Time scales of the movements of phospholipids and permeants in a fluid phospholipid bilayer. More details are given in the text.
Atomistic MD simulations belong to the most informative computational approaches for studying bilayers. The maximum duration of published force-field-based MD studies of hydrated patches of phospholipids, currently a hundred(722) or more nanoseconds,(723) is by a wide margin too short to cover many bilayer-related phenomena with significant time-scale (Figure 8) and length-scale differences. In addition, the slower phenomena usually require the use of a larger phospholipid ensemble than those currently utilized.(724) Modified approaches are needed for the situations where atomistic simulations cannot provide answers.
A broad palette of available computational approaches can be classified based on (i) the used lipid molecule representations—simplified particles or all-atom molecules; (ii) solvation—approximated by a continuum or explicit solvent molecules; (iii) interactions between molecules—varying levels of approximation; (iv) spatial arrangement of the molecular representations—one-dimensional to study, e.g., some aspects of the ripple phase, two-dimensional to study surface phenomena, or three-dimensional to study complete structures; (v) movement of molecules—free, confined to a lattice, or none, i.e. the use of average equilibrium positions; and (vi) the used simulation method − in the order of increasing complexity: energy minimization, Self-Consistent Mean-Field Theory (SCMFT), MC simulations to study mainly equilibria, and Brownian dynamics (BD) and MD to study both kinetics and equilibria. Selection of the approach is dependent on the structures and/or processes to be modeled. The approaches are frequently combined and the multiscale methods are of special promise.725–735
2.1.5.1. Bilayer Formation and Rearrangements
Collective phenomena requiring the use of large systems and long time scales are usually simulated using approximate molecular representations of lipids and other participating molecules. The mesoscale approaches can be classified as faster lattice-based models677,736–741 and more-detailed off-lattice models.(742) The lateral rearrangements are frequently studied in two dimensions: on-lattice MC models were used to simulate the ripple phase formation743–745 and de-mixing of lipids differing in chain flexibility caused by cholesterol746,747 (also analyzed by SCMFT(748)) or membrane proteins.234,236,746,749 The off-lattice models were utilized to analyze complex phenomena proceeding at long time scales such as lipoprotein nanodisk359–361,750 and micelle742,751 formation, bilayer formation,752–754 dissolution and rearrangements(755) (also analyzed by SCMFT(756)), mechanical properties,757–759 pore formation,(752) vesicle shape fluctuations760–763 (also analyzed by SCMFT(764)), vesicle tubulation upon interaction with a protein,(765) vesicle fusion,766,767 lipid interactions with transmembrane proteins,(733) lipid−cholesterol interactions,241,768–770 lipid lateral diffusion,241,768,769,771 and lipid demixing.241,768,769,772 These approaches, also known as coarse-grained simulations, use the representations of small groups of atoms by beads or particles with specified interaction potentials. The particle consistency can be either hard(742) or penetrable and interacting via soft potentials such as in dissipative particle dynamics (DPD).755,766 Coarse-grained models with particles corresponding to individual phospholipid substructures, which interact through potentials resembling atomistic force fields,359–361,750,752–754,773–779 provide more quantitative results. Mesoscale modeling methods have become more available after the release of specialized software suites, such as Mesodyn(780) and DPD.(781)
2.1.5.2. Bilayer Structure and Dynamics
Early approaches to characterization of bilayer structure focused on the acyl chains using an SCMFT approach782–785 and BD simulations of a single lipid molecule786,787 with a continuous version of the mean-field potential.(782) The force-field energy minimization of the monolayer and bilayer models788,789 was used to analyze the intrinsic dipole potential,790–792 acyl chain packing,793–797 and the mode of insertion of drugs641–643 and proteins.(798)
More-extensive sampling of the simulated systems can be obtained using either the probabilistic MC methods or the deterministic MD methods. The explicit time dependencies resulting from the integration of equations of motion for all atoms in a system made MD the preferred technique in most applications. In MC approaches, time-dependent properties can be, at best, obtained indirectly. For some situations, especially for the analysis of the equilibrium characteristics of processes, which are slow at the time scale covered by the MD simulations, MC can be the method of choice.(799)
The first MC studies of the bilayer structure in three dimensions focused on the chains that were anchored at the interface, with the surface density corresponding to that of the respective phospholipid bilayer. The dihedrals were initially restricted to three states(746) or to a three-dimensional diamond lattice,(800) to allow the simulations to finish in a reasonable time. The order of chains was studied, both for neat chains and under the influence of cholesterol,(801) a simple cylindrical protein model,(749) and gramicidin.(802) United-atom phospholipid models, confined to the diamond lattice and used in fast MC simulations of bilayer structure,(803) reproduced all states of the bilayer along the temperature scale, from the crystal phase through liquid-crystal and quasi-hexagonal phases to an isotropic liquid. United-atom representation of phospholipid molecules was used in the MD simulations combined with: (i) an MC procedure to analyze the distribution and shapes of the free volumes(804) and (ii) the mean-field potential BD approach to study the chain properties.(805)
An early full-atom MC simulation of the bilayer patch of 36 phospholipid molecules(806) analyzed the chain-order parameters and hydration differences between the gel and liquid-ordered phases. The used Metropolis procedure did not approach the recommended thousand moves per a degree of freedom.(807) To enhance the sampling, several techniques were proposed to replace the random moves in the studied system, including configurational bias(808) and extension-based rotation sampling.(799) The techniques enabled full-atom studies that analyzed orientation of water molecules,(809) provided structural information on pure bilayers and on the effects of cholesterol,810–812 and simulated voids in the bilayers of pure saturated(813) and unsaturated phospholipids,(814) as well as phospholipid−cholesterol mixtures.(815) The analysis of voids is relevant for the hypothetical transport mechanism of small molecules, as described in section .
Availability of the general-purpose molecular simulation suites such as CHARMM,(816) AMBER,(817) GROMOS,(818) OPLS,(819) and later NAMD722,820,821 promoted the use of MD methods. Two monolayers separated by a pool of water molecules were used to analyze the repulsive hydration force,822–824 as well as the structure of hydrating water,823–826 the headgroup region,823,824,826,827 and the core region.826–828 Structure and dynamics of hydrating water were also analyzed in monolayer systems.829,830
For the bilayer, the initial coarse-grained models utilizing particles,804,831 hexadecane chains confined on one end,(832) and other surfactants, with either continuum solvent approximation833–835 or with explicit solvent,513,616,836–838 were soon replaced by atomistic models of fully hydrated phospholipid molecules.839–843 Several technical issues, including the refinement of force fields,838,844–853 the choice of simulated ensembles maintaining the invariability of selected traits (pressure, box dimensions, surface area per lipid),846,854–858 the choice of periodic boundary conditions,(859) and effective calculation of electrostatic840,860–862 and van der Waals interactions(842) had to be resolved before the force-field MD simulations could be deployed for the analyses of structural and dynamical issues in bilayers. Regarding the use of surface tension,553,863,864 it seemed that this need was diminished for the simulations of larger systems.(865) The equilibration times were significantly shortened by augmenting MD simulations with carefully chosen MC steps.866–868
The MD simulations of bilayer patches were mostly applied to liquid-ordered phases but the behavior of the gel phase was also analyzed.(869) Among individual phospholipids, PC as the most abundant mammalian phospholipid was the most frequently studied molecule. Behaviors of other zwitterionic phospholipids, phosphatidylethanolamine302,826 and sphingomyelin,(870) and anionic phosphatidylserine871–875 and phosphatidylglycerol,(443) were also analyzed. As admixtures, cholesterol810,876–884,884–886 and sphingomyelin887–889 were studied most frequently; the former because of its abundance and physiological significance, and the latter because of the role as a component of the canonical mixture forming the lipid rafts.(890) The most popular tails were myristoyl and palmitoyl chains but MD simulations for lauroyl chains,825,826 stearoyl chains,(891) branched chains(892) as components of archaeal and thermostable bacterial membranes, unsaturated oleoyl chains,294,849,875,893,894 (also analyzed by the molecular-level SCMFT(895)), di-unsaturated linoleoyl chains,(896) and poly-unsaturated docosahexaenoyl chains891,897,898 were performed.
The simulated bilayer patch usually contained 64 lipids842,854,891,899–901 or 72 lipids;716,856,857,897,902 however, in some cases, larger systems294,553,903–905 up to 1600 lipid molecules,(870) were analyzed. The size of the patch is important for lateral lipid diffusion and does not affect internal dynamics of the lipid molecules.(906) The duration of simulation extended up to several hundred nanoseconds.(723) The studied aspects included fluidity of the fatty acyl chains,839,842,907–911 structure of the four bilayer subregions differing in density (section ),292,912 structure of the headgroup region,(913) hydration,294,302,902,914–916 free volume distribution,(912) lateral diffusion of phospholipids,(917) and the bilayer structure changes under the influence of ions,872–875,918–921 cholesterol and other sterols,876,878,880–883,885,886 and acyl chain unsaturation.849,886,896,897,922–924 Extended MD simulations were used to analyze slower phenomena, such as undulatory and thickness fluctuations;(904) the evolution of hydrophobic defects;(925) the development of lipid domains,888,889,926 the hexagonal phase,(927) and the ripple phase;(928) the pore opening by mechanical929–931 and electrostatic forces;929,930 the formation of the bilayer(905) and a vesicle(932) from diluted phospholipid solutions; and bilayer fusion.933,934
The MD simulations involve details that are frequently beyond the reach of current analytical and imaging techniques, so that the match of simulations with experiments can only be tested in a limited number of cases. Special protocols have been developed to compare the MD results with some NMR data,716,723,856,935,936 IR data,(911) and with the results of diffraction experiments.(937)
As common in the modeling research, plausibility of the simulations is judged by their ability to imitate known phenomena. The situation that is selected for testing the suitability of an approach should imitate, as closely as possible, the studied structure, event, or effect. Although atomistic simulations are able to reproduce several bilayer-related phenomena, they are under continuous development. For instance, the two most frequently used MD simulation packages, GROMOS and CHARMM, in then-current phospholipid parameterization, were not able to completely reproduce experimental bilayer structure from diffraction experiments.(937) The largest deviations were seen in the distribution of terminal methyl groups of acyl chains.(937) These analyses led to the development of constraints ensuring the agreement of the observed and simulated bilayer structure,(938) as well as to further developments in the phospholipid force fields.(853)
The limitations of the used approach with respect to the simulated event should always be kept in mind to avoid over-interpretation of obtained results. In particular, the simulations are based on numerous parameters, including, for the most detailed MD approaches, partial charges,(844) the force-field parameters for van der Waals interactions, and the equilibrium values and force constants for bonds, angles, and dihedrals. Many of these parameters are obtained from either experimental or quantum mechanical results for simpler compounds. The simplified forms of the common potential functions currently do not include polarizability of atoms and induced electrostatic effects, although a vigorous development is noticeable in this area.939–948 The most common force fields can only be used for systems exhibiting weak interactions and no covalent bond changes. Even for such systems, there is no guarantee that the simulations will provide quantitatively correct results. For instance, free binding energies of ligand−protein interactions are best-described by the methods that recalibrate the simulation results by optimization of additional coefficients.949,950 This approach could work also for the interactions of chemicals with bilayers but the experimental data required for such recalibration are scarce.
2.1.5.3. Equilibrium Partitioning of Chemicals
The interactions of chemicals with bilayers are affected by fine structural details. Mesoscale models do not work well in this context,(753) and atomistic simulations are the methods of choice. The overall partitioning is often the only experimentally measured quantity, and the intra-bilayer distribution of the studied compounds remains largely unknown. The compounds with known bilayer locations are listed in section , in Tables 2−4 and Charts 1−3.
The importance of partitioning is occasionally underestimated because the concentrations of compounds in the bilayer are believed to be too low to play significant roles. As summarized in section , some chemicals accumulate in the bilayer regions in much-higher concentrations than in the aqueous phases. Although the volume of all bilayers in the cell is 2−3 orders of magnitude smaller than the cell volume, the amount in the bilayer is higher than that in water for all chemicals with the bilayer/water partition coefficient being >1000. The transfers between the bilayer subregions (section ) and between the headgroup region and surrounding water proceed at widely different time scales. The fast processes can be described as pseudo-equilibria in the kinetic models of slow processes. Under such circumstances, the equilibrium partitioning in the bilayer regions is of interest, even for kinetic transport studies.
Early approaches to partitioning of chemicals used a continuum representation of the bilayer, most frequently in the form of a hydrophobic slab wetted by two water phases. Conformational analysis, utilizing different dielectric constants for the aqueous and nonpolar phases, and atomic transfer energies between the two phases were used to estimate the overall energetics. Selected conformations were optimized by energy minimization.951–953 The initial insertion of chemicals and peptides was guided by the hydrophobic and hydrophilic centers of the molecules that were determined using the atomic energies of transfer between water and a nonpolar solvent.(641) The binding mode was refined by a force-field geometry optimization of a complex with several phospholipids.(642) The approach was extended to membrane proteins,(954) utilizing a combination with hydrophobicity profiles,(955) molecular lipophilicity potential,956–958 and an MC procedure with a continuous lipophilicity function.959–962
A continuum approach with a smooth function describing the bilayer properties was devised for geometry optimization and MD simulation of bilayer interactions with solutes(640) and for insertion of proteins into bilayer.(963) The location of comparatively complex chemicals in the bilayer was studied by the molecular-level SCMFT models.964,965 United-atom phospholipid models, with the movements restricted to a diamond lattice, were deployed in MC simulations of peptide insertion into bilayers.966,967 The partitioning of six noble gases was analyzed using MD simulations and the particle insertion method(968) for the united-atom representations of phospholipids, with the simplified headgroups tethered to a mobile plane by a harmonic potential.(969) Solute−lipid interaction energies increased more rapidly with the molecular size than the reversible work for cavity creation, leading to a more-favorable partitioning of larger noble gas molecules in the bilayer core.
The atomistic models with frozen bond length and angles were used to obtain free-energy profiles for small molecules such as H2O, O2, CO, CO2, NO, NH3, chloroform, and formamide, using the particle insertion(968) MC simulations in the bilayers of pure DMPC(970) and phospholipid−cholesterol mixtures.(971) This approach utilizes pre-existent voids in the bilayer for the placement of the solute molecules. In this way, it counteracts the nonphysical assumption of the particle-insertion method that the lipid environment remains unperturbed by the inserted solute molecules. The use of voids is relevant because small molecules may utilize them for trans-bilayer transport, which is faster than predicted from the permeation models (section ). The approach can be guided by detailed analyses of the size distribution of the voids in individual bilayer regions.813,815 The method could possibly be extended to larger molecules using the idea of a gradual growth of the solute molecules.972,973
Estimates of partitioning from the MD-simulated trajectories have been attempted by monitoring the time that the solute molecules spent in different regions616,969 or probability distribution of molecules.(973) Taking into account that the experimental values of the partition coefficients of chemicals span a dozen orders of magnitude, these direct approaches seem to be limited to the chemicals with comparable affinities for water and the bilayer.
Partitioning between water and a given region of the bilayer is quantitatively characterized by the difference in chemical potentials in these two phases representing the reversible work to bring the solute from bulk water to a given position in the bilayer. To sample the interactions in different bilayer regions in reasonable time, the permeants can be dragged through the bilayer. This approach was used in the steered MD simulations of oxygen diffusion in a monolayer of hexadecane and cholesterol molecules, with one end constrained by a harmonic potential to imitate the headgroup attachment.(832) The techniques for reconstruction of the free-energy profiles from these experiments are being developed.974–976 The placement of the permeants’ molecules at various positions in the bilayer at the start of the simulation and subsequent monitoring of the trajectories, or confinement of the permeants to certain depths have also been used.(616)
For the majority of studied chemicals, MD simulations provided mostly qualitative results because of the complications arising from the size and interactions of permeants with the bilayer. A possible solution to the size problem is a gradual grow of the permeants’ molecules in the bilayer.972,973
The first atomistic MD simulations of non-physiological molecules514,977,978 in the fully hydrated DMPC bilayer patch used the mean displacement method to calculate the diffusion coefficients. The role of the void size in transport of small molecules was illustrated for benzene on one hand, and adamantane and a nifedipine derivative (skeleton 27 in Chart 2) on the other hand.(979) Benzene exhibits faster diffusion in subregion 4 of the bilayer, where the voids are concentrated. Curiously, the two larger molecules did not differ in the diffusion rates in individual bilayer subregions. Benzene was mainly found in subregion 4 at 310 K, and in subregion 3 at 340 K (the subregions are specified in section ). A preference of rigid aromatic molecules for subregion 3 was also observed in other simulations (see below). This phenomenon was explained by a lower entropy loss for intercalation in the ordered subregion 3, compared to the loss of chain mobility that would be elicited by partitioning in subregion 4.
A fluorescent probe, 1,6-diphenyl-1,3,5-hexatriene (DPH, 33 with R = H in Table 3 and Chart 2) in a dipalmitoylphosphatidylcholine (DPPC) bilayer patch was studied in 250-ps(980) and 50-ns simulations.(515) The latter study showed that DPH assumes the position with the mass center in subregion 3, and the long axis approximately parallel with the acyl chains. This orientation affects chain packing and lateral diffusion of DPPC molecules.(981) Fluorescence anisotropy(96) indicated a wide bimodal orientational distribution along the molecular axis that was pronounced for angles that are parallel and perpendicular to the bilayer normal. A fraction of DPH associated with headgroups has also been detected.(982) As shown by 2H NMR experiments, the order parameter of deuterated DPH decreases with temperature and increases with the cholesterol content.(111)
MD simulations placed pyrene into subregions 3 and 4 (section ) of the 1-palmitoyl-2-oleoylphosphatidylcholine (POPC) bilayer.(983) Pyrene molecules adopted, presumably for entropic reasons, the orientation parallel with the chains, similar to that of DPH, as shown by solid-state 2H NMR of oriented bilayers and extended MD simulations.(516)
Numerous MD simulations focused on finding the preferred locale and the effects of chemicals on the bilayer. Dihydropyridine calcium channel antagonists nifedipine and lacidipine (skeleton 27 in Table 3 and Chart 2) were shown to interact with subregions 2 and 3 (section ) of a DMPC bilayer patch.(984) The changes in the dioleoylphosphatidylcholine (DOPC) bilayer caused by trichloroethane(985) were monitored. The MD simulations of benzyl alcohol (2 in Table 2 and Chart 1) in a DMPC bilayer patch(986) placed this molecule in subregion 3, in contrast to experimental data indicating the headgroup subregion 2 as the main locale.(647)
In the POPC bilayer, indole and N-methylindole (10 in Table 2 and Chart 1) were located in subregion 2, using the interaction energies, or in subregion 3, based on the free energies.(718) Experimental data indicate that subregion 2 is the preferred locale.123,124 This study was undertaken to help explain frequent observations of the placement of tryptophan in trans-membrane helices in the headgroup region,(987) although the partitioning studies using surrogate solvents indicate Trp’s lipophilicity, which would indicate partitioning in subregions 3 and 4. The authors assumed that rigidity of Trp prevents its partitioning into the core for entropic reasons, and the quadrupole potential facilitates electrostatic interactions in the headgroup region.
Anesthetics exhibit interesting interactions with the bilayer. Halothane (3 in Table 2 and Chart 1) was found mainly in subregion 3 of the DPPC bilayer,973,988 in accordance with the 2H and NOE NMR studies,(119) and contrary to the FT-IR ATR spectroscopy results(97) and the 19F NMR experiments in combination with the use of paramagnetic shifting agents.(106) Hexafluoroethane accumulated in subregions 3 and 4, with a slight preference for the latter.(989) An anesthetic bicyclo[4.1.0]heptan-3-one derivative, KP-23, was found to associate mainly with the interface of the POPC bilayer. The protonated form preferred the headgroup region and the neutral form intercalated deeper and protruded into the hydrocarbon region.(990)
Two pools of β-carotene were identified in the POPC bilayer patch, both having the cyclohexene rings localized in the headgroup regions, and differing in the chain conformations, with bent and stretched geometries.(991) Salicylate in the DPPC bilayer assumed the position typical for amphiphilic compounds, led to a decreased area per lipid and a higher chain order, and affected the electrical properties of the bilayer.(992) Pentachlorophenol mostly diffused into the positions in subregions 2 and 3, with the hydroxy group interacting with the water and lipid oxygens, regardless the starting position in bulk water or in the core of the 1-palmitoyl-2-oleoylphosphatidylethanolamine and POPC bilayers.(993)
Large anions were found to intercalate deeper than small anions into the POPC bilayer patch, below the level of phosphate groups,(921) presumably as a consequence of the easier shedding of the less-structured hydration shell, and the influence of the dipole potential. This observation is in accordance with the experiments showing that chaotropic, water-structure-perturbing anions affect the conformations of the PC headgroups(994) and reduce the dipole potential.(995) Both phenomena behave as the Hofmeister effects,(996) i.e., exhibit the magnitudes proportional to the surface charge density of anions.
A detailed MD study of partitioning of n-hexane into the DOPC bilayer at three temperatures provided the profiles of free energy, enthalpy, entropy, and heat capacity. The prevalent partitioning into the core was entropy-driven, while the partitioning into the dense alkyl chain subregion is dominated by a favorable enthalpy change.(997)
To study the effects of membrane potential, the standard setup with one bilayer and two aqueous phases in the unit cell was replaced by three aqueous phases and two bilayers, allowing a different salt composition in the central aqueous phase.998,999
2.1.5.4. Transport of Chemicals
The trans-bilayer transport of chemicals is most frequently characterized experimentally by the permeability coefficients, which are valid for the steady-state situation in the bilayer. The steady state can only be achieved if the permeant concentration in the entry compartment remains constant over the time interval when permeability is determined. This condition is not routinely satisfied in many experimental permeability measurements and may affect the accuracy of the determined permeability coefficient values. Under physiological conditions, the need for an invariant entry concentration of the permeant is satisfied by continuous dosing (e.g., intravenous drug infusions and the exposure of aqueous fauna and flora to pollutants in large basins with balanced input and output). However, other transport processes are more completely characterized by individual transport rate constants as described in section .
Trans-bilayer transport of any permeant is too slow (section ) to be simulated completely from the relevant starting state on one side of the bilayer to a steady state or equilibrium. Shorter transfers between individual bilayer regions have a better chance to be sufficiently sampled in simulations. The permeability estimates have been made by the application of the solubility-diffusion theory, combining estimates of free energy and diffusion coefficients at given depths of the bilayer.292,513,1000
The first full-atom MD simulations of transport of water(292) provided a detailed view of the PC bilayer structure. The diffusion coefficient profile was obtained by the mean-squared-displacement method in the headgroup regions, and by the force correlation method for the molecules confined at a given depth in the chain subregions. The diffusion was found almost isotropic except subregion 3, where the transversal movement was faster. The free-energy profile of water molecules in the bilayer was calculated directly from water concentrations in the headgroup subregions 1 and 2, from the average force acting on confined molecules in subregion 3, and by the particle insertion method(968) in subregion 4. The diffusion and partitioning data were used to calculate the permeabilities, which reached their maximum magnitudes in the center of the bilayer and were minimal in subregion 3.
Similar studies were performed for oxygen and ammonia,(1001) which are assumed to exhibit the same void-based transport mechanism as water. The highest permeabilities were observed in the core subregion 4, and depended greatly on the polarity of the molecules. Oxygen diffusion was actually faster in the bilayer than in bulk water. Proton transport properties were inferred from the formation of transient water pores(1002) and from direct simulations.(1003) In another study of small permeants, diffusion of nitric oxide was found to be isotropic.(1004) The transport of small molecules was shown to be associated with chain dynamics: the rate significantly decreased for diphytanoylphosphatidylcholine, because of a denser packing of branched acyl chains,(892) compared to the straight-chain phospholipids.
The permeability coefficients of eight small organic compounds (acetamide, non-ionized acetic acid, benzene, ethane, methanol, methyl acetate, methylamine, and water) were systematically analyzed using the depth-dependent free energies of partitioning and diffusion coefficients on the basis of the solubility-diffusion theory.(1005) The approach allows identification of the rate-limiting steps and has a potential to provide a better correlation with experimental data thanks to the use of the relevant, depth-specific partition coefficients instead of the experimental partition coefficients, which are based on space-averaged concentrations in the heterogeneous bilayers. The rate-limiting steps or resistances were identified in subregions 1 and 2 for ethane, methylacetate, and benzene; subregions 3 and 4 for water; and subregion 4 for methanol, methylamide, non-ionized acetic acid, and acetamide.(1005) The calculated permeability coefficients were about one order of magnitude larger than the experimental values but the relative trends were correctly reproduced. Interestingly, the partition coefficients of the small studied compound series in individual bilayer subregions 3 and 4 correlated well with both hexadecane/water and 1-octanol/water partition coefficients.(588) The diffusion coefficients decreased with the molecular weight and the cross-sectional area of the permeants.
Inclusion of larger drug molecules (β-blockers alprenolol, atenolol, and pindolol, with the skeleton similar to propranolol: 34 in Table 3 and Chart 2)(1006) provided the opportunity to examine other aspects of the transport process. When crossing the bilayer, the drug molecules tended to keep their long axis parallel with the bilayer normal, and deviated from this orientation only when it was advantageous for the formation of hydrogen bonds. The polar vectors of permeants were more ordered than in liquid water. These tendencies were most pronounced in the dense bilayer subregions 2 and 3. The conformer populations in individual subregions differed: the molecules were more packed near the interface, to form hydrogen bonds, and more elongated in the core.
2.2. Vesicle-Mediated Transport
Absorption, distribution, and excretion of some chemicals can be affected by mechanisms that evolved in specialized cells for transport of physiological molecules and macromolecules, to enable the cells to perform key functions of living organisms. The transport of chemicals can be mediated by vesicles pinching off the membranes and delivering the enclosed cargo into or across the cell, or by membrane proteins transferring molecules across the membranes (section ).
The rates of trans-bilayer transport of chemicals vary in a broad range (section ). The mediated mechanisms play meaningful roles only in the situations when they significantly contribute to the actual trans-bilayer transport rates, regardless of the direction. While the mediated transport can affect the trans-bilayer transport rates in the specialized cells, the interactions with the bilayers (section ) determine the transport rates for crossing other membranes. The overall absorption and distribution of chemicals are determined by both passive diffusion and mediated transport, and cannot be understood without a proper consideration of both contributions. Currently, we do not know the detailed transport mechanisms for many chemicals,but this picture will gradually change, because of intense research in the area of mediated transport.
Formation of the vesicles can be spontaneous (pinocytosis) or initialized by the binding of macromolecules or small molecules to the receptors in cell membranes. Pinocytosis was first studied for amoeba in the 1950s.(1007) The concept of receptor-based endocytosis was elaborated about two decades later for the intracellular entry of proteins(1008) and low-density lipoproteins.1009,1010 The receptors are encountered in two distinct domains of cell membranes, for which the mechanisms and the cargo differ. The receptors for both small molecules and macromolecules are located in caveolae and the respective mechanism is termed potocytosis (section ).
The exclusively macromolecular receptors that are found in clathrin-coated pits elicit receptor-mediated endocytosis (section ). Other vesicle-mediated transport mechanisms are used for secretion of proteins or signaling molecules from the cells (exocytosis) and will not be described here. This mechanism can also play a role in drug efflux via shedding of vesicles and leading to multiple drug resistance.(1011)
Vesiculo-vacuolar organelles are clusters of vesicles and vacuoles in endothelial cells of lymphatic vessels and small veins in normal tissue and tumors, which can interconnect and create irregular trans-cellular channels.(1012) Their openings at the levels of the cell membrane are either unobstructed or covered by a glycoprotein diaphragm. The channels can serve for transport of chemicals and macromolecules.(1013)
2.2.1. Potocytosis
Caveolae are 50−100 nm wide invaginations of the cell membrane that are rich in cholesterol, and coated, on the cytoplasmic side, by caveolins and other proteins.(1014) The most frequently observed invaginations have the flask form, but tubules and grapelike shapes have also been observed.(1015) The flask-shaped invaginations can remain anchored to the cell membrane or detach and form separate vesicles. The caveolae are flattened by cholesterol depletion, and the exposure to polyene, cholesterol-binding antibiotics, such as filipin and nystatin.
The caveolae are found in many cell types, most frequently in capillary endothelium, Type I pneumocytes, adipocytes, as well as in skeletal, cardiac, and smooth muscles.1016,1017 Potocytosis was discovered as a mechanism for pre-concentration of nutrients and signaling molecules, enabling their faster carrier-mediated transport.1018,1019 The intracellular vesicle, which forms upon binding of the ligand to the receptor, encapsulates the ligand molecules in a higher concentration than that which is in the cell surroundings. The vesicle remains attached near the cellular membrane, and the concentrated molecules are transported into the cell interior by a carrier-mediated mechanism.
The term potocytosis has been extended to encompass all types of vesicular transport, which involve caveolae.(1020) Chemicals, macromolecules, and viruses may be transported by one of four possible mechanisms. The original pre-concentration mechanism, with the carrier-mediated release of small molecules from the surface-anchored vesicles into cytoplasm, was observed for folate, retinol, iron, and calcium. Cholesterol and some proteins are delivered by endocytosis to the endoplasmic reticulum or other organelles, and the unloaded vesicles return to the surface. Transcytosis, the transfer of ligands through the epithelial cell, with relocation of the caveolae to the opposite surface, was documented for plasma proteins including albumin and transthyretin. Lastly, the formation of vesicles functioning as depots for temporary sequestration, without an immediate intracellular release, were observed for acetylcholine and several proteins.(1020)
2.2.2. Receptor-Mediated Endocytosis
Clathrin-coated pits are cell membrane invaginations containing several receptors. Clathrin is a large coat protein that associates with the membrane surface. Upon binding of macromolecular ligands, the invaginations transform into vesicles, which detach from the cell membrane, with the help of dynamin and other proteins. Protons are pumped into the vesicles, causing the ligands to dissociate from the receptors, and the coat protein to separate from the bilayer. The bilayer vesicles then associate with endosomes.(1015) Endocytosis can proceed at a significant speed; some cultured mammalian cells can internalize 50% of their surface per hour.(1021) The process is inhibited by ammonium chloride, chlorpromazine,(1015) and monodansylcadaverine.(1022) Endocytosis was described in detail in the Peyer’s patches in the mucose of ileum,1022–1028 in renal tubules,1029–1031 and for the cases involving the receptors for low-density lipoprotein, transferrin,(1032) and scavenger receptors in the blood-brain barrier(1033) and liver.(1034)
2.2.3. Structure-Permeability Considerations
The vesicle-mediated transport has been considered as a route for targeting of drugs to specific cells.1034–1041 Chemicals can participate in the vesicle-mediated transport via direct binding to the receptors in potocytosis, binding to the internalized membrane, or piggy-backing on the internalized macromolecules. These interactions can affect the rate of both potocytosis and receptor-mediated endocytosis. Currently, no QSARs are available for the vesicle-mediated transport. It can be expected that the respective QSARs will be rather complex. In addition to the direct interactions with the receptors, the accumulation of chemicals in the internalized part of the membrane, and the binding of chemicals to the internalized macromolecules must be included.
2.3. Carrier-Mediated Transport
Some chemicals are transported in specialized tissues via carrier systems that have evolved for physiological reasons. The transporters are expressed at the highest levels in absorptive and excretory tissues such as intestinal epithelia, renal tubules, and liver;(1042) in the blood-tissue barriers in the brain (also at the surfaces facing cerebrospinal fluid),(1043) testis,(1044) ovaries,(1045) and placenta;1046,1047 as well as in tumors.1048,1049 In the past decade, hundreds of transporter proteins have been cloned and characterized,(1050) and this number is expected to grow. The mechanisms of carrier-mediated transport include (i) facilitated transport along the concentration gradient; (ii) facilitated influx of cations and efflux of anions due to the inside-negative membrane potential, which can lead to some pre-concentration; and (iii) active transport that proceeds against the concentration gradient on the account of the energy of coupled processes, most frequently adenosine triphosphate (ATP) hydrolysis and the co-transport of Na+, H+, and HCO3−.(1051)
Physiological ions, such as Na+, K+, Ca2+, HCO3−, and Cl−, are much smaller than the majority of organic molecules. Consequently, ion channels are not known to be directly involved in the transport of chemicals, except for their roles in co-transport. The channels can be blocked by chemicals and are targeted by several drugs.(1052)
The transporters are integral membrane proteins that are difficult to crystallize due to the structural changes, which can be elicited by the extraction from the natural membrane environment. Therefore, detailed structural information is scarcer than for other protein types, despite significant progress made in recent years, especially in elucidation of the structure of the efflux pumps (section ).
Chemicals utilizing the protein-mediated mechanisms must resemble the native substrates to the extent that is dependent on the specificity of the carrier proteins. Nutrient transporters seem to have a higher specificity than other transporter proteins, which are usually rather promiscuous.
In contrast to facilitated transport,1053–1055 active transporters seem to be strictly unidirectional and mediating either cellular influx or efflux.(1051) This feature leads to the typical polarity of epithelia forming the monolayers of tightly packed cells separating the bloodstream from non-tissue body compartments. The apical side of most epithelia, frequently rich in glycolipids and having villi, is oriented toward the nontissue compartments, such as the lumen of the gastrointestinal tract, air in the lungs, the glomerular filtrate in the tubule of nephrons, bile in the liver canaliculi, or urine in the urinary bladder. The basolateral side, facing the bloodstream, is separated from the apical side by tight junctions, which prevent both the paracellular transport of chemicals and the lateral diffusion of transporters.(1056) To ensure a flux through an epithelial layer, the influx and efflux transporters are co-expressed(1057) and properly arranged on the apical and basolateral sides of the cells. Combinations of active and passive processes (influx and efflux, respectively, for some anions;(1050) efflux and influx, respectively, for Type II cations,(1058) both described in section ) were observed in the tubular epithelium of the nephron. In the intestinal enterocytes, some transporters (GLUT and SGLT; see section ) are expressed in both apical and basolateral cell membranes.
Most transporters are encoded by the solute carrier (SLC) family genes. The exceptions are the efflux pumps that belong to the ATP-binding cassette (ABC) family. The used nomenclature is recommended by the Gene Nomenclature Committee of the Human Genome Organisation.(1059) By convention, the gene symbols are italicized and the respective protein symbols are not. Human genes and gene products are shown in upper case, while rodent genes and gene products are shown in lower case. Additional specifications and numbers denoting subfamilies and individual members complete the characterization of the transporters. All transporter subfamilies include several members. Although the roles of subfamily members frequently differ, the structural similarity of transporters within families complicates structure−activity analyses at the cellular and higher levels.
Several physiological processes are of significance in the context of transport of chemicals: (i) active transport of nutrients to the bloodstream, with absorption from the intestine and reabsorption from the tubular filtrate as typical examples; (ii) active renal and biliary secretion; and (iii) active transport of nutrients from the blood and the backward transport of metabolites and signaling molecules through the barrier tissues in the brain, testis, ovaries, and placenta. Although the participating transporters are not exclusively expressed in the aforementioned tissues and perform significant roles in other tissues as well, it is convenient to review the carrier-mediated transport along the themes of individual physiological processes, because the processes determine the overall disposition of chemicals in the body. This approach also leads to an intuitive recognition of the tissues with prevalent expression of individual transporters. To maintain the focus on the transport in individual cells, the influx and efflux carriers are covered separately, despite the fact that there is some overlap between the two groups.
2.3.1. Influx Transporters
Protein carrier systems have evolved in all organisms for the uptake of sugars,1060–1062 amino acids, dipeptides and tripeptides,1063,1064 nucleosides,(1065) lipids,(1062) vitamins,1062,1066 and hormones in higher organisms.(1067) This route has been considered a possible mechanism for the increase in bioavailability of the drugs bearing, at least in some parts, some similarity to the nutrient molecules such as antiviral nucleosides(1065) and peptidomimetic drugs including β-lactam antibiotics and inhibitors of angiotensin converting enzyme, renin, and thrombin.(1068)
2.3.1.1. Intestinal Absorption
The apical brush-border membrane of the enterocytes in the small intestine contains a plethora of carriers for glucose, amino acids, peptides, nucleosides and other nutrients. Usually, the transporters from the same families, and, for glucose and amino acids, sometimes the same transporters, are expressed at both sides of epithelial cells, supporting enhanced specificity of nutrient transport processes.
The carriers for the universal mammalian cell fuel, d-glucose, belong to the most intensively studied nutrient transporters. They include1069,1070 facilitative glucose transporters (GLUT) belonging to the SLC2A (facilitated glucose transporter) family,(1059) and concentrative sodium glucose co-transporters (SGLT) from the SLC5A (sodium/glucose co-transporter) family.(1059) Glucose transport in individual tissues is regulated, in addition to other factors, by specific distribution of transporters. For instance, GLUT1 is found in the erythrocytes, placenta, brain, kidney, and colon; GLUT2 in the liver; GLUT3 in the brain; insulin-sensitive GLUT4 in the adipose tissue and the skeletal and cardiac muscles; GLUT5 in the small intestine, and others in the kidneys. Selectivity of the GLUT carriers is high: GLUT1 in erythrocytes transports d-deoxyglucose and 3-O-methylglucose, but not l-glucose.(1071) Insulin elicits recruitment of intracellular GLUT4 to the cytoplasmic membrane, resulting in an increase in the d-glucose transport.1072,1073
The results of protein cleavage studies, immunologic epitope mapping experiments, chemical modifications, and glycosylation-scanning and cystein-scanning mutagenesis allowed for the construction of a working structural model of GLUTs that is based on 12 transmembrane segments.1061,1074 Similar steps are being made toward the elucidation of the SGLT structure.(1075) These results represent an important foundation for a rational development of GLUT and SGLT effectors promoting absorption and inhibiting reabsorption of glucose, as potential anti-diabetic drugs.1076–1079
Amino acids are absorbed via a complex system of amino acid transporters.(1080) The peptide transporters, the low-affinity PEPT1 and high-affinity PEPT2 carriers, which are members of the SLC15A (oligopeptide transporter) family,(1059) perform H+-coupled influx of dipeptides and tripeptides across apical membranes in the intestine and renal tubules.(1081) PEPT1 has a broader substrate specificity and was examined as a means to enhance intestinal absorption of ester prodrugs.(1082) The influx of nucleosides into the enterocytes(1080) is facilitated by equilibrative nucleoside transporters ENT1 and ENT2, belonging to the SLC29A (nucleoside transporter) family,(1059) and by other carriers.(1065) Specific transporters are available for phosphate, monocarboxylates, fatty acids, bile acids, and vitamins. Some transporters participating in renal secretion and reabsorption (OATPs and OCTNs; see the next section) were also found in the apical membrane of the enterocytes.(1080)
2.3.1.2. Renal Excretion
Renal excretion consists of glomerular filtration, tubular secretion, and reabsorption, which proceed in the nephrons of the kidneys. While filtration and some reabsorption steps are passive processes, transporters are involved in secretion and reabsorption of some physiologic molecules and chemicals.
Active renal secretion was discovered ∼80 years ago,(1083) using an anionic substrate, phenolsulfonphthalein, and proved for organic cations two decades later.(1084) Active secretion can be extremely fast: p-aminohippuric acid is almost completely secreted in one pass of the blood through the kidney.(1085) Some of the transporters involved in renal excretion are also expressed in the intestine, hepatocytes,(1086) brain capillaries,(1087) and the choroid plexus.(1088) The involved protein carriers include(1089) organic anion transporters (OATs) and organic cation transporters (OCTs and OCTN, where N stands for novel), all belonging to the SLC22A family,(1059) as well as organic anion transporting peptides (OATPs), which form the SLCO (organic anion transporter) family.(1059) The carriers have 12 transmembrane domains, with the N- and C-terminals located in the cytoplasm.1058,1090,1091 From the mechanistic viewpoint, they mostly act as exchangers: OATs exchange organic anions for dicarboxylates,(1091) OATPs exchange organic anions for bicarbonate, glutathione, or glutathione-S-conjugates,(1092) and OCTs and OCTN1 exchange organic cations for protons.(1093) In contrast, OCTN2 supports Na+ co-transport and electrogenic-facilitated diffusion.(1058) The substrates are mostly amphiphilic organic anions and cations, usually having distinct hydrophobic parts. Notably, amphiphilic compounds frequently exhibit slow passive trans-bilayer transport (section ), so the transporters significantly affect the overall transport rates.
The influx transporters are involved in the first step of active and facilitated tubular secretion, the uptake from the bloodstream through the basolateral cell membrane into the renal proximal tubule cells.(1094) The systems have evolved for numerous organic anions and cations as a means to clean the organisms of endogenous metabolites, toxins, and hormone and neurotransmitter metabolites, and to maintain homeostasis.(1050) Concentrations of chemicals and their metabolites are also affected by the secretion process.
Influx of organic anions into the tubular cells proceeds through a series of three coupled processes. The Na+/K+-ATPase uses the energy released by the ATP hydrolysis to support the Na+ efflux that drives the α-ketoglutarate (α-KG) influx via the Li+-blocked Na+-dicarboxylate co-transporter 3 (NaDC3), member 3 of the SLC13A (sodium-dependent dicarboxylate transporter) family.(1059) Organic anions are exchanged for α-KG by the probenecid-blocked dicarboxylate/organic anion exchangers OAT1,(1095) OAT3, and possibly OAT2.(1050) Organic cations are classified as Type I (smaller, monovalent cations, e.g. tetraethylammonium) and Type II (bulkier, often polyvalent cations, e.g. daunomycin). Type I cations enter the tubular cells via facilitated diffusion mediated by OCT1, OCT2, and OCT3, while Type II cations probably use passive transport.(1058)
Many endogenous compounds (e.g., glucose, electrolytes, and amino acids) and some drugs are subject to active reabsorption, which occurs predominantly in the proximal tubule. Glucose transport in the proximal tubules is mediated by several transporters, e.g., GLUT1 and SGLT2.1078,1094 OATs are involved in reabsorption of anionic substances.(1096) Peptides and peptidomimetics are reabsorbed via PEPT1 and PEPT2 transporters.(1097)
2.3.1.3. Biliary Excretion
Metabolic and excretion functions of the liver are facilitated by a comparatively dense network of capillaries, with each capillary serving several hepatocyte layers (Table 1, section 1.2), and by the direct contact of practically each hepatocyte with a bile canaliculus. The fenestrated liver capillaries (sinusoids) allow the entry of plasma proteins with bound ligands into the interstitial space (the space of Disse), where they can come into a direct contact with the basolateral membrane of hepatocytes.(171)
Biliary excretion plays an important physiological role in the bile formation, by the excretion of synthesized bile salts from hepatocytes into the bile, and by the concentrative transport of bile salts from the blood into the bile, contributing to the bile salts conservation, in a process called enterohepatic cycling. After synthesis from cholesterol in hepatocytes, bile salts are excreted into the bile, in which they enter the digestive tract, and help in emulsification of lipid substances. Bile salts are absorbed in the small intestine, and retrieved from the bloodstream in hepatocytes. The influx through the basolateral (sinusoidal) hepatocyte membrane is mediated by OAT1 and OATPs,1098–1102 which are described in section . The excretion from hepatocytes into bile is mediated by specific biliary transporters and multidrug resistance related proteins (MRPs), which are described in section .
The bile salt excretion mechanism can be used by some chemicals to get from the bloodstream into bile and then to the intestinal content. If the chemicals exhibit slow intestinal absorption, they will be eliminated in feces. The chemicals undergoing both intestinal absorption and biliary excretion can get trapped in enterohepatic cycling.(1103)
Some data suggest that albumin-bound compounds are preferentially excreted into bile and metabolized.1100,1104–1106 The mechanism of this interesting phenomenon could include albumin endocytosis through caveolae (section ) or clathrin-coated regions (section ) of the basolateral membrane of hepatocytes.(1107) The significance of the endocytotic contribution to biliary excretion of chemicals remains to be assessed.
Biliary excretion is dependent on the structure of chemicals,(1108) although no structure-dependent QSAR have been formulated so far. In a limited series of compounds, lipophilicity was seen as the rate-determining factor.1109,1110 An atypical convex parabolic dependences of the excretion rate on lipophilicity were observed for a series of nitroimidazoles.(1111)
2.3.1.4. Blood-Brain Barrier Permeation
The blood-brain barrier, represented by the brain capillary endothelium with tight junctions and low endocytotic activity, contains glucose, amino acid,(1112) and nucleoside(1033) transporters for the nutrient uptake. Secretion of organic anions through the brain capillary endothelial cells and the choroids plexus epithelial cells is mediated by OAT3 (section ) at the brain side, and by the efflux via MRP1 (section ) at the basolateral membrane.(1091)
Thanks to therapeutic importance of the blood-brain barrier permeation, numerous attempts have been made to correlate structure of chemicals with distribution into the brain. The models are mostly based on the assumption of passive diffusion, although the transporters can contribute to permeation of some chemicals.1033,1087,1113 In smaller datasets, lipophilicity,1114,1115 hydrogen-bonding ability,550,1116–1118 polarizability, size and shape,1119–1125 cross-sectional area,656,1126 polar surface area,1127–1129 and solvation free energies(1130) were found as the rate- or extent-determining properties. For larger datasets, numerous topological,(1131) electrotopological,(1132) and 3-D descriptors based on the VolSurf1133,1134 and GRID(1135) approaches were used, and processed by the PLS and principal component analysis (PCA)(1136) techniques. Simple rules were formulated for the CNS-active compounds: the number of O and N atoms should be less than five and smaller than the logP value.(1118)
2.3.1.5. QSAR Approaches
In the absence of structural data on the influx transporters, qualitative pharmacophore mapping methods(1088) and quantitative 3D-QSAR techniques(1137) were used to infer the binding site models of human OATP1B1 and rodent Oatp1a5, respectively. These approaches provide a spatial arrangement of electrostatic and steric regions that represent a putative binding site. Assignment of the site model to a specific state of the protein is complicated by large-scale protein motions, which are anticipated for a functioning transporter. Among several possible interpretations, the simplest one would be that the protein is in the resting unliganded state, to which the ligands bind quickly before the molecular machinery is set into motion.
A descriptor-based QSAR and a pharmacophore model were derived for human OCT1.(1138) The binding to human dipeptide/tripeptide transporter PEPT1, determined in the Caco-2 monolayers (section ), was analyzed(1139) using the Volsurf descriptors.655,1140–1142
2.3.2. Efflux Transporters
Extrusion of physiologic molecules and chemicals from cells has mainly been studied for (i) renal and biliary excretion of physiological molecules, and (ii) cellular efflux of drugs, especially in association with the multidrug resistance. The two research streams have discovered two groups of transporters, which are frequently co-expressed in tissues and have overlapping functions and specificities, but differ in structures and mechanisms. The latter attributes represent the basis for naming the carriers. Some names, which were initially assigned based on the location and function (e.g., canalicular multispecific organic anion transporter, CMOAT), have been changed to reflect the structure and mechanism (CMOAT is now multidrug resistance protein 2—MRP2, which is described in section ). The excretion-associated transporters are usually coupled with compatible influx transporters in the opposite cellular membrane, while the proteins involved in multidrug resistance may lack this attribute. During intestinal absorption of nutrients, the efflux from enterocytes into the bloodstream is often performed by the same transporters as the influx (GLUT, SGLT), reflecting the higher specificity of the nutrient transporters, compared to other transporters.
2.3.2.1. Tubular Secretion
Excretion of organic ions from the renal proximal tubule cells into the urine through the apical membrane proceeds via facilitated diffusion that is sensitive to membrane potential, and via active exchange transport.1050,1058 The anion exchange is performed by the urate transporter (URAT1, SLC22A12),(1059) member 12 of the SLC22A (organic anion/cation transporter) family, OAT4, and possibly OAT5,1050,1094 as well as by OATP1,(1089) and multidrug resistance associated proteins, such as MRP2,1089,1143 MRP4,(1144) and others,(1145) which are described in section .
Anion transporters with apical location, such as MRP2, MRP4, PEPT2, OATP1, and some OATs, contain the PDZ domains, consisting of 70−90 amino acid sequences.(1089) The Type I organic cations (section ) are excreted into urine by OCTN1 and OCTN2, while the Type II cations (section ) utilize ATP-dependent P-glycoprotein (PGP or MDR1), which is described in section .(1058) If the chemicals, which are actively transported into the tubular cells, exhibit slow efflux, a tubular injury may evolve as a result of the increase of intracellular concentrations.(1094)
2.3.2.2. Biliary Excretion
Efflux through the apical (canalicular) membrane of hepatocytes is performed1100–1102 by MRPs, characterized in section , which are also expressed in other organs, and specific biliary transporters such as Na+-taurocholate co-transporting polypeptide (NTCP, SLC10A1),(1059) member 1 of the SLC10 (sodium/bile acid cotransporter) family, and the bile salt export pump (BSEP, ABCB11),(1059) member 11 of the ATP-binding cassette family, sub-family B MDR/TAP.
2.3.2.3. Efflux Pumps
The ATP-binding cassette (ABC) transporters form a large, ancient family of active transporters that are widely expressed across species. In bacteria, the ABC transporters mediate both inward and outward transport, and they represent up to 4% of all proteins in some proteomes. Eukaryotes express several dozen ABC transporters each, all of them acting as efflux pumps.(1146) They rank among the most extensively studied transporters, because of their impact on drug disposition, especially in the connection with the resistance of bacteria1147,1148 and tumors1149–1154 to drug treatments. The range of affected compounds is wide and includes lipids, bile salts, drugs, and toxic and environmental agents, which are transported against the concentration gradient across the cell or organelle(1155) membranes, using ATP hydrolysis as the driving force.(1044) Human ABC transporters (49 genes) are divided into seven subfamilies: ABC1 (subfamily A, 12 members), MDR/TAP (subfamily B, 11 members), MRP/CFTR (subfamily C, 13 members), ALD (subfamily D, 4 members), OABP (subfamily E, 1 member), GCN20 (subfamily F, 3 members), and White (subfamily G, 5 members).(1156) Subfamilies B and C seem to be most relevant for the transport of chemicals.(1157) Human ABC transporters typically consist of 1200−1700 amino acids. Polypeptides shorter than 800 amino acids, which are frequently found in subfamilies B and D−G, are mostly half-transporters, which require dimerization to function. Some members of subfamily A contain 2100−2600 amino acids, and the predicted member 13 is even supposed to have more than 5000 amino acids.(1156)
The ABC transporters are integral membrane proteins that are folded into several trans-membrane domains (TMDs), each composed of fewer than 10, and most commonly 6 trans-membrane helices. They frequently contain extracellular N-glycosylation branches that do not seem to be associated with transport function.(1158) The minimal functional unit of an ABC transporter involved in the efflux of chemicals consists of two TMDs and two nucleotide binding domains (NBDs or ABCs),1159,1160 as illustrated in Figure 9. The TMDs contain one or more substrate binding sites. The TMDs exhibit some sequence similarity only if they mediate transport in the same direction.(1161) The NBD sequences are much more conserved than those of TMDs, because each contains the conserved Walker A and B motifs, which are shared by numerous ATP-binding proteins,(1162) and an ABC signature motif (ALSGGQ) that is also called the C motif. The bound nucleotides (Figure 9) are sandwiched between the C motif and the Walker A motif from the opposite NBD.1161,1163,1164 The NBDs act as molecular motors, initiating conformational changes of TMDs.(1160) The same motor seems to be connected to differing translocation channels. Therefore, a common mechanism may be shared by the majority of ABC transporters.(1161)
Figure 9.
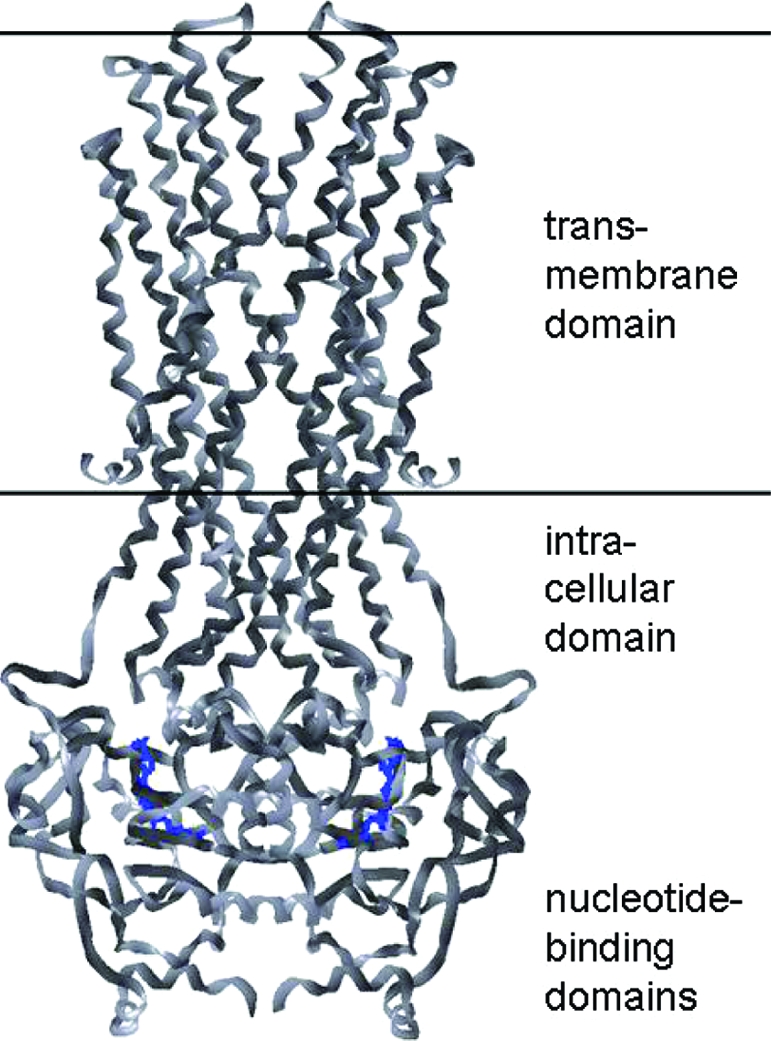
Structure of the bacterial ABC transporter Sav18661171,1172 (PDB(1173) file 2ONJ) is shown in the ribbon mode (gray). The bound molecules of the ATP analog adenosine-5′-(β,γ-imido)triphosphate are shown in blue.(1291) The approximate position of the bilayer is indicated by horizontal lines.
Large conformational changes, expected in both the TMDs and NBDs during the transport process, were confirmed by diverse conformations that were observed in experimentally determined structures of transporters. Structural information for eukaryotic ABC transporters is limited to low-resolution, electron cryo-microscopy structures of PGP1165–1167 and MRP1 (see below).(1168) For bacterial transporters, structures of NBDs1169,1170 as well as those of complete transporters are available. The latter category includes multidrug transporter Sav1866 from Staphylococcus aureus(1171,1172) (Protein Data Bank (PDB)(1173) files 2ONJ, shown in Figure 9, and 2HYD); molybdate/tungstate transporter ModBC-A from Archaeoglobus fulgidus(1174) (2ONK), vitamin B12 permease BtuCD from Escherichia coli(1163) (1L7V), putative metal-chelate importer HI147Q/71 from Haemophilus influenzae(1175) (2NQ2); multidrug transporter MsbA from E. coli (3B5W), Vibrio cholerae (3B5X), Salmonella typhimurium (3B5Y, 3B5Z, 3B60), and multidrug transporter EmrE from E. coli(1176) (3B5W, 3B61, 3B62). The MsbA1177–1179 and EmrE1180,1181 structures were retracted after publication of the Sav1866 structure(1171) and recalculated from original data to correct the handedness and topology. This structural insight, as well as the results of the uptake, cross-linking, labeling, and mutational experiments, and conformational changes of the NBD domains studied by molecular dynamics simulations,(1182) represent the basis for mechanistic hypotheses. The majority of data in the last four categories are available for PGP, so most mechanisms are being developed for this transporter. Various mechanisms for the extrusion step have been recently proposed.1160,1182,1183 Flipping of the amphiphilic substrate, i.e., the change of the inward-facing orientation of the hydrophilic part to the opposite orientation, is a key step in the extrusion process. A synthesis of experimental and structural data delineates the following features of the mechanism, which starts with free PGP in basal conformation, i.e., with the disengaged NBD domains: (i) the substrate binds from the inner leaflet to a high-affinity binding site and increases the affinity of the NBDs for ATP; (ii) binding of ATP brings the NBDs to a closed position and causes the “power stroke”, which is a movement of the TMDs leading to the expulsion and flip of the substrate through the cavity between TMDs to a low-affinity binding site at the level of the outer leaflet; and (iii) ATP hydrolysis and the release of ADP and phosphate are required to bring the ABC transporter to the basal state with open NBDs.1159,1184
The most frequently studied efflux pump is PGP, also known as multiple drug resistance protein 1 (MDR1, ABCB1), which is member 1 of the subfamily B (MDR/TAP) of ATP-binding cassette family.(1059) PGP was discovered as the cause of the resistance of normal(1185) and cancer cells1149–1153 to several cytotoxic agents. The 170-kDa single polypeptide, consisting of 1280 amino acids, is the most studied ABC transporter, because of its early discovery and medicinal importance. The expression in apical membranes of the intestine,1186–1188 hepatocytes, and renal proximal tubular cells underlines the actions of PGP in reducing absorption and enhancing renal and biliary excretion of chemicals. PGP can also be found in the capillary cells of barrier systems, especially in the brain, testes, and ovaries,(1045) as well as in a variety of stem cells(1189) and tumors. (1149–1153) Its localization in the cytoplasmic and intracellular membranes leads to the efflux from the cells and changes in the intracellular distribution of chemicals.(1190)
The activity of PGP,1191–1193 and probably all transporter proteins, is heavily dependent on the lipid environment. The substrate binding sites seem to be in contact with the inner leaflet of the apical cytoplasmic membrane,1194–1196 requiring special considerations for the description of transport kinetics.(1197) The transport kinetics of rhodamine and Hoechst 33342 was explained by a model containing two distinct binding sites, termed R- and H-sites according to the increased affinity for the respective substrate.(1198) The sites exhibited positive cooperativity, i.e., binding of one substrate stimulated the efflux of the other substrate, and the efflux was maximal when both sites were modified. Many but not all tested substrates could be classified as either H- or R-substrates. The H-substrates stimulate the transport of rhodamine and inhibit the transport of Hoechst 33342, while the R-substrates exhibit an inverse pattern.(1158)
In addition to the induction by many chemicals, PGP is up-regulated in some in vitro models of tissue regeneration, and in cells treated by growth factors, as recently reviewed.(1189) The PGP expression prevents apoptosis via a direct interference with the caspase-dependent pathway.1199,1200 These facts have been put together in a hypothesis stating that PGP protects the stem cells from apoptosis during natural stress conditions, aside from the effects of chemicals.(1189) Tumors seem to contain a subpopulation of cells with the stem cell characteristics, which are capable of initiating new tumor growth.(1201) Through PGP induction, some chemotherapies could actually enrich the stem cell subpopulation in tumors and promote their resistance.(1189) These facts may help analyze the failures of clinical trials with the PGP inhibitors.(1202)
Extensive photoaffinity labeling studies on binding of different substrates to PGP and single-point mutagenesis analyses, as recently reviewed,(1045) delineate the associations between structure and function. The PGP ability to bind a variety of structures was hypothesized to result from the presence of a large binding site with a set of hydrophobic and hydrogen-bonding points, which can anchor substrate molecules in different binding modes.(1203)
The pregnane X receptor, exhibiting a broad substrate specificity, is involved in transcriptional regulation of PGP and multiple CYPs.(1204) A broad substrate specificity of PGP overlaps remarkably, although not completely, with that of CYP3A4 (described in section ). This fact, along with the co-localization of the two proteins, indicates the existence of a concerted defense mechanism against exogeneous compounds.1157,1205–1207
Multidrug-resistance-related proteins (MRPs), the subfamily C (CFTR/MRP)(1059) of the ABC family, have a potential to confer resistance to chemotherapy. Clinically, this role has been proven(1044) for MRP1 (ABCC1(1059)). MRPs act as efflux pumps for glutathione, glucuronate, and sulfate conjugates of many drugs and chemicals.(1208) The efflux caused by several MRPs was inhibited by plant flavonoids.(1209) MRP1 is expressed in all tissues except the liver, and it is localized at the basolateral pole of the cells. MRP2 is found mainly in the apical membranes in the liver, intestine, and kidney.(1044) MRPs 1−3 contain ∼1530 amino acids(1156) in three TMDs and two NBDs. The N-terminus protrudes, in contrast to other ABC transporters, outside the cell, and the C-terminus is located in the cytoplasm.1158,1210 MRPs 4 and 5 are shorter; they contain 1325 and 1437 amino acids, respectively, which are organized in two TMDs and two NBDs.(1158) For MRP1, the low-resolution structural data were obtained by electron microscopy of negatively stained particles and 2D crystals of the protein in a lipid bilayer.(1211) The X-ray structure of MRP1 NBD shows a nonproductive conformation that may be associated with low ATPase activity.(1212)
Breast cancer resistance protein (BCRP, ABCG2), member 2 of subfamily G of the ABC family,(1059) is a half-transporter that contains 655 amino acids in only one six-helix TMD, and one NBD.(1044) BCRP needs to dimerize for function.(1213) Its substrate spectrum is similar to those of MRPs, but is more restricted. In addition to the breast tissues, BCRP is found in the liver, intestine, placenta, and also in stem cells.(1213) Its expression is promoted by hypoxia;(1214)therefore, it is found in venous and capillary endothelia, but not in arteries.(1215)
2.3.2.4. QSAR Approaches
Medical significance and comparatively early discovery have made PGP a prominent target of the studies between structure of chemicals and their efflux. The algorithms for prediction of efflux transport of chemicals by PGP and other transporters, and the effects of chemicals on this process were recently reviewed.1216,1217
Homology between bacterial efflux transporters and PGP(1149) was used to obtain structural models of PGP. Kinetic parameters of the interactions between PGP and 34 drugs were correlated with the partition coefficients and surface areas.(1218) Structural requirements for binding of chemicals to PGP, such as amphiphilicity and certain volumes, were generalized based on a structural comparison of known PGP substrates.(1219) More-detailed information about structural requirements for binding and, indirectly, the geometry of the binding site was obtained1220,1221 by classification analysis,(1222) pharmacophore studies,1203,1223,1224 pseudo-receptor modeling,(1225) GRIND descriptor studies,(1226) and 3D-QSAR analyses,1227–1232 which were combined with the homology models, based on the available bacterial ABC transporter structures.1233,1234 Diverse PGP substrates and modulators were shown to be predominantly localized at the headgroup/core interface.(1235) In addition to binding affinity, the interface concentration is an important determinant of the overall efflux rate and must be considered when making quantitative predictions. In this context, the knowledge of solvation energies in individual headgroup regions (section ) may help in a quantitative understanding of the efflux by PGP and other transporters.
The QSAR studies for MRPs and BCRP were performed less frequently and generally are less sophisticated than those for PGP. In many cases, the conclusions are limited to the importance of individual substructures for efflux or its inhibition in MRP1236–1242 and BCRP.(1243) The MRP1 and MRP2 efflux characteristics were correlated with fragment occurrences(1244) and topological indices.(1245) A spatial pharmacophore was generated for MRP1(1246) and MRP2,(1247) and compared with that for PGP. Ligand-based 3D-QSAR studies were performed for MRP2.(1248)
2.4. Binding to Body Constituents
Along with lipids, proteins are the most probable candidates for binding of chemicals, because of their significant physiological concentrations and the ability to participate in practically all types of intramolecular and intermolecular interactions. Strong (covalent) and weak (noncovalent) interactions differ in the magnitude of the liberated interaction energy (more or less than 40 kJ/mol, respectively), and the distance between the most closely located atoms of the molecules of the chemical and the cell constituent (more or less than 2 Å, respectively).(1249) The weak, noncovalent interactions include hydrogen bonding, electrostatic interactions, dispersion (van der Waals) interaction, and the charge-transfer interaction. The weak complexes are created quickly and spontaneously when two molecules come into contact, while the covalent bond formation requires certain activation energy to overcome the energy barrier. These facts are closely related to the kinetics of the processes: while a weak interaction is completed within milliseconds,(1250) formation of a strong covalent bond often requires minutes, hours, or days.
Protein binding is usually reversible and equilibrium is reached, as a rule, within milliseconds, unless covalent bonds are formed or large protein motions are elicited. Proteins can be classified into four types, according to the consequences of the binding event: receptors, metabolizing enzymes, protein carriers, and the remainder, which are sometimes called silent receptors. The receptor binding elicits bioactivity, the binding to enzymes is a prerequisite for metabolism, and the binding to protein carriers facilitates some transport steps. In these three structure-specific events (see section ), the binding of chemicals usually exhibits high affinity, limited capacity, and a tendency toward one major orientation and conformation (binding mode) of the bound molecules of chemicals.
The majority of proteins belong to the category of silent receptors. They frequently contain several binding sites within each molecule. From the pharmacokinetic viewpoint, the silent receptors serve as a depot: they bind free molecules of chemicals, decrease their concentration that is accessible to other processes, and liberate the bound molecules when the concentration has dropped. The binding to silent receptors is typically linked with low affinity, high capacity, and multiple binding modes.
For the formulation of the QSAR models for large sets of diverse compounds eliciting the effects at the cell to organism levels, binding to the most abundant proteins must be analyzed and correlated with structures. The preponderant human proteins are albumin in plasma, collagen in interstitial space, and G-actin in cells. Analyses of a broader set of proteins would lead to more-precise predictions.
2.4.1. Serum Albumin
Albumin is present in human plasma in concentrations of ∼0.6 mM or 40 g/L.(1251) The binding of sulfonamides to human serum albumin (HSA) and the consequent drug inactivation were observed in the early 1940s.(1252) At the end of that decade, the first comprehensive review on drug interactions with plasma proteins was published.(1253) The binding to HSA is usually determined after protein separation by dialysis, ultracentrifugation, or ultrafiltration.(1254) For compounds absorbing or emitting UV-Vis light, direct methods avoiding the separation of HSA are available.(1255) HSA, immobilized in a way that maintains accessibility of the binding sites, was used to determine the binding of chemicals by liquid chromatography,1256–1267 capillary electrophoresis,(1268) and surface plasmon resonance analysis.(1269) These methods are reproducible and fast, especially the last technique in combination with microfluidics.(1270) However, there is a concern regarding the artifacts, which may originate from the inability of the immobilized HSA to undergo large-scale movements, possibly induced in free HSA molecules by the binding event.1271–1273 While the studies usually document that the HSA binding affinities for the studied set are comparable with those determined by other techniques, the role of large-scale HSA movements may become important for the next analyzed ligand. The PAMPA setup (section ) with HSA present in the acceptor compartment allows the binding affinity determinations at an intermediate throughput, and without the potential for aforementioned complications.(1274)
HSA is composed of three homologous α-helical domains, differing in structural and functional characteristics. Each domain contains subdomains A and B, which share common structural elements. HSA undergoes three pH-dependent structural transitions: two in the acidic range and one in the slightly alkaline range.(1275) Among the up to 11 binding sites for fatty acids,(1276) there are at least 3 high-affinity sites for small molecules: the primary hemin site in domain 1,(1277) the primary warfarin site (drug site I) in domain 2A, and the primary diazepam site (drug site II) in domain 3A.(1278) Displacement of fluorescent markers by studied chemicals can be used to distinguish between associations with some sites: warfarin and dansyl-sulfonamide mark the 2A site, and dansyl-sarcosine points to the 3A site.(1279)
Structural details of the binding sites were clarified after the 3D structures of albumin1280–1284 and its complexes with small molecules1276,1285–1290 had become available. One of the X-ray structures of HSA, with binding cavities and ligands1287,1290 (structures in Chart 4) bound in sites I and II visualized using the Sybyl module SiteID,(1291) is shown in Figure 10. Interestingly, the drugs prefer the drug sites I and II, although each ligand also has at least one secondary site in other domains.(1277) Binding of a fatty acid changes the polarity and volume of the drug site I.(1290) These facts, along with the pH-dependent transitions, underline the difficulty of the HSA binding predictions under physiologic conditions. Binding in multiple sites was detected by diffusion NMR and transferred nuclear Overhauser effect (NOE).(1272) The NOE spectroscopy was combined with conformational analysis to identify the binding modes of some site I ligands.(1292) Restricted MD simulations were used to analyze the ionization states, water bridges, and functional roles of lysines in the 2A binding site.(1293) The binding of transition metal ions to the N-terminal of HSA was examined by both classical mechanics and quantum mechanics.(1294)
Chart 4. Albumin Ligands with X-Ray Structural Data of the Complexes (Also See Figure 10).

Figure 10.
The X-ray structure of human serum albumin (HSA) with ligands bound in the drug sites I (right) and II (left). Structures of the ligands (tubes shown in colors) are summarized in Chart 4 (number, the Protein Databank (PDB) file): 3-carboxy-4-methyl-5-propyl-2-furanopropionic acid, CMPF (46, 2BXA);(1290) propofol (47, 1E7A); halothane (48, 1E7B);(1287) diflunisal (49, 2BXE); diazepam (50, 2BXF); ibuprofen (51, 2BXG); indoxyl sulfate (52, 2BXH); azapropazone (53, 2BX8); oxyphenbutazone (54, 2BXB); phenylbutazone (55, 2BXC); warfarin (56, 2BXD); 3,5-diiodosalicylic acid, DIS (57, 2BXL);(1290) and 2,3,5-trioiodobenzoic acid, TIB (58, 1BKE).(1285) The PDB(1173) files were superimposed with respect to C-α carbon atoms. The cyan spheres(1290) represent potential binding cavities identified using the Sybyl module SiteID.(1291) Many of the cavities serve as binding sites for fatty acids and thyroxine and as secondary sites for some of the shown drugs. Individual domains are marked.
The development of prediction approaches to the HSA binding affinities follows the general trend in the area of QSAR modeling: qualitative identification of binding-enhancing structural features(1295) was developed into the dependencies, within homologous series, on physicochemical properties, such as lipophilicity1263,1266,1296,1297 and ionization;(1254) followed by the fragment-based approaches for diverse chemicals;1278,1298–1300 and the PLS or neural networks models with numerous global1263,1301–1303 and 3D variables.1304,1305 The most advanced step, the estimation of free energies of binding to HSA based on the atom-level simulations using the flexible 3D structure of albumin1276,1280–1290 is yet to appear. A reliable prediction of HSA binding is important for estimation of the overall fate of chemicals because, at the high HSA concentration in plasma, even the modest affinities result in a significant decrease of the free concentrations of chemicals. This task is complicated by two factors: (i) the low association constants represent a challenge for the prediction methods; and (ii) the low-affinity binding to the abundant protein results in an easy replacement of the bound chemical by another, only slightly better ligand, with subsequent significant effects on the disposition of both compounds.
2.4.2. Extracellular Matrix
The basement membranes, which are parts of the extracellular matrix (ECM), are formed by Type IV collagen in association with laminin, entactin, and heparan sulfate proteoglycans.(1306) ECM is the most abundant protein mixture in the extracellular space. Recently, we described a simple method for monitoring of the binding of chemicals to ECM.(1307) The simple dependencies of the association constants on lipophilicity hold only for rather small homologous series.(1307) To obtain predictions of ECM binding for large and diverse series, the 3D-QSAR methods(1308) that capture the averaged shapes and properties of the binding sites have been applied.(1309)
2.4.3. Intracellular Proteins
G-actin represents 10%−20% of intracellular protein of mammalian cells. Analytical techniques to be used for the determination of the association constants are similar to those utilized for albumin (section ). The G-actin association constants can be analyzed by receptor-based QSAR methods, using the available X-ray structure.(1310) There seem to be no QSAR studies available yet for the binding of chemicals to G-actin.
2.4.4. Binding versus Structure and Properties
The QSAR models for binding to albumin and other proteins are summarized in the preceding sections. There are some unique aspects of the QSAR models of nonspecific protein binding that deserve attention.
The specificity of binding to nonreceptor proteins is much lower than that of the association with the receptors. As a consequence, multiple binding modes of a compound can be encountered much more often than in the case of the receptors. The formulation of QSARs for this situation requires special treatment, because the observed association constant is the sum of the association constants for individual modes. Based on this premise, we have been developing a ligand-based multimode 3D-QSAR procedure to model unknown receptor sites for several years.(146) The multimode approach is also useful for the receptors with known structures, because the standard MD simulations do not sample the configurations too different from the starting situations, and the use of several starting conformation improves the sampling.(1311)
Among different weak interactions of ligands in the binding sites, hydrophobic interactions exhibit the lowest structural specificity. The reason is that the substructures forming hydrophobic interactions, i.e., nonpolar amino acid residues and alkyl or aromatic ligand parts are quite flexible. Consequently, within a homologous series, the association constants K quantifying binding to silent receptors frequently are dependent on lipophilicity, characterized by the reference partition coefficient P,(1312) as
Here, B and β are empirical coefficients, which are described in more detail in eq 43 in section . Equation 4 is also valid for the reciprocal values of the Michaelis−Menten constants (eq 45) in some cases.(1312) The empirical coefficients B (for binding) and β are specific for the chemicals and the protein. For the binding of various series of chemicals to the same protein, the Collander exponent β often remains identical for all series, and the linear coefficient B varies to reflect the involvement of other than hydrophobic interactions participating in the binding of the parent molecule.(1313) It is not clear if this fact might play a role in the QSAR models of complex biosystems, where many different proteins function as silent receptors, and the coefficients B and β in eq 4 are their averaged values.
Depending on the diversity of the binding sites, the protein binding to a mixture of proteins can still exhibit specificity, as we recently showed for ECM.(1309) In such cases, the 3D-QSAR techniques must be applied, which must be modified to describe the averaging process for binding to multiple proteins. The observed association constant, shown in eq 41 in section , must be described by the expression containing the elementary association constants in the right-most term of eq 39. The reason is that the standard 3D-QSAR equations can only be applied to the elementary association constants for the binding to one site. The proper equation for derivation of a conceptual 3D-QSAR study is obtained from eq 41 by back-substitution from eqs 40 and 39. The resulting 3D-QSAR equation is nonlinear with regard to the optimized coefficients, and it contains the fractions of individual binding sites and the fractions of individual species. While the former fractions are usually unknown and must be fitted as the optimized coefficients, the latter fractions may be estimated using the known dissociation constants and ion-pairing association constants of chemicals in aqueous media.
Unbound fractions are used in the practical pharmacokinetic models for individual drugs to adjust the dosing regimens. The typical ranges of this parameter are tabulated for individual drugs in drug monographs. However, the unbound fraction is not a true drug characteristic, because it is dependent on the protein concentration. Consequently, a direct use of this parameter in the QSAR models1304,1314,1315 is not warranted, unless the data are measured under the same conditions. Rather, the unbound fraction values should be recalculated to the association constants, which are related to the free energy of binding.
2.5. Metabolism
Covalent reactions, along with excretion, represent the route for elimination of chemicals from biosystems. The reactions are either spontaneous or enzymatically catalyzed. Metabolism in organisms proceeds predominantly in the hepatocytes(1316) but other cell types also contribute. Metabolism can affect the outcomes of in vitro experiments in preparations of subcellular organelles and cell suspensions, as well as in isolated tissues, organs, and organisms.
2.5.1. Enzymatic Reactions
Generally, a distinction is made between two phases of biotransformation. Phase-I reactions are mostly catalyzed by cytochromes P450 and include hydrolytic, oxidative, and reductive processes.(1317) The products of the Phase-I reactions or the unchanged molecules are conjugated with hydrophilic endogenous molecules (e.g., amino acids, glutathione, glucuronide, sulfates) in the Phase-II reactions.
2.5.1.1. Cytochromes P450
The largest fraction of metabolism, up to 90% of the Phase-I metabolism of current drugs,(1318) is performed by cytochromes P450 (CYPs). The 40−50 kD enzymes with a single heme group(1319) catalyze the hydroxylation of numerous substrates, increasing their aqueous solubility and accelerating renal excretion.(1319) Originally discovered in mammalian liver microsomal preparations (section )(1320) and named after the distinct UV-Vis absorption band at 450 nm,1321,1322 CYP mono-oxygenases have subsequently been found in almost all organisms. The counter-examples are rare and include, e.g., Escherichia coli and Salmonella typhimurium.(1323) In the superfamily of more than 1000 CYPs, 57 CYPs are expressed in humans.(1324)
Human CYPs metabolize more than a thousand known substrates, and ∼15 CYPs are involved in the degradation of xenobiotics.(1323) The CYP isoforms 3A4 and 2D6 are responsible for the metabolism of 50% and 30%, respectively, of current clinically used drugs.(1325) The levels of some CYPs in humans are highly regulated, whereas others (e.g., CYP2D6) exhibit considerable differences,(1326) leading to significant individual variation in pharmacokinetics of drugs. CYP3A4 is upregulated by the nuclear hormone receptor PXR (pregnane X receptor). Both proteins bind a similar broad range of lipophilic chemicals(1327) and represent the sensor and executioner of the axis of defense against xenobiotics.(1328) The CYP isoforms 1A1, 1B1, and 1A2 are regulated by the aryl hydrocarbon receptor.1329,1330 Monitoring the actual CYP isoform levels in individual patients is one of the first components of modern approaches to personalized medicine.
In mammals, CYPs are located mainly in the liver; however, they can also be encountered in the intestine and tumors. While CYPs of eukaryotes are mainly anchored in the endoplasmic reticulum, some are also found in mitochondrial membranes.1331,1332 All CYPs in the endoplasmic reticulum receive electrons from a single flavoprotein: NADPH−P450 reductase. The mitochondrial CYPs use an electron transport chain with the iron−sulfur protein adrenodoxin and the flavoprotein adrenodoxin reductase.(1323) The electron transfer proteins bind on the side of the CYP that is proximal to the heme.1333,1334 Although the structures of individual components have been determined, only structural models of the complex are available.(1335) CYPs can be induced by various chemicals and this ability, in limited series of compounds, increases with lipophilicity.(1336) The studies of CYP interactions with model bilayers355,1337 are important for understanding the CYP mechanism and the prediction of CYP metabolism.
Bacterial CYPs are soluble; therefore, they were crystallized and analyzed using XRD much earlier1338–1350 than rabbit CYP2C5,1333,1333,1351,1352 and human CYP3A41353,1354 (Figure 11) and CYP2C9.1355,1356 The structures show that CYPs have large, flexible binding sites, which can accommodate more than one substrate molecule,1357,1358 and allow for mutual influences of chemicals, which can act as substrates, activators, or inhibitors. The size of the binding cavity can explain the majority of complicating factors of CYP-mediated metabolism, which include broad specificity of individual subtypes,(1359) atypical allosteric kinetics,1360–1362 activation, substrate inhibition,(1362) stereoselectivity,(1362) regioselectivity,1363–1368 cooperativity,1369,1370 and formation of multiple metabolites of some chemicals.1369,1371–1373
Figure 11.

X-ray structure of human CYP3A4, with bound metyrapone (blue) shown close to the heme (red) and progesterone (blue) at the top of the protein.1353,1354 The PDB(1173) files 1W0F and 1W0G were superimposed with respect to C-α carbon atoms.(1291) The cyan spheres(1290) represent the binding cavity identified using the Sybyl module SiteID.(1291)
The active site of CYPs is buried deep in the protein next to the heme co-factor that plays an important role in activating the molecular oxygen.(1374) In the absence of substrate, the Fe3+ ion of the heme is hexacoordinated with four heme-pyrrole nitrogens in a planar arrangement, the sulfur of a cysteine below the heme, and a water molecule on the opposite side.(1339) When ligated to water, the Fe3+ ion prefers the hexacoordinated, low-spin state (S = 1/2), in which the five 3d electrons are maximally paired. The spin state remains low when the inhibitors are bound, forming a coordination bond with the iron. The substrates interacting with the binding pocket above the heme and expulsing the aqua ligand without a substantial interaction with the iron, which then becomes pentacoordinated, induce a transition to the high-spin state (S = 5/2), when the five 3d electrons are maximally unpaired.(1375) The spin changes are reflected in the UV-Vis spectra and frequently are used for a convenient measurement of binding affinities of inhibitors and substrates. The absorbances at 390 and 420 nm have been linked to the low-spin iron and the high-spin, pentacoordinated iron, respectively, using a range of quantum-chemical techniques.1375–1377
The high-spin Fe3+ ion moves out of the heme plane and becomes more prone to reduction to the Fe2+ ion by the electron transfer from NAD(P)H. Dioxygen binds to the Fe2+ ion, where it is stabilized by a network of hydrogen-bonded water molecules and acidic amino acid residues,(1378) and cleaved, following the second reduction. The resulting iron-linked O atom is a potent oxidant. The substrate is bound in the way ensuring proximity of the pertinent substrate atom to the iron-linked O atom for regioselective and stereoselective hydroxylation. The overall stoichiometry of the hydroxylation reaction includes the chemical, dioxygen, two protons, and two electrons on the substrate side, and the hydroxylated metabolite and water on the product side. The efficiency of the process can be diminished by reduction of the intermediateds to hydrogen peroxide or water. The futile reactions are facilitated by the presence of water in the active site. The elementary steps of CYP-catalyzed metabolism have been studied in great detail,(1379) and only the most important steps have been summarized above.
The CYP active site is often completely isolated from the medium. The protein must undergo dynamic motions to allow substrates to enter and products to leave the active site. These movements are accompanied by complementary water diffusion, for which a water channel was postulated.1340,1380 This route can also be used by protons.(1334) The replacement of water by a substrate shifts the redox potential toward the Fe2+ form.1381,1382 Three routes for substrates and products were identified, based on Random Expulsion(1383) and Steered MD simulations,(1384) and an analysis of the thermal motion factors in the X-ray structures.(1385) The preference for a route is dependent on properties of the transported chemical and varies among CYPs.(1386) For mammalian CYPs, two routes were suggested: a one-way route from the bilayer for membrane-bound substrates, associated with a one-way route into the aqueous surroundings for the products; and a two-way route connecting the active site with the aqueous medium for more water-soluble substrates and products.(1387) CYPs are rather flexible, and the X-ray structures with different geometries of the channels (closed, open just for water molecules, and open for larger substrates(1343)) were observed.(1334) Several steps in hydroxylation of camphor by CYPcam, revealing structures of the intermediates, conformational movements of the amino acid residues, and changes in the water network, were captured using cryocrystallography.(1388)
Reactive intermediates produced by CYPs may bind to proteins or react with water and other low-molecular-weight constituents. The protein adducts have long been implicated in organ toxicity, especially in the liver.1389–1393 Although some nontoxic compounds also form protein adducts, the potential for toxicity leads to a close monitoring of the adduct formation in the development of bioactive compounds.1394,1395 The approaches include binding of labeled compounds to microsomal fractions,(1394) reaction of chemicals with glutathione that is monitored using a fluorogenic competitor, and 2D NMR analysis (ALARM NMR) of binding to isotopically labeled human La antigen, whose two cysteins can react with chemicals or change the oxidation state in the presence of chemicals.(1396)
NMR can potentially play an important role in monitoring the binding modes of compounds in the active site of CYPs, thanks to the presence of a natural paramagnetic iron of the heme. The relaxation induced by unpaired electrons provides information about the distance to the iron,1397,1398 in addition to the identification of the interacting parts of the molecule. The proton−iron distances can be used as constraints for the docking of ligands to CYPs.(1399)
The importance of CYP metabolism in the development of bioactive compounds has led to a continuous interest in prediction of all aspects of CYP interactions with chemicals,(1400) including the induction of different CYP isoforms.(1401) The induction process in rat hepatocytes was analyzed as a function of structure of 4-n-alkyl-methylenedioxybenzenes.(1402)
Metabolic rates and their inhibition were often analyzed as a function of structure or properties of substrates and inhibitors. In a limited series of substrates and inhibitors, CYP binding or metabolism were frequently found to correlate with electronic indices(1403) or lipophilicity.1404–1409 The correlations are series-specific, and different correlations for the same CYP are not uncommon.(1410) Empirical correlations with large numbers of descriptors, processed by soft-modeling techniques, such as linear regression,(1411) neural networks,(1412) PLS regression,1413,1414 and k-nearest-neighbor variable selection(1415) extended the scope to larger sets of chemicals. Predictions for more-diverse chemicals required a 3D approach. The long absence of the mammalian CYP structures resulted in numerous ligand-based 3D-QSAR models of the CYP binding for both substrates1416–1422 and inhibitors.1423–1428 The 3D-QSAR models were combined with comparative structural models, which used bacterial,1425,1429–1432 rabbit,1433–1437 and human(1438) CYP structures as templates. Peculiar properties of the CYP binding sites, including sizes that often exceed the size of common ligands, conformational changes, and the possibility of accommodation of two bound molecules, led to less-predictive docking than for those with other receptors.1438,1439 The success rates were improved by re-parameterization of the ligand−iron interactions by knowledge-based potentials, and a new parameterization of lipophilicity contributions of the planar heme nitrogens.(1440) The estimation of binding free energies1378,1441 is complicated by the isolation of the binding site from the medium. The problematic exchange of the water molecules displaced by ligand binding with the medium was emulated by coupling of MD and Grand Canonical MC simulations.(1442) A more precise description of the CYP metabolism and prediction of structures and formation rates of individual metabolites is a formidable task.
Predictions of the sites of metabolism in the structures of metabolized molecules1443,1444 were performed using quantum-mechanical (QM) reactivity calculations,1403,1445QSAR approaches,(1446) and pharmacophore-based procedures1420,1430,1431,1433,1447 on substrate molecules, and docking(1439) utilizing the structural information about the binding site. Practical software applications use either knowledge-based rules or a simplified simulation of the interaction between the pertinent CYP and the metabolized molecule. The former category includes the commercial expert systems MetabolExpert,(1448) METEOR1449–1452 and META.(1453) The software tool MetaSite(1454) is the only representative of the second category. The binding sites in the homology models of several CYP isoforms are characterized by the GRID-based descriptors and the substrate molecules are described by clustered atom distances.(1455) A comparison of the resulting binding site and substrate fingerprints provides the accessibilities of individual hydrogens in the substrate molecule. The accessibilities are multiplied by estimated, semi-quantitative hydrogen abstraction energies to provide a probability of metabolism for each position.(1456)
2.5.1.2. Other Enzymes
Phase-II enzymes, hydrolases, and other metabolizing enzymes were studied less extensively than CYPs. For a limited series of compounds, the biotransformation rate parameter can be dependent linearly on a single property, such as the reactivity in the reaction imitating the pertinent process in the reaction center of the enzyme(1457) (section ), relevant QM indices,1458–1462 or the partition coefficient,(1463) if the noncovalent binding to the enzyme is dependent on lipophilicity and all the other parameters are invariant. For diverse compounds, the ligand-based1464,1465 or receptor-based 3D-QSAR methods(1466) were applied. If the prediction of disposition of diverse chemicals in organisms is of interest, the in-vitro-measured metabolism parameters often may be the best choice, considering the complexity of the QSAR prediction of metabolism. The scale-up procedures1467–1475 can be improved using the SBSP models. Kinetic issues are addressed in section .
2.5.2. Spontaneous Reactions
This category of elimination processes is comprised of hydrolysis, as well as the reactions of chemicals with low-molecular-weight body constituents and macromolecules. Abundant are the reactions of chemicals with cellular nucleophiles such as free amino acids, glutathione, and proteins.(1476) Spontaneous reactions do not require a preceding binding of the chemical to a macromolecule, and they are comparatively easy to imitate in vitro. The rate constants measured in the reaction mixtures mimicking biological conditions belong to the most-precise reactivity parameters.1477,1478 The experimentally determined reaction rate parameters for a reference reactant (k) can be used to assess the parameters for other reactants (kx) in similar reactions, as, e.g., in nucleophilic additions to different acceptors:
The empirical coefficients Ar and βr are dependent on the reactants and the reaction conditions. The reference reactants used include 4-(p-nitrobenzyl)pyridine, thiourea,(1479) glycine,(1480) cysteine,(1481) and thioglycolic acid.1482–1484 Empirical substituent constants and QM indices related to the electron density at the reaction centers in intact molecules and transition intermediates1485–1489 can be used to substitute the rate parameters of the spontaneous biological reactions.
The reactivity parameters were mainly applied in the QSAR studies of toxicities or other effects of alkylating agents. In these studies, the dependence of the effect on reactivity frequently exhibits an optimum.1457,1490,1491 This behavior reflects the interplay of two phenomena, binding to the receptors and metabolism, which may both involve similar chemical reactions behaving according to the nonlinear dependence described in eq 5. While the receptor binding increases with reactivity, the disposition in the receptor surroundings is independent of low reactivity and decreases after reactivity exceeds a certain limit (see eq 13 in section ). The increase in reactivity of chemicals leads to more-pronounced effects up to the point when high reactivity causes a fast elimination of the compounds, which are no longer able to reach the receptors in meaningful concentrations.
3. Disposition of Chemicals
The disposition of chemicals in biological systems has been experimentally analyzed at different levels of complexity, starting with membrane vesicles, subcellular organelles, cells, cell monolayers, biological sheets, up to organs and organisms. In this section, a brief overview of the most frequently studied biosystems and used techniques is provided. The coverage could not keep up with the abundance of published studies in all mentioned areas, but especially for the perfused organs (section ) and organisms (section ), and lists only the most important developments. The relationships between the transport or accumulation and physicochemical properties are analyzed, where available. In contrast to section , which focused on individual steps, in complex systems, several interactions between chemicals and biosystem components participate in the overall outcome.
3.1. Membrane Vesicles
The vesicles form spontaneously under appropriate experimental conditions from complete membranes in tissue homogenates and are isolated from other cellular material by differential centrifugation. Historically, availability of these systems was associated with the progress in the biochemical separation techniques: the microsomal fractions were first prepared in the 1940s, while membrane vesicles appeared in the 1970s.
Some of the membrane proteins in these preparations function as transporters or metabolizing enzymes, while others play more-passive roles. Integral and peripheral membrane proteins diminish the surface area available for passive transport, causing the process to become slower. The binding of chemicals to both integral and peripheral membrane proteins leads to a decrease in the free concentration of chemicals, thus reducing all types of transport.
3.1.1. Endoplasmic Reticulum Vesicles
Microsomal vesicles are derived mainly from endoplasmic reticulum, but the cell membrane and the membranes from lysozomes, mitochondria, and Golgi apparatus are also present.(1492) The majority of these minor microsomal components sediments out, along with intact cells, mitochondria, and nuclei, during centrifugation of a tissue homogenate at 9000g. The resulting supernatant, which is called the S-9 fraction, contains mainly endoplasmic reticulum and soluble enzymes and is sometimes used for metabolic activation of chemicals to test biological effects of metabolites.1493,1494 More-intense centrifugation of the supernatant at ∼100 000g pelletizes the microsomal membranes, which are usually of a reddish-brown color because of the high content of the CYP heme.(1495) The vesicles were prepared from the liver,1496–1501 brain,1502,1503 lungs,(1504) heart,1505,1506 kidney,1505,1507 intestine,1508,1509 skeletal muscle,1510,1511 placenta,(1512) leukemic cells,(1513) and other sources. The vesicular structure of the microsomes was demonstrated by electron microscopy.(1514) The preparations were first used to characterize subcellular location of enzymes,(1498) other macromolecules,(1515) and metabolic processes.1503,1504,1512 Microsomes have been mainly applied to the study of metabolism of chemicals.1472,1475,1494,1516–1518 Although the microsomal data serve as the input for prediction of hepatic metabolism,1469,1519,1520 the uptake and transport studies in the microsomes focused on physiological molecules,1521–1524 and the data on chemicals are rather scarce. Because the substrates may access the CYP binding sites from the membranes,(1387) the concentrations of chemicals in individual bilayer regions (sections and ) are important determinants of the overall kinetics.
The assessment of stability of chemicals in microsomes or S-9 fractions with different levels of expressed CYP isozymes belongs to the standard components of metabolic(1525) and drug−drug interaction(1526) tests in preclinical drug-development process. The data have been processed by three categories of models: (i) predictions of in vivo metabolic clearance of individual chemicals,1469,1519,1520 (ii) QSAR analyses of the rate of metabolism, and (iii) predictions of the site of metabolism. The QSAR models in the second category usually utilize empirical approaches based on numerous descriptors, which are calibrated by regression analyses1411,1527 or by the variable selection based on the model-free k-nearest-neighbor intrapolation.(1415) The predictions of metabolic sites in the third category are listed in section , because they are based on the structure of pertinent CYPs.
3.1.2. Cell Membrane Vesicles
Different techniques were used to prepare vesicles from cell membranes of epithelial cells, either from apical, brush border membranes from the intestine,1528–1530 renal tubules,1531,1532 and placenta,1533–1535 or from renal basolateral membranes.1529,1536,1537 The brush border membrane vesicle form spontaneously by segmentation of the microvilli in the preparation process, so the correct polarity of the membrane is maintained.(1538) They were mainly used for the monitoring of the active transport of physiologic molecules. Studies on the transport of chemicals are rare.(1539)
Red blood cells, which represent ∼45% (hematocrit) of the blood volume, lack a nucleus and organelles. Their membranes are sometimes isolated and resealed to generate vesicles for the study of transport and accumulation of chemicals. Erythrocyte ghosts are prepared by hypotonic(1540) or isotonic(1541) lysis at 4° C, washing the membranes, and subsequent resealing at 37° C, whereby lipid asymmetry is aborted.(1542) Membrane proteins of erythrocytes include several MRP efflux pumps (section ),(1543) and the vesicles were used to measure the outward transport of amphiphilic molecules.(1544) The ease of preparation makes the vesicles a popular system for the study of asymmetry of distribution of proteins(1545) and lipids,708,714,1546–1548 as well as for the measurement of the interactions of nonpolar(77) and amphiphilic chemicals82,103,335,441,1549–1553 with membranes. Accumulation in erythrocyte ghosts was analyzed via the PCA approach, using Volsurf descriptors.(1136)
3.2. Subcellular Organelles
The complexity of subcellular organelles is higher than that of membrane vesicles. Although both are covered by complete membranes, the organelles contain additional structures, which may bind the chemicals and complicate the interpretation of the disposition data.
The most often used subcellular organelle is mitochondrion, which became the object of interest for the study of energy metabolism and the effects of compounds affecting this process, as well as for the analysis of the mechanism of apoptosis. Mitochondria are isolated by differential centrifugation from the cell homogenate, which is prepared by mechanic disruption or nitrogen pulsation,(1554) most frequently from liver,1555–1557 heart,(1558) brown adipose tissue,(1559) skeletal muscle,(1560) neurons,1554,1561 protozoa,(1562) as well as from yeasts and other sources.(1563) The preparations were used to measure the uptake78,1564 and effects of uncouplers of oxidative phosphorylation and respiratory inhibitors.78,1564–1576 The uptake into rat liver mitochondria was described by a conceptual pseudo-equilibrium model (section ) and used to infer membrane concentrations of the uncouplers and to generate conceptual QSARs for their effects.78,1564
For DNA intercalators, the interactions with isolated nuclei were studied and compared to the binding of the chemicals to DNA in intact cells.1577,1578 The results contribute to the analysis of subcellular distribution of studied chemicals.
3.3. Cells
The accumulation of chemicals in cells has been studied since the end of the 19th century. The reports amply illustrate the thinking process, based on sparse and imprecise data. In the following, the most important milestones are listed in chronological order, with each paragraph covering about two decades.
Plant cells and protoplasts, and the eggs of marine invertebrates were the first systems to be studied from the viewpoint of transport of chemicals, and the permeabilities were monitored as osmotic volume changes.188,192 The Overton’s theory on the association between the permeability and lipophilicity(1579) that led to the hypothesis on the lipid character of the membranes, was developed using the tadpole irritability data.(192) In histology, uneven intracellular distribution of dyes, which were used to visualize various cell components for microscopic observation, was considered a consequence of different solubilities in cellular material (cited in ref (1580)).
The partitioning of acids and bases in surrogate solvent/water systems exhibited increasing or decreasing sigmoidal dependences on the pH values of the aqueous phase.(1580) With the objective of monitoring the transport of ionizable compounds,(1581) the pH indicators were introduced into cells and used to measure the uptake of bases by sea urchin eggs.(1582) To improve survival of the test material, cells containing natural pigments serving as the pH indicators were used.1183,1583–1585 However, these studies did not use single cells; therefore, they are described in section . In large cells of algae (Valonia, Laminara, Chara, Nitella and others), conductivity was monitored as a measure of the overall ion movement, and the effects of various chemicals on this parameter were studied.1586,1587
The size of the algae cells allowed a direct chemical analysis of the cellular content.1588,1589 A similar analysis of cellular content was also applied to the protoplasts of oyster plant (Rhoeo discolor).1590,1591 Micromanipulation of amoeba cells in the microscope field allowed for the analysis of anion and cation effects, not only by the cell immersion but also by microinjection.1592,1593 Transport of a series of acids was tested as hemolytic activity in the author’s own blood,(679) and as the threshold concentrations for the sour taste.(1594) The comparison with physicochemical properties of chemicals became more quantitative: the oil/water partition coefficients were used in the former study, and published data on charcoal adsorption in the second study. The first real correlations of permeabilities with oil/water partition coefficients were published by Collander(521) for the transport of nonelectrolytes in algae Chara ceratophila and other cells. This work is remarkable also for other conclusions: the transport process was described by Fick’s law, and some small molecules permeated at a faster rate than expected, based on their partition coefficient. The latter observation formed the basis of the lipoid-sieve hypothesis, which assumes the presence of pores, allowing the passage of smaller molecules. The nonelectrolyte data in algae Nitella mucronata(680) were fitted by a linear permeability−lipophilicity relationship and later were shown to adhere better to a parabolic equation.(1595)
The availability of radiolabeled chemicals opened the door for autoradiography.1596–1604 For visualization of subcellular distribution, autoradiographic images were enlarged by light microscopy.1605,1606 The use of electron microscopy in the transmission mode1607,1608 or scanning mode(1609) increased the resolution from ∼0.2 μm, which is typical for light microscopy, by three orders of magnitude. The technique enabled important discoveries related to DNA, RNA, and proteins, but the location of diffusible, noncovalently bound chemicals could not be analyzed precisely, because of the artifacts introduced by sample preparation. To monitor the distribution of chemicals and drugs, the precision was greatly improved1610,1611 by the introduction of precise cryosection(1612) of freeze-dried samples,(1613) which avoided fixation and embedding of the samples.
Other techniques for monitoring membrane accumulation, including potentiometry, equilibrium dialysis, and 1H NMR spectrometry, have been recently reviewed.(307) Confocal fluorescence microscopy allows monitoring of the kinetics of subcellular distribution of fluorescent compounds.1614–1618 The high shutter speeds are of special importance for capturing the detailed kinetics of the transport process.1619,1620 The rich spatial and temporal data represent an ideal test bed for the analyses by SBSP models.
Partitioning into erythrocytes and leukocytes was used to provide a picture about the binding of chemicals in the blood. Notably, in classical pharmacokinetics the blood corpuscles represent the components of tissues as opposed to those of plasma. This anomaly is caused by the definition of the distribution volume, which is given as the ratio of the actual drug amount in the body and the total drug concentration in plasma. As a consequence, a drug that, after intravascular administration, does not leave the bloodstream but binds to erythrocytes or leukocytes will have a distribution volume that is larger than the plasma volume. Blood corpuscles are readily available, are easy to purify, have comparatively simple structures, and their shapes can be comfortably monitored. Their interactions with chemicals have different consequences, depending on the used concentrations of chemicals: simple partitioning,1552,1621–1623 shape changes,1624–1628 aggregation,(1629) and even hemolysis.1630,1631 Hemolytic activity of homologous N-alkylpiperidines and α-monoglycerides exhibit a bilinear dependence on lipophilicity,8,64,706 indicating non-equilibrium transport, as decribed in detail in section . A linear lipophilicity−concentration profile was observed for the binding to erythrocytes for a series of barbiturates.(1314)
Understanding the uptake into epithelial cells or enterocytes lining the luminal surface of the intestine is important for orally dosed drugs. Enterocytes are highly polarized and feature completely different apical and basolateral membranes. Germinal cells in the crypts between the villi differ from nonproliferative columnar cells that cover the villi, through which the absorption proceeds. These cell populations(1632) can be separated after the cells are isolated.(1633) Upon isolation, enterocytes will round and depolarize. The cells respond extensively to environmental factors, are difficult to maintain in primary cultures, and have a short lifetime. Therefore, the uptake experiments are mostly performed using freshly isolated enterocytes.(1634) Alternatively, everted rings of the entire intestine are used to measure the drug uptake, with the risk that the uptake into the cells other than enterocytes will affect the results. With proper oxygenation and nutrition, the rings can be maintained for several hours before the changes to the mucosa become observable.(1635) The transport through the entire intestine in the form of everted sacs or sheets is described in section . Continuous monolayers of human intestinal adenocarcinoma Caco-2 cells, which are more comfortable to maintain and use, found widespread utility in assessing intestinal absorption in the drug-development process (section ).(1636)
The interest in the use of hepatocytes in disposition and metabolism studies stems from the importance of metabolism for drug pharmacokinetics. The uptake into hepatocytes1474,1637 was studied frequently, because of its importance for the prediction of metabolism.(1638) The importance of transporters in hepatocytes, as opposed to microsomes, for the prediction of transport in humans was emphasized.1639,1640 Human hepatocytes can be engrafted into immunochemically compromised mice, where they repopulate up to 90% of the liver.(1641) The animals can be used as a source of fresh human hepatocytes or directly for toxicity studies of metabolites.
The uptake of chemicals into various cell types(1642) was measured using the analysis of the medium after the cell separation by ultracentrifugation, ultrafiltration, gel filtration, and dialysis. The disadvantage of the uptake monitoring, the uncertainty about the localization of the chemical interacting with the cell either on the cell membrane or inside the cell, can be alleviated using the SBSP models (see sections −). This approach was used to estimate the subcellular distribution and resolve the mechanism of uncoupling of oxidative phosphorylation for a series of carbonylcyanide phenylhydrazones in rat liver mitochondria,(78) bacteria Paracoccus denitrificans, and leukemic P-388 cells.(1643) Advanced kinetic approaches were applied to the modeling of the cellular uptake data,1644,1645 although accumulation in the bilayers, which is an important process for the majority of chemicals, was not included in the analysis.
3.4. Cell Monolayers
Some cell lines form confluent cell monolayers, imitating biological epithelia, when grown on porous supports. The monolayer cultures were initially used to study physiological processes occurring in renal1646,1647 and intestinal1648–1650 epithelia, and later adapted for the analysis of the distribution of chemicals. Transport across the layer separating two aqueous compartments is conveniently examined by measuring the compound concentration in the receptor compartment. Sometimes, accumulation in the cells is also assessed. The initial use of radiolabeled chemicals was largely replaced by UV-Vis absorption spectrometry and liquid chromatography analyses. The latter technique, especially when coupled with mass spectrometry, allows the detection of metabolites, and provides the opportunity to increase the throughput by simultaneous testing of several chemicals in a mixture.(1651)
The most frequently used monolayer system is represented by human colorectal adenocarcinoma cell line Caco-2 that undergoes epithelial differentiation and forms monolayers(1652) of polarized cells(1653) on porous supports. The process may take up to three weeks, and attempts were made to shorten this period to seven(1654) or even fewer days.(1655) Initial studies, focusing on the uptake of physiologic compounds,1656,1657 were soon extended to the transport setup utilizing the porous supports.(1658) The differentiated monolayer imitates intestinal epithelium and is widely used to assess the oral absorption potential of chemicals.1659–1671 The unstirred aqueous layers affect permeation through the Caco-2 monolayer.(1672)
In several studies, the Caco-2 permeability was shown to be a predictor of intestinal absorption.1673–1681 An approximate sigmoidal relationship between intestinal and Caco-2 permeabilities was observed for a limited set of compounds.1682,1683 The relevance of the results for the prediction of intestinal absorption can be improved through the use of media that imitate the gut content in the apical compartment.(1684)
The throughput of experiments was increased by introducing multiwell systems(1654) and cassette dosing of several chemicals.(1685) The latter modification works well for passively transported chemicals, but saturable and competitive binding may invalidate the results if any of the protein-mediated mechanisms (section ) is involved. The Caco-2 monolayer system was used to study the transport of simple organic molecules (urea and mannitol as the markers of paracellular transport,(1686) creatinine, erythritol, phosphonomethanoic acid,(1687) amines,1686,1688 amino acids,1689,1690 carboxylates,(1686) oxazolidin-2-ones(1691)), adrenergic receptor antagonists,1686,1692,1693 β-lactams,(1694) peptides,1664,1691,1695–1699,1699–1702 peptide prodrugs, (1703–1706) and peptidomimetics,1707,1708 as well as numerous other drugs1683,1709–1714 and drug candidates.1680,1711,1715,1716 The impact on absorption of chemicals of permeation enhancers,1717–1720 excipients for drug dosage forms,1710,1721–1724 and polymeric nanoparticles as drug delivery vehicles(1725) was also analyzed.
Although the Caco-2 cells in monolayer attain several morphological and functional characteristics of mature enterocytes, some differences do exist, primarily because of the colonic origin of the cell line. Those most important for the transport studies include very low expression of cytochrome P450,(1726) especially that of the 3A4 isoenzyme that is important in drug metabolism,(1651) a lower content of efflux pumps and transporters for active absorption of vitamins, bile acids, amino acids, nucleosides and peptides,(1727) and tighter cell junctions reminiscent of those in the colon, as opposed to those in the small intestine.1667,1728
Variability of the Caco-2 monolayers, caused by cellular factors and culture conditions, complicates the interlaboratory comparisons. The parental cell line is heterogeneous, and different subpopulations may become prevalent, depending on the cultivation conditions.(1729) Circumstances such as the number of passages,(1730) the nature of support,1731,1732 and the presence of nutrients(1733) and induction factors1651,1706 affect the characteristics of the prepared monolayers. Standardization of the experimental setup, including the cell source, cultivation conditions, and the use of media and solvents, would contribute greatly to the availability of reference-quality permeability data of chemicals, leading to more-general models and increased understanding of the structural dependence of the intestinal transport process.
Depending on the structure of transported chemicals, the Caco-2 monolayer permeation is affected by any or all of the following processes: passive transcellular transport,1692,1734 active transport1733,1735 and efflux,(1697) transport through tight junctions,1688,1722,1736 endocytosis,(1737) transcytosis,(1663) and metabolism.1666,1738 This variety, although providing the opportunity to study any of the involved mechanisms, complicates the correlation of the permeability data with physicochemical properties of transported chemicals. Several attempts have been made to discern the fluxes through individual routes.1686–1688,1692,1728,1739,1740
The polarity of the Caco-2 monolayers provides a simple means to detect active transport mechanisms, especially the efflux mediated by P-glycoprotein. For this purpose, the trans-monolayer fluxes in the apical-to-basolateral direction and backwards are compared. Identity of both fluxes between two identical aqueous phases indicates passive permeation. If the backward flux is significantly higher, energy-dependent, saturable, and the difference is abolished by a P-glycoprotein inhibitor such as verapamil, the efflux probably affects the permeation.1700,1741 Mechanistic studies of active transport are performed preferably in the Caco-2 monolayers, because isolated intestinal enterocytes are fragile and have limited viability.(1742)
Thanks to the importance of oral absorption in the drug development process, there has been considerable interest in the elucidation of the relationships between structure or properties of chemicals and their Caco-2 permeability. The correlations are obscured by the multitude of processes, which contribute to the overall outcome of the transport experiment. Obviously, any mechanistic knowledge is of great value for the development of reliable descriptions. Unfortunately, this knowledge was mostly used for explanation of outliers,(1691) rather than for a systematic classification of the permeants for the correlations. In addition, the analyses are complicated by the fact that the determined permeability coefficient is not a complete characteristics of the transport process (section ), and the information about the lag time(1743) and accumulation is not considered.
The simplest models were applied to the data for compounds permeating without the use of protein carriers (i.e., by passive transport through enterocytes or through tight junctions). The former process received more attention because it can be applied to the majority of tested chemicals. The descriptors used in mostly linear correlations of permeability were the 1-octanol/water partition coefficients (P) for neutral molecules, the distribution coefficient representing the P values for ionized molecules at the pH of experiment,1691,1743,1744 the number of hydrogen bonds (1695) or more quantitative hydrogen-bond parameters,(1745) polar surface area,(1746) as calculated from all low-energy conformations,(1747) from 1000 conformations obtained by an MD simulation,(1748) from a single conformation,(1749) or by a fast fragment-based method,(1127) molecular size,1702,1743–1745 charge,1702,1712,1743,1744 and the partition coefficients for IAMs (section ).536,1702,1707,1711 For several peptide series, the 1-octanol/water distribution coefficient Pi was not a good predictor of permeability, especially if the transport was significantly affected by efflux.(1700) The correlations were mostly linear, with an exception of a sigmoidal relationship on log Pi, shifted by the contribution of paracellular transport parametrized by molecular weight.1743,1744 Similar but more-scattered sigmoidal relationships were observed for the log Pi values measured in the n-hexadecane/water and PGDP/water systems for 51 diverse drugs.(1683) Numerous descriptors (surface-related,1750–1752 hydrogen bonding with log P(1128) and charges(1679)) were simultaneously correlated as a linear combination using PLS or multiple linear regression with the permeability of diverse compounds, demonstrating a large negative impact of hydrogen bonding, and positive impacts of lipophilicity and sometimes polarizability. A linear combination of a hydrogen-bond descriptor, log Pi, molecular weight, and an indicator variable for charge was used in a permeability−property correlation.(1679) Volsurf descriptors were correlated with Caco-2 permeabilities using the PLS655,1140–1142 and PCA(1136) methods. The 3D-QSAR approaches, which are usually suitable for structure-specific protein-mediated interactions, were applied to the passive permeation of 38 inhibitors of rhinovirus replication.(1753) Detailed simulations were performed for passive uptake and permeation of cells(1754) and cell layers,(1755) respectively, as affected by physicochemical properties. The models accommodated the ionization of chemicals and varying acidities of individual compartments, but they did not include accumulation of chemicals in the bilayers and used rather simplified relationships between the permeability coefficients and lipophilicities.
The cooperation between P-glycoprotein (section ) and CYP3A4 (section ) at the apical membrane in creating a defense mechanism to prevent the entry of some chemicals into the body is an interesting concept that has important consequences for oral administration of drugs.1756–1758 The idea is supported by similar broad substrate specificities of both systems. To improve the suitability of the Caco-2 monolayers for studying this mechanism and intestinal metabolism in general, the CYP3A4 expression can be elevated using vitamin D31651,1706 or other inducers. The metabolizing enzyme levels are generally higher in earlier passages of the cells in the process of preparing the monolayers.(1730) However, if vitamin D3 is used for CYP3A4 induction, the cultures with higher passage numbers contain more enzymes.(1759) The Caco-2 cells transfected with CYP3A4 represent an alternative option.1760–1762 Other cell lines forming the polarized monolayer and having a high expression of both transporters and enzymes can be used to study their cooperation.(1726)
Among other than Caco-2 cellular monolayers, the Madin-Darby canine kidney (MDCK) cell line(1763) has been deployed most frequently. The initial focus on tissue cultures that were suitable for the monitoring of viral propagation,1764,1765 oncogenesis,(1766) and physiological processes1647,1767 was extended to the monolayers grown on a porous support, which were used to study the transport of drugs1768,1769 and ions, imitating tubular reabsorption.1770,1771 The MDCK cells, transfected with CYP3A4, are available.(1762) Transport studies using MDCK cells have been performed less frequently than those with Caco-2 monolayers. The QSAR studies of drug(1772) and peptide1773,1774 permeation through the MDCK cell monolayer are of rather preliminary character.
Other studied cellular monolayer systems include intestinal cell lines HT29-18-C1(1775) and IEC-18,(1776) as well as 2/4/A1 cells with reduced transporter activities(1777) for the study of intestinal absorption, LLC PK1 renal epithelial cells(1778) as an in vitro system for studying tubular reabsorption, brain microvessel endothelial cells1779,1780 imitating the blood-brain barrier, and Hep G2 cells(1781) for studying aspects of hepatobiliary clearance. The ability of the human intestinal goblet cell line HT29-H to produce mucus was deployed to assess the barrier properties of the mucus.(1782) Diffusion of drugs in the mucus was analyzed and compared to that in water.1783,1784 Cellular systems imitating skin(1785) did not provide relevant absorption data,1786,1787 but may find a more successful application in reconstructive surgery.(1788) The use of these monolayers was much less frequent than that of Caco-2 cells and no QSAR studies were performed.
3.5. Biological Sheets
Studies of transport through planar biological systems consisting of several different cell layers are briefly summarized in this section. The sheets include human, porcine, rhesus monkey,1789,1790 and mouse skin,1791,1792 dog and cat pericardia,(1793) the walls of the intestine(1794) and urinary bladder,(1795) frog skin, goldfish gills,(1796) and other planar objects that are suitable for the study of the transport of chemicals, because they can be mounted as barriers separating two media in diffusion chambers of Ussing1797,1798 and Franz1799,1800 designs. An air-lift system normally is used to provide a proper gas balance, and stirring is applied to reduce the diffusion barrier thickness and ensure homogenization of bulk media. Transport through the skin and the small intestine, although not fully representative of the real situation, are by far the most frequently studied processes in this category. The majority of the experiments with other sheets was made in the first half of the last century, focusing on general information about biological transport without placing much emphasis on the practical importance of the sheets that were used, such as frog skin,1801–1803 toad urinary bladder,(1795) or goldfish gills.(1796)
In quantitative studies, the transport was characterized by the permeability coefficients PC. Several relationships for the dependence of the PC on the structure of transported chemicals are noted below. They usually make simplifying assumption about the structure of the studied sheet. The expression that takes the multilamellar sheet structure into account is given as eq 51 in section .
3.5.1. Skin
Transdermal absorption can be studied in various skin preparations, which provide a more realistic picture of events than the phospholipid systems (section ); although some steps are still missing and some may be superfluous (section ). The process was studied in subcutaneous rat tissue(1804) and hairless mouse skin,1791,1792 as well as in human skin obtained from amputations and cadavers. Macroscopic dimensions allow mechanical separation of individual skin layers,1805,1806 tape stripping,(1807) sectioning of the skin layers at different depths,1808,1809 and visualization of permeant positions in the cross sections.1809–1812 The main barrier for percutaneous absorption of most chemicals is the stratum corneum1813–1815—the surface lamina consisting of several layers of corneocytes, which are embedded in a lipid matrix represented by multiple bilayers separating the cells.161,1816–1819 The corneocytes are filled with water and microfibrillar keratin, and they are surrounded by a keratinized envelope,1820,1821 which is covered by a monolayer of covalently bound lipids(1822) that play a role in cell adhesion.(1823)
Chemicals could permeate the stratum corneum via diffusion through the corneocytes,(1824) the voids in continuous lipid bilayers (as described in section ), aqueous pores resulting from either imperfections in lipid bilayers163,397 or reorganization of the bilayer forming the transitional opening lined with the headgroups,1825,1826 and shunts represented by hair follicles and sweat ducts, as well as by lateral diffusion along the plane of the bilayer.(163) Relative contributions of these parallel mechanisms to transdermal transport of a specific chemical are dependent on its physicochemical properties, although the latter mechanism is considered dominant for drugs.(1820) Impermeable keratinized corneocytes in the topmost layers of the stratum corneum impede the transport by creating a tortuous diffusion path for the lipid-based mechanisms. This fact makes percutaneous absorption a unique process, with the paracellular mechanism prevailing for most chemicals. The rate of absorption can be increased using chemical enhancers(1827) or iontophoresis.1828,1829
The importance of the transdermal route of administration for some drugs(1830) led to the development of modeling approaches for single compounds1805,1831,1832 and QSARs for sets of chemicals. Simple rules regarding molecular weight, aqueous solubility, melting point, and the number of hydrogen-bonding atoms were formulated to classify the permeants as fast and slow.(1833) Additive contributions of structural fragments to transdermal permeability were determined.(1834) For chemicals traversing the skin via the lipid-based paracellular route, the permeability coefficient was expressed as the product of the partition coefficient and diffusion coefficient,(1813) as known for the solubility-diffusion mechanism for transport through a single bilayer (section ). The simplest property-based QSARs reflect this fact and describe the permeability coefficient as a power function of the partition coefficient (see the Collander equation 1 in section ) in surrogate systems,(1835) including the stratum corneum/water system,(1836) for compounds of similar sizes. For more-diverse compounds, the fits improved via the addition of molecular weight,1837,1838 molecular volume,(1839) hydrogen-bond parameters,1836,1840,1841 and atomic charges,(1842) which should characterize the structural dependencies of the diffusion coefficients. Importance of the difference between logarithms of 1-octanol/water and alkane/water partition coefficients, formally equal to the 1-octanol/alkane partition coefficient and characterizing hydrogen bonding (section ), was also examined.(1843) Acceptable correlations for heterogeneous chemicals were obtained using Abraham’s solvatochromic equation.1844–1846 The interesting observation that asymmetric positions of hydroxyl groups in polyphenols led to a slower transdermal transport than a symmetric distribution(1847) might be related to the role of amphiphilicity of the asymmetric molecules in the trans-bilayer transport (section ). Complexity of the process was indicated by the need to break down the sets of compounds to obtain good linear relationships.(1848) Additional equations were summarized in detailed reviews of simple QSAR approaches to transdermal permeability.1838,1849
More-detailed models that take into account the structure of the stratum corneum and tortuosity of the lipid route were developed,163,1850 describing not only the steady-state transdermal flux but also the lag time. Simplified two-dimensional (2D) models164,1851,1852 were extended to more-realistic 3D representations. The presence of several pathways, each traversed to the extent given by the properties of chemicals, was considered.1853–1855
3.5.2. Small Intestine
The second-most frequently studied sheet was the small intestine, thanks to the importance of intestinal absorption for chemotherapy. Experiments in this area started at the beginning of the last century.1856,1857 The intestine was used in the form of everted sacs(1858) or mounted as a barrier separating two solutions in side-by-side diffusion chambers.1859–1863 The latter setup utilized either the entire intestine1864–1866 or mucosal sheets.1867–1873 The viability of the tissue must be carefully monitored.(1874) The in vitro systems were used to study the absorption of small sets of chemicals.1687,1868,1875–1877 Other intestine-based test systems include brush-border vesicles (section ), isolated enterocytes, and the rings of the entire intestine (section ), which are used to measure the uptake of chemicals, as opposed to the transport across the intestine.
Intestinal absorption of a chemical requires that the molecules cross only a small part of the intestinal structures, namely, from enterocytes to blood capillaries and lymphatic capillaries (lacteals) embedded in the villi of the mucosa, not the entire intestinal wall. Therefore, the relevance, for intestinal absorption, of the in vitro studies of transport through the complete intestinal wall is questionable. The use of mucosal sheets isolated from the small intestine only reduces the presence of the intestinal structures, which do not need to be crossed by absorbed molecules, but does not solve the problem. On the other hand, the intestine-based systems allow the study of the effects of unstirred layers(1878) and intestinal metabolism, as well as a comparison of absorption in different segments of the intestine.(1676)
The QSAR studies of intestinal absorption usually utilize the convenient Caco-2 monolayer permeabilities (section ) or the more-relevant in situ and in vivo data (section ), and the data obtained in the in vitro intestinal preparations are used less often. The permeabilities of a small set of diverse drugs in rat intestinal sacks were analyzed using the linear regression analysis and neural networks with the retention factors on the IAM columns (section ), molecular volume, and other physicochemical properties as the descriptors.(1879) Human jejunum permeabilities were correlated with the polar surface area of a small set of permeants.(1127)
3.6. Tissues
In SBSP, tissues are viewed as spatially arranged collections of similar cells that contain some extracellular fluid. Historically, the first studies were performed with the tissues containing natural pigments changing colors in response to the internal pH. Examples include the pieces of mantle edge of the hermaphroditic nudibranch Chromodoris zebra,(1584) and the testis filaments of a sea cucumber.(1583) The testis and intestines, after being expulsed in a defense act or harvested, regenerate upon the return of the animal to sea water. The importance of the complete dose-response curve in the tissue transport studies, as opposed to the measurements at a single concentration, for the characterization of relative transport rates of individual acids was noted.(1584) Permeabilities of acids and bases were measured using the flower petals containing pH-sensitive pigments.(1585) With the availability of radiolabeled chemicals, tracking the distribution at the levels of tissues, organs, or organisms was made possible by autoradiography.1596–1604 The first attempts at monitoring the distribution in entire animals were made using natural isotopes.(1880) Further developments of autoradiography at cellular level are described in section . Tissues have been used in two forms: slices for the uptake and metabolism studies1881,1882 and tissue homogenates for equilibrium binding studies.(1883)
The most frequently used tissue slices are the fresh or vitrified precision-cut slices from the rat liver, which were used to study metabolism of chemicals.1881,1882 Metabolism in these systems differs from that in hepatocytes, because of the diffusion limitations in the former system.(1884) This phenomenon could provide a basis for the analysis of the distribution process. The data from both slices and hepatocytes were used for prediction of in vivo metabolic clearance.(1519) Brain slices were used to analyze metabolism(1885) and the mechanisms of learning and addiction.1886–1889
Tissue homogenates were first used by anesthesiologists who wanted to understand the partitioning of anesthetic gases between blood and tissues, especially the brain.(1883) The technique was also used in occupational toxicology to assess the exposure risks of industrial chemicals(1890) and in environmental toxicology to understand disposition of chemicals in fish.(1891)
Depending on the structures and properties of chemicals, the transport rates into tissues vary over a broad range. For quickly transported chemicals, the distribution is characterized by the tissue/plasma or tissue/blood partition coefficients, which are measured using tissue and blood samples. For slower transport, which is observed most frequently in perfused organs, more-sophisticated, nonequilibrium descriptions are required. In the following, we will focus on equilibrium binding to tissue constituents.
For compounds quickly transported through membranes, tissue/plasma partition coefficients (PTP) are important characteristics, which are defined as the ratio of the average concentration of a chemical in the respective tissue and the concentration in plasma. The PTP values represent pseudo-equilibrium inputs in physiologically-based pharmacokinetics (PBPK) models of quickly transported chemicals. The relation between PTP and elementary processes is given by eq 57 or eq 58 in section . A sum of the PTP values for all tissues in the body, each weighted by the tissue volume, is equal to the volume of distribution.(1892) Therefore, the product of the PTP value and the volume of the tissue can be viewed as the distribution volume of the tissue.
Historically, the PTP values were first measured(1883) and utilized in QSAR studies for simple chemicals such as rare and other gases (H2, N2, CO, CO2, NO),(1893) gaseous narcotics, alkanes,(1894) and alcohols. These data correlated well with properties of the chemicals,1895–1897 especially using a model-based nonlinear function of lipophilicity expressed as a surrogate partition coefficient (section ).(1898) Several approaches were developed for modeling the data for more-diverse compounds.
The solvatochromic equation, which describes solvation-related phenomena as a linear combination of solute excess molar refractivity, dipolarity/polarizability, hydrogen-bond donor and acceptor abilities, and McGowan characteristic volume, was frequently applied to PTP or similar parameters.1119,1121,1124,1125,1846,1899–1904 Generally, the correlations were worse than those for one-phase(1905) or two-phase systems,1906,1907 for which the solvatochromic equation was originally devised. In addition to more-pronounced data variability in biological samples, there are two other potential sources of error. First, protein binding for diverse sets cannot be described as a structure-nonspecific process, using the same approach as for solvation and partitioning. Second, the equation that is used applies to one- or two- phase systems and cannot be used for tissues that contain multiple phases. The correlation equation should contain similar terms as eq 57 or eq 58 in section , and the solvatochromic equation should be applied to each phase separately. These changes would make the correlation equation more complex and nonlinear in optimized coefficients, but the number of independent variables would not increase. The situation is even more complicated for the tissue/blood partition coefficients, which were also frequently modeled using the solvatochromic equation.1901,1902 As compared with plasma, the blood contains additional phases in the corpuscles.
A series of publications advocated the use of the vegetable oil/water partition coefficients in modeling the tissue distribution.665,1908,1909 Vegetable oil, although composed mainly of triglycerides, contains other lipid species, varying by the source and preparation conditions, which may significantly affect the partitioning process. A better-defined triglyceride phase, such as triolein192,562,563 or trioctanoylglycerol (see section ) would help standardize the results and provide a good surrogate for adipose tissue. The partitioning to phospholipids was treated as if they were composed of 30% vegetable oil (triglycerides) and 70% water. This combination does not seem to create a good surrogate system, because the fatty acid chains differ in structure from triglycerides and water is not a good surrogate for the headgroup region, despite its high water content (see section ). The estimates of the accumulation in neutral phospholipids, assuming the 30:70 lipid/water composition, were also used in otherwise-sound models for tissue partitioning of basic drugs.(1910) Later, in a study of the volume of distribution,(559) vegetable oil was replaced by 1-octanol for nonadipose tissues. Interactions with proteins were treated by using the measured fraction unbound in plasma,(1911) and assuming that a similar protein binding is taking place in tissues. Albumin, although present in tissues, does not represent their major component (sections and ). Consequently, this assumption could affect the descriptive and predictive abilities of the models. The used equations are mostly conceptual and similar in form to eqs 57 and 58. A major drawback is the use of the implausible assumption regarding the proportionality or even equal magnitudes of the partition coefficients in surrogate solvents and biological systems. An application of the Collander equation to relate the partitioning in surrogate and biological phases via adjustable coefficients usually provides better results.
Conceptual equations reminiscent of eqs 57 and 58 were also used for the correlation of the tissue/blood partition coefficients of both neutral(1912) and ionizable compounds.1913–1915 While the tissues were decomposed into lipids, proteins, and water, the blood was considered as a homogeneous phase, thus neglecting the partitioning into the membranes of blood corpuscles and lipoproteins, as well as plasma protein binding. This omission could affect the results, especially for compounds with higher affinities to blood components. Extrathermodynamic relationships were used to describe the partition coefficients and proteins association constants. In the latter case, this description could be problematic for more-diverse compounds.
The approach was further refined by inclusion of neutral lipids, as well as neutral and acidic phospholipids, and was applied to tissue/plasma partition coefficients.(543) The experimental values of fraction unbound in plasma were used to describe protein binding in plasma and also in interstitial fluid, which has a similar protein composition. The binding to intracellular proteins was correlated with experimental partition coefficients for phospholipids,(1916) which limits the application to the studied chemicals.
All published approaches to the correlation of tissue partitioning of chemicals with their structures and properties treat the interactions with proteins as structure-nonspecific processes, which are related to the properties of entire molecules. This assumption limits the applicability of the techniques to sets of homologues. For larger and more-diverse sets of chemicals, protein binding must be treated as a structure-specific process, using 3D-QSAR techniques, as we recently demonstrated for the binding to the proteins of extracellular matrix (section ).1307,1309
3.7. Organs
When transport rates are comparable with those of metabolism or excretion, the distribution models become more complex and require kinetic data for characterization. These experiments are usually performed in perfused organs, because they have longer lifetimes than isolated tissues. Perfused organs such as frog legs;(1917) frog, cat, and dog hearts;1918–1921 dog livers,1922–1927 and rabbit and pig ears,1928,1929 have been used to study the effects of physiological molecules since the beginning of the last century. Currently, the organ preparations are used in situations when simpler models provide a very limited picture of the disposition events. Typical examples are the skin and intestine, which can be mounted as sheet barriers in the diffusion chambers (sections and ), but the results are not completely representative of the situation in vivo. In both cases, more rigorous results are obtained with perfused preparations, which better imitate the actual transport routes. On the other hand, the cost and labor of these preparations limits the number of available data.
Organ preparations can be used in either ex vivo or in situ conditions, the difference being the disconnection from the natural bloodstream of the test animal in the former case. The ex vivo setup allows a better control of the vascular perfusion but often results in a shorter lifetime of the preparation. The in situ preparation preserves a natural environment of the studied organ and maintains many in vivo features, while providing controlled inputs and outputs, which allow application of precise analytical techniques and the use of the mass balance in the description.
Intestinal absorption rate is one of the key parameters of orally dosed drugs. The absorption route from the intestinal content through the epithelial cells and connective tissue into the capillaries inside the villi cannot be precisely duplicated in in vitro intestine-based systems, such as the brush-border membrane vesicles (part ), isolated enterocytes, everted rings of the entire intestine (section ), and the entire intestinal wall or mucosal sheets (section ). Instead, it requires the use of comparatively complex ex vivo and in situ preparations, or in vivo conditions. The ex vivo, isolated intestine with adjacent vasculature, perfused both luminally and vascularly though the superior mesenteric artery and the portal vein, provides a better control of the experimental conditions, although the viability is shorter.1930–1934 A preparation of the ex vivo perfused rat intestine and liver was described.(1935) Different models were developed and evaluated for the description of absorption from the perfused intestine.(1936) A structure−permeability relationship was described for steroid absorption in perfused rabbit ileum.(1937)
A similar controversy between simple in vitro systems and reality applies to transdermal absorption, because the bilayer stacks of skin phospholipids (section ), cell monolayers (section ), and sheet-like skin preparations (section ) do not capture all details of the in vivo situation. To reproduce the absorption route from the surface of stratum corneum into the capillaries, several perfused skin preparations were described,1938–1942 of which the perfused skin flap technique for pigs,1943–1946 weaning pigs,(1947) or horses(1948) received the most attention. Small regions of skin, sometimes with tumors,(1947) which are primarily perfused by a single superficial artery, are incised and formed into a tubelike shape around the artery. Later, the artery is cannulated, the flap is removed from the body and placed into a tissue chamber, where the venous perfusate is collected and analyzed. Specialized models for the description of the recorded data have been developed for single chemicals(1949) and mixtures.(1950)
In situ preparations, where the artificially perfused organ remains attached to the systemic circulation, include the rat intestine,1864,1951–1959 kidney,(1960) and liver.1961–1969 The small intestine can be perfused in situ in humans, using stoppers to define a perfused segment.(1970) The externalized rat kidney was used for a direct monitoring of renal clearance of fluorescent compounds.(1971) The methods utilizing perfused organs have been used extensively in the drug-development process.1636,1972–1977
Intestinal permeability in rats was studied using in situ techniques for small sets of compounds,(1978) and a sigmoidal relationship on lipophilicity, containing a molecular weight term, was formulated.(1979)
Quantitative studies of distribution in terms of structure and properties of compounds are limited to small sets of homologous chemicals,1967,1980–1986 because of the cost and labor associated with the use of perfusion techniques. Because of the importance of the process in the metabolism of chemicals, distribution in the liver has been modeled using advanced techniques, which take into account spatial differences of the compound’s and enzymes’ concentrations in individual organ parts.1638,1961,1962,1964,1987–1996 Interestingly, the heterogeneity at the cellular level, which is caused by different accumulation in the membranes and aqueous phases, was not considered, although the importance of the binding to endoplasmic reticulum for the overall metabolism rate has been recognized.(1472)
3.8. Organisms
Historically, the first technique that allowed the distribution of chemicals at the level of organisms to be tracked was autoradiography,1596–1604 which was initially performed using natural isotopes.(1880) In addition to autoradiography,1603,1997–1999 the distribution of chemicals in entire organisms can be directly and noninvasively monitored by magnetic resonance imaging based on 7Li,(2000)13C,(2001)19F,(2002) and proton nuclei,2003–2006 positron emission tomography,(2007) and specific imaging techniques for fluorescent compounds in nude mice.2008–2012
Indirect techniques, based on the extraction of chemicals from the body fluid and tissue samples, have been used much more extensively, especially in the areas of drug development and environmental toxicology. The resulting data have been processed by pharmacokinetic modeling methods. A brief overview of the development of these approaches(2013) is provided below.
Pseudo-equilibrium accumulation of a chemical in an aquatic organism2014,2015 is usually characterized by the bioconcentration factor (BCF),(2016) given as the ratio of the chemical concentrations in an organism and the surrounding aqueous phase. BCFs were frequently and successfully correlated with the reference partition coefficients,1891,2017–2027 aqueous solubilities,2017,2021,2023 and acidities.(2028) For very lipophilic compounds, nonlinearities were observed in the relationships with lipophilicities2029–2033 and reactivities.(2034) Nonlinear dependences on the reference partition coefficients were also observed for the uptake and release rate constants.60,2035,2036 Clearances were inversely related to the reference partition coefficients.2024,2035 For reactive chemicals, the toxicities were conceptually correlated with the reference partition coefficients and reactivies.(1478) Because the chemicals studied in environmental toxicology usually have much simpler structures than drugs, research in this area contributed several interesting ideas to SBSP.
The disposition of chemicals in animals and humans has been studied mainly in relation to chemotherapy. The history of classical pharmacokinetics is summarized in Figure 12, and more details are given in the following text. The notion of a relationship between an anesthetic effect and the concentration of ether in the brain dates back to the middle of the 19th century.(2037) The kinetics of disposition of chemicals has been studied quantitatively in animals and humans and documented since the second decade of the 20th century.2038–2048 During the same time period, the Michaelis−Menten description of saturable kinetics(2049) and the one-compartment model(2050) were developed. The 1930s brought the two-compartment models(2051) and the key concepts of the mean residence time,(2052) clearance,2053,2054 and the volume of distribution.(2055) The one-compartment model and two-compartment models were later applied to the disposition kinetics of drugs2056–2058 and radioactive substances2059–2064 in the organisms. Since the 1950s, kinetic models of multiple dosage regimens2056,2065–2068 have had a broad impact on how therapeutic interventions are performed. The research area was called pharmacokinetics(2058) and developed(2013) into a key pharmacy discipline(75) that is taught in all pharmacy programs around the world. More-detailed descriptions of the fates of chemicals in the body, the PBPK models, were spearheaded by physiologists, who routinely monitored the blood flows and the concentrations of oxygen, carbon dioxide, and anesthetic gases in individual organs.(2069) The compartments in PBPK models are individual organs or their specified sets, in contrast to the models of classical pharmacokinetics, which utilize loosely defined compartments, the number and function of which is dictated by the fit of the model to the data. Principles of PBPK modeling were formulated in the 1960s.2070–2073
Figure 12.

Milestones in the history of classical pharmacokinetics. More details and references are given in the text.
The pseudo-equilibrium models of SBSP in section provide useful descriptions for clearance, the elimination rate, and the volume of distribution. The QSAR models for individual pharmacokinetic parameters are summarized and compared with the pseudo-equilibrium models in sections −.
4. Disposition Function in QSAR
The fates and effects of chemicals in biosystems are governed by a myriad of interactions. As outlined in section , some interactions can be described using global, conformation-averaged properties of the molecules, and the descriptions of other interactions require the knowledge of precise conformations. SBSP attempts to sort the interactions, lump together kinetically similar processes, identify the rate- and extent-determining steps, and systematically formulate model-based QSARs and their time-dependent forms (QSTARs, where T represents time), which we call the SBSP models. The SBSP models serve two main purposes: prediction of distribution and/or bioactivity of untested chemicals in biosystems, and elucidation of the mechanism of action. In the drug development process, the predictions are used in lead finding and mainly in the optimization of lead structures by derivatization.
Regardless of the size and complexity of the biosystem, formulation of a model-based QSTAR for an effect of chemicals requires appropriate descriptions for three steps of action of a chemical as shown in Figure 13: (i) disposition in the receptor surroundings, (ii) the interaction with receptor, and (iii) transformation of the receptor modification into biological response. In most cases, the loss of the chemical caused by its interaction with the receptors is negligibly small. Consequently, step (i) can be described separately. Linearity of the processes is an important concept that greatly simplifies the description of the fates of chemicals in biosystems. Linear (or, in other words, first-order) processes have the rate and/or extent proportional to the concentration of the chemical (denoted as [C]). Linearity is a plausible approximation, because the concentrations of chemicals in living biosystems are generally low. If the processes that control the distribution (such as transport, membrane accumulation, binding to body constituents, and elimination) are linear, the time course of [C] in the receptor surroundings can be expressed symbolically as
where c0 is the initial concentration, proportional to the dose, of the chemical in the entry compartment, and A(pp,t) is the disposition function, with the time of exposure (t) and physicochemical properties (pp) of both the chemicals and the biosystem as variables.(143) The term “disposition function” has been used in classical pharmacokinetics,(2074) in a sense similar to that illustrated by eq 6, but without the property variables. The actual form of the disposition function is dependent on the complexity of the studied biosystem and on the properties of analyzed chemicals.
Figure 13.

Phases of drug action. Under certain conditions (details in the text), the phases can be described by separate models, with little feedback from the subsequent process, as indicated by the dotted backward-facing arrows. Structure-based subcellular pharmacokinetics explicitly addresses disposition; however, the results are used to formulate the descriptions for the other two phases.
Using eq 6, the kinetics of the interaction between the receptor and a chemical can be described for various mechanisms of this step.54,143,2075 For the simplest mechanism of the chemical−receptor interaction that (i) consists of a fast and reversible 1:1 interaction, (ii) is characterized by the association constant K, and (iii) leads to a primary effect that (iv) is an immediate consequence of the receptor modification and (v) is proportional to the fraction of the occupied receptors,(143) the effects are related to K and the disposition function as follows:
The predefined relative intensity of the chemical’s effect (X) is a number between 0 and 1 (frequently, X = 0.5). The experimentally determined isoeffective concentration cX, eliciting the fraction X of the maximum effect, serves as a measure of the potency of individual chemicals. Equation 7 can be easily converted to a model-based QSTAR equation, if the chemical−receptor association constant and the disposition function are expressed via the structures and physicochemical properties of the chemicals. The bioactivity directly mirrors the disposition of a set of chemicals in the receptor surroundings, if K in eq 7 is invariant for all studied chemicals. In the rest of this communication, when comparing the derived form of the disposition function with biological effect data, as opposed to the disposition data, we will assume that these conditions are met, and that bioactivity and disposition are proportional at each moment. The relationships correlating potency with the chemical−receptor association constant K and the disposition function A(ppi,t) for more-complicated mechanisms of the chemical−receptor interaction can be found elsewhere.54,143,2075
The form of the disposition function is heavily affected by the size and complexity of the biosystem, as well as by the kinetics and types of the interactions of chemicals with the components of the biosystem. The construction of the disposition functions for various scenarios is described in the following parts, starting with simple cases, and gradually progressing to more-complex situations.
5. Numerical Simulations for Multi-Bilayer Systems
Some of the simplest model systems that imitate small biosystems are represented by a series of the alternating aqueous phases and the bilayer cores, in which the chemicals, which do not interact with the headgroups, are transported passively. Movement of molecules in a multimembrane system can be envisaged as a series of partitioning steps through alternating aqueous and lipid compartments,(2076) as shown in Figure 14. Protein binding and elimination that influence the disposition of chemicals are also depicted. The majority of experimental systems used for the monitoring of the interactions with chemicals are actually multimembrane systems: some subcellular organelles; suspensions of cells from organisms and some microorganisms, such as gram-negative bacteria, yeasts, and fungi (section ); cell monolayers (section ); and various physiological sheets that form the walls of the blood capillaries, urinary bladder, gallbladder, stomach, and the intestine (section ).
Figure 14.

Schematic outline of the distribution of the chemicals, which do not interact with the headgroups, in a morphologically compartmentalized system consisting of alternating aqueous phases and membranes (represented by the cores). The chemicals can be present as free non-ionized molecules (Ci), free molecules ionized to the jth degree (Cij), or ion pairs with the kth counterion (CZijk); each species can be bound to proteins (P) or enzymes (E), and eliminated by spontaneous and/or enzymatic reactions. The subscripts i, j, and k are typically used throughout the paper, in the meanings that are shown here. Two-sided arrows represent fast processes, whereas one-sided arrows represent time-dependent processes.
The approaches to the description of the disposition of chemicals via the disposition function, defined by eq 6, can be classified as numerical simulations and explicit descriptions, and are summarized in sections and , respectively. Note that all the results presented below are valid for chemical disposition following a single dose, except those in section , which is devoted to continuous dosing. The situations after repeated doses would be completely different and could be described by applying the principle of superposition, using the results for the single dose, as known in classical pharmacokinetics.2056,2065–2068
To understand the contributions of the individual processes to chemical disposition quantitatively, it is advantageous to examine them separately.
5.1. Pure Transport
From the viewpoint of chemical distribution, biosystems are generally open, because of irreversible elimination processes. Under certain conditions, however, the concept of a closed biosystem is useful. Providing that the reversible processes (transport and protein binding) are much faster than irreversible elimination, a closed model properly represents the in vivo situation for the time interval until elimination becomes observable. Among the processes summarized in Figure 14, transport has long been studied intensively as the primary cause of nonlinearities in the concentration−lipophilicity profiles of chemicals.(2077)
Hansch and Fujita(56) assumed, with remarkable intuition, that the probability of the occurrence of molecules inside a biosystem after a predetermined time interval could follow a Gaussian distribution, with respect to lipophilicity, expressed as the logarithm of the 1-octanol/water partition coefficient, P. The resulting parabolic dependence of the intracellular concentration or bioactivity on lipophilicity,
proved to be rather robust and usable. Here, c is the actual concentration in an intracellular compartment, cX is the isoeffective concentration that characterizes the bioactivity, and A, B, and C are adjustable coefficients that are optimized by linear regression analysis. Coefficient A is usually negative, so the function has a concave shape, with the maximum marking the optimum lipophilicity. Hansch and Clayton(6) compiled almost 200 parabolic dependencies of bioactivity on lipophilicity. A searchable QSAR database that includes 553 concave dependencies is available.(1)
The first mathematical treatment of the distribution versus properties problem was performed by Penniston et al.(58) Because the experimentally verified dependencies of the transfer rate parameters li and lo (Figure 7 in section ) on the partition coefficient (eqs 3 in section ) were not available at that time, the authors used the assumption lilo = 1. Their system consisted of the aqueous and lipid compartments with identical volumes and interfacial areas. The intuitively assumed fast equilibration of chemicals in the compartment volumes proved to be one of the most important simplifying steps in the SBSP modeling. This assumption is justified by the small distances (x), which the molecules of chemicals cross in the subcellular compartments. The typical distance, considering the average cell size, is smaller than δ = 10−5 m and reaches the nanometer dimensions in the bilayer headgroup and core regions (see section ). The diffusion coefficients of the low-molecular-weight compounds are of the order of 10−9 m2/s in most liquids.(2078) Fluorescence recovery after photobleaching and other experiments showed that small molecules and macromolecules up to 100 kDa exhibited diffusion coefficients in the cytoplasm that are only 4−5 times smaller than those in saline.(2079) The time t that the molecules require to cross the small subcellular aqueous compartments then can be estimated(2080) as t = δ2/(2D), i.e., t = 0.2 s, under these conditions. The equilibration times inside the bilayer regions are shorter by several orders of magnitude. The equilibration process in the miniature compartments is practically instantaneous on the timescale of most experiments. This fact warrants the representation of a biosystem by a set of alternating aqueous phases and membranes, with the molecules of chemicals diffusing quickly in bulk water and in the bilayer headgroup and core regions. The three phases have much lower density than subregions 2 and 3 in the bilayers (section , Figure 3). The stochastic kinetic effects of the crowded environment(2081) seem to be far more important for larger macromolecules than for chemicals. The differential equations for the fast intracompartmental equilibration, similar to eqs 35 in section , were integrated numerically.(58) The concave concentration−lipophilicity dependencies, consisting of two linear parts connected by a curve, observed after a constant distribution period, were interpreted as a support for the already known parabola.(56)
McFarland(59) used a probabilistic approach to describe the movement of chemicals in the Penniston model.(58) For constant transport periods, symmetrical concave concentration−lipophilicity relationships with linear ascending and descending parts were obtained. The relationships were not suitable for fitting to real data, because the maximum concentration was always attained by the chemical with equal affinities for both core and aqueous phases, i.e., having the core/water partition coefficient P = 1.
Kubinyi(64) re-examined the McFarland’s approach and considered the substantial differences in the volumes of aqueous and lipid phases in biosystems. The resulting concave concentration−lipophilicity profiles for aqueous phases were also symmetrical, but they were not restricted in the peak position.(59) Assuming that the receptor binding is also lipophilicity-dependent and making all the coefficients A, B, C, and D freely adjustable, Kubinyi obtained the asymmetrical bilinear equation:
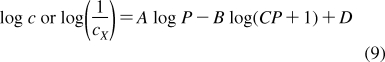 |
Equation 9 has been shown to fit the concentration−lipophilicity profiles generated by numerical simulations of chemical transport with the experimentally measured dependencies of the transfer rate parameters li and lo on lipophilicity, as described by eqs 3 in section . If a reference partition coefficient is used instead of the model partition coefficient, the Collander exponent β must be used with each P term in eq 9, according to eq 1 in section . The magnitude of β determines the sharpness of the curved stretch that joins the two linear parts in the concentration−lipophilicity plots(8) described by eq 9. The ability to describe the simulation results gives eq 9 the stature of a model-based description of the transport, although its original derivation started from rather intuitive concepts. As will be mentioned in sections and , eq 9 also describes the equilibrium distribution. This versatility makes the bilinear equation a valuable tool for describing the relationships between bioactivity and lipophilicity, which was used by many researchers.(30) A linearized biexponential expression that also describes the bilinear dependencies was recently introduced.(2082)
Dearden(65) further developed the Penniston model(58) and drew attention to the importance of the exposure time in the transport simulations and, generally, QSAR. The most-significant results include the behavior of the optimum lipophilicity (log P of the compound with the maximum concentration in the concentration−lipophilicity profiles) in dependence on the exposure time and on the number of the partitioning steps, as well as the dependence of the time to maximal response on lipophilicity.28,46
Van de Waterbeemd(2083) described the transport in a three-compartment model with equal compartment volumes analytically. He extended the results for the initial distribution period to multiple bilayer systems and showed that the concentrations are proportional to the product of the transfer rate parameters of involved steps.2084,2085
Some attempts67,2086 at description of the concentration−lipophilicity profiles were based on the use of a reduced time τ (where τ = lot, with lo being the transfer rate parameter for the membrane−water direction and t being time; see Figure 7 in section ). The reduced time τ is proportional to the real time t only when lo is a constant, which holds for P ≪ 1/Bt, as follows from eqs 3 in section . Thus, the presented results are valid just for hydrophilic compounds.
We examined the complete time course of the chemical transport in closed systems with identical lipid phases.2087–2089 For this purpose, the set of linear differential equations corresponding to the reduced equations described by eqs 47 (section ) was integrated numerically. The transfer rate parameters used are dependent on the partition coefficient as given by eqs 3 with the experimentally determined values(631) of the coefficients At and Bt. The results are illustrated in Figure 15, using the kinetics of the passive chemical transport in a 10-compartment system, composed of five aqueous phases, interspersed with five bilayer cores. Before the start of the simulation, the compounds are present only in the first aqueous compartment. The concentrations are plotted as a function of the reference partition coefficient (P) and the exposure time (t). The kinetics of distribution is dependent on the position and nature of the sampling compartment. In the third compartment, representing the aqueous phase immediately following the first bilayer core, the concentrations of the chemicals initially increase and later decrease to their equilibrium magnitudes, resulting, for a certain time period, in a remarkable accumulation of the chemicals above their equilibrium levels. This phenomenon was observed in the first half of the compartments (i.e., in compartments 2−5 in this case), regardless of their aqueous or lipid nature. The decline of the concentration caused by pure transport, taking the molecules to deeper compartments (no elimination was considered in this case), can be important in biosystems that contain many compartments, such as tissues, organs, and organisms (sections −, respectively). The situation in the second half of the compartments, which are more distant from the site of administration, is completely different, as illustrated by the transport kinetics in the sixth compartment, which is shown in Figure 16. The concentrations asymptotically approach the equilibrium magnitudes, and no accumulation above the equilibrium levels is observed.
Figure 15.

Transport kinetics in the third, aqueous compartment of a 10-compartment system (c is the concentration, the subscript “eq” denotes equilibrium, t is time), relative to lipophilicity.(2089) For compartment numbering, see Figure 14. The chemicals accumulate above the equilibrium level (c/ceq = 1) for a significant fraction of the distribution period. The data were obtained by numerical simulation of the pure transport of the compounds, which do not interact with the headgroups, in a system of alternating aqueous phases (5) and bilayers (5), with the transfer rate parameters li and lo related to the partition coefficient P according to eqs 3.(2089) The surface corresponds to eq 10 with the values of the coefficients specified in Table 5.
Figure 16.

Transport kinetics in the sixth, core compartment of a 10-compartment system, relative to lipophilicty.(2089) The system consists of alternating aqueous (5) and core (5) phases. The concentrations of chemicals do not exceed the equilibrium concentrations at any moment. The surface corresponds to eq 10 with the values of the coefficients specified in Table 5. Other details are as given in Figure 15.
The concentration−lipophilicity−time profiles, similar to those shown in Figures 15 and 16, can be described by semiempirical equations. We call these equations semiempirical, because they currently do not have an obvious mechanistic underpinning. In contrast to empirical equations, our equations are nonlinear in optimized coefficients and provide an almost-perfect fit to simulated data. Polynomials, which are typically used in empirical equations, do not perform well in SBSP models, because they are not able to properly fit planar areas in the surface plots as seen in Figures 15 and 16, and linear stretches in two-dimensional plots (see Figures 17−25, shown in this section through section ). The semiempirical equations represent an important step enabling a straightforward use of the simulations in quantitative characterization of biological data. The excellent fits to simulation data help select a proper form of the empirical equation for fitting the experimental data.
The simulated data subsets with constant P values in Figures 15 and 16 were first fitted by eq 11 (see below), and the optimized coefficients A, B, C, and D were subsequently fitted as a function of P and the exposure time t. This approach resulted in the following empirical equation:
 |
The overall fit to the data simulated for the third compartment of a 10-compartment system, shown in Figure 15, was characterized by the following coefficients. The exponents β1 and β3 were linearly dependent on log P, with slopes of (5.604 ± 4.3101) × 10−2 and (8.607 ± 2.982) × 10−2, respectively, and the same intercept (1.681 ± 0.0129). The values of the other adjustable coefficients, as optimized by nonlinear regression analysis,(443) are summarized in Table 5. All coefficients are statistically significant. The overall fit is very satisfactory, as shown in Figure 15 and by the values of statistical indices: number of experimental points, n = 1212; the squared correlation coefficient (equal to the percentage of explained variance), r2 = 0.992; the standard error of the fit, s = 0.063; and the Fisher test value, F = 18 210. These statistical indices will be used throughout the paper, to characterize the quality of a fit. For comparison, the best typical empirical equation (a polynomial with cross terms) with the same number of optimized coefficients as eq 10, achieved values of r2 = 0.743, s = 0.375, and F = 389.2.
Table 5. Optimized Values of Adjustable Coefficients in eq 10 Describing the Transport Kinetics in Compartments 3 and 6, as shown in Figures 15 and 16, Respectively.
| Compartment 3 |
Compartment 6 |
|||||
|---|---|---|---|---|---|---|
| parameter | value | standard error | t-value | value | standard error | t-value |
| A1 | 18.05 | 0.52 | 34.86 | |||
| A2 | 0.05701 | 0.00186 | 30.64 | |||
| A3 | 0.07373 | 0.00428 | 17.21 | 0.002719 | 0.000171 | 15.87 |
| A4 | 0.002358 | 0.000131 | 17.97 | 0.02691 | 0.00171 | 15.72 |
| B1 | 0.05003 | 0.00241 | 20.76 | |||
| B2 | 0.1407 | 0.0102 | 13.84 | 0.2955 | 0.0097 | 30.35 |
| β2 | 1.565 | 0.0148 | 105.7 | 1.571 | 0.019 | 83.54 |
| β3 | see text | 1.694 | 0.019 | 87.05 | ||
In the sixth compartment, no accumulation above the equilibrium concentrations was observed, as seen in Figure 16. The data were fitted with the second term in eq 10 (see Table 5). The fit was also very satisfactory, as illustrated by Figure 16 and the values of the statistical indices (n = 780, r2 = 0.996, s = 0.029, and F = 49 147). The best polynomial with the same number of optimized coefficients achieved values of r2 = 0.815, s = 0.196, and F = 1731.
The compounds with different lipophilicities reach the equilibrium at different times. Figures 15 and 16 show that the total period of distribution can be subdivided into three parts: the nonequilibrium period (log t < 2.5 in this case; no compounds have reached the equilibrium), the mixed period (2.5 < log t < 5), and the equilibrium period (log t > 5; all compounds have attained equilibrium).
The dependencies of the concentrations or effects on lipophilicity profiles are most frequently measured after a constant exposure time for all chemicals. The profiles are different for individual distribution periods and will be described in detail below. To facilitate the discussion, an empirical equation describing all simulated profiles(2087) is introduced at this point:
 |
Equation 11 describes the multilinear dependencies with up to four linear parts and can be considered an extended form of the bilinear equation described by eq 9. The model partition coefficient was substituted by a reference partition coefficient using the Collander equation 1 in section . The first term on the right side is written in a way that emphasizes the substitution of the partition coefficients. The connection between the adjustable coefficients A, B, and C and the shape of the corresponding curves is outlined in Figure 17. The coefficients A and Bi determine the slopes of linear parts, as shown in the upper panel of Figure 17. The coefficients Ci are associated with the position of the curvatures. The Collander exponent β modulates the sharpness of the curvatures. The upper curve, which is characteristic for the mixed period of distribution, is described using the integer values of the coefficients A and Bi, which are selected from available combinations, summarized in Table 6. The lower curve is typical for open systems. The leftmost and rightmost linear parts still maintain the integer values that are specified in Table 6. However, the slopes of two linear parts that characterize the convex dip are no longer confined to the integer values and, consequently, the coefficients B1 and B2 are freely adjustable. The adjustable coefficients of eq 11, for the dependencies shown in Figure 17 and other cases discussed in sections −, were fitted to the experimental data using nonlinear regression analysis, deploying the Fletcher and Powell algorithm.(2090)
Figure 17.

Meanings of the adjustable coefficients in eq 11 that describes the relationship between the subcellular concentrations c and lipophilicity expressed as the partition coefficients P. The slopes of the linear parts, relative to the coefficients A and Bi, are given in the upper part (all expressions are to be multiplied by the coefficient β). The positions of the curvatures are determined by the values of the coefficients Ci.(2089)
Table 6. Integer Slopes of Linear Parts of the Concentration−Lipophilicity Profiles (Figures 18−20) and the Values of the Coefficients A, B1, B2, and B3 in eq 11 for the ith Compartment during Individual Distribution Periods.
| Coefficients |
||||||||
|---|---|---|---|---|---|---|---|---|
| phase | Slopes(left to right) | A | B1 | B2 | B3 | |||
| Nonequilibrium Period | ||||||||
| aqueous | (i − 1)/2 | (1 − i)/2 | (i − 1)/2 | 1 − i | ||||
| lipid | i/2 | 1 − i/2 | i/2 | 1 − i | ||||
| Mixed Period | ||||||||
| aqueous | (i − 1)/2 | 0 | −1 | (1 − i)/2 | (i − 1)/2 | (1 − i)/2 | −1 | 3 − i/2 |
| lipid | i/2 | 1 | 0 | 1 − i/2 | i/2 | 1 − i/2 | −1 | 2 − i/2 |
| Equilibrium Period | ||||||||
| aqueous | 0 | −1 | 0 | −1 | ||||
| lipid | 1 | 0 | 1 | −1 | ||||
5.1.1. Nonequilibrium Period
The majority of previously published studies by other authors on the description of movement of chemicals through a series of alternating aqueous phases and membranes focus exclusively on the nonequilibrium period of transport when none of the studied chemicals have achieved the partitioning equilibrium. We also simulated this period and compared the results with those obtained in the most advanced studies by Kubinyi.8,64 The results of the simulations for the nonequilibrium period were practically identical, despite different integration methods. However, the interpretation of the results and their application to experimental data were somewhat different. Kubinyi made all the coefficients in eq 9 freely adjustable, whereas, in our approach, a tighter adherence to the simulation results had been chosen. The resulting concentration−lipophilicity profiles for individual compartments are given in Figure 18. For the first two compartments, the curves exhibit the zero slope values in some linear parts. Other curves consist of linear ascending and descending parts, connected with a rounded apical part. The curves are symmetrical for the aqueous phases (odd compartment numbers) and asymmetrical for the membranes (even compartment numbers). The slopes of the linear parts (Figures 18−20 and 22A) are integers, which are characteristic of the corresponding compartments (Table 6).8,2083,2087 This fact significantly promotes elucidation of the action mechanisms of bioactive chemicals, as the shape of the relationship between bioactivity and lipophilicity under the nonequilibrium conditions could indicate the sequential number and the nature of the compartment, where the receptors for the studied bioactivity are localized.
Figure 18.

Nonequilibrium concentration−lipophilicity profiles for individual phases of a 10-compartment system of alternating aqueous and core phases at a constant time of distribution.(2089) Other details as in Figure 15. The compartment numbers are shown near the respective curves. The aqueous phases are denoted by the odd numbers and the bilayers are marked by even numbers, as in Figure 14 in section .
Figure 20.
Concentration−lipophilicity profiles in the membranes of a 10-compartment system of alternating water and bilayer phases in the (A) nonequilibrium (10 time units) and (B) mixed (1000 time units) periods of distribution.(2089) Curves for compartments 2 (the first bilayer, red lines), 6 (the third bilayer, blue lines), and 10 (the fifth bilayer, green lines) are shown, along with the equilibrium curve (black lines). In compartment 2, chemicals accumulate above the equilibrium concentration in both nonequilibrium and mixed periods. The numbering is as given in Figure 14 in section , whereas other details are as shown in Figure 15 in section .
Figure 22.
Concentration−lipophilicity profiles of chemicals in the last compartment of the water−bilayer−water−bilayer system (A) without elimination, (B) with elimination from either both aqueous phases, or (C) with elimination from the intracellular aqueous phase after the following distribution periods (in time units): 0.1 (green), 1 (blue), 100 (red), and ∞ (black).(2089) The elimination rate constants are identical for all compounds and set to zero, except k1 = k3 = 1/(time unit) in panel B and k3 = 1/(time unit) in panel C. Other details are as given in Figure 15 in section .
This phenomenon(2091) is illustrated in Figure 19, showing the dependence of mutagenic activity of N-substituted alkyl amides of 3-(5-nitro-2-furyl)-acrylic acid (59 in Chart 5) after 1-h incubation with two strains of Salmonella typhimurium TA100 (rfa+ and rfa−) on lipophilicity. The strains differ in regard to the structure of the cell walls. In contrast to the rfa− strain, which has an exposed cell membrane, the cells of the rfa+ strain are covered by a lipopolysaccharidic complex that contains the second lipid bilayer, in addition to the cell membrane. The mutagenic effects of nitrofurans are likely caused by unstable metabolites formed in the course of enzymatic reduction of the nitro group.(2092) Because the compounds are n-alkyl derivatives, it is plausible that they do not differ in regard to the electronic properties of the nitro group and, consequently, in the rate of reduction. The data were fitted by eq 11 with i = 1; the optimized coefficient values are given in the first two rows of Table 7 and the corresponding lines are shown in Figure 19. The integer values of coefficients A and B1 in eq 11 indicate (cf. Table 6, nonequilibrium period) that the receptors are located in the fourth compartment of the rfa− strain and in the sixth compartment of the rfa+ strain (cf. Figure 19). The receptor compartment would correspond to the lipophilic DNA-core in both cases, because the cells of the rfa+ strain are covered by two membranes, and the rfa− strain only has the cell membrane.
Figure 19.

Mutagenicity of alkyl amides of 3-(5-nitro-2-furyl)-acrylic acid (59 in Chart 5) against Salmonella typhimurium TA100 rfa+ (full points) and rfa− (open points), as dependent on the 1-octanol/water partition coefficient P. The concentration cX (mol/l) elicits, under given conditions, 600 revertants per plate. The curves correspond to eq 11 with i = 1 and the optimized values of the coefficients given in the first two lines of Table 7.(2091)
Chart 5. Compounds Used in the Distribution or Bioactivity Studies.

Table 7. Optimized Values of the Adjustable Coefficients in eq 11 with i = 1 Describing the Dependencies between Bioactivity and Lipophilicity during the Nonequilibrium Period of Distribution.
| Coefficients |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| biosystem | process | compounda | A | B1 | β | C1 | D | n | r2 | s | F | ref |
| S. typhimurium rfa+ | mutagenicity | 5-NFA R-amides, 59 | 3 | −5 | 0.638 ± 0.073 | (4.390 ± 1.027) × 10−1 | 6.027 ± 0.421 | 7 | 0.976 | 0.120 | 20.5 | Balaz et al.(2091) |
| S. typhimurium rfa− | mutagenicity | 5-NFA R-amides, 59 | 2 | −3 | 0.650 ± 0.082 | (4.270 ± 1.135) × 10−1 | 5.221 ± 0.543 | 6 | 0.949 | 0.084 | 4.62 | Balaz et al.(2091) |
| C. vulgaris | growth | 4-R-semicarbazides, 60 | 1 | −2 | 0.983 ± 0.097 | 12.60 ± 2.345 | 6.265 ± 0.567 | 14 | 0.951 | 0.098 | 43.3 | Kramer et al.(2094) |
| S. aureus | growth | BzN+(R)(CH3)2, 61 | 2 | −3 | 0.604 ± 0.052 | (1.370 ± 0.221) × 10−1 | 4.395 ± 0.324 | 12 | 0.980 | 0.134 | 86.2 | Hansch and Clayton(6) |
| C. albicans | growth | BzN+(R)(CH3)2, 61 | 1 | −2 | 1.041 ± 0.874 | (4.396 ± 0.712) × 10−2 | 3.178 ± 0.146 | 11 | 0.962 | 0.219 | 38.3 | Hansch and Clayton(6) |
| S. typhosa | growth | BzN+(R)(CH3)2, 61 | 2 | −3 | 0.405 ± 0.097 | (2.371 ± 0.286) × 10−1 | 4.328 ± 0.259 | 10 | 0.910 | 0.232 | 12.7 | Hansch and Clayton(6) |
| M. tuberculosis | growth | 2-OR-3-NH2−Py, 62 | 2 | −3 | 1.165 ± 0.186 | (1.043 ± 0.102) × 10−1 | 2.385 ± 0.128 | 7 | 0.958 | 0.549 | 11.5 | Hansch and Clayton(6) |
| C. diphtheriae | growth | R-NH2 (C5H11−C18H37) | 1 | −2 | 0.915 ± 0.013 | (6.747 ± 1.112) × 10−7 | −0.225 ± 0.051 | 15 | 0.998 | 0.073 | 1250 | Kölzer and Büchi(2093) |
| B. subtilis | growth | R-NH2 (C5H11−C18H37) | 1 | −2 | 0.813 ± 0.007 | (2.338 ± 0.210) × 10−6 | 0.071 ± 0.028 | 15 | 1.000 | 0.041 | 3123 | Kölzer and Büchi(2093) |
| S. faecalis | growth | R-NH2 (C5H11−C18H37) | 1 | −2 | 0.773 ± 0.009 | (5.209 ± 0.586) × 10−6 | 0.385 ± 0.035 | 15 | 0.996 | 0.051 | 1250 | Kölzer and Büchi(2093) |
| erythrocytes | hemolysis | α-monoglycerides, 63 | 1 | −2 | 0.864 ± 0.067 | (2.768 ± 0.201) × 10−4 | 1.457 ± 0.083 | 8 | 0.996 | 0.056 | 187 | Kubinyi(706) |
| erythrocytes | hemolysisb | α-monoglycerides, 63 | 1 | −2 | 1.054 ± 0.075 | (3.689 ± 0.231) × 10−4 | 1.079 ± 0.073 | 7 | 0.994 | 0.096 | 83.0 | Kubinyi(706) |
| mice | neurotoxicity | R-OH (CH3−C10H21) | 1 | −2 | 0.904 ± 0.075 | (1.759 ± 0.154) × 10−2 | 1.606 ± 0.095 | 10 | 0.996 | 0.109 | 311 | Kubinyi(706) |
Some structures are shown in Chart 5. 5-NFA = 3-(5-nitro-2-furyl)-acrylic acid, Bz = benzyl, and Py = pyridine.
With 2% ethanol.
Table 7 also contains some other examples of eq 11 with i = 1 and the integer values of the coefficients A and B1 fitted to experimental dependencies of bioactivity2093,2094 on lipophilicity. The concave dependencies were also fitted quite well by the parabolic equation 8 and the bilinear equation 9 with freely adjustable coefficients A and B (the results not shown). Nevertheless, the adherence of our approach to the results of model simulations brings an additional advantage, especially in the possibility to locate the compartment, where the interactions of the chemicals with the receptor occur.
5.1.2. Equilibrium Period
In the closed system without elimination (as illustrated in Figure 14, if all elimination processes are omitted), the transport of chemicals will continue up to the achievement of the partitioning equilibrium (e.g., for log t > 5 in Figures 15 and 16).
The bilayer cores of individual membranes may have different solvation properties, and the differences are assumed to be minimal, as discussed in section . Nevertheless, this factor may lead to the multiple partition coefficients Pi (up to a total number M). For ionizable chemicals, differing compositions of the aqueous phases also generate multiple apparent partition coefficients, and this situation will be treated in section . The corresponding expressions for the concentration−lipophilicity profiles can be derived easily from the definition of the partition coefficient (Pi = cMi/cW):
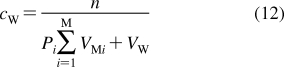 |
where n is the total amount, VM the total volume of the bilayers, and VW the total volume of water. The corresponding expression for the concentrations in the ith membrane is obtained as shown in eq 12, multiplied by Pi. Equation 12 truly describes the partitioning equilibrium and can be used to assess the accuracy of the integration method used for numerical simulations. The concentration−lipophilicity profiles resulting from eq 12 were approximated by the concave, bilinear curves, with the slopes of the linear parts having non-integer values.(8) If all bilayer cores have identical solvation properties (M = 1), eq 12 corresponds to a special form of the bilinear equation 9 and the multilinear equation described by eq 11. The equilibrium concentration−lipophilicity dependencies are bilinear on the bilogarithmic scale. The slopes in the linear parts are equal to 1 and 0 (left to right − hydrophilic compounds to lipophilic compounds) for the membranes, and 0 and −1 for the aqueous phases. The dependence for the membranes has a similar shape as the red curve in Figure 20A. This curve, flipped horizontally, defines the shape of the dependence for the aqueous phases.
5.1.3. Mixed Period
In this period, the fastest chemicals with the optimum partition coefficients have already reached the equilibrium, while other chemicals, which are either more lipophilic or more hydrophilic, are still diffusing. The concentration−lipophilicity profile in this case is a combination of the equilibrium and nonequilibrium profiles. As can be seen in Figure 20B, the mixed profiles consist of four linear parts, connected by the curved portions. The characteristic integer slopes of the linear parts are summarized in Table 6. The joining portions are mostly U-shaped. The S-shaped and Z-shaped parts are observed only for the first half of the compartments, where the chemicals temporarily accumulate in the concentrations exceeding their equilibrium values, as seen in Figure 15. Equation 11 with i ≤ 3 is suitable only for the description of the curves that contain the U-shaped connecting parts. A description of the curves with the S-shaped and Z-shaped parts would require the addition of more nonlinear terms to eq 11.
The experimental examples of the complete concentration−lipophilicity profiles typical for the mixed period of distribution are rather rare, because they are only observed if the tested chemicals cover a broad range of the lipophilicity scale. Nevertheless, the fragments consisting of more than two linear parts connected by curved portions were discernible in some published concave profiles. The data usually did not cover the sufficient range of lipophilicity, and only three linear parts were seen. Such profiles were observed among those for the growth inhibition of 17 fungal strains and 14 bacterial strains by n-alkylamines with 4−18 carbons.(2093) The resulting descriptions by the bilinear equation described by eq 9 and the trilinear equation described by eq 11 with i = 2 are summarized in Tables 8 and 9, respectively. The statistical indices in Table 8 deviate slightly from the published values,(27) because of the more-precise log P values(544) used in the present study. The differences in statistical indices between eqs 9 and 11 enable an assessment of the degree of trilinearity present in the experimental data. Both equations provide almost perfect fits. The data exhibit small but systematic deviations from the bilinear equation described by eq 9, as illustrated in Figure 21 for the growth inhibition of Ctenomyces mentagrophytes.(2093) The fact that this behavior occurred in 12 of the 17 fungal strains, as compared to only 1 of the 14 bacterial strains, speaks in favor of the hypothesis about the mixed period of distribution. In contrast to bacteria, fungi contain intracellular membranes, in addition to the cell membrane. Therefore, to achieve the partitioning equilibrium, the chemicals need more time in fungi than in bacteria. The mixed period of distribution has been observed in other experimental datasets.2087,2088
Table 8. Optimized Values of the Adjustable Coefficients in eq 9 Describing the Relationships between Antimicrobial Activity(2093) and Lipophilicity of n-Alkyl Amines (C4H9NH2−C18H37NH2) for the Nonequilibrium Period of Distribution(16)a.
| microorganism | A | −B | C × 107 | −D | nb | r2 | s | F |
|---|---|---|---|---|---|---|---|---|
| Candida albicans | 0.975 ± 0.014 | 2.942 ± 0.262 | 2.534 ± 0.548 | 0.816 ± 0.051 | 12 | 0.998 | 0.051 | 1331 |
| Candida tropicalis | 0.957 ± 0.026 | 2.195 ± 0.086 | 7.414 ± 1.613 | 0.265 ± 0.086 | 15 | 0.996 | 0.103 | 913.9 |
| Cryptococcus neoformans | 0.892 ± 0.026 | 2.372 ± 0.185 | 1.651 ± 0.512 | 0.027 ± 0.097 | 15 | 0.994 | 0.127 | 608.4 |
| Ctenomyces mentagrophytes | 0.927 ± 0.053 | 2.038 ± 0.102 | 18.14 ± 6.191 | 0.529 ± 0.175 | 14 | 0.984 | 0.155 | 205.8 |
| Fusarium moronei | 1.016 ± 0.041 | 2.749 ± 0.271 | 32.85 ± 11.65 | 0.652 ± 0.120 | 12 | 0.992 | 0.128 | 164.7 |
| Geotrichum candidum | 0.965 ± 0.022 | 2.401 ± 0.074 | 5.244 ± 0.869 | 0.605 ± 0.081 | 14 | 0.996 | 0.080 | 830.8 |
| Microsporum gypseum | 0.910 ± 0.039 | 2.131 ± 0.087 | 12.36 ± 3.221 | 0.434 ± 0.132 | 14 | 0.990 | 0.121 | 330.8 |
| Penicillium citrinum | 0.989 ± 0.040 | 2.084 ± 0.113 | 16.93 ± 5.072 | 0.885 ± 0.134 | 13 | 0.992 | 0.118 | 372.7 |
| Rhinocladium schenckii | 0.910 ± 0.029 | 2.274 ± 0.088 | 6.101 ± 1.304 | 0.378 ± 0.102 | 14 | 0.994 | 0.099 | 553.1 |
| Saccharomyces cerevisiae | 0.907 ± 0.012 | 2.350 ± 0.111 | 1.009 ± 0.168 | 0.181 ± 0.045 | 15 | 0.998 | 0.060 | 1830 |
| Saccharomyces fragilis | 0.813 ± 0.031 | 2.104 ± 0.177 | 2.351 ± 0.856 | 0.406 ± 0.110 | 15 | 0.990 | 0.141 | 363.9 |
| Staphylococcus aureus hosp | 0.846 ± 0.027 | 1.575 ± 0.180 | 1.818 ± 0.839 | −0.043 ± 0.098 | 15 | 0.994 | 0.127 | 608.4 |
| Trichophyton tonsurans | 0.911 ± 0.032 | 1.930 ± 0.089 | 7.798 ± 2.097 | 0.296 ± 0.114 | 14 | 0.994 | 0.108 | 553.1 |
The minimum inhibitory concentrations were recalculated to molar basis. The partition coefficients were found in the ClogP database for the first four congeners and calculated using the ClogP method(544) for the other compounds.
All compounds with determined minimum inhibitory concentrations are included.
Table 9. Optimized Values of the Adjustable Coefficients in eq 11 with i = 2, A = 2, B1= −1, B2= −3, Describing the Dependencies of Antimicrobial Activity of n-Alkyl Amines (C4H9NH2−C18H37NH2) on Lipophilicity during Mixed Period of Distribution(16)a.
| microorganism | β | C1 × 102 | C2 × 106 | −D | n | r2 | s | F |
|---|---|---|---|---|---|---|---|---|
| Candida albicans | 0.963 ± 0.012 | 12.95 ± 7.834 | 0.440 ± 0.073 | 1.637 ± 0.226 | 12 | 1.000 | 0.041 | 3331 |
| Candida tropicalis | 0.818 ± 0.022 | 4.785 ± 1.660 | 4.119 ± 1.381 | 1.008 ± 0.095 | 15 | 0.996 | 0.091 | 913.9 |
| Cryptococcus neoformans | 0.831 ± 0.011 | 6.965 ± 1.396 | 1.618 ± 0.269 | 0.812 ± 0.057 | 15 | 1.000 | 0.047 | 4580 |
| Ctenomyces mentagrophytes | 0.700 ± 0.018 | 1.223 ± 0.272 | 45.06 ± 13.50 | 1.381 ± 0.064 | 14 | 0.998 | 0.060 | 1664 |
| Fusarium moronei | 0.935 ± 0.034 | 8.821 ± 4.978 | 5.592 ± 2.366 | 1.397 ± 0.166 | 12 | 0.996 | 0.102 | 664.7 |
| Geotrichum candidum | 0.857 ± 0.015 | 2.564 ± 0.648 | 2.332 ± 0.532 | 1.712 ± 0.077 | 14 | 0.998 | 0.059 | 1664 |
| Microsporum gypseum | 0.765 ± 0.016 | 2.308 ± 1.070 | 14.69 ± 3.328 | 1.388 ± 0.085 | 14 | 0.998 | 0.064 | 1664 |
| Penicillium citrinum | 0.771 ± 0.030 | 1.314 ± 0.419 | 13.33 ± 6.071 | 1.821 ± 0.098 | 13 | 0.996 | 0.084 | 747.7 |
| Rhinocladium schenckii | 0.809 ± 0.117 | 2.879 ± 0.568 | 5.468 ± 0.995 | 1.446 ± 0.058 | 14 | 0.998 | 0.046 | 1664 |
| Saccharomyces cerevisiae | 0.877 ± 0.007 | 19.75 ± 4.643 | 0.477 ± 0.049 | 0.739 ± 0.082 | 15 | 1.000 | 0.032 | 4580 |
| Saccharomyces fragilis | 0.754 ± 0.014 | 7.671 ± 1.961 | 6.679 ± 1.490 | 0.376 ± 0.069 | 15 | 0.998 | 0.059 | 1830 |
| Staphylococcus aureus hosp | 0.732 ± 0.010 | 4.826 ± 0.688 | 3.278 ± 0.507 | 0.727 ± 0.034 | 15 | 1.000 | 0.036 | 4580 |
| Trichophyton tonsurans | 0.724 ± 0.021 | 1.609 ± 0.398 | 14.53 ± 4.852 | 1.218 ± 0.072 | 14 | 0.998 | 0.067 | 1664 |
Other details are given in Table 8.
Figure 21.

Growth inhibition of Ctenomyces mentagrophytes by alkyl amines (cX is the minimum inhibitory concentration)(2093), relative to the lipophilicity, expressed as the partition coefficient P. The dotted curve corresponds to the bilinear equation 9, and the full curve represents eq 11 for the mixed period of distribution (i = 2). The optimized coefficient values are given in Tables 8 and 9, respectively.
For the analyzed data of a homologous series of chemicals, the differences between bilinear and trilinear fits are small and play a minor role in the quality of the fit. They are more important from the mechanistic viewpoint, because they confirm the existence of the mixed period of distribution. As illustrated in the next part, dramatic changes have been observed in the simulated concentration−lipophilicity profiles during the mixed distribution period, if elimination has a role.
5.2. Transport with Elimination
In principle, biosystems must be treated as open systems, because of irreversible elimination. In multicellular organisms, elimination of chemicals includes metabolism and excretion, via specialized organs, into urine, sweat, expired air, or bile and feces. In the cell suspensions and tissues, elimination is represented by catalyzed or spontaneous metabolic reactions. Efflux, which could be viewed as a cell-level excretion process, is treated in SBSP as a part of carrier-mediated transport. Consequently, in this section, which involves cellular systems, elimination is synonymous with metabolism.
Elimination is one of the key processes in the disposition of chemicals, and it has a pronounced effect on the concentration−lipophilicity profiles. The phenomenon was first described by Dearden and Townend,(65) who studied the Penniston transport model(58) after prolonged periods of distribution. For a constant exposure time, they observed a distortion of the initially concave concentration-lipophilicity profiles in the region of optimum lipophilicity. The maximum of the initially bilinear curve decreased at longer times, forming a double-peaked dependence, as shown in the blue curve in Figure 22C. Similar dependencies were also observed in other simulations.(706) The minimum between the two maxima arises from the following situation. The compounds with optimum lipophilicity are transported at the highest rates and are present in the highest concentrations in the internal compartments where elimination occurs. Because the rate of elimination is proportional to the concentration, these chemicals are eliminated most rapidly, and their concentrations decrease faster than those of slowly transported compounds.
To compare behaviors of the open and corresponding closed models,(2089) the concentration−lipophilicity profiles are shown for a four-compartment closed system (see Figure 22A), as well as for the situations when the system is made open via the introduction of hydrolysis proceeding in both extracellular and intracellular aqueous phases (see Figure 22B) and metabolism, which is confined to the intracellular space (see Figure 22C). The dependencies in the closed and open systems have similar shapes in the initial period of distribution, corresponding to the nonequilibrium period in the closed system. The curves are bilinear with the integer values of the slopes, which are equal to those given in Table 6 for the nonequilibrium period. The influence of elimination becomes noticeable at the time when the mixed period of distribution starts in the corresponding closed system (the blue curve in Figure 22A). The lipophilicity of the most rapidly eliminated chemicals, determining the position of the deformation in the concentration−lipophilicity profile, is dependent on the compartment where elimination occurs. Elimination proceeding in both aqueous compartments, e.g., hydrolysis, affects preferentially hydrophilic compounds, and the concentration−lipophilicity profiles drop in the region of low lipophilicity (Figure 22B, blue and red curves). They remain more or less bilinear, with the slopes in the distorted part having non-integer values, which are much higher than the original integer values of the nonequilibrium profiles (Table 6). Equation 9, with the optimized coefficient A and the coefficient B, which is dependent on A to maintain the integer slope of the rightmost part of the profile (cf. Figure 17), describes each with sufficient precision. The situation is completely different when only intracellular elimination is considered. The maxima of the curves are distorted, because the chemicals with the optimum lipophilicity are eliminated at the fastest pace. The concentration−lipophilicity profiles have two maxima separated by a minimum (Figure 22C) and can be described by eq 11 with i = 3. Coefficients B1 and B2 are optimized, and coefficient B3 is dependent on B1 and B2 to maintain the integer slope value of the rightmost linear part (the association between the slope values and the magnitudes of coefficients A and B is depicted in Figure 17).
During the initial period of distribution, the overall shapes of the curves are similar, regardless of elimination. However, the curves are shifted along the log c-axis, depending on the rate and location of the elimination process. The simulation results were accurately described by the following modification of eq 11:
 |
where k is the elimination rate parameter. The results for the fourth compartment of a four-compartment system are presented in Figure 23. Evidently, elimination becomes important when the value of the rate parameter k exceeds a certain limit that decreases with prolongation of the time of distribution. The dependence of the intracellular concentration on the elimination rate parameter k is bilinear, with slopes of 0 and −1. For a real situation, the rate parameter k can be substituted by the reactivity of the chemical in a reference reaction mimicking the metabolic elimination process, or by other experimental or computational reactivity parameters, according to eq 5 in section .
Figure 23.

Reactivity dependence of the concentrations in compartment 4 of a four-compartment, water−bilayer−water−bilayer model that were corrected for the contribution of lipophilicity to show only the reactivity portion of this form of the disposition function (eq 13). The exposure time is increasing from top to bottom. Other details are as given in Figure 22.
6. Explicit Descriptions for Multi-Bilayer Systems
Some biologically relevant systems are sufficiently simple to be described by explicit solutions to the set of differential equations 47 (section ), which characterize the disposition of chemicals in multimembrane systems. Complexity can be reduced by considering a few compartments or by neglecting the reverse transport processes, if applicable. Both simplifications result in several useful descriptions. The advantage of the explicit descriptions is that the transfer rate parameters l do not need to be strictly limited to the passive diffusion and can be replaced by characteristics of active transport mechanisms in a linear regime. The explicit descriptions represent realistic yet comparatively simple SBSP models.
6.1. Transport through a Single Bilayer
A description of transport through the single membrane is relevant for simple systems such as gram-positive bacteria and unilamellar vesicles, as well as for various absorption simulators, if the studied sheet only consists of one lipophilic phase in a transport simulator(2095) or a single phospholipid bilayer, such as BLM (section ), if the chemicals do not significantly interact with the headgroups. The frequently published applications of single-membrane-based mathematical treatments to the description of transport through various physiological sheets of multimembrane nature (e.g., the intestine or skin; see section ) will hardly promote our understanding of the processes.
The rate of transport through a single membrane is usually characterized by the permeability coefficient PC (Figure 7, row 1, section ), relating the trans-membrane flux of the chemical to the surface area and the difference in the concentrations in the donor and acceptor aqueous phases. The magnitude of PC is equal to the reciprocal value of the total resistance, which can be calculated in a laminary system as the sum of the reciprocal values of the diffusional resistances in individual laminae. The classical solubility−diffusion model (section ) uses just a single lamina. The partial resistance in the ith lamina can be written as1000,2096Ri = hi/(DiPi), where h is the effective thickness of the lamina, D the diffusion coefficient, and P the partition coefficient for the ith lamina, with respect to the external phase (if both phases are identical, P = 1). This generalization has been frequently used for the construction of membrane transport models differing in the number and quality of the laminae,2097–2103 as well as for the calculation of the permeability coefficients from the MD simulation data.292,588,1005 The membrane transport models mostly result in the dependence of the permeability coefficient on the partition coefficient, which initially linearly increases and later becomes stable, such as, e.g., the black curve in Figure 22. Some reports document the effect of the solute size on permeability.2104,2105 A comprehensive treatment showed that the solubility−diffusion mechanism (section ) overpredicts the permeabilities through the liquid-ordered bilayers, and the correction factors were proportional to the surface density of phosholipids(506) and to the minimal cross-sectional area of the seven studied permeants.(73) It can be shown that, for the transport of non-amphiphilic chemicals through a symmetrical bilayer (Figure 7, rows 2 and 3), PC = li/2, where li is the water-to-bilayer transfer rate parameter.(2106)
The permeability coefficient PC (Figure 7, row 1) characterizes the steady-state rate of passage through the membrane. For most chemicals, the direct use of PC to calculate the time course of the concentration in the acceptor aqueous phase leads to substantial errors. The reason is that the concept resulting in the definition of the permeability coefficient considers the membrane an inert diaphragm. As illustrated in Figure 7, this assumption is not plausible for the majority of chemicals, which accumulate to some level in the membranes. The membranes represent a small volume fraction of the biosystem. Nevertheless, even considering the volume of the surrounding aqueous phases being a thousand times larger than that of the membranes, a chemical with the value of the membrane/water partition coefficient of ∼1000 will partition in equal amounts in the aqueous phases and in the membranes. The equilibrium concentrations calculated without the consideration of membrane accumulation would be ∼2 times higher than the experimentally observed quantities.
Membrane accumulation can be taken into account via the use of at least two transfer rate parameters: one for the transfer of the chemical into the membrane and one for its exit (li and lo, respectively). The transfer rate parameters are characterized in section ; their most important attributes are the dependencies on the partition coefficients, as described by eqs 3. The representation of the membrane by a single hydrocarbon core is a valid approximation for the chemicals, which do not interact with the headgroups. The explicit solutions to differential equations 47 were published for the simple water−core−water system (section ),(2095) and for the system containing an additional lipophilic phase.(2107) The concentration−lipophilicity profiles for individual distribution periods t, as generated by the explicit solutions, are similar to those given in Figure 20.
6.2. Unidirectional Transport
The disposition of chemicals involving only unidirectional transport often represents real situations, at least for a certain time period.(2108) Transport in any multimembrane system proceeds initially in one direction, away from the site of administration. The duration of the period of unidirectionality is primarily dependent on the length of the sequence of compartments, as well as on the rates and equilibria of the processes, which reduce the free concentrations in individual compartments either reversibly (noncovalent binding to macromolecules, section ) or irreversibly (metabolism and excretion, section ). The approximation may occasionally be sufficiently precise even for the complete period of distribution, especially in two borderline cases: (i) for large systems, where the total number of compartments approaches several hundred, as described in section for the muscles, connective tissues, and fat; and (ii) for rapidly biotransformed chemicals, even in the biosystems with a few bilayers, as in the cell suspensions.
6.2.1. Model Description
The model system depicted in Figure 14 in section , if the transport is considered unidirectional, is described by the set of linear differential equations 47 (section ) with a bi-diagonal matrix B. The elements above the diagonal represent the backward process and vanish for the unidirectional transport. If all compartments of the model are different (also known as the normal case), the free concentration in the mth compartment can be expressed2064,2109 as a multi-exponential function of the exposure time t:
 |
Here, n is the dose. The eigenvalue λi (eq 49, section ) comprises the traits of the ith compartment: the volume Vi, the interfacial area Si connecting it to the (i + 1)th compartment (in the flux terms Fi,i+1, c.f., eq 46), the pH value of the phase (in the dissociation terms dij, c.f., eqs 36 and 46), and the concentration of protein binding sites sij (in the binding terms bijk, c.f., eq 38), as well as the properties of the tested chemicals, such as the rate parameters for the transfer from water to the core and backwards (li and lo, respectively, in the terms Fi,i+1, c.f., eqs 35 and 46), the ionization constant to the jth degree (in dij), ion pairing with the kth counterion (in the pairing term pijk, c.f., eqs 36 and 46), the elimination rate parameters kijk (in Ei, defined in eq 46), and affinities for proteins Kij, (in bijk, c.f., eq 38).
The descriptions equivalent to eq 14 were published for simpler scenarios when protein binding or elimination was not significant.2110–2112 Equation 14 supports the previously published conclusion about the proportionality between the concentration at a given time and the product of the rate parameters of involved transport processes.(2083)
The cellular structure of tissues leads to the situation in which all membranes and all aqueous phases have the same characteristics (also known as the degenerate case). This phenomenon may also be observed in isolated mammalian cells and eukaryotic microbial cells, because they are subdivided into the sets of similarly sized compartments by endoplasmic reticulum and other intracellular membranes. The time courses of the drug concentrations in all compartments, preceding the cellular structure with repeated pairs of identical compartments, are described by eq 14. Inside the cellular structure, recurrent formulae should be applied, as has been shown for a reduced scenario.(2112)
Equation 14 and the recurrent formulae can be used in two ways: (i) for a direct construction of the SBSP models in smaller systems, such as suspensions of bacteria or mammalian cells, which comprise a few compartments; and (ii) as a basis for a derivation of more-manageable limit expressions, when the compartment number is large.
6.2.2. Concentration−Lipophilicity Profiles
Equation 14, with the transfer rate parameters expressed by eqs 3, which is valid for chemicals that are not accumulating in the headgroups and at the interface, describes transport(2113) as being combined with elimination(2111) and protein binding.(2112) For pure transport and transport with elimination, the generated concentration−lipophilicity profiles have been shown to be identical to those obtained by numerical simulations. The influence of protein binding on disposition of chemicals has only been investigated in SBSP models using the explicit relationship described by eq 14.(2110) As the first approximation, protein binding was assumed to be dependent on lipophilicity, according to eq 4 in section . More-precise models will require replacement of the association constants Kij, which are contained in the terms bijk in eq 14, by the 3D-QSAR models for protein binding, as reviewed in sections −. All adjustable coefficients, such as those in eq 14, as well as those in the 3D-QSAR model, may need to be optimized simultaneously.
The corresponding concentration−lipophilicity profiles for shorter periods, corresponding to the nonequilibrium distribution period in closed systems, are depicted in Figure 24. The bilinear profiles without the influence of protein binding have characteristic integer slopes (see Table 6 in section ). Lipophilicity-dependent protein binding changes the slope of the decreasing linear part, as can be seen in Figure 24. This fact could explain why the experimentally observed smooth concentration−lipophilicity profiles cannot always be described using eq 11 with the integer values of the adjustable coefficients, as given in Table 6. The dependencies for a plummeting right-hand part, which are similar to those in Figure 24, are accurately described using a simple empirical relation:(2114)
Here, A, B, and C are optimized coefficients. Equation 15 provided excellent fits to several published datasets.(2114)
Figure 24.
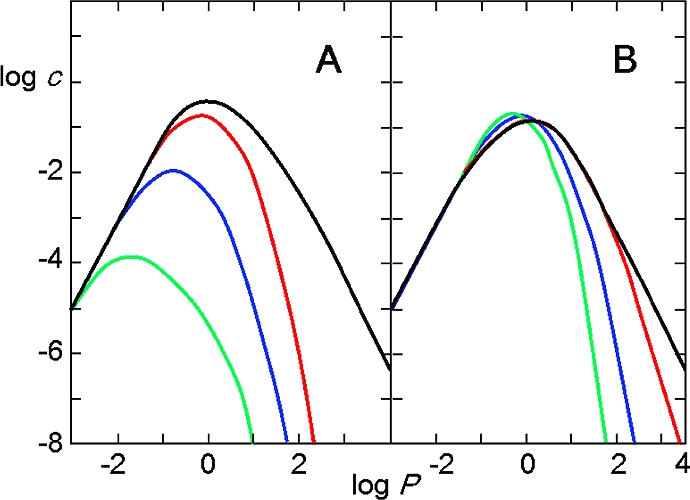
Influence of protein binding on the concentration−lipophilicity profiles of nonionizable chemicals in the nonequilibrium period in the fifth aqueous compartment of a system of alternating aqueous phases and bilayers, as calculated using eq 14. Protein binding is lipophilicity-dependent, according to eq 4 with bi00 = BiPβ, Bi = B1 = B3 = B5, and B2 = B4 = 0 (see eqs 38 and 43). The transfer rate parameters li and lo are dependent on the partition coefficient P, according to eq 3. Panel A illustrates the effect of the increasing protein concentration: β = 1 and Bi = 0 (black), 0.01 (red), 1 (blue), and 100 (green). Panel B illustrates the effect of the increase in the sensitivity of binding to lipophilicity: Bi = 0 (black); and Bi = 1 and β = 0.75 (red), 1.00 (blue), and 1.25 (green).(2110)
The results for the longer distribution periods are plotted in Figure 25. Analogous curves for pure transport (Figure 25A) have been described in sections and . The initially bilinear concentration−lipophilicity profiles are distorted after longer distribution periods in the region of optimum lipophilicity and form the double-peaked curves. Lipophilicity-dependent protein binding (Figure 25B) decreases the actual drug concentrations at shorter times.
Figure 25.

Dependencies(2112) of the concentration c in the fifth compartment of the system of alternating aqueous and bilayer phases on the partition coefficient P for (A) pure unidirectional transport, (B) transport combined with protein binding, and (C) transport influenced by protein binding and metabolism, after the following times of distribution (in time units): 3.2 (green), 10 (blue), 32 (red), and 100 (black). Calculated using eq 14, with the parameters as in Figure 24A (Bi = 1) and ki = 1/(time unit).
For the longer distribution periods, the depot function of proteins for chemicals with log P > 0 starts to play a role. The slowdown of the transport of the molecules with the optimum lipophilicities is the reason why the minima in Figure 25B become shallower than those without the protein influence (Figure 25A). The depot effect is more pronounced in the presence of elimination (Figure 25C). While hydrophilic compounds have been eliminated at longer times and the left-hand peaks have completely disappeared, lipophilic compounds are still present in the system, because of the depot function of protein binding.
6.3. Steady-State Distribution
Continuous dosing at a steady rate is frequently used for therapeutic purposes, in the form of intravenous infusions for critical drugs or via the steady-release dosage forms, producing long-lasting effects. Environmental scenarios with practically invariant pollution levels also provide a steady input. If the continuous input of chemicals is maintained at the same rate for a sufficiently long period, the system will ultimately attain the steady state, manifested by the time-invariant concentrations in all compartments, which are depicted in Figure 14 in section . Consequently, all the left-hand-side derivatives in SBSP models, which are represented by eqs 46 in section , vanish. To describe the situation, eqs 47 are not needed in this case, because the factors that are used to convert eqs 46 to eqs 47 would cancel each other in the solutions of the algebraic equations resulting from eqs 46 for the steady-state conditions. Because of a sustained concentration pressure from the site of dosing, reverse transport is negligible, and the back-flux terms Fi,i−1 and Fi+1,i in eqs 46 can be omitted. This simplification affords a compact solution for the steady-state concentration of the free non-ionized molecules in the ith compartment (see the numbering in Figure 14 in section ), obtained by back-substitution:(2115)
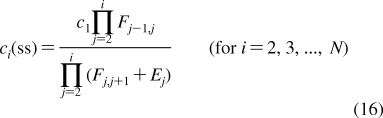 |
Here, c1 is the invariant concentration of the free non-ionized chemical in entry (aqueous) compartment 1, and the flux and elimination terms F and E are defined in eqs 46. The last compartment is usually an aqueous phase, so the sequence number of this compartment (N in eq 16) is an odd number, according to the numbering scheme in Figure 14 (section ).
If the studied biosystem, such as a tissue, consists of the cells of the same type with similar intracellular compartments, the flux terms F are practically identical for the transport from all aqueous phases (FA, odd initial subscript i) and from all bilayers (FM, even initial subscript i). For this common situation, eq 16 can be simplified to eq 17 for the aqueous phases and eq 18 for the bilayers,(2115) with the compartment numbering given in Figure 14 in section :
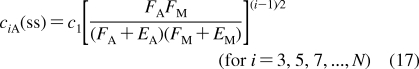 |
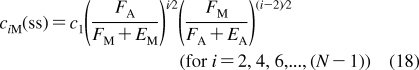 |
The dependencies on the lipophilicity of the free, non-ionized steady-state concentrations, which were attained during continuous dosing, are illustrated in Figures 26 and 27. The transfer rate parameters li and lo contained in the flux terms were expressed using eqs 3 in section , so the results are valid for the chemicals, which do not accumulate in the headgroups and at the interfaces. Interestingly, no peaks are apparent in the bilogarithmic dependencies; rather, the concentrations of chemicals are, after an initial linear increase for hydrophilic chemicals, independent of lipophilicity for aqueous phases, and rise at a lower rate for membranes. The slopes of the leftmost linear parts are dependent on the compartment number (see Figure 26). The break-point log P value is affected less by the compartment number (see Figure 26) than by the magnitude of the elimination rate (Figure 27).
Figure 26.

Steady-state concentrations versus lipophilicity in (A) individual aqueous phases and (B) individual bilayers for the invariant elimination rate constants. The aqueous phases are represented by compartments 3−23 and membranes are represented by compartments 2−22, both with step 4 (curves 1−6, respectively; compartment numbering is as shown in Figure 14 in section ). The curves correspond to eqs 17 and 18 with c1 = 1 unit and EM = 1 (time unit)−1. The flux terms in eqs 17 and 18 are defined in eqs 46 and only contain the surface areas and the transfer rate parameters. The transfer rate parameters are expressed by eqs 3.(2115) The dotted line indicates c1 in A and c1P in B.
Figure 27.

Steady-state concentration−lipophilicity profiles for (A) aqueous compartment 7 and (B) bilayer 8 of a catenary system of alternating aqueous and bilayer phases, relative to the elimination rate constant k.(2115) The elimination terms only contain k, in (time unit)−1: 10−4 (curve 1), 10−2 (curve 2), 1 (curve 3), 10 (curve 4), 102 (curve 5), and 103 (curve 6). Other details are as shown in Figure 26.
Apparently, the peaks in the concentration−lipophilicity profiles after single doses (e.g., Figures 17−24) are caused by a slow transport of lipophilic compounds. As can be seen in Figures 26 and 27, this phenomenon does not exist under the continuous dosing regimens, after the distribution reaches the steady state. However, there is a string attached to the last phrase. The duration of the transient period, before the steady state is established, varies with lipophilicity, and can be very long for extremely hydrophilic and extremely lipophilic compounds. Achievement of the steady state for chemicals with a broad range of lipophilicities requires long exposure periods, which are typical for environmental scenarios but may be much longer than the common infusion periods of drugs. Consequently, in real situations, a type of concave concentration−lipophilicity profile can also be expected for the continuous dosing of drugs, if the lipophilicity range is sufficiently broad. The concentrations of very hydrophilic and very lipophilic drugs would be lower than expected on the basis of the steady-state profiles, which are shown in Figures 26 and 27.
The concentrations in individual phases, as expressed by eqs 17 and 18, are of interest, when the studied chemicals act by a specific mechanism on well-defined receptors, which are localized in spatially limited compartments. However, environmental pollutants frequently elicit their effects in all phases by mechanisms, which cannot be tracked back to the modification of a single receptor, such as polar and nonpolar narcosis, and alkylation of body constituents.1487,2116,2117 These effects are caused by interactions proceeding in all aqueous phases or in all membranes; consequently, the average concentrations in all aqueous phases and all membranes are important for their descriptions. If individual steady-state concentrations in membranes are enumerated according to eq 18 and used to calculate the average concentrations, the result contains a practically infinite (N may reach several hundred; see section ) geometric sequence with the argument x = FAFM/(FA + EA)(FM + EM) < 1 and the powers 0, 1,..., (N − 3)/2. The sum of such a sequence is equal to 1/(1 − x) and can be used to calculate the average concentrations for all (N − 1)/2 membranes (eq 20).(2115) For the aqueous phases, the first term with the power of 0 (i.e., term = 1) is missing and the sum is then [1/(1 − x)] − 1. The average concentration for all (N − 1)/2 aqueous phases, except the first phase, which maintains the constant concentration c1, is given by eq 19:(2115)
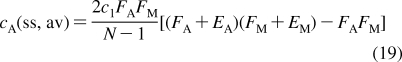 |
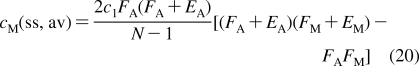 |
Here, N is the number of compartments. The dependencies of the average concentrations in the aqueous phases and membranes on lipophilicity are shown in Figure 28. In aqueous phases, saturation-type curves are observed, whereas biphasic increasing dependencies result for the membranes.
Figure 28.

Average steady-state concentration−lipophilicity profiles in (A) all aqueous phases and (B) all bilayers of a system of alternating aqueous and bilayer phases, as a function of the elimination rate constant. Curves correspond to eqs 19 and 20, with other details as given in Figure 26. The elimination rates constants k, in (time unit)−1, are 10−4 (curve 1 in both parts), 10−2 (curve 2), 1 (curve 3), 102 (curves 4 and 3 in parts A and B, respectively), 104 (curves 5 and 3 in parts A and B, respectively), and 106 (curves 6 and 3 in parts A and B, respectively).(2115)
The elimination rate affects the average aqueous concentrations much more significantly than the average membrane concentrations. This fact is consistent with experimental observations regarding pollutants acting by narcosis, which is presumably elicited by a membrane perturbation. The toxicity of these chemicals is linearly dependent on lipophilicity, with a slope that approaches unity. The dependencies are perfectly linear for persistent pollutants, and reactive chemicals exhibit only modest deviations from the line.2118–2121
7. Pseudo-Equilibrium Disposition
The SBSP models, utilizing the pseudo-equilibrium approximation to describe the disposition, include all processes, which chemicals undergo in biosystems, besides absorption. Absorption is usually much faster than elimination and is considered a fast process under the pseudo-equilibrium approximation. The models capture essential features of the disposition process, including a substantial portion of its kinetics, exhibit a low complexity of the description, and allow for incorporation of external 3D-QSARs for protein binding and metabolism where necessary. From this viewpoint, the pseudo-equilibrium models belong to the most advanced tools of SBSP. The pseudo-equilibrium models evolved from equilibrium models, which represent the first non-empirical models of SBSP.
The equilibrium approach is applicable to the distribution of the chemicals, with the transport being much faster than elimination. The equilibrium models describe the disposition accurately in the time interval after the transport has been finished and before elimination is measurable. The first equilibrium model was formulated by Higuchi and Davis.(2122) Their expressions, after a later conversion to QSAR equations,706,2123 were equivalent to the bilinear equation 9 in section , under some circumstances. The addition of ionization by Martin34,35,63 to the equilibrium models represents an important advance because many chemicals and most drugs ionize under physiologic conditions.
The extension from the equilibrium to pseudo-equilibrium approach was accomplished by a conceptual inclusion of elimination.(2124) As a result, the elimination period, representing usually the major portion of the residence time of chemicals in the biosystems after a single dose, has become a part of the time interval covered by the model. In contrast to the selection of special model structures and reduced scenarios, as used in sections −, here, the time hierarchy of the distribution processes was deployed to reduce mathematical complexity. The number of the time-dependent variables decreased dramatically, because of the approximate representation of the fast processes as instantaneous events. The objective was to introduce a new type of the disposition function, which would be sufficiently robust (i.e., describing the main features yet simple) for a direct application in the formulation of model-based QSTARs (where T represents time), and advance SBSP modeling.
7.1. Model Construction
Interactions with bilayer regions determine the trans-bilayer transport rates of chemicals, as summarized in sections and . For a single bilayer under physiological conditions and dimensions, the rates vary from milliseconds for chemicals with intermediate strength of interactions with bilayer regions, to minutes or hours for substances with extreme lipophilicities, to days for some amphiphilic compounds. The overall distribution rates are dependent on the complexity of biosystems, in addition to the properties of the chemicals. If only transport is considered, the complexity of a biosystem can be characterized by the number of membranes. The time a chemical requires to achieve the lipo-hydrophilic equilibrium increases with the number of the membranes it must cross.
The real rate of transport, encompassing both the absorption and distribution phases, is dependent on the interplay of all these factors. For non-amphiphilic chemicals with optimal lipophilicity, the process is completed within an interval ranging from milliseconds to minutes in simple biosystems such as liposomes,82,704,2125 subcellular organelles, and cells.78,2126–2128 For amphiphilic permeants,710,711,2129 very hydrophilic or lipophilic permeants(2130) in simple biosystems, or non-amphiphilic and optimally lipophilic chemicals in large biosystems such as organs or organisms,(2131) the period needed to achieve the lipo-hydrophilic equilibrium may be as long as several minutes,2132,2133 hours,(709) or even days710–712 after the administration. Ionization of a chemical, which makes it more amphiphilic, also significantly slows its trans-bilayer transport.711,2132
The transport of chemicals is usually faster than elimination. Provided that the rates of the two processes differ sufficiently, transport can maintain the actual concentrations in individual cellular phases permanently near the lipo-hydrophilic equilibrium. This is the premise of the pseudo-equilibrium disposition model. A corresponding morphologically compartmentalized biosystem was shown in Figure 14 in section . The only modification that must be made in Figure 14 is to depict the transport as an instantaneous process. In the aqueous phases, both ionized and non-ionized molecules are present, but only the latter are assumed to readily cross the hydrocarbon core of the bilayers readily, according to the pH-partition hypothesis.(2134) Provided that it is much faster than elimination, the transport can be considered instantaneous and reversible, as are the intracompartmental processes, which include ionization and noncovalent binding to enzymes and proteins. Irreversible elimination then represents the only time-dependent step. Under such conditions, all the phases of the biosystem become kinetically undistinguishable. From the viewpoint of the chemicals’ disposition, it means that the originally N-compartment system loses its structure and collapses to a one-compartment open system. The free non-ionized species move quickly in such a system; therefore, their concentrations are identical at each moment in all aqueous phases and in all membranes. The aqueous and membrane concentrations are time-dependent, and are, at any moment, related by the partition coefficient, according to the Nernst distribution law. A similar time hierarchy has been used in the development of the model-based equilibrium dependencies of bioactivity on lipophilicity and acidity.34,35
The decisive criterion for applicability of the pseudo-equilibrium approach is the ratio between the rates of transport and elimination. If this ratio is higher than 5−10, the transport is able to maintain the actual concentrations of chemicals in individual cellular phases permanently near the lipo-hydrophilic equilibrium. Then transport joins ionization, ion pairing, and protein binding in the category of fast, reversible processes, and elimination remains the only time-dependent step.
The pseudo-equilibrium disposition is characterized in a standard way using the Law of Mass Action. The concentrations of individual species in the multiple, rapid equilibria are mutually inter-related at each moment; therefore, any of them can serve as a reference variable for the expression of all of the other concentrations, as documented in section . The free non-ionized concentration cA was selected as the reference concentration, because it has the same magnitude in all aqueous compartments under the conditions of the adopted time hierarchy. The concentrations of the free non-ionized molecules in all membranes are also equal and related to cA via the membrane/water partition coefficient PM.
The molecules are present in each aqueous phase in nine different states: free and bound to proteins and enzymes, all three states comprising non-ionized molecules, ionized molecules, and ion pairs (see Figure 14 in section and consider only monovalent acids or bases and one type of counterions). The scenario is characterized by 13 equations (6 definitions of the association constants for the binding of the non-ionized, ionized, and ion-paired molecules to proteins and enzymes, plus 6 definitions of the ionization constants and ion-pair formation constants for the free, protein-bound, and enzyme-bound molecules, plus the mass balance equation). Four equations are redundant. The definitions of the ionization constants for the complexes of the chemical C with proteins (CPij) and enzymes (CEij), and the definitions of the ion-pairing association constants for CZPijk and CZEijk for the counterion Z were set aside and not used (the processes are not indicated by the double-head arrows in Figure 14 in section ), because they do not comprise the reference concentration of free non-ionized chemicals, cA.
7.1.1. Accumulation in Membranes
As the first approximation, the hydrocarbon cores of the bilayers are considered as having practically identical solvation properties in all membranes. For the chemicals, which do not interact with the headgroups and only accumulate in the core of the bilayer, as shown in rows 2 and 3 of Figure 7 in section , the partition equilibrium is described by the apparent partition coefficient Pi that can be expressed as
Here, PC is the core/water partition coefficient and the proportionality constant ai is defined by eq 56 in section . The aqueous phase that is more distant from the site of entry of chemicals was chosen for the definition of Pi by eq 21. The composition of this phase (pH, the concentrations of the counterions) determines, along with other factors (see section ), the magnitude of the accumulation factor ai. The partition coefficient PC is an additive-constitutive property that can be expressed as the product of the hypothetical partition coefficients (fragment contributions) of substructures, and the factors correcting for the interactions among the substructures.649,2135 This principle enables a fast prediction of the PC values for new structures.
Partitioning of amphiphilic and cephalophilic chemicals is more complicated, and its quantitative description requires the use of the headgroups/water partition coefficients. However, these chemicals are transported slowly(20) (see section ), and their behavior may not conform to the assumptions of the pseudo-equilibrium model.
7.1.2. Ionization in Aqueous Phases
Extracellular and intracellular aqueous phases of biosystems differ in composition, including the proton concentrations, as illustrated by the following examples. The stomach content has different acidities in fasted and fed states. A pH gradient exists along the intestine. The acidity of the urine is dependent on physiological factors. Among the intracellular compartments, lysozomes and mitochondria exhibit more-acidic aqueous milieu than other organelles. The differences in the pH values of individual aqueous phases affect both the rate and the extent of transport processes, especially for ionizable chemicals.
The pH-partition hypothesis states the non-ionized species of a ionizable molecule are transported at a faster rate than their ionized counterparts.(2134) This well-known concept was developed for intestinal absorption, although the influences of the pH value of the medium on intracellular uptake and biological effects were studied earlier, first qualitatively1580–1582,2136–2144 and later quantitatively.2145–2147 In 1942, a comparatively smooth dependence of the bacteriostatic activity of sulfanilamides against Escherichia coli on the pKa values, exhibiting a maximum at pKa ≈ 6.5, was observed and explained by a conceptual quantitative model.(2148) The peak in bioactivity results from an interplay between the ionization equilibrium affecting the uptake and the intrinsic activity,(2149) which exhibit opposite tendencies with regard to the pKa values. The effects of substituents on the acidity of organic acids2150,2151 and other reactions(2152) led to the formulation of the Hammett equation, which greatly contributed to the QSAR development.
The pH-related variability in the accumulation of ionizable chemicals is one of the factors that affects pseudo-equilibrium distribution. A typical example are cationic drugs, for which ion-trapping in lysozomes and mitochondria,(1754) along with other interactions, leads to the increase in distribution volumes.2153,2154 The accumulation is pronounced in lysozome-rich organs, including lungs, kidneys, and the liver.(2155)
Ionization has long been recognized as an important factor that affects the disposition of chemicals. The experimental determination2156,2157 and prediction of the dissociation constants2158–2171 belong to standard procedures in preclinical drug development. The presence of several ionizable groups with the pKa values closer than 2−3 units requires a special attention in both experimental2172–2176 and computational2177,2178 approaches. Four programs for estimating the pKa values—PALLAS/pKalc,(2179) MARVIN,(2180) ACD/pKa DB,2180,2181 and SPARC(2182) —were compared, using three datasets.(2183)
In the context of pseudo-equilibrium distribution, the ionization equilibria in the aqueous phases are described using the factors dij, given by eq 36 in section . The concentration of the jth ionized species is obtained as the product of the factor dij and the free concentration of the neutral species. The formation of the ion pairs is characterized by the factor pijk, which is defined in eq 37.
7.1.3. Elimination
The loss of chemicals via elimination is the summation of excretion, reactions with low-molecular-weight body constituents, and enzymatic biotransformation of all low-molecular-weight species (neutral molecules, ions, ion pairs) in all compartments. The first-order elimination rate constant k includes the contributions from all the first-order processes, i.e., excretion and spontaneous and enzymatic reactions, affecting each low-molecular-weight drug species in the ith compartment, as defined in eq 45 in section . The overall elimination rate constant can be partitioned into its components: spontaneous reactions r, enzymatic reactions e (the subscript “P” indicates lipophilicity-dependent reactions), and excretion processes x, as shown in eq 45.
There is usually no need to differentiate between enzymatically catalyzed biotransformation to inactive metabolites (eijk) and covalent binding to proteins (typical, e.g., for alkylating agents), if the latter proceeds in a small, nontoxic extent. Both processes play, from the viewpoint of distribution, the same role: they cause the irreversible loss of the molecules of chemicals, which are originally noncovalently bound to them. The covalent protein binding must be taken into account only if it is significant and leads to toxicity.
7.2. Model Description
The amount (n) of a chemical is, at any moment, proportional to its free non-ionized concentration in water (cA). The proportionality factor is the unbound non-ionized volume of distribution, Vdf (eq 60 in section ), that accounts for accumulation in membranes and aqueous phases, and protein binding of all species of the chemical. The total rate of loss of the chemical from the biosystem is the summation of the elimination processes proceeding in all N subcellular compartments with the first-order and zero-order kinetics (the respective clearances Cl are specified in eqs 62 and 63 in section ). The eliminated amount per time unit is converted to the free concentration, cA, using Vdf:
The elimination rate constants are equal to the clearances divided by Vdf. Integration of eq 22 gives
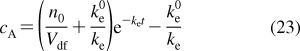 |
The first term in eq 23 represents the initial concentration of the free non-ionized compound in the aqueous phases as given by eq 60, with the dose n0, instead of the actual amount n. In eq 23, the resulting concentration (cA) would become negative for the infinitely long exposure time t. This anomaly is explained by the fact that eq 23 is not valid for the complete duration of the residence of chemicals in the biosystem. During elimination, when the concentration drops below a certain limit, the zero-order character of the process will change to the first order.
Equation 23 is suitable not only for the cases with combined zero- and first-order elimination, but also for the first-order elimination alone (simply k0e = 0). For the opposite case, when only the zero-order elimination is encountered in a limited time interval (i.e., ke = 0), eq 23 results in an undefined expression. The corresponding description (eq 24) can be derived either de novo or from the limit of eq 23 for ke approaching zero using the LʼHospital rule:
At any moment, the free non-ionized concentrations (cA) are identical in all aqueous phases. However, the total concentrations in individual phases differ, because of ionization, ion pairing, and protein binding. The actual concentration of any molecular species in the given compartment can be easily calculated as the product of cA and the respective proportionality factor as shown in eqs 36−38 in section . The concentrations of metabolites and excreted molecules can be described using the integral of the time dependence of the corresponding precursor concentration, multiplied by the respective rate parameter.
Note that the global elimination rate parameters ke and k0e in eqs 23 and 24, as specified in eqs 62 and 63 (section ), are not plain summations: the rate parameters in individual compartments are weighted by the respective volumes, Vi, to scale the contributions to the total elimination according to the size of the compartments. The unbound non-ionized volume of distribution, Vdf, accounts for the effect of protein binding and membrane accumulation of chemicals on the available concentrations (eqs 23 and 24), and on the decrease in the elimination rates (eqs 62 and 63). An illustration of the latter phenomenon is given later in Figure 29 in section .
Figure 29.

Relationship(2124) between the concentration c of the protein-bound chemical, the partition coefficient P, and the exposure time of distribution t for a pseudo-equilibrium situation: (A) projection into the plane c−P, (B) projection into the plane c−t, and (C) overall view. The values were calculated using eq 25 multiplied by P, with c0 = 1 unit, β = A = 1, B = 0.1, C = 0.01, and D = 0. Individual curves in projection A are valid for log t = −4 (curve 1), −3 (curve 2), −1.75 (curve 3), −0.5 (curve 4), 0.75 (curve 5), 1.25 (curve 6), 1.75 (curve 7), 2.75 (curve 8), 3.75 (curve 9), and 4.75 (curve 10). Individual curves in projection B are valid for log P = −4 (curve 10), −3 (curve 9), −2 (curve 8), −1 (curve 7), 0 (curve 6), 1.25 (curve 5), 2.5 (curve 4), 3.75 (curve 3), 5 (curve 2), and 6 (curve 1).(2124)
Equations 22−24, with further specifications given in eqs 60−63 in sections and , can be used to derive model-based expressions relating pharmacokinetic parameters or bioactivities to the structure of chemicals, for interspecies scaling, and for population pharmacokinetics. The application in the QSAR field is illustrated in the next section. The biosystem parameters (membrane volumes, protein concentrations, the pH values of the aqueous phases) must be specified for interspecies scaling and expressed as a function of conveniently measurable characteristics of individuals for population pharmacokinetics.
7.3. Use in QSAR
In the following, we will only address the first-order elimination (eq 23 with k0e = 0), which is more typical for low concentrations of chemicals, such as those used in drug therapy and environmental toxicology. To convert eq 23 to a QSTAR expression, the model parameters should be substituted by the structural and physicochemical properties of the chemicals. The association constants for the structure-dependent processes (binding to prevalent proteins, transporters, enzymes) in ke = Cl/Vdf and Vdf (eqs 62 and 60) must be replaced by the appropriate 3D-QSAR relations where necessary. The parameters for property-related processes in ke and Vdf (partitioning, ionization, reactivity) are replaced by physicochemical properties using plausible extrathermodynamic assumptions, as introduced by the pioneers of the QSAR field.(56) The core/water partition coefficient, PC, is related to the reference partition coefficient, P, according to the Collander equation (eq 1) in section . The reactivity to body constituents can be related to the reference reactivity using eq 5 in section . Ionization is usually characterized by the pKa values for diluted aqueous solutions. The quantities for individual chemicals can be disproportionately affected by the ionic strength of the cell environment, but this effect is usually deemed insignificant. We will analyze, in detail, the situation when the observed disposition or bioactivity, and, consequently, all participating processes, are smooth functions of physicochemical properties of the chemicals. In such cases, elimination proceeds via property-related processes, and the nonspecific noncovalent binding of homologous series of chemicals to proteins is structure-independent and obeys eq 4 in section .
A combination of eq 1 in section and eq 4 in section with eq 23 results in the disposition function (eq 6 in section ), with lipophilicity, acidity, ion pairing, and elimination rate parameters as variables, and a large number of adjustable coefficients. To adapt the expression to a real situation, many simplifications are available, based either on information about the used biosystem and chemicals, or on a hypothesis to be tested. The possibilities are numerous and the procedure is comparatively straightforward. Therefore, an exhaustive overview of all equations is not given; rather, the concept is illustrated by the application to some simple cases.
The number of coefficients can be reduced(2075) by classification of the fate-determining processes into two groups: lipophilicity-dependent processes (membrane accumulation, protein binding, binding to some metabolizing enzymes, as described by eqs 45 and 4 in sections and , respectively), and lipophilicity-independent processes (ionization, ion pairing, spontaneous reactions, some enzymatic reactions, and excretion). We will further assume that all lipophilicity-dependent processes have practically identical magnitudes of the Collander exponent β in eqs 1 and 4, when correlated with the 1-octanol/water partition coefficient P. Equation 23, with k0e = 0, then can be rewritten for the distribution or for the effects that are proportional to receptor modification (see eq 7 in section for the explanation of symbols):
 |
The complete expressions for terms A, B, C, and D are listed in section . The terms describe the individual processes that affect the chemicals’ disposition: A describes membrane accumulation and protein binding, B describes distribution in the aqueous phases, C describes lipophilicity-dependent elimination processes (based on eqs 45 and 4), and D describes other first-order elimination processes. The terms A, B, C, and D can be either constant within the series tested or dependent on global physicochemical properties of the chemicals (e.g., the pKa values, which are contained in the parameters dij, which are defined in eq 36), and of the biosystem (e.g., the pH values of aqueous compartments, which also appear in the parameters dij). Moreover, the terms C and D may also be dependent on the exact 3-D geometry of individual molecules, as well as on the spatial distribution of their properties. Similar complex dependencies are to be expected for the chemical−receptor association constant K in eq 25. In all three cases, the terms can be substituted by 3D-QSAR expressions, if necessary. In a restricted series of homologous compounds, K, C, and D may be dependent on the global properties of tested chemicals.
Equation 25 can be used to construct a model-based QSTAR expression or an SBSP model for various scenarios differing in diversity of studied compounds and biosystems. In the following sections, the application of eq 25 will be illustrated for some simple situations.
7.3.1. Lipophilicity−Bioactivity Profiles
Membrane accumulation and protein binding of chemicals can affect the elimination rates and lead to the frequently observed concave bioactivity−lipophilicity profiles, in addition to transport56,58,59,64,65,2087–2089 and equilibrium distribution,34,35,63,706,2122,2123 which have been considered as the causes of this behavior previously. This phenomenon will be illustrated in more detail with the following simple yet realistic example for a set of compounds with invariant acidity, lipophilicity-dependent binding to membranes, inert proteins, and metabolizing enzymes according to eq 25 with the Collander exponent β = 1, and metabolized solely by lipophilicity-dependent enzymatic reactions (i.e., D = 0 in eq 25).(2124) We are interested in the concentrations of the chemicals bound to proteins (which may well represent the receptors); therefore, eq 25 must be multiplied by the term BPβ, as expressed in eq 4 in section .
The concentration−lipophilicity profiles at a constant exposure time, which are commonly measured for the QSAR studies, can be seen in Figure 29A. For short times (log t < −1.5), there is no significant difference between the initial and actual concentrations. The relationships have the bilinear form that is characteristic of the lipo-hydrophilic equilibrium (curves 1−3). Later (log t > −1), the equilibrium dependencies become distorted in the region of high lipophilicity and adopt trilinear shapes (curves 5−10).
The time courses of the concentrations of the chemical−protein complexes for varying lipophilicities of chemicals are presented in Figure 29B. They have comparatively simpler shapes. The concentrations (c) are initially independent of the exposure time (t) and suddenly decrease after t reaches a certain value. The limiting value of log t is constant for lipophilic compounds (log P > 1, curves 1−4) and is inversely proportional to lipophilicity for other compounds (curves 6−10).
Figure 29C depicts the combined influence of both the exposure time and lipophilicity on distribution. The low apparent concentrations of the complexes of hydrophilic compounds (logP < 0) bound to the proteins remain practically intact, even after comparatively long incubation periods: the lower the logP values, the longer the stability period. The comparatively slow elimination of hydrophilic solutes is caused by the low affinity for the biotransformation enzymes. Moreover, the aqueous compartment, containing a prevailing part of the hydrophilic molecules, might serve as a depot that supplies the molecules for the binding processes, after the concentrations of bound chemicals decrease. This compensation mechanism works until the eliminated amount is no longer negligible in comparison with the total amount. In contrast, the concentrations of lipophilic solutes significantly decrease when the exposure is longer than log t > 0, because of the stronger binding to metabolizing enzymes, resulting in higher elimination rate constants.
The plots similar to Figure 29C can be constructed for any particular example with specific assumptions about the terms A, B, C, and D in eq 25. Their main features can be described semiquantitatively as follows. The concentration−lipophilicity profiles initially have the equilibrium shapes similar to curves 1−3 in Figure 29A. At longer times after the administration, the concentrations of the chemicals start to decrease, but the decrease is not equal for all compounds. Preferably lipophilic compounds are eliminated, if lipophilicity is a prerequisite for the binding to metabolizing enzymes (curves 5−10, Figure 29A) and mainly hydrophilic solutes disappear during elimination of the free molecules (e.g., as in excretion or spontaneous hydrolysis). The locale of the elimination process is reflected in the nonlinear shape of the relationships between the concentrations and time (Figure 29B), which can be characterized by the moment when the concentrations start to decrease. The break-point values are dependent on increasing lipophilicity in two different ways. If hydrophilic chemicals are preferably eliminated, the break-point value of log t is initially constant and then increases; for elimination that affects predominantly hydrophobic chemicals, the break-point first declines and then levels off (see Figure 29B).
The dependencies in Figure 29 were calculated using eq 25. The coefficient C, characterizing lipophilicity-dependent elimination, appears only once in eq 25, as the product with the exposure time t. Therefore, the aforementioned qualitative conclusions made for t also apply for the elimination rate constants ke or its product with the exposure time. For this reason, the exposure time t was plotted on the logarithmic scale in Figure 29.
For homologous series of non-ionizable chemicals or ionizable chemicals with approximately identical pKa values, bioactivity at a predetermined exposure time is frequently a smooth, nonlinear function of lipophilicity.6,2184 In such cases, distribution must be property-related and is probably dependent on lipophilicity. Equation 25 indicates that smooth relations can be obtained only if all the terms A, B, C, and D are either zero or constant. The terms dij and pijk (see eqs 36 and 38 in section ), accounting for ionization and ion pairing in eq 25, then can be expected to have either zero or constant values.
For a restricted series of homologous chemicals, the chemical−receptor association constant K in eq 25 sometimes is dependent on the reference partition coefficient P, and can be substituted by eq 4 (section ) with the exponent. The lipophilicity−activity profiles at a given exposure time then can be described by the following modification of eq 25, where all terms were divided by B:
 |
The coefficients C and D contain the fixed exposure time t. They both describe elimination: C characterizes the lipophilicity-dependent process and D characterizes the lipophilicity-independent process. Usually, one of the processes dominates, and eq 26 can be simplified by presetting the coefficient characterizing the minor process to zero. Selection of the term to neglect can be made by a visual comparison of the experimental lipophilicity−activity profiles with the model curves. A pragmatic rule of thumb can be formulated as follows: if the left-hand portion of the experimental lipophilicity−bioactivity profile has the strictly equilibrium shape (as curves 5−10 in Figure 29A), then D = 0; if this comparison holds for the right-hand side, then C = 0.
Equation 26 generates the lipophilicity−bioactivity profiles with a maximum, if both the receptor binding and elimination are either lipophilicity-dependent (D = 0) or invariant for the tested compounds (the first term β log P is omitted and C = 0). Otherwise, eq 26 generates the lipophilicity−bioactivity profiles with plateaus.
With regard to the limited number of compounds in the series exhibiting lipophilicity-dependent distribution and bioactivity, eq 26, with its five adjustable coefficients at a maximum, seems to be a reasonable compromise between the exactness of the description and accessible experimental information. Equation 26 with D = 0 was used to fit the published data,2124,2185 and the results are summarized in Table 10. Satisfactory fits were obtained in all analyzed cases. Within the framework of the proposed model, this fact indicates that the binding to the receptors and to biotransformation enzymes are both lipophilicity-dependent processes in each case. Inspection of the statistical indices, which were summarized for the pseudo-equilibrium, parabolic, and bilinear models in Table 10, shows that the first model provides the best fit to the analyzed data, not only with regard to the correlation coefficients, but also in terms of the standard deviations. Some systematic deviations for the parabolic and bilinear models can be seen in the smooth lipophilicity−bioactivity profile presented in Figure 30.
Table 10. Fits of eq 26 with D = 0 to Published Lipophilicity−Bioactivity Profiles and a Comparison with the Parabolic and Bilinear Models.
| model |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pseudo-equilibrium |
parabolic |
bilinear |
||||||||||||
| compounda | bioactivityb | A × 103 | β | C × 103 | E | n | F | r2 | s | r2 | s | r2 | s | ref |
| C6H5CH2N+R(CH3)2, 61c | Candida albicans, MKC | 0.719 ± 0.381 | 1.056 ± 0.040 | 2.014 ± 0.381 | 3.144 ± 0.049 | 11 | 146 | 0.990 | 0.095 | 0.923 | 0.243 | 0.968 | 0.170 | d |
| C6H5CH2N+R(CH3)2, 61 | Pseudomonas aeruginosa, MKC | 2.831 ± 0.473 | 0.929 ± 0.035 | 7.534 ± 1.078 | 2.614 ± 0.039 | 12 | 155 | 0.988 | 0.090 | 0.925 | 0.219 | 0.980 | 0.122 | d |
| C6H5CH2N+R(CH3)2, 61 | Pseudomonas aeruginosa, MIC | 21.34 ± 7.384 | 0.821 ± 0.108 | 35.75 ± 9.846 | 2.867 ± 0.081 | 9 | 11 | 0.920 | 0.136 | 0.774 | 0.208 | 0.878 | 0.168 | d |
| C6H5CH2N+R(CH3)2, 61 | Clostridium welchii, MKC | 2.813 ± 0.734 | 0.999 ± 0.048 | 4.393 ± 0.805 | 3.690 ± 0.046 | 12 | 152 | 0.990 | 0.107 | 0.960 | 0.190 | 0.984 | 0.125 | d |
| C6H5CH2N+R(CH3)2, 61 | Clostridium welchii, MIC | 3.456 ± 1.299 | 0.922 ± 0.068 | 5.441 ± 1.416 | 3.758 ± 0.066 | 12 | 64 | 0.974 | 0.154 | 0.933 | 0.230 | 0.966 | 0.172 | d |
| C6H5CH2N+R(CH3)2, 61 | Proteus vulgaris, MKC | 5.958 ± 1.730 | 0.697 ± 0.067 | 17.13 ± 4.619 | 2.896 ± 0.076 | 10 | 27 | 0.956 | 0.138 | 0.922 | 0.172 | 0.947 | 0.153 | d |
| chlorinated phenyl methacrylates, 64 | Hansenula anomala | 2.335 ± 1.647 | 0.650 ± 0.123 | 2.267 ± 1.655 | 1.656 ± 0.309 | 10 | 11 | 0.895 | 0.078 | 0.795 | 0.101 | 0.884 | 0.082 | e |
| n-alkanes, C5H12−C17H36 | mice, LD100 | 0.482 ± 0.214 | 0.639 ± 0.043 | 1.042 ± 0.447 | 0.217 ± 0.016 | 13 | 94 | 0.980 | 0.065 | 0.748 | 0.217 | 0.947 | 0.104 | f |
Some structures are given in Chart 5.
MKC, minimum killing concentration; MIC, minimum inhibitory concentration, LD100, dose that is lethal in 100% of cases.
Chlorides.
The data and the parabolic equation are taken from ref (6); the bilinear equation is taken from ref (64).
Figure 30.

Inhibitory activity of n-alkanes in mice (LD100 in mol/kg)(64) versus lipophilicity, expressed as the 1-octanol/water partition coefficient P. Data were fitted with eq 26 (D = 0, the values of other adjustable coefficients are given in the bottom line in Table 10) (solid line),(2124) the bilinear equation described by eq 9 (dashed line), and the parabolic equation described by eq 8 (dotted line).(2124)
The concave lipophilicity−bioactivity profiles were most frequently explained by the equilibrium accumulation and transport phenomena.(6) The pseudo-equilibrium model offers another alternative: the influence of a lipophilicity-governed accumulation of chemicals in the membranes and lipophilic binding sites of proteins on the actual rate of elimination (Figure 29A). For simple biosystems such as cellular suspensions, the pseudo-equilibrium model, assuming a fast attainment of the lipo-hydrophilic equilibrium, is more realistic than the transport-based models. The concave lipophilicity−activity profiles are frequently observed several hours after drug administration, but the results of uptake experiments indicate that the transport and distribution are finished within a few seconds or minutes.2126–2128
The description of experimental data by concave lipophilicity−activity profiles serves mainly as an elementary validation of the pseudo-equilibrium model. In the case of one independent variable, the model helps in the interpretation of the results, and in the prediction of the behavior for longer exposure times. The real value of the pseudo-equilibrium model becomes apparent in more-complex situations with the interplay of several properties, where finding the optimum magnitudes of properties is much more difficult.
7.3.2. Influence of Ionization
The majority of drugs and many other chemicals ionize under physiological conditions. This process significantly affects their disposition in biosystems. Ionization is characterized by the dissociation factors dij in the terms A, B, C, and D in eq 25 (details are given in section ). According to eq 36 in section , each factor dij is the product of two terms: one contains only the pKa values, and the other term contains only the pH values of the subcellular compartments. The order of summations in section can be changed so that the jth summation with the terms dij becomes the leftmost summation. Each term A, B, C, and D (collectively represented as Y) can be deconvoluted into the sum of terms that are associated with each ionization degree:
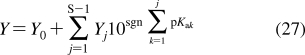 |
The terms Y can be treated as adjustable coefficients, if the terms they contain do not vary for the tested series. As in eq 33 (section ), the sign function (sgn) has the value of the ion charge: sgn = −1 for anions and sgn = +1 for cations. The subscripts 0 and j indicate the coefficients, which are valid for the non-ionized compounds and for the jth ionization species (total number S−1), respectively.
Equation 27 can be used for a series of compounds that have the same ionizable groups, with the pKa values separated by 2−3 units. More-similar macroscopic pKa values are no longer equal to the microscopic pKa values, and the population of the molecular species becomes more complex. Different species may have the same charge (e.g., neutral molecules and zwitterions), and their prevalence cannot be calculated using the macroscopic pKa values. Under such conditions, the ionization fractions αij of individual species must be calculated before the optimization of the coefficients in the QSAR equations. The microconstants and the ionization fractions can be calculated for the given pH value, e.g., by the SPARC (SPARC Performs Automated Reasoning in Chemistry) approach,2159,2160,2177,2186,2187 which performs well for druglike compounds(2168) and is also available as a web-based service.(2188) The approach utilizes the linear free-energy relationships(51) for prediction of thermodynamic effects and perturbation molecular orbital theory(2189) to describe delocalization and polarizability. For compounds with close pKa values, the deconvolution of individual terms A, B, C, and D in eq 25 or eq 26 is preferably made as
 |
The ionization fractions αij are related to the quantities dij (defined by eq 36 in section ) as dij = αij/αi0, where αi0 is the fraction of the neutral species. Note that the terms Y have different meanings in eqs 27 and 28.
Equation 25 with the coefficients A, B, C, and D deconvoluted according to eq 27, was applied to the inhibition of Sarcina lutea by 32 lincomycin analogs(2190) (65 in Chart 5), with the inhibitory concentrations cX expressed as the ratios to the lincomycin activity. The following form, with the indicator variable Itrans used for the trans-derivatives, generated the best fit:
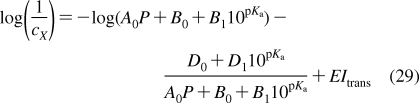 |
The nonlinear regression analysis provided the following optimized values of adjustable coefficients: A0 = (1.183 ± 0.017) × 10−2, B0 = (1.123 ± 0.011) × 10−2, B1 = (2.077 ± 0.016) × 10−10, D0 = (5.145 ± 0.049) × 10−2, D1 = (5.504 ± 0.032) × 10−10, and E = 0.283 ± 0.056. The satisfactory quality of the fit can be seen in Figure 31, and it is also indicated by the low values of the standard errors of coefficients and the following statistical indices: n = 31, r2 = 0.968, s = 0.125, F = 103.2.
Figure 31.

Growth-inhibiting activity of lincomycin derivatives(2190) against Sarcina lutea in the agar diffusion test, as a function of acidity, given as the negative logarithm of the dissociation constant Ka, and lipophilicity, parametrized by the 1-octanol/water partition coefficient P.(34) The surface corresponds(20) to eq 29. The fitted activities of the trans derivatives (not shown) form a similar surface, raised by 0.283 logarithmic units. The outlier did not conform to the smooth surface. More details are given in the text.
In the context of the pseudo-equilibrium model, the results, including the optimal form of eq 29 and the optimized values of the adjustable coefficients, can be interpreted as follows. The values of the lincomycin−receptor association constants K (eq 25) are identical for all cis derivatives and all trans derivatives, the latter set showing a higher affinity, as indicated by the optimized value of coefficient E. If the cis−trans isomerism would affect processes other than receptor binding, eq 29 would show a better fit if the term EItrans would be combined with the term A in the case of binding to inert proteins, or the term D in the case of binding to metabolizing enzymes. The remaining variation in the antimicrobial potency of lincomycins seems to be explained by their distribution in the microbial suspension. Non-ionized and ionized molecules are eliminated at two different rates, which are contained in the coefficients D0 and D1, respectively. Neither elimination rate is dependent on lipophilicity, indicating that the responsible processes are either spontaneous reactions or lipophilicity-independent enzymatic reactions. Protein binding is exclusively lipophilicity-related, and the ionized and non-ionized molecules do not differ in this aspect, as indicated by the absence of the coefficient A1 from eq 27 in eq 29. The distribution of lincomycins in the aqueous phases is influenced by ionization, because eq 29 contains both B0 and B1 terms from eq 27.
Equation 25 was used to correlate, with lipophilicity and acidity, the published toxicities against Tetrahymena pyriformis of 129 monoprotic phenols(2191) and 7 phenols containing two or three ionizable groups.(2192) The Tetrahymena toxicities were frequently analyzed by other approaches.2193–2222 For the monoprotic phenols, the following modification of eq 25, with the values of the coefficients A, B, C, and D deconvoluted according to eq 27 and the entire expression divided by B0, provided the best fit:
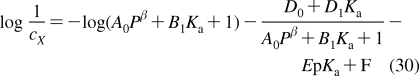 |
The toxicity cX is expressed as the concentration (mmol/L) of the tested phenolic compounds causing 50% reduction in the growth of the protozoa after the 96-h exposure. The first two terms in eq 30 come from the disposition function, as given by eqs 25 and 27, and the third term represents nonspecific binding to the receptors (K in eq 7 in section or in eq 25). The values of individual adjustable coefficients were optimized as follows: A0 = 0.1830 ± 0.1033, β = 0.2665 ± 0.0631, B1 = 6407 ± 3805, D0 = 5.471 ± 0.929, D1 = 16020 ± 8630, E = 0.1758 ± 0.0143, and F = 5.393 ± 0.330. Some coefficients were preset (B0 = 1 and A1 = C0 = C1 = 0), because their optimization did not improve the fit. Statistical indices of the fit for n = 122 experimental points were r2 = 0.908, s = 0.257, and F = 191.0. One compound was omitted due to an additional mechanism of action, and six compounds were inactive. All inactive compounds were predicted to have lower activities than the highest tested concentrations. The agreement between the experimental and calculated toxicities is satisfactory, as shown in Figure 32A.
Figure 32.

Acidity−lipophilicity−toxicity profiles for phenols against Tetrahymena pyriformis(55) generated by the model-based eq 30 (panels A and C) and the best empirical model (panels B and D) for the complete set of compounds (panels A and B) and for the reduced set of compounds (panels C and D) as used in leave-extremes-out cross validation. The omitted points in the reduced set are indicated by empty circles.(55) The model-based equations are able to predict beyond the used parameter space, whereas the empirical models fail in this aspect. Toxicity (T) values are the inverse values of the isoeffective concentrations (in mmol/L) causing 50% growth inhibition after 96 h of exposure. More details are given in the text.
The model described by eq 30 can be interpreted as follows. The differences in toxicity are essentially ascribable to distribution, although a small effect of the receptor binding is apparent. Significantly lower toxicities are observed for compounds with (i) pKa > 6 and log P < 4, and (ii) pKa < 2, as seen in Figure 32A. The low toxicities are caused by metabolism that occurs in aqueous phases of the cells and proceeds with approximately equal rate (micro)parameters for all non-ionized and all ionized molecules, as expressed by the coefficients D0 and D1 in eq 30, respectively. The faster metabolism of the ionized species (D1 ≫ D0) causes a rapid decrease in the toxicity of very acidic derivatives (pKa < 2), regardless of their lipophilicity (Figure 32A). The hydrophilic compounds are metabolized faster because they are present in the aqueous phases in higher concentrations than lipophilic derivatives, which are preserved from being metabolized by accumulation in the membranes, and by nonspecific binding to proteins. Membrane accumulation and protein binding of ionized species do not contribute significantly to the total distribution of the compounds in the protozoan suspension, as indicated by A1 = 0 in eq 30. The term EpKa reflects the association of phenol molecules with the receptors. The sign of the optimized value of the coefficient E is negative, i.e., the strength of the association rises with the increase in the electron withdrawal from the phenol group. According to the model, the receptors seem to be localized in the aqueous phases. Phenols probably bind in one ionization state, because a multispecies binding would require a more-complex expression. The data do not provide enough information to identify the binding species. Other plots in Figure 32 will be analyzed in section , which compares the performance of the pseudo-equilibrium approach with that of empirical models.
For all phenolic compounds, including those capable of multiple ionizations, the best results were obtained using the following equation:
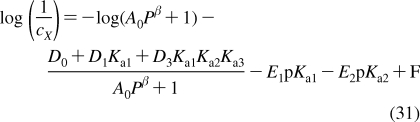 |
Individual adjustable coefficients were optimized as follows: A0 = (9.269 ± 3.838)×10−3, β = 0.6106 ± 0.0422, D0 = 3.582 ± 0.118, D1 = 3.860 ± 1.171, D3 = (1.250 ± 0.286) × 10−25, E1 = 0.1248 ± 0.0158, E2 = (4.789 ± 1.246) × 10−2 and F = 4.437 ± 0.173. Statistical indices of the fit were r2 = 0.870, s = 0.277, and F = 121.9. To the best of our knowledge, eq 31 is the first QSAR correlation that conceptually includes multiple ionizations.
7.3.3. Influence of pH of the Medium
In the case of ionizable compounds, the in vitro bioactivity determination can be done under conditions of varying acidity of the external medium. A study of the pH dependence of the QSAR coefficients may provide a deeper insight into the role of ionizable groups in distribution and receptor binding. The buffering capacity of the intracellular compartments in biosystems is heavily dependent on the presence of inhibitors, as well as on the metabolic and physiologic state of the cells.(2223) As the first approximation, the intracellular pHi changes under the influence of external pHe can be expressed, assuming identical sensitivity (ϑ) in all compartments, as
Despite its simplicity, eq 32 could describe the situation with sufficient precision, because the variation in pHe in the experiment usually cannot be greater than ∼3 units, to preserve the proper function of the cells. The robust eq 32 allows for some flexibility: it allows pHi to respond proportionally to the change in pHe, as well as keeping pHi independent of pHe, if ϑ = 0.
The extracellular and intracellular aqueous compartments differ in the pH values. Therefore, it is useful to distinguish them also in the expressions for the coefficients A, B, C, and D in eqs 25 and 27. Equation 27 then can be extended by deconvoluting each term Yj for ionizable compounds into two terms—Yje for the external aqueous phase and Yji for the intracellular aqueous phases—as
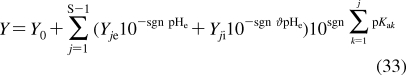 |
The model was applied to the data on antimicrobial effects and lipophilicity of the homologous series of α-bromoalkanoic acids,(6) which were measured in media with pH values 6.0, 6.5, 7.5, and 8.5. The compounds have approximately identical dissociation constants and belong to the group of alkylating agents, which are readily eliminated, even in vitro, by reactions with the nucleophilic groups of the cell components. Satisfactory fits were obtained with the following modification(2224) of eq 25, with all terms divided by B0:
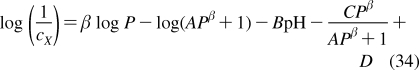 |
Here, cX is the minimal killing concentration. The first term reflects the dependence of the acid−receptor association constants K (eqs 7 and 25) on the P values. The linear pH term originated from a graphical analysis(2224) of the behavior of eq 33. The application of the concept of varying pH in the intracellular compartments was an important step in the model development, which allowed for a successful fit to the data. Otherwise, if the internal pH would be considered invariant, the magnitude of the slope B in the pH term could only be 1 or 0, while the fitted values varied in the range of 0.3−0.4.
The optimized values of the adjustable coefficients are given in Table 11. As follows from the values of the standard deviations of the coefficients and those of statistical indices in Table 11, as well as from Figure 33, the fit is satisfactory. All coefficients are statistically significant, despite the fact that the ratio between the value and the standard deviation of the coefficient C is somewhat larger than are those of the other linear coefficients B and D. This fact is due to the small number of lipophilic compounds (log P > 3) in the dataset.
Table 11. Fits of eq 34 to the Data on Inhibitory Potency and Lipophilicity of α-Bromo-Alkanoic acids(6) (RCHBrCOOH, where R = C4H9−C20H41 with even numbers of carbons) in Media with pH = 6.0, 6.5, 7.5, or 8.5.
| microorganism | β | A | B | C | D | n | r2 | s | F |
|---|---|---|---|---|---|---|---|---|---|
| V. cholerae | 0.692 ± 0.050 | (5.214 ± 1.362) × 10−3 | 0.389 ± 0.045 | (1.612 ± 0.365) × 10−2 | 5.356 ± 0.335 | 19 | 0.951 | 0.195 | 51 |
| D. pneumoniae | 0.849 ± 0.053 | (1.117 ± 0.495) × 10−4 | 0.339 ± 0.079 | (4.733 ± 2.008) × 10−4 | 5.364 ± 0.601 | 24 | 0.933 | 0.315 | 50 |
| S. haemolyticus | 0.847 ± 0.046 | (8.445 ± 3.457) × 10−5 | 0.357 ± 0.075 | (4.029 ± 1.595) × 10−4 | 5.083 ± 0.569 | 24 | 0.947 | 0.299 | 63 |
Figure 33.

Dependence of the inhibitory activity of α-bromoalkanoic acids against V. cholerae on the 1-octanol/water partition coefficient P and the pH values of the medium. Toxicities (T) are given as the inverse values of the minimum killing concentrations. The surface corresponds to eq 34, with the optimized values of adjustable coefficients given in Table 11.(2224)
The fit of eq 34 to the experimental data highlights several characteristics of the process.(2224) The pH dependencies of the coefficients A, B, and C in eq 34, which represent the accumulation of chemicals in the membranes and protein binding, the pure distribution in aqueous phases, and enzymatic biotransformation, respectively, are parallel. The dependence on lipophilicity of the chemical−receptor association constant, the membrane/water partition coefficient, and the chemical−protein association constant can be described by the Collander equation (eq 1 in section ), with the averaged value of the exponent given in Table 11. Unfortunately, the linearity of the pH term in eq 34 precludes the identification of the species (ionized or non-ionized molecules), which participate in the receptor binding.
The potential number of adjustable coefficients is rather high in eq 25, which must be combined with eq 27 in the case of chemicals with differing pKa values, or with eq 33 if media with varying pH values were used. Therefore, each QSAR application of these equations should carefully examine, which of the adjustable coefficients A, B, C, and D really must be deconvoluted, according to eqs 27 or 33. Initial estimates can be based on a graphical examination of the model behavior for a realistic range of coefficient values,(2224) as well as on the comparison of the plotted experimental data with the model plots. The guesses must be confirmed by coefficient optimization. A more rigorous approach is to automatically examine all possible combinations of the adjustable coefficients in the fits.
8. Comparison with Other Approaches
SBSP involves the interactions of chemicals with organisms at all complexity levels. These interactions are also a subject of other techniques, methods, and approaches. The following two sections compare SBSP with two widely used approaches: empirical QSAR analysis and classical pharmacokinetics.
8.1. SBSP versus Empirical QSAR Approaches
Model-free fitting methods have always been popular, because of their ease of use. In drug design, neural networks, as well as empirical models consisting of polynomials with or without cross terms, with the coefficients optimized by linear regression analysis, PLS, and genetic algorithms, have been used routinely. If a simple linear combination of lipophilicity and acidity does not provide a good fit, some modelers resort to a search for new descriptors, instead of looking for the true causes of the nonlinearity or the scatter. As a result, hundreds of descriptors have been suggested, without showing that they are related to the kinetics or thermodynamics of any of the processes, which chemicals undergo in biosystems. Instead of the quest for conceptual models, construction of a QSAR model has been reduced to the deployment of hundreds of descriptors, the number of which frequently widely exceeds the number of tested chemicals. The aforementioned methods always find many of the numerous solutions in these underdetermined systems. The best solutions are then picked based on a cross validation. In contrast to conceptual SBSP models, there is no mechanistic information utilized or tested by the empirical approaches. Conclusions based on the presence or absence of certain descriptors in the final selection of descriptors are of limited value, because of the incorrect, oversimplified form of the fitting equations. The main outcome of the model-free techniques is the prediction of disposition or effects of untested chemicals.
How do the prediction abilities of conceptual SBSP models and empirical models compare? Let us illustrate this issue by fitting the phenol toxicities against Tetrahymena pyriformis (section ). Polynomials with cross terms have been shown to perform equally well or better than several neural network architectures with a higher number of adjustable weights.(2225) The polynomials with cross terms of log P and pKa, containing up to 10 optimized coefficients, were chosen as representative empirical models and were fitted to the data.(55) The fit of the best polynomial model was satisfactory, as seen in Figure 32B. The statistical indices were slightly better than those for the model-based equation described in eq 30. A similar trend was also observed for the standard cross-validation approaches using either the omission of one compound or a random omission of several compounds (see rows 1−3 in Table 12). By all common criteria, the empirical model was carefully cross-validated and would be recommended as a reliable prediction tool by any QSAR practitioner.
Table 12. Cross Validation of the QSAR Models for Toxicity of Phenolic Compounds against Tetrahymena pyriformis(2191,2192).
| PRESSa |
|||
|---|---|---|---|
| method | number of groups | eq 30 | empirical model |
| leave-one-out | 122 | 9.577 | 7.981 |
| leave-several-out | 20 | 9.458 | 7.486 |
| leave-several-out | 7 | 9.821 | 7.242 |
| leave-extremes-out | 1 | 6.055 | 30.40 |
PRESS is the predictive sum of squares of deviations between the calculated and experimental values of the omitted points. The optimized coefficients of the SBSP-based equation 30 are given in the text, and the corresponding surfaces are shown in Figures 32A and 32C. The best empirical model is described in the text, and the corresponding surfaces are shown in Figures 32B and 32D.
The models are mainly needed for predictions outside the parameter space. The reason is that predictions for new compounds, which have the parameter values in the ranges defined by the parameter values of tested compounds, can essentially be done using various model-free intrapolations.1415,2226,2227 Cross validation based on a random omission of data points does not rigorously address predictions outside the tested parameter space. To address this issue, a cross-validation technique that we call leave-extremes-out (LEO cross validation) was devised.(55) As the name indicates, the data points with high and low values of both independent variables were omitted. For the phenol toxicities, the dataset then included 97 compounds with logP values of 1.0−5.3, and pKa values in the range of 5.0−11.0. These limits were chosen arbitrarily to leave out at least four compounds beyond each side of the log P−pKa property rectangle. The calculated predictive sum of squares of deviations (PRESS) values (see Table 12, last row) for 25 omitted compounds show that the LEO cross validation provides significant discrimination between the SBSP and empirical models. The model-based eq 30 predicts outside the parameter space much better than the best empirical model. As seen in Figure 32D, the omission of compounds with extreme log P and pKa values causes the empirical equations to dramatically change the surface shape in the region of omission. The flexible polynomials are not able to approximate the flat surfaces, which are typical for the SBSP models. In contrast, the model-based eq 30 maintains the shape of the associated surface, even if optimized for the reduced parameter ranges, as shown in Figure 32C.
The authors working with empirical approaches frequently state that the models cannot be used for predictions outside the tested parameter space. The results of the LEO cross validation show that they are correct, as long as they talk about empirical models.
8.2. SBSP and Classical Pharmacokinetics
Among the attempts to describe the relationship between chemical structure and the kinetics of disposition, two main directions can be distinguished, based on either classical or subcellular pharmacokinetics. Although having similar objectives, both approaches have developed separately, and the mutual connections are rather loose. This situation is partially related to different purposes, for which the resulting expressions are used. Classical pharmacokinetics focuses on a description of the time course of a single drug in the body. The primary objective of SBSP is describing the differences in the kinetics of disposition of sets of chemicals in subcellular compartments.
Classical pharmacokinetics involves a phenomenological description of the time course of the drug concentration in tissues, organs, and organisms. The biosystem is represented by a variably structured set of kinetically distinct macroscopic compartments, which may correspond to the blood plasma and organs; however, more often, they have very loosely defined morphological basis. Drug disposition is expressed in terms of the space-averaged drug concentrations in the nonhomogeneous macroscopic compartments. Although these concentrations are of great value for therapeutic purposes, the analysis of the drug effects at the molecular level requires the use of the actual drug concentrations in the immediate surroundings of the receptors. The phenomenological coefficients in the models of classical pharmacokinetics are not inherently related to drug structure. The relations are sought additionally from partial models for individual pharmacokinetic parameters,45,68,1313,2228–2232 such as the absorption rate constants (section ),34,35,57,69,1747,1979,2097,2099,2100,2102,2233–2250 volumes of distribution (section ),665,1965,2251 and elimination rate constants (section ).1404,1980,1981,1984,2252–2254
The objective of SBSP is to describe the time course of the chemicals’ concentrations in individual physically distinct subcellular compartments such as the extracellular and intracellular aqueous phases and membranes. This fact allows for broadening the scope to the fates of chemicals in microorganisms, suspensions of cells, or subcellular organelles. Numerical simulations of the fates of chemicals in biosystems provide insight into distribution-based QSARs (section ). Complexity of the mathematical description in the explicit SBSP models has been reduced in the solution of the pertinent differential equations using simplified scenarios (section ) or the experimentally verified time hierarchy of the processes that determine the disposition of chemicals (section ).
The form of expressions describing the time courses of drug distribution has been repeatedly proven by the compartmental models of classical pharmacokinetics. Therefore, a proper application of the SBSP concepts to the chemical disposition in organisms should result in expressions that provide similar time dependencies as the compartmental equations of classical pharmacokinetics, the difference being that the global model parameters of classical pharmacokinetics are now given in terms of structure of both the biosystem and drugs. Equation 25, which is equivalent to the one-compartment open model of classical pharmacokinetics, is a good example of this premise.
The most advanced models of classical pharmacokinetics are the entire-body physiological models, which provide a more-detailed picture of the concentrations of chemicals in individual organs and different parts of the bloodstream.(2255) The use of physiological models, currently hampered by the need for more-detailed experimental data,(2256) is expected to expand. The SBSP principles can be applied to physiologic models to the same extent as the compartmental models.
There is an overlap between individual approaches to the modeling of the interactions of chemicals with biosystems. For instance, the terms A, B, C, and D of eq 25 can help correlate pharmacokinetic parameters with the properties of drugs (quantitative structure−pharmacokinetics relationships) and biosystems (interspecies scaling, population pharmacokinetics). A comparison of individual features of distribution-based QSAR (section ), classical pharmacokinetics, and SBSP (sections and ) is given in Table 13.
Table 13. Some Differences between Distribution-Based QSAR, Subcellular Pharmacokinetics, and Classical Pharmacokinetics.
| pharmacokinetics |
|||
|---|---|---|---|
| aspect | distribution-based QSAR | subcellular | classical |
| compartment nature | microscopic, homogeneous | microscopic, homogeneous | macroscopic, nonhomogeneous |
| model simplification | fixed time | time−hierarchy-based | phenomenological |
| concentrations | actual | actual | space-averaged |
| relation “parameters vs. properties” of | |||
| chemicals | LFER-based | LFER-based | loose |
| biosystem | loose | defined | loose |
| kinetics included | no | yes | yes |
8.2.1. Absorption
Absorption denotes the transport of chemicals from the site of their first contact with a multicellular organism into the bloodstream. The main routes in humans are represented by intestinal, transdermal, and pulmonary absorption. For drug administration, the buccal, ocular, rectal, and vaginal routes are used when appropriate.
Transdermal absorption can be conveniently studied using various skin preparations, and is described for the systems of increasing complexity, including the available QSAR models, in sections , , and . SBSP can contribute to the improvement of these models by taking into account the cellular nature of the subdermal tissue and capillary walls.
Intestinal absorption, proceeding mainly in the small intestine, is more difficult to study than transdermal absorption, because of the roles of carrier-mediated transport (section ), vesicle-mediated transport (section ), differences in physiology of individual intestinal segments, and the transit of the changing intestinal content through the intestine. The systems for studying some aspects of intestinal absorption range in complexity from brush border membrane vesicles (section ), isolated enterocytes (section ), everted intestinal rings (section ), Caco-2 cell monolayers (section ), the entire intestine used as everted sacs or a barrier in diffusion chambers (section ), to the ex vivo or in situ perfused intestine (section ). The structure−transport relationships are covered in the respective parts.
Here, we will only review the approaches that utilize the human or animal data and attempt to predict the fraction absorbed as a function of properties of chemicals. The organism-level data capture all details of the intestinal absorption process, but they are also affected by possible interactions of chemicals with other body components. Consequently, the data have significant potential for errors if treated as purely absorption data. The fraction absorbed adopts a limited range of values; therefore, it should not be used as a dependent variable in linear models, because they cannot be properly calibrated in this situation.(2257) A transformation of the fraction absorbed values to the first-order absorption rate constants69,2249,2258–2260 is a better approach, which, however, may not always lead to a proper representation of the absorption process. Numerous reviews,25,46,2231,2257,2261–2279 as well as some book chapters1313,2228,2280−2283 and books,(2284) examine the relationship between oral bioavailability and the structures and properties of chemicals. Intestinal absorption of drugs is affected by many factors, such as dosage forms, physiological and psychological status, food, physical activity, and differs even if a person is lying on the left side or right side. The data for the model calibration must be standardized or, otherwise, only fuzzy and approximate models can be generated.
We will first focus on the models for water-soluble compounds. Early models of intestinal absorption were often plagued by the use of oversimplified biosystem representations, based on a hydrophobic slab, which is a single bilayer or membrane, whereas the data used for the model calibration were measured in an organism. As an example, linear1937,2233,2285 and parabolic(2286) dependences of the absorption rate constants of several chemicals in rabbits and rats on lipophilicity can be mentioned.
Conceptual nonlinear expressions on lipophilicity were formulated for the equilibrium extraction as the rate-limiting process of absorption(2235) and for the effect of diffusion in the unstirred layers on the overall process.57,560,2099,2101,2287,2288 A series of models with lipophilicity and dissociation constants as variables was created(69) based on the pH-partition hypothesis.(2134) The models explained the experimentally observed pH shift, as the consequence of the diffusion in unstirred layers. A size exclusion limit of 250 Da was suggested for intestinal absorption by the paracellular route, giving rise to further nonlinearities in the rate−lipophilicity dependence.2238,2289–2291 Similar considerations led to the formulation of the permeability model consisting of a molecular-weight-dependent set of sigmoids on lipophilicity.2292,2293
Sigmoidal dependences were found for the measures of hydrogen-bonding ability of molecules, the sum of the hydrogen-bond donor and acceptor fragment values,(2294) the sum of the molecular hydrogen-bonding potentials,(2295) and the sum of hydrogen-bond donors and acceptors.(2296) The predictions were improved if the chemicals were classified according to similarity and the sigmoidal dependence was applied to the subsets.(2297)
Similar sigmoidal dependences of the fraction absorbed were found for the polar surface area (PSA).(2245) Although the initially used PSA was obtained as a Boltzmann-weighted average of all low-energy conformations, a much simpler approach based on a single low-energy conformation,(1749) even using a fast fragment-based approach,(1127) provided similar results for the studied datasets. For more-diverse datasets, the sigmoidal dependence still held but contained much more scatter.1749,2269,2272 Limiting both PSA and molecular flexibility was claimed to improve absorption of chemicals in rats,(2298) but this conclusion could not be generalized.(2299) The PSA combined with lipophilicity were able to distinguish, to some extent, between compounds with poor and good human intestinal absorption.(2300)
In a more-complex quantitative model of fraction absorbed, PSA was augmented by the partition coefficient at pH 6.5, the square of the hydrogen-bond count, and the number of Rule of Five2301–2303 (see below) violations.(2304) A PLS model of human oral bioavailability, combined with Caco-2 permeability, was derived using easy-to-calculate parameters.(2305) A classification scheme for four categories of human oral bioavailability identified the 1-octanol/water partition coefficient at pH 6.5 and its difference with that at pH 7.4 as important parameters, capturing the overall trend of increasing bioavailability: bases < neutral compounds < acids.(2248) Neural network models with numerous descriptors were used to predict oral bioavailability.2306–2308 The solvatochromic approach, valid for solvation of molecules in one or two phases, was applied to human absorption data,2249,2258–2260 subject to limitations described in section . Fragment-based approaches, which work well for the prediction of individual partition coefficients, were applied to more-complex human permeability data, with limited success.2309–2311 Molecular interaction fields as used in the 3D-QSAR techniques were generated for the inertial field superposition of chemicals and related to human absorption by the principal component analysis (PCA).(2312) The PLS regression on a set of lipophilic and electronic descriptors was used to models human absorption.(1315) Topological descriptors, with loosely defined relations to the processes, which the chemicals undergo during absorption, were used for a correlation with human absorption data via fuzzy adaptive regression,(2313) neural networks,2314,2315 and linear discriminant analysis,(2316) and for the classification-based descriptor selection.(2317)
Semiquantitative ranges of properties that are typical for drugs entering Phase II clinical trials, presumably having a good absorption, were summarized as the Rule of Five:2301–2303 molecular weight < 500 Da, the estimated 1-octanol/water partition coefficient < 105, number of hydrogen-bond donors/acceptors (sum of OH and NH) < 5, number of hydrogen-bond acceptors (sum of O and N) < 10. A compound violating any two rules has an increased probability of absorption problems. The ranges were reduced and summarized as the Rule of Three for fragments, which serve as the building blocks of drug candidates and have the structures further developed and optimized.(2318) The original authors2301–2303 observed that the Rule of Five does not hold for actively transported chemicals. Other exception for families of drugs acting against specific receptors was highlighted.(2319) A comparison of the Rule of Five properties between orally and intravenously administered drugs indicated significant differences.(2320) A more-detailed scheme, titled “A Bioavailability Score” (ABS), used the ranges of different properties for different overall charges of the permeants.(2321)
Intestinal absorption of sparingly soluble drugs and chemicals is further complicated by the availability of free molecules for the transport process.(2322) The effect of solubility was first addressed by the use of the Absorption Potential, which was defined as the product of the 1-octanol/water partition coefficient, the fraction non-ionized at pH 6.5, aqueous solubility, the intestinal lumen volume, and the inverse value of the dose. For seven diverse drugs, a sigmoidal dependence of the fraction absorbed on this descriptor was observed.(2322) The Absorption Potential was later modified by the introduction of the liposome distribution coefficients(342) and simplified by the use of intrinsic aqueous solubilities.(2323) The veracity of the experimental solubilities for intestinal absorption in humans is increased by the use of media imitating the relevant physiological media.(2324)
A semiquantitative estimate of oral bioavailability can be made using the Biopharmaceutics Classification System, which divides drugs and drug products into four categories, using low and high magnitudes of permeability and solubility.(72) The classification is used to determine the type and extent of the tests, which are required for legal approval of drugs. Extensions of the system to include elimination criteria were proposed.74,180,2325
Physiology-based models have the best chance to capture all relevant factors affecting oral bioavailability: geometry of the intestine, intestinal transit times, dissolution and permeation of the given dose of a chemical from the intestinal content exhibiting a pH gradient, metabolism, and excretion.(2326) The small intestine is represented by a tube ∼280 cm long, which is subdivided into the duodenum, jejunum, and ileum, representing ∼5%, ∼50%, and ∼45%, respectively, of the overall length. The tube has the gradually decreasing radius, from 1.75 cm at the pyloric sphincter to 1 cm at the ileocecal sphincter. The surface area in contact with the lumen is increased by folds, villi, and microvilli, which are less abundant toward the ileocecal sphincter, resulting in the gradual decrease of the effective transfer area. Absorption of chemicals in physiological systems was simulated using compartment models,2327–2332 a heterogeneous tube model,(2333) a segmented-flow model,(2334) and an intestinal transit function.(2335) In addition to the modeling framework, individual models differ in the quality of the approximations, which are used to replace the model parameters, which would be too tedious to measure for each chemical. For instance, intestinal permeability can be replaced by in vitro rabbit or monkey data (section )2247,2336 or by predictions from QSAR models.
8.2.2. Elimination and Clearance
Elimination represents the summation of all processes contributing to the irreversible removal of chemicals from the biosystem. In the context of the pseudo-equilibrium model, the global first-order elimination rate parameter is defined by eq 23 in section as ke = Cl/Vdf, where Cl is given by eq 62 and Vdf by eq 60 (in sections and , respectively). The elimination rate constant is the sum of the apparent rate parameters of all eliminating processes, each weighted by the volume of the phase where the process is encountered and divided by the unbound non-ionized distribution volume (Vdf). Equations 23 and 24, if processed as described in section , define the relations between the elimination rate constant and properties of chemicals and biosystems. The approach was used to correlate microbial degradation rates of simple chemicals with lipophilicity.(2337) More-precise estimates for a broader series of compounds would require the use of the 3D-QSARs for structure-specific process such as metabolism and active secretion, replacing the rate parameters e and x in eq 62 in section .
The QSAR models for overall elimination and clearance can be considered to be in an early stage of development. The dependence of the dose-dependent elimination in rabbits on lipophilicity was studied for a small set of barbiturates.(1981) The clearance of barbituric acids in rats exhibited a nonlinear dependence on lipophilicity.(1314) The clearance of 14 β-adrenergic antagonists was deconvoluted into the renal and nonrenal contributions, both depending in a parabolic way on lipophilicity.(1296) A classification scheme for high and low urinary excretion of drugs was created using numerous descriptors processed by linear discriminant analysis and recursive partitioning.(2338) The kth-nearest neighbor technique was used to predict clearance of 44 antibiotics.(2339) Renal clearance was correlated with 3D descriptors,(1304) with electronic and lipophilicity descriptors(1315) and with Volsurf descriptors,(2340) using either multivariate or PLS regression, and with topological descriptors using neural networks.(1303)
8.2.3. Volume of Distribution
In classical pharmacokinetics, the volume of distribution Vd relates the administered dose of a drug or the actual drug amount at a given moment, to the total plasma drug concentration ciT, which can be easily measured. The pseudo-equilibrium model uses the unbound non-ionized distribution volume Vdf (eq 60 in section ) that is equal to the ratio of the dose to the free non-ionized drug concentration cA. The conversion of Vdf to Vd is described by eq 61 in section . The resulting equations are in accordance with the expression for the volume of distribution being equal to the plasma volume and the summation of products of the tissue/plasma partition coefficients and the tissue volumes.(2341) A combination of eqs 60 and 61 provides a recipe for the correlation of the distribution volume with the properties of chemicals and biosystems. For nonionizable compounds or congeneric acids, the fragments of the dependencies on lipophilicity that result from eqs 60 and 61 were observed.68,1296,1314,2230,2251,2342–2344 For ionizable drugs, the first estimates have been made without fitting, using the 1-octanol/water and olive oil/water partition coefficients,(559) the distribution coefficients and fractions ionized at pH 7.4,(2345) and the experimentally determined fraction unbound in plasma as the input parameters. Numerous descriptors were utilized in the stepwise regression analysis,2346,2347 the fuzzy adaptive regression analysis,(2348) the variable selection by the genetic algorithms,(2349) the PLS regression,1304,1315 and neural networks1303,2350 to create the empirical models of the volume of distribution. The IAM partitioning and albumin binding data were used for the same purpose.(2351) The kth-nearest neighbor technique in the space of numerous topological and physicochemical descriptors was used to predict the distribution volumes of 44 antibiotics.(2339) Lipophilicity, ionization, molecular weights, and various drug fragments served as the variables in a discriminant analysis and the Random Forest model for the human distribution volume of 384 drugs.(2352)
For more-precise correlations, eqs 60 and 61 must be processed analogously as was done for the derivation of model-based QS(T)ARs in section . Further increase in the quality of description can be expected with the use of more appropriate surrogate phases for phospholipids and triglycerides, and the replacement of the parameters for protein binding, which are contained in the parameter bijk (eqs 60 and 61), by the 3D-QSARs for binding to preponderant proteins, as described in section .
9. Conclusions and Outlook
SBSP models the behavior and, in some cases, the effects of chemicals in biosystems, as determined by the physicochemical properties and structures of both chemicals and biosystems. Absorption, distribution, metabolism, excretion, and toxicity (ADMET) are, in practice, often predicted using empirical modeling with molecular descriptors selected from large pools using exclusively statistical criteria, without paying much attention to the mechanisms of involved steps. These approaches try to capture the complexity of the ADMET processes via the unjustified use of a plethora of descriptors, rather than by focusing on the relevant chemico-biological interactions and building mechanistic models, which are often nonlinear in optimized coefficients. SBSP, in contrast, relies on the model-based descriptions of the underlying processes, including their kinetics. The conceptual SBSP models include transport and accumulation in a series of membranes, ionization, protein binding, hydrolysis, and other interactions with body constituents. The resulting differential equations are solved either numerically or explicitly, after appropriate simplifications. Individual rate and equilibrium parameters are related either to the properties of the chemicals, using extra-thermodynamic LFERs, or to the structures of the chemicals, using 3D-QSARs. The kinetics of disposition is described as a nonlinear disposition function of the lipophilicity, acidity, reactivity, and 3D structure of chemicals. Once calibrated for a given biosystem, the SBSP models provide a significantly more accurate prediction of ADMET than empirical models, especially outside the tested parameter space. The calibrated SBSP models are excellent tools for tailoring the properties of drug candidates to ensure optimum pharmacokinetics, understanding bioaccumulation of chemical pollutants, and advancing physiologic pharmacokinetic models, population pharmacokinetics, allometric scaling, and other disciplines studying chemico-biological interactions.
SBSP is in early stages of the development. The pseudo-equilibrium SBSP models treat absorption as a fast process. A more-detailed treatment is required for the chemicals, which exhibit slower transport rates. Current SBSP models predominantly involve the compounds that do not significantly interact with the headgroup regions of phospholipid bilayers. The future development of SBSP must focus more on cephalophilic and amphiphilic chemicals, which may exhibit lower transport rates than other compounds. To enable a rigorous treatment of these chemicals, a reliable system is required for the prediction of the solvation energies of chemicals in individual bilayer regions.
Current SBSP applications correlate the behavior or effects of chemicals mainly with their global properties such as lipophilicity, acidity, and reactivity. The use of structural features of chemicals in the SBSP models is conceptually feasible. The inclusion of 3D-QSARs for structure-specific processes is necessary to broaden the application domain of the SBSP models to more-diverse chemicals and biological systems. This step is paramount for the development of SBSP toward the ultimate goal: an integrated description of human pharmacokinetics and toxicokinetics dependent on structures and properties of chemicals.
10. Modeling Details
To facilitate the reading, the details of the model construction, including the used approximations and their justifications, and the equations used for the description of the SBSP models were concentrated in this section.
10.1. Model Construction Principles
In most cases, chemicals cross biological membranes by consecutive passive diffusion steps through different regions of the bilayer (section ). The properties of the bilayer, composed of physically distinct regions (sections and ), change discontinuously in space. Therefore, the concentration gradients are discontinuous at the interfaces. The exact description of the chemical diffusion in the nonhomogeneous space would require the use of the Fick’s Second Law for each phase separately, because of the different solvation energies of chemicals in individual phases. Moreover, the diffusion coefficients in individual phases should be space-dependent, because of the inhomogeneity of the phases, caused by structured layers at both sides of the interfaces, represented by subregions 2 and 3 (see section ). Such a description would probably be too tedious for any practical use; therefore, the application of some simplifying assumptions seems unavoidable.
10.1.1. Diffusion inside Compartments
The intracompartmental diffusion of chemicals is so fast that it can be treated as instantaneous in the SBSP models. Although, in classical pharmacokinetics that involves macroscopic compartments, this concept represents a crude approximation, in SBSP, it belongs to plausible assumptions, because of small distances that the molecules of chemicals must cross in the subcellular compartments (more details are given in section ). This fact warrants the representation of a biosystem by a set of alternating aqueous phases and membranes, with the molecules of chemicals diffusing quickly in bulk water, headgroup region, and bilayer core. The approximation of the fast diffusion inside the miniature intracellular compartments greatly simplifies the descripion of the transport of chemicals. The second-order partial differential equations based on the Fick’s Second Law can be replaced by a set of first-order linear differential equations. Consequently, the exact intracellular localization of molecules, in terms of coordinates, is substituted by a coarse mesh of compartments. The real shape of biological compartments is losing its significance in the approximation, as the geometry of the system is fully specified by the volumes and interfacial areas of individual compartments.
The actual number of compartments and their assignment to the individual parts of the biosystem is crucial for preserving physical soundness of the model. Each part of the biosystem, where the chemical is at least temporarily accumulated during disposition in the amount contributing significantly to the mass balance, represents a compartment. Aqueous phases containing proteins and other macromolecules, which can bind chemicals, qualify as compartments in practically all situations. Individual bilayer regions may or may not represent individual compartments, depending on the properties of distributed chemicals. As a result, SBSP depicts a biosystem as a set of homogeneous, morphologically based compartments, representing the bilayer regions, and extracellular and intracellular aqueous compartments.
10.1.2. Bilayer Representation
Based on the solvation properties, three main layers can be distinguished in the phospholipid bilayer: two headgroup regions and the hydrocarbon core. The bilayers can be further divided into subregions that are characterized by (i) low headgroup density, (ii) high headgroup density, (iii) high tail density, and (iv) low tail density292,699,912 (see Figure 3 in section ). Subregions 2 and 3 represent structured laminae at both sides of the headgroups/core interface,(698) which are also known as the polar and nonpolar diffusion layers. A more-detailed classification of bilayer subregions was used to analyze the distribution of intermolecular voids.(814)
The actual number of compartments representing the bilayer in a phenomenological description of interactions with chemicals is dependent on the accumulation of chemicals in individual regions. The use of seven compartments—represented by the outer headgroup/water interface, the outer headgroup region, the outer core/headgroup interface, the hydrocarbon core, the inner core/headgroup interface, the inner headgroup region, and the inner headgroup/water interface—seems appropriate, given the current knowledge about interactions of chemicals with the bilayers (section ). Because the accumulation is dependent on the properties of chemicals, this is the maximum setup that can be reduced for specific chemicals. For compounds with low lipophilicity, which do not accumulate in the core, the core is not considered a compartment. If the studied compounds exhibit low cephalophilicity, they do not bind in the headgroup regions, and the headgroup regions can be omitted in the model. For nonamphiphilic compounds, the interfaces do not represent compartments. For compounds that do not interact with phospholipids, the bilayer is treated as an inert barrier and does not need to be represented in the compartment model. The situation is depicted in Figure 7 in section .
10.1.3. Strong and Weak Interactions
In individual compartments, the chemicals undergo numerous strong (covalent) and weak (noncovalent) interactions with the low-molecular-weight molecules and macromolecular components of the cell (more details are given in section ). For SBSP, the most important difference between weak and strong interactions lies in the kinetics of the complex formation. While the weak interactions are completed within milliseconds, the formation of strong covalent bonds often proceeds with half-times on the order of minutes or hours.(1249) Because the shortest biological tests usually last at least a minute, weak interactions can obviously be treated as practically instantaneous. This approach greatly simplifies the mathematical description of the fates of chemicals in biosystems.
10.1.4. Lumping
The number of variables in a realistic model is, strictly speaking, identical to the number of different states of the molecules of chemicals (e.g., dissolved in the extracellular and intracellular aqueous phases, accumulated in different membrane regions, covalently or noncovalently bound to various proteins in individual compartments, etc.). All processes in the same compartment that proceed on similar time scales, and are dependent on the same physicochemical properties, can be lumped together and described by one variable. This procedure is well known in the theory of dynamic modeling.(53) The reduction in the number of variables can be applied to ionization, ion pairing, and nonspecific binding of chemicals to various proteins (section ), and the first-order elimination processes (section ).
10.2. Model Description
Using the aforementioned approximations, the distribution of a chemical in a biosystem can be described by the scheme given in Figure 14 in section . The scheme is valid for the chemicals, which do not interact with the headgroups (see Figure 7, rows 2 and 3, in section ), because most SBSP results were obtained for this class of compounds. This simplified scenario illustrates all aspects of the model description. The scheme can be easily modified for other classes of compounds (Figure 7, rows 1 and 4−6), via the replacement of the core compartment by a proper combination of the intrabilayer compartments.
Absorption and distribution are represented as diffusion in a catenary chain of alternating aqueous phases and membranes, with rapid equilibration within the bulks of the compartments. In the membranes, only the cores are included, because the model is only valid for the compounds, which do not interact with the headgroups. The sizes of interfacial areas connecting individual compartments may differ significantly, unless they represent two sides of the same membrane. The extracellular aqueous phase represents the incubation medium for the biosystem consisting of a suspension of subcellular organelles, cells, tissues, or organs. For organisms, the assignment of individual compartments to their physiological counterparts is dependent on the route of administration. If a chemical is taken orally, the first aqueous compartment corresponds to the part of the gastrointestinal tract from which the absorption proceeds, i.e., to the content of the small intestine in most cases. The molecules must cross several membranes to get into the bloodstream, which is represented by one of the internal aqueous compartments. For intravenous administration, the blood represents the first aqueous compartment. The effect of the blood flow on the rate of diffusion through the first capillary membrane can be taken into account via the dependence of the transfer rate parameters (section ) on the flow rate. In organs with porous capillary walls, such as the liver or spleen, most chemicals use the paracellular route to pass easily into the interstitial fluid, which can be therefore considered a part of the same aqueous compartment as the blood. The cells of capillary walls in other organs and the cells in tissues must be decomposed into individual bilayer regions and aqueous phases in the model, as shown in Figure 14 in section .
Chemicals may exist in each compartment in several states, comprising the low-molecular-weight species, such as the free non-ionized and ionized molecules, and the ion pairs with low-molecular-weight counterions, which can be noncovalently or covalently bound to macromolecules or eliminated. The last category includes the molecules undergoing the zero-order or first-order elimination, via metabolism (biotransformation) and excretion. For the mathematical description of the system, linear differential equations of the first order are suitable. Because the applied doses of specific drugs and contamination levels of environmental pollutants are generally low, it can be assumed that chemical activities are equal to the concentrations.
10.2.1. Transport in Multimembrane Systems
The first model of subcellular distribution of chemicals(58) described the pure transport of free molecules. Variations in the volumes of the compartments and in the interfacial areas between them were not considered. To take the geometry of the system into account, the mass balance for the ith compartment can be written as
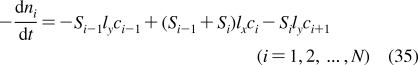 |
where n is the amount and c are the concentrations of the chemical in the compartment specified by the subscript, Si is the interfacial area between the ith and (i + 1)th compartment (S0 = SN = 0), l are the transfer rate parameters, and t is time. For l, the subscripts are x = i and y = o for the odd compartment numbers, and x = o and y = i for the even compartment numbers. Here, the subscript i indicates the water-to-membrane direction (only when associated with l, otherwise the subscript i indicates the compartment number), and the subscript o denotes the backward process (section ). The total number of compartments is N. To place eq 35 on the concentration basis, it needs to be divided by the volume of the ith compartment (Vi).
The majority of models used are based on the assumption that diffusion inside all subcellular compartments is practically instantaneous, because of the small dimensions of the compartments (more details are given in sections and ).
10.2.2. Fast Intracompartmental Processes
Now let us consider the processes in which the molecules of chemicals participate in each of the compartments. Noncovalent protein binding, ionization, and ion pairing are much faster than transport and spontaneous or enzymatically catalyzed chemical reactions(1250) and are treated as instantaneous. This fact allows for simplification of the model by lumping together the concentrations of all molecular species that are present inside an aqueous compartment. From the viewpoint of the pH-partition hypothesis(2134) stating that non-ionized molecules diffuse passively through the hydrocarbon core of membranes much faster than ionized molecules, it is advantageous to choose the concentration of free non-ionized molecules in the ith compartment (ci) as the representative variable. The concentrations of other species can be calculated from ci by multiplication with a proper factor resulting from the definition of the respective equilibrium constants. This generalization is based on the assumption that the concentrations of the interaction partners of chemicals (protons, counterions, and protein binding sites) are not significantly affected by the interactions and practically do not change upon binding of chemicals. This assumption is plausible because, in most cases, the compounds are present in living biosystems in comparatively low concentrations. Moreover, homeostasis and the buffering capacity of intracellular aqueous compartments contribute to the stability of proton and counterion concentrations.(2344)
For an ionizable chemical encompassing S species, one of them being neutral and (S − 1) species being ionized, the indices j are assigned in the following way: the most protonated species has j = 1, and the least protonated species has j = S. The position of the neutral species in this sequence is marked as n (1 < n < S), although, formally, in the equations, we will use the subscript j = 0 for the neutral species. For this special notation, the sum or product of j running from 0 to S has S members, because the term with j = n is omitted. The concentration of the molecules ionized to the jth degree in the ith compartment, [C]ij, is equal to the free non-ionized concentration ci multiplied by the factor dij (where d represents dissociation). The factor dij is calculated in different ways for the positively charged species with j < n and for negatively charged species with j > n, respectively:
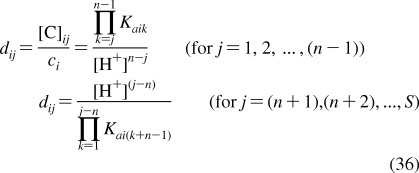 |
Equations 36 are derived from the definition of the ionization constant Kaik = [Ci(k+1)]/[Cik][H+], characterizing the equilibrium between species k and k + 1, and using the proton concentration in the ith compartment. This treatment is applicable only if the pKak values are at least 3−4 units apart and, consequently, the Kak values are equal to the microconstants. Otherwise, the number of species is higher than the number of pKak values plus one, because of the presence of two or more species with the same charge (e.g., neutral molecules and zwitterions for the overall zero charge). In this case, the factors dij must be calculated using the microconstants(2177) (more details are given in sections and ).
The formation of ion pairs with low-molecular-weight counterions leads to charge neutralization of the ionized species and is important mainly for membrane accumulation. For simplicity, only the 1:1 ion pairs will be considered, with the counterions Z (total number of Z) and the association constant Kijk. The proportionality factor pijk = Kijk[Z]ijk (p as in pairing) relates the concentration of the kth ion pair, [CZ]ijk, to the concentration [C]ij of chemicals ionized to the jth degree in the ith compartment. The ion-pair concentration is proportional to the free non-ionized concentration ci:
with dij being defined by eqs 36.
The proportionality factor between the protein-bound concentration and the concentration of the respective low-molecular-weight species is derived in a similar way, using the association constant Kijkl for the binding to the lth binding site, and assuming linear binding (i.e., a negligible loss of the binding site concentration on binding). The concentration of the compound ionized to the jth degree, paired with the kth counterion, and bound to the lth binding site on proteins in the ith compartment can be calculated as the product of the corresponding ion-pair concentration, Kijkl and the concentration of the binding sites sil of the lth type, as follows from the definition of Kijkl, if sil is practically unaffected by the binding and can be considered constant. The total bound concentration of the species in the ith compartment is then the sum over all T types of binding sites in the ith compartment:
The ion-pair concentration [CZ]ijk is expressed as the product of the free non-ionized concentration ci and the proportionality factors dij and pijk, as defined in eqs 36 and 37. The proportionality factor bijk (b for binding) is equal to the l-summation in the middle term of eq 38 and relates the free and protein-bound concentrations of the chemical in the jth ionization degree and kth ion pair, which are present in the ith compartment. The proportionality factor bijk can be used to calculate the protein-bound concentration of any species in the ith compartment, as the product of bijk and the free concentration of the species.
The observed association constant for the protein binding of the kth ion pair of the jth species in the ith compartment can be written for linear binding as
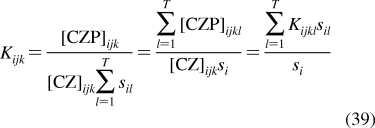 |
The concentration of the protein-bound ion pair in the third term of eq 39 was replaced using the second term in eq 38. The l-summation over all types of binding sites in the second term was, in the subsequent terms, written as the overall concentration of the binding sites in the ith compartment, si. The association constant for an ion pair is a weighted average of the association constants for individual binding sites, where the weights are the fractions of individual types of binding sites. The comparison of eqs 38 and 39 shows that the proportionality factor bijk can be viewed as the product of the association constant Kijk and the overall concentration of the binding sites, si. Equation 39 also applies to the protein binding of neutral and ionized species.
The association constant for the protein binding of the jth species in the ith compartment is
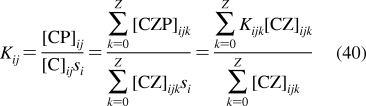 |
The concentration of the bound ion pairs in the third term of eq 40 was replaced using an expression resulting from the first three terms in eq 39. Obviously, Kij is the weighted average of the association constants for individual ion pairs and the free species (subscript k = 0), with the weight equal to the fractions of ion pairs and the free species.
The protein binding of a chemical in the ith compartment is characterized by the association constant that can be written as
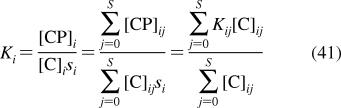 |
Here, Ki is the weighted average of the association constants of individual species, with the weights given as the fractions of individual species. In the third term, the protein-bound concentration of the jth species was expressed using the first two terms of eq 40.
For the pseudo-equilibrium distribution (section ), protein binding in the biosystem comprising N compartments is of interest, because it appears in term A of eq 25 in section . The process is characterized by the overall association constant, K:
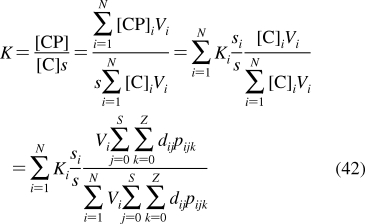 |
In the third term, the free and bound concentrations of the chemical from the second term were expressed as the weighted averages of the respective concentrations in individual compartments, with the weights given by the volume fraction Vi/V of the compartment, whereby the V terms mutually cancel in the numerator and the denominator. In the fourth term, the protein-bound concentration in the ith compartment, [CP]i, was expressed from the first two terms of eq 41. The pool of free species includes the non-ionized species, ionized species, and ion pairs, with the concentrations expressed as cA, cAdij, and cAdijpijk, as shown in eqs 36 and 37. The appropriate summations, discussed in more detail in eq 44 with bijk = 0 below, replace the overall concentration of free species [C]i in the fourth term of eq 42. Under the pseudo-equilibrium conditions, the free non-ionized concentration cA is the same in all compartments and mutually cancels in the numerator and denominator of the fifth term. The overall association constant K is the weighted average of the association constants Ki in individual compartments, with the weights given by the product of two fractions in individual compartments: the concentration of the binding sites, si/s, and the factor accounting for ionization and ion pairing of the chemical, multiplied by the volume.
Application of a 3D-QSAR technique for the binding of a diverse set of chemicals to silent receptors in the biosystems consisting of several compartments, such as mitochondria, eukaryotic cells, tissues, organs, and organisms, requires that the overall association constant given by eq 42 be expressed in terms of the elementary association constant Kijkl from eq 39, using the back-substitution based on eqs 40 and 41. The reason is that the 3D-QSAR equations can only be applied to Kijkl and not to the composite association constants Kijk, Kij,Ki, or K.
Within a homologous series, the association constant Kijkl that characterizes the binding of a chemical to the lth silent receptor, is frequently dependent on lipophilicity, expressed by the reference partition coefficient P, as given by eq 4 in section :
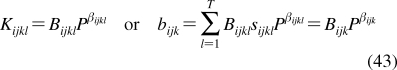 |
The second equality is valid for the binding of one molecular species to T protein sites in the ith compartment (see eq 38 for symbol definitions), if the Collander exponents β do not significantly differ for the proteins in the ith compartment.
For generality, we assume that all ion pairs can bind to proteins. Obviously, the unpaired molecules (k = 0) with charge j may have high association constants Kij0l. Their binding factor will be denoted bij0. The neutral molecules (j = 0) do not form ion pairs (k = 0). They bind to protein sites with the association constants Ki00l and their binding factor is bi00 = [CP]i0/ci.
The overall concentration of the chemical in the ith compartment can be expressed using eqs 36−38 as the sum of concentrations of (i) free non-ionized molecules; (ii) molecules that are ionized to various degrees (j), encompassing (S − 1) species, (iii) ion pairs with up to Z counterions, each denoted by the subscript k, which can originate from each ionized molecule, giving rise to (S − 1) × Z species; and (iv) molecules bound to the proteins, which can originate from each species, i.e., S + (S − 1) × Z species, if binding of each species in the given compartment is characterized by one averaged binding parameter bijk:
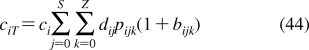 |
To keep eq 44 compact: (i) the j-summation runs from zero to include the non-ionized molecules (with the subscript j = 0), but the term with j = n is omitted, so that the overall number of the terms is S; (ii) the k-summation runs from zero to include the nonpaired ionized molecules (with k = 0); and (iii) the following definitions, which do not directly follow from eqs 36−38, are introduced. For neutral molecules, di0 = 1, pi00 = 1, pi0k = bi0k = 0 (k = 1, 2,..., Z). For ionized molecules, which are not paired with a counterion, pij0 = 1 (j = 1, 2,..., S, for j ≠ n).
In any subcellular compartment, the k- and l-summations for the ion pairs in eq 44 and for the chemical−protein complexes in eq 38 formally go up to the overall maximum number of counterions (Z) and protein binding sites (T), respectively. If a particular counterion or binding site is not present in a particular compartment, the corresponding factors p or b are set to zero. In this way, the use of subscripts to the numbers Z and T is avoided, although both quantities are compartment-specific.
10.2.3. Elimination
Chemicals can be eliminated in each compartment via spontaneous and enzymatically catalyzed reactions and, from some compartments, by excretion. In the ith compartment, spontaneous reactions of a chemical in the jth ionization degree and kth ion pair are characterized by the products of the actual rate constants ρijk and the concentrations of the reaction partners [R]i, which do not substantially change on the reactions with chemicals, because of low concentrations of the chemicals and homeostasis.
The kinetics of a saturable metabolic process of the same species in the ith compartment can be conveniently described by the Michaelis−Menten equation:(2049)
Here, m is the enzyme concentration, v is the reaction rate, and the subscript “m” refers to its maximum saturated value. The Michaelis−Menten constant Km can represent, under certain assumptions, the reciprocal value of the chemical−enzyme association constant. To avoid nonlinearity, two borderline cases are usually considered: for [C]ijk≪Kmijk, the enzymatic reaction is a first-order process,
and for [C]ijk≫Kmijk, the reaction is of the zero order,
All the first-order elimination processes (i.e., spontaneous reactions, enzymatic reactions, and excretion that affect a molecular species in one compartment) are characterized by the overall first-order rate parameter, kijk:
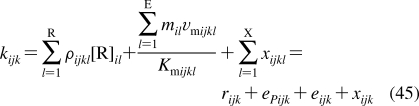 |
Here, R, E, and X are the total numbers of spontaneous reactions, enzymatic reactions, and excretion processes (with the individual rate constants xijkl), respectively. The subscripts refer to the ith compartment, the jth ionization degree, the kth ion pair, and the lth process. The second equality in eq 45 will be used in a pseudo-equilibrium description of disposition (see sections and ). The term e for the enzymatic reactions has been split in two parts: lipophilicity-dependent reactions have been singled out and marked with the subscript P (indicating the relation to the reference partition coefficient P).
10.2.4. Differential Equations
Disposition of the chemicals, which do not interact with the headgroups, includes bidirectional transport of free molecules (both non-ionized and ionized) and ion pairs, ionization, binding to proteins and enzymes, and the unidirectional, first-order elimination of all low-molecular-weight species, as illustrated in Figure 14 in section . The mass balance for the ith compartment can be written using the flux terms F (the first subscript indicates the originating compartment, and the second subscript represents the receiving compartment) and the elimination term E (the subscript indicates the compartment where elimination occurs):
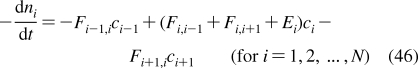 |
where
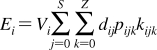 |
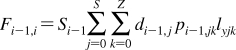 |
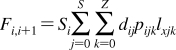 |
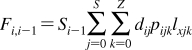 |
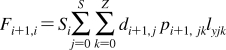 |
Equations 46 are extensions of eqs 35 and describe the distribution scheme given in Figure 14 in section . All the flux parameters F must be defined simultaneously for the given compartment number i, because the number will determine the values of the subscripts x and y, as introduced in eqs 35. Other symbols are defined in eqs 36−45.
In eqs 46, n is the total amount and c are the free non-ionized concentrations of the chemical. To obtain the concentration c on the left side, eqs 46 must be divided by Vi∑Sj=0 ∑Zk=0dijpijk(1 + bijk) as follows from eq 44. The final set of linear differential equations can then be written in matrix notation as
where c is the column vector of the chemical concentrations in individual compartments (their total number is N), and B is the tridiagonal matrix with dimensions N × N with the elements:
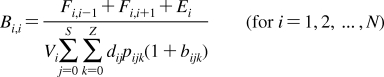 |
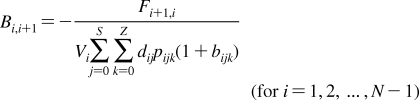 |
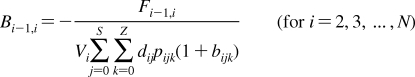 |
Generally, the differential equations described by eq 47 do not have explicit solutions and must be solved numerically (see section ). Explicit solutions can only be found for special, reduced scenarios (see section ).
10.2.5. Explicit Solutions for Single Bilayer
The differential equations described by eq 47 can be solved explicitly for the transport through one bilayer, providing the chemicals bind neither to the interfaces nor to proteins (bijk = 0) and do not ionize (dij = pijk = 0). The transport is then described by two rate parameters, li and lo (Figure 7, rows 2 and 3, in section ). If the chemicals are initially present in the first (donor) aqueous compartment in the concentration c0, the time course of the drug concentration in the ith phase of the water−core−water system (i = 1−3, respectively) can be described by the solution to the set of three differential equations (eq 47 with N = 3) corresponding to the scheme given in Figure 14 in section :
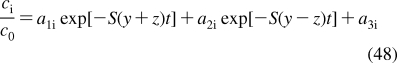 |
where
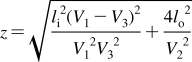 |
Equations 48 can be simplified for some real-life situations. For dilute suspensions of gram-positive bacteria or liposomes V1 ≫ V2 ≈ V3 and then y ≅ 2(lo/V2) + (li/V3) and z ≅ [(li2/V32) + 4(lo2/V22)]1/2. In an absorption simulator, V1 = V3 and, consequently, y = 2[(li/V1) + (lo/V2)] and z = 2lo/V2.
The rate parameters li and lo are dependent on the membrane/water partition coefficient P = li/lo, according to eqs 3 in section . For the constant exposure time t, lipophilicity is the only property of the chemicals governing their membrane transport under the given conditions. The concentration−lipophilicity profiles for individual distribution periods t as generated by eq 48 are equivalent to those generated by numerical simulations (curves 1−3 in Figure 18 in section ). Obviously, the simple three-compartment model provides the concave dependences of biological activity or concentration on lipophilicity, which are so often encountered in the QSAR practice (section ). This is in contrast with the statement that the three-compartment system (Figure 14 with N = 3) with elimination from the first compartment is the simplest system, which is able to generate the concave concentration−lipophilicity profiles.(2086) These profiles result also from closed three-compartment system, with reversible transport as shown in eq 48 with i = 3, and even with unidirectional transport (not shown).
10.2.6. Equations for Unidirectional Transport
For unidirectional transport, the set of differential equations 47 has the bidiagonal coefficient matrix B with Bi,i+1 = 0 and Fi,i−1 = 0 in Bi,i. The eigenvalues λi of the coefficient matrix B are simply the diagonal elements:
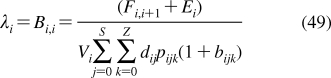 |
The flux terms Fi,i+1 and the elimination terms Ei are defined in eq 46.
The differential equations 47 with the bidiagonal matrix B can be solved easily by standard methods.2108,2109 The solutions2110–2112 differ according to whether all the eigenvalues λi defined by eq 49 are different (the normal case) or at least two eigenvalues have identical values (the degenerate case). The solution for the normal case is represented by eq 14 in section . Some solutions for the degenerate case have been published.(2112)
10.2.7. Steady-State Solutions
If the concentration in the entry compartment is maintained at a constant level, the system will attain the steady state, when the concentrations in individual compartments do not change. The corresponding differential equations 46 or 47 will become algebraic equations, by nullifying the left-hand time derivatives. The steady-state concentrations can be found by solving the algebraic equations using standard determinant-based techniques. An especially compact solution exists for the system without elimination, when all Ei = 0 in eqs 46 or 47. This situation is relevant for the description of the permeability coefficients in terms of individual steps. Under the steady-state conditions, the net fluxes through each interface are equal to the overall flux φ:
The overall flux can be expressed as a function of individual steps, the constant concentration c1 in the entry (donor) compartment, and the concentration cN in the last (acceptor) compartment. Back-substitution can be conveniently used, as demonstrated in the classical work on the rate theory:(2096)
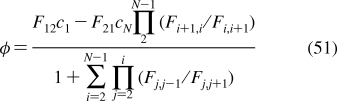 |
For the steady-state assumption to hold, the concentrations c1 and cN must be time-independent. For the application of eq 51 to a permeation through a biological sheet (section ), the only choice is to set c1 ≅ c1(0), and cN → 0, causing the second term in the numerator to vanish. The overall flux then is the product of the permeability coefficient PC and the constant concentration c1. PC is equal to the flux from the donor compartment, divided by the term containing a summation of products of ratios of the forward and backward fluxes from individual compartment. If the two interfaces surrounding the compartment are identical to the point of having the same transfer rate parameters l (eq 46), the respective flux ratio is reduced to a constant number given as the ratio of the surface areas S.
10.3. Pseudo-Equilibrium Disposition
This type of disposition originates if absorption is either fast or absent, distribution is fast, and elimination is the only time-dependent process. The scenario works best for chemicals that exhibit fast trans-bilayer transport. As the overall amount of the chemical decreases in time because of elimination, the fast transport is able to maintain, at any given moment, approximately the same free concentrations of the chemical in all aqueous phases (cA) and in all membranes (cM), and these two concentrations are related by the apparent partition coefficient (see below). The resulting description does not include absorption, which is present if the chemical is not dosed directly into the bloodstream. The overall time course of the chemical’s concentration consists of the absorption and elimination phases, which are characterized by the increase and decrease in the concentration, respectively. The duration of the absorption phase is dependent on the size of the biosystem, in addition to the transport rates of chemicals. In the pseudo-equilibrium models, absorption is considered instantaneous.
10.3.1. Apparent Partition Coefficient
The apparent core/water partition coefficient Pi accounts for partitioning of non-ionized molecules, ionized molecules (subscript j indicates the jth ionization degree), and ion pairs (subscript k indicates the kth counterion). Two aqueous phases contacting the membrane may have a different composition, so the phase that is more distant from the site of entry of chemicals was chosen for the definition of Pi. For the chemicals, which do not interact with the headgroups, Pi is defined as the ratio of the total concentration of the chemical in the membrane core and in the aqueous phase:
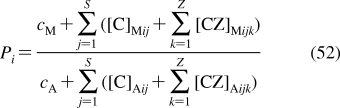 |
Subscripts indicate the places where the concentrations are measured: M and A refer to the membrane core and the aqueous phase, respectively.
Using eq 44 with ci = cA or cM, cM = PCcA, and bijk = 0 (no protein binding), eq 52 can be rearranged as
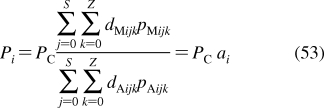 |
The proportionality factors relating the free non-ionized concentrations cA and cM to the concentrations of ionized species (dij) and ion pairs (pijk) in the membrane and the aqueous phase (subscripts M and A were added to distinguish the phases) were defined in eqs 36−44. The accumulation factor ai is dependent on the properties of both the tissue and the chemical.
The factor ai can also be looked at in a different way. Each of the present molecular species has its own partition coefficient that is given as the ratio of the species’ concentrations in the core and in the aqueous phase. The membrane concentration of each species can be expressed as the product of the respective partition coefficient and the aqueous concentration of the species:
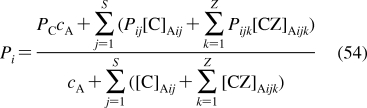 |
The use of proportionality factors for ionized species (dAij) and ion pairs (pAijk) in the aqueous phase, as defined in eqs 36−44, leads to
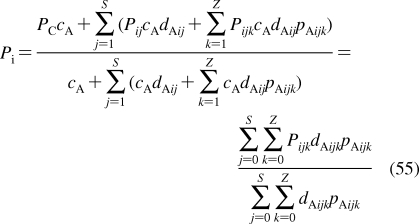 |
Equation 55 was made compact by the extensions of the j- and k-summations to the index zero, to include non-ionized molecules (j = 0) and free (non-paired) ionized molecules (k = 0), in a similar way as in eq 44. For neutral molecules, dAi0 = 1, pAi00 = 1, pAi0k = PAi0k = 0 for k = 1, 2,..., Z (this notation is valid solely in section ). For ionized molecules, which are not paired with a counterion, pAij0 = 1 (j = 1, 2,..., S; j ≠ n). The core/water partition coefficients of non-ionized molecules (PC = Pi00), molecules ionized to the jth degree (Pij0), and ion pairs with the kth counterion (Pijk) have the same magnitudes for all membranes. Equation 55 can frequently be simplified using the following comparisons: (i) the partition coefficients of the ionized species Pij will be much lower than the partition coefficients of the ion pairs (Pijk) and occasionally can be neglected; and (ii) ion pairing in water can be neglected, i.e., in the denominator, all pAijk = 0, except pAij0 = 1 (j = 0, 1,..., S; j ≠ n).
The partition coefficient is an additive-constitutive property that can be expressed as the product of the fragment contributions of properly defined substructures(649) and the factors correcting for the interactions among the substructures,501,2135 jointly denoted as f (this definition of f slightly differs from that in original literature, to keep the expressions compact). Applying this principle, the partition coefficients of each ionized species or ion pair can be expressed as the product of the partition coefficient of non-ionized molecules (PC) and the ratio π of the factors f for the ionized substructure and the respective counterion on one side, and the non-ionized substructure on the other. The apparent partition coefficient Pi is proportional to the core/water partition coefficient for non-ionized molecules PC as(2344)
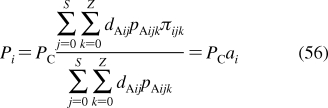 |
The magnitude of the apparent partition coefficient Pi is dependent on the composition (pH, concentration of counterions) of the ith aqueous phase that is more distant from the site of entry of chemicals, the nature of the ionizable substructures, and the concentration of counterions. This dependence is described by the accumulation factor ai that can be larger or smaller than unity. The terms dijk and pijk were defined in eqs 36−44; the subscripts for the factors f for free non-ionized and ionized molecules are defined analogously, with fi00 = 1.
10.3.2. Tissue/Plasma Partition Coefficient
The tissue/plasma partition coefficient PTP is defined as the ratio of the total concentrations of a chemical in tissue and in plasma under equilibrium conditions. For these conditions, the tissue is represented by a set of M membranes and A aqueous phases, which are organized in pairs represented by a membrane and the aqueous phase that is more distant from the site of application. This arrangement is necessary to account for a different composition of individual phases. The chemical can be present in any or all of these phases as neutral, ion-paired, or ionized molecules. The total concentrations of chemicals in each membrane and each aqueous phases were given in eq 44 in section (ci = cM and cA, cM = PCcA). Multiplication by the volumes of membranes and aqueous phases (VMi and VAi, respectively) gives the respective amounts, which can be used to define PTP as(1898)
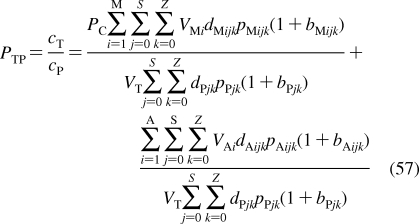 |
The free non-ionized concentration cA appeared in each term and was cancelled. The concentrations of all low-molecular-weight species in each membrane/water pair can be assumed to be given by the respective partition coefficient. Following the approach given in eqs 54−56 for the apparent partition coefficient, we get
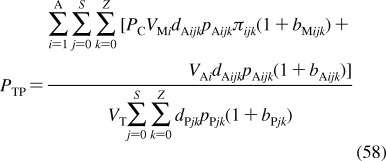 |
All membrane-related terms except the protein-binding term (bMijk) were converted to the terms for the aqueous phase. The i-summation runs through all membrane/water pairs.
10.3.3. Distribution Volume
For the pseudo-equilibrium conditions, the distribution volume relates the dose to the initial plasma concentration or the actual drug amount in the body to actual plasma concentration. For fast distribution, the concentration of free non-ionized molecules is equal to cA in all aqueous compartments, and equal to cM in all membranes. The definition of the core/water partition coefficient PC for the non-ionized molecules was used to find the relation between the total amount n and cA:
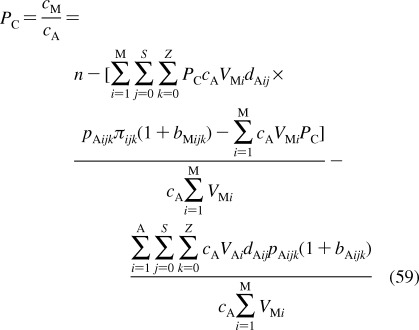 |
In the numerator, the non-ionized amount in the membranes was expressed as the difference between the actual amount n, the amount in membranes (total number M) in the form of all species except free non-ionized molecules (the term in the square brackets), and the amount of all species in aqueous phases (total number A). The volumes V of the aqueous phases and the membranes are indicated by the subscripts A and M, respectively. The amounts are expressed in a similar way as in eq 58 for the tissue/plasma partition coefficient. Separating cA = n/Vdf, we get an expression for the free non-ionized volume of distribution, Vdf:(2344)
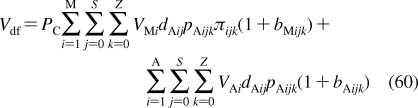 |
The two summations could have been combined into one summation going through all membrane/water pairs, as used in eq 58. The separate summations allow for the following interpretation of Vdf. The second term represents the volume of the aqueous phases corrected for ionization, ion pairing, and protein binding. The first term represents the volume of membranes corrected for the same processes, and membrane accumulation.
In classical pharmacokinetics, the volume of distribution Vd relates the dose of a drug administered to the total drug concentration ciT in plasma. Vdf in eq 60 is based on the free non-ionized concentration cA instead of ciT. Using eq 44, which relates ciT and cA (identical to ci) in plasma, the expression for the distribution volume is
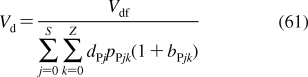 |
The summations in eq 61 run through all species of the given chemical and all counterions, which are available in plasma (denoted by the subscript “P”). The term bPjk describes protein binding of the jth species and the kth ion pair in plasma.
A comparison of eqs 60 and 61 with eq 58 shows that they have similar forms, and there are only two differences: (i) the summations are through different sets of membranes and aqueous phases corresponding to the entire organism and a tissue, respectively; and (ii) eq 58 contains the tissue volume VT in the denominator. This similarity means that the volume of distribution can be viewed as a weighted summation of the tissue/plasma partition coefficients, where the weights are the volumes of individual tissues.(2341)
10.3.4. Clearance
The total loss of the chemical from the biosystem is the summation of the first-order elimination processes encountered in all N macroscopic, extracellular, and subcellular compartments with the volumes Vi. The rate of each process is given as the product of the rate parameter kijk defined in eq 45 in section , and the respective concentration of the chemical as given by eqs 36 and 37 in section :
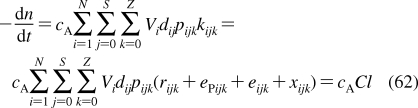 |
The term that is multiplied by cA in all equalities is the clearance (Cl) of the first-order processes. The summations run through N compartments; (S − 1) ionized species of the chemical, plus one uncharged species; and Z counterions (the parameters for k = 0 have preset values to keep eq 62 compact).
The zero-order elimination processes in the ith compartment will be lumped together, and characterized by the zero-order rate parameter k0ijk for the species ionized to the jth degree (j = 0 for non-ionized molecules) and bound to the kth counterion (k = 0 for the free ionized molecules). Because the zero-order processes will only be included in the pseudo-equilibrium model (see section ), which assumes a parallel elimination of chemicals from all phases, the overall zero-order clearance for the entire biosystem is of interest:
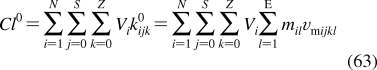 |
The symbols are explained in eq 45. The volumes of individual phases V are used to convert the concentration-based rate constants k0ijk to amount-based quantities, which constitute clearance.
10.3.5. Pseudo-Equilibrium QSAR Model
As specified in section , the combination of eqs 1, 4, and 43 with eq 23 provides the pseudo-equilibrium QSAR model that is described by eq 25. The terms A, B, C, and D in eq 25 are defined as
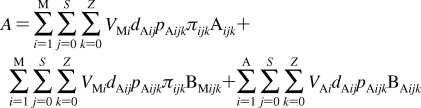 |
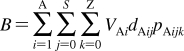 |
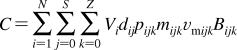 |
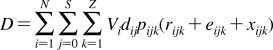 |
The summations run through all membranes (the total number M), all aqueous phases (A), all compartments (N), all ionization species (S), and all ion-pair species (Z; the parameters for k = 0 have preset values to keep the terms A, B, C, and D compact). Other parameters are defined in eqs 36−45, 55, and 56. The subscripts A and M were added, where it was necessary to distinguish the dissociation terms d or the ion-pairing terms p for aqueous phases and membranes, respectively.
12. Acknowledgments
Although the research described in this article has been funded in part by the USEPA (R82-6652-011) and NIH NCRR (1 P20 RR 15566 and 1 P20 RR 16741), it has not been subjected to the agencies’ peer and policy reviews and therefore does not necessarily reflect the views of the agencies and no official endorsement should be inferred.
Biography

Stefan Balaz is a tenured professor at the North Dakota State University (NDSU) College of Pharmacy in Fargo, ND. Since his early undergraduate years, his experimental and computational research has been focused on subcellular pharmokinetics, the modelling of drug−receptor interaction, and the combination of the outcomes from these two areas into conceptual cell-QSAR models. His research results have been summarized in ∼140 papers and recognized by the 1993 Ebert Prize of the American Pharmaceutical Association (now the American Pharmacists Association). Dr. Balaz was born and educated in Slovakia. He received his M.S. degree in Physical Chemistry from the Slovak University of Technology (SUT), Bratislava, in 1979; he received his Ph.D. degree in Biochemistry in 1986 and his D.Sc. degree in Pharmaceutical Chemistry in 1994, both from the Comenius University, Bratislava. He did postdoctoral work as a Humboldt Fellow with Joachim K. Seydel at the Research Institute for Experimental Biology and Medicine in Borstel, Germany (1988−1989), and as a Fulbright Fellow with Ronald J. Sawchuk at the University of Minnesota in Minneapolis (1996). Prior to joining NDSU in 1996, he had been a member of the faculty of SUT since 1986 and served as Associate Dean for Graduate Studies in Chemistry (1991−1994). He is an elected board member of the Cheminformatics and QSAR Society, serves on the editiorial boards of several journals, and reviews for NIH and other agencies.
11. Abbreviations and Symbols
Abbreviations
- α-KG
α-ketoglutarate
- ABC
ATP-binding cassette (family)
- ABS
a bioavailability score
- ADME(T)
absorption, distribution, metabolism, excretion (toxicity)
- ADP/ATP
adenosine diphosphate/adenosine triphosphate
- ALARM
La assay to detect reactive metabolites (by NMR)
- ATR
attenuated total reflection
- BCF
bioconcentration factor
- BCRP
breast cancer resistance protein
- BD
Brownian dynamics (simulations)
- BSEP
bile salt export pump
- Bz
benzyl
- C
chemical
- C16
n-hexadecane
- CD
circular dichroism
- Cl
clearance
- CMOAT
canalicular multispecific organic anion transporter
- CMPF
3-carboxy-4-methyl-5-propyl-2-furanopropionic acid
- CP/CE
complex of a chemical with a protein/enzyme
- CYP
cytochrome P450
- CZP/CZE
complex of CP/CE with a counterion Z
- DAcPC
diacetylphosphatidylcholine
- DIS
3,5-diiodosalicylic acid
- DMPC
dimyristoylphosphatidylcholine
- DOPC
dioleoylphosphatidylcholine
- DPD
Dissipative Particle Dynamics (simulations)
- DPH
1,6-diphenyl-1,3,5-hexatriene
- DPPC
dipalmitoylphosphatidylcholine
- ECM
extracellular matrix
- ENT
equilibrative nucleoside transporter
- EPR
electron paramagnetic resonance
- FT-IR
Fourier-transform infrared (spectroscopy)
- GLUT
glucose transporter (facilitative)
- HSA
human serum albumin
- IAMs
immobilized artificial membranes
- LEO
leave-extremes-out (cross-validation)
- LFER
linear free-energy relationship
- MAS
magic-angle spinning
- MC
Monte Carlo (simulations)
- MD
molecular dynamics (simulations)
- MDR
multiple drug resistance protein
- MIC
minimum inhibitory concentration
- MKC
minimum killing concentration
- MRP
multidrug resistance related protein
- MSAB
4-(methylsulfonylamid)-butyl
- NAD(P)H
nicotinamide adenine dinucleotide (phosphate) - reduced
- NBD
nucleotide binding domain
- 5-NFA
3-(5-nitro-2-furyl)-acrylic acid
- NMR
nuclear magnetic resonance
- NOE
nuclear Overhauser effect
- NOESY
nuclear Overhauser enhancement spectroscopy
- NTCP
Na+-taurocholate cotransporting polypeptide
- OAT
organic anion transporter
- OATP
organic anion transporting peptide
- OCT/OCTN
organic cation transporter (N stands for novel)
- ODS
octadecylated silica
- PAMPA
Parallel Artificial Membrane Permeation Assay
- PBPK
physiology-based pharmacokinetic (models)
- PC
phosphatidylcholine
- PCA
principal component analysis
- PDB
Protein Data Bank
- PE
phosphatidylethanolamine
- PEPT
peptide transporter
- PFG
pulsed field gradient
- PGDP
propyleneglycol dipelargonate
- PGP
P-glycoprotein
- PLS
partial least squares projection to latent structures
- POPC
1-palmitoyl-2-oleoylphosphatidylcholine
- POPE
1-palmitoyl-2-oleoylphosphatidylethanolamine
- PRESS
predictive sum of squares of deviations
- PSA
polar surface area
- PXR
pregnane X receptor
- Py
pyridine
- QSAR
quantitative structure−activity relationship
- QSTAR
quantitative structure−time−activity relationship
- R
reactant; substituent
- ss
solid-state (NMR)
- SBSP
structure-based subcellular pharmacokinetics
- SCMFT
Self-Consistent Mean-Field Theory
- SGLT
sodium glucose co-transporter
- SLC
solute carrier (family)
- SLCO
organic anion transporter (family)
- TAG
triacylglycerol
- TIB
2,3,5-triiodobenzoic acid
- TMD
trans-membrane domain
- URAT
urate transporter
- UV-Vis
ultraviolet and visible (light)
- XRD
X-ray diffraction substituent
Parameters
- a
accumulation factor
- A
total number of aqueous phases
- Ax
membrane accumulation coefficient
- At
interface transfer coefficient
- Ar
reactivity coefficient
- A, B, C, D, E, F
adjustable coefficients optimized by regression analysis
- A(pp,t)
disposition function with t and pp as variables
- b
binding factor
- B
aqueous distribution coefficient in pseudo-equilibrium models
- B
coefficient relating protein binding to lipophilicity
- Bt
coefficient relating interfacial transfer to lipophilicity
- c
concentration
- cX
isoeffective concentration, eliciting the fraction X of the maximum effect
- C
lipophilicity-dependent elimination coefficient in pseudo-equilibrium models
- d
dissociation factor
- D
lipophilicity-independent elimination coefficient in pseudo-equilibrium models
- D
diffusion coefficient
- e
rate parameter of enzymatic reactions
- E
elimination term
- E
total number of enzymatic reactions in a compartment
- f
hypothetical log P value of a substructure or a correction factor
- F
flux term
- F
Fisher test value
- g
standard gravity
- h
effective thickness
- k
rate parameter of metabolism or elimination
- K
association constant for binding to proteins or receptors
- Km
Michaelis constant of an enzymatic reaction
- l
rate parameter for transport between bilayer regions and water
- m
enzyme concentration
- M
total number of membranes (in eq 12, with different solvation properties of the cores)
- n
amount
- n
position of neutral species in the sequence of species ordered by protonation level
- N
number of subcellular compartments
- p
ion-pairing factor
- P
reference partition coefficient
- Pi
apparent partition (distribution) coefficient for the given pH value of the aqueous phase, which may be indicated in the subscript
- PB/W
bilayer/water partition coefficient
- PC16/W
hexadecane/water partition coefficient
- PO/W
1-octanol/water partition coefficient (sometimes referred to as P)
- PTP
tissue/plasma partition coefficient
- PC
permeability coefficient
- pp
physicochemical properties
- r
rate parameter of spontaneous reactions in a compartment
- r2
squared correlation coefficient
- R
resistance of a stratum to transport
- R
total number of spontaneous reactions in a compartment
- s
concentration of protein binding sites in a biosystem
- si
concentration of protein binding sites in a compartment
- sl
concentration of protein binding sites representing a specific type
- s
standard error of the fit
- S
interfacial area; number of species of a chemical
- τ
reduced time
- t
exposure time
- T
number of types of protein binding sites in a compartment
- v
rate of an enzymatic reaction
- vm
maximum rate of an enzymatic reaction
- V
volume
- Vd
distribution volume
- Vdf
distribution volume of free non-ionized molecules
- Zdf
the total number of countions in a compartment
- x
rate parameter of excretion
- X
relative intensity of the chemical’s effect
- X
total number of excretion processes in a compartment
- y,z
terms in the description of the transport through a single bilayer
- Y
one of the terms A, B, C, D describing pseudo-equilibrium distribution
- Z
the total number of counterions in a compartment
- Z
counterion
Greek Symbols
- α
ionized fraction
- β
Collander equation exponent
- δ
diffusion distance
- λ
eigenvalue
- ϑ
sensitivity of a compartment to a change in external pH value
- π
ratio of factors f for ionized and non-ionized substructures
- ρ
rate parameter of a spontaneous reaction
Subscripts
- A
subscript referring to the aqueous phase
- B
subscript referring to the bilayer
- m
subscript denoting the maximum rate or the Michaelis constant
- O
subscript referring to 1-octanol
- P
subscript referring the plasma
- T
subscript referring to tissues
- W
subscript denoting the aqueous phase
Funding Statement
National Institutes of Health, United States
13. References
- Bio-Loom Database and Software; Biobyte Corp.: Claremont, CA, 2006. (www.biobyte.com)
- Kurup A. J. Comput. Aid. Mol. Des. 2003, 17, 187. [DOI] [PubMed] [Google Scholar]
- Hansch C.; Steward A. R.; Iwasa J.; Deutsch E. W. Mol. Pharmacol. 1965, 1, 205. [PubMed] [Google Scholar]
- Hansch C. Farmaco 1968, 23, 293. [PubMed] [Google Scholar]
- Hansch C.; Dunn W. J. J. Pharm. Sci. 1972, 61, 1. [DOI] [PubMed] [Google Scholar]
- Hansch C.; Clayton J. M. J. Pharm. Sci. 1973, 62, 1. [DOI] [PubMed] [Google Scholar]
- Hansch C. J. Med. Chem. 1976, 19, 1. [DOI] [PubMed] [Google Scholar]
- Kubinyi H. Progr. Drug Res. 1979, 23, 97. [DOI] [PubMed] [Google Scholar]
- Martin Y. C. J. Med. Chem. 1981, 24, 229. [DOI] [PubMed] [Google Scholar]
- Hansch C. Drug Develop. Res. 1981, 1, 267. [Google Scholar]
- Hansch C. Drug Metab. Rev. 1984, 15, 1279. [DOI] [PubMed] [Google Scholar]
- Hansch C. In Molecular Structure and Energetics; Liebman J. F., Greenberg A., Eds.; VCH Publishers: New York, 1987; Chapter 11, p 341. [Google Scholar]
- Hansch C.; Kim D.; Leo A. J.; Novellino E.; Silipo C.; Vittoria A. Crit. Rev. Toxicol. 1989, 19, 185. [DOI] [PubMed] [Google Scholar]
- Hansch C.; Klein T. E. Meth. Enzymol. 1991, 202, 512. [DOI] [PubMed] [Google Scholar]
- Hansch C. Acc. Chem. Res. 1993, 26, 147. [Google Scholar]
- Balaz S. Quant. Struct. Act. Relat. 1994, 13, 381. [Google Scholar]
- Hansch C.; Telzer B. R.; Zhang L. Crit. Rev. Toxicol. 1995, 25, 67. [DOI] [PubMed] [Google Scholar]
- Hansch C.; Hoekman D.; Gao H. Chem. Rev. 1996, 96, 1045. [DOI] [PubMed] [Google Scholar]
- Balaz S. In Lipophilicity in Drug Design and Toxicology; Pliska V., Testa B., van de Waterbeemd H., Eds.; VCH Publishers: Weinheim, Germany, 1996; p 295. [Google Scholar]
- Balaz S. Perspect. Drug Discovery Des. 2000, 19, 157. [Google Scholar]
- Hansch C.; Kurup A.; Garg R.; Gao H. Chem. Rev. 2001, 101, 619. [DOI] [PubMed] [Google Scholar]
- van de Waterbeemd H.; Smith D. A.; Jones B. C. J. Comput. Aid. Mol. Des. 2001, 15, 273. [DOI] [PubMed] [Google Scholar]
- van de Waterbeemd H.; Smith D. A.; Beaumont K.; Walker D. K. J. Med. Chem. 2001, 44, 1313. [DOI] [PubMed] [Google Scholar]
- Hansch C.; Hoekman D.; Leo A.; Weininger D.; Selassie C. D. Chem. Rev. 2002, 102, 783. [DOI] [PubMed] [Google Scholar]
- van de Waterbeemd H.; Gifford E. Nat. Rev. Drug. Discovery 2003, 2, 192. [DOI] [PubMed] [Google Scholar]
- Martin Y. C. In Quantitative Structure−Activity Analysis; Franke R., Oehme P., Eds.; Akademie-Verlag: Berlin, Germany, 1978; p 351. [Google Scholar]
- Kubinyi H. In QSAR in Design of Bioactive Compounds; Kuchar M., Ed.; Prous: Barcelona, Spain, 1984; p 321. [Google Scholar]
- Dearden J. C. Environ. Health Perspect. 1985, 61, 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Waterbeemd H.Hydrophobicity of Organic Compounds; CompuDrug: Vienna, Austria, 1986. [Google Scholar]
- Kubinyi H.QSAR: Hansch Analysis and Related Approaches; VCH Publishers: Weinheim, Germany,1993. [Google Scholar]
- Hansch C.; Leo A.. Exploring QSAR: Fundamentals and Applications in Chemistry and Biology; American Chemical Society: Washington, DC, 1995. [Google Scholar]
- Kubinyi H. Quant. Struct. Act. Relat. 2002, 21, 348. [Google Scholar]
- Hansch C. In Drug Design; Ariens E. J., Ed.; Academic Press: New York, 1971; Vol. 1, p 271. [Google Scholar]
- Martin Y. C.Quantitative Drug Design: A Critical Introduction; Marcel Dekker: New York, 1978. [Google Scholar]
- Martin Y. C. In Drug Design; Ariens E. J., Ed.; Academic Press: New York, 1979; Vol. 8, p 1. [Google Scholar]
- Valvani S. C.; Yalkowsky S. H. In Physical Chemical Properties of Drugs; Yalkowsky S. H., Sinkula A. A., Valvani S. C., Eds.; Marcel Dekker: New York, 1980; p 201. [Google Scholar]
- Martin Y. C. In Physical Chemical Properties of Drugs; Yalkowsky S. H., Sinkula A. A., Valvani S. C., Eds.; Marcel Dekker: New York,1980; p 49. [Google Scholar]
- Yalkowsky S. H.; Morozowich W. In Drug Design; Ariens E. J., Ed.; Academic Press: New York, 1981; Vol. 9, p 121. [Google Scholar]
- Hansch C.; Blaney J. M. In Drug Design: Fact or Fantasy?; Jolles G., Wooldridge K. R. H., Eds.; Academic Press: London, U.K., 1984; Chapter 10, p 185. [Google Scholar]
- van de Waterbeemd H.; Testa B. In Advances in Drug Research; Testa B., Ed.; Academic Press: London, U.K., 1987; p 87. [Google Scholar]
- van de Waterbeemd H., Ed. Structure−Property Correlations in Drug Research; R. G. Landes: Austin, TX, 1996. [Google Scholar]
- Hansch C.; Fujita T., Eds. Classical and Three-Dimensional QSAR in Agrochemistry; American Chemical Society: Washington, DC, 1995. [Google Scholar]
- Hansch C. In Classical and Three-Dimensional QSAR in Agrochemistry; Hansch C., Fujita T., Eds.; American Chemical Society: Washington, DC, 1995; Chapter 19, p 254. [Google Scholar]
- van de Waterbeemd H. In Experimental Pharmacokinetics; Aiache J. M., Hirtz J., Eds.; Jahr: Freiburg, Germany, 1978; p 24. [Google Scholar]
- Martin Y. C. In Modern Drug Research: Paths to Better and Safer Drugs; Martin Y. C., Kutter E., Austel V., Eds.; Marcel Dekker: New York, 1989; p 161. [Google Scholar]
- Dearden J. C. In Comprehensive Medicinal Chemistry; Hansch C., Ed.; Pergamon Press: Oxford, U.K., 1990; Vol. 4, p 375. [Google Scholar]
- Martin Y. C.; Kim K. H.; Bures M. G. In Medicinal Chemistry for the 21st Century; Wermuth C. G., Koga N., Konig H., Metcalf B. W., Eds.; Blackwell: Oxford, U.K., 1992; Chapter 20, p 295. [Google Scholar]
- Kubinyi H. In Burger’s Medicinal Chemistry; Wolff M. E., Ed.; John Wiley & Sons: New York, 1995; p 497. [Google Scholar]
- van de Waterbeemd H. In The Practice of Medicinal Chemistry; Wermuth C. G., Ed.; Academic Press: London, U.K., 1996; Chapter 19, p 367. [Google Scholar]
- Kubinyi H. In Computational Medicinal Chemistry for Drug Discovery; Bultinck P., de Winter H., Langenaeker W., Tollenaere J. P., Eds.; Marcel Dekker: New York, 2003; p 539. [Google Scholar]
- Hammett L. P.Physical Organic Chemistry; McGraw−Hill: New York, 1970. [Google Scholar]
- Collander R. Acta Chem. Scand. 1951, 5, 774. [Google Scholar]
- Zeigler B. P.Theory of Modelling and Simulation; John Wiley & Sons: New York, 1976. [Google Scholar]
- Balaz S. In Trends in QSAR and Molecular Modelling; Wermuth C. G., Ed.; Escom: Leiden, The Netherlands, 1993; p 137. [Google Scholar]
- Balaz S.; Lukacova V. J. Mol. Graph. Model. 2002, 20, 479. [DOI] [PubMed] [Google Scholar]
- Hansch C.; Fujita T. J. Am. Chem. Soc. 1964, 86, 1616. [Google Scholar]
- Stehle R. G.; Higuchi W. I. J. Pharm. Sci. 1967, 56, 1367. [DOI] [PubMed] [Google Scholar]
- Penniston J. T.; Beckett L.; Bentley D. L.; Hansch C. Mol. Pharmacol. 1969, 5, 333. [PubMed] [Google Scholar]
- McFarland J. W. J. Med. Chem. 1970, 13, 1192. [DOI] [PubMed] [Google Scholar]
- Yalkowsky S. H.; Carpenter O. S.; Flynn G. L.; Slunick T. G. J. Pharm. Sci. 1973, 62, 1949. [DOI] [PubMed] [Google Scholar]
- Yalkowsky S. H.; Flynn G. L. J. Pharm. Sci. 1973, 62, 210. [DOI] [PubMed] [Google Scholar]
- Leo A.; Jow P. Y.; Silipo C.; Hansch C. J. Med. Chem. 1975, 18, 865. [DOI] [PubMed] [Google Scholar]
- Martin Y. C.; Hackbarth J. J. J. Med. Chem. 1976, 19, 1033. [DOI] [PubMed] [Google Scholar]
- Kubinyi H. J. Med. Chem. 1977, 20, 625. [DOI] [PubMed] [Google Scholar]
- Dearden J. C.; Townend M. S. In Quantitative Structure−Activity Analysis; Franke R., Oehme P., Eds.; Akademie−Verlag: Berlin, Germany,1978; p 387. [Google Scholar]
- van de Waterbeemd H.; van Bakel H.; Jansen A. J. Pharm. Sci. 1981, 70, 1081. [DOI] [PubMed] [Google Scholar]
- Cooper E. R.; Berner B.; Bruce R. D. J. Pharm. Sci. 1981, 70, 57. [DOI] [PubMed] [Google Scholar]
- Seydel J. K. Method. Find. Exp. Clin. Pharmacol. 1984, 6, 571. [PubMed] [Google Scholar]
- Schaper K. J. Quant. Struct. Act. Relat. 1982, 1, 13. [Google Scholar]
- Franke R.Theoretical Drug Design Methods; Akademie−Verlag: Berlin, Germany, 1984. [Google Scholar]
- Berner B.; Cooper E. R. J. Pharm. Sci. 1984, 73, 102. [DOI] [PubMed] [Google Scholar]
- Amidon G. L.; Lennernäs H.; Shah V. P.; Crison J. R. Pharm. Res. 1995, 12, 413. [DOI] [PubMed] [Google Scholar]
- Xiang T. X.; Anderson B. D Biophys. J. 1998, 75, 2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benet L. Z.; Amidon G. L.; Barends D. M.; Lennernäs H.; Polli J. E.; Shah V. P.; Stavchansky S. A.; Yu L. X. Pharm. Res. 2008, 25, 483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland M.; Tozer T. N.. Clinical Pharmacokinetics: Concepts and Applications; Williams & Wilkins: Media, PA, 1995. [Google Scholar]
- Bean R. C.; Shepherd W. C.; Chan H. J. Gen. Physiol. 1968, 52, 495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung C. Y.; Carlson L. M.; Balzer C. J. Biochim. Biophys. Acta 1973, 298, 101. [DOI] [PubMed] [Google Scholar]
- Balaz S.; Sturdik E.; Durcova E.; Antalik M.; Sulo P. Biochim. Biophys. Acta 1986, 851, 93. [DOI] [PubMed] [Google Scholar]
- Cano G. D.; Busche R.; Tummler B.; Riordan J. R. Biochem. Biophys. Res. Commun. 1990, 167, 48. [DOI] [PubMed] [Google Scholar]
- Blakey G. E.; Ballard P.; Leahy D. E.; Rowland M. J. Pharm. Biomed. Anal. 1999, 18, 927. [DOI] [PubMed] [Google Scholar]
- Chou C. H.; Rowland M. J. Pharm. Sci. 1997, 86, 1310. [DOI] [PubMed] [Google Scholar]
- Welti R.; Mullikin L. J.; Yoshimura T.; Helmkamp G. M. Jr Biochemistry 1984, 23, 6086. [DOI] [PubMed] [Google Scholar]
- Gruszecki W. I.; Sujak A.; Strzalka K.; Radunz A.; Schmid G. H. J. Biosci. C 1999, 54, 517. [DOI] [PubMed] [Google Scholar]
- Sujak A.; Gabrielska J.; Milanowska J.; Mazurek P.; Strzalka K.; Gruszecki W. I. BBA—Biomembranes 2005, 1712, 17. [DOI] [PubMed] [Google Scholar]
- Gruszecki W. I.; Strzalka K. BBA—Mol. Basis Dis. 2005, 1740, 108. [DOI] [PubMed] [Google Scholar]
- Sujak A.; Gabrielska J.; Grudzinski W.; Borc R.; Mazurek P.; Gruszecki W. I. Arch. Biochem. Biophys. 1999, 371, 301. [DOI] [PubMed] [Google Scholar]
- Ho C.; Stubbs C. D. Biochemistry 1997, 36, 10630. [DOI] [PubMed] [Google Scholar]
- Asuncion-Punzalan E.; London E. Biochemistry 1995, 34, 11460. [DOI] [PubMed] [Google Scholar]
- Kachel K.; Asuncion-Punzalan E.; London E. Biochemistry 1995, 34, 15475. [DOI] [PubMed] [Google Scholar]
- Hutterer R.; Kramer K.; Schneider F. W.; Hof M. Chem. Phys. Lipids 1997, 90, 11. [Google Scholar]
- Kachel K.; Asuncion-Punzalan E.; London E. Biochim. Biophys. Acta 1998, 1374, 63. [DOI] [PubMed] [Google Scholar]
- Kaiser R. D.; London E. Biochemistry 1998, 37, 8180. [DOI] [PubMed] [Google Scholar]
- Vazquez J. L.; Montero M. T.; Merino S.; Domenech O.; Berlanga M.; Vinas M.; Hernandez-Borrell J. Langmuir 2001, 17, 1009. [DOI] [PubMed] [Google Scholar]
- de Castro B.; Gameiro P.; Lima J. L. F. C.; Matos C.; Reis S. Colloid. Surf. A 2001, 190, 205. [Google Scholar]
- Klymchenko A. S.; Duportail G.; Ozturk T.; Pivovarenko V. G.; Mely Y.; Demchenko A. P. Chem. Biol. 2002, 9, 1199. [DOI] [PubMed] [Google Scholar]
- Mitchell D. C.; Litman B. J. Biophys. J. 1998, 74, 879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai Y. S.; Ma S. M.; Nishimura S.; Ueda I. BBA−Biomembranes 1990, 1022, 245. [DOI] [PubMed] [Google Scholar]
- Gicquaud C.; Auger M.; Wong P. T. T.; Poyet P.; Boudreau N.; Gaudreault R. Arch. Biochem. Biophys. 1996, 334, 193. [DOI] [PubMed] [Google Scholar]
- Phadke R. S.; Kumar N. V.; Hosur R. V.; Saran A.; Govil G. Int. J. Quantum Chem. 1981, 20, 85. [Google Scholar]
- Ondrias K.; Ondriasova E.; Stasko A. Chem. Phys. Lipids 1992, 62, 11. [DOI] [PubMed] [Google Scholar]
- Strzalka K.; Gruszecki W. I. BBA−Biomembranes 1994, 1194, 138. [DOI] [PubMed] [Google Scholar]
- Bartucci R.; Mollica P.; Sapia P.; Sportelli L. Appl. Magn. Reson. 1998, 15, 181. [Google Scholar]
- de Matos Alves Pinto L.; Yokaichiya D. K.; Fraceto L. F.; de Paula E. Biophys. Chem. 2000, 87, 213. [DOI] [PubMed] [Google Scholar]
- Hendrich A. B.; Wesolowska O.; Komorowska M.; Motohashi N.; Michalak K. Biophys. Chem. 2002, 98, 275. [DOI] [PubMed] [Google Scholar]
- Yeagle P. L.; Hutton W. C.; Martin R. B. Biochim. Biophys. Acta 1977, 465, 173. [DOI] [PubMed] [Google Scholar]
- Koehler L. S.; Fossel E. T.; Koehler K. A. Biochemistry 1977, 16, 3700. [DOI] [PubMed] [Google Scholar]
- Boulanger Y.; Schreier S.; Leitch L. C.; Smith I. C. Can. J. Biochem 1980, 58, 986. [DOI] [PubMed] [Google Scholar]
- Boulanger Y.; Schreier S.; Smith I. C. Biochemistry 1981, 20, 6824. [DOI] [PubMed] [Google Scholar]
- Westman J.; Boulanger Y.; Ehrenberg A.; Smith I. C. Biochim. Biophys. Acta 1982, 685, 315. [DOI] [PubMed] [Google Scholar]
- Kelusky E. C.; Smith I. C. P. Can. J. Biochem. Cell Biol. 1984, 62, 178. [DOI] [PubMed] [Google Scholar]
- Kintanar A.; Kunwar A. C.; Oldfield E. Biochemistry 1986, 25, 6517. [DOI] [PubMed] [Google Scholar]
- Jacobs R. E.; White S. H. J. Am. Chem. Soc. 1984, 106, 6909. [Google Scholar]
- Bäuerle H. D.; Seelig J. Biochemistry 1991, 30, 7203. [DOI] [PubMed] [Google Scholar]
- Barry J. A.; Gawrisch K. Biochemistry 1994, 33, 8082. [DOI] [PubMed] [Google Scholar]
- Henderson J. M.; Iannucci R. M.; Petersheim M. Biophys. J. 1994, 67, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda Y.; Ogawa M.; Nasu H.; Terashima M.; Kasahara M.; Kiyama Y.; Wakita M.; Fujiwara Y.; Fujii N.; Nakagawa T. Biophys. J. 1994, 71, 1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jezowska I.; Wolak A.; Gruszecki W. I.; Strzalka K. Biochim. Biophys. Acta 1994, 1194, 143. [DOI] [PubMed] [Google Scholar]
- Peng X.; Jonas A.; Jonas J. Chem. Phys. Lipids 1995, 75, 59. [DOI] [PubMed] [Google Scholar]
- Baber J.; Ellena J. F.; Cafiso D. S. Biochemistry 1995, 34, 6533. [DOI] [PubMed] [Google Scholar]
- Tang P.; Yan B.; Xu Y Biophys. J. 1997, 72, 1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North C.; Cafiso D. S Biophys. J. 1997, 72, 1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavromoustakos T.; Theodoropoulou E. Chem. Phys. Lipids 1998, 92, 37. [DOI] [PubMed] [Google Scholar]
- Yau W. M.; Wimley W. C.; Gawrisch K.; White S. H. Biochemistry 1998, 37, 14713. [DOI] [PubMed] [Google Scholar]
- White S. H.; Wimley W. C. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 319. [DOI] [PubMed] [Google Scholar]
- Mavromoustakos T.; Daliani I. Biochim. Biophys. Acta 1999, 1420, 252. [DOI] [PubMed] [Google Scholar]
- Okamura E.; Nakahara M. In Liquid Interfaces in Chemical, Biological, and Pharmaceutical Applications; Volkov A. G., Ed.; Marcel Dekker: New York, 2001; Chapter 32, p 775. [Google Scholar]
- Feller S. E.; Brown C. A.; Nizza D. T.; Gawrisch K. Biophys. J. 2002, 82, 1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernabeu A.; Shapiro S.; Villalain J. Chem. Phys. Lipids 2002, 119, 33. [DOI] [PubMed] [Google Scholar]
- Gaede H. C.; Gawrisch K. Biophys. J. 2003, 85, 1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokono S.; Ogli K.; Miura S.; Ueda I. BBA−Biomembranes 1989, 982, 300. [DOI] [PubMed] [Google Scholar]
- Xu Y.; Tang P. Biochim. Biophys. Acta 1997, 1323, 154. [DOI] [PubMed] [Google Scholar]
- White S. H.; King G. I.; Cain J. E. Nature 1981, 290, 161. [Google Scholar]
- Herbette L.; Katz A. M.; Sturtevant J. M. Mol. Pharmacol. 1983, 24, 259. [PubMed] [Google Scholar]
- King G. I.; Chao N. M.; White S. H. Basic Life Sci. 1984, 27, 159. [Google Scholar]
- King G. I.; Jacobs R. E.; White S. H. Biochemistry 1985, 24, 4637. [DOI] [PubMed] [Google Scholar]
- Herbette L. G.; Mason P. E.; Sweeney K. R.; Trumbore M. W.; Mason R. P. Neuropharmacology 1994, 33, 241. [DOI] [PubMed] [Google Scholar]
- Balgavy P.; Uhrikova D.; Gallova J.; Kucerka N.; Madaraszova I.; Islamov A.; Gordeliy V. Chem. Preprint Server, Phys. Chem. 2002, 1. [Google Scholar]
- Hauss T.; Dante S.; Dencher N. A.; Haines T. H. BBA−Bioenergetics 2002, 1556, 149. [DOI] [PubMed] [Google Scholar]
- Trumbore M.; Chester D. W.; Moring J.; Rhodes D.; Herbette L. G Biophys. J. 1988, 54, 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan G. E.; Paul I. A.; Fassberg J. B.; Powell K. R.; Stumpf W. E.; Breese G. R. Pharmacol. Biochem. Behav. 1991, 38, 621. [DOI] [PubMed] [Google Scholar]
- Baumgart T.; Offenhausser A. Biophys. J. 2002, 83, 1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr T. E.; Thompson N. L. Biophys. Chem. 2002, 97, 29. [DOI] [PubMed] [Google Scholar]
- Balaz S.; Sturdik E.; Tichy M. Quant. Struct. Act. Relat. 1985, 4, 77. [Google Scholar]
- Ferguson J. Proc. R. Soc. B 1939, 127, 387. [Google Scholar]
- Cody V.; Chan D.; Galitsky N.; Rak D.; Luft J. R.; Pangborn W.; Queener S. F.; Laughton C. A.; Stevens M. F. Biochemistry 2000, 39, 3556. [DOI] [PubMed] [Google Scholar]
- Lukacova V.; Balaz S. J. Chem. Inf. Comput. Sci. 2003, 43, 2093. [DOI] [PubMed] [Google Scholar]
- Ohhashi T.; Mizuno R.; Ikomi F.; Kawai Y. Pharmacol. Ther. 2005, 105, 165. [DOI] [PubMed] [Google Scholar]
- Charman W. N.; Porter C. J. H. Adv. Drug Delivery Rev. 1996, 19, 149. [Google Scholar]
- Porter C. J. H.; Charman W. N. Adv. Drug Delivery Rev. 2001, 50, 61. [DOI] [PubMed] [Google Scholar]
- Trevaskis N. L.; Charman W. N.; Porter C. J. H. Adv. Drug Delivery Rev. 2008, 60, 702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellershohn C.; Benichoux R. Minerva Nucleare 1958, 2, 159. [PubMed] [Google Scholar]
- Myers R. A.; Stella V. J Int. J. Pharm. 1992, 80, 51. [Google Scholar]
- Stella V. J.; Pochopin N. L. In Lymphatic Transport of Drugs; Charman W. N., Stella V. J., Eds.; CRC Press: Boca Raton, FL, 1992; p 181. [Google Scholar]
- Nankervis R.; Davis S. S.; Day N. H.; Shaw P. N. Int. J. Pharm. 1995, 119, 173. [Google Scholar]
- Nankervis R.; Davis S. S.; Day N. H.; Shaw P. N. Int. J. Pharm. 1996, 130, 57. [Google Scholar]
- Hauss D. J.; Fogal S. E.; Ficorilli J. V.; Price C. A.; Roy T.; Jayaraj A. A.; Keirns J. J. J. Pharm. Sci. 1998, 87, 164. [DOI] [PubMed] [Google Scholar]
- Nielsen P. B.; Mullertz A.; Norling T.; Kristensen H. G. J. Pharm. Pharmacol. 2001, 53, 1439. [DOI] [PubMed] [Google Scholar]
- Shackleford D. M.; Faassen W. A.; Houwing N.; Lass H.; Edwards G. A.; Porter C. J. H.; Charman W. N. J. Pharmacol. Exp. Therap. 2003, 306, 925. [DOI] [PubMed] [Google Scholar]
- Yuan H.; Chen J.; Du Y. Z.; Hu F. Q.; Zeng S.; Zhao H. L. Colloids Surf. B 2007, 58, 157. [DOI] [PubMed] [Google Scholar]
- Porter C. J. H.; Trevaskis N. L.; Charman W. N. Nat. Rev. Drug Discovery 2007, 6, 231. [DOI] [PubMed] [Google Scholar]
- Bouwstra J. A.; Honeywell-Nguyen P. L.; Gooris G. S.; Ponec M. Prog. Lipid Res. 2003, 42, 1. [DOI] [PubMed] [Google Scholar]
- McIntosh T. J. Biophys. J. 2003, 85, 1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M. E.; Blankschtein D.; Langer R. J. Pharm. Sci. 1997, 86, 1162. [DOI] [PubMed] [Google Scholar]
- Frasch H. F.; Barbero A. M. J. Pharm. Sci. 2003, 92, 2196. [DOI] [PubMed] [Google Scholar]
- Burton P. S.; Ho N. F. H.; Conradi R. A.; Hilgers A. R. Pharm. Res. 1993, 10, 636. [Google Scholar]
- Hwa C.; Aird W. C. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H2667. [DOI] [PubMed] [Google Scholar]
- Freitas R. A., Jr.Nanomedicine; Landes Bioscience: Georgetown, TX, 1999. [Google Scholar]
- Mehta D.; Malik A. B. Physiol. Rev. 2006, 86, 279. [DOI] [PubMed] [Google Scholar]
- Wisse E.; de Zanger R. B.; Charels K.; van der Smissen P.; McCuskey R. S. Hepatology 1985, 5, 683. [DOI] [PubMed] [Google Scholar]
- Horn T.; Henriksen J. H.; Christoffersen P. Liver 1986, 6, 98. [DOI] [PubMed] [Google Scholar]
- Wisse E.; Braet F.; Luo D.; de Zanger R.; Jans D.; Crabbe E.; Vermoesen A. Toxicol. Pathol. 1996, 24, 100. [DOI] [PubMed] [Google Scholar]
- Jang S. H.; Wientjes M. G.; Lu D.; Au J. L. S. Pharm. Res. 2003, 20, 1337. [DOI] [PubMed] [Google Scholar]
- Matsumoto K.; Nishi K.; Tokutomi Y.; Irie T.; Suenaga A.; Otagiri M. Biol. Pharm. Bull. 2003, 26, 123. [DOI] [PubMed] [Google Scholar]
- Boyd W.; Sheldon H.. Boyd’s Introduction to the Study of Disease; Lea & Febiger: Philadelphia, PA, 1992. [Google Scholar]
- Lentner C., Ed. Geigy Scientific Tables, 8th ed.; Ciba−Geigy Corporation: West Caldwell, NJ, 1986. [Google Scholar]
- Snyder W. S.; Cook M. J.; Nasset E. S.; Karhausen L. R.; Howells G. P.; Tipton I. H.. Report of the Task Group on Reference Man; Pergamon Press: Oxford, U.K., 1975. [Google Scholar]
- Mizuno N.; Sugiyama Y. Drug Metabol. Pharmacokin. 2002, 17, 93. [DOI] [PubMed] [Google Scholar]
- van Tellingen O. Toxicol. Lett. 2001, 120, 31. [DOI] [PubMed] [Google Scholar]
- Zhang E. Y.; Phelps M. A.; Cheng C.; Ekins S.; Swaan P. W. Adv. Drug Delivery Rev. 2002, 54, 329. [DOI] [PubMed] [Google Scholar]
- Custodio J. M.; Wu C. Y.; Benet L. Z. Adv. Drug. Delivery Rev. 2008, 60, 717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson P. D.; Kell D. B. Nat. Rev. Drug Discovery 2008, 7, 205. [DOI] [PubMed] [Google Scholar]
- Denison M. S.; Nagy S. R. Annu. Rev. Pharmacol. 2003, 43, 309. [DOI] [PubMed] [Google Scholar]
- Poland A.; Glover E.; Kende A. S. J. Biol. Chem. 1976, 251, 4936. [PubMed] [Google Scholar]
- Pollenz R. S.; Sattler C. A.; Poland A. Mol. Pharmacol. 1994, 45, 428. [PubMed] [Google Scholar]
- Seydel J. K.; Wiese M.. Drug−Membrane Interactions: Analysis, Drug Distribution, Modeling; Wiley−VCH: Weinheim, Germany, 2003. [Google Scholar]
- DiGirolamo M.; Owens J. L. Am. J. Physiol. 1976, 231, 1568. [DOI] [PubMed] [Google Scholar]
- Naegeli C.; Cramer C.. Pflanzenphysiologische Untersuchungen; F. Schultess: Zurich, Switzerland, 1855. [Google Scholar]
- Pfeffer W.Osmotische Untersuchungen; W. Engelmann: Zurich, Switzerland, 1877. [Google Scholar]
- Smith H. W. Circulation 1962, 26, 987. [DOI] [PubMed] [Google Scholar]
- Wilhelmy L. Ann. Phys. (Leipzig) 1863, 119, 177. [Google Scholar]
- Pockels A. Nature 1891, 43, 437. [Google Scholar]
- Overton E. Vierteljahresschr. Naturforsch. Ges. Zuerich 1899, 44, 88. [Google Scholar]
- Langmuir I. J. Am. Chem. Soc. 1917, 39, 1848. [Google Scholar]
- Gorter E.; Grendel F. J. Exp. Med. 1925, 41, 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielli J. F.; Davson H. J. Cell Comput. Physiol. 1935, 5, 495. [Google Scholar]
- Robertson J. D. J. Biophys. Biochem. Cytol. 1957, 3, 1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer S. J.; Nicholson G. L. Science 1972, 175, 720. [DOI] [PubMed] [Google Scholar]
- Robertson J. D. In Cellular Membranes in Development; Academic Press: New York, 1964; p 1. [Google Scholar]
- Luzzati V.In Biological Membranes; Chapman D., Ed.; Academic Press: London, U.K., 1968, p 71. [Google Scholar]
- de Kruyff B.; van Dijck P. W. M.; Goldbach R. W.; Demel R. A.; van Deenen L. L. M. BBA−Biomembranes 1973, 330, 269. [DOI] [PubMed] [Google Scholar]
- Levine Y. K.; Wilkins M. H. F. Nature 1971, 230, 69. [DOI] [PubMed] [Google Scholar]
- Frank H. S.; Evans M. W. J. Chem. Phys. 1945, 13, 507. [Google Scholar]
- Labes R.; Jansen E. Arch. Exp. Pathol. Pharmakol. 1930, 158, 1. [Google Scholar]
- Tamamushi B. Kolloid-Z. 1957, 150, 44. [Google Scholar]
- Lange H. Kolloid-Z. 1960, 169, 124. [Google Scholar]
- Nathanson A.German Patent525300, 1929. Chem. Abstr. 1929, 25, 2860.
- Scheraga H. A. J. Phys. Chem. 1961, 65, 1071. [Google Scholar]
- Tanford C. J. Am. Chem. Soc. 1962, 84, 4240. [Google Scholar]
- Tanford Ch.The Hydrophobic Effect: Formation of Micelles and Biological Membranes; Wiley: New York, 1973. [Google Scholar]
- Büldt G.; Gally H. U.; Seelig A.; Seelig J.; Zaccai G. Nature 1978, 271, 182. [DOI] [PubMed] [Google Scholar]
- Büldt G.; Gally H. U.; Seelig J.; Zaccai G. J. Mol. Biol. 1979, 134, 673. [DOI] [PubMed] [Google Scholar]
- Zaccai G.; Büldt G.; Seelig A.; Seelig J. J. Mol. Biol. 1979, 134, 693. [DOI] [PubMed] [Google Scholar]
- Seelig J.; Macdonald P. M.; Scherer P. G. Biochemistry 1987, 26, 7535. [DOI] [PubMed] [Google Scholar]
- Unwin N.; Henderson R. Sci. Am. 1984, 250, 78. [DOI] [PubMed] [Google Scholar]
- Mouritsen O. G.; Bloom M. Biophys. J. 1984, 46, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiener M. C.; White S. H. Biophys. J. 1991, 59, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiener M. C.; White S. H. Biophys. J. 1991, 59, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiener M. C.; King G. I.; White S. H. Biophys. J. 1991, 60, 568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiener M. C.; White S. H. Biophys. J. 1992, 61, 428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiener M. C.; White S. H. Biophys. J. 1992, 61, 434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagle J. F.; Zhang R.; Tristram-Nagle S.; Sun W.; Petrache H. I.; Suter R. M. Biophys. J. 1996, 70, 1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen L. K.; Bjornholm T.; Mouritsen O. G. Nature 2000, 404, 352. [DOI] [PubMed] [Google Scholar]
- Leonard A.; Escrive C.; Laguerre M.; Pebay-Peyroula E.; Neri W.; Pott T.; Katsaras J.; Dufourc E. J. Langmuir 2001, 17, 2019. [Google Scholar]
- Seeger H. M.; Fidorra M.; Heimburg T. Macromol. Symp. 2004, 219, 85. [Google Scholar]
- Hill W. G.; Zeidel M. L. J. Biol. Chem. 2000, 275, 30176. [DOI] [PubMed] [Google Scholar]
- Hill W. G.; Almasri E.; Ruiz W. G.; Apodaca G.; Zeidel M. L. Am. J. Physiol. 2005, 289, C33. [DOI] [PubMed] [Google Scholar]
- Redelmeier T. E.; Hope M. J.; Cullis P. R. Biochemistry 1990, 29, 3046. [DOI] [PubMed] [Google Scholar]
- Holthuis J. C. M.; Van Meer G.; Huitema K. Mol. Membr. Biol. 2003, 20, 231. [PubMed] [Google Scholar]
- Cooper G. J.; Zhou Y.; Bouyer P.; Grichtchenko I. I.; Boron W. F. J. Physiol. 2002, 542, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwaal R. F. A.; Comfurius P.; Bevers E. M. Cell. Mol. Life Sci. 2005, 62, 971. [DOI] [PubMed] [Google Scholar]
- Selinsky B. S.; Yeagle P. L. Biochemistry 1984, 23, 2281. [DOI] [PubMed] [Google Scholar]
- Jain M. K.; Yu B. Z.; Kozubek A. Biochim. Biophys. Acta 1989, 980, 23. [DOI] [PubMed] [Google Scholar]
- Gawrisch K.; Barry J. A.; Holte L. L.; Sinnwell T.; Bergelson L.; Ferretti J. A. Mol. Membr. Biol. 1995, 12, 83. [DOI] [PubMed] [Google Scholar]
- Hinderliter A. K.; Almeida P. F. F.; Biltonen R. L.; Creutz C. E. Biochim. Biophys. Acta 1998, 1448, 227. [DOI] [PubMed] [Google Scholar]
- Sabra M. C.; Mouritsen O. G Biophys. J. 1998, 74, 745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinderliter A. K.; Almeida P. F. F.; Creutz C. E.; Biltonen R. L. Biochemistry 2001, 40, 4181. [DOI] [PubMed] [Google Scholar]
- Jensen M. O.; Mouritsen O. G. BBA−Biomembranes 2004, 1666, 205. [DOI] [PubMed] [Google Scholar]
- Almeida P. F. F.; Vaz W. L. C.; Thompson T. E. Biochemistry 1992, 31, 6739. [DOI] [PubMed] [Google Scholar]
- Almeida P. F. F.; Vaz W. L. C.; Thompson T. E. Biochemistry 1992, 31, 7198. [DOI] [PubMed] [Google Scholar]
- Almeida P. F. F.; Vaz W. L. C.; Thompson T. E. Biophys. J. 1993, 64, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens M. J. J. Am. Chem. Soc. 2005, 127, 15330. [DOI] [PubMed] [Google Scholar]
- Almeida P. F. F.; Pokorny A.; Hinderliter A. Biochim. Biophys. Acta 2006, 1720, 1. [DOI] [PubMed] [Google Scholar]
- Dowhan W. Annu. Rev. Biochem. 1997, 66, 199. [DOI] [PubMed] [Google Scholar]
- Hinderliter A. K.; Dibble A. R. G.; Biltonen R. L.; Sando J. J. Biochemistry 1997, 36, 6141. [DOI] [PubMed] [Google Scholar]
- Brown D. A.; London E. Annu. Rev. Cell Develop. Biol. 1998, 14, 111. [DOI] [PubMed] [Google Scholar]
- Brown D. A.; London E. J. Membr. Biol. 1998, 164, 103. [DOI] [PubMed] [Google Scholar]
- Brown D. A.; London E. J. Biol. Chem. 2000, 275, 17221. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan A.; Anderson T. G.; McConnell H. M. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 12422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodin S.; Tronchere H.; Payrastre B. BBA−Biomembranes 2003, 1610, 247. [DOI] [PubMed] [Google Scholar]
- Edidin M. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 257. [DOI] [PubMed] [Google Scholar]
- Opekarova M.; Tanner W. BBA−Biomembranes 2003, 1610, 11. [DOI] [PubMed] [Google Scholar]
- Hinderliter A.; Biltonen R. L.; Almeida P. F. F. Biochemistry 2004, 43, 7102. [DOI] [PubMed] [Google Scholar]
- Mouritsen O. G.; Jorgensen K. Pharm. Res. 1998, 15, 1507. [DOI] [PubMed] [Google Scholar]
- Xiang T. X.; Anderson B. D. Biochim. Biophys. Acta 1998, 1370, 64. [DOI] [PubMed] [Google Scholar]
- Pokorny A.; Almeida P. F. F.; Melo E. C. C.; Vaz W. L. C. Biophys. J. 2000, 78, 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokorny A.; Almeida P. F. F.; Vaz W. L. C. Biophys. J. 2001, 80, 1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S. S.; Gong K.; Chew J. Langmuir 2002, 18, 4061. [Google Scholar]
- Heerklotz H.; Szadkowska H.; Anderson T.; Seelig J. J. Mol. Biol. 2003, 329, 793. [DOI] [PubMed] [Google Scholar]
- Davies M. A.; Hubner W.; Blume A.; Mendelsohn R. Biophys. J. 1992, 63, 1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimburg T. In Planar Lipid Bilayers (BLMs) and Their Applications; Tien H. T., Ottova A., Eds.; Elsevier: Amsterdam, The Netherlands, 2003; p 269. [Google Scholar]
- Nagle J. F.; Tristram-Nagle S. BBA−Rev. Biomembr. 2000, 1469, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimburg T.; Jackson A. D. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 9790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebel H.; Grabitz P.; Heimburg T. J. Phys. Chem. B 2001, 105, 7353. [Google Scholar]
- Grabitz P.; Ivanova V. P.; Heimburg T. Biophys. J. 2002, 82, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieleman D. P.; Marrink S. J.; Berendsen H. J. Biochim. Biophys. Acta 1997, 1331, 235. [DOI] [PubMed] [Google Scholar]
- Nagle J. F.; Wilkinson D. A. Biophys. J. 1978, 23, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh D.CRC Handbook of Lipid Bilayers; CRC Press: Boca Raton, FL, 1990. [Google Scholar]
- Cevc G.; Marsh D.. Phospholipid Bilayers: Physical Principles and Models; Wiley: New York, 1989. [Google Scholar]
- Brumm T.; Naumann C.; Sackmann E.; Rennie A. R.; Thomas R. K.; Kanellas D.; Penfold J.; Bayerl T. M. Eur. Biophys. J. 1994, 23, 289. [Google Scholar]
- Westlund P. O. J. Phys. Chem. B 2000, 104, 6059. [Google Scholar]
- Costigan S. C.; Booth P. J.; Templer R. H. Biochim. Biophys. Acta 2000, 1468, 41. [DOI] [PubMed] [Google Scholar]
- De Young L. R.; Dill K. A. Biochemistry 1988, 27, 5281. [DOI] [PubMed] [Google Scholar]
- Balgavy P.; Dubnickova M.; Kucerka N.; Kiselev M. A.; Yaradaikin S. P.; Uhrikova D. Biochim. Biophys. Acta 2001, 1512, 40. [DOI] [PubMed] [Google Scholar]
- Ho C.; Slater S. J.; Stubbs C. D. Biochemistry 1995, 34, 6188. [DOI] [PubMed] [Google Scholar]
- Nagle J. F.; Tristram-Nagle S. Curr. Opin. Struct. Biol. 2000, 10, 474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand R. P.; Parsegian V. A. BBA−Rev. Biomembr. 1989, 988, 351. [Google Scholar]
- King G. I.; White S. H. Biophys. J. 1986, 49, 1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armen R. S.; Uitto O. D.; Feller S. E. Biophys. J. 1998, 75, 734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelig J. Q. Rev. Biophys. 1977, 10, 353. [DOI] [PubMed] [Google Scholar]
- Seelig J.; MacDonald P. M. Acc. Chem. Res. 1987, 20, 221. [Google Scholar]
- Mason J. T. Meth. Enzymol. 1998, 295, 468. [DOI] [PubMed] [Google Scholar]
- Mendelsohn R.; Moore D. J. Chem. Phys. Lipids 1998, 96, 141. [DOI] [PubMed] [Google Scholar]
- Borle F.; Seelig J. BBA−Biomembranes 1983, 735, 131. [DOI] [PubMed] [Google Scholar]
- Seelig J. BBA−Rev. Biomembr. 1978, 515, 105. [DOI] [PubMed] [Google Scholar]
- Garidel P.; Blume A.; Hübner W. BBA−Biomembranes 2000, 1466, 245. [DOI] [PubMed] [Google Scholar]
- Hübner W.; Blume A. J. Phys. Chem. 1990, 94, 7726. [Google Scholar]
- Hauser H.; Guyer W.; Levine B.; Skrabal P.; Williams R. J. P. Biochim. Biophys. Acta 1978, 508, 450. [DOI] [PubMed] [Google Scholar]
- Smith S. O.; Kustanovich I.; Bhamidipati S.; Salmon A.; Hamilton J. A. Biochemistry 1992, 31, 11660. [DOI] [PubMed] [Google Scholar]
- Blume A.; Hübner W.; Messner G. Biochemistry 1988, 27, 8239. [DOI] [PubMed] [Google Scholar]
- Yeagle P. L.; Hutton W. C.; Huang C. H.; Martin R. B. Proc. Natl. Acad. Sci. U.S.A. 1975, 72, 3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallova J.; Uhrikova D.; Hanulova M.; Teixeira J.; Balgavy P. Colloid. Surf. B 2004, 38, 11. [DOI] [PubMed] [Google Scholar]
- Marrink S. J.; Berendsen H. J. C. J. Phys. Chem. 1994, 98, 4155. [Google Scholar]
- Hristova K.; White S. H. Biophys. J. 1998, 74, 2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashl R. J.; Scott H. L.; Subramaniam S.; Jakobsson E. Biophys. J. 2001, 81, 3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin B.; Hopfinger A. J. Pharm. Res. 1996, 13, 1786. [DOI] [PubMed] [Google Scholar]
- Dill K. A.; Flory P. J. Proc. Natl. Acad. Sci. U.S.A. 1980, 77, 3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelstein A. J. Gen. Physiol. 1976, 68, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huster D.; Jin A. J.; Arnold K.; Gawrisch K. Biophys. J. 1997, 73, 855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon S. A.; McIntosh T. J. Meth. Enzymol. 1986, 127, 511. [DOI] [PubMed] [Google Scholar]
- Meier E. M.; Schummer D.; Sandhoff K. Chem. Phys. Lipids 1990, 55, 103. [DOI] [PubMed] [Google Scholar]
- Jacobs R. E.; White S. H. Biochemistry 1989, 28, 3421. [DOI] [PubMed] [Google Scholar]
- Suits F.; Pitman M. C.; Feller S. E. J. Chem. Phys. 2005, 122, 244714. [DOI] [PubMed] [Google Scholar]
- Gurtovenko A. A. J. Chem. Phys. 2005, 122, 1. [DOI] [PubMed] [Google Scholar]
- Dachary-Prigent J.; Dufourcq J.; Lussan C.; Boisseau M. Thromb. Res. 1979, 14, 15. [DOI] [PubMed] [Google Scholar]
- Surewicz W. K.; Leyko W. Biochim. Biophys. Acta 1981, 643, 387. [DOI] [PubMed] [Google Scholar]
- Kubo M.; Gardner M. F.; Hostetler K. Y. Biochem. Pharmacol. 1986, 35, 3761. [DOI] [PubMed] [Google Scholar]
- van Balen G. P.; Martinet C.; Caron G.; Bouchard G.; Reist M.; Carrupt P. A.; Fruttero R.; Gasco A.; Testa B. Med. Res. Rev. 2004, 24, 299. [DOI] [PubMed] [Google Scholar]
- Makino K.; Yamada T.; Kimura M. Biophys. Chem. 1991, 41, 175. [DOI] [PubMed] [Google Scholar]
- Burns S. T.; Khaledi M. G. J. Pharm. Sci. 2002, 91, 1601. [DOI] [PubMed] [Google Scholar]
- Clarke R. J. Biochim. Biophys. Acta 1997, 1327, 269. [DOI] [PubMed] [Google Scholar]
- Clarke R. J. Adv. Colloid Interface 2001, 89−90, 263. [DOI] [PubMed] [Google Scholar]
- Gawrisch K.; Ruston D.; Zimmerberg J.; Parsegian V. A.; Rand R. P.; Fuller N. Biophys. J. 1992, 61, 1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis S. S.; James M. J.; Anderson N. H. Faraday Disc. Chem. Soc. 1986, 81, 313. [Google Scholar]
- Betageri G. V.; Rogers J. A. Pharm. Res. 1989, 6, 399. [DOI] [PubMed] [Google Scholar]
- Escher B. I.; Schwarzenbach R. P. Environ. Sci. Technol. 1996, 30, 260. [Google Scholar]
- Vaes W. H.; Ramos E. U.; Verhaar H. J.; Cramer C. J.; Hermens J. L. Chem. Res. Toxicol. 1998, 11, 847. [DOI] [PubMed] [Google Scholar]
- Vaes W. H. J.; Ramos E. U.; Verhaar H. J. M.; Cramer C. J.; Hermens J. L. M. In Molecular Modeling and Prediction of Bioactivity; Gundertofte K., Jorgensen S. F., Eds.; Plenum Press: New York, 2000. [Google Scholar]
- Bangham A. D.; Standish M. M.; Watkins J. C. J. Mol. Biol. 1965, 13, 238. [DOI] [PubMed] [Google Scholar]
- Beschiaschvili G.; Seelig J. Biochemistry 1992, 31, 10044. [DOI] [PubMed] [Google Scholar]
- Lepore L. S.; Ellena J. F.; Cafiso D. S. Biophys. J. 1992, 61, 767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oku N.; Scheerer J. F.; Macdonald R. C. Biochim. Biophys. Acta 1982, 692, 384. [DOI] [PubMed] [Google Scholar]
- Marassi F. M.; Shivers R. R.; Macdonald P. M. Biochemistry 1993, 32, 9936. [DOI] [PubMed] [Google Scholar]
- Oku N.; MacDonald R. C. BBA−Biomembranes 1983, 734, 54. [Google Scholar]
- Saito Y.; Hirashima N.; Kirino Y. Biochem. Biophys. Res. Commun. 1988, 154, 85. [DOI] [PubMed] [Google Scholar]
- Akashi K.; Miyata H.; Itoh H.; Kinosita K. Jr Biophys. J. 1996, 71, 3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi K.; Miyata H.; Itoh H.; Kinosita K. Jr Biophys. J. 1998, 74, 2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscho A.; Orwar O.; Chiu D. T.; Modi B. P.; Zare R. N. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 11443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes D. J.; Mayer M. BBA−Biomembranes 2005, 1712, 152. [DOI] [PubMed] [Google Scholar]
- Hotani H.; Nomura F.; Suzuki Y. Curr. Opin. Colloid Interface Sci. 1999, 4, 358. [Google Scholar]
- Korlach J.; Schwille P.; Webb W. W.; Feigenson G. W. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 8461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahya N.; Schwille P.; Brown D. A. Biochemistry 2005, 44, 7479. [DOI] [PubMed] [Google Scholar]
- Needham D.; Evans E. Biochemistry 1988, 27, 8261. [DOI] [PubMed] [Google Scholar]
- Sapper A.; Janshoff A. Langmuir 2006, 22, 10869. [DOI] [PubMed] [Google Scholar]
- Riquelme G.; Lopez E.; Garcia-Segura L. M.; Ferragut J. A.; Gonzalez-Ros J. M. Biochemistry 1990, 29, 11215. [DOI] [PubMed] [Google Scholar]
- Luxnat M.; Galla H. J. Biochim. Biophys. Acta 1986, 856, 274. [DOI] [PubMed] [Google Scholar]
- Zachowski A.; Durand P. Biochim. Biophys. Acta 1988, 937, 411. [DOI] [PubMed] [Google Scholar]
- Kitamura K.; Imayoshi N.; Goto T.; Shiro H.; Mano T.; Nakai Y. Anal. Chim. Acta 1995, 304, 101. [Google Scholar]
- Takegami S.; Kitamura K.; Takahashi K.; Kitade T. J. Pharm. Sci. 2002, 91, 1568. [DOI] [PubMed] [Google Scholar]
- Pola A.; Michalak K.; Burliga A.; Motohashi N.; Kawase M. Eur. J. Pharm. Sci. 2004, 21, 421. [DOI] [PubMed] [Google Scholar]
- Casals E.; Galan A. M.; Escolar G.; Gallardo M.; Estelrich J. Chem. Phys. Lipids 2003, 125, 139. [DOI] [PubMed] [Google Scholar]
- Avdeef A.; Box K. J.; Comer J. E. A.; Hibbert C.; Tam K. Y. Pharm. Res. 1998, 15, 209. [DOI] [PubMed] [Google Scholar]
- Balon K.; Riebesehl B. U.; Müller B. W. Pharm. Res. 1999, 16, 882. [DOI] [PubMed] [Google Scholar]
- Liu X. Y.; Yang Q.; Hara M.; Nakamura G.; Miyake J. Mater. Sci. Eng. C 2001, 17, 119. [Google Scholar]
- Zhang Y.; Zeng C. M.; Li Y. M.; Hjerten S.; Lundahl P. J. Chromatogr. A 1996, 749, 13. [DOI] [PubMed] [Google Scholar]
- Yang Q.; Liu X. Y.; Ajiki S. I.; Hara M.; Lundahl P.; Miyake J. J. Chromatogr. B 1998, 707, 131. [DOI] [PubMed] [Google Scholar]
- Danelian E.; Karlen A.; Karlsson R.; Winiwarter S.; Hansson A.; Lofas S.; Lennernäs H.; Hamalainen M. D. J. Med. Chem. 2000, 43, 2083. [DOI] [PubMed] [Google Scholar]
- Baird C. L.; Courtenay E. S.; Myszka D. G. Anal. Biochem. 2002, 310, 93. [DOI] [PubMed] [Google Scholar]
- Frostell-Karlsson A.; Widegren H.; Green C. E.; Haemaelaeinen M. D.; Westerlund L.; Karlsson R.; Fenner K.; van de Waterbeemd H. J. Pharm. Sci. 2005, 94, 25. [DOI] [PubMed] [Google Scholar]
- Cimitan S.; Lindgren M. T.; Bertucci C.; Danielson U. H. J. Med. Chem. 2005, 48, 3536. [DOI] [PubMed] [Google Scholar]
- Bayburt T. H.; Carlson J. W.; Sligar S. G. J. Struct. Biol. 1998, 123, 37. [DOI] [PubMed] [Google Scholar]
- Bayburt T. H.; Grinkova Y. V.; Sligar S. G. Nano Lett. 2002, 2, 853. [Google Scholar]
- Nieh M. P.; Raghunathan V. A.; Glinka C. J.; Harroun T.; Katsaras J. Macromol. Symp. 2004, 219, 135. [Google Scholar]
- Nieh M. P.; Raghunathan V. A.; Glinka C. J.; Harroun T. A.; Pabst G.; Katsaras J. Langmuir 2004, 20, 7893. [DOI] [PubMed] [Google Scholar]
- Sanders C. R.; Landis G. C. Biochemistry 1995, 34, 4030. [DOI] [PubMed] [Google Scholar]
- Bayburt T. H.; Sligar S. G. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 6725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard K. P.; Opella S. J. J. Magn. Reson. B 1996, 112, 91. [DOI] [PubMed] [Google Scholar]
- Bayburt T. H.; Sligar S. G. Protein Sci. 2003, 12, 2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih A.Y.; Denisov I. G.; Phillips J. C.; Sligar S. G.; Schulten K. Biophys. J. 2005, 88, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih A.Y.; Freddolino P. L.; Arkhipov A.; Schulten K. J. Struct. Biol. 2007, 157, 579. [DOI] [PubMed] [Google Scholar]
- Shih A.Y.; Arkhipov A.; Freddolino P. L.; Sligar S. G.; Schulten K. J. Phys. Chem. B 2007, 111, 11095. [DOI] [PubMed] [Google Scholar]
- Shih A.Y.; Freddolino P. L.; Sligar S. G.; Schulten K. Nano Lett. 2007, 7, 1692. [DOI] [PubMed] [Google Scholar]
- Raedler J.; Strey H.; Sackmann E. Langmuir 1995, 11, 4539. [Google Scholar]
- Reimhult E.; Höök F.; Kasemo B. Phys. Rev. E 2002, 66, 051905. [DOI] [PubMed] [Google Scholar]
- Loidl-Stahlhofen A.; Eckert A.; Hartmann T.; Schottner M. J. Pharm. Sci. 2001, 90, 599. [DOI] [PubMed] [Google Scholar]
- Leonenko Z. V.; Carnini A.; Cramb D. T. Biochim. Biophys. Acta 2000, 1509, 131. [DOI] [PubMed] [Google Scholar]
- Tamm L. K.; Boehm C.; Yang J.; Shao Z.; Hwang J.; Edidin M.; Betzig E. Thin Solid Films 1996, 284−285, 813. [Google Scholar]
- Cremer P. S.; Boxer S. G. J. Phys. Chem. B 1999, 103, 2554. [Google Scholar]
- Bayerl T. M.; Bloom M. Biophys. J. 1990, 58, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng C. C.; Cheng Y. L.; Pennefather P. S. Biophys. J. 2004, 87, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinem C.; Janshoff A.; Ulrich W. P.; Sieber M.; Galla H. J. Biochim. Biophys. Acta 1996, 1279, 169. [DOI] [PubMed] [Google Scholar]
- Stephens S. M.; Dluhy R. A. Thin Solid Films 1996, 284−285, 381. [Google Scholar]
- Katz M.; Ben Shlush I.; Kolusheva S.; Jelinek R. Pharm. Res. 2006, 23, 580. [DOI] [PubMed] [Google Scholar]
- Kew S. J.; Hall E. A. H. Anal. Chem. 2006, 78, 2231. [DOI] [PubMed] [Google Scholar]
- Collander R. Protoplasma 1927, 3, 213. [Google Scholar]
- Collander R. Kolloidchem. Beih. 1924, 19, 72. [Google Scholar]
- Collander R. Kolloidchem. Beih. 1925, 20, 273. [Google Scholar]
- Collander R. Soc. Sci. Fenn., Commentat. Biol. 1926, 2, 1. [Google Scholar]
- Müller P.; Rudin D. O.; Tien H. T. T.; Wescott W. C. Nature 1962, 194, 979. [DOI] [PubMed] [Google Scholar]
- Müller P.; Rudin D. O. J. Theor. Biol. 1963, 4, 268. [DOI] [PubMed] [Google Scholar]
- Müller P.; Rudin D. O.; Tien H. T.; Wescott W. C. J. Phys. Chem. 1963, 67, 534. [Google Scholar]
- Montal M.; Mueller P. Proc. Natl. Acad. Sci. U.S.A. 1972, 69, 3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funakoshi K.; Suzuki H.; Takeuchi S. Anal. Chem. 2006, 78, 8169. [DOI] [PubMed] [Google Scholar]
- Thompson M.; Lennox R. B.; McClelland R. A. Anal. Chem. 1982, 54, 76. [Google Scholar]
- Thompson M.; Krull U. J. Anal. Chim. Acta 1982, 141, 33. [Google Scholar]
- Thompson M.; Krull U. J. Anal. Chim. Acta 1982, 142, 207. [Google Scholar]
- Hanai T.; Haydon D. A.; Taylor J. Proc. R. Soc. A 1964, 281, 377. [Google Scholar]
- Walter A.; Gutknecht J. J. Membr. Biol. 1984, 77, 255. [DOI] [PubMed] [Google Scholar]
- Yamada H.; Matsue T.; Uchida I. Biochem. Biophys. Res. Commun. 1991, 180, 1330. [DOI] [PubMed] [Google Scholar]
- Xiang T. X.; Chen X.; Anderson B. D. Biophys. J. 1992, 63, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang T. X.; Anderson B. D. Pharm. Res. 1993, 10, 1654. [DOI] [PubMed] [Google Scholar]
- Troiano G. C.; Tung L.; Sharma V.; Stebe K. J. Biophys. J. 1998, 75, 880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugao N.; Sugawara M.; Minami H.; Uto M.; Umezawa Y. Anal. Chem. 1993, 65, 363. [DOI] [PubMed] [Google Scholar]
- Antonenko Y. N.; Yaguzhinsky L. S. FEBS Lett. 1983, 163, 42. [DOI] [PubMed] [Google Scholar]
- Shi B.; Tien H. T. Biochim. Biophys. Acta 1986, 859, 125. [DOI] [PubMed] [Google Scholar]
- Troiano G. C.; Stebe K. J.; Raphael R. M.; Tung L. Biophys. J. 1999, 76, 3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuempel D.; Swartzendruber D. C.; Squier C. A.; Wertz P. W. BBA−Biomembranes 1998, 1372, 135. [DOI] [PubMed] [Google Scholar]
- Yoneto K.; Li S. K.; Ghanem A. H.; Crommelin D. J.; Higuchi W. I. J. Pharm. Sci. 1995, 84, 853. [DOI] [PubMed] [Google Scholar]
- Yoneto K.; Li S. K.; Higuchi W. I.; Jiskoot W.; Herron J. N. J. Pharm. Sci. 1996, 85, 511. [DOI] [PubMed] [Google Scholar]
- Jager M.; Groenink W.; Guivernau R.; Andersson E.; Angelova N.; Ponec M.; Bouwstra J. Pharm. Res. 2006, 23, 951. [DOI] [PubMed] [Google Scholar]
- Setaka M.; Yamamoto T.; Sato N.; Yano M.; Kwan T. J. Biochem. 1982, 91, 79. [DOI] [PubMed] [Google Scholar]
- Coronado R.; Latorre R. Biophys. J. 1983, 43, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson S. J.; Bayerl T. M.; McDermott D. C.; Adam G. W.; Rennie A. R.; Thomas R. K.; Sackmann E. Biophys. J. 1991, 59, 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger S.; Koenig B. W.; Orts W. J.; Berk N. F.; Majkrzak C. F.; Gawrisch K. Basic Life Sci. 1996, 64, 205. [DOI] [PubMed] [Google Scholar]
- Ziegler W.; Gaburjakova J.; Gaburjakova M.; Sivak B.; Rehacek V.; Tvarozek V.; Hianik T. Colloid. Surf. A 1998, 140, 357. [Google Scholar]
- Ekeroth J.; Konradsson P.; Höök F. Langmuir 2002, 18, 7923. [Google Scholar]
- Hianik T.; Snejdarkova M.; Rehak M.; Passechnik V. I.; Sokolikova L.; Sivak B.; Ivanov S. A. Thin Solid Films 1996, 284−285, 817. [Google Scholar]
- Hianik T.; Dlugopolsky J.; Gyeppessova M.; Sivak B.; Tien H. T.; Ottova-Leitmannova A. Bioelectrochem. Bioenerg. 1996, 39, 299. [Google Scholar]
- Hianik T.; Dlugopolsky J.; Passechnik V. I.; Sargent D. F.; Ivanov S. A. Colloid. Surf. A 1996, 106, 109. [Google Scholar]
- Naumann C. A.; Prucker O.; Lehmann T.; Ruehe J.; Knoll W.; Frank C. W. Biomacromolecules 2002, 3, 27. [DOI] [PubMed] [Google Scholar]
- Yoshikawa K.; Hayashi H.; Shimooka T.; Terada H.; Ishii T. Biochem. Biophys. Res. Commun. 1987, 145, 1092. [DOI] [PubMed] [Google Scholar]
- Nikolelis D. P.; Siontorou C. G. Anal. Chem 1995, 67, 936. [DOI] [PubMed] [Google Scholar]
- Weng K. C.; Stalgren J. J. R.; Risbud S. H.; Frank C. W. J. Non-Cryst. Solids 2004, 350, 46. [Google Scholar]
- Kansy M.; Senner F.; Gubernator K. J. Med. Chem. 1998, 41, 1007. [DOI] [PubMed] [Google Scholar]
- Kansy M.; Fischer H.; Kratzat K. Senner F.; Wagner B.; Parrilla I. In Pharmacokinetic Optimization in Drug Research; Testa B., van de Waterbeemd H., Folkers G., Guy R., Eds.; Wiley−VCH: Weinheim, Germany, 2001; p 448. [Google Scholar]
- Kansy M.; Avdeef A.; Fischer H. Drug Discovery Today 2004, 1, 349. [DOI] [PubMed] [Google Scholar]
- Avdeef A.; Nielsen P. E.; Tsinman O. Eur. J. Pharm. Sci. 2004, 22, 365. [DOI] [PubMed] [Google Scholar]
- Zhu C.; Jiang L.; Chen T. M.; Hwang K. K. Eur. J. Med. Chem. 2002, 37, 399. [DOI] [PubMed] [Google Scholar]
- Loftsson T.; Konradsdottir F.; Masson M. Int. J. Pharm. 2006, 326, 60. [DOI] [PubMed] [Google Scholar]
- Corti G.; Maestrelli F.; Cirri M.; Furlanetto S.; Mura P. Eur. J. Pharm. Sci 2006, 27, 346. [DOI] [PubMed] [Google Scholar]
- Sugano K.; Hamada H.; Machida M.; Ushio H. J. Biomol. Screen. 2001, 6, 189. [DOI] [PubMed] [Google Scholar]
- Teksin Z. S.; Hom K.; Balakrishnan A.; Polli J. E. J. Controlled Release 2006, 116, 50. [DOI] [PubMed] [Google Scholar]
- Di L.; Kerns E. H.; Fan K.; McConnell O. J.; Carter G. T. Eur. J. Med. Chem 2003, 38, 223. [DOI] [PubMed] [Google Scholar]
- Ottaviani G.; Martel S.; Carrupt P. A. J. Med. Chem. 2006, 49, 3948. [DOI] [PubMed] [Google Scholar]
- Wohnsland F.; Faller B. J. Med. Chem. 2001, 44, 923. [DOI] [PubMed] [Google Scholar]
- Ruell J. A.; Tsinman K. L.; Avdeef A. Eur. J. Pharm. Sci. 2003, 20, 393. [DOI] [PubMed] [Google Scholar]
- Faller B.; Grimm H. P.; Loeuillet-Ritzler F.; Arnold S.; Briand X. J. Med. Chem. 2005, 48, 2571. [DOI] [PubMed] [Google Scholar]
- Verma R. P.; Hansch C.; Selassie C. D. J. Comput. Aid. Mol. Des. 2007, V21, 3. [DOI] [PubMed] [Google Scholar]
- Peake C.; Box K.; Mole J.; Huque F.; Platts J.; Allen R.; Cimpan G.; Comer J. In QSAR and Molecular Modelling in Rational Design of Bioactive Molecules; Aki E., Yalcin I., Eds.; Turkey CADDD Society: Ankara, Turkey, 2006; p 261. [Google Scholar]
- Avdeef A. Curr. Top. Med. Chem. 2001, 1, 277. [DOI] [PubMed] [Google Scholar]
- Avdeef A.; Strafford M.; Block E.; Balogh M. P.; Chambliss W.; Khan I. Eur. J. Pharm. Sci. 2001, 14, 271. [DOI] [PubMed] [Google Scholar]
- Blok M. C.; van Dam K. BBA−Biomembranes 1978, 507, 48. [DOI] [PubMed] [Google Scholar]
- Blok M. C.; van Dam K. BBA−Biomembranes 1979, 550, 527. [DOI] [PubMed] [Google Scholar]
- Flanagan M. T. Thin Solid Films 1983, 99, 133. [Google Scholar]
- Kobatake Y.; Irimajiri A.; Matsumoto N. Biophys. J. 1970, 10, 728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M.; Kobatake Y.; Hashimoto M.; Morita S. J. Membr. Biol. 1971, 5, 185. [DOI] [PubMed] [Google Scholar]
- Yoshida M.; Kamo N.; Kobatake Y. J. Membr. Biol. 1972, 8, 389. [Google Scholar]
- Flaten G. E.; Dhanikula A. B.; Luthman K.; Brandl M. Eur. J. Pharm. Sci. 2006, 27, 80. [DOI] [PubMed] [Google Scholar]
- Flaten G. E.; Bunjes H.; Luthman K.; Brandl M. Eur. J. Pharm. Sci. 2006, 28, 336. [DOI] [PubMed] [Google Scholar]
- Flaten G. E.; Skar M.; Luthman K.; Brandl M. Eur. J. Pharm. Sci. 2007, 30, 324. [DOI] [PubMed] [Google Scholar]
- Jakobsson E.; Subramaniam S.; Scott H. L. In Biological Membranes: A Molecular Perspective from Computation and Experiment; Merz K. M. Jr., Roux B., Eds.; Birkhauser: Boston, MA, 1996; Chapter 4, p 105. [Google Scholar]
- Brockman H. L.; Momsen M. M.; Knudtson J. R.; Miller S. T.; Graff G.; Yanni J. M. Ocul. Immunol. Inflamm. 2003, 11, 247. [DOI] [PubMed] [Google Scholar]
- Tanford C.Ben Franklin Stilled the Waves: An Informal History of Pouring Oil on Water with Reflections on the Ups and Downs of Scientific Life in General; Duke University Press: Durham, NC, 1989. [Google Scholar]
- Kaznessis Y. N.; Kim S.; Larson R. G. Biophys. J. 2002, 82, 1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordi F.; Cametti C.; Motta A.; Diociaiuti M.; Molinari A. Bioelectrochem. Bioenerg. 1999, 49, 51. [DOI] [PubMed] [Google Scholar]
- Borissevitch G. P.; Tabak M.; Oliveira O. N. BBA−Biomembranes 1996, 1278, 12. [DOI] [PubMed] [Google Scholar]
- Borissevitch G. P.; Tabak M.; Borissevitch I. E.; Oliveira O. N. Jr Colloid. Surf. B 1996, 7, 69. [Google Scholar]
- Weis R. M. Chem. Phys. Lipids 1991, 57, 227. [DOI] [PubMed] [Google Scholar]
- Yu H.; Hui S. W. Chem. Phys. Lipids 1992, 62, 69. [DOI] [PubMed] [Google Scholar]
- Evert L. L.; Leckband D.; Israelachvili J. N. Langmuir 1994, 10, 303. [Google Scholar]
- Teissie J.; Tocanne J. F.; Baudras A. Eur. J. Biochem. 1978, 83, 77. [DOI] [PubMed] [Google Scholar]
- Bayerl T. M.; Thomas R. K.; Penfold J.; Rennie A.; Sackmann E. Biophys. J. 1990, 57, 1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flach C. R.; Brauner J. W.; Mendelsohn R. Biophys. J. 1993, 65, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoma M.; Schwendler M.; Baltes H.; Pfohl T.; Riegler H.; Moehwald H. Langmuir 1996, 12, 1722. [Google Scholar]
- Brezesinski G.; Thoma M.; Struth B.; Moehwald H. J. Phys. Chem. 1996, 100, 3126. [Google Scholar]
- Rey Gomez-Serranillos I.; Minones J.; Dynarowicz-Latka P.; Iribarnegaray E.; Casas M. Phys. Chem. Chem. Phys. 2004, 6, 1580. [Google Scholar]
- Kimizuka H.; Koketsu K. Nature 1962, 196, 995. [DOI] [PubMed] [Google Scholar]
- Read B. D.; Demel R. A.; Wiegandt H.; van Deenen L. L. M. Biochim. Biophys. Acta 1977, 470, 325. [DOI] [PubMed] [Google Scholar]
- Le Compte M. F.; Rubinstein I.; Miller I. R. J. Colloid Interface Sci. 1983, 91, 12. [Google Scholar]
- Mingins J.; Pethica B. A. Langmuir 2004, 20, 7493. [DOI] [PubMed] [Google Scholar]
- Pickard W. F.; Sehgal K. C.; Jackson C. M. Biochim. Biophys. Acta 1979, 552, 1. [DOI] [PubMed] [Google Scholar]
- Mingins J.; Stigter D.; Dill K. A. Biophys. J. 1992, 61, 1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoma M.; Moehwald H. J. Colloid Interface Sci. 1994, 162, 340. [Google Scholar]
- Harvey E. N. Am. J. Physiol. 1913, 31, 335. [Google Scholar]
- Pidgeon C.; Venkataram U. V. Anal. Biochem. 1989, 176, 36. [DOI] [PubMed] [Google Scholar]
- Pidgeon C.; Ong S. W.; Liu H. L.; Qiu X. X.; Pidgeon M.; Dantzig A. H.; Munroe J.; Hornback W. J.; Kasher J. S.; Glunz L.; Szczerba T. J. Med. Chem. 1995, 38, 590. [DOI] [PubMed] [Google Scholar]
- Ong S.; Liu H.; Qiu X.; Bhat G.; Pidgeon C. Anal. Chem. 1995, 67, 755. [DOI] [PubMed] [Google Scholar]
- Barbato F.; Larotonda M. I.; Quaglia F. Eur. J. Med. Chem. 1996, 31, 311. [Google Scholar]
- Barbato F.; La Rotonda M. I.; Quaglia F. J. Pharm. Sci. 1997, 86, 225. [DOI] [PubMed] [Google Scholar]
- Taillardat-Bertschinger A.; Carrupt P.; Barbato F.; Testa B. J. Med. Chem. 2003, 46, 655. [DOI] [PubMed] [Google Scholar]
- Yang C. Y.; Cai S. J.; Liu H. L.; Pidgeon C. Adv. Drug Delivery Rev. 1996, 23, 229. [Google Scholar]
- Alifrangis L. H.; Christensen I. T.; Berglund A.; Sandberg M.; Hovgaard L.; Frokjaer S. J. Med. Chem. 2000, 43, 103. [DOI] [PubMed] [Google Scholar]
- Taillardat-Bertschinger A.; Martinet C. A. M.; Carrupt P. A.; Reist M.; Caron G.; Fruttero R.; Testa B. Pharm. Res. 2002, 19, 729. [DOI] [PubMed] [Google Scholar]
- Ong S.; Pidgeon C. Anal. Chem. 1995, 67, 2119. [DOI] [PubMed] [Google Scholar]
- Escher B. I.; Schwarzenbach R. P.; Westall J. C. Environ. Sci. Technol. 2000, 34, 3962. [Google Scholar]
- Sheng Q.; Schulten K.; Pidgeon C. J. Phys. Chem. 1995, 99, 11018. [Google Scholar]
- Ong S.; Qiu X.; Pidgeon C. J. Phys. Chem. 1994, 98, 10189. [Google Scholar]
- Ong S.; Liu H.; Pidgeon C. J. Chromatogr. A 1996, 728, 113. [DOI] [PubMed] [Google Scholar]
- Ong S.; Cai S. J.; Bernal C.; Rhee D.; Qiu X.; Pidgeon C. Anal. Chem. 1994, 66, 782. [DOI] [PubMed] [Google Scholar]
- Brechling A.; Sundermann M.; Kleineberg U.; Heinzmann U. Thin Solid Films 2003, 433, 281. [Google Scholar]
- Smith D. P. E.; Bryant A.; Quate C. F.; Rabe J. P.; Gerber Ch.; Swalen J. D. Proc. Natl. Acad. Sci. U.S.A. 1987, 84, 969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuse C. W.; Krüger S.; Majkrzak C. F.; Dura J. A.; Fu J.; Connor J. T.; Plant A. L. Biophys. J. 1998, 74, 1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winger T. M.; Chaikof E. L. Langmuir 1998, 14, 4148. [Google Scholar]
- Siekmann B.; Westesen K. Pharm. Pharmacol. Lett. 1992, 1, 123. [Google Scholar]
- Müller R. H.; Schwarz C.; Mehnert W.; Lucks J. S. In Controlled Release of Bioactive Materials; Roseman T. J., Peppas N. A., Gabelnick H. L., Eds.; Controlled Release Society: Deerfield, IL, 1993; p 480. [Google Scholar]
- Schwarz C.; Mehnert W.; Lucks J. S.; Müller R. H. J. Controlled Release 1994, 30, 83. [Google Scholar]
- zur Muehlen A.; Schwarz C.; Mehnert W. Eur. J. Pharm. Biopharm. 1998, 45, 149. [DOI] [PubMed] [Google Scholar]
- Ahlin P.; Kristl J.; Sentjurc M. Farm. Vestnik 1999, 50, 323. [Google Scholar]
- Mehnert W.; Mader K. Adv. Drug Delivery Rev. 2001, 47, 165. [DOI] [PubMed] [Google Scholar]
- Maia C. S.; Mehnert W.; Schaller M.; Korting H. C.; Gysler A.; Haberland A.; Schafer-Korting M. J. Drug Target. 2002, 10, 489. [DOI] [PubMed] [Google Scholar]
- Jores K.; Haberland A.; Wartewig S.; Maeder K.; Mehnert W. Pharm. Res. 2005, 22, 1887. [DOI] [PubMed] [Google Scholar]
- Lombardi Borgia S.; Regehly M.; Sivaramakrishnan R.; Mehnert W.; Korting H. C.; Danker K.; Roeder B.; Kramer K. D.; Schaefer-Korting M. J. Controlled Release 2005, 110, 151. [DOI] [PubMed] [Google Scholar]
- Lukacova V.; Peng M.; Fanucci G.; Tandlich R.; Hinderliter A.; Maity B.; Mannivanan E.; Cook G. R.; Balaz S. J. Biomol. Screen. 2007, 12, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer H. Arch. Exp. Pathol. Pharmacol. 1899, 42, 109. [Google Scholar]
- Overton E.Studien über die Narkose zugleich ein Betrag zur allgemeinen Pharmakologie; G. Fischer: Jena, Germany,1901. [Google Scholar]
- Bodansky M.; Meigs A. V. J. Phys. Chem. 1932, 36, 814. [Google Scholar]
- Tabern D. L.; Shelberg E. F. J. Am. Chem. Soc. 1933, 55, 328. [Google Scholar]
- Macy R. J. Ind. Hygiene Toxicol. 1948, 30, 140. [PubMed] [Google Scholar]
- Davies J. T. J. Phys. Colloid Chem. 1950, 54, 185. [Google Scholar]
- Diamond J. M.; Katz Y. J. Membr. Biol. 1974, 17, 121. [DOI] [PubMed] [Google Scholar]
- Davis S. S.; Higuchi T.; Rytting J. H. In Advances in Pharmaceutical Sciences; Bean H. S., Beckett A. H., Carless J. E., Eds.; Academic Press: London, U.K., 1974; Vol. 4, p 73. [Google Scholar]
- Rekker R. F.The Hydrophobic Fragmental Constant; Elsevier: Amsterdam, The Netherlands, 1977. [Google Scholar]
- Dix J. A.; Kivelson D.; Diamond J. M. J. Membr. Biol. 1978, 40, 315. [DOI] [PubMed] [Google Scholar]
- Leahy D. E.; Taylor P. J.; Wait A. R. Quant. Struct. Act. Relat. 1989, 8, 17. [Google Scholar]
- De Young L. R.; Dill K. A. J. Phys. Chem. 1990, 94, 801. [Google Scholar]
- Xiang T. X.; Anderson B. D. J. Membr. Biol. 1995, 148, 157. [DOI] [PubMed] [Google Scholar]
- Xiang T. X.; Anderson B. D. Biophys. J. 1997, 72, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang T.; Xu Y.; Anderson B. D. J. Membr. Biol. 1998, 165, 77. [DOI] [PubMed] [Google Scholar]
- Mayer P. T.; Anderson B. D. J. Pharm. Sci. 2002, 91, 640. [DOI] [PubMed] [Google Scholar]
- Suomalainen P.; Johans C.; Soederlund T.; Kinnunen P. K. J. J. Med. Chem. 2004, 47, 1783. [DOI] [PubMed] [Google Scholar]
- Leo A.; Hansch C.; Jow P. Y. J. Med. Chem. 1976, 19, 611. [DOI] [PubMed] [Google Scholar]
- Testa B.; Seiler P. Drug Res. 1981, 31, 1053. [PubMed] [Google Scholar]
- Funasaki N.; Hada S.; Neya S.; Machida K. J. Colloid Interface Sci. 1985, 106, 255. [Google Scholar]
- Pohorille A.; Wilson M. A. J. Chem. Phys. 1996, 104, 3760. [DOI] [PubMed] [Google Scholar]
- Bassolino-Klimas D.; Alper H. E.; Stouch T. R. J. Am. Chem. Soc. 1995, 117, 4118. [Google Scholar]
- Repakova J.; Capkova P.; Vattulainen I.; Holopainen J. M. J. Phys. Chem. B 2004, 108, 13438. [Google Scholar]
- Hoff B.; Strandberg E.; Ulrich A. S.; Tieleman D. P.; Posten C. Biophys. J. 2005, 88, 1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collander R. Acta Physiol. Scand. 1947, 13, 363. [PubMed] [Google Scholar]
- Leo A.; Hansch C.. Substituent Constants for Correlation Analysis in Chemistry and Biology; Wiley: New York, 1979. [DOI] [PubMed] [Google Scholar]
- Giesen D. J.; Hawkins G. D.; Liotard D. A.; Cramer C. J.; Truhlar D. G. Theor. Chem. Acc. 1997, 98, 85. [Google Scholar]
- White S. H.; Ladokhin A. S.; Jayasinghe S.; Hristova K. J. Biol. Chem. 2001, 276, 32395. [DOI] [PubMed] [Google Scholar]
- Collander R. Trans. Faraday Soc. 1937, 33, 985. [Google Scholar]
- Collander R. Acta Chem. Scand. 1949, 3, 717. [Google Scholar]
- Collander R. Acta Chem. Scand. 1950, 4, 1085. [Google Scholar]
- Leo A.; Hansch C.; Elkins D. Chem. Rev. 1971, 71, 525. [Google Scholar]
- Margolis S. A.; Levenson M.; Fresen J. Anal. Chem 2000, 367, 1. [DOI] [PubMed] [Google Scholar]
- Wimley W. C.; White S. H. Nat. Struct. Biol. 1996, 3, 842. [DOI] [PubMed] [Google Scholar]
- Franks N. P.; Abraham M. H.; Lieb W. R. J. Pharm. Sci. 1993, 82, 466. [DOI] [PubMed] [Google Scholar]
- Sassi P.; Paolantoni M.; Cataliotti R. S.; Palombo F.; Morresi A. J. Phys. Chem. B 2004, 108, 19557. [DOI] [PubMed] [Google Scholar]
- DeBolt S. E.; Kollman P. A. J. Am. Chem. Soc. 1995, 117, 5316. [Google Scholar]
- MacCallum J. L.; Tieleman D. P. J. Am. Chem. Soc. 2002, 124, 15085. [DOI] [PubMed] [Google Scholar]
- Dallas A. J.; Carr P. W. J. Chem. Soc. Perkin Trans. 2 1992, 2155. [Google Scholar]
- Dulfer W. J.; Govers H. A. J. Environ. Sci. Technol. 1995, 29, 2548. [DOI] [PubMed] [Google Scholar]
- Carrozzino J. M.; Khaledi M. G. Pharm. Res. 2004, 21, 2327. [DOI] [PubMed] [Google Scholar]
- White S. H. Science 1980, 207, 1075. [DOI] [PubMed] [Google Scholar]
- Gobas F. A. P. C.; Lahittete J. M.; Garofalo G.; Shiu W. Y.; Mackay D. J. Pharm. Sci. 1988, 77, 265. [DOI] [PubMed] [Google Scholar]
- Osterberg T.; Svensson M.; Lundahl P. Eur. J. Pharm. Sci 2001, 12, 427. [DOI] [PubMed] [Google Scholar]
- Fruttero R.; Caron G.; Fornatto E.; Boschi D.; Ermondi G.; Gasco A.; Carrupt P. A.; Testa B. Pharm. Res. 1998, 15, 1407. [DOI] [PubMed] [Google Scholar]
- Vaes W. H. J.; Ramos E. U.; Hamwijk C.; van Holsteijn I.; Blaauboer B. J.; Seinen W.; Verhaar H. J. M.; Hermens J. L. M. Chem. Res. Toxicol. 1997, 10, 1067. [DOI] [PubMed] [Google Scholar]
- Patel H.; Schultz T. W.; Cronin M. T. D. J. Mol. Struct. THEOCHEM 2002, 593, 9. [Google Scholar]
- Herbette L. G.; Chester D. W.; Rhodes D. G. Biophys. J. 1986, 49, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes D. H.; Simkowitz P. J. Membr. Biol. 1977, 33, 63. [DOI] [PubMed] [Google Scholar]
- Katagi T. J. Pest. Sci. 2001, 26, 354. [Google Scholar]
- Schmitt W. Toxicol. in Vitro 2008, 22, 457. [DOI] [PubMed] [Google Scholar]
- ClogP Software, Biobyte Corp.: Claremont, CA, 1999. (www.biobyte.com)
- Xiang T. X.; Anderson B. D. J. Pharm. Sci. 1994, 83, 1511. [DOI] [PubMed] [Google Scholar]
- Mayer P. T.; Xiang T. X.; Anderson B. D.. Pharm. Sci. 2000, 2, Article 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M.; Fukuda H.; Nagai T. Chem. Pharm. Bull. 1978, 26, 9. [Google Scholar]
- el-Tayar N.; Tsai R. S.; Testa B.; Carrupt P. A.; Leo A. J. Pharm. Sci. 1991, 80, 590. [DOI] [PubMed] [Google Scholar]
- Burton P. S.; Conradi R. A.; Hilgers A. R.; Ho N. F. H.; Maggiora L. L. J. Controlled Release 1992, 19, 87. [Google Scholar]
- Chikhale E. G.; Ng K. Y.; Burton P. S.; Borchardt R. T. Pharm. Res. 1994, 11, 412. [DOI] [PubMed] [Google Scholar]
- Paterson D. A.; Conradi R. A.; Hilgers A. R.; Vidmar T. J.; Burton P. S. Quant. Struct. Act. Relat. 1994, 13, 4. [Google Scholar]
- Lukacova V.; Peng M.; Tandlich R.; Hinderliter A.; Balaz S. Langmuir 2006, 22, 1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu S. W.; Clark M.; Balaji V.; Subramaniam S.; Scott H. L.; Jakobsson E. Biophys. J. 1995, 69, 1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler P. Eur. J. Med. Chem. 1974, 9, 473. [Google Scholar]
- Caron G.; Ermondi G. J. Med. Chem. 2005, 48, 3269. [DOI] [PubMed] [Google Scholar]
- Young R. C.; Mitchell R. C.; Brown T. H.; Ganellin C. R.; Griffiths R.; Jones M.; Rana K. K.; Saundners D. D.; Smith I. R.; Sore N. E.; Wilks T. J. J. Med. Chem. 1988, 31, 656. [DOI] [PubMed] [Google Scholar]
- Origin, Version 7; OriginLab Software: Northampton, MA, 2002. [Google Scholar]
- Pratt L. R.; Pohorille A. Chem. Rev. 2002, 102, 2671. [DOI] [PubMed] [Google Scholar]
- Poulin P.; Theil F. P. J. Pharm. Sci. 2002, 91, 129. [DOI] [PubMed] [Google Scholar]
- Stehle R. G.; Higuchi W. I. J. Pharm. Sci. 1972, 61, 1931. [DOI] [PubMed] [Google Scholar]
- Patton J. S.; Stone B.; Papa C.; Abramowitz R.; Yalkowsky S. H. J. Lipid Res. 1984, 25, 189. [PubMed] [Google Scholar]
- Chiou C. T.; Manes M. J. Chem. Soc. Faraday Trans. 1 1986, 82, 243. [Google Scholar]
- Niimi A. J. Water Res. 1991, 25, 1515. [Google Scholar]
- Johansson J. S.; Zou H. Biophys. Chem. 1999, 79, 107. [DOI] [PubMed] [Google Scholar]
- Wolosin J. M.; Ginsburg H.; Lieb W. R.; Stein W. D. J. Gen. Physiol. 1978, 71, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyauchi S.; Ono A.; Yoshimoto M.; Kamo N. J. Pharm. Sci. 1993, 82, 27. [DOI] [PubMed] [Google Scholar]
- Xiang T. X.; Anderson B. D. J. Membr. Biol. 1994, 140, 111. [DOI] [PubMed] [Google Scholar]
- Cohen B. E.; Bangham A. D. Nature 1972, 236, 173. [DOI] [PubMed] [Google Scholar]
- Chowhan Z. U.; Higuchi W. I. J. Pharm. Sci. 1974, 63, 1428. [DOI] [PubMed] [Google Scholar]
- Blok M. C.; van Deenen L. L.; de Gier J. Biochim. Biophys. Acta 1976, 433, 1. [DOI] [PubMed] [Google Scholar]
- Verkman A. S. Biochim. Biophys. Acta 1980, 599, 370. [DOI] [PubMed] [Google Scholar]
- Ahmed M.; Burton J. S.; Hadgraft J.; Kellaway I. W. J. Membr. Biol. 1981, 58, 181. [DOI] [PubMed] [Google Scholar]
- Bochain A.; Estey L.; Haronian G.; Reale M.; Rojas C.; Cramer J. J. Membr. Biol. 1981, 60, 73. [DOI] [PubMed] [Google Scholar]
- Viscio D. B.; Prestegard J. H. Proc. Natl. Acad. Sci. U.S.A.. 1981, 78, 1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke T. G.; Tritton T. R. Biochemistry 1985, 24, 1768. [DOI] [PubMed] [Google Scholar]
- Clarke R. J.; Apell H. J. Biophys. Chem. 1989, 34, 225. [DOI] [PubMed] [Google Scholar]
- Subczynski W. K.; Hyde J. S.; Kusumi A. Proc. Natl. Acad. Sci. U.S.A. 1989, 86, 4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke R. J. Biophys. Chem. 1991, 39, 91. [DOI] [PubMed] [Google Scholar]
- Pezeshk A.; Pezeshk V.; Firlej A.; Wojas J.; Subczynski W. K. Life Sci. 1993, 52, 1071. [DOI] [PubMed] [Google Scholar]
- Clerc S. G.; Thompson T. E. Biophys. J. 1995, 68, 2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang T. X.; Anderson B. D. J. Pharm. Sci. 1995, 84, 1308. [DOI] [PubMed] [Google Scholar]
- Eytan G. D.; Regev R.; Oren G.; Assaraf Y. G. J. Biol. Chem. 1996, 271, 12897. [DOI] [PubMed] [Google Scholar]
- Kleinfeld A. M.; Chu P.; Romero C. Biochemistry 1997, 36, 14146. [DOI] [PubMed] [Google Scholar]
- Regev R.; Eytan G. D. Biochem. Pharmacol. 1997, 54, 1151. [DOI] [PubMed] [Google Scholar]
- Mayer P. T.; Xiang T. X.; Niemi R.; Anderson B. D. Biochemistry 2003, 42, 1624. [DOI] [PubMed] [Google Scholar]
- Kuzelova K.; Brault D. Biochemistry 1994, 33, 9447. [DOI] [PubMed] [Google Scholar]
- Kuzelova K.; Brault D. Biochemistry 1995, 34, 11245. [DOI] [PubMed] [Google Scholar]
- Bemporad D.; Luttmann C.; Essex J. W. Biophys. J. 2004, 87, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulaboski R.; Borges F.; Pereira C. M.; Cordeiro M. N.; Garrido J.; Silva A. F. Comb. Chem. High Throughput Screen. 2007, 10, 514. [DOI] [PubMed] [Google Scholar]
- Conboy J. C.; Messmer M. C.; Walker R. A.; Richmond G. L. Prog. Colloid. Polymer Sci. 1997, 103, 10. [Google Scholar]
- Smiley B. L.; Walker R. A.; Gragson D. E.; Hannon T. E.; Richmond G. L. Proc. SPIE 1998, 3273, 134. [Google Scholar]
- Richmond G. L. Annu. Rev. Phys. Chem. 2001, 52, 357. [DOI] [PubMed] [Google Scholar]
- Scatena L. F.; Brown M. G.; Richmond G. L. Science 2001, 292, 908. [DOI] [PubMed] [Google Scholar]
- Watry M. R.; Brown M. G.; Richmond G. L. Appl. Spectrosc. 2001, 55, 321A. [Google Scholar]
- Richmond G. L. Chem. Rev. 2002, 102, 2693. [DOI] [PubMed] [Google Scholar]
- Buch V. J. Phys. Chem. B 2005, 109, 17771. [DOI] [PubMed] [Google Scholar]
- Mitrinovic D. M.; Zhang Z.; Williams S. M.; Huang Z.; Schlossman M. L. J. Phys. Chem. B 1999, 103, 1779. [Google Scholar]
- Schlossman M. L.; Li M.; Mitrinovic D. M.; Tikhonov A. M. High Perform. Polym. 2000, 12, 551. [Google Scholar]
- Schlossman M. L. Curr. Opin. Colloid Interface Sci. 2002, 7, 235. [Google Scholar]
- Luo G.; Malkova S.; Pingali S. V.; Schultz D. G.; Lin B.; Meron M.; Graber T. J.; Gebhardt J.; Vanysek P.; Schlossman M. L. Electrochem. Commun. 2005, 7, 627. [DOI] [PubMed] [Google Scholar]
- Malkova S.; Long F.; Stahelin R. V.; Pingali S. V.; Murray D.; Cho W.; Schlossman M. L. Biophys. J. 2005, 89, 1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pingali S. V.; Takiue T.; Luo G.; Tikhonov A. M.; Ikeda N.; Aratono M.; Schlossman M. L. J. Phys. Chem. B 2005, 109, 1210. [DOI] [PubMed] [Google Scholar]
- Schlossman M. L. Physica B 2005, 357, 98. [Google Scholar]
- Benjamin I. Annu. Rev. Phys. Chem. 1997, 48, 407. [DOI] [PubMed] [Google Scholar]
- Benjamin I. In Molecular Dynamics Methods for Studying Liquid Interfacial Phenomena; Thompson D. L., Ed.; World Scientific Publishing Company: Singapore, 1998; p 101. [Google Scholar]
- Benjamin I. J. Chem. Phys. 2004, 121, 10223. [DOI] [PubMed] [Google Scholar]
- Benjamin I. J. Phys. Chem. B 2005, 109, 13711. [DOI] [PubMed] [Google Scholar]
- Mitrinovic D. M.; Tikhonov A. M.; Li M.; Huang Z.; Schlossman M. L. Phys. Rev. Lett. 2000, 85, 582. [DOI] [PubMed] [Google Scholar]
- Tikhonov A. M.; Mitrinovic D. M.; Li M.; Huang Z.; Schlossman M. L. J. Phys. Chem. B 2000, 104, 6336. [Google Scholar]
- Walker D. S.; Brown M. G.; McFearin C. L.; Richmond G. L. J. Phys. Chem. B 2004, 108, 2111. [Google Scholar]
- Tikhonov A. M.; Schlossman M. L. J. Phys. Chem. B 2003, 107, 3344. [Google Scholar]
- Tikhonov A. M.; Pingali S. V.; Schlossman M. L. J. Chem. Phys. 2004, 120, 11822. [DOI] [PubMed] [Google Scholar]
- Gragson D. E.; Richmond G. L. J. Phys. Chem. B 1998, 102, 569. [Google Scholar]
- Walker R. A.; Gragson D. E.; Richmond G. L. Colloid. Surf. A 1999, 154, 175. [Google Scholar]
- Pohorille A.; Wilson M. A.; Chipot C. Prog. Coll. Polymer Sci. 1997, 103, 29. [Google Scholar]
- Pohorille A.; Cieplak P.; Wilson M. A. Chem. Phys. 1996, 204, 337. [DOI] [PubMed] [Google Scholar]
- de Bruijn J.; Busser F.; Seinen W.; Hermens J. Environ. Toxicol. Chem. 1989, 8, 499. [Google Scholar]
- Byron P. R.; Notari R. E.; Tomlinson E. J. Pharm. Sci. 1980, 69, 527. [DOI] [PubMed] [Google Scholar]
- Rosano H. L.; Schulman J. H.; Weisbuch J. B. Ann. New York Acad. Sci. 1961, 92, 457. [DOI] [PubMed] [Google Scholar]
- Lippold B. C.; Schneider G. F. Drug Res. 1975, 25, 843. [PubMed] [Google Scholar]
- Dearden J. C.; Patel K. D. J. Pharm. Pharmacol. 1978, 30, 51P. [DOI] [PubMed] [Google Scholar]
- Miller D. M. BBA−Biomembranes 1986, 856, 27. [Google Scholar]
- Reymond F.; Steyaert G.; Carrupt P. A.; Testa B.; Girault H. H. Helv. Chim. Acta 1996, 79, 101. [Google Scholar]
- Reymond F.; Brevet P. F.; Carrupt P. A.; Girault H. J. Electroanal. Chem. 1997, 424, 121. [Google Scholar]
- Reymond F.; Carrupt P. A.; Girault H. H. J. Electroanal. Chem. 1998, 449, 49. [Google Scholar]
- Reymond F.; Lagger G.; Carrupt P. A.; Girault H. H. J. Electroanal. Chem. 1998, 451, 59. [Google Scholar]
- Reymond F.; Chopineaux-Court Steyaert G.; Bouchard G.; Carrupt P. A.; Testa B.; Girault H. H. J. Electroanal. Chem. 1999, 462, 235. [Google Scholar]
- Maelkiae A.; Liljeroth P.; Kontturi A. K.; Kontturi K. J. Phys. Chem. B 2001, 105, 10884. [Google Scholar]
- Gobry V.; Ulmeanu S.; Reymond F.; Bouchard G.; Carrupt P. A.; Testa B.; Girault H. H. J. Am. Chem. Soc. 2001, 123, 10684. [DOI] [PubMed] [Google Scholar]
- Malkia A.; Liljeroth P.; Kontturi K. Electrochem. Commun. 2003, 5, 473. [Google Scholar]
- Kubinyi H. J. Pharm. Sci. 1978, 67, 262. [DOI] [PubMed] [Google Scholar]
- Flynn G. L.; Yalkowsky S. H.; Roseman T. J. J. Pharm. Sci. 1974, 63, 479. [DOI] [PubMed] [Google Scholar]
- Leahy D. E.; de Meere A. L. J.; Wait A. R.; Taylor P. J.; Tomenson J. A.; Tomlinson E. Int. J. Pharm. 1989, 50, 117. [Google Scholar]
- Albery W. J.; Burke J. F.; Leffler E. B.; Hadgraft J. J. Chem. Soc. Faraday Trans. 1976, 72, 1618. [Google Scholar]
- de Haan F. H.; Jansen A. C. Pharm. Weekbl. 1983, 5, 222. [DOI] [PubMed] [Google Scholar]
- Fleming R.; Guy R. H.; Hadgraft J. J. Pharm. Sci. 1983, 72, 142. [DOI] [PubMed] [Google Scholar]
- Leahy D. E.; Wait A. R. J. Pharm. Sci. 1986, 75, 1157. [DOI] [PubMed] [Google Scholar]
- Byron P. R.; Rathbone M. J. Int. J. Pharm. 1986, 29, 103. [Google Scholar]
- Horspool K. R.; Lipinski C. A. Drug Delivery Technol. 2003, 3, 34. [Google Scholar]
- Sanders C. R.; Schwonek J. P. Biophys. J. 1993, 65, 1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasseur R.; Laurent G.; Ruysschaert J. M.; Tulkens P. Biochem. Pharmacol. 1984, 33, 629. [DOI] [PubMed] [Google Scholar]
- Brasseur R.; Chatelain P.; Goormaghtigh E.; Ruysschaert J. M. BBA−Biomembranes 1985, 814, 227. [Google Scholar]
- Brasseur R.; Goormaghtigh E.; Ruysschaert J. M.; Duquenoy P. H.; Marichal P.; van den Bossche H. J. Pharm. Pharmacol. 1991, 43, 167. [DOI] [PubMed] [Google Scholar]
- Simon S. A.; McDaniel R. V.; McIntosh T. J. J. Phys. Chem. 1982, 86, 1449. [Google Scholar]
- Gusev D. G.; Vasilenko I. A.; Evstigneeva R. P. Biol. Membr. 1986, 3, 685. [Google Scholar]
- Minobe M.; Sakurai I.; Shibata T.; Kawamura Y. Mol. Cryst. Liq. Cryst. A 1998, 319, 75. [Google Scholar]
- Okamura E.; Nakahara M. J. Phys. Chem. B 1999, 103, 3505. [Google Scholar]
- Katsaras J.; Stinson R. H.; Davis J. H.; Kendall E. J. Biophys. J. 1991, 59, 645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita T.; Iwasa J.; Hansch C. J. Am. Chem. Soc. 1964, 86, 5175. [Google Scholar]
- Ghose A. K.; Crippen G. M. J. Comput. Chem. 1986, 7, 565. [Google Scholar]
- Suzuki T.; Kudo Y. J. Comput. Aid. Mol. Des. 1990, 4, 155. [DOI] [PubMed] [Google Scholar]
- Mannhold R.; van de Waterbeemd H. J. Comput. Aid. Mol. Des. 2001, 15, 337. [DOI] [PubMed] [Google Scholar]
- Eisenberg D.; Weiss R. M.; Wilcox W.; Terwilliger T. C. Faraday Symp. Chem. Soc. 1982, 17, 109. [Google Scholar]
- Fischer H.; Kansy M.; Bur D. Chimia 2000, 54, 640. [Google Scholar]
- Cruciani G.; Crivori P.; Carrupt P. A.; Testa B. J. Mol. Struct. THEOCHEM 2000, 503, 17. [Google Scholar]
- Gerebtzoff G.; Seelig A. J. Chem. Inf. Model. 2006, 46, 2638. [DOI] [PubMed] [Google Scholar]
- Wasan K. M.; Brocks D. R.; Lee S. D.; Sachs-Barrable K.; Thornton S. J. Nat. Rev. Drug Discovery 2008, 7, 84. [DOI] [PubMed] [Google Scholar]
- Gershkovich P.; Hoffman A. Eur. J. Pharm. Sci 2005, 26, 394. [DOI] [PubMed] [Google Scholar]
- Holm R.; Hoest J. Int. J. Pharm. 2004, 272, 189. [DOI] [PubMed] [Google Scholar]
- Unruh T.; Bunjes H.; Westesen K.; Koch M. H. J. J. Phys. Chem. B 1999, 103, 10373. [Google Scholar]
- Unruh T.; Westesen K.; Bosecke P.; Lindner P.; Koch M. H. J. Langmuir 2002, 18, 1796. [Google Scholar]
- Heinrich J.; Addona G. H.; Miller K. W. Biochim. Biophys. Acta 1997, 1346, 158. [DOI] [PubMed] [Google Scholar]
- Sum A. K.; Biddy M. J.; de Pablo J. J.; Tupy M. J. J. Phys. Chem. B 2003, 107, 14443. [Google Scholar]
- Rane S. S.; Anderson B. D. Adv. Drug Delivery Rev. 2008, 60, 638. [DOI] [PubMed] [Google Scholar]
- Poulin P.; Theil F. P. J. Pharm. Sci. 2000, 89, 16. [DOI] [PubMed] [Google Scholar]
- Chiou C. T. Environ. Sci. Technol. 1985, 19, 31. [DOI] [PubMed] [Google Scholar]
- Fettiplace R.; Haydon D. A. Physiol. Rev. 1980, 60, 510. [DOI] [PubMed] [Google Scholar]
- Lande M. B.; Donovan J. M.; Zeidel M. L. J. Gen. Physiol. 1995, 106, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill W. G.; Rivers R. L.; Zeidel M. L. J. Gen. Physiol. 1999, 114, 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krylov A. V.; Pohl P.; Zeidel M. L.; Hill W. G. J. Gen. Physiol. 2001, 118, 333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G. J.; Liu H. W.; Yang L.; Zhong Y. G.; Zheng Y. Z. J. Membr. Biol. 2000, 175, 53. [DOI] [PubMed] [Google Scholar]
- Dix J. A.; Diamond J. M.; Kivelson D. Proc. Natl. Acad. Sci. U.S.A. 1974, 71, 474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papahadjopoulos D.; Jacobson K.; Nir S.; Isac T. Biochim. Biophys. Acta 1973, 311, 330. [DOI] [PubMed] [Google Scholar]
- Herbette L.; Katz A. M.; Sturtevant J. M. Mol. Pharmacol. 1983, 24, 259. [PubMed] [Google Scholar]
- Mason R. P.; Gonye G. E.; Chester D. W.; Herbette L. G. Biophys. J. 1989, 55, 769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janse M.; Blume A. Biophys. J. 1995, 68, 997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagle J. F.; Scott H. L. Jr BBA−Biomembranes 1978, 513, 236. [DOI] [PubMed] [Google Scholar]
- Cruzeiro H. L.; Mouritsen O. G. Biochim. Biophys. Acta 1988, 944, 63. [DOI] [PubMed] [Google Scholar]
- Bodansky M. J. Biol. Chem. 1928, 79, 241. [Google Scholar]
- Collander R. Physiol. Plant. 1954, 7, 420. [Google Scholar]
- Finkelstein A.; Cass A. J. Gen. Physiol. 1968, 52, 145. [PMC free article] [PubMed] [Google Scholar]
- Wolosin J. M.; Ginsburg H. BBA−Biomembranes 1975, 389, 20. [DOI] [PubMed] [Google Scholar]
- Orbach E.; Finkelstein A. J. Gen. Physiol. 1980, 75, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paula S.; Volkov A. G.; Van Hoek A. N.; Haines T. H.; Deamer D. W. Biophys. J. 1996, 70, 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paula S.; Volkov A. G.; Deamer D. W. Biophys. J. 1998, 74, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subczynski W. K.; Hyde J. S.; Kusumi A. Biochemistry 1991, 30, 8578. [DOI] [PubMed] [Google Scholar]
- Subczynski W. K.; Lomnicka M.; Hyde J. S. Free Radical Res. 1996, 24, 343. [DOI] [PubMed] [Google Scholar]
- Walter A.; Gutknecht J. J. Membr. Biol. 1986, 90, 207. [DOI] [PubMed] [Google Scholar]
- Träuble H. J. Membr. Biol. 1971, 4, 193. [DOI] [PubMed] [Google Scholar]
- Potts R. O.; Francoeur M. L. Proc. Natl. Acad. Sci. U.S.A. 1990, 87, 3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carruthers A.; Melchior D. L. Biochemistry 1983, 22, 5797. [Google Scholar]
- Chen P. J.; Liu Y.; Chen S. H.; Weiss T. M.; Huang H. W.; Sinn H.; Alp E. E.; Alatas A.; Said A. Biophys. Chem. 2003, 105, 721. [DOI] [PubMed] [Google Scholar]
- Zhelev D. V.; Needham D. Biochim. Biophys. Acta 1993, 1147, 89. [DOI] [PubMed] [Google Scholar]
- Tepper H. L.; Voth G. A. J. Phys. Chem. B 2006, 110, 21327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ipsen J. H.; Jorgensen K.; Mouritsen O. G. Biophys. J. 1990, 58, 1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton R. T.; Kaler E. W. J. Membr. Sci. 1990, 54, 259. [Google Scholar]
- Garrick R. A.; Ryan U. S.; Bower V.; Cua W. O.; Chinard F. P. Biochim. Biophys. Acta 1993, 1148, 108. [DOI] [PubMed] [Google Scholar]
- Sears D. F.Biological Horizons in Surface Sciences; Academic Press: New York, 1977. [Google Scholar]
- Benjamin I. Chem. Rev. 1996, 96, 1449. [DOI] [PubMed] [Google Scholar]
- Kedem O.; Katchalsky A. Biochim. Biophys. Acta 1958, 27, 229. [DOI] [PubMed] [Google Scholar]
- Chowhan Z. U.; Yotsuyanagi T.; Higuchi W. I. Biochim. Biophys. Acta 1972, 266, 320. [DOI] [PubMed] [Google Scholar]
- Chowhan Z. U.; Yotsuyanagi T.; Higuchi W. I. J. Pharm. Sci. 1973, 62, 221. [DOI] [PubMed] [Google Scholar]
- Chowhan Z. U.; Higuchi W. I. J. Pharm. Sci. 1973, 62, 443. [DOI] [PubMed] [Google Scholar]
- Balaz S.; Kuchar A.; Drevojanek J.; Adamcova J.; Vrbanova A. J. Biochem. Biophys. Method. 1988, 16, 75. [DOI] [PubMed] [Google Scholar]
- Kubinyi H. Drug Res. 1976, 26, 1991. [Google Scholar]
- Kubinyi H. Drug Res. 1979, 29, 1067. [PubMed] [Google Scholar]
- Cabrini G.; Verkman A. S. Biochim. Biophys. Acta 1986, 862, 285. [DOI] [PubMed] [Google Scholar]
- Marx U.; Lassmann G.; Wimalasena K.; Müller P.; Herrmann A. Biophys. J. 1997, 73, 1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Classen J.; Deuticke B.; Haest C. W. J. Membr. Biol. 1989, 111, 169. [DOI] [PubMed] [Google Scholar]
- Middelkoop E.; Lubin B. H.; Op den Kamp J. A.; Roelofsen B. Biochim. Biophys. Acta 1986, 855, 421. [DOI] [PubMed] [Google Scholar]
- Serra M. V.; Kamp D.; Haest C. W. Biochim. Biophys. Acta 1996, 1282, 263. [DOI] [PubMed] [Google Scholar]
- Matsuzaki K.; Murase O.; Fujii N.; Miyajima K. Biochemistry 1996, 35, 11361. [DOI] [PubMed] [Google Scholar]
- Haest C. W.; Oslender A.; Kamp D. Biochemistry 1997, 36, 10885. [DOI] [PubMed] [Google Scholar]
- Koefoed P.; Brahm J. BBA−Biomembranes 1994, 1195, 55. [DOI] [PubMed] [Google Scholar]
- Gawrisch K. In The Structure of Biological Membranes; Yeagle P. L., Ed.; CRC Press: Boca Raton, FL, 2005; p 147. [Google Scholar]
- Feller S. E.; Huster D.; Gawrisch K. J. Am. Chem. Soc. 1999, 121, 8963. [Google Scholar]
- Hirn R.; Benz R.; Bayerl T. M. Phys. Rev. E 1999, 59, 5987. [DOI] [PubMed] [Google Scholar]
- Grossfield A.; Woolf T. B. Langmuir 2002, 18, 198. [Google Scholar]
- Wimley W. C.; Thompson T. E. Biochemistry 1991, 30, 1702. [DOI] [PubMed] [Google Scholar]
- Raphael R. M.; Waugh R. E. Biophys. J. 1996, 71, 1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J.; Pagano R. E. Biochemistry 1997, 36, 8840. [DOI] [PubMed] [Google Scholar]
- Gumbart J.; Wang Y.; Aksimentiev A.; Tajkhorshid E.; Schulten K. Curr. Opin. Struct. Biol. 2005, 15, 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl E.; Edholm O. J. Chem. Phys. 2001, 115, 4938. [Google Scholar]
- Feller S. E.; Pastor R. W. Pac. Symp. Biocomput. 1997, 142. [PubMed] [Google Scholar]
- Ayton G.; Bardenhagen S. G.; McMurtry P.; Sulsky D.; Voth G. A. J. Chem. Phys. 2001, 114, 6913. [Google Scholar]
- Ayton G.; Voth G. A. Biophys. J. 2002, 83, 3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayton G.; Smondyrev A. M.; Bardenhagen S. G.; McMurtry P.; Voth G. A. Biophys. J. 2002, 83, 1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izvekov S.; Parrinello M.; Bumham C. J.; Voth G. A. J. Chem. Phys. 2004, 120, 10896. [DOI] [PubMed] [Google Scholar]
- Izvekov S.; Voth G. A. J. Phys. Chem. B 2005, 109, 2469. [DOI] [PubMed] [Google Scholar]
- Shi Q.; Voth G. A. Biophys. J. 2005, 89, 2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang R.; Ayton G. S.; Voth G. A. J. Chem. Phys 2005, 122, 244716/1. [DOI] [PubMed] [Google Scholar]
- Ayton G. S.; Noid W. G.; Voth G. A. Curr. Opin. Struct. Biol. 2007, 17, 192. [DOI] [PubMed] [Google Scholar]
- Ayton G. S.; Voth G. A. J. Struct. Biol. 2007, 157, 570. [DOI] [PubMed] [Google Scholar]
- Chu J. W.; Ayton G. S.; Izvekov S.; Voth G. A. Mol. Phys. 2007, 105, 167. [Google Scholar]
- Noid W. G.; Chu J. W.; Ayton G. S.; Voth G. A. J. Phys. Chem. B 2007, 111, 4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittington S. G.; Chapman D. Trans. Faraday Soc. 1966, 62, 3319. [Google Scholar]
- Nagle J. F. J. Chem. Phys. 1973, 58, 252. [Google Scholar]
- Caille A.; Pink D.; De Verteuil F.; Zuckermann M. J. Can. J. Phys. 1980, 58, 581. [Google Scholar]
- Mouritsen O. G.; Boothroyd A.; Harris R.; Jan N.; Lookman T.; MacDonald L.; Pink D. A.; Zuckermann M. J. J. Chem. Phys. 1983, 79, 2027. [Google Scholar]
- Eggl P.; Pink D.; Quinn B.; Ringsdorf H.; Sackmann E. Macromolecules 1990, 23, 3472. [Google Scholar]
- Mouritsen O. G. Curr. Opin. Colloid Interface Sci. 1998, 3, 78. [Google Scholar]
- Smit B.; Hilbers P. A. J.; Esselink K.; Rupert L. A. M.; van Os N. M.; Schlijper A. G. Nature 1990, 348, 624. [Google Scholar]
- Scott H. L. Comments Mol. Cell. Biophys. 1984, 2, 197. [Google Scholar]
- Scott H. L.; Pearce P. A. Biophys. J. 1989, 55, 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimburg T. Biophys. J. 2000, 78, 1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott H. L.; Cherng S. L. BBA−Biomembranes 1978, 510, 209. [DOI] [PubMed] [Google Scholar]
- Scott H. L.; Kalaskar S. Biochemistry 1989, 28, 3687. [DOI] [PubMed] [Google Scholar]
- Khelashvili G. A.; Pandit S. A.; Scott H. L. J. Chem. Phys. 2005, 123, 34910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott H. L. Biochemistry 1986, 25, 6122. [DOI] [PubMed] [Google Scholar]
- Shih A. Y.; Arkhipov A.; Freddolino P. L.; Schulten K. J. Phys. Chem. B 2006, 110, 3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit B.; Esselink K.; Hilbers P. A. J.; van Os N. M.; Rupert L. A. M.; Szleifer I. Langmuir 1993, 9, 9. [Google Scholar]
- Marrink S. J.; de Vries A. H.; Mark A. E. J. Phys. Chem. B 2004, 108, 750. [Google Scholar]
- Shelley J. C.; Shelley M. Y.; Reeder R. C.; Bandyopadhyay S.; Moore P. B.; Klein M. L. J. Phys. Chem. B 2001, 105, 9785. [Google Scholar]
- Shelley J. C.; Shelley M. Y.; Reeder R. C.; Bandyopadhyay S.; Klein M. L. J. Phys. Chem. B 2001, 105, 4464. [Google Scholar]
- Kranenburg M.; Vlaar M.; Smit B. .Biophys. J. 2004, 87, 1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leermakers F. A. M.; Scheutjens J. M. H. M. J. Chem. Phys. 1988, 89, 6912. [Google Scholar]
- Goetz R.; Gompper G.; Lipowsky R. Phys. Rev. Lett. 1999, 82, 221. [Google Scholar]
- Ayton G.; Smondyrev A. M.; Bardenhagen S. G.; McMurtry P.; Voth G. A. Biophys. J. 2002, 82, 1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illya G.; Lipowsky R.; Shillcock J. C. J. Chem. Phys. 2005, 122, 244901. [DOI] [PubMed] [Google Scholar]
- Dobereiner H. G.; Selchow O.; Lipowsky R. Eur. Biophys. J. 1999, 28, 174. [Google Scholar]
- Breidenich M.; Netz R. R.; Lipowsky R. Europhys. Lett. 2000, 49, 431. [Google Scholar]
- Linke G. T.; Lipowsky R.; Gruhn T. Phys. Rev. E 2005, 71, 051602/1. [DOI] [PubMed] [Google Scholar]
- Imparato A.; Shillcock J. C.; Lipowsky R. Europhys. Lett. 2005, 69, 650. [Google Scholar]
- Leermakers F. A. M.; Scheutjens J. M. H. M. J. Phys. Chem. 1989, 93, 7417. [Google Scholar]
- Ayton G. S.; Blood P. D.; Voth G. A. Biophys. J. 2007, 92, 3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S.; Maruyama Y.; Hyodo S. J. Chem. Phys. 2002, 116, 5842. [Google Scholar]
- Shillcock J. C.; Lipowsky R. Nat. Mater. 2005, 4, 225. [DOI] [PubMed] [Google Scholar]
- Lipowsky R. J. Biol. Phys. 2002, 28, 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipowsky R.; Dimova R. J. Phys. Condens. Mat. 2003, 15, S31. [Google Scholar]
- Izvekov S.; Voth G. A. J. Chem. Theory Comput. 2006, 2, 637. [DOI] [PubMed] [Google Scholar]
- Ayton G. S.; Voth G. A. Biophys. J. 2004, 87, 3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q.; Voth G. A. Biophys. J. 2005, 89, 2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez C. F.; Nielsen S. O.; Moore P. B.; Shelley J. C.; Klein M. L. J. Phys. Condens. Matter 2002, 14, 9431. [Google Scholar]
- Murtola T.; Falck E.; Patra M.; Karttunen M.; Vattulainen I. J. Chem. Phys. 2004, 121, 9156. [DOI] [PubMed] [Google Scholar]
- Srinivas G.; Klein M. L. Nanotechnology 2004, 15, 1289. [Google Scholar]
- Elezgaray J.; Laguerre M. Comput. Phys. Commun. 2006, 175, 264. [Google Scholar]
- Murtola T.; Falck E.; Karttunen M.; Vattulainen I. J. Chem. Phys. 2007, 126, 075101/1. [DOI] [PubMed] [Google Scholar]
- Periole X.; Huber T.; Marrink S. J.; Sakmar T. P. J. Am. Chem. Soc. 2007, 129, 10126. [DOI] [PubMed] [Google Scholar]
- Orsi M.; Haubertin D. Y.; Sanderson W. E.; Essex J. W. J. Phys. Chem. B 2008, 112, 802. [DOI] [PubMed] [Google Scholar]
- Fraaije J. G. E. M.; Zvelindovsky A. V.; Sevink G. J. A. Mol. Simulat. 2004, 30, 225. [Google Scholar]
- DPD Software; Accelrys: San Diego, CA, 2003. [Google Scholar]
- Marcelja S. Biochim. Biophys. Acta 1974, 367, 165. [DOI] [PubMed] [Google Scholar]
- Wiegel F. W.; Kox A. J. Phys. Lett. A 1978, 68A, 286. [Google Scholar]
- Wiegel F. W.; Kox A. J. Adv. Chem. Phys. 1980, 41, 195. [Google Scholar]
- Leermakers F. A. M.; Scheutjens J. M. H. M. J. Chem. Phys. 1988, 89, 3264. [Google Scholar]
- Pastor R. W.; Venable R. M.; Karplus M. J. Chem. Phys. 1988, 89, 1112. [Google Scholar]
- Pastor R. W.; Venable R. M.; Karplus M. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderkooi G. Chem. Phys. Lipids 1990, 55, 253. [DOI] [PubMed] [Google Scholar]
- Wang J.; Pullman A. Int. J. Quantum Chem. 1991, 18, 317. [Google Scholar]
- Wang J.; Pullman A. Biochim. Biophys. Acta 1990, 1024, 10. [DOI] [PubMed] [Google Scholar]
- Zheng C.; Vanderkooi G. Biophys. J. 1992, 63, 935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabdoulline R. R.; Zheng C.; Vanderkooi G. Chem. Phys. Lipids 1996, 84, 139. [Google Scholar]
- Wang J.; Pullman A. Chem. Phys. Lipids 1991, 57, 1. [Google Scholar]
- Brasseur R.; Goormaghtigh E.; Ruysschaert J. M. Biochem. Biophys. Res. Commun. 1986, 103, 301. [DOI] [PubMed] [Google Scholar]
- Vanderkooi G. Biochemistry 1991, 30, 10760. [DOI] [PubMed] [Google Scholar]
- Wang J.; Pullman A. J. Mol. Struct. THEOCHEM 1991, 73, 379. [Google Scholar]
- Vanderkooi G. Biophys. J. 1994, 66, 1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasseur R.; Vandenbranden M.; Cornet B.; Burny A.; Ruysschaert J. M. BBA−Biomembranes 1990, 1029, 267. [DOI] [PubMed] [Google Scholar]
- Jedlovszky P.; Mezei M. J. Chem. Phys. 1999, 111, 10770. [Google Scholar]
- Milik M.; Kolinski A.; Skolnick J. J. Chem. Phys. 1990, 93, 4440. [Google Scholar]
- Scott H. L. Biophys. J. 1991, 59, 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing J.; Scott H. L. Biochim. Biophys. Acta 1992, 1106, 227. [DOI] [PubMed] [Google Scholar]
- Milik M.; Skolnick J.; Kolinski A. J. Phys. Chem. 1992, 96, 4015. [Google Scholar]
- Xiang T. X. Biophys. J. 1993, 65, 1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang T. X.; Anderson B. D. J. Chem. Phys. 1995, 103, 8666. [Google Scholar]
- Taga T.; Masuda K. J. Comput. Chem. 1995, 16, 235. [Google Scholar]
- Scott H. L. In Biological Membranes: A Molecular Perspective from Computation and Experiment; Merz K. M. Jr., Roux B., Eds.; Birkhauser: Boston, MA, 1996; Chapter 3, p 83. [Google Scholar]
- Siepmann J. I.; Frenkel D. Mol. Phys. 1992, 75, 59. [Google Scholar]
- Jedlovszky P.; Mezei M. J. Phys. Chem. B 2001, 105, 3614. [Google Scholar]
- Chiu S. W.; Jakobsson E.; Mashl R. J.; Scott H. L. Biophys. J. 2002, 83, 1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedlovszky P.; Mezei M. J. Phys. Chem. B 2003, 107, 5311. [Google Scholar]
- Jedlovszky P.; Medvedev N. N.; Mezei M. J. Phys. Chem. B 2004, 108, 465. [DOI] [PubMed] [Google Scholar]
- Alinchenko M. G.; Anikeenko A. V.; Medvedev N. N.; Voloshin V. P.; Mezei M.; Jedlovszky P. J. Phys. Chem. B 2004, 108, 19056. [Google Scholar]
- Rabinovich A. L.; Balabaev N. K.; Alinchenko M. G.; Voloshin V. P.; Medvedev N. N.; Jedlovszky P. J. Chem. Phys. 2005, 122, 084906. [DOI] [PubMed] [Google Scholar]
- Alinchenko M. G.; Voloshin V. P.; Medvedev N. N.; Mezei M.; Partay L.; Jedlovszky P. J. Phys. Chem. B 2005, 109, 16490. [DOI] [PubMed] [Google Scholar]
- Brooks B. R.; Bruccoleri R. E.; Olafson B. D.; States D. J.; Swaminathan S.; Karplus M. J. Comput. Chem. 1983, 4, 187. [Google Scholar]
- Weiner S. J.; Kollman P. A.; Case D. A.; Singh U. C.; Ghio C.; Alagona G.; Profeta S. J.; Weiner P. J. Am. Chem. Soc 1984, 106, 765. [Google Scholar]
- Koehler J. E.; Saenger W.; van Gunsteren W. F. Eur. Biophys. J. 1987, 15, 197. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Tirado-Rives J. J. Am. Chem. Soc. 1988, 110, 1657. [DOI] [PubMed] [Google Scholar]
- Nelson M.; Humphrey W.; Kufrin R.; Gursoy A.; Dalke A.; Kale L.; Skeel R.; Schulten K. Comput. Phys. Commun. 1995, 91, 111. [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. J. Mol. Graphics 1996, 14, 33. [DOI] [PubMed] [Google Scholar]
- Berkowitz M. L.; Raghavan K. Langmuir 1991, 7, 1042. [Google Scholar]
- Damodaran K. V.; Merz K. M. Jr Langmuir 1993, 9, 1179. [Google Scholar]
- Marrink S. J.; Berkowitz M.; Berendsen H. J. C. Langmuir 1993, 9, 3122. [Google Scholar]
- Raghavan K.; Reddy M. R.; Berkowitz M. L. Langmuir 1992, 8, 233. [Google Scholar]
- Damodaran K. V.; Merz K. M. Jr.; Gaber B. P. Biochemistry 1992, 31, 7656. [DOI] [PubMed] [Google Scholar]
- Ahlstrom P.; Berendsen H. J. C. J. Phys. Chem. 1993, 97, 13691. [Google Scholar]
- Berendsen H. J. C.; Egberts B.; Marrink S. J.; Ahlstrom P. In Membrane Proteins: Structures, Interactions and Models; Pullman B., Jortner J., Pullman A., Eds.; Kluwer: Dordrecht, The Netherlands,1992; p 457. [Google Scholar]
- Alper H. E.; Bassolino D.; Stouch T. R. J. Chem. Phys. 1993, 98, 9798. [Google Scholar]
- Alper H. E.; Bassolino-Klimas D.; Stouch T. R. J. Chem. Phys. 1993, 99, 5547. [DOI] [PubMed] [Google Scholar]
- Kox A. J.; Michels J. P. J.; Wiegel F. W. Nature 1980, 287, 317. [Google Scholar]
- McKinnon S. J.; Whittenburg S. L.; Brooks B. J. Phys. Chem. 1992, 96, 10497. [Google Scholar]
- van der Ploeg P.; Berendsen H. J. C. J. Chem. Phys. 1982, 76, 3271. [Google Scholar]
- Edholm O.; Berendsen H. J. C.; van der Ploeg P. Mol. Phys. 1983, 48, 379. [Google Scholar]
- van der Ploeg P.; Berendsen H. J. C. Mol. Phys. 1983, 49, 233. [Google Scholar]
- Berendsen H. J.; Egberts, C B. In Structure, Dynamics, and Function of Biomolecules; Ehrenberg A., Rigler R., Gräalund A., Nilsson L., Eds.; Springer: Berlin, Germany, 1987; p 275. [Google Scholar]
- Egberts E.; Berendsen H. J. C. J. Chem. Phys. 1988, 89, 3718. [Google Scholar]
- Stouch T. R.; Ward K. B.; Altieri A.; Hagler A. T. J. Comput. Chem. 1991, 12, 1033. [Google Scholar]
- Venable R. M.; Zhang Y.; Hardy B. J.; Pastor R. W. Science 1993, 262, 223. [DOI] [PubMed] [Google Scholar]
- Heller H.; Schaefer M.; Schulten K. J. Phys. Chem. 1993, 97, 8343. [Google Scholar]
- Stouch T. R. Mol. Simulat. 1993, 10, 335. [Google Scholar]
- Egberts E.; Marrink S. J.; Berendsen H. J. Eur. Biophys. J. 1994, 22, 423. [DOI] [PubMed] [Google Scholar]
- Huang P.; Perez J. J.; Loew G. H. J. Biomol. Struct. Dynam. 1994, 11, 927. [DOI] [PubMed] [Google Scholar]
- Stouch T. R.; Williams D. E. J. Comput. Chem. 1992, 13, 622. [Google Scholar]
- Williams D. E.; Stouch T. R. J. Comput. Chem. 1993, 14, 1066. [Google Scholar]
- Schlenkrich M.; Brickmann J.; MacKerell A.D. Jr.; Karplus M. In Biological Membranes: A Molecular Perspective from Computation and Experiment; Merz K. M. Jr., Roux B., Eds.; Birkhauser: Boston, MA, 1996; Chapter 2, p 31. [Google Scholar]
- Berger O.; Edholm O.; Jahnig F. Biophys. J. 1997, 72, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobias D. J.; Tu K.; Klein M. L. J. Chim. Phys. Phys. Chim. Biol. 1997, 94, 1482. [Google Scholar]
- Feller S. E.; Yin D.; Pastor R. W.; MacKerell A. D. Jr Biophys. J. 1997, 73, 2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller S. E.; MacKerell A. D. Jr J. Phys. Chem. B 2000, 104, 7510. [Google Scholar]
- MacKerell A. D., Jr. In Computational Biochemistry and Biophysics; Becker O. M., Ed.; Marcel Dekker: New York, 2001; p 7. [Google Scholar]
- Chandrasekhar I.; Kastenholz M.; Lins R. D.; Oostenbrink C.; Schuler L. D.; Tieleman D. P.; van Gunsteren W. F. Eur. Biophys. J. 2003, 32, 67. [DOI] [PubMed] [Google Scholar]
- Klauda J. B.; Brooks B. R.; MacKerell A. D. Jr.; Venable R. M.; Pastor R. W. J. Phys. Chem. B 2005, 109, 5300. [DOI] [PubMed] [Google Scholar]
- Tu K.; Tobias D. J.; Klein M. L. Biophys. J. 1995, 69, 2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller S. E.; Zhang Y. H.; Pastor R. W.; Brooks B. R. J. Chem. Phys. 1995, 103, 4613. [Google Scholar]
- Feller S. E.; Venable R. M.; Pastor R. W. Langmuir 1997, 13, 6555. [Google Scholar]
- Feller S. E.; Pastor R. W. J. Chem. Phys. 1999, 111, 1281. [Google Scholar]
- Patel R. Y.; Balaji P. V. J. Phys. Chem. B 2005, 109, 14667. [DOI] [PubMed] [Google Scholar]
- Dolan E. A.; Venable R. M.; Pastor R. W.; Brooks B. R. Biophys. J. 2002, 82, 2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden T.; York D.; Pedersen L. J. Chem. Phys. 1993, 98, 10089. [Google Scholar]
- Feller S. E.; Pastor R. W.; Rojnuckarin A.; Bogusz S.; Brooks B. R. J. Phys. Chem. 1996, 100, 17011. [Google Scholar]
- Patra M.; Karttunen M.; Hyvonen M. T.; Falck E.; Lindqvist P.; Vattulainen I. Biophys. J. 2003, 84, 3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller S. E.; Zhang Y. H.; Pastor R. W. J. Chem. Phys. 1995, 103, 10267. [Google Scholar]
- Sankararamakrishnan R.; Weinstein H. J. Phys. Chem. B 2004, 108, 11802. [Google Scholar]
- Feller S. E.; Pastor R. W. Biophys. J. 1996, 71, 1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott H. L.; Jakobsson E.; Subramaniam S. Comput. Phys. 1998, 12, 328. [Google Scholar]
- Chiu S. W.; Jakobsson E.; Scott H. L. Biophys. J. 2001, 80, 1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu S. W.; Jakobsson E.; Scott H. L. J. Chem. Phys. 2001, 114, 5435. [Google Scholar]
- Venable R. M.; Brooks B. R.; Pastor R. W. J. Chem. Phys. 2000, 112, 4822. [Google Scholar]
- Chiu S. W.; Jakobsson E.; Mashl R. J.; Vasudevan S.; Scott J. Biophys. J. 2003, 85, 3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez Cascales J. J.; Berendsen H. J. C.; Garcia de la Torre J. J. Phys. Chem. 1996, 100, 8621. [Google Scholar]
- Lopez Cascales J.; Garcia de la Torre J. Biochim. Biophys. Acta 1997, 1330, 145. [DOI] [PubMed] [Google Scholar]
- Pandit S. A.; Berkowitz M. L. Biophys. J. 2002, 82, 1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit S. A.; Bostick D.; Berkowitz M. L. Biophys. J. 2003, 85, 3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P.; Monticelli L.; Tieleman D. P. Biophys. J. 2004, 86, 1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabdoulline R. R.; Vanderkooi G.; Zheng C. J. Phys. Chem. 1996, 100, 15942. [Google Scholar]
- Tu K.; Klein M. L.; Tobias D. J. Biophys. J. 1998, 75, 2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smondyrev A. M.; Berkowitz M. L. Biophys. J. 1999, 77, 2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasenkiewicz-Gierula M.; Rog T.; Kitamura K.; Kusumi A. Biophys. J. 2000, 78, 1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rog T.; Pasenkiewicz-Gierula M. Biophys. J. 2001, 81, 2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smondyrev A. M.; Berkowitz M. L. Biophys. J. 2001, 80, 1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofsass C.; Lindahl E.; Edholm O. Biophys. J. 2003, 84, 2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rog T.; Pasenkiewicz-Gierula M. Biophys. J. 2003, 84, 1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K.; Hata M.; Neya S.; Hoshino T. Chem-Bio Informatics J. 2004, 4, 15. [Google Scholar]
- Pandit S. A.; Bostick D.; Berkowitz M. L. Biophys. J. 2004, 86, 1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitman M. C.; Suits F.; MacKerell A. D. Jr.; Feller S. E. Biochemistry 2004, 43, 15318. [DOI] [PubMed] [Google Scholar]
- Hyvönen M. T.; Kovanen P. T. J. Phys. Chem. B 2003, 107, 9102. [Google Scholar]
- Pandit S. A.; Jakobsson E.; Scott H. L. Biophys. J. 2004, 87, 3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit S. A.; Vasudevan S.; Chiu S. W.; Mashl R. J.; Jakobsson E.; Scott H. L. Biophys. J. 2004, 87, 1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch S. L.; Keller S. L. Biophys. J. 2003, 84, 725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiz L.; Klein M. L. J. Chem. Phys. 2002, 116, 3052. [Google Scholar]
- Shinoda W.; Mikami M.; Baba T.; Hato M. J. Phys. Chem. B 2004, 108, 9346. [Google Scholar]
- Tieleman D. P.; Berendsen H. J. Biophys. J. 1998, 74, 2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich A. L.; Ripatti P. O.; Balabaev N. K.; Leermakers F. A. M. Phys. Rev. E 2003, 67, 011909. [DOI] [PubMed] [Google Scholar]
- Leermakers F. A. M.; Rabinovich A. L.; Balabaev N. K. Phys. Rev. E 2003, 67, 011910/1. [DOI] [PubMed] [Google Scholar]
- Hyvönen M. T.; Rantala T. T.; Ala K. M. Biophys. J. 1997, 73, 2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller S. E.; Gawrisch K.; MacKerell A. D. Jr. J. Am. Chem. Soc. 2002, 124, 318. [DOI] [PubMed] [Google Scholar]
- Eldho N. V.; Feller S. E.; Tristram-Nagle S.; Polozov I. V.; Gawrisch K. J. Am. Chem. Soc. 2003, 125, 6409. [DOI] [PubMed] [Google Scholar]
- Tieleman D. P.; Berendsen H. J. C. J. Chem. Phys. 1996, 105, 4871. [Google Scholar]
- Berger O.; Jahnig F.; Edholm O. Biophys. J. 1997, 72, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smondyrev A. M.; Berkowitz M. L. J. Comput. Chem. 1999, 20, 531. [Google Scholar]
- Pasenkiewicz-Gierula M.; Takaoka Y.; Miyagawa H.; Kitamura K.; Kusumi A. J. Phys. Chem. A 1997, 101, 3677. [Google Scholar]
- Chiu S. W.; Subramaniam S.; Jakobsson E. Biophys. J. 1999, 76, 1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl E.; Edholm O. Biophys. J. 2000, 79, 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrink S. J.; Lindahl E.; Edholm O.; Mark A. E. J. Am. Chem. Soc. 2001, 123, 8638. [DOI] [PubMed] [Google Scholar]
- Klauda J. B.; Brooks B. R.; Pastor R. W. J. Chem. Phys. 2006, 125, 144710/1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin B.; Hopfinger A. J. Biopolymers 1997, 41, 37. [DOI] [PubMed] [Google Scholar]
- Pasenkiewicz-Gierula M.; Rog T. Acta Biochim. Pol. 1997, 44, 607. [PubMed] [Google Scholar]
- Murzyn K.; Rog T.; Pasenkiewicz-Gierula M. Curr. Top. Biophys. 1999, 23, 87. [Google Scholar]
- Smondyrev A. M.; Berkowitz M. L. J. Chem. Phys. 1999, 110, 3981. [Google Scholar]
- Snyder R. G.; Tu K.; Klein M. L.; Mendelssohn R.; Strauss H. L.; Sun W. J. Phys. Chem. B 2002, 106, 6273. [Google Scholar]
- Marrink S. J.; Sok R. M.; Berendsen H. J. C. J. Chem. Phys. 1996, 104, 9090. [Google Scholar]
- Pasenkiewicz-Gierula M.; Takaoka Y.; Miyagawa H.; Kitamura K.; Kusumi A. Biophys. J. 1999, 76, 1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrink S. J.; Tieleman D. P.; van Buuren A. R.; Berendsen H. J. C. Faraday Discuss. 1996, 103, 191. [Google Scholar]
- Rog T.; Murzyn K.; Pasenkiewicz-Gierula M. Chem. Phys. Lett. 2002, 352, 323. [Google Scholar]
- Marrink S. J.; Berkowitz M.; Berendsen H. J. C. Langmuir 2003, 9, 3122. [Google Scholar]
- Moore P. B.; Lopez C. F.; Klein M. L. Biophys. J. 2001, 81, 2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez Cascales J. J.; Garcia de la Torre J.; Marrink S. J.; Berendsen H. J. C. J. Chem. Phys. 1996, 104, 2713. [Google Scholar]
- Gambu I.; Roux B. J. Phys. Chem. B 1997, 101, 6066. [Google Scholar]
- Pandit S. A.; Bostick D.; Berkowitz M. L. Biophys. J. 2003, 84, 3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs J. N.; Woolf T. B. J. Am. Chem. Soc. 2003, 125, 8742. [DOI] [PubMed] [Google Scholar]
- Murzyn K.; Rog T.; Jezierski G.; Takaoka Y.; Pasenkiewicz-Gierula M. Biophys. J. 2001, 81, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachar M.; Brunelle P.; Tieleman D. P.; Rauk A. J. Phys. Chem. B 2004, 108, 7170. [Google Scholar]
- Roach C.; Feller S. E.; Ward J. A.; Shaikh S. R.; Zerouga M.; Stillwell W. Biochemistry 2004, 43, 6344. [DOI] [PubMed] [Google Scholar]
- Tieleman D. P.; Bentz J. Biophys. J. 2002, 83, 1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faller R.; Marrink S. J. Langmuir 2004, 20, 7686. [DOI] [PubMed] [Google Scholar]
- Marrink S. J.; Mark A. E. Biophys. J. 2004, 87, 3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries A. H.; Yefimov S.; Mark A. E.; Marrink S. J. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieleman D. P.; Leontiadou H.; Mark A. E.; Marrink S. J. J. Am. Chem. Soc. 2003, 125, 6382. [DOI] [PubMed] [Google Scholar]
- Tieleman D. P. BMC Biochem. 2004, 5, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leontiadou H.; Mark A. E.; Marrink S. J. Biophys. J. 2004, 86, 2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries A. H.; Mark A. E.; Marrink S. J. J. Am. Chem. Soc. 2004, 126, 4488. [DOI] [PubMed] [Google Scholar]
- Ohta-Iino S.; Pasenkiewicz-Gierula M.; Takaoka Y.; Miyagawa H.; Kitamura K.; Kusumi A. Biophys. J. 2001, 81, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrink S. J.; Tieleman D. P. Biophys. J. 2002, 83, 2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrache H. I.; Tu K.; Nagle J. F. Biophys. J. 1999, 76, 2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor R. W.; Venable R. M.; Feller S. E. Acc. Chem. Res. 2002, 35, 438. [DOI] [PubMed] [Google Scholar]
- Benz R. W.; Castro-Roman F.; Tobias D. J.; White S. H. Biophys. J. 2005, 88, 805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benz R. W.; Nanda H.; Castro-Roman F.; White S. H.; Tobias D. J. Biophys. J. 2006, 91, 3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren P.; Ponder J. W. J. Phys. Chem. B 2003, 107, 5933. [Google Scholar]
- Donchev A. G.; Galkin N. G.; Illarionov A. A.; Khoruzhii O. V.; Olevanov M. A.; Ozrin V. D.; Subbotin M. V.; Tarasov V. I. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 8613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S.; Brooks C. L. III Mol. Simulat. 2006, 32, 231. [Google Scholar]
- Piquemal J.; Perera L.; Cisneros G. A.; Ren P.; Pedersen L. G.; Darden T. A. J. Chem. Phys. 2006, 125, 054511. [DOI] [PubMed] [Google Scholar]
- Swart M.; van Duijnen P. T. Mol. Simulat. 2006, 32, 471. [Google Scholar]
- Dehez F.; Angyan J. G.; Soteras Gutierrez I.; Luque F. J.; Schulten K.; Chipot C. J. Chem. Theory Comput. 2007, 3, 1914. [DOI] [PubMed] [Google Scholar]
- Paesani F.; Iuchi S.; Voth G. A. J. Chem. Phys. 2007, 127, 074506/1. [DOI] [PubMed] [Google Scholar]
- Warshel A.; Kato M.; Pisliakov A. V. J. Chem. Theory Comput. 2007, 3, 2034. [DOI] [PubMed] [Google Scholar]
- Xie W.; Pu J.; MacKerell A. D. Jr.; Gao J. J. Chem. Theory Comput. 2007, 3, 1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder E.; Anisimov V. M.; Whitfield T.; MacKerell A. D. Jr.; Roux B. J. Phys. Chem. B 2008, 112, 3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Åqvist J.; Medina C.; Samuelsson J. E. Protein Eng. 1994, 7, 385. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L. Chemtracts Org. Chem. 1995, 8, 374. [Google Scholar]
- Brasseur R.; Dellers M.; Malaisse W. J.; Ruysschaert J. M. Proc. Natl. Acad. Sci. U.S.A. 1982, 79, 2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasseur R.; Vandenbosch C.; van den Bossche H.; Ruysschaert J. M. Biochem. Pharmacol. 1983, 32, 2175. [DOI] [PubMed] [Google Scholar]
- Brasseur R.; Ruysschaert J. M.; Chatelain P. Biochem. Pharmacol. 1984, 33, 1015. [DOI] [PubMed] [Google Scholar]
- Brasseur R.; de Loof H.; Ruysschaert J. M.; Rosseneu M. BBA−Biomembranes 1988, 943, 95. [DOI] [PubMed] [Google Scholar]
- de Loof H.; Rosseneu M.; Brasseur R.; Ruysschaert J. M. Proc. Natl. Acad. Sci. U.S.A. 1986, 83, 2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasseur R. J. Biol. Chem. 1991, 266, 16120. [PubMed] [Google Scholar]
- Brasseur R.; Lins L.; Vanloo B.; Ruysschaert J. M.; Rosseneu M. Proteins 1992, 13, 246. [DOI] [PubMed] [Google Scholar]
- Mingeot-Leclercq M. P.; Tulkens P. M.; Brasseur R. Biochem. Pharmacol. 1992, 44, 1967. [DOI] [PubMed] [Google Scholar]
- Ducarme P.; Rahman M.; Brasseur R. Proteins 1998, 30, 357. [PubMed] [Google Scholar]
- Lins L.; Ducarme P.; Breukink E.; Brasseur R. BBA−Biomembranes 1999, 1420, 111. [DOI] [PubMed] [Google Scholar]
- Vogt B.; Ducarme P.; Schinzel S.; Brasseur R.; Bechinger B. Biophys. J. 2000, 79, 2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basyn F.; Charloteaux B.; Thomas A.; Brasseur R. J. Mol. Graph. Model. 2001, 20, 235. [DOI] [PubMed] [Google Scholar]
- Milik M.; Skolnick J. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 9391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer L. A.; Leermakers F. A. M.; Lyklema J. J. Chem. Phys. 1999, 110, 6560. [Google Scholar]
- Leermakers F. A. M.; Kleijn J. M. In Physicochemical Kinetics and Transport at Biointerfaces; van Leeuwen H. P., Koester W. Eds.; John Wiley & Sons: New York, 2004; p 15. [Google Scholar]
- Skolnick J.; Milik M. In Biological Membranes: A Molecular Perspective from Computation and Experiment; Merz K. M. Jr., Roux B., Eds.; Birkhauser: Boston, MA, 1996; Chapter 16, p 535. [Google Scholar]
- Skolnick J.; Milik M. In Membrane Protein Assembly; von Heijne G., Ed.; Landes: Austin, TX, 1997; p 201. [Google Scholar]
- Widom B. J. Chem. Phys. 1963, 39, 2808. [Google Scholar]
- Xiang T. X.; Anderson B. D. J. Chem. Phys. 1999, 110, 1807. [Google Scholar]
- Jedlovszky P.; Mezei M. J. Am. Chem. Soc. 2000, 122, 5125. [Google Scholar]
- Jedlovszky P.; Mezei M. J. Phys. Chem. B 2003, 107, 5322. [Google Scholar]
- Tu K.; Tarek M.; Klein M. L.; Scharf D. Biophys. J. 1998, 75, 2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koubi L.; Tarek M.; Klein M. L.; Scharf D. Biophys. J. 2000, 78, 800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummer G.; Szabo A. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummer G.; Szabo A. Biophys. J. 2003, 85, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.; Khalili-Araghi F.; Tajkhorshid E.; Schulten K. J. Chem. Phys. 2003, 119, 3559. [Google Scholar]
- Bassolino-Klimas D.; Alper H. E.; Stouch T. R. Biochemistry 1993, 32, 12624. [DOI] [PubMed] [Google Scholar]
- Stouch T. R.; Alper H. E.; Bassolino D. In Computer-Aided Molecular Design: Applications in Agrochemicals, Materials, and Pharmaceuticals; Reynolds C. H., Holloway M. K., Cox H. K., Eds.; Oxford University Press: New York, 1995; p 127. [Google Scholar]
- Stouch T. R.; Bassolino D. In Biological Membranes: A Molecular Perspective from Computation and Experiment; Merz K. M. Jr., Roux B., Eds.; Birkhauser: Boston, MA, 1996; Chapter 8, p 255. [Google Scholar]
- Lopez Cascales J. J.; Huertas M. L.; Garcia de la Torre J. Biophys. Chem. 1997, 69, 1. [DOI] [PubMed] [Google Scholar]
- Repakova J.; Capkova P.; Vattulainen I.; Holopainen J. M.; Morrow M. R.; McDonald M. C. Biophys. J. 2005, 88, 3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopasek I.; Vecer J.; Strzalka K.; Amler E. Chem. Phys. Lipids 2004, 130, 135. [DOI] [PubMed] [Google Scholar]
- Herrenbauer M.; Tieleman D. P.; Posten C. In Computer Applications in Biotechnology 2001; Dochain D., Perrier M., Eds.; Pergamon Press: Oxford, U.K., 2002; p 311. [Google Scholar]
- Aiello M.; Moran O.; Pisciotta M.; Gambale F. Eur. Biophys. J. 1998, 27, 211. [DOI] [PubMed] [Google Scholar]
- Huang P.; Bertaccini E.; Loew G. H. J. Biomol. Struct. Dynam. 1995, 12, 725. [DOI] [PubMed] [Google Scholar]
- Lopez Cascales J. J.; Cifre J. G. H.; Garcia de la Torre J. J. Phys. Chem. B 1998, 102, 625. [Google Scholar]
- Landolt-Marticorena C.; Williams K. A.; Deber C. M.; Reithmeier R. A. F. J. Mol. Biol. 1993, 229, 602. [DOI] [PubMed] [Google Scholar]
- Saiz L.; Klein M. L. Acc. Chem. Res. 2002, 35, 482. [DOI] [PubMed] [Google Scholar]
- Koubi L.; Tarek M.; Bandyopadhyay S.; Klein M. L.; Scharf D. Anesthesiology 2002, 97, 848. [DOI] [PubMed] [Google Scholar]
- Pasenkiewicz-Gierula M.; Rog T.; Grochowski J.; Serda P.; Czarnecki R.; Librowski T.; Lochynski S. Biophys. J. 2003, 85, 1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemiola-Rzeminska M.; Pasenkiewicz-Gierula M.; Strzalka K. Chem. Phys. Lipids 2005, 135, 27. [DOI] [PubMed] [Google Scholar]
- Song Y.; Guallar V.; Baker N. A. Biochemistry 2005, 44, 13425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P.; Vogel H. J.; Tieleman D. P. Biophys. J. 2004, 86, 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydall J. R.; Macdonald P. M. Biochemistry 1992, 31, 1092. [DOI] [PubMed] [Google Scholar]
- Clarke R. J.; Lupfert C. Biophys. J. 1999, 76, 2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins K. D.; Washabaugh M. W. Q. Rev. Biophys. 1985, 18, 323. [DOI] [PubMed] [Google Scholar]
- MacCallum J. L.; Tieleman D. P. J. Am. Chem. Soc. 2006, 128, 125. [DOI] [PubMed] [Google Scholar]
- Sachs J. N.; Crozier P. S.; Woolf T. B. J. Chem. Phys. 2004, 121, 10847. [DOI] [PubMed] [Google Scholar]
- Gurtovenko A. A. J. Chem. Phys. 2005, 122, 244902/1. [DOI] [PubMed] [Google Scholar]
- Scheuplein R. J. J. Theor. Biol. 1968, 18, 72. [DOI] [PubMed] [Google Scholar]
- Marrink S. J.; Berendsen H. J. C. J. Phys. Chem. 1996, 100, 16729. [Google Scholar]
- Marrink S. J.; Jahnig F.; Berendsen H. J. Biophys. J. 1996, 71, 632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn D.; Brickmann J. Phys. Chem. Chem. Phys. 2001, 3, 848. [Google Scholar]
- Pasenkiewicz-Gierula M.; Subczynski W. K. Curr. Top. Biophys. 1996, 20, 93. [Google Scholar]
- Bemporad D.; Essex J. W.; Luttmann C. J. Phys. Chem. B 2004, 108, 4875. [Google Scholar]
- Bemporad D.; Luttmann C.; Essex J. W. BBA—Biomembranes 2005, 1718, 1. [DOI] [PubMed] [Google Scholar]
- Holter H.; Marshall J. M. Jr Compt. Rend. Trav. Lab. Carlsberg 1954, 29, 7. [PubMed] [Google Scholar]
- Goldstein J. L.; Anderson R. G.; Brown M. S. Nature 1979, 279, 679. [DOI] [PubMed] [Google Scholar]
- Brown M. S.; Goldstein J. L. Proc. Natl. Acad. Sci. U.S.A. 1979, 76, 3330.226968 [Google Scholar]
- Brown M. S.; Goldstein J. L. Science 1986, 232, 34. [DOI] [PubMed] [Google Scholar]
- Shedden K.; Xie X. T.; Chandaroy P.; Chang Y. T.; Rosania G. R. Cancer Res. 2003, 63, 4331. [PubMed] [Google Scholar]
- Feng D.; Nagy J. A.; Dvorak A. M.; Dvorak H. F. Microvasc. Res. 2000, 59, 24. [DOI] [PubMed] [Google Scholar]
- Feng D.; Nagy J. A.; Dvorak H. F.; Dvorak A. M. Microscopy Res. Techn. 2002, 57, 289. [DOI] [PubMed] [Google Scholar]
- Couet J.; Li S.; Okamoto T.; Scherer P. E.; Lisanti M. P. Trends Cardiovasc. Med. 1997, 7, 103. [DOI] [PubMed] [Google Scholar]
- Matveev S.; Li X.; Everson W.; Smart E. J. Adv. Drug Delivery Rev. 2001, 49, 237. [DOI] [PubMed] [Google Scholar]
- Gumbleton M.; Abulrob A. N.; Campbell L. Pharm. Res. 2000, 17, 1035. [DOI] [PubMed] [Google Scholar]
- Page E.; Iida H.; Doyle D. D. In Handbook of Physiology; Page E., Fozzard H. A., Solaro R. J., Eds.; Oxford University Press: New York, 2002; p 145. [Google Scholar]
- Anderson R. G. W.; Kamen B. A.; Rothberg K. G.; Lacey S. W. Science 1992, 255, 410. [DOI] [PubMed] [Google Scholar]
- Anderson R. G. W. Trends Cell Biol. 1993, 3, 69. [DOI] [PubMed] [Google Scholar]
- Mineo C.; Anderson R. G. W. Histochem. Cell Biol. 2001, 116, 109. [DOI] [PubMed] [Google Scholar]
- Gumbleton M. Adv. Drug Delivery Rev. 2001, 49, 217. [DOI] [PubMed] [Google Scholar]
- Green B. T.; Brown D. R. Vet. Microbiol. 2006, 113, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beahon S. J.; Woodley J. F. Biochem. Soc. Trans. 1984, 12, 1088. [Google Scholar]
- Madara J. L.; Bye W. A.; Trier J. S. Gastroenterology 1984, 87, 1091. [PubMed] [Google Scholar]
- Heyman M.; Bonfils A.; Fortier M.; Crain-Denoyelle A. M.; Smets P.; Desjeux J. F. Int. J. Pharm. 1987, 37, 33. [Google Scholar]
- Ho N. F. H.; Day J. S.; Barsuhn C. L.; Burton P. S.; Raub T. J. J. Controlled Release 1990, 11, 3. [Google Scholar]
- Haseto S.; Ouchi H.; Isoda T.; Mizuma T.; Hayashi M.; Awazu S. Pharm. Res. 1994, 11, 361. [DOI] [PubMed] [Google Scholar]
- Isoda T.; Watanabe E.; Haga M.; Haseto S.; Awazu S.; Hayashi M. Eur. J. Pharm. Biopharm. 1997, 44, 133. [Google Scholar]
- Gekle M.; Freudinger R.; Mildenberger S. J. Physiol. 2001, 531, 619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hryciw D. H.; Ekberg J.; Lee A.; Lensink I. L.; Kumar S.; Guggino W. B.; Cook D. I.; Pollock C. A.; Poronnik P. J. Biol. Chem. 2004, 279, 54996. [DOI] [PubMed] [Google Scholar]
- Caruso-Neves C.; Kwon S. H.; Guggino W. B. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 17513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones A. T.; Gumbleton M.; Duncan R. Adv. Drug Delivery Rev. 2003, 55, 1353. [DOI] [PubMed] [Google Scholar]
- de Boer A. G.; van der Sandt I. C. J.; Gaillard P. J. Annu. Rev. Pharmacol. 2003, 43, 629. [DOI] [PubMed] [Google Scholar]
- Yamasaki Y.; Hisazumi J.; Yamaoka K.; Takakura Y. Eur. J. Pharm. Sci. 2003, 18, 305. [DOI] [PubMed] [Google Scholar]
- Carpentier M.; Descamps L.; Allain F.; Denys A.; Durieux S.; Fenart L.; Kieda C.; Cecchelli R.; Spik G. J. Neurochem. 1999, 73, 260. [DOI] [PubMed] [Google Scholar]
- Masereeuw R.; Terlouw S. A.; van Aubel R. A. M. H.; Russel F. G. M.; Miller D. S. Mol. Pharmacol. 2000, 57, 59. [PubMed] [Google Scholar]
- Yang C.; Tirucherai G. S.; Mitra A. K. Expert Opin. Biol. Therap. 2001, 1, 159. [DOI] [PubMed] [Google Scholar]
- Qian Z. M.; Li H.; Sun H.; Ho K. Pharmacol. Rev. 2002, 54, 561. [DOI] [PubMed] [Google Scholar]
- Tuan Giam Chuang V.; Kragh-Hansen U.; Otagiri M. Pharm. Res. 2002, 19, 569. [DOI] [PubMed] [Google Scholar]
- Chan W. Y.; Huang H.; Tam S. C. Toxicology 2003, 186, 191. [DOI] [PubMed] [Google Scholar]
- Nishikawa M. Biol. Pharm. Bull. 2005, 28, 195. [DOI] [PubMed] [Google Scholar]
- Ayrton A.; Morgan P. Xenobiotica 2001, 31, 469. [DOI] [PubMed] [Google Scholar]
- Rao V. V.; Dahlheimer J. L.; Bardgett M. E.; Snyder A. Z.; Finch R. A.; Sartorelli A. C.; Piwnica-Worms D. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie E. M.; Deeley R. G.; Cole S. P. C. Toxicol. Appl. Pharmacol. 2005, 204, 216. [DOI] [PubMed] [Google Scholar]
- Ambudkar S. V.; Kimchi-Sarfaty C.; Sauna Z. E.; Gottesman M. M. Oncogene 2003, 22, 7468. [DOI] [PubMed] [Google Scholar]
- Ganapathy V.; Prasad P. D. Toxicol. Appl. Pharmacol. 2005, 207, S381. [DOI] [PubMed] [Google Scholar]
- Myllynen P.; Pasanen M.; Pelkonen O. Placenta 2005, 26, 361. [DOI] [PubMed] [Google Scholar]
- Choi C. H. Cancer Cell Int. 2005, 5, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs B. C.; Bode B. P. Semin. Cancer Biol. 2005, 15, 254. [DOI] [PubMed] [Google Scholar]
- Sweet D. H. Toxicol. Appl. Pharmacol. 2005, 204, 198. [DOI] [PubMed] [Google Scholar]
- Gerloff T. N. Schmied. Arch. Pharmacol. 2004, 369, 69. [DOI] [PubMed] [Google Scholar]
- Hermsmeyer R. K. In Cell Physiology; Sperelakis N., Ed.; Academic Press: London, U.K., 1995; Chapter 29, p404. [Google Scholar]
- Csala M.; Senesi S.; Banhegyi G.; Mandl J.; Benedetti A. Arch. Biochem. Biophys. 2005, 440, 173. [DOI] [PubMed] [Google Scholar]
- Bevers E. M.; Comfurius P.; Dekkers D. W. C.; Zwaal R. F. A. BBA—Mol. Cell. Biol. Lipids 1999, 1439, 317. [DOI] [PubMed] [Google Scholar]
- LaManna J. C.; Harrington J. F.; Vendel L. M.; Abi-Saleh K.; Lust W. D.; Harik S. I. In Role of Neurotransmitters in Brain Injury; Globus M. Y. T., Dietrich W. D., Eds.; Plenum Press: New York, 1992; p 293. [Google Scholar]
- Putnam R. W. In Cell Physiology; Sperelakis N., Ed.; Academic Press: London, U.K., 1995; Chapter 17, p 230. [Google Scholar]
- Eraly S. A.; Hamilton B. A.; Nigam S. K. Biochem. Biophys. Res. Commun. 2003, 300, 333. [DOI] [PubMed] [Google Scholar]
- Wright S. H. Toxicol. Appl. Pharm. 2005, 204, 309. [DOI] [PubMed] [Google Scholar]
- Genew, HUGO Gene Nomenclature Committee (http://www.gene.ucl.ac.uk/cgi-bin/nomenclature/searchgenes.pl);Department of Biology, University College: London, U.K., February 2006. [Google Scholar]
- Gemmecker G.; Erni B. In NMR in Microbiology; Barbotin J. N., Portais J. C., Eds.; Horizon Scientific Press: Wymondham, U.K., 2000; p 191. [Google Scholar]
- Hruz P. W.; Mueckler M. M. Mol. Membr. Biol. 2001, 18, 183. [DOI] [PubMed] [Google Scholar]
- Rubin D. C. Curr. Opin. Gastroen. 2004, 20, 65. [DOI] [PubMed] [Google Scholar]
- Meredith D.; Boyd C. A. R. Cell. Mol. Life Sci. 2000, 57, 754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen C. U.; Brodin B.; Jorgensen F. S.; Frokjaer S.; Steffansen B. Expert Opin. Ther. Pat. 2002, 12, 1329. [Google Scholar]
- Wang J.; Schaner M. E.; Thomassen S.; Su S. F.; Piquette-Miller M.; Giacomini K. M. Pharm. Res. 1997, 14, 1524. [DOI] [PubMed] [Google Scholar]
- Prasad P. D.; Ganapathy V. Curr. Opin. Clin. Nutr. 2000, 3, 263. [DOI] [PubMed] [Google Scholar]
- Friesema E. C. H.; Jansen J.; Visser T. J. Curr. Opin. Endocrin. Diabetes 2005, 12, 371. [Google Scholar]
- Yang C. Y.; Dantzig A. H.; Pidgeon C. Pharm. Res. 1999, 16, 1331. [DOI] [PubMed] [Google Scholar]
- Wood I. S.; Trayhurn P. Brit. J. Nutr. 2003, 89, 3. [DOI] [PubMed] [Google Scholar]
- Scheepers A.; Joost H. G.; Schurmann A. J. Parenter. Enter. Nutr. 2004, 28, 364. [DOI] [PubMed] [Google Scholar]
- Cuppoletti J. In Cell Physiology; Sperelakis N., Ed.; Academic Press: London, U.K., 1995; Chapter 11, p 139. [Google Scholar]
- Thirone A.; Rudich A.; Klip A. Recent Res. Develop. Physiol. 2004, 2, 367. [Google Scholar]
- Ver M. R.; Chen H.; Quon M. J. Curr. Med. Chem. Immunol. Endocr. Metab. Agents 2005, 5, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naftalin R. J. In Red Cell Membrane Transport in Health and Disease; Bernhardt I., Ellory J. C., Eds.; Springer: Berlin, 2003; p 339. [Google Scholar]
- Raja M. M.; Tyagi N. K.; Kinne R. K. H. J. Biol. Chem. 2003, 278, 49154. [DOI] [PubMed] [Google Scholar]
- Asano T.; Kurihara H.; Uchijima Y.; Ogihara T.; Katagiri H.; Sakoda H.; Ono H.; Anai M.; Fujishiro M. Curr. Med. Chem. 2004, 11, 2717. [DOI] [PubMed] [Google Scholar]
- Asano T.; Kurihara H.; Uchijima Y.; Anai M.; Ono H.; Sakoda H.; Fujishiro M. Drugs Future 2004, 29, 461. [DOI] [PubMed] [Google Scholar]
- Handlon A. L. Expert Opin. Ther. Pat. 2005, 15, 1531. [Google Scholar]
- Cunningham P.; Afzal-Ahmed I.; Naftalin R. J. J. Biol. Chem. 2006, 281, 5797. [DOI] [PubMed] [Google Scholar]
- Steffansen B.; Nielsen C. U.; Brodin B.; Eriksson A. H.; Andersen R.; Frokjaer S. Eur. J. Pharm. Sci. 2004, 21, 3. [DOI] [PubMed] [Google Scholar]
- Inui K.; Masuda S.; Saito H. Kidney Int. 2000, 58, 944. [DOI] [PubMed] [Google Scholar]
- Han H.; de Vrueh R. L.; Rhie J. K.; Covitz K. M.; Smith P. L.; Lee C. P.; Oh D. M.; Sadee W.; Amidon G. L. Pharm. Res. 1998, 15, 1154. [DOI] [PubMed] [Google Scholar]
- Marshall E. K.; Vickers J. L. Bull. Johns Hopkins Hosp. 1923, 34, 1. [Google Scholar]
- Rennick B. R.; Moe G. K.; Lyons R. H.; Hoobler S. W.; Neligh R. J. Pharmacol. Exp. Ther. 1947, 91, 210. [PubMed] [Google Scholar]
- Smeets P. H. E.; van Aubel R. A. M. H.; Wouterse A. C.; van den Heuvel J. J. M. W.; Russel F. G. M. J. Am. Soc. Nephrol. 2004, 15, 2828. [DOI] [PubMed] [Google Scholar]
- van Montfoort J. E.; Hagenbuch B.; Groothuis G. M. M.; Koepsell H.; Meier P. J.; Meijer D. K. F. Curr. Drug Metab. 2003, 4, 185. [DOI] [PubMed] [Google Scholar]
- Hagenbuch B.; Gao B.; Meier P. J. News Physiol. Sci. 2002, 17, 231. [DOI] [PubMed] [Google Scholar]
- Chang C.; Pang K. S.; Swaan P. W.; Ekins S. J. Pharmacol. Exp. Ther. 2005, 314, 533. [DOI] [PubMed] [Google Scholar]
- Russel F. G. M.; Masereeuw R.; van Aubel R. A. M. H. Annu. Rev. Physiol. 2002, 64, 563. [DOI] [PubMed] [Google Scholar]
- Hagenbuch B.; Meier P. J. BBA−Biomembranes 2003, 1609, 1. [DOI] [PubMed] [Google Scholar]
- Miyazaki H.; Sekine T.; Endou H. Trends Pharmacol. Sci. 2004, 25, 654. [DOI] [PubMed] [Google Scholar]
- Hagenbuch B.; Meier P. J. Pflüg. Arch. 2004, 447, 653. [DOI] [PubMed] [Google Scholar]
- Köpsell H. Annu. Rev. Physiol. 1998, 60, 243. [DOI] [PubMed] [Google Scholar]
- Masereeuw R.; Russel F. G. M. Drug Metab. Rev. 2001, 33, 299. [DOI] [PubMed] [Google Scholar]
- Burckhardt G.; Wolff N. A.; Bahn A. Cell Biochem. Biophys. 2002, 36, 169. [DOI] [PubMed] [Google Scholar]
- Sekine T.; Cha S. H.; Endou H. Pflüg. Arch. 2000, 440, 337. [DOI] [PubMed] [Google Scholar]
- Inui K.; Terada T.; Masuda S.; Saito H. Nephrol. Dial. Transpl. 2000, 15, 11. [DOI] [PubMed] [Google Scholar]
- Elferink R. P. J. O.; Jansen P. L. M. Pharmacol. Therap. 1994, 64, 77. [DOI] [PubMed] [Google Scholar]
- van Montfoort J. E.; Hagenbuch B.; Fattinger K. E.; Müller M.; Groothuis G. M. M.; Meijer D. K. F.; Meier P. J. J. Pharmacol. Exp. Therap. 1999, 291, 147. [PubMed] [Google Scholar]
- Meier P. J.; Stieger B. Annu. Rev. Physiol. 2002, 64, 635. [DOI] [PubMed] [Google Scholar]
- Kullak-Ublick G. A.; Stieger B.; Meier P. J. Gastroenterology 2004, 126, 322. [DOI] [PubMed] [Google Scholar]
- Pauli-Magnus C.; Stieger B.; Meier Y.; Kullak-Ublick G. A.; Meier P. J. J. Hepatol. 2005, 43, 342. [DOI] [PubMed] [Google Scholar]
- Kullak-Ublick G. A.; Ismair M. G.; Hagenbuch B.; Stieger B.; Meier P. J. Falk Symp. 2001, 117, 5. [DOI] [PubMed] [Google Scholar]
- Pardridge W. M. In Handbook of Physiology; Conn P. M., Ed.; Oxford University Press: New York, 1998; Section 7, p 335. [Google Scholar]
- Reichen J. News Physiol. Sci. 1999, 14, 117. [DOI] [PubMed] [Google Scholar]
- Tang C.; Lin Y.; Rodrigues A. D.; Lin J. H. Drug Metab. Dispos. 2002, 30, 648. [DOI] [PubMed] [Google Scholar]
- Adachi Y.; Kobayashi Y. Hepatol. Res. 2005, 31, 193. [DOI] [PubMed] [Google Scholar]
- Hauptmann J.; Steinmetzer T.; Vieweg H.; Wikstroem P.; Stürzebecher J. Pharm. Res. 2002, 19, 1027. [DOI] [PubMed] [Google Scholar]
- Anderson R. P.; Butt T. J.; Chadwick V. S. Digest. Dis. Sci. 1992, 37, 248. [DOI] [PubMed] [Google Scholar]
- Proost J. H.; Roggeveld J.; Wierda J. M.; Meijer D. K. F. J. Pharmacol. Exp. Ther. 1997, 282, 715. [PubMed] [Google Scholar]
- Biagi G. L.; Cantelli-Forti G.; Barbaro A. M.; Guerra M. C.; Hrelia P.; Borea P. A. J. Med. Chem. 1987, 30, 420. [DOI] [PubMed] [Google Scholar]
- Jolliet-Riant P.; Tillement J. P. Fundam. Clin. Pharmacol. 1999, 13, 16. [DOI] [PubMed] [Google Scholar]
- Ecker G. F.; Noe C. R. Curr. Med. Chem. 2004, 11, 1617. [DOI] [PubMed] [Google Scholar]
- Maksay G.; Tegyey Z.; Kemeny V.; Lukovits I.; Ötvös L. J. Med. Chem. 1979, 22, 1436. [DOI] [PubMed] [Google Scholar]
- Meulemans A.; Vicart P.; Mohler J.; Vulpillat M. Chemotherapy 1988, 34, 90. [DOI] [PubMed] [Google Scholar]
- van de Waterbeemd H.; Kansy M. Chimia 1992, 46, 299. [Google Scholar]
- Abraham M. H.; Chadha H. S.; Mitchell R. C. J. Pharm. Sci. 1994, 83, 1257. [DOI] [PubMed] [Google Scholar]
- Norinder U.; Haeberlein M. Adv. Drug Delivery Rev. 2002, 54, 291. [DOI] [PubMed] [Google Scholar]
- Chadha H. S.; Abraham M. H.; Mitchell R. C. Bioorg. Med. Chem. Lett. 1994, 4, 2511. [Google Scholar]
- Abraham M. H.; Chadha H. S.; Mitchell R. C. Drug Des. Discovery 1995, 13, 123. [PubMed] [Google Scholar]
- Gratton J. A.; Abraham M. H.; Bradbury M. W.; Chadha H. S. J. Pharm. Pharmacol. 1997, 49, 1211. [DOI] [PubMed] [Google Scholar]
- van de Waterbeemd H.; Camenisch G.; Folkers G.; Chretien J. R.; Raevsky O. A. J. Drug Target. 1998, 6, 151. [DOI] [PubMed] [Google Scholar]
- Abraham M. H.; Chadha H. S.; Martins F.; Mitchell R. C.; Bradbury M. W.; Gratton J. A. Pestic. Sci. 1999, 55, 78. [Google Scholar]
- Platts J. A.; Abraham M. H.; Zhao Y. H.; Hersey A.; Ijaz L.; Butina D. Eur. J. Med. Chem. 2001, 36, 719. [DOI] [PubMed] [Google Scholar]
- Abraham M. H. Eur. J. Med. Chem. 2004, 39, 235. [DOI] [PubMed] [Google Scholar]
- Fischer H.; Gottschlich R.; Seelig A. J. Membr. Biol. 1998, 165, 201. [DOI] [PubMed] [Google Scholar]
- Ertl P.; Rohde B.; Selzer P. J. Med. Chem. 2000, 43, 3714. [DOI] [PubMed] [Google Scholar]
- Österberg T.; Norinder U. J. Chem. Inf. Comput. Sci. 2000, 40, 1408. [DOI] [PubMed] [Google Scholar]
- Kaznessis Y. N.; Snow M. E.; Blankley C. J. J. Comput. Aid. Mol. Des. 2001, 15, 697. [DOI] [PubMed] [Google Scholar]
- Keserü G. M.; Molnar L. J. Chem. Inf. Comput. Sci. 2001, 41, 120. [DOI] [PubMed] [Google Scholar]
- Luco J. M. J. Chem. Inf. Comput. Sci. 1999, 39, 396. [DOI] [PubMed] [Google Scholar]
- Rose K.; Hall L. H.; Kier L. B. J. Chem. Inf. Comput. Sci. 2002, 42, 651. [DOI] [PubMed] [Google Scholar]
- Crivori P.; Cruciani G.; Carrupt P. A.; Testa B. J. Med. Chem. 2000, 43, 2204. [DOI] [PubMed] [Google Scholar]
- Ooms F.; Weber P.; Carrupt P. A.; Testa B. Biochim. Biophys. Acta 2002, 1587, 118. [DOI] [PubMed] [Google Scholar]
- Segarra V.; Lopez M.; Ryder H.; Palacios J. M. Quant. Struct. Act. Relat. 1999, 18, 474. [Google Scholar]
- Oprea T. I.; Zamora I.; Ungell A. L. J. Comb. Chem. 2002, 4, 258. [DOI] [PubMed] [Google Scholar]
- Yarim M.; Moro S.; Huber R.; Meier P. J.; Kaseda C.; Kashima T.; Hagenbuch B.; Folkers G. Bioorg. Med. Chem. 2004, 13, 463. [DOI] [PubMed] [Google Scholar]
- Bednarczyk D.; Ekins S.; Wikel J. H.; Wright S. H. Mol. Pharmacol. 2003, 63, 489. [DOI] [PubMed] [Google Scholar]
- Andersen R.; Jorgensen F. S.; Olsen L.; Vabeno J.; Thorn K.; Nielsen C. U.; Steffansen B. Pharm. Res. 2006, 23, 483. [DOI] [PubMed] [Google Scholar]
- Cruciani G.; Pastor M.; Guba W. Eur. J. Pharm. Sci 2000, 11, S29. [DOI] [PubMed] [Google Scholar]
- Cruciani G.; Clementi S.; Crivori P.; Carrupt P. A.; Testa B. In Pharmacokinetic Optimization in Drug Research; Testa B., Ed.; Verlag Helvetica Chimica Acta: Zurich, Switzerland, 2001; p 539. [Google Scholar]
- Cruciani G.; Meniconi M.; Carosati E.; Zamora I.; Mannhold R. In Drug Bioavailability: Estimation of Solubility, Permeability, Absorption, and Bioavailability; van de Waterbeemd H., Lennernäs H., Artursson P., Eds.; Wiley−VCH: Weinehim, Germany, 2003; p 406. [Google Scholar]
- Masereeuw R.; Notenboom S.; Smeets P. H. E.; Wouterse A. C.; Russel F. G. M. J. Am. Soc. Nephrol. 2003, 14, 2741. [DOI] [PubMed] [Google Scholar]
- van Aubel R. A. M. H.; Smeets P. H. E.; van den Heuvel J. J. M. W.; Russel F. G. M. Am. J. Physiol. 2005, 288, F327. [DOI] [PubMed] [Google Scholar]
- van de Water F. M.; Masereeuw R.; Russel F. G. M. Drug Metab. Rev. 2005, 37, 443. [DOI] [PubMed] [Google Scholar]
- Goffeau A.; de Hertogh B.; Baret P. V. In Encyclopedia of Biological Chemistry; Lennarz W. J., Lane M. D., Eds.; Elsevier: Oxford, U.K., 2004; p 1. [Google Scholar]
- Davidson A. L.; Chen J. Annu. Rev. Biochem. 2004, 73, 241. [DOI] [PubMed] [Google Scholar]
- Jones P. M.; George A. M. Int. J. Parasitol. 2005, 35, 555. [DOI] [PubMed] [Google Scholar]
- Gerlach J. H.; Endicott J. A.; Juranka P. F.; Henderson G.; Sarangi F.; Deuchars K. L.; Ling V. Nature 1986, 324, 485. [DOI] [PubMed] [Google Scholar]
- Bradley G.; Juranka P. F.; Ling V. BBA Rev. Cancer 1988, 948, 87. [DOI] [PubMed] [Google Scholar]
- Juranka P. F.; Zastawny R. L.; Ling V. FASEB J. 1989, 3, 2583. [DOI] [PubMed] [Google Scholar]
- Ambudkar S. V.; Lelong I. H.; Zhang J.; Cardarelli C. O.; Gottesman M. M.; Pastan I. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breier A.; Barancik M.; Sulova Z.; Uhrik B. Curr. Cancer Drug Targets 2005, 5, 457. [DOI] [PubMed] [Google Scholar]
- Seelig A.; Gatlik-Landwojtowicz E. Mini-Rev. Med. Chem. 2005, 5, 135. [DOI] [PubMed] [Google Scholar]
- Rajagopal A.; Simon S. M. Mol. Biol. Cell 2003, 14, 3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human ABC Transporters; Wageningen University; http://nutrigene.4t.com/humanabc.htm, February 2006. [Google Scholar]
- Schwab M.; Eichelbaum M.; Fromm M. F. Annu. Rev. Pharmacol. 2003, 43, 285. [DOI] [PubMed] [Google Scholar]
- Schinkel A. H.; Jonker J. W. Adv. Drug Delivery Rev. 2003, 55, 3. [DOI] [PubMed] [Google Scholar]
- Higgins C. F.; Linton K. J. Nat. Struct. Mol. Biol. 2004, 11, 918. [DOI] [PubMed] [Google Scholar]
- Jones P. M.; George A. M. Cell. Mol. Life Sci. 2004, 61, 682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locher K. P.; Borths E. FEBS Lett. 2004, 564, 264. [DOI] [PubMed] [Google Scholar]
- Walker J. E.; Saraste M.; Runswick M. J.; Gay N. J. EMBO J. 1982, 1, 945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locher K. P.; Lee A. T.; Rees D. C. Science 2002, 296, 1091. [DOI] [PubMed] [Google Scholar]
- Locher K. P. Curr. Opin. Struct. Biol. 2004, 14, 426. [DOI] [PubMed] [Google Scholar]
- Rosenberg M. F.; Callaghan R.; Ford R. C.; Higgins C. F. J. Biol. Chem. 1997, 272, 10685. [DOI] [PubMed] [Google Scholar]
- Rosenberg M. F.; Kamis A. B.; Callaghan R.; Higgins C. F.; Ford R. C. J. Biol. Chem. 2003, 278, 8924. [DOI] [PubMed] [Google Scholar]
- Rosenberg M. F.; Callaghan R.; Modok S.; Higgins C. F.; Ford R. C. J. Biol. Chem. 2005, 280, 2857. [DOI] [PubMed] [Google Scholar]
- Rosenberg M. F.; Mao Q.; Holzenburg A.; Ford R. C.; Deeley R. G.; Cole S. P. J. Biol. Chem. 2001, 276, 16076. [DOI] [PubMed] [Google Scholar]
- Karpowich N.; Martsinkevich O.; Millen L.; Yuan Y. R.; Dai P. L.; MacVey K.; Thomas P. J.; Hunt J. F. Structure 2001, 9, 571. [DOI] [PubMed] [Google Scholar]
- Gaudet R.; Wiley D. C. EMBO J. 2001, 20, 4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson R. J. P.; Locher K. P. Nature 2006, 443, 180. [DOI] [PubMed] [Google Scholar]
- Dawson R. J. P.; Locher K. P. FEBS Lett. 2007, 581, 935. [DOI] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. Nucleic Acids Res. 2000, 28, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenstein K.; Frei D. C.; Locher K. P. Nature 2007, 446, 213. [DOI] [PubMed] [Google Scholar]
- Pinkett H. W.; Lee A. T.; Lum P.; Locher K. P.; Rees D. C. Science 2007, 315, 373. [DOI] [PubMed] [Google Scholar]
- Chen Y. J.; Pornillos O.; Lieu S.; Ma C.; Chen A. P.; Chang G. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 18999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang G.; Roth C. B. Science 2001, 293, 1793. [DOI] [PubMed] [Google Scholar]
- Reyes C. L.; Chang G. Science 2005, 308, 1028. [DOI] [PubMed] [Google Scholar]
- Chang G. J. Mol. Biol. 2003, 330, 419. [DOI] [PubMed] [Google Scholar]
- Ma C.; Chang G. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 2852. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Pornillos O.; Chen Y. J.; Chen A. P.; Chang G. Science 2005, 310, 1950. [DOI] [PubMed] [Google Scholar]
- Jones P. M.; George A. M. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 12639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Does C.; Tampe R. Biol. Chem. 2004, 385, 927. [DOI] [PubMed] [Google Scholar]
- Callaghan R.; Ford R. C.; Kerr I. D. FEBS Lett. 2006, 580, 1056. [DOI] [PubMed] [Google Scholar]
- Juliano R. L.; Ling V. Biochim. Biophys. Acta 1976, 455, 152. [DOI] [PubMed] [Google Scholar]
- Lindell M.; Karlsson M. O.; Lennernäs H.; Pahlman L.; Lang M. A. Eur. J. Clin. Invest. 2003, 33, 493. [DOI] [PubMed] [Google Scholar]
- Lindell M.; Lang M.; Lennernäs H. Eur. J. Drug Metab. Pharmacokin. 2003, 28, 41. [DOI] [PubMed] [Google Scholar]
- Berggren S.; Gall C.; Wollnitz N.; Ekelund M.; Karlbom U.; Hoogstraate J.; Schrenk D.; Lennernäs H. Mol. Pharm. 2007, 4, 252. [DOI] [PubMed] [Google Scholar]
- Israeli D.; Ziaei S.; Gonin P.; Garcia L. J. Theor. Biol. 2004, 232, 41. [DOI] [PubMed] [Google Scholar]
- Larsen A. K.; Escargueil A. E.; Skladanowski A. Pharmacol. Therap. 2000, 85, 217. [DOI] [PubMed] [Google Scholar]
- Romsicki Y.; Sharom F. J. Biochemistry 1999, 38, 6887. [DOI] [PubMed] [Google Scholar]
- Ferte J. Eur. J. Biochem. 2000, 267, 277. [DOI] [PubMed] [Google Scholar]
- Luker G. D.; Pica C. M.; Kumar A. S.; Covey D. F.; Piwnica-Worms D. Biochemistry 2000, 39, 7651. [DOI] [PubMed] [Google Scholar]
- Shapiro A. B.; Ling V. Eur. J. Biochem. 1997, 250, 122. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Pant A. C.; Simon S. M. Cancer Res. 2001, 61, 7763. [PubMed] [Google Scholar]
- Lugo M. R.; Sharom F. J. Biochemistry 2005, 44, 643. [DOI] [PubMed] [Google Scholar]
- Bentz J.; Tran T. T.; Polli J. W.; Ayrton A.; Ellens H. Pharm. Res. 2005, 22, 1667. [DOI] [PubMed] [Google Scholar]
- Shapiro A. B.; Ling V. Eur. J. Biochem. 1997, 250, 130. [DOI] [PubMed] [Google Scholar]
- Pallis M.; Turzanski J.; Grundy M.; Seedhouse C.; Russell N. Br. J. Haematol. 2003, 120, 1009. [DOI] [PubMed] [Google Scholar]
- Pallis M.; Turzanski J.; Higashi Y.; Russell N. Leuk. Lymphoma 2002, 43, 1221. [DOI] [PubMed] [Google Scholar]
- Al Hajj M.; Wicha M. S.; Benito-Hernandez A.; Morrison S. J.; Clarke M. F. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polgar O.; Bates S. E. Biochem. Soc. Trans. 2005, 33, 241. [DOI] [PubMed] [Google Scholar]
- Pajeva I. K.; Wiese M. J. Med. Chem. 2002, 45, 5671. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Erickson J. A. Drug Metab. Dispos. 2002, 30, 96. [DOI] [PubMed] [Google Scholar]
- Wacher V. J.; Wu C. Y.; Benet L. Z. Mol. Carcinog. 1995, 13, 129. [DOI] [PubMed] [Google Scholar]
- Watkins P. B. Adv. Drug Delivery Rev. 1997, 27, 161. [DOI] [PubMed] [Google Scholar]
- van de Waterbeemd H. Eur. J. Pharm. Sci. 2000, 12, 1. [DOI] [PubMed] [Google Scholar]
- Konig J.; Nies A. T.; Cui Y.; Leier I.; Keppler D. BBA−Biomembranes 1999, 1461, 377. [DOI] [PubMed] [Google Scholar]
- Wu C. P.; Calcagno A. M.; Hladky S. B.; Ambudkar S. V.; Barrand M. A. FEBS J. 2005, 272, 4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann U.; Kroemer H. K. Drug Metab. Rev. 2004, 36, 669. [DOI] [PubMed] [Google Scholar]
- Rosenberg M. F.; Mao Q.; Holzenburg A.; Ford R. C.; Deeley R. G.; Cole S. P. J. Biol. Chem. 2001, 276, 16076. [DOI] [PubMed] [Google Scholar]
- Ramaen O.; Leulliot N.; Sizun C.; Ulryck N.; Pamlard O.; Lallemand J. Y.; van Tilbeurgh H.; Jacquet E. J. Mol. Biol. 2006, 359, 940. [DOI] [PubMed] [Google Scholar]
- Ejendal K. F. K.; Hrycyna C. A. Curr. Protein Pept. Sci. 2002, 3, 503. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy P.; Schuetz J. D. BioMetals 2005, 18, 349. [DOI] [PubMed] [Google Scholar]
- Maliepaard M.; Scheffer G. L.; Faneyte I. F.; van Gastelen M. A.; Pijnenborg A. C. L. M.; Schinkel A. H.; van de Vijver M. J.; Scheper R. J.; Schellens J. H. M. Cancer Res. 2001, 61, 3458. [PubMed] [Google Scholar]
- Wiese M.; Pajeva I. K. In Virtual ADMET Assessment in Target Selection and Maturation; Testa B., Turski L., Eds.; IOS Press: Amsterdam, The Netherlands, 2006; p. 187. [Google Scholar]
- Ecker G. F.; Chiba P. In Computational Toxicology; Ekins S., Ed.; Wiley: Hoboken, NJ, 2007; p 295. [Google Scholar]
- Litman T.; Zeuthen T.; Skovsgaard T.; Stein W. D. BBA−Mol. Basis Dis. 1997, 1361, 159. [DOI] [PubMed] [Google Scholar]
- Ueda K.; Taguchi Y.; Morishima M. Semin. Cancer Biol. 1997, 8, 151. [DOI] [PubMed] [Google Scholar]
- Wiese M.; Pajeva I. K. Curr. Med. Chem. 2001, 8, 685. [DOI] [PubMed] [Google Scholar]
- Stouch T. R.; Gudmundsson O. Adv. Drug Delivery Rev. 2002, 54, 315. [DOI] [PubMed] [Google Scholar]
- Didziapetris R.; Japertas P.; Avdeef A.; Petrauskas A. J. Drug Target. 2003, 11, 391. [DOI] [PubMed] [Google Scholar]
- Seelig A. Int. J. Clin. Pharmacol. Therap. 1998, 36, 50. [PubMed] [Google Scholar]
- Seelig A. Eur. J. Biochem. 1998, 251, 252. [DOI] [PubMed] [Google Scholar]
- Pajeva I. K.; Wiese M. Quant. Struct. Act. Relat. 2001, 20, 130. [Google Scholar]
- Cianchetta G.; Singleton R. W.; Zhang M.; Wildgoose M.; Giesing D.; Fravolini A.; Cruciani G.; Vaz R. J. J. Med. Chem. 2005, 48, 2927. [DOI] [PubMed] [Google Scholar]
- Pajeva I. K.; Wiese M. In Molecular Modeling and Prediction of Bioactivity; Gundertofte K., Jorgensen F. S., Eds.; Kluwer: New York, 2000; p 414. [Google Scholar]
- Ekins S.; Kim R. B.; Leake B. F.; Dantzig A. H.; Schuetz E. G.; Lan L. B.; Yasuda K.; Shepard R. L.; Winter M. A.; Schuetz J. D.; Wikel J. H.; Wrighton S. A. Mol. Pharmacol. 2002, 61, 964. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Kim R. B.; Leake B. F.; Dantzig A. H.; Schuetz E. G.; Lan L. B.; Yasuda K.; Shepard R. L.; Winter M. A.; Schuetz J. D.; Wikel J. H.; Wrighton S. A. Mol. Pharmacol. 2002, 61, 974. [DOI] [PubMed] [Google Scholar]
- Fleischer R.; Wiese M. J. Med. Chem. 2003, 46, 4988. [DOI] [PubMed] [Google Scholar]
- Globisch C.; Pajeva I. K.; Wiese M. Bioorg. Med. Chem. 2006, 14, 1588. [DOI] [PubMed] [Google Scholar]
- Müller H.; Pajeva I. K.; Globisch C.; Wiese M. Bioorg. Med. Chem. 2008, 16, 2448. [DOI] [PubMed] [Google Scholar]
- Pajeva I. K.; Globisch C.; Wiese M. J. Med. Chem. 2004, 47, 2523. [DOI] [PubMed] [Google Scholar]
- Seigneuret M.; Garnier-Suillerot A. J. Biol. Chem. 2003, 278, 30115. [DOI] [PubMed] [Google Scholar]
- Siarheyeva A.; Lopez J. J.; Glaubitz C. Biochemistry 2006, 45, 6203. [DOI] [PubMed] [Google Scholar]
- Leslie E. M.; Bowers R. J.; Deeley R. G.; Cole S. P. C. J. Pharmacol. Exp. Therap. 2003, 304, 643. [DOI] [PubMed] [Google Scholar]
- Boumendjel A.; Baubichon-Cortay H.; Trompier D.; Perrotton T.; di Pietro A. Med. Res. Rev. 2005, 25, 453. [DOI] [PubMed] [Google Scholar]
- Lania-Pietrzak B.; Michalak K.; Hendrich A. B.; Mosiadz D.; Grynkiewicz G.; Motohashi N.; Shirataki Y. Life Sci. 2005, 77, 1879. [DOI] [PubMed] [Google Scholar]
- Norman B. H.; Lander P. A.; Gruber J. M.; Kroin J. S.; Cohen J. D.; Jungheim L. N.; Starling J. J.; Law K. L.; Self T. D.; Tabas L. B.; Williams D. C.; Paul D. C.; Dantzig A. H. Bioorg. Med. Chem. Lett. 2005, 15, 5526. [DOI] [PubMed] [Google Scholar]
- Bobrowska-Hagerstrand M.; Lillas M.; Mrowczynska L.; Wrobel A.; Shirataki Y.; Motohashi N.; Hagerstrand H. Anticancer Res. 2006, 26, 2081. [PubMed] [Google Scholar]
- Katayama K.; Masuyama K.; Yoshioka S.; Hasegawa H.; Mitsuhashi J.; Sugimoto Y. Cancer Chemother. Pharmacol. 2007, 60, 789. [DOI] [PubMed] [Google Scholar]
- Lai Y.; Xing L.; Poda G. I.; Hu Y. Drug Metab. Dispos. 2007, 35, 937. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Yang X.; Coburn R. A.; Morris M. E. Biochem. Pharmacol. 2005, 70, 627. [DOI] [PubMed] [Google Scholar]
- van Zanden J. J.; Wortelboer H. M.; Bijlsma S.; Punt A.; Usta M.; van Bladeren P. J.; Rietjens I. M. C. M.; Cnubben N. H. P. Biochem. Pharmacol. 2005, 69, 699. [DOI] [PubMed] [Google Scholar]
- Lather V.; Madan A. K. Bioorg. Med. Chem. Lett. 2005, 15, 4967. [DOI] [PubMed] [Google Scholar]
- Seelig A.; Li Blatter X.; Wohnsland F. Int. J. Clin. Pharmacol. Therap. 2000, 38, 111. [DOI] [PubMed] [Google Scholar]
- Ng C.; Xiao Y. D.; Lum B. L.; Han Y. H. Eur. J. Pharm. Sci. 2005, 26, 405. [DOI] [PubMed] [Google Scholar]
- Hirono S.; Nakagome I.; Imai R.; Maeda K.; Kusuhara H.; Sugiyama Y. Pharm. Res. 2005, 22, 260. [DOI] [PubMed] [Google Scholar]
- Hobza P.; Zahradnik R.. Weak Intermolecular Interactions in Chemistry and Biology; Academia: Prague, Czech Republic, 1980. [Google Scholar]
- Eigen M. In Nobel Symposium on Fast Reactions and Primary Processes in Chemical Kinetics; Almquist and Wiksel: Stockholm, Sweden, 1968; p 333. [Google Scholar]
- Peters T.All about Albumin: Biochemistry, Genetics, and Medical Applications; Academic Press: San Diego, CA, 1996. [Google Scholar]
- Davis B. D. Science 1942, 95, 78. [DOI] [PubMed] [Google Scholar]
- Goldstein A. J. Pharmacol. Exp. Ther. 1949, 95 Pt. 2, 102. [PubMed] [Google Scholar]
- Ermondi G.; Lorenti M.; Caron G. J. Med. Chem. 2004, 47, 3949. [DOI] [PubMed] [Google Scholar]
- Liu J.; Tian J.; He W.; Xie J.; Hu Z.; Chen X. J. Pharm. Biomed. Anal. 2004, 35, 671. [DOI] [PubMed] [Google Scholar]
- Kaliszan R.; Noctor T. A. G.; Wainer I. W. Chromatographia 1992, 33, 546. [Google Scholar]
- Kaliszan R.; Noctor T. A.; Wainer I. W. Mol. Pharmacol. 1992, 42, 512. [PubMed] [Google Scholar]
- Noctor T. A. G.; Wainer I. W. J. Chromatogr. 1992, 577, 305. [DOI] [PubMed] [Google Scholar]
- Noctor T. A.; Diaz-Perez M. J.; Wainer I. W. J. Pharm. Sci. 1993, 82, 675. [DOI] [PubMed] [Google Scholar]
- Russeva V. N.; Zhivkova Z. D. Int. J. Pharm. 1998, 168, 23. [DOI] [PubMed] [Google Scholar]
- Sengupta A.; Hage D. S. Anal. Chem. 1999, 71, 3821. [DOI] [PubMed] [Google Scholar]
- Hage D. S.; Austin J. J. Chromatogr. Biomed. 2000, 739, 39. [DOI] [PubMed] [Google Scholar]
- Colmenarejo G.; Alvarez-Pedraglio A.; Lavandera J. L. J. Med. Chem. 2001, 44, 4370. [DOI] [PubMed] [Google Scholar]
- Hage D. S. J. Chromatogr. B 2002, 768, 3. [DOI] [PubMed] [Google Scholar]
- Talbert A. M.; Tranter G. E.; Holmes E.; Francis P. L. Anal. Chem. 2002, 74, 446. [DOI] [PubMed] [Google Scholar]
- Valko K.; Nunhuck S.; Bevan C.; Abraham M. H.; Reynolds D. P. J. Pharm. Sci. 2003, 92, 2236. [DOI] [PubMed] [Google Scholar]
- Liu R.; Meng Q.; Xi J.; Yang J.; Ha C. E.; Bhagavan N. V.; Eckenhoff R. G. Biochem. J. 2004, 380, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertucci C.; Bartolini M.; Gotti R.; Andrisano V. J. Chromatogr. B 2003, 797, 111. [DOI] [PubMed] [Google Scholar]
- Rich R. L.; Day Y. S. N.; Morton T. A.; Myszka D. G. Anal. Biochem. 2001, 296, 197. [DOI] [PubMed] [Google Scholar]
- Day Y. S. N.; Myszka D. G. J. Pharm. Sci. 2003, 92, 333. [DOI] [PubMed] [Google Scholar]
- Kragh-Hansen U. Dan. Med. Bull. 1990, 37, 57. [PubMed] [Google Scholar]
- Lucas L. H.; Price K. E.; Larive C. K. J. Am. Chem. Soc. 2004, 126, 14258. [DOI] [PubMed] [Google Scholar]
- Trynda-Lemiesz L. Bioorg. Med. Chem. 2004, 12, 3269. [DOI] [PubMed] [Google Scholar]
- Lazaro E.; Lowe P. J.; Briand X.; Faller B. J. Med. Chem. 2008, 51, 2009. [DOI] [PubMed] [Google Scholar]
- Dockal M.; Carter D. C.; Ruker F. J. Biol. Chem. 2000, 275, 3042. [DOI] [PubMed] [Google Scholar]
- Bhattacharya A. A.; Grune T.; Curry S. J. Mol. Biol. 2000, 303, 721. [DOI] [PubMed] [Google Scholar]
- Dockal M.; Carter D. C.; Ruker F. J. Biol. Chem. 1999, 274, 29303. [DOI] [PubMed] [Google Scholar]
- Hajduk P. J.; Mendoza R.; Petros A. M.; Huth J. R.; Bures M.; Fesik S. W.; Martin Y. C. J. Comput. Aid. Mol. Des. 2003, 17, 93. [DOI] [PubMed] [Google Scholar]
- Epps D. E.; Raub T. J.; Kezdy F. J. Anal. Biochem. 1995, 227, 342. [DOI] [PubMed] [Google Scholar]
- Carter D. C.; He X. M.; Munson S. H.; Twigg P. D.; Gernert K. M.; Broom M. B.; Miller T. Y. Science 1989, 244, 1195. [DOI] [PubMed] [Google Scholar]
- Carter D. C.; He X. M. Science 1990, 249, 302. [DOI] [PubMed] [Google Scholar]
- He X. M.; Carter D. C. Nature 1992, 358, 209. [DOI] [PubMed] [Google Scholar]
- Carter D. C.; Chang B.; Ho J. X.; Keeling K.; Krishnasami Z. Eur. J. Biochem. 1994, 226, 1049. [DOI] [PubMed] [Google Scholar]
- Carter D. C.; Ho J. X. Adv. Protein Chem. 1994, 45, 153. [DOI] [PubMed] [Google Scholar]
- Curry S.; Mandelkow H.; Brick P.; Franks N. Nat. Struct. Biol. 1998, 5, 827. [DOI] [PubMed] [Google Scholar]
- Sugio S.; Kashima A.; Mochizuki S.; Noda M.; Kobayashi K. Protein Eng. 1999, 12, 439. [DOI] [PubMed] [Google Scholar]
- Bhattacharya A. A.; Curry S.; Franks N. P. J. Biol. Chem. 2000, 275, 38731. [DOI] [PubMed] [Google Scholar]
- Petitpas I.; Grüne T.; Bhattacharya A. A.; Curry S. J. Mol. Biol. 2001, 314, 955. [DOI] [PubMed] [Google Scholar]
- Petitpas I.; Bhattacharya A. A.; Twine S.; East M.; Curry S. J. Biol. Chem. 2001, 276, 22804. [DOI] [PubMed] [Google Scholar]
- Ghuman J.; Zunszain P. A.; Petitpas I.; Bhattacharya A. A.; Otagiri M.; Curry S. J. Mol. Biol. 2005, 353, 38. [DOI] [PubMed] [Google Scholar]
- Sybyl Software; Tripos: St. Louis,2008. [Google Scholar]
- Matsushita Y.; Gouda H.; Tsujishita H.; Hirono S. J. Pharm. Sci. 1998, 87, 379. [DOI] [PubMed] [Google Scholar]
- Diaz N.; Suarez D.; Sordo T. L.; Merz K. M. Jr J. Med. Chem 2001, 44, 250. [DOI] [PubMed] [Google Scholar]
- de Silva S.; de Silva R. M.; de Silva N. K. M. J. Mol. Struct. THEOCHEM 2004, 711, 73. [Google Scholar]
- Wanwimolruk S.; Birkett D. J.; Brooks P. M. Mol. Pharmacol. 1983, 24, 458. [PubMed] [Google Scholar]
- Hinderling P. H.; Schmidlin O.; Seydel J. K. J. Pharmacokinet. Biopharm. 1984, 12, 263. [DOI] [PubMed] [Google Scholar]
- Hersey A.; Hyde R. M.; Livingstone D. J.; Rahr E. J. Pharm. Sci. 1991, 80, 333. [DOI] [PubMed] [Google Scholar]
- Saiakhov R. D.; Stefan L. R.; Klopman G. Persp. Drug Discovery Des. 2000, 19, 133. [Google Scholar]
- Kratochwil N. A.; Huber W.; Müller F.; Kansy M.; Gerber P. R. Biochem. Pharmacol. 2002, 64, 1355. [DOI] [PubMed] [Google Scholar]
- Kratochwil N. A.; Huber W.; Müller F.; Kansy M.; Gerber P. R. Curr. Opin. Drug Discovery 2004, 7, 507. [PubMed] [Google Scholar]
- Gobburu J. V. S.; Shelver W. H. J. Pharm. Sci. 1995, 84, 862. [DOI] [PubMed] [Google Scholar]
- Colmenarejo G. Med. Res. Rev. 2003, 23, 275. [DOI] [PubMed] [Google Scholar]
- Turner J. V.; Maddalena D. J.; Cutler D. J. Int. J. Pharm. 2004, 270, 209. [DOI] [PubMed] [Google Scholar]
- van der Graaf P. H.; Nilsson J.; Van Schaick E. A.; Danhof M. J. Pharm. Sci. 1999, 88, 306. [DOI] [PubMed] [Google Scholar]
- Aureli L.; Cruciani G.; Cesta M. C.; Anacardio R.; De Simone L.; Moriconi A. J. Med. Chem. 2005, 48, 2469. [DOI] [PubMed] [Google Scholar]
- Kleinman H. K.; McGarvey M. L.; Hassell J. R.; Star V. L.; Cannon F. B.; Laurie G. W.; Martin G. R. Biochemistry 1986, 25, 312. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Lukacova V.; Reindl K.; Balaz S. J. Biochem. Biophys. Meth. 2006, 67, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprea T. I. In Computational Medicinal Chemistry for Drug Discovery; Bultinck P., de Winter H., Langenäker W., Tollenaere J. P., Eds.; Marcel Dekker: New York, 2004; p 571. [Google Scholar]
- Zhang Y.; Lukacova V.; Bartus V.; Balaz S. Chem. Res. Toxicol. 2006, 20, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otterbein L. R.; Graceffa P.; Dominguez R. Science 2001, 293, 708. [DOI] [PubMed] [Google Scholar]
- Khandelwal A.; Lukacova V.; Kroll D. M.; Raha S.; Comez D.; Balaz S. J. Phys. Chem. A 2005, 109, 6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansch C. Drug Metab. Rev. 1973, 5, 1. [Google Scholar]
- Austel V.; Kutter E. In Quantitative Structure−Activity Relationships of Drugs; Topliss J. G., Ed.; Academic Press: New York, 1983; p 437. [Google Scholar]
- Toon S.; Rowland M. J. Pharmacol. Exp. Ther. 1983, 225, 752. [PubMed] [Google Scholar]
- Mager D. E.; Jusko W. J. J. Pharm. Sci. 2002, 91, 2441. [DOI] [PubMed] [Google Scholar]
- Jacoby W. B.Enzymatic Basis of Detoxification; Academic Press: New York, 1980. [Google Scholar]
- Thummel K. E.; Kunze K. L.; Shen D. D. Adv. Drug Delivery Rev. 1997, 27, 99. [DOI] [PubMed] [Google Scholar]
- Rendic S. Drug Metab. Rev. 2002, 34, 83. [DOI] [PubMed] [Google Scholar]
- Poulos T. L. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 13121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie B.; Axelrod J.; Cooper J. R.; Gaudette L.; LaDu B. N.; Mitoma C.; Udenfriend S. Science 1955, 121, 603. [DOI] [PubMed] [Google Scholar]
- Omura T.; Sato R. J. Biol. Chem. 1964, 239, 2370. [PubMed] [Google Scholar]
- Omura T.; Sato R. J. Biol. Chem. 1964, 239, 2378. [PubMed] [Google Scholar]
- Guengerich F. P. Mol. Interv. 2003, 3, 194. [DOI] [PubMed] [Google Scholar]
- Ingelman-Sundberg M. Toxicol. Appl. Pharmacol. 2005, 207, S52. [DOI] [PubMed] [Google Scholar]
- Ansede J. H.; Thakker D. R. J. Pharm. Sci. 2004, 93, 239. [DOI] [PubMed] [Google Scholar]
- de Groot M. J.; Vermeulen N. P.; Kramer J. D.; van Acker F. A.; Donne-Op den Kelder G. M. Chem. Res. Toxicol. 1996, 9, 1079. [DOI] [PubMed] [Google Scholar]
- Redinbo M. R. Drug Discovery Today 2004, 9, 431. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Mason J. S.; Overington J. P. Curr. Opin. Struct. Biol. 2006, 16, 127. [DOI] [PubMed] [Google Scholar]
- Ramadoss P.; Marcus C.; Perdew G. H. Expert Opin. Drug Metab. Toxicol. 2005, 1, 9. [DOI] [PubMed] [Google Scholar]
- Chiaro C. R.; Patel R. D.; Marcus C. B.; Perdew G. H. Mol. Pharmacol. 2007, 72, 1369. [DOI] [PubMed] [Google Scholar]
- Anandatheerthavarada H. K.; Addya S.; Dwivedi R. S.; Biswas G.; Mullick J.; Avadhani N. G. Arch. Biochem. Biophys. 1997, 339, 136. [DOI] [PubMed] [Google Scholar]
- Anandatheerthavarada H. K.; Vijayasarathy C.; Bhagwat S. V.; Biswas G.; Mullick J.; Avadhani N. G. J. Biol. Chem. 1999, 274, 6617. [DOI] [PubMed] [Google Scholar]
- Williams P. A.; Cosme J.; Sridhar V.; Johnson E. F.; McRee D. E. Mol. Cell 2000, 5, 121. [DOI] [PubMed] [Google Scholar]
- Wade R. C.; Winn P. J.; Schlichting I.; Sudarko. J. Inorg. Biochem. 2004, 98, 1175. [DOI] [PubMed] [Google Scholar]
- Wade R. C.; Motiejunas D.; Schleinkofer K.; Sudarko; Winn P. J.; Banerjee A.; Kariakin A.; Jung C.. BBA−Proteins Proteom. 2005, 1754, 239. [DOI] [PubMed] [Google Scholar]
- Sinclair J.; Cornell N. W.; Zaitlin L.; Hansch C. Biochem. Pharmacol. 1986, 35, 707. [DOI] [PubMed] [Google Scholar]
- Shank-Retzlaff M. L.; Raner G. M.; Coon M. J.; Sligar S. G. Arch. Biochem. Biophys. 1998, 359, 82. [DOI] [PubMed] [Google Scholar]
- Poulos T. L.; Finzel B. C.; Gunsalus I. C.; Wagner G. C.; Kraut J. J. Biol. Chem. 1985, 260, 16122. [PubMed] [Google Scholar]
- Poulos T. L.; Finzel B. C.; Howard A. J. Biochemistry 1986, 25, 5314. [DOI] [PubMed] [Google Scholar]
- Poulos T. L.; Finzel B. C.; Howard A. J. J. Mol. Biol. 1987, 195, 687. [DOI] [PubMed] [Google Scholar]
- Hasemann C. A.; Ravichandran K. G.; Peterson J. A.; Deisenhofer J. J. Mol. Biol. 1994, 236, 1169. [DOI] [PubMed] [Google Scholar]
- Li H.; Poulos T. L. Acta Crystallogr., Sect. D 1995, 51, 21. [DOI] [PubMed] [Google Scholar]
- Dunn A. R.; Dmochowski I. J.; Bilwes A. M.; Gray H. B.; Crane B. R. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 12420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Christopher A.; Jones J. P.; Bell S. G.; Guo Q.; Xu F.; Rao Z.; Wong L. L. J. Biol. Chem. 2002, 277, 37519. [DOI] [PubMed] [Google Scholar]
- Leys D.; Mowat C. G.; McLean K. J.; Richmond A.; Chapman S. K.; Walkinshaw M. D.; Munro A. W. J. Biol. Chem. 2003, 278, 5141. [DOI] [PubMed] [Google Scholar]
- Nagano S.; Li H.; Shimizu H.; Nishida C.; Ogura H.; de Montellano P. R. O.; Poulos T. L. J. Biol. Chem. 2003, 278, 44886. [DOI] [PubMed] [Google Scholar]
- Nagano S.; Tosha T.; Ishimori K.; Morishima I.; Poulos T. L. J. Biol. Chem. 2004, 279, 42844. [DOI] [PubMed] [Google Scholar]
- Nagano S.; Cupp-Vickery J. R.; Poulos T. L. J. Biol. Chem. 2005, 280, 22102. [DOI] [PubMed] [Google Scholar]
- Nagano S.; Poulos T. L. J. Biol. Chem. 2005, 280, 31659. [DOI] [PubMed] [Google Scholar]
- Kuznetsov V. Y.; Blair E.; Farmer P. J.; Poulos T. L.; Pifferitti A.; Sevrioukova I. F. J. Biol. Chem. 2005, 280, 16135. [DOI] [PubMed] [Google Scholar]
- Scott E. E.; He Y. A.; Wester M. R.; White M. A.; Chin C. C.; Halpert J. R.; Johnson E. F.; Stout C. D. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 13196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott E. E.; White M. A.; He Y. A.; Johnson E. F.; Stout C. D.; Halpert J. R. J. Biol. Chem. 2004, 279, 27294. [DOI] [PubMed] [Google Scholar]
- Williams P. A.; Cosme J.; Vinkovic D. M.; Ward A.; Angove H. C.; Day P. J.; Vonrhein C.; Tickle I. J.; Jhoti H. Science 2004, 305, 683. [DOI] [PubMed] [Google Scholar]
- Yano J. K.; Wester M. R.; Schoch G. A.; Griffin K. J.; Stout C. D.; Johnson E. F. J. Biol. Chem. 2004, 279, 38091. [DOI] [PubMed] [Google Scholar]
- Williams P. A.; Cosme J.; Ward A.; Angove H. C.; Vinkovic D. M.; Jhoti H. Nature 2003, 424, 464. [DOI] [PubMed] [Google Scholar]
- Wester M. R.; Yano J. K.; Schoch G. A.; Yang C.; Griffin K. J.; Stout C. D.; Johnson E. F. J. Biol. Chem. 2004, 279, 35630. [DOI] [PubMed] [Google Scholar]
- Shou M.; Grogan J.; Mancewicz J. A.; Krausz K. W.; Gonzalez F. J.; Gelboin H. V.; Korzekwa K. R. Biochemistry 1994, 33, 6450. [DOI] [PubMed] [Google Scholar]
- Shou M.; Mei Q.; Ettore M. W. J.; Dai R.; Baillie T. A.; Rushmore T. H. Biochem. J. 1999, 340, 845. [PMC free article] [PubMed] [Google Scholar]
- Graham-Lorence S.; Peterson J. A. FASEB J. 1996, 10, 206. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Ring B. J.; Binkley S. N.; Hall S. D.; Wrighton S. A. Int. J. Clin. Pharmacol. Ther. 1998, 36, 642. [PubMed] [Google Scholar]
- Miller V. P.; Stresser D. M.; Blanchard A. P.; Turner S.; Crespi C. L. Ann. New York Acad. Sci. 2000, 919, 26. [DOI] [PubMed] [Google Scholar]
- Korzekwa K. R.; Krishnamachary N.; Shou M.; Ogai A.; Parise R. A.; Rettie A. E.; Gonzalez F. J.; Tracy T. S. Biochemistry 1998, 37, 4137. [DOI] [PubMed] [Google Scholar]
- Korzekwa K. R.; Swinney D. C.; Trager W. F. Biochemistry 1989, 28, 9019. [DOI] [PubMed] [Google Scholar]
- Korzekwa K. R.; Trager W. F.; Gillette J. R. Biochemistry 1989, 28, 9012. [DOI] [PubMed] [Google Scholar]
- Korzekwa K. R.; Trager W. F.; Mancewicz J.; Osawa Y. J. Steroid Biochem. Mol. Biol. 1993, 44, 367. [DOI] [PubMed] [Google Scholar]
- Korzekwa K. R.; Gillette J. R.; Trager W. F. Drug. Metab. Rev. 1995, 27, 45. [DOI] [PubMed] [Google Scholar]
- Jones J. P.; Korzekwa K. R. Meth. Enzymol. 1996, 272, 326. [DOI] [PubMed] [Google Scholar]
- Jones J. P.; Shou M.; Korzekwa K. R. Adv. Exp. Med. Biol. 1996, 387, 355. [DOI] [PubMed] [Google Scholar]
- Hosea N. A.; Miller G. P.; Guengerich F. P. Biochemistry 2000, 39, 5929. [DOI] [PubMed] [Google Scholar]
- He Y. A.; Roussel F.; Halpert J. R. Arch. Biochem. Biophys. 2002, 409, 92. [DOI] [PubMed] [Google Scholar]
- Collins J. R.; Loew G. H. J. Biol. Chem. 1988, 263, 3164. [PubMed] [Google Scholar]
- Gillette J. R.; Darbyshire J. F.; Sugiyama K. Biochemistry 1994, 33, 2927. [DOI] [PubMed] [Google Scholar]
- Zeng Z.; rew N. W.; Arison B. H.; Luffer A. D.; Wang R. W. Xenobiotica 1998, 28, 313. [DOI] [PubMed] [Google Scholar]
- Erman J. E.; Hager L. P.; Sligar S. G. Adv. Inorg. Biochem. 1994, 10, 71. [PubMed] [Google Scholar]
- Harris D.; Loew G.; Waskell L. J. Inorg. Biochem. 2001, 83, 309. [DOI] [PubMed] [Google Scholar]
- Harris D.; Loew G. H.; Komornicki A. J. Phys. Chem. A 1997, 101, 3959. [Google Scholar]
- Harris D. L.; Park J. Y.; Gruenke L.; Waskell L. Proteins 2004, 55, 895. [DOI] [PubMed] [Google Scholar]
- Blobaum A. L.; Harris D. L.; Hollenberg P. F. Biochemistry 2005, 44, 3831. [DOI] [PubMed] [Google Scholar]
- Loew G. H.; Harris D. L. Chem. Rev. 2000, 100, 407. [DOI] [PubMed] [Google Scholar]
- Oprea T. I.; Hummer G.; Garcia A. E. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sligar S. G.; Cinti D. L.; Gibson G. G.; Schenkman J. B. Biochem. Biophys. Res. Commun. 1979, 90, 925. [DOI] [PubMed] [Google Scholar]
- Raag R.; Poulos T. L. Front. Biotransform. 1992, 7, 1. [Google Scholar]
- Winn P. J.; Ludemann S. K.; Gauges R.; Lounnas V.; Wade R. C. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludemann S. K.; Lounnas V.; Wade R. C. J. Mol. Biol. 2000, 303, 813. [DOI] [PubMed] [Google Scholar]
- Ludemann S. K.; Carugo O.; Wade R. C. J. Mol. Model. 1997, 3, 369. [Google Scholar]
- Scott E. E.; He Y. Q.; Halpert J. R. Chem. Res. Toxicol. 2002, 15, 1407. [DOI] [PubMed] [Google Scholar]
- Schleinkofer K.; Sudarko Winn P. J.; Luedemann S. K.; Wade R. C. EMBO Rep. 2005, 6, 584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlichting I.; Berendzen J.; Chu K.; Stock A. M.; Maves S. A.; Benson D. E.; Sweet R. M.; Ringe D.; Petsko G. A.; Sligar S. G. Science 2000, 287, 1615. [DOI] [PubMed] [Google Scholar]
- Brodie B. B.; Cho A. K.; Krishna G.; Reid W. D. Ann. New York Acad. Sci. 1971, 179, 11. [DOI] [PubMed] [Google Scholar]
- Jollow D. J.; Mitchell J. R.; Potter W. Z.; Davis D. C.; Gillette J. R.; Brodie B. B. J. Pharmacol. Exp. Ther. 1973, 187, 195. [PubMed] [Google Scholar]
- Mitchell J. R.; Jollow D. J.; Potter W. Z.; Davis D. C.; Gillette J. R.; Brodie B. B. J. Pharmacol. Exp. Ther. 1973, 187, 185. [PubMed] [Google Scholar]
- Potter W. Z.; Davis D. C.; Mitchell J. R.; Jollow D. J.; Gillette J. R.; Brodie B. B. J. Pharmacol. Exp. Ther. 1973, 187, 203. [PubMed] [Google Scholar]
- Gillette J. R.; Mitchell J. R.; Brodie B. B. Annu. Rev. Pharmacol. 1974, 14, 271. [Google Scholar]
- Evans D. C.; Watt A. P.; Nicoll-Griffith D. A.; Baillie T. A. Chem. Res. Toxicol. 2004, 17, 3. [DOI] [PubMed] [Google Scholar]
- Evans D. C.; Baillie T. A. Curr. Opin. Drug Discovery 2005, 8, 44. [PubMed] [Google Scholar]
- Huth J. R.; Mendoza R.; Olejniczak E. T.; Johnson R. W.; Cothron D. A.; Liu Y.; Lerner C. G.; Chen J.; Hajduk P. J. J. Am. Chem. Soc. 2005, 127, 217. [DOI] [PubMed] [Google Scholar]
- Koerts J.; Rietjens I. M.; Boersma M. G.; Vervoort J. FEBS Lett. 1995, 368, 279. [DOI] [PubMed] [Google Scholar]
- Modi S.; Paine M. J.; Sutcliffe M. J.; Lian L. Y.; Primrose W. U.; Wolf C. R.; Roberts G. C. Biochemistry 1996, 35, 4540. [DOI] [PubMed] [Google Scholar]
- Yao H.; Costache A. D.; Sem D. S. J. Chem. Inf. Comput. Sci. 2004, 44, 1456. [DOI] [PubMed] [Google Scholar]
- de Groot M. J. Curr. Top. Med. Chem. 2004, 4, 1731. [DOI] [PubMed] [Google Scholar]
- Meyer U. A.; Blattler S.; Gnerre C.; Oscarson M.; Peyer A. K.; Rencurel F.; Rifki O.; Roth A. In Pharmacokinetic Profiling in Drug Research; Testa B., Ed., Verlag Helvetica Chimica Acta: Zurich, Switzerland, 2006; p 93. [Google Scholar]
- Sidhu J. S.; Marcus C. B.; Parkinson A.; Omiecinski C. J. J. Biochem. Mol. Toxicol. 1998, 12, 253. [DOI] [PubMed] [Google Scholar]
- Korzekwa K. R.; Grogan J.; DeVito S.; Jones J. P. Adv. Exp. Med. Biol. 1996, 387, 361. [DOI] [PubMed] [Google Scholar]
- Gaudette L.; Brodie B. B. Biochem. Pharmacol. 1959, 2, 89. [DOI] [PubMed] [Google Scholar]
- Hansch C.; Zhang L. Drug Metab. Rev. 1993, 25, 1. [DOI] [PubMed] [Google Scholar]
- Lewis D. F. V.; Dickins M. Toxicology 2002, 170, 45. [DOI] [PubMed] [Google Scholar]
- Lewis D. F. V.; Modi S.; Dickins M. Drug Metab. Rev. 2002, 34, 69. [DOI] [PubMed] [Google Scholar]
- Lewis D. F. V. Inflammopharmacology 2003, 11, 43. [DOI] [PubMed] [Google Scholar]
- Hansch C.; Mekapati S. B.; Kurup A.; Verma R. P. Drug Metab. Rev. 2004, 36, 105. [DOI] [PubMed] [Google Scholar]
- Lewis D. F. V.; Jacobs M. N.; Dickins M. Drug Discovery Today 2004, 9, 530. [DOI] [PubMed] [Google Scholar]
- Ethell B. T.; Ekins S.; Wang J. B.; Burchell B. Drug Metab. Dispos. 2002, 30, 734. [DOI] [PubMed] [Google Scholar]
- Molnar L.; Keseru G. M. Bioorg. Med. Chem. Lett. 2002, 12, 419. [DOI] [PubMed] [Google Scholar]
- Afzelius L.; Masimirembwa C. M.; Karlen A.; Andersson T. B.; Zamora I. J. Comput. Aid. Mol. Des. 2002, 16, 443. [DOI] [PubMed] [Google Scholar]
- Crivori P.; Zamora I.; Speed B.; Orrenius C.; Poggesi I. J. Comput. Aid. Mol. Des. 2004, 18, 155. [DOI] [PubMed] [Google Scholar]
- Shen M.; Xiao Y.; Golbraikh A.; Gombar V. K.; Tropsha A. J. Med. Chem. 2003, 46, 3013. [DOI] [PubMed] [Google Scholar]
- Waller C. L.; Evans M. V.; McKinney J. D. Drug Metab. Dispos. 1996, 24, 203. [PubMed] [Google Scholar]
- Ekins S.; Bravi G.; Wikel J. H.; Wrighton S. A. J. Pharmacol. Exp. Ther. 1999, 291, 424. [PubMed] [Google Scholar]
- Ekins S.; Obach R. S. J. Pharmacol. Exp. Ther. 2000, 295, 463. [PubMed] [Google Scholar]
- Ekins S.; de Groot M. J.; Jones J. P. Drug Metab. Dispos. 2001, 29, 936. [PubMed] [Google Scholar]
- de Groot M. J.; Ekins S. Adv. Drug Delivery Rev. 2002, 54, 367. [DOI] [PubMed] [Google Scholar]
- Snyder R.; Sangar R.; Wang J. B.; Ekins S. Quant. Struct. Act. Relat. 2002, 21, 357. [Google Scholar]
- Haji-Momenian S.; Rieger J. M.; Macdonald T. L.; Brown M. L. Bioorg. Med. Chem. 2003, 11, 5545. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Bravi G.; Binkley S.; Gillespie J. S.; Ring B. J.; Wikel J. H.; Wrighton S. A. Pharmacogenetics 1999, 9, 477. [PubMed] [Google Scholar]
- Ekins S.; Bravi G.; Binkley S.; Gillespie J. S.; Ring B. J.; Wikel J. H.; Wrighton S. A. J. Pharmacol. Exp. Ther. 1999, 290, 429. [PubMed] [Google Scholar]
- Afzelius L.; Zamora I.; Ridderstrom M.; ersson T. B.; Karlen A.; Masimirembwa C. M. Mol. Pharmacol. 2001, 59, 909. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Stresser D. M.; rew Williams J. Trends Pharmacol. Sci. 2003, 24, 161. [DOI] [PubMed] [Google Scholar]
- Liu J.; Pan D.; Tseng Y.; Hopfinger A. J. J. Chem. Inf. Comput. Sci. 2003, 43, 2170. [DOI] [PubMed] [Google Scholar]
- Afzelius L.; Zamora I.; Masimirembwa C. M.; Karlen A.; Andersson T. B.; Mecucci S.; Baroni M.; Cruciani G. J. Med. Chem. 2004, 47, 907. [DOI] [PubMed] [Google Scholar]
- Braatz J. A.; Bass M. B.; Ornstein R. L. J. Comput. Aid. Mol. Des. 1994, 8, 607. [DOI] [PubMed] [Google Scholar]
- de Groot M. J.; Ackland M. J.; Horne V. A.; Alex A. A.; Jones B. C. J. Med. Chem. 1999, 42, 1515. [DOI] [PubMed] [Google Scholar]
- de Groot M. J.; Ackland M. J.; Horne V. A.; Alex A. A.; Jones B. C. J. Med. Chem. 1999, 42, 4062. [DOI] [PubMed] [Google Scholar]
- Vermeulen N. P. E.; de Groot M. J.; Meerman J. H. N.; Venhorst J.. Medicinal Chemistry into the Millennium; Royal Society of Chemistry: London, U.K., 2001; p 374. [Google Scholar]
- de Groot M. J.; Alex A. A.; Jones B. C. J. Med. Chem. 2002, 45, 1983. [DOI] [PubMed] [Google Scholar]
- Kirton S. B.; Kemp C. A.; Tomkinson N. P.; St.-Gallay S.; Sutcliffe M. J. Proteins 2002, 49, 216. [DOI] [PubMed] [Google Scholar]
- Masimirembwa C. M.; Ridderstrom M.; Zamora I.; ersson T. B. Meth. Enzymol. 2002, 357, 133. [DOI] [PubMed] [Google Scholar]
- Lewis D. F. V.; Lake B. G.; Bird M. G.; Loizou G. D.; Dickins M.; Goldfarb P. S. Toxicol. in Vitro 2003, 17, 93. [DOI] [PubMed] [Google Scholar]
- Lewis D. F. V.; Lake B. G.; Dickins M.; Goldfarb P. S. Toxicol. in Vitro 2003, 17, 179. [DOI] [PubMed] [Google Scholar]
- Keizers P. H. J.; de Graaf C.; de Kanter F. J. J.; Oostenbrink C.; Feenstra K. A.; Commandeur J. N. M.; Vermeulen N. P. E. J. Med. Chem. 2005, 48, 6117. [DOI] [PubMed] [Google Scholar]
- de Graaf C.; Pospisil P.; Pos W.; Folkers G.; Vermeulen N. P. E. J. Med. Chem. 2005, 48, 2308. [DOI] [PubMed] [Google Scholar]
- Kirton S. B.; Murray C. W.; Verdonk M. L.; Taylor R. D. Proteins 2005, 58, 836. [DOI] [PubMed] [Google Scholar]
- Harris D. L. J. Inorg. Biochem. 2002, 91, 568. [DOI] [PubMed] [Google Scholar]
- Deng Y.; Roux B. J. Chem. Phys 2008, 128, 115103/1. [DOI] [PubMed] [Google Scholar]
- Smith D. A.; Jones B. C. Biochem. Pharmacol. 1992, 44, 2089. [DOI] [PubMed] [Google Scholar]
- de Groot M. J.; Kirton S. B.; Sutcliffe M. J. Curr. Top. Med. Chem. 2004, 4, 1803. [DOI] [PubMed] [Google Scholar]
- Singh S. B.; Shen L. Q.; Walker M. J.; Sheridan R. P. J. Med. Chem. 2003, 46, 1330. [DOI] [PubMed] [Google Scholar]
- Sheridan R. P.; Korzekwa K. R.; Torres R. A.; Walker M. J. J. Med. Chem. 2007, 50, 3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones B. C.; Hawksworth G.; Horne V. A.; Newlands A.; Morsman J.; Tute M. S.; Smith D. A. Drug Metab. Dispos. 1996, 24, 260. [PubMed] [Google Scholar]
- Darvas F. J. Mol. Graphics 1988, 6, 80. [Google Scholar]
- Greene N.; Judson P. N.; Langowski J. J.; Marchant C. A. SAR QSAR Environ. Res. 1999, 10, 299. [DOI] [PubMed] [Google Scholar]
- Button W. G.; Judson P. N.; Long A.; Vessey J. D. J. Chem. Inf. Comput. Sci. 2003, 43, 1371. [DOI] [PubMed] [Google Scholar]
- Testa B.; Balmat A. L.; Long A. Pure Appl. Chem. 2004, 76, 907. [Google Scholar]
- Testa B.; Balmat A. L.; Long A.; Judson P. Chem. Biodivers. 2005, 2, 872. [DOI] [PubMed] [Google Scholar]
- Klopman G.; Sedykh A. Predict. Toxicol. 2005, 415. [Google Scholar]
- Zamora I.; Afzelius L.; Cruciani G. J. Med. Chem. 2003, 46, 2313. [DOI] [PubMed] [Google Scholar]
- Goodford P. J. J. Med. Chem. 1985, 28, 849. [DOI] [PubMed] [Google Scholar]
- Cruciani G.; Carosati E.; de Boeck B.; Ethirajulu K.; Mackie C.; Howe T.; Vianello R. J. Med. Chem. 2005, 48, 6970. [DOI] [PubMed] [Google Scholar]
- Balaz S.; Sturdik E.; Sitkey V.; Rosenberg M.; Augustin J. Stud. Biophys. 1986, 114, 23. [Google Scholar]
- Bentzien J.; Müller R. P.; Florian J.; Warshel A. J. Phys. Chem. B 1998, 102, 2293. [Google Scholar]
- Bentzien J.; Florian J.; Glennon T. M.; Warshel A.. Combined Quantum Mechanical and Molecular Mechanical Methods; Gao J., Thompson M. A., Eds.; Oxford University Press: New York, 1998; p 16. [Google Scholar]
- Warshel A.; Parson W. W. Q. Rev. Biophys. 2001, 34, 563. [DOI] [PubMed] [Google Scholar]
- Massova I.; Kollman P. A. J. Comput. Chem. 2002, 23, 1559. [DOI] [PubMed] [Google Scholar]
- Truhlar D. G.; Gao J.; Alhambra C.; Garcia-Viloca M.; Corchado J. S. M. L.; Villa J. Acc. Chem. Res. 2002, 35, 341. [DOI] [PubMed] [Google Scholar]
- Carrieri A.; Altomare C.; Barreca M. L.; Contento A.; Carotti A.; Hansch C. Farmaco 1995, 50, 573. [Google Scholar]
- Altomare C.; Carrupt P. A.; Gaillard P.; El-Tayar N.; Testa B.; Carotti A. Chem. Res. Toxicol. 1992, 5, 366. [DOI] [PubMed] [Google Scholar]
- Dearden J. C.; Stott I. P. SAR QSAR Environ. Res. 1995, 4, 189. [DOI] [PubMed] [Google Scholar]
- Tomic S.; Kojic-Prodic B. J. Mol. Graphics Modell. 2002, 21, 241. [DOI] [PubMed] [Google Scholar]
- Obach R. S. Drug Metab. Dispos 1996, 24, 1047. [PubMed] [Google Scholar]
- Obach R. S. Drug Metab. Dispos. 1997, 25, 1359. [PubMed] [Google Scholar]
- Ito K.; Iwatsubo T.; Kanamitsu S.; Nakajima Y.; Sugiyama Y. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 461. [DOI] [PubMed] [Google Scholar]
- Obach R. S. Drug Metab. Dispos. 1999, 27, 1350. [PubMed] [Google Scholar]
- Lave T.; Coassolo P.; Reigner B. Clin. Pharmacokinet. 1999, 36, 211. [DOI] [PubMed] [Google Scholar]
- Austin R. P.; Barton P.; Cockroft S. L.; Wenlock M. C.; Riley R. J. Drug Metab. Dispos. 2002, 30, 1497. [DOI] [PubMed] [Google Scholar]
- Riley R. J.; McGinnity D. F.; Austin R. P. Drug Metab. Dispos. 2005, 33, 1304. [DOI] [PubMed] [Google Scholar]
- Hallifax D.; Houston J. B. Drug Metab. Dispos. 2006, 34, 1829. [DOI] [PubMed] [Google Scholar]
- Lu C.; Li P.; Gallegos R.; Uttamsingh V.; Xia C. Q.; Miwa G. T.; Balani S. K.; Gan L. S. Drug Metab. Dispos. 2006, 34, 1600. [DOI] [PubMed] [Google Scholar]
- Drobnica L.; Kristian P.; Augustin J. In Chemistry of Cyanates and their Thio Derivatives; Patai S., Ed.; John Wiley & Sons: Chichester, UK, 1977; Vol 2, p 1003. [Google Scholar]
- Drobnica L.; Ondrejickova O.; Augustin J. In Progress in Antimicrobial and Anticancer Chemotherapy; University Park Press: Baltimore, MD, 1970; p 129. [Google Scholar]
- Hermens J.; Busser F.; Leeuwanch P.; Musch A. Toxicol. Environ. Chem. 1985, 9, 219. [Google Scholar]
- Deneer J. W.; Sinnige T. L.; Pauly J.; Seinen W.; Hermens J. L. M. Chemosphere 1987, 16, 2365. [Google Scholar]
- Drobnica L.; Augustin J. Collect. Czechoslov. Chem. Commun. 1965, 30, 99. [Google Scholar]
- Drobnica L.; Augustin J. Collect. Czechoslov. Chem. Commun. 1965, 30, 1221. [Google Scholar]
- Drobnica L.; Augustin J. Collect. Czechoslov. Chem. Commun. 1965, 30, 1618. [Google Scholar]
- Balaz S.; Sturdik E.; Drobnica L. Collect. Czechoslov. Chem. Commun. 1982, 47, 1659. [Google Scholar]
- Balaz S.; Vegh D.; Sturdik E.; Augustin J.; Liptaj T.; Kovac J. Collect. Czechoslov. Chem. Commun. 1987, 52, 431. [Google Scholar]
- Weinstein H.; Osman R.; Green J. P.; Topiol S. In Chemical Applications of Atomic and Molecular Electrostatic Potentials; Politzer P., Truhlar D. G. Eds.; Plenum Press: New York,1981; p 309. [Google Scholar]
- Weinstein H.; Rabinowitz J.; Liebman M. N.; Osman R. Environ. Health Persp. 1985, 61, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaar H. J. M.; Rorije E.; Borkent H.; Seinen W.; Hermens J. L. M. Environ. Toxicol. Chem. 1996, 15, 1011. [Google Scholar]
- Soffers A. E.; Ploemen J. H.; Moonen M. J.; Wobbes T.; van Ommen B.; Vervoort J.; van Bladeren P. J.; Rietjens I. M. Chem. Res. Toxicol. 1996, 9, 638. [DOI] [PubMed] [Google Scholar]
- Smiesko M.; Benfenati E. J. Chem. Inf. Comput. Sci. 2004, 44, 976. [DOI] [PubMed] [Google Scholar]
- Balaz S.; Sturdik E.; Augustin J.; Ilavsky D.; Vegh D.; Stibranyi L.; Kovac J. Chem. Biol. Interact. 1987, 63, 195. [DOI] [PubMed] [Google Scholar]
- Balaz S.; Sturdik E.; Rosenberg M.; Augustin J.; Skarka B. J. Theor. Biol. 1988, 131, 115. [DOI] [PubMed] [Google Scholar]
- Eriksson L. C. BBA−Biomembranes 1978, 508, 155. [DOI] [PubMed] [Google Scholar]
- Ellenberger J.; Mohn G. Arch. Toxicol. 1975, 33, 225. [DOI] [PubMed] [Google Scholar]
- Li A. P. Curr. Top. Med. Chem. 2004, 4, 701. [DOI] [PubMed] [Google Scholar]
- Paigen K. Biochim. Biophys. Acta 1956, 19, 297. [DOI] [PubMed] [Google Scholar]
- Claude A. J. Exp. Med. 1946, 84, 51. [PubMed] [Google Scholar]
- Claude A. J. Exp. Med. 1946, 84, 61. [PubMed] [Google Scholar]
- Schneider W. C.; Claude A.; Hogeboom G. J. Biol. Chem. 1948, 172, 451. [PubMed] [Google Scholar]
- Jackson K. L.; Walker E. L.; Pace N. Science 1953, 118, 136. [DOI] [PubMed] [Google Scholar]
- Nelson E. B.; Masters B. S.; Peterson J. A. Anal. Biochem. 1971, 39, 128. [DOI] [PubMed] [Google Scholar]
- Tangen O.; Jonsson J.; Orrenius S. Anal. Biochem. 1973, 54, 597. [DOI] [PubMed] [Google Scholar]
- Vignais P.; Gallagher C. H.; Zabin I. J. Neurochem. 1958, 2, 283. [DOI] [PubMed] [Google Scholar]
- Hokin M. R.; Hokin L. E. J. Biol. Chem. 1959, 234, 1381. [PubMed] [Google Scholar]
- Dobiasova M.; Hahn P. Physiol. Bohemoslov. 1968, 17, 26. [PubMed] [Google Scholar]
- Dallam R. D. J. Histochem. Cytochem. 1957, 5, 178. [DOI] [PubMed] [Google Scholar]
- Hulsmans H. A. M. Biochim. Biophys. Acta 1961, 54, 1. [DOI] [PubMed] [Google Scholar]
- Hanson H.; Kleine R.; Blech W. Z. Physiol. Chem. 1959, 316, 127. [DOI] [PubMed] [Google Scholar]
- Harris E. S.; Bergren W.; Bavetta L. A.; Mehl J. W. Proc. Soc. Exp. Biol. Med. 1952, 81, 593. [DOI] [PubMed] [Google Scholar]
- Morton R. K. Biochem. J. 1954, 57, 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florini J. R. Biochem. Biophys. Res. Commun. 1962, 8, 125. [DOI] [PubMed] [Google Scholar]
- Sampson S. R.; Karler R. J. Cell. Comput. Physiol. 1963, 62, 303. [DOI] [PubMed] [Google Scholar]
- Morato T.; Hayano M.; Dorfman R. I.; Axelrod L. R. Biochem. Biophys. Res. Commun. 1961, 6, 334. [DOI] [PubMed] [Google Scholar]
- Claude A. J. Exp. Med. 1944, 80, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trump B. F.; Duttera S. M.; Byrne W. L.; Arstila A. U. Proc. Natl. Acad. Sci. U.S.A. 1970, 66, 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siekewitz P. J. Biophys. Biochem. Cytol. 1955, 1, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatakrishnan K.; von Moltke L. L.; Obach R. S.; Greenblatt D. J. Curr. Drug Metab. 2003, 4, 423. [DOI] [PubMed] [Google Scholar]
- Singh S. S. Curr. Drug Metab. 2006, 7, 165. [DOI] [PubMed] [Google Scholar]
- van de Kerkhof E. G.; de Graaf I. A. M.; Groothuis G. M. M. Curr. Drug Metab. 2007, 8, 658. [DOI] [PubMed] [Google Scholar]
- Houston J. B.; Carlile D. J. Drug Metab. Rev. 1997, 29, 891. [DOI] [PubMed] [Google Scholar]
- Obach R. S. Curr. Opin. Drug Discovery Dev. 2001, 4, 36. [PubMed] [Google Scholar]
- Moore L.; Pastan I. Ann. New York Acad. Sci. 1978, 307, 177. [DOI] [PubMed] [Google Scholar]
- De Meis L.; Inesi G. Membr. Transp. Calcium 1982, 141. [Google Scholar]
- Friedmann N. K. Trends Biomembr. Bioenerg. 1990, 1, 171. [Google Scholar]
- Burchell A. Biochem. Soc. Trans. 1994, 22, 658. [DOI] [PubMed] [Google Scholar]
- Rodrigues A. D. Pharm. Res. 1997, 14, 1504. [DOI] [PubMed] [Google Scholar]
- Gao F.; Johnson D. L.; Ekins S.; Janiszewski J.; Kelly K. G.; Mayer R. D.; West M. J. Biomol. Screen. 2002, 7, 373. [DOI] [PubMed] [Google Scholar]
- Lee P. H.; Cucurull-Sanchez L.; Lu J.; Du Y. J. J. Comput. Aid. Mol. Des. 2007, 21, 665. [DOI] [PubMed] [Google Scholar]
- Hopfer U.; Nelson K.; Perrotto J.; Isselbacher K. J. J. Biol. Chem. 1973, 248, 25. [PubMed] [Google Scholar]
- Hopfer U. Am. J. Physiol. 1977, 233, E445. [DOI] [PubMed] [Google Scholar]
- Hopfer U. Am. J. Physiol. 1978, 234, F89. [DOI] [PubMed] [Google Scholar]
- Beck J. C.; Sacktor B. J. Biol. Chem. 1975, 250, 8674. [PubMed] [Google Scholar]
- Murer H.; Hopfer U.; Kinne R. Biochem. J. 1976, 154, 597. [PMC free article] [PubMed] [Google Scholar]
- Asai M.; Keino H.; Kashiwamata S. Biochem. Int. 1982, 4, 377. [Google Scholar]
- Ganapathy M. E.; Mahesh V. B.; Devoe L. D.; Leibach F. H.; Ganapathy V. Am. J. Obst. Gynecol. 1985, 153, 83. [DOI] [PubMed] [Google Scholar]
- Grassl S. M. J. Pharmacol. Exp. Therap. 2001, 298, 695. [PubMed] [Google Scholar]
- Sacktor B.; Rosenbloom I. L.; Liang C. T.; Cheng L. J. Membr. Biol. 1981, 60, 63. [DOI] [PubMed] [Google Scholar]
- Grassl S. M.; Aronson P. S. J. Biol. Chem. 1986, 261, 8778. [PubMed] [Google Scholar]
- Hearn P. R.; Russell R. G.; Farmer J. J. Cell Sci. 1981, 47, 227. [DOI] [PubMed] [Google Scholar]
- Alcorn C. J.; Simpson R. J.; Leahy D.; Peters T. J. Biochem. Pharmacol. 1991, 42, 2259. [DOI] [PubMed] [Google Scholar]
- Schwoch G.; Passow H. Mol. Cell. Biochem. 1973, 2, 197. [DOI] [PubMed] [Google Scholar]
- Jausel-Hüsken S.; Deuticke B. J. Membrane Biol. 1981, 63, 61. [DOI] [PubMed] [Google Scholar]
- Bucki R.; Bachelot-Loza C.; Zachowski A.; Giraud F.; Sulpice J. C. Biochemistry 1998, 37, 15383. [DOI] [PubMed] [Google Scholar]
- Wu C. P.; Klokouzas A.; Hladky S. B.; Ambudkar S. V.; Barrand M. A. Biochem. Pharmacol. 2005, 70, 500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekkers D. W. C.; Comfurius P.; Schroit A. J.; Bevers E. M.; Zwaal R. F. A. Biochemistry 1998, 37, 14833. [DOI] [PubMed] [Google Scholar]
- Yeagle P. L.; Selinsky B. S.; Albert A. D. Biophys. J. 1984, 45, 1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelusky E. C.; Dufourc E. J.; Smith I. C. Biochim. Biophys. Acta 1983, 735, 302. [DOI] [PubMed] [Google Scholar]
- Rodgers W.; Glaser M. Biochemistry 1993, 32, 12591. [DOI] [PubMed] [Google Scholar]
- Pomorski T.; Herrmann A.; Zachowski A.; Devaux P. F.; Müller P. Mol. Membr. Biol. 1994, 11, 39. [DOI] [PubMed] [Google Scholar]
- Korten K.; Miller K. W. Can. J. Physiol. Pharmacol. 1979, 57, 325. [DOI] [PubMed] [Google Scholar]
- Tamura A.; Yoshikawa K.; Fujii T.; Ohki K.; Nozawa Y.; Sumida Y. BBA−Biomembranes 1986, 855, 250. [DOI] [PubMed] [Google Scholar]
- Aki H.; Yamamoto M. J. Pharm. Pharmacol. 1990, 42, 637. [DOI] [PubMed] [Google Scholar]
- Scheufler E.; Vogelgesang R.; Wilffert B.; Pegram B. L.; Hunter J. B.; Wermelskirchen D.; Peters T. J. Pharmacol. Exp. Therap. 1990, 252, 333. [PubMed] [Google Scholar]
- Kuroda Y.; Wakita M.; Fujiwara Y.; Nakagawa T. Spec. Publ. R. Soc. Chem. 1991, 94, 171. [Google Scholar]
- Brown M. R.; Sullivan P. G.; Dorenbos K. A.; Modafferi E. A.; Geddes J. W.; Steward O. J. Neurosci. Methods 2004, 137, 299. [DOI] [PubMed] [Google Scholar]
- Amoore J. E.; Bartley W. Biochem. J. 1958, 69, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham J. M. Methods Mol. Biol. (Totowa, NJ, U.S.) 1993, 19, 29. [DOI] [PubMed] [Google Scholar]
- Frezza C.; Cipolat S.; Scorrano L. Nat. Protocols 2007, 2, 287. [DOI] [PubMed] [Google Scholar]
- Norseth J.; Normann P. T.; Flatmark T. Biochim. Biophys. Acta 1982, 719, 569. [DOI] [PubMed] [Google Scholar]
- Gaikwad A. S.; Ramasarma T.; Kurup C. K. R. Mol. Cell. Biochem. 1991, 105, 119. [DOI] [PubMed] [Google Scholar]
- Rasmussen H. N.; ersen A. J.; Rasmussen U. F. Anal. Biochem. 1997, 252, 153. [DOI] [PubMed] [Google Scholar]
- Kristian T.; Hopkins I. B.; McKenna M. C.; Fiskum G. J. Neurosci. Methods 2006, 152, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allemann N.; Schneider A. Mol. Biochem. Parasitol. 2000, 111, 87. [DOI] [PubMed] [Google Scholar]
- Diekert K.; de Kroon A. I. P. M.; Kispal G.; Lill R. Meth. Cell Biol. 2001, 65, 37. [DOI] [PubMed] [Google Scholar]
- Escher B. I.; Snozzi M.; Schwarzenbach R. P. Environ. Sci. Technol. 1996, 30, 3071. [Google Scholar]
- Bakker E. P.; Arents J. C.; Hoebe J. P. M.; Terada H. Biochim. Biophys. Acta 1975, 387, 491. [DOI] [PubMed] [Google Scholar]
- Cunarro J.; Weiner M. W. Biochim. Biophys. Acta 1975, 387, 234. [DOI] [PubMed] [Google Scholar]
- Terada H.; Van Dam K. Biochim. Biophys. Acta 1975, 387, 507. [DOI] [PubMed] [Google Scholar]
- Motais R.; Sola F.; Cousin J. L. Biochim. Biophys. Acta 1978, 510, 201. [DOI] [PubMed] [Google Scholar]
- Kim K. H.; Hansch C. Farmaco. Sci. 1979, 34, 588. [PubMed] [Google Scholar]
- Topaly E. E.; Topaly V. P.; Kotojaru A. F.; Rakul A. D. Biofizika 1982, 27, 535. [PubMed] [Google Scholar]
- Ravanel P. Phytochemistry 1986, 25, 1015. [Google Scholar]
- Miyoshi H.; Nishioka T.; Fujita T. Biochim. Biophys. Acta 1987, 891, 293. [DOI] [PubMed] [Google Scholar]
- Cajina-Quezada M.; Schultz T. W. Aquat. Toxicol. 1990, 17, 239. [Google Scholar]
- Escher B. I.; Snozzi M.; Haberli K.; Schwarzenbach R. P. Environ. Toxicol. Chem. 1997, 16, 405. [Google Scholar]
- Escher B. I.; Hunziker R.; Schwarzenbach R. P. Environ. Sci. Technol. 1999, 33, 560. [DOI] [PubMed] [Google Scholar]
- Spycher S.; Escher B. I.; Gasteiger J. Chem. Res. Toxicol. 2005, 18, 1858. [DOI] [PubMed] [Google Scholar]
- Foka M.; Belehradek J. Jr.; Paoletti J. Biochem. Pharmacol. 1988, 37, 3467. [DOI] [PubMed] [Google Scholar]
- Haj H. T. B.; Salerno M.; Priebe W.; Kozlowski H.; Garnier-Suillerot A. Chem. Biol. Interact. 2003, 145, 349. [DOI] [PubMed] [Google Scholar]
- Overton C. E.Studies of Narcosis; Lipnick R. L., Ed.; Chapman and Hall: London, U.K.,1991. [Google Scholar]
- Robertson T. B. J. Biol. Chem. 1908, 4, 1. [Google Scholar]
- Bethe A. Arch. Ges. Physiol. 1909, 127, 219. [Google Scholar]
- Warburg O. Z. Physiol. Chem. 1909, 66, 305. [Google Scholar]
- Harvey E. N. Science 1914, 39, 947. [DOI] [PubMed] [Google Scholar]
- Crozier W. J. J. Biol. Chem. 1916, 24, 256. [Google Scholar]
- Haas A. R. J. Biol. Chem. 1916, 27, 225. [Google Scholar]
- Osterhout W. J. V. J. Biol. Chem. 1914, 19, 335. [Google Scholar]
- Osterhout W. J. V. J. Biol. Chem. 1914, 19, 493. [Google Scholar]
- Wodehouse R. P. J. Biol. Chem. 1917, 29, 453. [Google Scholar]
- Irwin M. J. Gen. Physiol. 1922, 5, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collander R. Jahrb. Wiss. Bot. 1921, 60, 354. [Google Scholar]
- Collander R.; Barlund H. Soc. Sci. Fenn. Commentat. Biol. 1926, 2, 12. [Google Scholar]
- Chambers R.; Reznikoff P. J. Gen. Physiol. 1926, 8, 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reznikoff P. J. Gen. Physiol. 1926, 10, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor N. W. J. Gen. Physiol. 1928, 11, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milborrow B. V.; Williams D. A. Physiol. Plant. 1968, 21, 902. [Google Scholar]
- Hamilton J. G.; Soley M. H.; Eichhorn K. B. Univ. Calif. Berkeley Publ. Pharmacol. 1940, 1, 339. [Google Scholar]
- MacDonald A. M.; Cobb J. Proc. Soc. Exp. Biol. Med. 1949, 72, 117. [DOI] [PubMed] [Google Scholar]
- Lux R. E.; Scott P. M.; Christian J. E. Science 1950, 112, 205. [DOI] [PubMed] [Google Scholar]
- Harris J. E.; Sloane J. F.; King D. T. Nature 1950, 166, 25. [DOI] [PubMed] [Google Scholar]
- Holt M. W.; Warren S. Proc. Soc. Exp. Biol. Med. 1950, 73, 545. [DOI] [PubMed] [Google Scholar]
- Winteringham F. P. W.; Harrison A.; Hammond J. H. Nature 1950, 165, 149. [DOI] [PubMed] [Google Scholar]
- Boyd G. A.; Levi H. Science 1950, 111, 58. [DOI] [PubMed] [Google Scholar]
- Solon E. G.; Balani S. K.; Lee F. W. Curr. Drug Metab. 2002, 3, 451. [DOI] [PubMed] [Google Scholar]
- Solon E. G. Expert Opin. Drug Discovery 2007, 2, 503. [DOI] [PubMed] [Google Scholar]
- Hammarstroem L.; Appelgren L. E.; Ullberg S. Exp. Cell Res. 1965, 37, 608. [DOI] [PubMed] [Google Scholar]
- Blumcke S.; Backmann R. Naturwissenschaften 1966, 53, 362. [DOI] [PubMed] [Google Scholar]
- Harford C. G.; Hamlin A. Nature 1961, 189, 505. [DOI] [PubMed] [Google Scholar]
- Wolfe D. E.; Potter L. T.; Richardson K. C.; Axelrod J. Science 1962, 138, 440. [DOI] [PubMed] [Google Scholar]
- Paul D.; Groebe A.; Zimmer F. Nature 1970, 227, 488. [DOI] [PubMed] [Google Scholar]
- Brown D. A.; Roth L. J.; Stumpf W. E. Naunyn Schmiedebergs Arch. Exp. Pathol. Pharmakol. 1968, 259, 204. [DOI] [PubMed] [Google Scholar]
- Stumpf W. E. J. Histochem. Cytochem. 1970, 18, 21. [DOI] [PubMed] [Google Scholar]
- Stumpf W. E.; Roth L. J. Nature 1965, 205, 712. [DOI] [PubMed] [Google Scholar]
- Stumpf W. E.; Roth L. J. J. Histochem. Cytochem. 1967, 15, 243. [DOI] [PubMed] [Google Scholar]
- Stone G.; Wood P.; Dixon L.; Keyhan M.; Matin A. Antimicrob. Agents Chemotherap. 2002, 46, 2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shedden K.; Brumer J.; Chang Y. T.; Rosania G. R. J. Chem. Inf. Comput. Sci. 2003, 43, 2068. [DOI] [PubMed] [Google Scholar]
- Rosania G. R.; Lee J. W.; Ding L.; Yoon H. S.; Chang Y. T. J. Am. Chem. Soc. 2003, 125, 1130. [DOI] [PubMed] [Google Scholar]
- Chen V. Y.; Khersonsky S. M.; Shedden K.; Chang Y. T.; Rosania G. R. Mol. Pharm. 2004, 1, 414. [DOI] [PubMed] [Google Scholar]
- Rani S. A.; Pitts B.; Stewart P. S. Antimicrob. Agents Chemotherap. 2005, 49, 728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petty H. R. Proc. SPIE 2005, 5580, 81. [Google Scholar]
- Petty H. R. Biosystems 2006, 83, 217. [DOI] [PubMed] [Google Scholar]
- Kivman G. I.; Kostiuk I. N. Antibiotiki 1967, 12, 471. [PubMed] [Google Scholar]
- Olaisen B.; Oeye I. Eur. J. Pharmacol. 1973, 22, 112. [DOI] [PubMed] [Google Scholar]
- Müller S.; Denet S.; Candiloros H.; Barrois R.; Wiernsperger N.; Donner M.; Drouin P. Eur. J. Pharmacol. 1997, 337, 103. [DOI] [PubMed] [Google Scholar]
- Sheetz M. P.; Singer S. J. Proc. Natl. Acad. Sci. U.S.A. 1974, 71, 4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J. E.; Mohandas N.; Shohet S. B. Am. J. Vet. Res. 1982, 43, 1041. [PubMed] [Google Scholar]
- Malheiros S. V. P.; Brito M. A.; Brites D.; Meirelles N. C. Chem. Biol. Interact. 2000, 126, 79. [DOI] [PubMed] [Google Scholar]
- Ahyayauch H.; Gallego M.; Casis O.; Bennouna M. J. Physiol. Biochem. 2006, 62, 199. [DOI] [PubMed] [Google Scholar]
- Suwalsky M.; Mennickent S.; Norris B.; Villena F.; Sotomayor C. P. Toxicol. in Vitro 2006, 20, 1363. [DOI] [PubMed] [Google Scholar]
- Kitagawa S.; Hiyama F.; Kato M.; Watanabe R. J. Pharm. Pharmacol. 2002, 54, 773. [DOI] [PubMed] [Google Scholar]
- Malheiros S. V. P.; Meirelles N. C.; Volpe P. L. O. Thermochim. Acta 1999, 328, 121. [Google Scholar]
- Ahyayauch H.; Goni F. M.; Bennouna M. Int. J. Pharm. 2004, 279, 51. [DOI] [PubMed] [Google Scholar]
- Hartmann F.; Owen R.; Bissell D. M Am. J. Physiol. 1982, 242, G147. [DOI] [PubMed] [Google Scholar]
- Osiecka I.; Porter P. A.; Borchardt R. T.; Fix J. A.; Gardner C. R. Pharm. Res. 1985, 2, 284. [DOI] [PubMed] [Google Scholar]
- Quaroni A.; Hochman J. Adv. Drug Delivery Rev. 1996, 22, 3. [Google Scholar]
- Leppert P. S.; Fix J. A. J. Pharm. Sci. 1994, 83, 976. [DOI] [PubMed] [Google Scholar]
- Stewart B. H.; Chan O. H.; Jezyk N.; Fleisher D. Adv. Drug Delivery Rev. 1997, 23, 27. [Google Scholar]
- Austin R. P.; Barton P.; Mohmed S.; Riley R. J. Drug Metab. Dispos. 2005, 33, 419. [DOI] [PubMed] [Google Scholar]
- Roberts M. S.; Rowland M. J. Pharm. Sci. 1985, 74, 585. [DOI] [PubMed] [Google Scholar]
- Benet L. Z.; Cummins C. L.; Wu C. Y. Curr. Drug Metab. 2003, 4, 393. [DOI] [PubMed] [Google Scholar]
- Lam J. L.; Benet L. Z. Drug Metab. Dispos. 2004, 32, 1311. [DOI] [PubMed] [Google Scholar]
- Azuma H.; Paulk N.; Ranade A.; Dorrell C.; Al Dhalimy M.; Ellis E.; Strom S.; Kay M. A.; Finegold M.; Grompe M. Nat. Biotechnol. 2007, 25, 903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drobnica L.; Augustin J.; Nemec P. In Quantitative Structure−Activity Relationships; Tichy M., Ed.; Akademiai Kiado: Budapest, Hungary, 1976; Chapter 10, p 65. [Google Scholar]
- Sturdik E.; Balaz S.; Durcova E.; Sturdikova M.; Sulo P.; Antalik M.; Mikes V.; Dadak V. Quant. Struct. Act. Relat. 1988, 7, 221. [Google Scholar]
- Ho N. F. H.; Turi J.; Shipman C. Jr.; Higuchi W. I. J. Theor. Biol. 1972, 34, 451. [DOI] [PubMed] [Google Scholar]
- Ando H. Y.; Ho N. F. H.; Higuchi W. I.; Turi J.; Shipman C. Jr. J. Theor. Biol. 1976, 62, 211. [DOI] [PubMed] [Google Scholar]
- Chambers R.; Kempton R. T. J. Cell. Compar. Physiol. 1933, 3, 131. [Google Scholar]
- Handler J. S.; Perkins F. M.; Johnson J. P. Am. J. Physiol. Renal. Physiol. 1980, 238, F1. [DOI] [PubMed] [Google Scholar]
- Rousset M. Biochimie 1986, 68, 1035. [DOI] [PubMed] [Google Scholar]
- Blais A.; Bissonnette P.; Berteloot A. J. Membr. Biol. 1987, 99, 113. [DOI] [PubMed] [Google Scholar]
- Hughes T. E.; Sasak W. V.; Ordovas J. M.; Forte T. M.; Lamon-Fava S.; Schaefer E. J. J. Biol. Chem. 1987, 262, 3762. [PubMed] [Google Scholar]
- van Breemen R. B.; Li Y. Expert Opin. Drug Metab. Toxicol. 2005, 1, 175. [DOI] [PubMed] [Google Scholar]
- Pinto M.; Robine-Leon S.; Appay M. D.; Kedinger M.; Tradou N.; Dussaulx E.; Lacroix B.; Simon-Assmann P.; Haffen K.; Fogh J.; Zwiebaum A. Biol. Cell 1983, 47, 323. [Google Scholar]
- Grasset E.; Pinto M.; Dussaulx E.; Zweibaum A.; Desjeux J. F. Am. J. Physiol. 1984, 247, C260. [DOI] [PubMed] [Google Scholar]
- Alsenz J.; Haenel E. Pharm. Res. 2003, 20, 1961. [DOI] [PubMed] [Google Scholar]
- Miret S.; Abrahamse L.; de Groene E. M. J. Biomol. Screen. 2004, 9, 598. [DOI] [PubMed] [Google Scholar]
- Vincent M. L.; Russell R. M.; Sasak V. Hum. Nutr. Clin. Nutr. 1985, 39, 355. [PubMed] [Google Scholar]
- Mohrmann I.; Mohrmann M.; Biber J.; Murer H. Am. J. Physiol. 1986, 250, G323. [DOI] [PubMed] [Google Scholar]
- Dix C. J.; Obray H. Y.; Hassan I. F.; Wilson G. Biochem. Soc. Trans. 1987, 15, 439. [Google Scholar]
- Hidalgo I. J.; Raub T. J.; Borchardt R. T. Gastroenterology 1989, 96, 736. [PubMed] [Google Scholar]
- Artursson P. J. Pharm. Sci. 1990, 79, 476. [DOI] [PubMed] [Google Scholar]
- Hilgers A. R.; Conradi R. A.; Burton P. S. Pharm. Res. 1990, 7, 902. [DOI] [PubMed] [Google Scholar]
- Wilson G. Eur. J. Drug Metab. Pharmacokinet. 1990, 15, 159. [DOI] [PubMed] [Google Scholar]
- Luzio J. P.; Jackman M. R.; Ellis J. A. Biochem. Soc. Trans. 1992, 20, 717. [DOI] [PubMed] [Google Scholar]
- Conradi R. A.; Hilgers A. R.; Burton P. S.; Hester J. B. J. Drug Target. 1994, 2, 167. [DOI] [PubMed] [Google Scholar]
- Artursson P.; Palm K.; Luthman K. Adv. Drug Delivery Rev. 1996, 22, 67. [DOI] [PubMed] [Google Scholar]
- Artursson P.; Borchardt R. T. Pharm. Res. 1997, 14, 1655. [DOI] [PubMed] [Google Scholar]
- Barthe L.; Woodley J.; Houin G. Fundam. Clin. Pharmacol. 1999, 13, 154. [DOI] [PubMed] [Google Scholar]
- Wagner D.; Spahn-Langguth H.; Hanafy A.; Koggel A.; Langguth P. Adv. Drug Delivery Rev. 2001, 50, S13. [DOI] [PubMed] [Google Scholar]
- Chen W.; Tang F.; Horie K.; Borchardt R. T. In Cell Culture Models of Biological Barriers; Lehr C. M., Ed.; Taylor & Francis: London, U.K., 2002; p 143. [Google Scholar]
- Polli J. E.; Balakrishnan A.; Seo P. R. In Cellular Drug Delivery; Lu D. R., Oeie S., Eds.; Humana: Totowa, NJ, 2004; p 163. [Google Scholar]
- Sun D.; Yu L. X.; Hussain M. A.; Wall D. A.; Smith R. L.; Amidon G. L. Curr. Opin. Drug Discovery Develop. 2004, 7, 75. [PubMed] [Google Scholar]
- Hidalgo I. J.; Hillgren K. M.; Grass G. M.; Borchardt R. T. Pharm. Res. 1991, 8, 222. [DOI] [PubMed] [Google Scholar]
- Artursson P. Crit. Rev. Therap. Drug Carrier Syst. 1991, 8, 305. [PubMed] [Google Scholar]
- Hillgren K. M.; Kato A.; Borchardt R. T. Med. Res. Rev. 1995, 15, 83. [DOI] [PubMed] [Google Scholar]
- Artursson P.; Palm K.; Luthman K. Adv. Drug Delivery Rev. 1996, 22, 67. [DOI] [PubMed] [Google Scholar]
- Jezyk N.; Rubas W.; Grass G. M. Pharm. Res. 1992, 9, 1580. [DOI] [PubMed] [Google Scholar]
- Lennernäs H. J. Pharm. Pharmacol. 1997, 49, 627. [DOI] [PubMed] [Google Scholar]
- Lennernäs H. J. Pharm. Sci. 1998, 87, 403. [DOI] [PubMed] [Google Scholar]
- Ren S.; Lien E. J. Progr. Drug Res. 2000, 54, 1. [DOI] [PubMed] [Google Scholar]
- Artursson P.; Palm K.; Luthman K. Adv. Drug Delivery Rev. 2001, 46, 27. [DOI] [PubMed] [Google Scholar]
- Haemaelaeinen M. D.; Frostell-Karlsson A. Drug Discovery Today 2004, 1, 397. [DOI] [PubMed] [Google Scholar]
- Artursson P.; Karlsson J. Biochem. Biophys. Res. Commun. 1991, 175, 880. [DOI] [PubMed] [Google Scholar]
- Yazdanian M.; Glynn S. L.; Wright J. L.; Hawi A. Pharm. Res. 1998, 15, 1490. [DOI] [PubMed] [Google Scholar]
- Ingels F. M.; Augustijns P. F. J. Pharm. Sci. 2003, 92, 1545. [DOI] [PubMed] [Google Scholar]
- Markowska M.; Oberle R.; Juzwin S.; Hsu C. P.; Gryszkiewicz M.; Streeter A. J. J. Pharm. Toxicol. Methods 2001, 46, 51. [DOI] [PubMed] [Google Scholar]
- Knipp G. T.; Ho N. F.; Barsuhn C. L.; Borchardt R. T. J. Pharm. Sci. 1997, 86, 1105. [DOI] [PubMed] [Google Scholar]
- Karlsson J.; Ungell A. L.; Grasjo J.; Artursson P. Eur. J. Pharm. Sci. 1999, 9, 47. [DOI] [PubMed] [Google Scholar]
- Adson A.; Raub T. J.; Burton P. S.; Barsuhn C. L.; Hilgers A. R.; Audus K. L.; Ho N. F. J. Pharm. Sci. 1994, 83, 1529. [DOI] [PubMed] [Google Scholar]
- Hu M.; Borchardt R. T. Biochim. Biophys. Acta 1992, 1135, 233. [DOI] [PubMed] [Google Scholar]
- Chen J.; Zhu Y.; Hu M. J. Nutr. 1994, 124, 1907. [DOI] [PubMed] [Google Scholar]
- Ranaldi G.; Seneci P.; Guba W.; Islam K.; Sambuy Y. Antimicrob. Agents Chemotherap. 1996, 40, 652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adson A.; Burton P. S.; Raub T. J.; Barsuhn C. L.; Audus K. L.; Ho N. F. H. J. Pharm. Sci. 1995, 84, 1197. [DOI] [PubMed] [Google Scholar]
- Neuhoff S.; Ungell A. L.; Zamora I.; Artursson P. Pharm. Res. 2003, 20, 1141. [DOI] [PubMed] [Google Scholar]
- Hu M.; Chen J.; Zhu Y.; Dantzig A. H.; Stratford R. E. Jr.; Kuhfeld M. T. Pharm. Res. 1994, 11, 1405. [DOI] [PubMed] [Google Scholar]
- Conradi R. A.; Hilgers A. R.; Ho N. F.; Burton P. S. Pharm. Res. 1991, 8, 1453. [DOI] [PubMed] [Google Scholar]
- Conradi R. A.; Hilgers A. R.; Ho N. F.; Burton P. S. Pharm. Res. 1992, 9, 435. [DOI] [PubMed] [Google Scholar]
- Burton P. S.; Conradi R. A.; Hilgers A. R.; Ho N. F. Biochem. Biophys. Res. Commun. 1993, 190, 760. [DOI] [PubMed] [Google Scholar]
- Conradi R. A.; Wilkinson K. F.; Rush B. D.; Hilgers A. R.; Ruwart M. J.; Burton P. S. Pharm. Res. 1993, 10, 1790. [DOI] [PubMed] [Google Scholar]
- Tamura K.; Lee C. P.; Smith P. L.; Borchardt R. T. Pharm. Res. 1996, 13, 1752. [DOI] [PubMed] [Google Scholar]
- Lang V. B.; Langguth P.; Ottiger C.; Wunderli A. H.; Rognan D.; Rothen R. B.; Perriard J. C.; Lang S.; Biber J.; Merkle H. P. J. Pharm. Sci. 1997, 86, 846. [DOI] [PubMed] [Google Scholar]
- Okumu F. W.; Pauletti G. M.; Vander V. D.; Siahaan T. J.; Borchardt R. T. Pharm. Res. 1997, 14, 169. [DOI] [PubMed] [Google Scholar]
- Pauletti G. M.; Okumu F. W.; Borchardt R. T. Pharm. Res. 1997, 14, 164. [DOI] [PubMed] [Google Scholar]
- Gangwar S.; Jois S. D.; Siahaan T. J.; Vander V. D.; Stella V. J.; Borchardt R. T. Pharm. Res. 1996, 13, 1657. [DOI] [PubMed] [Google Scholar]
- Gudmundsson O. S.; Nimkar K.; Gangwar S.; Siahaan T.; Borchardt R. T. Pharm. Res. 1999, 16, 16. [DOI] [PubMed] [Google Scholar]
- Gudmundsson O. S.; Pauletti G. M.; Wang W.; Shan D.; Zhang H.; Wang B.; Borchardt R. T. Pharm. Res. 1999, 16, 7. [DOI] [PubMed] [Google Scholar]
- Wang B.; Wang W.; Camenisch G. P.; Elmo J.; Zhang H.; Borchardt R. T. Chem. Pharm. Bull. 1999, 47, 90. [DOI] [PubMed] [Google Scholar]
- Sudoh M.; Pauletti G. M.; Yao W.; Moser W.; Yokoyama A.; Pasternak A.; Sprengeler P. A.; Smith A. B.; Hirschmann R.; Borchardt R. T. Pharm. Res. 1998, 15, 719. [DOI] [PubMed] [Google Scholar]
- Kamm W.; Hauptmann J.; Behrens I.; Sturzebecher J.; Dullweber F.; Gohlke H.; Stubbs M.; Klebe G.; Kissel T. Pharm. Res. 2001, 18, 1110. [DOI] [PubMed] [Google Scholar]
- Hu M.; Borchardt R. T. Pharm. Res 1990, 7, 1313. [DOI] [PubMed] [Google Scholar]
- Shima M.; Yohdoh K.; Yamaguchi M.; Kimura Y.; Adachi S.; Matsuno R. Biosci. Biotechnol. Biochem. 1997, 61, 1150. [DOI] [PubMed] [Google Scholar]
- Stewart B. H.; Chung F. Y.; Tait B.; John C.; Chan O. H. Pharm. Res. 1998, 15, 1401. [DOI] [PubMed] [Google Scholar]
- Palm K.; Luthman K.; Ros J.; Grasjo J.; Artursson P. J. Pharmacol. Exp. Therap. 1999, 291, 435. [PubMed] [Google Scholar]
- Lo Y. L. Biochem. Pharmacol. 2000, 60, 1381. [DOI] [PubMed] [Google Scholar]
- Pachot J. I.; Botham R. P.; Haegele K. D.; Hwang K. J. Pharm. Pharm. Sci. 2003, 6, 1. [PubMed] [Google Scholar]
- Gan L. S. L.; Thakker D. R. Adv. Drug Delivery Rev. 1997, 23, 77. [Google Scholar]
- Yamashita S.; Tanaka Y.; Endoh Y.; Taki Y.; Sakane T.; Nadai T.; Sezaki H. Pharm. Res. 1997, 14, 486. [DOI] [PubMed] [Google Scholar]
- Lindmark T.; Nikkila T.; Artursson P. J. Pharmacol. Exp. Ther. 1995, 275, 958. [PubMed] [Google Scholar]
- Schipper N. G.; Olsson S.; Hoogstraate J. A.; de Boer A. G.; Varum K. M.; Artursson P. Pharm. Res. 1997, 14, 923. [DOI] [PubMed] [Google Scholar]
- Lindmark T.; Schipper N.; Lazorova L.; de Boer A. G.; Artursson P. J. Drug Target. 1998, 5, 215. [DOI] [PubMed] [Google Scholar]
- Lindmark T.; Kimura Y.; Artursson P. J. Pharmacol. Exp. Ther. 1998, 284, 362. [PubMed] [Google Scholar]
- Anderberg E. K.; Nystrom C.; Artursson P. J. Pharm. Sci. 1992, 81, 879. [DOI] [PubMed] [Google Scholar]
- Anderberg E. K.; Artursson P. J. Pharm. Sci. 1993, 82, 392. [DOI] [PubMed] [Google Scholar]
- Hovgaard L.; Brondsted H. Pharm. Res. 1995, 12, 1328. [DOI] [PubMed] [Google Scholar]
- Rubas W.; Jezyk N.; Grass G. M. J. Drug Target. 1995, 3, 15. [DOI] [PubMed] [Google Scholar]
- Garcia-Fuentes M.; Prego C.; Torres D.; Alonso M. J. Bull. Tech. Gattefosse 2004, 97, 51. [Google Scholar]
- Brimer-Cline C.; Schuetz E. G. Meth. Enzymol. 2002, 357, 321. [DOI] [PubMed] [Google Scholar]
- Hsu C. P.; Hilfinger J. M.; Walter E.; Merkle H. P.; Roessler B. J.; Amidon G. L. Pharm. Res. 1998, 15, 1376. [DOI] [PubMed] [Google Scholar]
- Saitoh R.; Sugano K.; Takata N.; Tachibana T.; Higashida A.; Nabuchi Y.; Aso Y. Pharm. Res. 2004, 21, 749. [DOI] [PubMed] [Google Scholar]
- Sambuy Y.; de Angelis I.; Ranaldi G.; Scarino M. L.; Stammati A.; Zucco F. Cell Biol. Toxicol. 2005, 21, 1. [DOI] [PubMed] [Google Scholar]
- Yu H. S.; Cook T. J.; Sinko P. J. Pharm. Res. 1997, 14, 757. [DOI] [PubMed] [Google Scholar]
- Yu H. S.; Sinko P. J. J. Pharm. Sci. 1997, 86, 1448. [DOI] [PubMed] [Google Scholar]
- Delie F.; Rubas W. Crit. Rev. Therap. Drug Carrier Syst. 1997, 14, 221. [PubMed] [Google Scholar]
- Hu M.; Borchardt R. T. Pharm. Res. 1990, 7, 1313. [DOI] [PubMed] [Google Scholar]
- Artursson P. J. Pharm. Sci. 1990, 79, 476. [DOI] [PubMed] [Google Scholar]
- Khoursandi S.; Scharlau D.; Herter P.; Kuhnen C.; Martin D.; Kinne R. K. H.; Kipp H. Am. J. Physiol. 2004, 287, C1041. [DOI] [PubMed] [Google Scholar]
- Artursson P.; Magnusson C. J. Pharm. Sci. 1990, 79, 595. [DOI] [PubMed] [Google Scholar]
- Matter K.; Stieger B.; Klumperman J.; Ginsel L.; Hauri H. P. J. Biol. Chem. 1990, 265, 3503. [PubMed] [Google Scholar]
- Artursson P.; Borchardt R. T. Pharm. Res. 1997, 14, 1655. [DOI] [PubMed] [Google Scholar]
- Ho N. F.; Burton P. S.; Conradi R. A.; Barsuhn C. L. J. Pharm. Sci. 1995, 84, 21. [DOI] [PubMed] [Google Scholar]
- Pade V.; Stavchansky S. Pharm. Res. 1997, 14, 1210. [DOI] [PubMed] [Google Scholar]
- Makhey V. D.; Guo A. L.; Norris D. A.; Hu P. D.; Yan J. S.; Sinko P. J. Pharm. Res. 1998, 15, 1160. [DOI] [PubMed] [Google Scholar]
- Hidalgo I. J.; Li J. Adv. Drug Delivery Rev. 1996, 22, 53. [Google Scholar]
- Camenisch G.; Folkers G.; van de Waterbeemd H. Int. J. Pharm. 1997, 147, 61. [Google Scholar]
- Camenisch G.; Alsenz J.; van de Waterbeemd H.; Folkers G. Eur. J. Pharm. Sci. 1998, 6, 313. [PubMed] [Google Scholar]
- van de Waterbeemd H.; Camenisch G.; Folkers G.; Raevsky O. A. Quant. Struct. Act. Relat. 1996, 15, 480. [Google Scholar]
- Stenberg P.; Norinder U.; Luthman K.; Artursson P. J. Med. Chem. 2001, 44, 1927. [DOI] [PubMed] [Google Scholar]
- Palm K.; Luthman K.; Ungell A. L.; Strandlund G.; Artursson P. J. Pharm. Sci. 1996, 85, 32. [DOI] [PubMed] [Google Scholar]
- Krarup L. H.; Christensen I. T.; Hovgaard L.; Frokjaer S. Pharm. Res. 1998, 15, 972. [DOI] [PubMed] [Google Scholar]
- Clark D. E. J. Pharm Sci. 1999, 88, 807. [DOI] [PubMed] [Google Scholar]
- Norinder U.; Österberg T.; Artursson P. Pharm. Res. 1997, 14, 1786. [DOI] [PubMed] [Google Scholar]
- Bergström C. A. S.; Strafford M.; Lazorova L.; Avdeef A.; Luthman K.; Artursson P. J. Med. Chem. 2003, 46, 558. [DOI] [PubMed] [Google Scholar]
- Bergström C. A. S. Basic Clin. Pharmacol. Toxicol. 2005, 96, 156. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Durst G. L.; Stratford R. E.; Thorner D. A.; Lewis R.; Loncharich R. J.; Wikel J. H. J. Chem. Inf. Comput. Sci. 2001, 41, 1578. [DOI] [PubMed] [Google Scholar]
- Trapp S.; Horobin R. W. Eur. Biophys. J. 2005, 34, 959. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Shedden K.; Rosania G. R. Mol. Pharm. 2006, 3, 704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacher V. J.; Salphati L.; Benet L. Z. Adv. Drug Delivery Rev. 1996, 20, 99. [DOI] [PubMed] [Google Scholar]
- Benet L. Z.; Cummins C. L. Adv. Drug Delivery Rev. 2001, 50, S3. [DOI] [PubMed] [Google Scholar]
- Benet L. Z.; Cummins C. L.; Wu C. Y. Int. J. Pharm. 2004, 277, 3. [DOI] [PubMed] [Google Scholar]
- Engman H. A.; Lennernäs H.; Taipalensuu J.; Otter C.; Leidvik B.; Artursson P. J. Pharm. Sci. 2001, 90, 1736. [DOI] [PubMed] [Google Scholar]
- Crespi C. L.; Penman B. W.; Hu M. Pharm. Res. 1996, 13, 1635. [DOI] [PubMed] [Google Scholar]
- Hu M.; Li Y.; Davitt C. M.; Huang S. M.; Thummel K.; Penman B. W.; Crespi C. L. Pharm. Res. 1999, 16, 1352. [DOI] [PubMed] [Google Scholar]
- Crespi C. L.; Fox L.; Stocker P.; Hu M.; Steimel D. T. Eur. J. Pharm. Sci. 2000, 12, 63. [DOI] [PubMed] [Google Scholar]
- Gaush C. R.; Hard W. L.; Smith T. F. Proc. Soc. Exp. Biol. Med. 1966, 122, 931. [DOI] [PubMed] [Google Scholar]
- Tajima M.; Motohashi T.; Samejima T. Am. J. Vet. Res. 1961, 22, 236. [PubMed] [Google Scholar]
- Green I. J. Science 1962, 138, 42. [DOI] [PubMed] [Google Scholar]
- Leighton J.; Brada Z.; Estes L. W.; Justh G. Science 1969, 163, 472. [DOI] [PubMed] [Google Scholar]
- Leighton J.; Estes L. W.; Mansukhani S.; Brada Z. Cancer 1970, 26, 1022. [DOI] [PubMed] [Google Scholar]
- Horio M.; Chin K. V.; Currier S. J.; Goldenberg S.; Williams C.; Pastan I.; Gottesman M. M.; Handler J. J. Biol. Chem. 1989, 264, 14880. [PubMed] [Google Scholar]
- Cho M. J.; Thompson D. P.; Cramer C. T.; Vidmar T. J.; Scieszka J. F. Pharm. Res. 1989, 6, 71. [DOI] [PubMed] [Google Scholar]
- Rabito C. A.; Tchao R.; Valentich J.; Leighton J. J. Membr. Biol. 1978, 43, 351. [DOI] [PubMed] [Google Scholar]
- Simmons N. L. J. Physiol. 1978, 276, 28P. [PubMed] [Google Scholar]
- Chen L. I.; Yao J.; Yang J. B.; Yang J. Acta Pharmacol. Sin. 2005, 26, 1322. [DOI] [PubMed] [Google Scholar]
- Christiaens B.; Grooten J.; Reusens M.; Joliot A.; Goethals M.; Vandekerckhove J.; Prochiantz A.; Rosseneu M. Eur. J. Biochem. 2004, 271, 1187. [DOI] [PubMed] [Google Scholar]
- Cook G. A.; Prakash O.; Zhang K.; Shank L. P.; Takeguchi W. A.; Robbins A.; Gong Y. X.; Iwamoto T.; Schultz B. D.; Tomich J. M. Biophys. J. 2004, 86, 1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wils P.; Phung-Ba V.; Warnery A.; Lechardeur D.; Raeissi S.; Hidalgo I. J.; Scherman D. Biochem. Pharmacol. 1994, 48, 1528. [DOI] [PubMed] [Google Scholar]
- Versantvoort C. H. M.; Ondrewater R. C. A.; Duizer E.; van de Sandt J. J. M.; Gilde A. J.; Groten J. P. Environ. Toxicol. Pharmacol. 2002, 11, 335. [DOI] [PubMed] [Google Scholar]
- Tavelin S.; Taipalensuu J.; Hallbook F.; Vellonen K. S.; Moore V.; Artursson P. Pharm. Res. 2003, 20, 373. [DOI] [PubMed] [Google Scholar]
- Takakura Y.; Morita T.; Fujikawa M.; Hayashi M.; Sezaki H.; Hashida M.; Borchardt R. T. Pharm. Res. 1995, 12, 1968. [DOI] [PubMed] [Google Scholar]
- Raeissi S.; Audus K. L. J. Pharm. Pharmacol. 1989, 41, 848. [DOI] [PubMed] [Google Scholar]
- Shah M. V.; Audus K. L.; Borchardt R. T. Pharm. Res. 1989, 6, 624. [DOI] [PubMed] [Google Scholar]
- Fearn R. A.; Hirst B. H. Environ. Toxicol. Pharmacol. 2006, 21, 168. [DOI] [PubMed] [Google Scholar]
- Wikman A.; Karlsson J.; Carlstedt I.; Artursson P. Pharm. Res. 1993, 10, 843. [DOI] [PubMed] [Google Scholar]
- Larhed A. W.; Artursson P.; Grasjo J.; Bjork E. J. Pharm. Sci. 1997, 86, 660. [DOI] [PubMed] [Google Scholar]
- Larhed A. W.; Artursson P.; Bjork E. Pharm. Res. 1998, 15, 66. [DOI] [PubMed] [Google Scholar]
- Rein G.; Karasek M. A. Int. Arch. Allergy Immunol. 1992, 98, 211. [DOI] [PubMed] [Google Scholar]
- Ademola J. I.; Bloom E.; Maczulak A. E.; Maibach H. I. J. Toxicol. Cutan. Ocul. Toxicol. 1993, 12, 129. [Google Scholar]
- Flynn G. L. In Advances in Animal Alternatives for Safety and Efficacy Testing; Salem H., Katz S. A., Eds.; Taylor & Francis: Washington, DC, 1998; p 37. [Google Scholar]
- Coulomb B.; Dubertret L. Wound Repair Regen. 2002, 10, 109. [DOI] [PubMed] [Google Scholar]
- Wester R. C.; Maibach H. I. J. Invest. Dermatol. 1976, 67, 518. [DOI] [PubMed] [Google Scholar]
- Wester R. C.; Maibach H. I. Toxicol. Appl. Pharmacol. 1975, 32, 394. [DOI] [PubMed] [Google Scholar]
- Durrheim H.; Flynn G. L.; Higuchi W. I.; Behl C. R. J. Pharm. Sci. 1980, 69, 781. [DOI] [PubMed] [Google Scholar]
- Jetzer W. E.; Huq A. S.; Ho N. F.; Flynn G. L.; Duraiswamy N.; Condie L. J. Pharm. Sci. 1986, 75, 1098. [DOI] [PubMed] [Google Scholar]
- Mudd S. J. Gen. Physiol. 1926, 9, 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grass G. M.; Sweetana S. A. Pharm. Res. 1988, 5, 372. [DOI] [PubMed] [Google Scholar]
- Rosen H.; Leaf A.; Schwartz W. B. J. Gen. Physiol. 1964, 48, 379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A. Z. Vgl. Physiol. 1937, 25, 335. [Google Scholar]
- Ussing H. H. Physiol. Rev. 1949, 29, 127. [DOI] [PubMed] [Google Scholar]
- Ussing H. H. Acta Physiol. Scand. 1949, 17, 1. [DOI] [PubMed] [Google Scholar]
- Franz T. J. J. Invest. Dermatol. 1975, 64, 190. [DOI] [PubMed] [Google Scholar]
- Franz T. J. Curr. Probl. Dermatol. 1978, 7, 58. [DOI] [PubMed] [Google Scholar]
- Huf E. Pflügers Arch. Gesamte Physiol. Menschen Tiere 1936, 237, 143. [DOI] [PubMed] [Google Scholar]
- Ussing H. H. Acta Physiol. Scand. 1949, 19, 43. [DOI] [PubMed] [Google Scholar]
- Franz T. J.; Van Bruggen J. T. J. Gen. Physiol. 1967, 50, 933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy R. H.; Rowland M. J. Pharmacokinet. Biopharm. 1974, 2, 313. [DOI] [PubMed] [Google Scholar]
- Scheuplein R. J. J. Invest. Dermatol. 1965, 45, 334. [DOI] [PubMed] [Google Scholar]
- Behl C. R.; Barrett M.; Flynn G. L.; Kurihara T.; Walters K. A.; Gatmaitan O. G.; Harper N.; Higuchi W. I.; Ho N. F. H.; Pierson C. L. J. Pharm. Sci. 1982, 71, 229. [DOI] [PubMed] [Google Scholar]
- Marttin E.; Neelissen-Subnel M. T.; de Haan F. H.; Bodde H. E. Skin Pharmacol. 1996, 9, 69. [DOI] [PubMed] [Google Scholar]
- Flynn G. L.; Dürrheim H.; Higuchi W. I. J. Pharm. Sci. 1981, 70, 52. [DOI] [PubMed] [Google Scholar]
- Grams Y. Y.; Whitehead L.; Cornwell P.; Bouwstra J. A. Pharm. Res. 2004, 21, 851. [DOI] [PubMed] [Google Scholar]
- de Haan F. H. N.; Bodde H. E.; de Bruijn W. C.; Ginsel L. A.; Junginger H. E. Int. J. Pharm. 1989, 56, 75. [Google Scholar]
- Bodde H. E.; Van den Brink I.; Koerten H. K.; de Haan F. H. N. J. Controlled Release 1991, 15, 227. [Google Scholar]
- Grams Y. Y.; Whitehead L.; Cornwell P.; Bouwstra J. A. J. Controlled Release 2004, 98, 367. [DOI] [PubMed] [Google Scholar]
- Blank I. H.; Scheuplein R. J. Br. J. Dermatol. 1969, 81, 4. [Google Scholar]
- Cevc G.; Blume G.; Schatzlein A.; Gebauer D.; Paul A. Adv. Drug Delivery Rev. 1996, 18, 349. [Google Scholar]
- Fartasch M. Adv. Drug Deliv. Rev. 1996, 18, 273. [Google Scholar]
- Swartzendruber D. C.; Wertz P. W.; Kitko D. J.; Madison K. C.; Downing D. T. J. Invest. Dermatol. 1989, 92, 251. [DOI] [PubMed] [Google Scholar]
- Bouwstra J. A.; Gooris G. S.; van der Spek J. A.; Bras W. J. Invest. Dermatol. 1991, 97, 1005. [DOI] [PubMed] [Google Scholar]
- Swartzendruber D. C.; Manganaro A.; Madison K. C.; Kremer M.; Wertz P. W.; Squier C. A. Cell Tissue Res. 1995, 279, 271. [DOI] [PubMed] [Google Scholar]
- Wertz P. W. Adv. Drug Delivery Rev. 1996, 18, 283. [Google Scholar]
- Gooris G. S.; Bouwstra J. A. Biophys. J. 2007, 92, 2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice R. H.; Green H. Cell 1977, 11, 417. [DOI] [PubMed] [Google Scholar]
- Swartzendruber D. C.; Wertz P. W.; Madison K. C.; Downing D. T. J. Invest. Dermatol. 1987, 88, 709. [DOI] [PubMed] [Google Scholar]
- Wertz P. W.; Swartzendruber D. C.; Kitko D. J.; Madison K. C.; Downing D. T. J. Invest. Dermatol. 1989, 93, 169. [DOI] [PubMed] [Google Scholar]
- Menon G. K.; Lee S. H.; Roberts M. S. In Dermal Absorption and Toxicity Assessment; Roberts M. S., Walters K. A., Eds.; Marcel Dekker: New York,1998; p 727. [Google Scholar]
- Blank I. H.; Scheuplein R. J.; MacFarlane D. J. J. Invest. Dermatol. 1967, 49, 582. [PubMed] [Google Scholar]
- Li S. K.; Suh W.; Parikh H. H.; Ghanem A. H.; Mehta S. C.; Peck K. D.; Higuchi W. I. Int. J. Pharm. 1998, 170, 93. [Google Scholar]
- Walker R. B.; Smith E. W. Adv. Drug Delivery Rev. 1996, 18, 295. [Google Scholar]
- Singh P.; Maibach H. I. Adv. Drug Delivery Rev. 1996, 18, 379. [Google Scholar]
- Prausnitz M. R. Adv. Drug Delivery Rev. 1996, 18, 395. [DOI] [PubMed] [Google Scholar]
- Hadgraft J.; Lane M. E. Int. J. Pharm. 2005, 305, 2. [DOI] [PubMed] [Google Scholar]
- Guy R. H.; Hadgraft J. Dermatology 1985, 6, 3. [Google Scholar]
- Guy R. H.; Hadgraft J.; Maibach H. I. Toxicol. Appl. Pharmacol. 1985, 78, 123. [DOI] [PubMed] [Google Scholar]
- Magnusson B. M.; Pugh W. J.; Roberts M. S. Pharm. Res. 2004, 21, 1047. [DOI] [PubMed] [Google Scholar]
- Pugh W. J.; Hadgraft J. Int. J. Pharm. 1994, 103, 163. [DOI] [PubMed] [Google Scholar]
- Scheuplein R. J.; Blank I. H. Physiol. Rev. 1971, 51, 702. [DOI] [PubMed] [Google Scholar]
- Roberts M. S.; Pugh W. J.; Hadgraft J. Int. J. Pharm. 1996, 132, 23. [Google Scholar]
- Potts R. O.; Guy R. H. Pharm. Res. 1992, 9, 663. [DOI] [PubMed] [Google Scholar]
- Moss G. P.; Dearden J. C.; Patel H.; Cronin M. T. D. Toxicol. in Vitro 2002, 16, 299. [DOI] [PubMed] [Google Scholar]
- Poulin P.; Krishnan K. J. Toxicol. Environ. Health A 2001, 62, 143. [DOI] [PubMed] [Google Scholar]
- Pugh W. J.; Roberts M. S.; Hadgraft J. Int. J. Pharm. 1996, 138, 149. [Google Scholar]
- Patel H.; Berge W.; Cronin M. T. D. Chemosphere 2002, 48, 603. [DOI] [PubMed] [Google Scholar]
- Pugh W. J.; Degim I. T.; Hadgraft J. Int. J. Pharm. 2000, 197, 203. [DOI] [PubMed] [Google Scholar]
- El-Tayar N.; Tsai R. S.; Testa B.; Carrupt P. A.; Hansch C.; Leo A. J. Pharm. Sci. 1991, 80, 744. [DOI] [PubMed] [Google Scholar]
- Roberts M. S.; Pugh W. J.; Hadgraft J.; Watkinson A. C. Int. J. Pharm. 1995, 126, 219. [Google Scholar]
- Abraham M. H.; Martins F.; Mitchell R. C. J. Pharm. Pharmacol. 1997, 49, 858. [DOI] [PubMed] [Google Scholar]
- Abraham M. H.; Martins F. J. Pharm. Sci. 2004, 93, 1508. [DOI] [PubMed] [Google Scholar]
- du Plessis J.; Pugh W. J.; Judefeind A.; Hadgraft J. Eur. J. Pharm. Sci. 2002, 16, 107. [DOI] [PubMed] [Google Scholar]
- Magee P. S. In Dermatotoxicology; Marzulli F. N., Maibach H. I., Eds.; Taylor & Francis: Washington, DC, 1996; p 49. [Google Scholar]
- Geinoz S.; Guy R. H.; Testa B.; Carrupt P. A. Pharm. Res. 2004, 21, 83. [DOI] [PubMed] [Google Scholar]
- Cussler E. L.; Hughes S. E.; Ward W. J. III; Aris R. J. Membr. Sci. 1988, 38, 161. [Google Scholar]
- Heisig M.; Lieckfeldt R.; Wittum G.; Mazurkevich G.; Lee G. Pharm. Res. 1996, 13, 421. [DOI] [PubMed] [Google Scholar]
- Frasch H. F.; Barbero A. M. J. Pharm. Res. 2004, 93, 1940. [Google Scholar]
- Scheuplein R. J. Biophys. J. 1966, 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A. J.; King J. R.; Barrett D. A. J. Controlled Release 1997, 45, 141. [Google Scholar]
- Mitragotri S. J. Controlled Release 2003, 86, 69. [DOI] [PubMed] [Google Scholar]
- Reid W. E. J. Physiol. 1901, 26, 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noll A. Pflügers Arch. Gesamte Physiol. Menschen Tiere 1911, 136, 208. [Google Scholar]
- Wilson T. H.; Wiseman G. J. Physiol. 1954, 123, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukie B. E.; Westergaard H.; Dietschy J. M. Gastroenterology 1974, 67, 652. [PubMed] [Google Scholar]
- Westergaard H.; Dietschy J. M. J. Clin. Invest. 1974, 54, 718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braybrooks M. P.; Barry B. W.; Abbs E. T. J. Pharm. Pharmacol. 1975, 27, 508. [DOI] [PubMed] [Google Scholar]
- Sutton S. C.; Forbes A. E.; Cargill R.; Hochman J. H.; LeCluyse E. L. Pharm. Res. 1992, 9, 316. [DOI] [PubMed] [Google Scholar]
- Grass G. M.; Bozarth C. A.; Vallner J. J. J. Drug Target. 1994, 2, 23. [DOI] [PubMed] [Google Scholar]
- Sugiyama T.; Yamamoto A.; Kawabe Y.; Uchiyama T.; Quan Y. S.; Muranishi S. Biol. Pharm. Bull. 1997, 20, 812. [DOI] [PubMed] [Google Scholar]
- Wu-Pong S.; Livesay V.; Dvorchik B.; Barr W. H. Biopharm. Drug Dispos. 1999, 20, 411. [DOI] [PubMed] [Google Scholar]
- Gotoh Y.; Kamada N.; Momose D. J. Biomol. Screen. 2005, 10, 517. [DOI] [PubMed] [Google Scholar]
- Drews R. E.; Takaori K.; Burton J.; Donowitz M. In Peptides: Structure and Function; Hruby V. J., Rich D. H., Eds.; Pierce: Rockford, IL, 1983; p 895. [Google Scholar]
- Grass G. M.; Sweetana S. A. Pharm. Res. 1989, 6, 857. [DOI] [PubMed] [Google Scholar]
- Amerongen H. M.; Mack J. A.; Wilson J. M.; Neutra M. R. J. Cell Biol. 1989, 109, 2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riviere P. J.; Sheldon R. J.; Malarchik M. E.; Burks T. F.; Porreca F. J. Pharmacol. Exp. Ther. 1990, 253, 778. [PubMed] [Google Scholar]
- Poonyachoti S.; Portoghese P. S.; Brown D. R. J. Pharmacol. Exp. Therap. 2001, 297, 672. [PubMed] [Google Scholar]
- Townsend D. W. IV; Brown D. R. Br. J. Pharmacol. 2003, 140, 691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkova K.; Keith J. C. Jr.; Greenwood-van Meerveld B. J. Pharmacol. Exp. Therap. 2004, 308, 206. [DOI] [PubMed] [Google Scholar]
- Soderholm J. D.; Hedman L.; Artursson P.; Franzen L.; Larsson J.; Pantzar N.; Permert J.; Olaison G. Acta Physiol. Scand. 1998, 162, 47. [DOI] [PubMed] [Google Scholar]
- Saitoh H.; Aungst B. J. Pharm. Res. 1995, 12, 1304. [DOI] [PubMed] [Google Scholar]
- Collett A.; Walker D.; Sims E.; He Y. L.; Speers P.; Ayrton J.; Rowland M.; Warhurst G. Pharm. Res. 1997, 14, 767. [DOI] [PubMed] [Google Scholar]
- Lennernäs H.; Nylander S.; Ungell A. L. Pharm. Res. 1997, 14, 667. [DOI] [PubMed] [Google Scholar]
- Karlsson J.; Artursson P. Biochim. Biophys. Acta 1992, 1111, 204. [DOI] [PubMed] [Google Scholar]
- Genty M.; Gonzalez G.; Clere C.; Desangle-Gouty V.; Legendre J. Y. Eur. J. Pharm. Sci. 2001, 12, 223. [DOI] [PubMed] [Google Scholar]
- Lomholt S. J. Pharmacol. Exp. Therap. 1930, 40, 235. [Google Scholar]
- Ekins S.; Williams J. A.; Murray G. I.; Burke M. D.; Marchant N. C.; Engeset J.; Hawksworth G. M. Drug Metab. Dispos. 1996, 24, 990. [PubMed] [Google Scholar]
- Ekins S. Drug Metab. Rev. 1996, 28, 591. [DOI] [PubMed] [Google Scholar]
- Kety S. S.; Harmel M. H.; Broomell H. T.; Rhode C. B. J. Biol. Chem. 1948, 173, 487. [PubMed] [Google Scholar]
- Ekins S.; Murray G. I.; Burke M. D.; Williams J. A.; Marchant N. C.; Hawksworth G. M. Drug Metab. Dispos. 1995, 23, 1274. [PubMed] [Google Scholar]
- Christians U.; Gottschalk S.; Miljus J.; Hainz C.; Benet L. Z.; Leibfritz D.; Serkova N. Br. J. Pharmacol. 2004, 143, 388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J. J.; Etemad L. R.; Thompson A. M. J. Pharmacol. Exp. Therap. 2001, 296, 776. [PubMed] [Google Scholar]
- Thompson A. M.; Gosnell B. A.; Wagner J. J. Neuropharmacology 2002, 42, 1039. [DOI] [PubMed] [Google Scholar]
- Thompson A. M.; Swant J.; Gosnell B. A.; Wagner J. J. Neuroscience 2004, 127, 177. [DOI] [PubMed] [Google Scholar]
- Thompson A. M.; Swant J.; Wagner J. J. Neuropharmacology 2005, 49, 185. [DOI] [PubMed] [Google Scholar]
- Fiserova-Bergerova V.; Tichy M.; Di-Carlo F. J. Drug Metab. Rev 1984, 15, 1033. [DOI] [PubMed] [Google Scholar]
- Bertelsen S. L.; Hoffman A. D.; Gallinat C. A.; Elonen C. M.; Nichols J. W. Environ. Toxicol. Chem. 1998, 17, 1447. [Google Scholar]
- Sawada Y.; Hanano M.; Sugiyama Y.; Harashima H.; Iga T. J. Pharmacokinet. Biopharm. 1984, 12, 587. [DOI] [PubMed] [Google Scholar]
- Abraham M. H.; Kamlet M. J.; Taft R. W.; Doherty R. M.; Weathersby P. K. J. Med. Chem. 1985, 28, 865. [DOI] [PubMed] [Google Scholar]
- Kamlet M. J.; Abraham D. J.; Doherty R. M.; Taft R. W.; Abraham M. H. J. Pharm. Sci. 1986, 75, 350. [DOI] [PubMed] [Google Scholar]
- Poulin P.; Krishnan K. Toxicol. Appl. Pharmacol. 1996, 136, 126. [DOI] [PubMed] [Google Scholar]
- Poulin P.; Krishnan K. Toxicol. Appl. Pharmacol. 1996, 136, 131. [DOI] [PubMed] [Google Scholar]
- DeJongh J.; Verhaar H. J. M.; Hermens J. L. M. Arch. Toxicol. 1997, 72, 17. [DOI] [PubMed] [Google Scholar]
- Balaz S.; Lukacova V. Quant. Struct. Act. Relat. 1999, 18, 361. [Google Scholar]
- Kamlet M. J.; Doherty R. M.; Fiserova B., V; Carr P. W.; Abraham M. H.; Taft R. W. J. Pharm. Sci. 1987, 76, 14. [DOI] [PubMed] [Google Scholar]
- Abraham M. H.; Ibrahim A. Eur. J. Med. Chem. 2006, 41, 1430. [DOI] [PubMed] [Google Scholar]
- Abraham M. H.; Ibrahim A.; Acree W. E. Jr Eur. J. Med. Chem. 2006, 41, 494. [DOI] [PubMed] [Google Scholar]
- Abraham M. H.; Ibrahim A.; Acree W. E. Jr Chem. Res. Toxicol. 2006, 19, 801. [DOI] [PubMed] [Google Scholar]
- Abraham M. H.; Ibrahim A.; Acree W. E. Jr Eur. J. Med. Chem. 2007, 42, 743. [DOI] [PubMed] [Google Scholar]
- Abraham M. H.; Ibrahim A.; Acree W. E. Jr Eur. J. Med. Chem. 2008, 43, 478. [DOI] [PubMed] [Google Scholar]
- Kamlet M. J.; Doherty R. M.; Abboud J. L.; Abraham M. H.; Taft R. W. J. Pharm. Sci. 1986, 75, 338. [DOI] [PubMed] [Google Scholar]
- Taft R. W.; Abraham M. H.; Famini G. R.; Doherty R. M.; Abboud J. L.; Kamlet M. J. J. Pharm. Sci. 1985, 74, 807. [DOI] [PubMed] [Google Scholar]
- Abraham M. H.; Chadha H. S.; Whiting G. S.; Mitchell R. C. J. Pharm. Sci. 1994, 83, 1085. [DOI] [PubMed] [Google Scholar]
- Poulin P.; Krishnan K. J. Toxicol. Environ. Health 1995, 46, 117. [DOI] [PubMed] [Google Scholar]
- Poulin P.; Krishnan K. Human Exp. Toxicol. 1995, 14, 273. [DOI] [PubMed] [Google Scholar]
- Rodgers T.; Leahy D.; Rowland M. J. Pharm. Sci. 2005, 94, 1259. [DOI] [PubMed] [Google Scholar]
- Poulin P.; Schoenlein K.; Theil F. P. J. Pharm. Sci. 2001, 90, 436. [DOI] [PubMed] [Google Scholar]
- Zhang H. J. Pharm. Sci. 2004, 93, 1595. [DOI] [PubMed] [Google Scholar]
- Zhang H. J. Chem. Inf. Comput. Sci. 2005, 45, 121. [Google Scholar]
- Zhang H. QSAR Combin. Sci. 2006, 25, 15. [Google Scholar]
- Zhang H.; Zhang Y. J. Med. Chem. 2006, 49, 5815. [DOI] [PubMed] [Google Scholar]
- Keldenich J.; Schmitt W.; Willmann S. In Biological and Physicochemical Profiling in Drug Research; Testa B., Kraemer S. D., Wunderli-Allenspach H., Folkers G., Eds.; VCH: Weinheim, Germany, 2004. [Google Scholar]
- Trendelenburg P. Z. Biol. 1912, 57, 90. [Google Scholar]
- Backmann E. L. Skand. Arch. Physiol. 1906, 18, 323. [Google Scholar]
- Busquet H.; Pachon V. C. R. Seances Soc. Biol. Ses Fil. 1909, 66, 384. [Google Scholar]
- Burridge W. Quart. J. Exp. Physiol. 1913, 5, 347. [Google Scholar]
- Yanagawa H. J. Pharmacol. 1916, 8, 89. [Google Scholar]
- Friedrich K. Jr Pflügers Arch. 1903,, 98, 452. [Google Scholar]
- Grube K. J. Physiol. 1903, 29, 276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuigan H. Am. J. Physiol. 1908,, 21, 334. [Google Scholar]
- Bezzola C.; Izar G.; Preti L. Z. Physiol. Chem. 1910, 62, 229. [Google Scholar]
- Embden G.; Wirth J. Biochem. Z. 1910, 27, 1. [Google Scholar]
- Lattes L. Turin. Biochem. Z. 1910, 20, 215. [Google Scholar]
- Ortmann P. Arch. Exp. Pathol. Pharmakol. 1938, 191, 12. [Google Scholar]
- Meier R.; Müller R. Schweiz. Med. Wochenschr. 1941, 71, 554. [Google Scholar]
- Mattila M. J.; Takki S. Ann. Med. Exp. Biol. Fenn. 1967, 45, 357. [PubMed] [Google Scholar]
- Hartmann F.; Vieillard-Baron D.; Heinrich R. Adv. Exp. Med. Biol. 1984, 180, 711. [DOI] [PubMed] [Google Scholar]
- Andlauer W.; Kolb J.; Furst P. FEBS Lett. 2000, 475, 127. [DOI] [PubMed] [Google Scholar]
- Andlauer W.; Stumpf C.; Furst P. Biochem. Pharmacol. 2001, 62, 369. [DOI] [PubMed] [Google Scholar]
- Andlauer W.; Kolb J.; Furst P. Clin. Nutr. 2004, 23, 989. [DOI] [PubMed] [Google Scholar]
- Hirayama H.; Xu X.; Pang K. S. Am. J. Physiol. 1989, 257, G249. [DOI] [PubMed] [Google Scholar]
- Amidon G. L.; Kou J.; Elliott R. L.; Lightfoot E. N. J. Pharm. Sci. 1980, 69, 1369. [DOI] [PubMed] [Google Scholar]
- Aldini R.; Roda A.; Montagnani M.; Cerre C.; Pellicciari R.; Roda E. Steroids 1996, 61, 590. [DOI] [PubMed] [Google Scholar]
- Eldridge E.; Paton W. D. J. Physiol. 1954, 124, 27. [PubMed] [Google Scholar]
- Johnson K. G. J. Physiol. 1975, 250, 633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K. G.; Creed K. E. Comput. Biochem. Physiol. C 1982, 73, 265. [DOI] [PubMed] [Google Scholar]
- Johnson K. G.; Creed K. E. Comput. Biochem. Physiol. C 1982, 73, 259. [DOI] [PubMed] [Google Scholar]
- Riviere J. E.; Bowman K. F.; Monteiro-Riviere N. A. Br. J. Dermatol. 1987, 116, 739. [DOI] [PubMed] [Google Scholar]
- Riviere J. E.; Bowman K. F.; Monteiro-Riviere N. A. In Swine in Biomedical Research; Tubleson M. E., Ed.; Plenum Press: New York, 1986; p 657. [Google Scholar]
- Riviere J. E.; Monteiro-Riviere N. A.; Inman A. O. In Vitro Toxicol. 1997, 10, 169. [Google Scholar]
- Riviere J. E. In Dermatotoxicology Methods, Marzulli F. N., Maibach H., Eds.; Taylor & Francis: Washington, DC, 1998; p 41. [Google Scholar]
- Riviere J. E. In Dermal Absorption Models in Toxicology and Pharmacology, Riviere J. E., Ed.; CRC Press: Boca Raton, FL, 2006; p 29. [Google Scholar]
- Vaden S. L.; Page R. L.; Riviere J. E. J. Pharmacol. Toxicol. Methods 1996, 35, 173. [DOI] [PubMed] [Google Scholar]
- Bristol D. G.; Riviere J. E.; Monteiro-Riviere N. A.; Bowman K. F.; Rogers R. A. Vet. Surg. 1991, 20, 424. [DOI] [PubMed] [Google Scholar]
- Williams P. L.; Riviere J. E. J. Pharm. Sci. 1995, 84, 599. [DOI] [PubMed] [Google Scholar]
- Williams P. L.; Thompson D.; Qiao G.; Monteiro-Riviere N.; Riviere J. E. Toxicol. Appl. Pharmacol. 1996, 141, 487. [DOI] [PubMed] [Google Scholar]
- Neil M. W. Biochem. J. 1958,, 69, 15P. [Google Scholar]
- Doluisio J. T.; Billups N. F.; Dittert L. W.; Sugita E. T.; Swintosky J. V. J. Pharm. Sci. 1969, 58, 1196. [DOI] [PubMed] [Google Scholar]
- Doluisio J. T.; Tan G. H.; Billups N. F.; Diamond L. J. Pharm. Sci. 1969, 58, 1200. [DOI] [PubMed] [Google Scholar]
- Komiya I.; Park J. Y.; Kamani A.; Ho N. F. H.; Higuchi W. I. Int. J. Pharm. 1980,, 4, 249. [Google Scholar]
- Amidon G. E.; Ho N. F. H.; French A. B.; Higuchi W. I. J. Theor. Biol. 1981, 89, 195. [DOI] [PubMed] [Google Scholar]
- Park J. Y.; Ho N. F. H.; Morozowich W. J. Pharm. Sci. 1984, 73, 1588. [DOI] [PubMed] [Google Scholar]
- Leahy D. E.; Lynch J.; Finney R. E.; Taylor D. C. J. Pharmacokinet. Biopharm. 1994, 22, 411. [DOI] [PubMed] [Google Scholar]
- Sinko P. J.; Hu P.; Waclawski A. P.; Patel N. R. J. Pharm. Sci. 1995, 84, 959. [DOI] [PubMed] [Google Scholar]
- Fagerholm U.; Johansson M.; Lennernäs H. Pharm. Res. 1996, 13, 1336. [DOI] [PubMed] [Google Scholar]
- Hall S.; Rowland M. J. Pharmacol. Exp. Ther. 1984, 228, 174. [PubMed] [Google Scholar]
- Roberts M. S.; Rowland M. J. Pharmacokinet. Biopharm. 1986, 14, 261. [DOI] [PubMed] [Google Scholar]
- Roberts M. S.; Rowland M. J. Pharmacokinet. Biopharm. 1986, 14, 289. [DOI] [PubMed] [Google Scholar]
- Roberts M. S.; Rowland M. J. Pharm. Pharmacol. 1986, 38, 177. [DOI] [PubMed] [Google Scholar]
- Roberts M. S.; Rowland M. J. Pharmacokinet. Biopharm. 1986, 14, 227. [DOI] [PubMed] [Google Scholar]
- Blakey G. E.; Nestorov I. A.; Arundel P. A.; Aarons L. J.; Rowland M. J. Pharmacokinet. Biopharm. 1997, 25, 277. [DOI] [PubMed] [Google Scholar]
- Hussein Z.; Evans A. M.; Rowland M. J. Pharm. Sci. 1993, 82, 880. [DOI] [PubMed] [Google Scholar]
- Chou C. H.; Evans A. M.; Fornasini G.; Rowland M. Drug Metab. Dispos. 1993, 21, 933. [PubMed] [Google Scholar]
- Hung D. Y.; Mellick G. D.; Whitehead B. D.; Roberts M. S. J. Pharm. Pharmacol. 1998, 50, 63. [DOI] [PubMed] [Google Scholar]
- Hung D. Y.; Mellick G. D.; Anissimov Y. G.; Weiss M.; Roberts M. S. J. Pharm. Sci. 1998, 87, 943. [DOI] [PubMed] [Google Scholar]
- Fagerholm U.; Nilsson D.; Knutson L.; Lennernäs H. Acta Physiol. Scand. 1999, 165, 315. [DOI] [PubMed] [Google Scholar]
- Yu W.; Sandoval R. M.; Molitoris B. A. Am. J. Physiol. 2007, 292, F1873. [DOI] [PubMed] [Google Scholar]
- Lennernäs H. In Peptide and Protein Drug Delivery; Munksgaard: Copenhagen, Denmark, 1998; p 97. [Google Scholar]
- Stretch G. L.; Nation R. L.; Evans A. M.; Milne R. W. Drug Dev. Res. 1999, 46, 292. [Google Scholar]
- Barthe L.; Woodley J.; Houin G. Fundam. Clin. Pharmacol. 1999, 13, 154. [DOI] [PubMed] [Google Scholar]
- Balimane P. V.; Chong S.; Morrison R. A. J. Pharmacol. Toxicol. Methods 2001, 44, 301. [DOI] [PubMed] [Google Scholar]
- Bohets H.; Annaert P.; Mannens G.; Van Beijsterveldt L.; Anciaux K.; Verboven P.; Meuldermans W.; Lavrijsen K. Curr. Top. Med. Chem. 2001, 1, 367. [DOI] [PubMed] [Google Scholar]
- Chaturvedi P. R.; Decker C. J.; Odinecs A. Curr. Opin. Chem. Biol. 2001, 5, 452. [DOI] [PubMed] [Google Scholar]
- Westergaard H. BBA−Biomembranes 1987, 900, 129. [DOI] [PubMed] [Google Scholar]
- Bermejo M.; Merino V.; Garrigues T. M.; Delfina J. M. P.; Mulet A.; Vizet P.; Trouiller G.; Mercier C. J. Pharm. Sci. 1999, 88, 398. [DOI] [PubMed] [Google Scholar]
- Tong G. L.; Lien E. J. J. Pharm. Sci. 1976, 65, 1651. [DOI] [PubMed] [Google Scholar]
- Kaneniwa N.; Hiura M.; Nakagawa S. Chem. Pharm. Bull. 1979, 27, 1501. [DOI] [PubMed] [Google Scholar]
- Chou C.; McLachlan A. J.; Rowland M. J. Pharmacol. Exp. Ther. 1995, 275, 933. [PubMed] [Google Scholar]
- Proost J. H.; Roggeveld J.; Wierda J. M.; Meijer D. K. J. Pharmacol. Exp. Ther. 1997, 282, 715. [PubMed] [Google Scholar]
- Hung D. Y.; Mellick G. D.; Anissimov Y. G.; Weiss M.; Roberts M. S. Br. J. Pharmacol. 1998, 124, 1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung D. Y.; Mellick G. D.; Anissimov Y. G.; Weiss M.; Roberts M. S. J. Pharm. Sci. 1998, 87, 943. [DOI] [PubMed] [Google Scholar]
- Cao X.; Gibbs S. T.; Fang L.; Miller H. A.; Landowski C. P.; Shin H. C.; Lennernas H.; Zhong Y.; Amidon G. L.; Yu L. X.; Sun D. Pharm. Res. 2006, 23, 1675. [DOI] [PubMed] [Google Scholar]
- Pang K. S.; Rowland M. J. Pharmacokinet. Biopharm. 1977, 5, 625. [DOI] [PubMed] [Google Scholar]
- Pang K. S.; Rowland M. J. Pharmacokinet. Biopharm. 1977, 5, 655. [DOI] [PubMed] [Google Scholar]
- Pang K. S.; Rowland M. J. Pharmacokinet. Biopharm. 1977, 5, 681. [DOI] [PubMed] [Google Scholar]
- Ahmad A. B.; Bennett P. N.; Rowland M. J. Pharm. Pharmacol. 1983, 35, 219. [DOI] [PubMed] [Google Scholar]
- Sawada Y.; Sugiyama Y.; Miyamoto Y.; Iga T.; Hanano M. Chem. Pharm. Bull. 1985, 33, 319. [DOI] [PubMed] [Google Scholar]
- Tirona R. G.; Pang K. S. Drug Metab. Dispos. 1996, 24, 821. [PubMed] [Google Scholar]
- Andersen M. E.; Eklund C. R.; Mills J. J.; Barton H. A.; Birnbaum L. S. Toxicol. Appl. Pharmacol. 1997, 144, 135. [DOI] [PubMed] [Google Scholar]
- Weiss M.; Stedtler C.; Roberts M. S. Bull. Math. Biol. 1997, 59, 911. [DOI] [PubMed] [Google Scholar]
- Chou C. H.; Aarons L.; Rowland M. J. Pharmacokinet. Biopharm. 1998, 26, 595. [DOI] [PubMed] [Google Scholar]
- Tirona R. G.; Schwab A. J.; Geng W. P.; Pang K. S. Drug Metab. Dispos. 1998, 26, 465. [PubMed] [Google Scholar]
- Klivenyi G.; Schuhmacher J.; Patzelt E.; Hauser H.; Matys R.; Moock M.; Regiert T.; Maier-Borst W. J. Nuclear Med. 1998, 39, 1769. [PubMed] [Google Scholar]
- Solon E. G.; Kraus L. J. Pharmacol. Toxicol. Methods 2001, 46, 73. [DOI] [PubMed] [Google Scholar]
- Whitby B. In Radiotracers in Drug Development; Lappin G., Temple S., Eds.; CRC Press: Boca Raton, FL, 2006; p 129. [Google Scholar]
- Kilts C. D. J. Clin. Psychiatry 2000, 61, 41. [PubMed] [Google Scholar]
- Henry P. G.; Tkac I.; Grütter R. Magn. Reson. Med. 2003, 50, 684. [DOI] [PubMed] [Google Scholar]
- Bachert P. Progress Nuclear Magn. Reson. Spectr. 1998, 33, 1. [Google Scholar]
- Tkac I.; Rao R.; Georgieff M. K.; Grütter R. Magn. Reson. Med. 2003, 50, 24. [DOI] [PubMed] [Google Scholar]
- van der Linden A.; Veryohe M.; Portner H. O.; Bock C. Magn. Reson. Mater. Phys. Biol. Med. 2004, 17, 236. [Google Scholar]
- Tkac I.; Henry P. G.; Andersen P.; Keene C. D.; Low W. C.; Grütter R. Magn. Reson. Med. 2004, 52, 478. [DOI] [PubMed] [Google Scholar]
- Tkac I.; Dubinsky J. M.; Keene C. D.; Grütter R.; Low W. C. J. Neurochem. 2007, 100, 1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt K. C.; Smith C. B. Nucl. Med. Biol. 2005, 32, 719. [DOI] [PubMed] [Google Scholar]
- Autiero M.; Celentano L.; Cozzolino R.; Laccetti P.; Marotta M.; Mettivier G.; Montesi M. C.; Riccio P.; Roberti G.; Russo P. IEEE Trans. Nucl. Sci. 2005, 52, 205. [Google Scholar]
- Turchin I. V.; Plehanov V. I.; Orlova A. G.; Kamenskiy V. A.; Kleshnin M. S.; Shirmanova M. V.; Shakhova N. M.; Balalaeva I. V.; Savitskiy A. P. Laser Phys. 2006, 16, 741. [Google Scholar]
- Autiero M.; Celentano L.; Cozzolino R.; Laccetti P.; Marotta M.; Mettivier G.; Montesi M. C.; Riccio P.; Roberti G.; Russo P. Proc. SPIE 2006, 6191, 61911F/1. [Google Scholar]
- Hoffman R. M.; Yang M. Nat. Protocols 2006, 1, 1429. [DOI] [PubMed] [Google Scholar]
- Evanko D. Nat. Methods 2006, 3, 240. [DOI] [PubMed] [Google Scholar]
- Wagner J. G. Pharmacol. Therap. 1981, 12, 537. [DOI] [PubMed] [Google Scholar]
- Metcalf R. L.; Kapoor I. P.; Lu P. Y.; Schuth C. K.; Sherman P. Environ. Health Persp. 1973, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P. Y.; Metcalf R. L. Environ. Health Pers. 1975, 10, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veith G. D.; DeFoe D. L.; Bergstedt B. V. J. Fish. Res. Board Can. 1979, 36, 1040. [Google Scholar]
- Kenaga E. E.; Goring C. A. I. In Aquatic Toxicology; Eaton J. G., Ed.; ASTM: West Conshohocken: PA, 1980; p 78. [Google Scholar]
- Veith G. D.; Macek K. J.; Petrocelli S. R.; Carroll J. In Aquatic Toxicology; Eaton J. G., Ed.; ASTM: West Conshohocken: PA, 1980; p 116. [Google Scholar]
- Mackay D. Environ. Sci. Technol. 1982, 16, 274. [DOI] [PubMed] [Google Scholar]
- Oliver B. G.; Niimi A. J. Environ. Sci. Technol. 1983, 17, 287. [DOI] [PubMed] [Google Scholar]
- Davies R. P.; Dobbs A. J. Water Res. 1984, 18, 1253. [Google Scholar]
- Chiou C. T. Environ. Sci. Technol. 1985, 19, 57. [DOI] [PubMed] [Google Scholar]
- van Gestel C. A.; Otermann K.; Canton J. H. Regul. Toxicol. Pharmacol. 1985, 5, 422. [DOI] [PubMed] [Google Scholar]
- Hawker D. W.; Connell D. W. Ecotoxicol. Environ. Saf. 1986, 11, 184. [DOI] [PubMed] [Google Scholar]
- Deneer J. W.; Sinnige T. L.; Seinen W.; Hermens J. L. M. Aquatic Toxicol. 1987, 10, 115. [Google Scholar]
- de Wolf W.; Seinen W.; Hermens J. L. M. Arch. Environ. Contam. Toxicol. 1993, 25, 110. [Google Scholar]
- Axelman J.; Broman D.; Naf C.; Pettersen H. Environ. Sci. Pollut. Res. 1995, 2, 33. [DOI] [PubMed] [Google Scholar]
- Saarikoski J.; Viluksela M. Ecotoxicol. Environ. Saf. 1982, 6, 501. [DOI] [PubMed] [Google Scholar]
- Connell D. W.; Bowman M.; Hawker D. W. Ecotoxicol. Environ. Saf. 1988, 16, 293. [DOI] [PubMed] [Google Scholar]
- Chessells M.; Hawker D. W.; Connell D. W. Ecotoxicol. Environ. Saf. 1992, 23, 260. [DOI] [PubMed] [Google Scholar]
- Govers H. A. J.; Krop H. B. Chemosphere 1998, 37, 2139. [Google Scholar]
- Meylan W. M.; Howard P. H.; Boethling R. S.; Aronson D.; Printup H.; Gouchie S. Environ. Toxicol. Chem. 1999, 18, 664. [Google Scholar]
- Hermens J. L. M. Pestic. Sci. 1986, 17, 287. [Google Scholar]
- Deneer J. W.; Seinen W.; Hermens J. L. M. Aquatic Toxicol. 1988, 12, 185. [Google Scholar]
- Connell D. W.; Hawker D. W. Ecotoxicol. Environ. Saf. 1988, 16, 242. [DOI] [PubMed] [Google Scholar]
- de Bruijn J.; Hermens J. Environ. Toxicol. Chem. 1991, 10, 791. [Google Scholar]
- Buchanan A. London Med. Gaz. 1847, 39, 715. [Google Scholar]
- Sollmann T.; Hanzlik P. J.; Pilcher J. D. J. Pharmacol. 1910, 1, 409. [Google Scholar]
- Hanzlik P. J. J. Biol. Chem. 1911, 7, 459. [Google Scholar]
- Hanzlik P. J. Pharmacol. 1912, 3, 387. [Google Scholar]
- Sollmann T.; Hanzlik P. J. Cleveland Med. J. 1912, 11, 192. [Google Scholar]
- Hanzlik P. J. Cleveland Med. J. 1913, 12, 274. [Google Scholar]
- Haggard H. W. J. Biol. Chem. 1924, 59, 737. [Google Scholar]
- Haggard H. W. J. Biol. Chem. 1924, 59, 753. [Google Scholar]
- Haggard H. W. J. Biol. Chem. 1924, 59, 771. [Google Scholar]
- Haggard H. W. J. Biol. Chem. 1924, 59, 783. [Google Scholar]
- Widmark E. Skand. Arch. Physiol. 1925, 46, 343. [Google Scholar]
- Moller E.; McIntosh J. F.; Van Slyke D. D. J. Clin. Invest. 1928, 6, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis L.; Menten M. L. Biochem. Z. 1913, 49, 333. [Google Scholar]
- Widmark E.; Tanberg J. Biochem. Z. 1924, 147, 358. [Google Scholar]
- Teorell T. Arch. Int. Pharmacodyn. 1937, 57, 205. [PubMed] [Google Scholar]
- Hamilton W. F.; Moore J. W.; Kinsman J. M.; Spurling R. G. Am. J. Physiol. 1932, 99, 534. [Google Scholar]
- Jolliffe N.; Smith H. W. Am. J. Physiol. 1931, 98, 572. [Google Scholar]
- Jolliffe N.; Smith H. W. Am. J. Physiol. 1931, 99, 101. [Google Scholar]
- Dominguez R.; Pomerene E. J. Biol. Chem. 1934, 104, 449. [Google Scholar]
- Boxer G. E.; Jelinek V. C.; Tompsett R.; DuBois R.; Edison A. O. J. Pharmacol. Exp. Therap. 1948, 92, 226. [PubMed] [Google Scholar]
- Dost F. H. Naunyn Schmiedebergs Arch. Exp. Pathol. Pharmakol. 1951, 213, 435. [PubMed] [Google Scholar]
- Dost F. H.Der Blutspiegel; Georg Thieme: Leipzig, Germany, 1953. [Google Scholar]
- Solomon A. K. J. Clin. Invest. 1949, 28, 1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon A. K. Adv. Biol. Med. Phys. 1953, 3, 65. [DOI] [PubMed] [Google Scholar]
- Rescigno A. Biochim. Biophys. Acta 1954, 15, 340. [DOI] [PubMed] [Google Scholar]
- Rescigno A. Biochim. Biophys. Acta 1956, 21, 111. [DOI] [PubMed] [Google Scholar]
- Riggs D. S.The Matematical Approach to Physiological Problems. A Critical Primer; Williams & Wilkins: Baltimore, MD, 1963. [Google Scholar]
- Rescigno A.; Segre G.. Drug and Tracer Kinetics; Blaisdell: Waltham, MA, 1966. [Google Scholar]
- Krüger-Thiemer E.; Bünger P. Chemotherapy 1965, 10, 61. [DOI] [PubMed] [Google Scholar]
- Krüger-Thiemer E.; Bünger P. Chemotherapy 1965, 10, 129. [DOI] [PubMed] [Google Scholar]
- Krüger-Thiemer E.; Eriksen S. P. J. Pharm. Sci. 1966, 55, 1249. [DOI] [PubMed] [Google Scholar]
- Krüger-Thiemer E. Kinetics of Drug Action; van Rossum J. M., Ed.; Springer: Berlin, Germany, 1977; Chapter 2, p 63. [Google Scholar]
- Kety S. S. Pharmacol. Rev. 1951, 3, 1. [PubMed] [Google Scholar]
- Jacquez J. A.; Bellman R.; Kalaba R. Bull. Math. Biophys. 1960, 22, 309. [Google Scholar]
- Bischoff K. B.; Brown R. G. Chem. Eng. Progr. Symp. Ser. 1966, 62, 33. [Google Scholar]
- Bischoff K. B.; Dedrick R. L. J. Pharm. Sci. 1968, 57, 1346. [DOI] [PubMed] [Google Scholar]
- Dedrick R. L.; Bischoff K. B. Chem. Eng. Progr. Symp. Ser. 1968, 64, 32. [Google Scholar]
- Wagner J. G.Fundamentals of Clinical Pharmacokinetics; Drug Intelligence Publishers: Hamilton, IL, 1975; p 231. [Google Scholar]
- Balaz S.; Wiese M.; Chi H. L.; Seydel J. K. Anal. Chim. Acta 1990, 235, 195. [Google Scholar]
- Perlovich G. L.; Bauer-Brandl A. Pharm. Res. 2003, 20, 471. [DOI] [PubMed] [Google Scholar]
- Leo A. J.; Hansch C. Persp. Drug Discovery Des. 1999, 17, 1. [Google Scholar]
- Glasstone S.; Laidler K. J.; Eyring H.. The Theory of Rate Processes; McGraw−Hill: New York, 1941. [Google Scholar]
- Verkman A. S. Trends Biochem. Sci. 2002, 27, 27. [DOI] [PubMed] [Google Scholar]
- Moore W. J.Physical Chemistry; Prentice−Hall: Englewood Cliffs, NJ, 1999. [Google Scholar]
- Schnell S.; Turner T. E. Prog. Biophys. Mol. Biol. 2004, 85, 235. [DOI] [PubMed] [Google Scholar]
- Buchwald P. J. Pharm. Sci. 2005, 94, 2355. [DOI] [PubMed] [Google Scholar]
- van de Waterbeemd J. T. M.; Jansen A. C. A.; Gerritsma K. W. Pharm. Weekbl. Sci. Ed. 1978, 113, 1097. [Google Scholar]
- van de Waterbeemd J. T. M.; van Boekel C. C. A. A.; de Sevaux R. L. F. M.; Gerritsma K. W. Pharm. Weekbl. Sci. Ed. 1981, 3, 12. [Google Scholar]
- van de Waterbeemd J. T. M.; Jansen A. C. A. Pharm. Weekbl. Sci. Ed. 1981, 3, 71. [Google Scholar]
- Aarons L.; Bell D.; Waigh R. J. Pharm. Pharmacol. 1982, 34, 746. [DOI] [PubMed] [Google Scholar]
- Balaz S.; Sturdik E.; Hrmova M.; Breza M.; Liptaj T. Eur. J. Med. Chem. 1984, 19, 167. [Google Scholar]
- Balaz S.; Sturdik E. In QSAR in Design of Bioactive Compounds; Kuchar M., Ed., Prous: Barcelona, Spain, 1984; p 289. [Google Scholar]
- Balaz S.; Sturdik E. In QSAR in Toxicology and Xenobiochemistry; Tichy M., Ed., Elsevier: Amsterdam, The Netherlands, 1985; p 257. [Google Scholar]
- Fletcher R.; Powell M. J. D. Comput. J. 1963, 6, 163. [Google Scholar]
- Balaz S.; Sturdik E.; Dibus I.; Ebringer L.; Stibranyi L.; Rosenberg M. Chem. Biol. Interact. 1985, 55, 93. [PubMed] [Google Scholar]
- McCalla D. R.; Arlett C. R.; Broughton B. Chem. Biol. Interact. 1978, 21, 89. [DOI] [PubMed] [Google Scholar]
- Kölzer P.; Büchi J. Drug Res. 1971, 21, 1721. [PubMed] [Google Scholar]
- Kramer C. R.; Beck L.; Arndt H. Biochem. Physiol. Pflanzen 1982, 177, 192. [Google Scholar]
- Balaz S.; Sturdik E. Gen. Physiol. Biophys. 1985, 4, 105. [PubMed] [Google Scholar]
- Zwolinski B. J.; Eyring H.; Reese C. E. J. Phys. Colloid Chem. 1949, 53, 1426. [Google Scholar]
- Koizumi T.; Arita T.; Kakemi K. Chem. Pharm. Bull. 1964, 12, 413. [DOI] [PubMed] [Google Scholar]
- Koizumi T.; Arita T.; Kakemi K. Chem. Pharm. Bull. 1964, 12, 421. [DOI] [PubMed] [Google Scholar]
- Suzuki A.; Higuchi W. I.; Ho N. F. H. J. Pharm. Sci. 1970, 59, 644. [DOI] [PubMed] [Google Scholar]
- Ho N. F. H.; Higuchi W. I.; Turi J. J. Pharm. Sci. 1972, 61, 192. [DOI] [PubMed] [Google Scholar]
- Stehle R. G.; Higuchi W. I. J. Pharm. Sci. 1972, 61, 1922. [DOI] [PubMed] [Google Scholar]
- Flynn G. L.; Yalkowsky S. H. J. Pharm. Sci. 1972, 61, 838. [DOI] [PubMed] [Google Scholar]
- Fürst U.; Neubert E.; Reppel L. Pharmazie 1980, 35, 106. [PubMed] [Google Scholar]
- Stein W. D.The Movement of Molecules Across Cell Membranes; Academic Press: New York, 1967. [Google Scholar]
- Leahy D. E.; de Meere A. L. J.; Wait A. R.; Taylor P. J.; Tomenson J. A.; Tomlinson E. In QSAR in Drug Design and Toxicology; Hadzi D., Jerman-Blazic B., Eds.; Elsevier: Amsterdam, The Netherlands, 1987; p 144. [Google Scholar]
- Miller D. M. Biochim. Biophys. Acta 1991, 1065, 75. [DOI] [PubMed] [Google Scholar]
- Dvorsky R.; Balaz S.; Sawchuk R. J. J. Theor. Biol. 1997, 185, 213. [DOI] [PubMed] [Google Scholar]
- Rescigno A.; Lambrecht R. M.; Duncan C. C. In Tracer Kinetics in Physiologic Modeling; Lambrecht R. M., Rescigno A., Eds.; Springer: Berlin, Germany, 1983; p 53. [Google Scholar]
- Wolf M.; Heinzel G.; Koss F. W.; Bozler G. Drug Res. 1977, 27, 900. [PubMed] [Google Scholar]
- Balaz S.; Sturdik E.; Augustin J. Biophys. Chem. 1986, 24, 135. [DOI] [PubMed] [Google Scholar]
- Balaz S.; Sturdik E.; Augustin J. Gen. Physiol. Biophys. 1987, 6, 65. [PubMed] [Google Scholar]
- Balaz S.; Sturdik E.; Augustin J. Bull. Math. Biol. 1988, 50, 367. [DOI] [PubMed] [Google Scholar]
- Balaz S.; Sturdik E.; Skarka B. Collect. Czechoslov. Chem. Commun. 1984, 49, 1382. [Google Scholar]
- Balaz S.; Wiese M.; Seydel J. K. Quant. Struct. Act. Relat. 1992, 11, 45. [Google Scholar]
- Balaz S. SAR QSAR Environ. Sci. 1995, 4, 177. [DOI] [PubMed] [Google Scholar]
- Verhaar H. J. M.; Ramos E. U.; Hermens J. L. M. J. Chemometr. 1996, 10, 149. [Google Scholar]
- Verhaar H. J. M.; Solbe J.; Speksnijder J.; Van Leeuwen C. J.; Hermens J. L. M. Chemosphere 2000, 40, 875. [DOI] [PubMed] [Google Scholar]
- Koenemann H. Toxicology 1981, 19, 209. [DOI] [PubMed] [Google Scholar]
- Veith G. D.; Broderius J. S. In QSAR in Environmental Toxicology; Kaiser K. L. E., Ed., Reidel: Dordrecht, The Netherlands, 1987; p 385. [Google Scholar]
- Turner L.; Choplin F.; Dugard P.; Hermens J.; Jaeckh R.; Marsmann M.; Roberts D. Toxicol. in Vitro 1987, 1, 143. [DOI] [PubMed] [Google Scholar]
- Jaworska J. S.; Schultz T. W. SAR QSAR Environ. Res. 1993, 1, 3. [DOI] [PubMed] [Google Scholar]
- Higuchi T.; Davis S. S. J. Pharm. Sci. 1970, 59, 1376. [DOI] [PubMed] [Google Scholar]
- Hyde R. M. J. Med. Chem. 1975, 18, 231. [DOI] [PubMed] [Google Scholar]
- Balaz S.; Wiese M.; Seydel J. K. J. Pharm. Sci. 1992, 81, 849. [DOI] [PubMed] [Google Scholar]
- D’Silva J. B.; Notari R. E. J. Pharm. Sci. 1982, 71, 1394. [DOI] [PubMed] [Google Scholar]
- Bikhazi A. B.; Baasiri G. M.; Boulos N. Z.; Khuri R. N. J. Pharm. Sci. 1983, 72, 296. [DOI] [PubMed] [Google Scholar]
- Brahm J. J. Gen. Physiol. 1983, 81, 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson U. G.; Tozer T. N.; Sosnovsky G.; Lukszo J.; Brasch R. C. J. Pharm. Sci. 1986, 75, 334. [DOI] [PubMed] [Google Scholar]
- Turi J. S.; Higuchi W. I.; Shipman C. J.; Ho N. F. H. J. Pharm. Sci. 1972, 61, 1618. [DOI] [PubMed] [Google Scholar]
- Chowhan Z. T.; Yotsuyanagi T.; Higuchi W. I. J. Pharm. Sci. 1973, 62, 221. [DOI] [PubMed] [Google Scholar]
- Lullmann H.; Timmermans P.; Weikert G.; Ziegler A. J. Med. Chem. 1980, 23, 560. [DOI] [PubMed] [Google Scholar]
- Kamp F.; Hamilton J. A. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 11367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinfeld A. M.; Chu P.; Storch J. Biochemistry 1997, 36, 5702. [DOI] [PubMed] [Google Scholar]
- Shore P. A.; Brodie P. P.; Hogben C. A. M. J. Pharmacol. Exp. Therap. 1957, 119, 361. [PubMed] [Google Scholar]
- Leo A. J. Chem. Rev. 1993, 93, 1281. [Google Scholar]
- Browning C. H.; Gulbransen R.; Kennaway E. L. J. Pathol. Bacteriol. 1919, 23, 106. [Google Scholar]
- Vermast P. G. Biochem. Z. 1921, 125, 106. [Google Scholar]
- Cruess W. V.; Richert P. H. J. Bacteriol. 1929, 17, 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruess W., V;; Irish J. H. J. Bacteriol. 1932, 23, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershenfeld L.; Perlstein D. Am. J. Pharm. 1941, 113, 237. [Google Scholar]
- Gershenfeld L.; Perlstein D. Am. J. Pharm. 1941, 113, 88. [Google Scholar]
- Eagle H. J. Pharmacol. 1945, 85, 265. [PubMed] [Google Scholar]
- Cowles P. B.; Klotz I. M. J. Bacteriol. 1948, 56, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eagle H.; Levy M.; Fleischman R. Antibiot. Chemother. 1952, 2, 563. [PubMed] [Google Scholar]
- Beevers H.; Simon E. W. Nature 1949, 163, 408. [DOI] [PubMed] [Google Scholar]
- Simon E. W. Nature 1950, 166, 343. [DOI] [PubMed] [Google Scholar]
- Simon E. W.; Beevers H. Science 1951, 114, 124. [DOI] [PubMed] [Google Scholar]
- Bell P. H.; Roblin R. O. Jr J. Am. Chem. Soc 1942, 64, 2905. [Google Scholar]
- Cowles P. B. Yale J. Biol. Med. 1942, 14, 599. [PMC free article] [PubMed] [Google Scholar]
- Bettman B.; Branch G. E. K.; Yabroff D. L. J. Am. Chem. Soc. 1934, 56, 1865. [Google Scholar]
- Hammett L. P. J. Am. Chem. Soc. 1937, 59, 96. [DOI] [PubMed] [Google Scholar]
- Hammett L. P. Trans. Faraday Soc. 1938, 34, 156. [Google Scholar]
- Daniel W. A.; Wojcikowski J.; Palucha A. Br. J. Pharmacol. 2001, 134, 807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel W. A. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2003, 27, 65. [DOI] [PubMed] [Google Scholar]
- Siebert G. A.; Hung D. Y.; Chang P.; Roberts M. S. J. Pharmacol. Exp. Therap. 2004, 308, 228. [DOI] [PubMed] [Google Scholar]
- Kaufman J. J.; Semo N. M.; Koski W. S. J. Med. Chem. 1975, 18, 647. [DOI] [PubMed] [Google Scholar]
- Wan H.; Ulander J. Expert Opin. Drug Metab. Toxicol. 2006, 2, 139. [DOI] [PubMed] [Google Scholar]
- Perrin D. D.; Dempsey B.; Serjeant E. P.. pKa Prediction for Organic Acids and Bases; Chapman and Hall: London, U.K., 1981; Chapter 2, p 10. [Google Scholar]
- Hilal S. H.; Carreira L. A.; Baughman G. L.; Karickhoff S. W.; Melton C. M. J. Phys. Org. Chem. 1994, 7, 122. [Google Scholar]
- Hilal S. H.; Karickhoff S. W.; Carreira L. A. Quant. Struct. Act. Relat. 1995, 14, 348. [Google Scholar]
- Potter M. J.; Gilson M. K.; McCammon J. A. J. Am. Chem. Soc. 1994, 116, 10298. [Google Scholar]
- Xing L.; Glen R. C.; Clark R. D. J. Chem. Inf. Comput. Sci. 2003, 43, 870. [DOI] [PubMed] [Google Scholar]
- Bender A.; Mussa H. Y.; Gill G. S.; Glen R. C. J. Med. Chem. 2004, 47, 6569. [DOI] [PubMed] [Google Scholar]
- Kogej T.; Muresan S. Curr. Drug Discovery Technol. 2005, 2, 221. [DOI] [PubMed] [Google Scholar]
- Zhang J. H.; Kleinoder T.; Gasteiger J. J. Chem. Inf. Model. 2006, 46, 2256. [DOI] [PubMed] [Google Scholar]
- Glen R. C.; Bender A.; Arnby C. H.; Carlsson L.; Boyer S.; Smith J. IDrugs 2006, 9, 199. [PubMed] [Google Scholar]
- Jelfs S.; Ertl P.; Selzer P. J. Chem. Inf. Model. 2007, 47, 450. [DOI] [PubMed] [Google Scholar]
- Lee P. H.; Ayyampalayam S. N.; Carreira L. A.; Shalaeva M.; Bhattachar S.; Coselmon R.; Poole S.; Gifford E.; Lombardo F. Mol. Pharm. 2007, 4, 498. [DOI] [PubMed] [Google Scholar]
- Milletti F.; Storchi L.; Sforna G.; Cruciani G. J. Chem. Inf. Model. 2007, 47, 2172. [DOI] [PubMed] [Google Scholar]
- Morgenthaler M.; Schweizer E.; Hoffmann-Roder A.; Benini F.; Martin R. E.; Jaeschke G.; Wagner B.; Fischer H.; Bendels S.; Zimmerli D.; Schneider J.; Diederich F.; Kansy M.; Müller K. ChemMedChem 2007, 2, 1100. [DOI] [PubMed] [Google Scholar]
- Ghasemi J.; Saaidpour S.; Brown S. D. J. Mol. Struct. THEOCHEM 2007, 805, 27. [Google Scholar]
- Takacs-Novak K.; Avdeef A.; Box K. J.; Podanyi B.; Szasz G. J. Pharm. Biomed. Anal. 1994, 12, 1369. [DOI] [PubMed] [Google Scholar]
- Szakacs Z.; Noszal B. J. Math. Chem. 2000, 26, 139. [Google Scholar]
- Avdeef A.Absorption and Drug Development: Solubility, Permeability, and Charge State; Wiley: Hoboken, NJ, 2003. [Google Scholar]
- Noszal B.; Szakacs Z. J. Phys. Chem. B 2003, 107, 5074. [Google Scholar]
- Szakacs Z.; Krasnzi M.; Noszal B. Anal. Bioanal. Chem. 2004, 378, 1428. [DOI] [PubMed] [Google Scholar]
- Hilal S. H.; Karickhoff S. W.; Carreira L. A. Talanta 1999, 50, 827. [DOI] [PubMed] [Google Scholar]
- Japertas P.; Didziapetris R.; Petrauskas A. Mini-Rev. Med. Chem. 2003, 3, 797. [DOI] [PubMed] [Google Scholar]
- Pallas Software; Compudrug International, Inc.: Sedona, AZ, 2008. [Google Scholar]
- Marvin Software; ChemAxon: Budapest, Hungary, 2008. [Google Scholar]
- ACD/pKa DB Software; Advanced Chemistry Development: Toronto, ON, Canada, 2008. [Google Scholar]
- SPARC Software; Department of Chemistry, University of Georgia: Athens, GA, 2008. [Google Scholar]
- Meloun M.; Bordovska S. Anal. Bioanal. Chem. 2007, 389, 1267. [DOI] [PubMed] [Google Scholar]
- Hansch C.; Lien E. J. J. Med. Chem. 1971, 14, 653. [DOI] [PubMed] [Google Scholar]
- Kubinyi H. J. Med. Chem. 1976, 19, 587. [DOI] [PubMed] [Google Scholar]
- Karickhoff S. W.; McDaniel V. K.; Melton C.; Vellino A. N.; Nute D. E.; Carreira L. A. Environ. Toxicol. Chem. 1991, 10, 1405. [Google Scholar]
- Hilal S. H.; El Shabrawy Y.; Carreira L. A.; Karickhoff S. W.; Toubar S. S.; Rizk M. Talanta 1996, 43, 607. [DOI] [PubMed] [Google Scholar]
- SPARC On-Line Calculator, Version 4.2, University of Georgia, http://ibmlc2.chem.uga.edu/sparc/index.cfm,2008. [Google Scholar]
- Dewar M. J. S.The Molecular Orbital Theory of Organic Chemistry; McGraw−Hill: New York, 1969. [Google Scholar]
- Magerlein B. J.; Birkenmeyer R. D.; Kagan F. J. Med. Chem. 1967, 10, 355. [DOI] [PubMed] [Google Scholar]
- Pirselova K.; Balaz S.; Schultz T. W. Arch. Environ. Contam. Toxicol. 1996, 30, 170. [DOI] [PubMed] [Google Scholar]
- Balaz S.; Pirselova K.; Schultz T. W.; Hermens J. J. Theor. Biol. 1996, 178, 7. [Google Scholar]
- Schultz T. W.; Holcombe G. W.; Phipps G. L. Ecotoxicol. Environ. Saf. 1986, 12, 146. [DOI] [PubMed] [Google Scholar]
- Schultz T. W.; Lin D. T.; Wiilke T. S.; Arnold L. M. In Practical Applications of Quantitative Structure-Activity Relationships in Enviromental Chemistry and Toxicology; Karcher W., Devillers J., Eds.; Kluwer: London, U.K., 1990; Chapter 14, p 241. [Google Scholar]
- Kaiser K. L.; Esterby S. R. Sci. Total Environ. 1991, 109−110, 499. [DOI] [PubMed] [Google Scholar]
- Schultz T. W.; Tichy M. Bull. Environ. Contam. Toxicol. 1993, 51, 681. [DOI] [PubMed] [Google Scholar]
- Cronin M. T.; Bryant S. E.; Dearden J. C.; Schultz T. W. SAR QSAR Environ. Res. 1995, 3, 1. [DOI] [PubMed] [Google Scholar]
- Cronin M. T. D.; Schultz T. W. Chemosphere 1996, 32, 1453. [DOI] [PubMed] [Google Scholar]
- Schultz T. W.; Comeaux J. L. Bull. Environ. Contam. Toxicol. 1996, 56, 638. [DOI] [PubMed] [Google Scholar]
- Jaworska J. S.; Hunter R. S.; Gobble J. R.; Schultz T. W. In Quantitative Structure-Activity Relationships in Enviromental Relationships in Enviromental Sciences; Chen F.; Schuurmann G., Eds.; SETAC: Pensacola, FL, 1997; Chapter 19, p 277. [Google Scholar]
- Schultz T. W.; Sinks G. D.; Cronin M. T. D. In Quantitative Structure-Activity Relationships in Enviromental Sciences; Chen F.; Schüürmann G., Eds.; SETAC: Pensacola, FL, 1997; Chapter 23, p 329. [Google Scholar]
- Schüürmann G.; Flemmig B.; Dearden J. C.; Cronin M. T. D.; Schultz T. W. In Quantitative Structure-Activity Relationships in Enviromental Sciences; Chen F.; Schüürmann G., Eds.; SETAC: Pensacola, FL, 1997; Chapter 22, p 315. [Google Scholar]
- Schultz T. W.; Sinks G. D.; Bearden A. P. In Comparative QSAR; Taylor & Francis: Washington, DC, 1998; Chapter 2, p 51. [Google Scholar]
- Muccini M.; Layton A. C.; Sayler G. S.; Schultz T. W. Bull. Environ. Contam. Toxicol. 1999, 62, 616. [DOI] [PubMed] [Google Scholar]
- Schultz T. W.; Seward J. R. Quant. Struct. Act. Relat. 2000, 19, 339. [Google Scholar]
- Cronin M. T. D.; Schultz T. W. Chem. Res. Toxicol. 2001, 14, 1284. [DOI] [PubMed] [Google Scholar]
- Aptula A. O.; Netzeva T. I.; Valkova I. V.; Cronin M. T. D.; Schultz T. W.; Kühne R.; Schüürmann G. Quant. Struct. Act. Relat. 2002, 21, 12. [Google Scholar]
- Cronin M. T. D.; Aptula A. O.; Duffy J. C.; Netzeva T. I.; Rowe P. H.; Valkova I. V.; Schultz T. W. Chemosphere 2002, 49, 1201. [DOI] [PubMed] [Google Scholar]
- Mekapati S. B.; Hansch C. J. Chem. Inf. Comput. Sci. 2002, 42, 956. [DOI] [PubMed] [Google Scholar]
- Netzeva T. I.; Aptula A. O.; Chaudary S. H.; Duffy J. C.; Schultz T. W.; Schüürmann G.; Cronin M. T. D. QSAR Comb. Sci. 2003, 22, 575. [Google Scholar]
- Smiesko M.; Benfenati E. J. Chem. Inf. Model. 2005, 45, 379. [DOI] [PubMed] [Google Scholar]
- Ren S. Agric. Soil Pollut. 2005, 191. [Google Scholar]
- Hewitt M.; Cronin M. T. D.; Madden J. C.; Rowe P. H.; Johnson C.; Obi A.; Enoch S. J. J. Chem. Inf. Model. 2007, 47, 1460. [DOI] [PubMed] [Google Scholar]
- Lagunin A. A.; Zakharov A. V.; Filimonov D. A.; Poroikov V. V. SAR QSAR Environ. Res. 2007, 18, 285. [DOI] [PubMed] [Google Scholar]
- Kahn I.; Maran U.; Benfenati E.; Netzeva T. I.; Schultz T. W.; Cronin M. T. D. ATLA-Altern. Lab. Anim. 2007, 35, 15. [DOI] [PubMed] [Google Scholar]
- Schultz T. W.; Hewitt M.; Netzeva T. I.; Cronin M. T. D. QSAR Combin. Sci. 2007, 26, 238. [Google Scholar]
- Schultz T. W.; Ralston K. E.; Roberts D. W.; Veith G. D.; Aptula A. O. SAR QSAR Environ. Res. 2007, 18, 21. [DOI] [PubMed] [Google Scholar]
- Laszlo T.; Beteringhe A. QSAR Combin. Sci. 2006, 25, 944. [Google Scholar]
- Duchowicz P. R.; Mercader A. G.; Fernandez F. M.; Castro E. A. Chemom. Intell. Lab. Syst. 2008, 90, 97. [Google Scholar]
- Martin T. M.; Harten P.; Venkatapathy R.; Das S.; Young D. M. Toxicol. Mech. Methods 2008, 18, 251. [DOI] [PubMed] [Google Scholar]
- Enoch S. J.; Cronin M. T. D.; Schultz T. W.; Madden J. C. Chemosphere 2008, 71, 1225. [DOI] [PubMed] [Google Scholar]
- Zhu H.; Tropsha A.; Fourches D.; Varnek A.; Papa E.; Gramatica P.; Oberg T.; Dao P.; Cherkasov A.; Tetko I. V. J. Chem. Inf. Model. 2008, 48, 766. [DOI] [PubMed] [Google Scholar]
- Anderson R. G. W.; Orci L. J. Cell Biol. 1988, 106, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaz S.; Cronin M. T. D.; Dearden J. C. Pharm. Sci. Commun. 1993, 4, 51. [Google Scholar]
- Lucic B.; Amic D.; Trinajstic N. J. Chem. Inf. Comput. Sci. 2000, 40, 403. [DOI] [PubMed] [Google Scholar]
- Zheng W.; Tropsha A. J. Chem. Inf. Comput. Sci. 2000, 40, 185. [DOI] [PubMed] [Google Scholar]
- Itskowitz P.; Tropsha A. J. Chem. Inf. Model. 2005, 45, 777. [DOI] [PubMed] [Google Scholar]
- Lien E. J. In Drug Design; Ariens E. J., Ed.; Academic Press: New York, 1975; Vol. 5, p 81. [Google Scholar]
- Lien E. J. Progr. Drug Res. 1985, 29, 67. [DOI] [PubMed] [Google Scholar]
- Seydel J. K.; Schaper K. J. Pharmacol. Therap. 1982, 15, 131. [DOI] [PubMed] [Google Scholar]
- Mayer J. M.; van de Waterbeemd H. Environ. Health Persp. 1985, 61, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozler G.; Schmid J. In Modern Drug Research: Paths to Better and Safer Drugs; Martin Y. C., Kutter E., Austel V., Eds.; Marcel Dekker: New York, 1989; p 77. [Google Scholar]
- Kakemi K.; Arita T.; Hori R.; Konishi R. Chem. Pharm. Bull. 1967, 15, 1883. [DOI] [PubMed] [Google Scholar]
- Vora K. R. M.; Higuchi W. I.; Ho N. F. H. J. Pharm. Sci. 1972, 61, 1785. [DOI] [PubMed] [Google Scholar]
- Wagner J. G.; Sedman A. J. J. Pharmacokinet. Biopharm. 1973, 1, 23. [Google Scholar]
- Winne D. J. J. Pharmacokinet. Biopharm. 1977, 5, 53. [DOI] [PubMed] [Google Scholar]
- Tsuji A.; Nakashima E.; Kagami I.; Yamana T. J. Pharm. Sci. 1981, 70, 768. [DOI] [PubMed] [Google Scholar]
- Pla-Delfina J. M.; Moreno J. J. Pharmacokinet. Biopharm. 1981, 9, 191. [DOI] [PubMed] [Google Scholar]
- Pla-Delfina J. M.; Perez Buendia M. D.; Casabo V. G.; Peris-Ribera J. E.; Sanches-Moyano E.; Martin-Villodre A. Int. J. Pharm. 1987, 37, 49. [Google Scholar]
- Schaper K. J. In Proceedings of the Third European Congress of Biopharmaceutics and Pharmacokinetics; Aiache J. M., Hirtz J., Eds.; University Press: Clermont-Ferrand, France, 1978; p 14. [Google Scholar]
- Iyer M.; Tseng Y. J.; Senese C. L.; Liu J.; Hopfinger A. J. Mol. Pharm. 2007, 4, 218. [DOI] [PubMed] [Google Scholar]
- Burton P. S.; Conradi R. A.; Ho N. F.; Hilgers A. R.; Borchardt R. T. J. Pharm. Sci. 1996, 85, 1336. [DOI] [PubMed] [Google Scholar]
- Sinko P. J.; Leesman G. D.; Waclawski A. P.; Yu H.; Kou J. H. Pharm. Res. 1996, 13, 570. [DOI] [PubMed] [Google Scholar]
- Grass G. M. Adv. Drug Delivery Rev. 1997, 23, 199. [Google Scholar]
- Palm K.; Stenberg P.; Luthman K.; Artursson P. Pharm. Res. 1997, 14, 568. [DOI] [PubMed] [Google Scholar]
- Tsuji A.; Miyamoto E.; Kagami I.; Sakaguchi H.; Yamana T. J. Pharm. Sci. 1999, 67, 1701. [DOI] [PubMed] [Google Scholar]
- Norris D. A.; Leesman G. D.; Sinko P. J.; Grass G. M. J. Controlled Release 2000, 65, 55. [DOI] [PubMed] [Google Scholar]
- Yoshida F.; Topliss J. G. J. Med. Chem. 2000, 43, 2575. [DOI] [PubMed] [Google Scholar]
- Abraham M. H.; Zhao Y. H.; Le J.; Hersey A.; Luscombe C. N.; Reynolds D. P.; Beck G.; Sherborne B.; Cooper I. Eur. J. Med. Chem 2002, 37, 595. [DOI] [PubMed] [Google Scholar]
- Egan W. J.; Lauri G. Adv. Drug Delivery Rev. 2002, 54, 273. [DOI] [PubMed] [Google Scholar]
- Watanabe J.; Kozaki A. Chem. Pharm. Bull. 1978, 26, 665. [DOI] [PubMed] [Google Scholar]
- Martin Y. C.; Hansch C. J. Med. Chem. 1971, 14, 777. [DOI] [PubMed] [Google Scholar]
- Janssen L. H. M. Eur. J. Med. Chem 1987, 22, 131. [Google Scholar]
- Wagner J. G. J. Clin. Pharmacol. 1999, 20, 89. [Google Scholar]
- Nestorov I. A.; Aarons L. J.; Arundel P. A.; Rowland M. J. Pharmacokinet. Biopharm. 1998, 26, 21. [DOI] [PubMed] [Google Scholar]
- Nestorov I. Clin. Pharmacokinet. 2003, 42, 883. [DOI] [PubMed] [Google Scholar]
- Seydel J. K.; Schaper K. J. Pharmacol. Ther. 1981, 15, 131. [DOI] [PubMed] [Google Scholar]
- Zhao Y. H.; Le J.; Abraham M. H.; Hersey A.; Eddershaw P. J.; Luscombe C. N.; Butina D.; Beck G.; Sherborne B.; Cooper I.; Platts J. A.; Boutina D. J. Pharm. Sci. 2001, 90, 749. [DOI] [PubMed] [Google Scholar]
- Zhao Y. H.; Abraham M. H.; Le J.; Hersey A.; Luscombe C. N.; Beck G.; Sherborne B.; Cooper I. Pharm. Res. 2002, 19, 1446. [DOI] [PubMed] [Google Scholar]
- Zhao Y. H.; Abraham M. H.; Hersey A.; Luscombe C. N. Eur. J. Med. Chem. 2003, 38, 939. [DOI] [PubMed] [Google Scholar]
- Camenisch G.; van de Waterbeemd H.; Folkers G. Pharm. Acta Helv. 1996, 71, 309. [DOI] [PubMed] [Google Scholar]
- Ekins S.; Waller C. L.; Swaan P. W.; Cruciani G.; Wrighton S. A.; Wikel J. H. J. Pharmacol. Toxicol. Methods 2000, 44, 251. [DOI] [PubMed] [Google Scholar]
- Podlogar B. L.; Mügge I.; Brice L. J. Curr. Opin. Drug Discovery Dev. 2001, 4, 102. [PubMed] [Google Scholar]
- Podlogar B. L.; Mügge I. Curr. Top. Med. Chem. 2001, 1, 257. [DOI] [PubMed] [Google Scholar]
- Bains W.; Gilbert R.; Sviridenko L.; Gascon J. M.; Scoffin R.; Birchall K.; Harvey I.; Caldwell J. Curr. Opin. Drug Discovery Dev. 2002, 5, 44. [PubMed] [Google Scholar]
- Butina D.; Segall M. D.; Frankcombe K. Drug Discovery Today 2002, 7, S83. [DOI] [PubMed] [Google Scholar]
- Stenberg P.; Bergström C. A. S.; Luthman K.; Artursson P. Clin. Pharmacokin. 2002, 41, 877. [DOI] [PubMed] [Google Scholar]
- van de Waterbeemd H. Curr. Opin. Drug Discovery Dev. 2002, 5, 33. [PubMed] [Google Scholar]
- Artursson P.; Bergström C. A. S. In Drug Bioavailability: Estimation of Solubility, Permeability, Absorption, and Bioavailability; van de Waterbeemd H., Lennernäs H., Artursson P., Eds.; Wiley−VCH: Weinehim, Germany, 2003; p 341. [Google Scholar]
- Leahy D. E. Curr. Top. Med. Chem. 2003, 3, 1257. [DOI] [PubMed] [Google Scholar]
- Lombardo F.; Gifford E.; Shalaeva M. Y. Mini-Rev. Med. Chem. 2003, 3, 861. [DOI] [PubMed] [Google Scholar]
- Clark D. E.; Grootenhuis P. D. Curr. Top. Med. Chem. 2003, 3, 1193. [DOI] [PubMed] [Google Scholar]
- Dickins M.; van de Waterbeemd H. Drug Discovery Today 2004, 2, 38. [Google Scholar]
- Lajiness M. S.; Vieth M.; Erickson J. Curr. Opin. Drug Discovery Dev. 2004, 7, 470. [PubMed] [Google Scholar]
- Hansch C.; Leo A.; Mekapati S. B.; Kurup A. Bioorg. Med. Chem. 2004, 12, 3391. [DOI] [PubMed] [Google Scholar]
- Leahy D. E. Drug Discovery Today 2004, 2, 78. [Google Scholar]
- Hou T.; Wang J.; Zhang W.; Wang W.; Xu X. Curr. Med. Chem. 2006, 13, 2653. [DOI] [PubMed] [Google Scholar]
- Gola J.; Obrezanova O.; Champness E.; Segall M. QSAR Combin. Sci. 2006, 25, 1172. [Google Scholar]
- Clark R. D.; Wolohan P. R. N. Curr. Top. Med. Chem. 2003, 3, 1269. [DOI] [PubMed] [Google Scholar]
- van de Waterbeemd H. In Drug Bioavailability: Estimation of Solubility, Permeability, Absorption, and Bioavailability; van de Waterbeemd H., Lennernäs H., Artursson P., Eds.; Wiley−VCH: Weinehim, Germany, 2003; p 3. [Google Scholar]
- Harter M. W.; Keldenich J.; Schmitt W. In Handbook of Combinatorial Chemistry, Vol. 2; Nicolaou K. C., Hanko R., Hartwig G., Eds.; Wiley−VCH: Weinheim, Germany, 2002; p 743. [Google Scholar]
- Duffy J. C. In Predicting Chemical Toxicity and Fate; Cronin M. T. D., Livingstone D. J., Eds.; CRC Press: Boca Raton, FL, 2004, p 229. [Google Scholar]
- Oprea T. I.; Benedetti P.; Berellini G.; Olah M.; Fejgin K.; Boyer S. In Molecular Interaction Fields: Applications in Drug Discovery and ADME Prediction; Cruciani G., Ed.; Wiley−VCH: Weinheim, Germany, 2006; p 249. [Google Scholar]
- van de Waterbeemd H., Lennernäs H., Artursson P., Eds. Drug Bioavailability: Estimation of Solubility, Permeability, Absorption, and Bioavailability; Wiley−VCH: Weinheim, Germany, 2003; p 1. [Google Scholar]
- Watari N.; Sugiyama Y.; Kaneniwa N.; Hiura M. J. Pharmacokin. Biopharm. 1988, 16, 279. [DOI] [PubMed] [Google Scholar]
- Houston J. B.; Upshall D. G.; Bridges J. W. J. Pharmacol. Exp. Ther. 1975, 195, 67. [PubMed] [Google Scholar]
- Ho N. F. H.; Higuchi W. I.; Turi J. J. Pharm. Sci. 1972, 61, 192. [DOI] [PubMed] [Google Scholar]
- Merino V.; Freixas J.; Val Bermejo M.; Garrigues T. M.; Moreno J.; Pla-Delfina J. M. J. Pharm. Sci. 1995, 84, 777. [DOI] [PubMed] [Google Scholar]
- Casabo V. G.; Nunez-Benito E.; Martinez-Coscolla A.; Miralles-Loyola E.; Martin-Villodre A.; Pla-Delfina J. M. J. Pharmacokin. Biopharm. 1987, 15, 633. [DOI] [PubMed] [Google Scholar]
- Collado E. F.; Fabra-Campos S.; Peris-Ribera J. E.; Casabo V. G.; Martin-Villodre A.; Pla-Delfina J. M. Int. J. Pharm. 1988, 44, 187. [Google Scholar]
- Lennernäs H. Pharm. Res. 1995, 12, 1573. [DOI] [PubMed] [Google Scholar]
- Camenisch G.; Folkers G.; van de Waterbeemd H. Eur. J. Med. Chem. 1998, 6, 321. [PubMed] [Google Scholar]
- Bermejo M.; Merino V.; Garrigues T. M.; Pla-Delfina J. M.; Mulet A.; Vizet P.; Trouiller G.; Mercier C. J. Pharm. Sci. 1999, 88, 398. [DOI] [PubMed] [Google Scholar]
- Raevsky O. A.; Fetisov V. I.; Trepalina E. P.; McFarland J. W.; Schaper K. J. Quant. Struct. Act. Relat. 2000, 19, 366. [Google Scholar]
- Rey S.; Caron G.; Ermondi G.; Gaillard P.; Pagliara A.; Carrupt P. A.; Testa B. J. Mol. Graph. Model. 2001, 19, 521. [DOI] [PubMed] [Google Scholar]
- Deretey E.; Feher M.; Schmidt J. M. Quant. Struct. Act. Relat. 2002, 21, 493. [Google Scholar]
- Raevsky O. A.; Schaper K. J.; Artursson P.; McFarland J. W. Quant. Struct. Act. Relat. 2002, 20, 402. [Google Scholar]
- Veber D. F.; Johnson S. R.; Cheng H. Y.; Smith B. R.; Ward K. W.; Kopple K. D. J. Med. Chem. 2002, 45, 2615. [DOI] [PubMed] [Google Scholar]
- Lu J. J.; Crimin K.; Goodwin J. T.; Crivori P.; Orrenius C.; Xing L.; Tandler P. J.; Vidmar T. J.; Amore B. M.; Wilson A. G. E.; Stouten P. F. W.; Burton P. S. J. Med. Chem. 2004, 47, 6104. [DOI] [PubMed] [Google Scholar]
- Egan W. J.; Merz K. M. Jr.; Baldwin J. J. J. Med. Chem. 2000, 43, 3867. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Adv. Drug Delivery Rev. 1997, 23, 3. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A. J. Pharmacol. Toxicol. Methods 2001, 44, 235. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A. Drug Discovery Today 2004, 1, 337. [DOI] [PubMed] [Google Scholar]
- Hou T.; Wang J.; Zhang W.; Xu X. J. Chem. Inf. Model. 2007, 47, 208. [DOI] [PubMed] [Google Scholar]
- Oprea T. I.; Gottfries J. J. Mol. Graph. Model. 1999, 17, 261. [DOI] [PubMed] [Google Scholar]
- Wessel M. D.; Jurs P. C.; Tolan J. W.; Muskal S. M. J. Chem. Inf. Comput. Sci. 1998, 38, 726. [DOI] [PubMed] [Google Scholar]
- Agatonovic-Kustrin S.; Beresford R.; Yusof A. P. M. J. Pharm. Biomed. Anal. 2001, 25, 227. [DOI] [PubMed] [Google Scholar]
- Gohlke H.; Dullweber F.; Kamm W.; Marz J.; Kissel T.; Klebe G. In Rational Approaches to Drug Design; Höltje H. D., Sippl W., Eds.; Prous: Barcelona, Spain, 2001; p 261. [Google Scholar]
- Andrews C. W.; Bennett L.; Yu L. X. Pharm. Res. 2000, 17, 639. [DOI] [PubMed] [Google Scholar]
- Klopman G.; Stefan L. R.; Saiakhov R. D. Eur. J. Pharm. Sci. 2002, 17, 253. [DOI] [PubMed] [Google Scholar]
- Zmuidinavicius D.; Didziapetris R.; Japertas P.; Avdeef A.; Petrauskas A. J. Pharm. Sci. 2003, 92, 621. [DOI] [PubMed] [Google Scholar]
- Wolohan P. R. N.; Clark R. D. J. Comput. Aid. Mol. Des. 2003, 17, 65. [DOI] [PubMed] [Google Scholar]
- Hirono S.; Nakagome I.; Hirano H.; Matsushita Y.; Yoshii F.; Moriguchi I. Biol. Pharm. Bull. 1994, 17, 306. [DOI] [PubMed] [Google Scholar]
- Niwa T. J. Chem. Inf. Comput. Sci. 2003, 43, 113. [DOI] [PubMed] [Google Scholar]
- Turner J. V.; Maddalena D. J.; Agatonovic-Kustrin S. Pharm. Res. 2004, 21, 68. [DOI] [PubMed] [Google Scholar]
- Perez M. A. C.; Sanz M. B.; Torres L. R.; Avalos R. G.; Gonzalez M. P.; Diaz H. G. D. Eur. J. Med. Chem. 2004, 39, 905. [DOI] [PubMed] [Google Scholar]
- Wegner J. K.; Froehlich H.; Zell A. J. Chem. Inf. Comput. Sci. 2004, 44, 931. [DOI] [PubMed] [Google Scholar]
- Congreve M.; Carr R.; Murray C.; Jhoti H. Drug Discovery Today 2003, 8, 876. [DOI] [PubMed] [Google Scholar]
- Vieth M.; Sutherland J. J. J. Med. Chem. 2006, 49, 3451. [DOI] [PubMed] [Google Scholar]
- Vieth M.; Siegel M. G.; Higgs R. E.; Watson I. A.; Robertson D. H.; Savin K. A.; Durst G. L.; Hipskind P. A. J. Med. Chem. 2004, 47, 224. [DOI] [PubMed] [Google Scholar]
- Martin Y. C. J. Med. Chem. 2005, 48, 3164. [DOI] [PubMed] [Google Scholar]
- Dressman J. B.; Amidon G. L.; Fleisher D. J. Pharm. Sci. 1985, 74, 588. [DOI] [PubMed] [Google Scholar]
- Sanghvi T.; Ni N.; Yalkowsky S. H. Pharm. Res. 2001, 18, 1794. [DOI] [PubMed] [Google Scholar]
- Kostewicz E. S.; Brauns U.; Becker R.; Dressman J. B. Pharm. Res. 2002, 19, 345. [DOI] [PubMed] [Google Scholar]
- Wu C. Y.; Benet L. Z. Pharm. Res. 2005, 22, 11. [DOI] [PubMed] [Google Scholar]
- Küntz M.; Nick S.; Parrott N.; Röthlisberger D. Eur. J. Pharm. Sci. 2006, 27, 91. [DOI] [PubMed] [Google Scholar]
- Yu L. X.; Crison J. R.; Amidon G. L. Int. J. Pharm. 1996, 140, 111. [Google Scholar]
- Yu L. X.; Amidon G. L. Int. J. Pharm. 1998, 171, 157. [Google Scholar]
- Yu L. X.; Amidon G. L. Eur. J. Pharm. Biopharm. 1998, 45, 199. [DOI] [PubMed] [Google Scholar]
- Yu L. X.; Amidon G. L. Int. J. Pharm. 1999, 186, 119. [DOI] [PubMed] [Google Scholar]
- Agoram B.; Woltosz W. S.; Bolger M. B. Adv. Drug Delivery Rev. 2001, 50, S41. [DOI] [PubMed] [Google Scholar]
- Bolger M. B.; Agoram B.; Fraczkiewicz R.; Steere B. In Drug Bioavailability: Estimation of Solubility, Permeability, Absorption, and Bioavailability; van de Waterbeemd H., Lennernäs H., Artursson P., Eds.; Wiley−VCH: Weinheim, Germany, 2003; p 420. [Google Scholar]
- Kalampokis A.; Argyrakis P.; Macheras P. Pharm. Res. 1999, 16, 1764. [DOI] [PubMed] [Google Scholar]
- Tam D.; Tirona R. G.; Pang K. S. Drug Metab. Dispos. 2003, 31, 373. [DOI] [PubMed] [Google Scholar]
- Willmann S.; Schmitt W.; Keldenich J.; Lippert J.; Dressman J. B. J. Med. Chem. 2004, 47, 4022. [DOI] [PubMed] [Google Scholar]
- Grass G. M.; Bozarth C. A.; Vallner J. J. J. Drug Target. 1994, 2, 23. [DOI] [PubMed] [Google Scholar]
- Balaz S.; Wiese M.; Kansy M.; Han-lin Chi Seydel J. K. Chemosphere 1989, 19, 1677. [Google Scholar]
- Manga N.; Duffy J. C.; Rowe P. H.; Cronin M. T. D. QSAR Combin. Sci. 2003, 22, 263. [Google Scholar]
- Ng C.; Xiao Y.; Putnam W.; Lum B.; Tropsha A. J. Pharm. Sci. 2004, 93, 2535. [DOI] [PubMed] [Google Scholar]
- Doddareddy M. R.; Cho Y. S.; Koh H. Y.; Kim D. H.; Pae A. N. J. Chem. Inf. Model. 2006, 46, 1312. [DOI] [PubMed] [Google Scholar]
- Jusko W. J. In Applied Pharmacokinetics; Evans W. E., Schentag J. J., Jusko W. J., Eds.; Applied Therapeutics: San Francisco, CA, 1980; p 639. [Google Scholar]
- Watanabe J.; Kozaki A. Chem. Pharm. Bull. 1978, 26, 3463. [DOI] [PubMed] [Google Scholar]
- Hinderling P. H. Progr. Pharmacol. 1988, 6, 1. [Google Scholar]
- Balaz S. Am. J. Pharm. Educ. 2002, 66, 66. [Google Scholar]
- Lombardo F.; Obach R. S.; Shalaeva M. Y.; Gao F. J. Med. Chem. 2002, 45, 2867. [DOI] [PubMed] [Google Scholar]
- Herman R. A.; Veng-Pedersen P. J. Pharm. Sci. 1994, 83, 423. [DOI] [PubMed] [Google Scholar]
- Ghafourian T.; Barzegar-Jalali M.; Hakimiha N.; Cronin M. T. D. J. Pharm. Pharmacol. 2004, 56, 339. [DOI] [PubMed] [Google Scholar]
- Hirono S.; Nakagome I.; Hirano H.; Yoshii F.; Moriguchi I. Biol. Pharm. Bull. 1994, 17, 686. [DOI] [PubMed] [Google Scholar]
- Ghafourian T.; Barzegar-Jalali M.; Dastmalchi S.; Khavari-Khorasani T.; Hakimiha N.; Nokhodchi A. Int. J. Pharm. 2006, 319, 82. [DOI] [PubMed] [Google Scholar]
- Gleeson M. P.; Waters N. J.; Paine S. W.; Davis A. M. J. Med. Chem. 2006, 49, 1953. [DOI] [PubMed] [Google Scholar]
- Hollosy F.; Valko K.; Hersey A.; Nunhuck S.; Keri G.; Bevan C. J. Med. Chem. 2006, 49, 6958. [DOI] [PubMed] [Google Scholar]
- Lombardo F.; Obach R. S.; DiCapua F. M.; Bakken G. A.; Lu J.; Potter D. M.; Gao F.; Miller M. D.; Zhang Y. J. Med. Chem. 2006, 49, 2262. [DOI] [PubMed] [Google Scholar]
- Woodside R.; Zief M.; Sumrell G. Antibiot. Chemother. 1959, 9, 470. [PubMed] [Google Scholar]