

Abstract
Minichromosome maintenance (MCM) proteins are believed to provide the replicative helicase activity in eukaryotes and archaea. The single MCM orthologue from Methanothermobacter thermautotrophicus (MthMCM) has been extensively characterised as a model of the eukaryotic heterohexameric MCM complex. MthMCM forms high molecular weight complexes in solution consistent with a dodecamer. Visualization of this complex by electron microscopy suggests that single and double heptameric or hexameric rings can form. We have mutated two arginine residues (R137, R160) in the N-terminal sub-domain B of MthMCM based on their apparent potential to form inter-ring hydrogen bonds. Both the single R137A and the double RR137,160AA mutants were characterised by a combination of biophysical, biochemical and electron microscopy (EM) techniques. Biophysical analysis coupled with EM studies shows that the R137A mutant forms a double heptameric ring, whereas the RR137,160AA protein assembles as a single heptamer. They both show a defect in DNA binding and a concomitant conformational change in sub-domain A, with the double mutant displaying significant defects in helicase activity as well. We propose a model in which MCM loading and the subsequent activation of the helicase activity involves a conformational transition that is connected to a DNA binding event.
The process of DNA replication in eukaryotic organisms involves a complicated interplay between a number of proteins required for licensing origins of replication and restricting the replication of the genome to only once per cell cycle (1-5). The putative replicative helicase consists of six discrete minichromosome maintenance (MCM) subunits, MCM2-7, that form a variety of sub-complexes as well as a complete heterohexamer (6-9). Archaea are a group of organisms with features of both eukaryotic and prokaryotic cells (10). Interestingly, archaeal DNA processing enzymes tend to be simplified homologues of the equivalent eukaryotic machinery rather than homologues of bacterial proteins. For this reason the archaeal MCM complex is a good model for understanding some of the mechanisms associated with eukaryotic helicase activity. The archaeal MCM protein can be divided into three distinct domains (11): an N-terminal domain (discussed in detail below), a central ATPase domain that contains the NTP binding and hydrolysis motifs essential for helicase activity (12), and a less well-defined C-terminal domain. A pre-sensor 1 β-hairpin and helix-2 insert in the ATPase domain have been shown to be essential for DNA unwinding (13,14). A role for the C-terminus in DNA binding has been proposed (14).
The crystal structure for the N-terminal domain of MthMCM has been solved, revealing that this domain forms two opposing hexameric rings, with each monomer consisting of three structural sub-domains (15). Sub-domain B encompasses a zinc-finger motif, sub-domain C provides the contact between the N-terminal and the ATPase domains and contains a highly conserved loop that functions to transmit signals between domains (16), and sub-domain A forms the remainder of the N-terminus. Sub-domains B and C were identified as essential for hexamerisation and dodecamerisation (17). Deletion of each sub-domain in turn demonstrated that sub-domain C was sufficient for hexamerisation and essential for helicase activity, while sub-domain B was found to interact with ssDNA. No specific function was assigned to sub-domain A (17), but recent structural and biochemical work has highlighted its role as a novel DNA binding site, possibly involved in the initial loading of the protein onto DNA (18).
The MthMCM protein is unusual among MCMs in forming stable double rings in solution (19-21). MCM complexes from other archaea, such as Sulfolobus solfataricus and Archaeoglobus fulgidus, have been reported to form single ring complexes in solution (14,22,23), although the A. fulgidus MCM can form double ring complexes in the presence of DNA in a concentration dependent manner (23). Electron microscopy (EM) studies have shown that MthMCM can produce a large variety of structures, including single and double rings, each containing either six or seven subunits (24-26) as well as helical fibres (27). A systematic study of the complex in the presence of various substrates showed that treatment with nucleotides and/or short stretches of dsDNA led to the stabilization of double-ring structures, whereas untreated protein tends to form single hexameric or heptameric rings. More specifically, the presence of nucleotide analogues triggers the formation of double heptamers, while a consistent shift from a double heptameric to a double hexameric arrangement was shown to be associated with DNA binding, which suggests that the double hexamer is an active form of the protein, with the heptameric complex possibly a configuration ready to load onto DNA (28,29).
The crystal structure of the N-terminal MthMCM domain shows that the head-to-head interface between the two rings involves the bottom face of sub-domain B, containing a Zn domain of the CXXCXnCXXC (C4) type. This motif is present in other archaeal MCMs and in a modified form (HC3) in S. solfataricus and Aeropyrum pernix MCMs (22). A C4 zinc motif is also conserved in eukaryotic MCMs apart from MCM3, which has an unconventional motif probably also capable of binding zinc with the sequence (C/A)XXTXn(S/T)X7D (N. Atassanova, pers. com.). The conservation of this motif suggests that it may play an important role in the function of these proteins.
Close examination of the crystal structure has allowed us to identify two arginine residues located at the hexamer-hexamer interface as potential sources of hydrogen bonding between the rings (R137 and R160, Fig. 1A). The R160 residue has been previously characterized as producing a single ring complex consistent with a hexamer (described as R161 in (30)). The R160A mutant protein showed wild-type (WT) ATPase activity in the absence of DNA, and WT ssDNA and dsDNA binding in the absence of ATP (30). We have used site directed mutagenesis to produce a R137A mutant and a RR137,160AA (RRAA) double mutant and characterised the resultant proteins. Electron microscopy and biophysical analysis are consistent with the single R137A mutant forming a double heptamer and the RRAA mutant a single heptamer. Three-dimensional reconstructions showed that both mutants are characterised by a large conformational change that is consistent with a swing-out movement of sub-domain A within the N-terminal domain. The R137A mutant shows reduced binding to short DNA substrates, but this defect does not significantly affect the ability of the mutant protein to function as a processive DNA helicase. In contrast, the RRAA double mutant shows a severely compromised ability to bind short linear DNA substrates and a reduction in processive DNA unwinding compared to WT (despite WT ATPase activity in the presence and absence of DNA).
Fig. 1. Identification of R137 and R160 as residues potentially involved in ring-ring interactions.

(A) Detail of the hexamer-hexamer interface from the crystal structure of the N-terminal domain of MthMCM ((15); Pdb reference: 1LTL) The α-carbon traces for two monomers from the top hexamer (green and yellow) and one from the bottom hexamer (blue) are shown; Zinc atoms are in magenta. The hydrogen bonding pattern involving R137 and R160 with main chain carbonyls is shown in red. The figure was generated with MacPyMol (DeLano Scientific, USA).
(B) ClustalX (40) alignment of zinc-finger regions of various MCM proteins. Zinc-finger residues are highlighted in orange, other conserved residues are highlighted in grey. R137 and R160, and corresponding arginines in other MCMs are indicated in white on red. Species names are abbreviated as follows: Mth, Methanothermobacter thermautotrophicus; Hal, Halobacterium sp. pNRC-1; Afu, Archaeoglobus fulgidus; Pfu, Pyrococcus furiosus; Ape, Aeropyrum pernix; Sso, Sulfolobus solfataricus; Sc, Saccharomyces cerevisiae.
Our results show that mutation of R137 appears to reduce the transient interaction of the complex with DNA, causes a swing-out movement of sub-domain A and potentially inhibits the loading of the complex perhaps by preventing the required remodelling of the ring-shaped complex. Our data support a regulatory mechanism whereby MCMs may be loaded onto DNA through a conformational transition that involves DNA binding and may cause an activation of helicase activity, consistent with previous work in this system (18).
Experimental Procedures
Cloning and Mutagenesis
pJC025 (19) was used as a template for site directed mutagenesis. Mutations were made using the Quikchange kit (Stratagene). R137 was mutated to alanine using a mutagenic primer 5′-GAG TGC CGT GGG TGT ATG GCG CAC CAT GCG GTA ACA CAG-3′ (mutation underlined) and its complementary partner. The resultant construct was further mutated to incorporate the R160 mutation using primer 5′-GCT CAG AGT GTG GTG GGG CAT CCT TCA GGC TCC TTC AGG-3′ and its complementary partner. Constructs were verified by sequencing and designated pJC098 and pJC104 respectively. Residues in MthMCM were counted from the second methionine.
Protein purification
E. coli expressing WT MthMCM or mutant protein was grown using a 3 litre fed batch culture and processed as previously described (13,19).
Gel filtration
15 μg of purified recombinant protein was loaded onto a 2.4 ml Superose 6 column (GE Biosciences). Samples were diluted prior to loading into 50 μl 30 mM Tris pH 7.5, 300 mM NaCl for WT protein or 30 mM Tris pH 7.5, 500 mM NaCl for RRAA. The column was pre-equilibrated in 30 mM Tris pH 7.5, 300 mM NaCl and run at 20 μl/min. The column was calibrated using native molecular weight markers (GE Biosciences).
Sucrose gradients
4.8 ml of 20% sucrose buffer (50 mM Hepes pH 7.6, 150 mM NaCl, 1 mM DTT and 20% sucrose) and 4.8 ml of 50% sucrose buffer (as for 20% but with 50% sucrose) were mixed to form a gradient. 100 μg protein was diluted to 400 μl in 10 mM Tris pH 7.5 and loaded onto the gradient. A control sample containing native molecular weight markers (GE Biosciences) was loaded in parallel. Samples were spun at 40000 rpm (Beckman L-60, SW41TI), for 24 hours at 4°C with no brake. 400 μl fractions were recovered from the gradients and the proteins precipitated using deoxycholate / trichloroacetic acid before SDS-PAGE analysis.
Analytical Ultracentrifugation
Samples were diluted in 20 mM Tris pH 7.5 to an OD of 0.4 at A280. Sedimentation equilibrium experiments using three dilutions for each protein were carried out on a Beckman Optima XL/I analytical ultracentrifuge at 20°C in 12 mm path length cells using an AN-50Ti rotor. 115-120 μl of each sample was spun at 3000, 4200, 5500 and 8000 rpm using an overspeed method (31) until sedimentation equilibrium was achieved. Sedimentation velocity ultracentrifugation experiments were carried out at 20000 rpm on samples at an OD of 0.3-0.35 at A280. Centrifugation was carried out using an AN-60Ti rotor with 416-420 μl sample at various dilutions.
DNA binding assay
Reactions used either 2.5 nM T4 PNK radiolabelled single stranded 30mer oligo (5′-GGG TAA CGC CAG GGT TTT CCC AGT CAC GAC-3′) or a 30mer duplex made by annealing a complementary oligo to the labelled strand. DNA templates were incubated with increasing amounts of protein (60-960 nM monomer), and processed as previously described (19).
Strand displacement
Helicase template was made using complementary oligos with a 24 nucleotide overhang. PM2 (5′-GGA CAT GCT GTC TAG AGA CTA TCG AT-3′) was labelled using T4 PNK. Unincorporated [γ32P] ATP was removed by a G25 spin column (GE Biosciences). The labelled oligo was annealed to RGL16 (5′-ATC GAT AGT CTC TAG ACA GCA TGT CCT AGC AAG CCA GAA TTC GGC AGC GT-3′). Reaction mix containing HDB, 7 mM MgCl2, 1 nM labelled template, increasing amounts of protein (0-300 nM monomer) and 4 mM ATP was prepared on ice. Reactions were processed as described (13).
Processivity
A population of closed circular molecules with increasingly long dsDNA regions was made as described previously (19). 6 nM substrate was incubated with 60-240 nM (dodecamer) MCM protein in 40 μl reactions containing 1xHDB (19), 7 mM MgCl2 and 4 mM ATP for 30 min at 50°C. Samples were processed (32), normalised and separated on a 5% polyacrylamide gel supplemented with 8 M urea. The gel was dried and displaced products were visualised using a phosphorimager.
Electron microscopy
A 5 μl sample was applied to continuous-carbon-coated copper grids (Agar Scientific, UK), and treated with 2% uranyl acetate. Data for the WT MthMCM protein were collected on a Tecnai12 microscope (FEI), while for the RRAA and R137A mutants data were collected on a Philips CM200 FEG electron microscope, at a nominal magnification of 50000x. Micrographs were scanned using a Nikon Super Coolscan 8000 at a pixel size of 1.3 Å on the specimen scale and image processing was carried out using the IMAGIC-5 package (33). Datasets for the various proteins were generated from digitised micrographs, according to the shape and dimensions of the protein complexes: a set of “ring shaped” datasets was created selecting particles which presented a circular arrangement and framing them in a 144×144 square box (2665 images for WT protein, 2438 images for RRAA). Single images were initially band-pass filtered with a low frequency cut-off of 150 Å and a high frequency cut off of 15 Å. After centring to their rotationally averaged total sum, the ring shaped particle images were treated with multivariate statistical analysis to determine the main symmetry components contained (MSA symmetry analysis, (34)). In order to suppress low frequency noise generated by the negative stain, images were band-pass filtered with a low frequency cut-off of 40 Å and a high frequency cut-off of 15 Å before inspection of the eigenimages. Class averages were produced containing either ~10 particles per class (RRAA mutant) or ~20 particles per class (R137A mutant). Further MSA mirror-symmetry analysis was performed on double-ring side view particles aligned to a low-passed filtered characteristic side view (35). Single particle reconstruction of the R137A mutant was performed using 1560 selected double-ring side and tilted views while ring-shaped top views were ignored. Seven-fold symmetry was imposed, based on the evidence derived from MSA mirror symmetry analysis. The resolution achieved was 30 Å, according to the Fourier shell correlation (1/2 bit criterion). Single particle reconstitution for the RRAA mutant was carried out using 1600 particles and by applying seven-fold symmetry based on the result of MSA symmetry analysis. The structure was refined to a resolution of 22 Å, according to the 1/2 bit criterion.
RESULTS
Identification of residues putatively required for ring dimerisation
By analysing the crystal structure of the dodecameric MthMCM N-terminal domain (15), (PDB: 1LTL), we identified arginine residues (R137, R160) on either side of the zinc finger motif with the potential to form hydrogen bonds across the ring-ring interface (Fig. 1A). Alignment of this region in archaeal and eukaryotic MCMs showed that neither of these residues is conserved. Most eukaryotic MCM3 orthologues possess an arginine (or lysine) corresponding to R137 and most MCM6 proteins possess an arginine (or lysine) residue corresponding to R160 (Fig. 1B). To determine whether R137 plays an additional role in MthMCM double ring formation, we generated a R137A mutant and a RR137,160AA double mutant (referred to as RRAA) for further characterization.
Biophysical characterization of the RRAA mutant is consistent with formation of a heptamer
Biophysical analysis to determine the size of the RRAA complex was carried out (Fig. 2). Size exclusion chromatography detected a complex smaller than the wild-type (WT), but larger than a single hexamer (Fig. 2A). Sucrose gradient sizing was consistent with WT protein forming double rings and the RRAA mutant forming smaller complexes (Fig. 2B,C). Calculations combining gel filtration and sucrose gradient data (36) resulted in an apparent molecular weight of 898 kDa for the WT complex (equivalent to 11.6 monomers) and a molecular weight of 592 kDa for the RRAA complex (equivalent to 7.6 monomers). Sedimentation equilibrium analytical ultracentrifugation analysis provided a WT molecular weight of 948 kDa, (or 12.2 subunits/complex) (Fig. 2D). A similar analysis of the RRAA complex provided a size estimate of 665 kDa (~8.6 subunits), with some evidence of complex heterogeneity (Fig. 2E). Neither of the samples showed a detectable change in apparent molecular weight associated with protein concentration, suggesting little in the way of dynamic equilibrium between different oligomeric states of significantly different sizes. Sedimentation velocity ultracentrifugation studies were also consistent with the WT protein forming a double ring complex (Fig. 2F), although a small peak corresponding to heptamers was also detected. The RRAA data were consistent with a heterogeneous mix of predominantly heptameric complexes (Fig. 2G).
Fig. 2. Biophysical characterisation of the RRAA mutant is consistent with the formation of heptameric complexes.

(A) Size estimation of WT (solid line) and RR137,160AA (broken line) proteins by gel filtration. Diamonds indicate the position of molecular weight markers with masses indicated in kDa. (B) WT and (C) RRAA mutant protein fractions subjected to SDS-PAGE from sucrose gradient centrifugation. Estimates of complex size were determined based on elution peak fractions compared to the fraction position of native molecular markers (in kDa) as indicated by arrows. Each lane is annotated with a fraction identity (numbered from the top of the gradient). SDS-PAGE size standards are indicated by “M” from top to bottom, markers are 250, 150, 100, 75, 50, 37, 25 and 20 kDa. (D) WT and (E) RRAA sedimentation equilibrium analytical ultracentrifugation traces from experiments performed at 4200 rpm. (F) WT and (G) RRAA transformation analyses of sedimentation velocity analytical ultracentrifugation performed using SEDFIT (41), showing some heterogeneity of samples and apparent molecular weights.
Electron microscopy of the RRAA protein confirms the formation of single heptameric rings
Samples of WT, R137A and RRAA protein were visualised using negative stain electron microscopy (Supp. Fig. 1A). The samples showed a heterogeneous population of molecules that fell into two main categories: particles consistent with ring-shaped molecules, and rod-like aggregates with helical characteristics.
The WT protein consisted mostly of single rings. MSA symmetry analysis shows a strong heptameric component (Supp. Fig. 1B). However, when top-view class averages are examined, a significant proportion of hexameric classes can be detected. Although the indication derived from MSA symmetry analysis is unbiased and statistically more significant, this probably suggests the presence of a degree of polymorphism within the sample. When 2438 “end-on” ring-shaped particles of the RRAA protein were selected and treated with multivariate statistical analysis (MSA), the resulting eigenimages showed a strong 7-fold symmetry component (Supp. Fig. 1B), while class averages showing heptameric single rings were obtained (Supp. Fig. 1C). In contrast, electron microscopy of the R137A mutant was consistent with this protein still readily forming double rings. Although the absence of end-on views hindered a canonical MSA symmetry analysis approach, the presence of anti-mirror symmetric eigenimages deriving from aligned double-ring side view particles (35) suggests that this mutant forms double heptamers (Supp. Fig. 1C).
Both mutant proteins reveal an unusual conformation of the N-terminal domain
A 3D reconstruction was carried out for the RRAA and R137A mutant proteins, showing heptameric single- and double-ring complexes, respectively (Fig. 3). When this model is compared with the heptameric double ring wild-type reconstruction previously obtained in the presence of an ATP transition state analogue (28), a striking conformational change can be observed, with large movements involving sub-domain A within the N-terminal domain (Fig. 3). The movement is reminiscent of the swing-out movement of sub-domain A that has been previously inferred based on biochemical and crystallography data (15,30) and visualised in EM reconstruction of the N-terminal domain alone (27) or the full-length protein bound to large segments of dsDNA (18).
Fig. 3. 3D reconstructions of the R137A and RRAA mutants show large conformational changes involving the N-terminal domain.

Three-dimensional models for WT double heptamer (left panel, (28)), the R137A (middle panel) and RRAA (right panel) mutants are compared. Red and green boxes highlight the location of the AAA+ and N-terminal domains respectively. A round yellow circle indicates the position of sub-domain A.
A DNA binding phenotype is associated with R137A
The MthMCM R160A mutant has previously been reported to display WT DNA binding activity (30). We compared the DNA-binding activities of WT, R137A and RRAA proteins using short linear ssDNA and dsDNA substrates in the presence and absence of hydrolysable ATP. In the absence of ATP, both mutants were indistinguishable from each other, showing a substantial decrease in their ability to bind ssDNA (Fig. 4A) and dsDNA (Fig. 4C) under these conditions. In the presence of ATP, a more complicated phenotype was observed: the RRAA mutant showed greatly decreased binding of ssDNA compared to WT (Fig. 4B), with the R137A mutation alone showing an intermediate effect. In the presence of ATP the R137A mutant appeared WT in its ability to bind dsDNA (Fig. 4D). In contrast, the RRAA mutant showed essentially WT ATP hydrolysis activity in the presence and absence of DNA (Supp. Fig. 2).
Fig. 4. Characterisation of DNA binding activities of R137 and R160.

Protein, indicated in nM (monomer), was incubated at room temperature in the presence of 32P-labelled 30mer ssDNA or dsDNA before being subjected to agarose gel electrophoresis. Protein-bound DNA was quantified following autoradiography as a proportion of the total labelled substrate. All data points are the average of at least three independent experiments. Error bars indicate standard deviation from the mean. WT, closed circles, continuous line; R137A, open diamonds, continuous line; RRAA, open circles, broken line. (A) ssDNA substrate in the absence of ATP, (B) ssDNA substrate in the presence of 4 mM ATP, (C) dsDNA substrate in the absence of ATP, (D) dsDNA substrate in the presence of 4 mM ATP .
The R137A and R160A mutations have a cumulative effect on DNA helicase activity
WT MthMCM shows 3′-5′ processive helicase activity (19-21), with linear duplexes requiring a 3′ tail for efficient activity (37). We used a standard strand displacement assay to determine the effect of the mutations on DNA helicase activity (Fig. 5). 30 nM (monomer) WT protein resulted in maximal unwinding of 1 nM forked substrate (Fig. 5A, D). Increasing WT protein concentrations above 60 nM resulted in a slight inhibition of duplex unwinding activity. Protein concentrations above 300 nM had a more dramatic inhibitory effect (not shown). The R137A mutant showed similar levels of unwinding to WT, but this required 60-120 nM protein (Fig. 5C, D). Similar amounts of the RRAA protein as the R137A protein were required for peak activity, but in this case only ~ 50% of the maximal levels of substrate strand displacement activity were observed (Fig. 5B, D). We investigated this defect in DNA unwinding further by employing a processivity assay (Fig. 6). In this assay the R137A protein performed as well as the WT protein, unwinding duplexes of up to ~350 bp when present in large excess over substrate (Fig. 6, compare lanes 12-14 to lanes 6- 8). In contrast, the RRAA mutant showed poorer processive helicase activity, unwinding only ~ 150 bp under the same conditions (Fig. 6, lanes 9-11).
Fig. 5. Strand displacement activity of RRAA mutant is substantially reduced compared to WT.
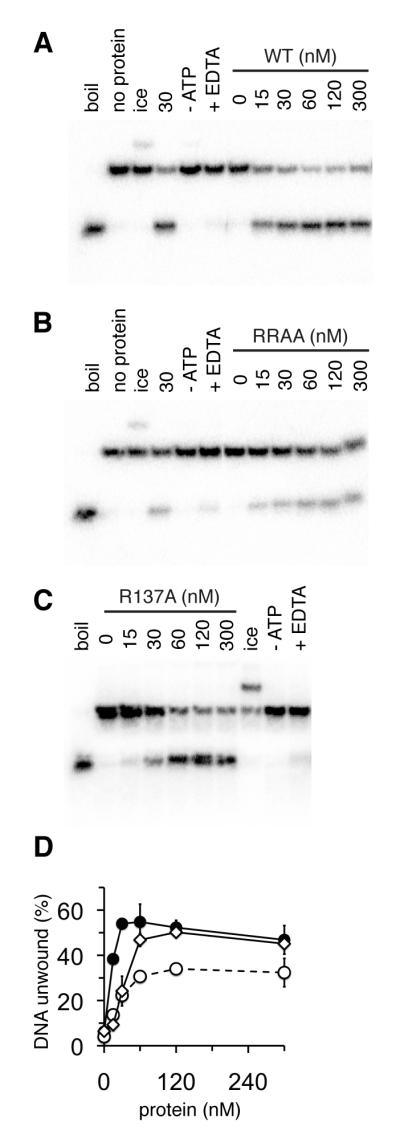
Autoradiographs showing strand displacement of a labelled 24mer annealed to a 50mer to produce a template with a 3′ ssDNA region. Lanes are: (“boil”), substrate boiled for 5 mins in the absence of protein; (“no protein”), substrate alone; (“ice”), reaction incubated on ice; (“- ATP’), reaction without ATP; (“+EDTA”), reaction in the presence of 10 mM EDTA. Remaining lanes are reactions containing increasing concentrations of MthMCM protein indicated in nM (monomer). (A) WT, (B) RRAA, (C) R137A. (D) Quantification of DNA unwinding activity from experiments shown in (A) – (C). Unwinding was calculated as a percentage of 24mer displaced from the 50mer template. All points are an average of at least three experiments and error bars show the standard deviation from the mean. WT, closed circles, continuous line; R137A, open diamond, continuous line; RRAA, open circles, broken line.
Fig. 6. Strand displacement activity is compromised in the RRAA mutant.

Autoradiograph showing products of a strand displacement assay. 6 nM substrate was incubated in the presence of various concentrations of protein (nM dodecamer, as indicated) for 30 min at 50°C. Lane 1, labelled oligonucleotide used in substrate extension reaction; lane 2, DNA ladder; lane 3, reaction incubated on ice; lane 4, boiled substrate; lane 5, reaction without protein; lanes 6-8, increasing concentrations of WT protein; lanes 9-11, increasing concentrations of RRAA protein; lanes 12-14, increasing concentrations of R137A protein.
DISCUSSION
A number of electron microscopy studies on MthMCM have highlighted a remarkable degree of polymorphism, raising the question of the physiological significance of the multiple conformations observed in vitro and their role in the mechanism of helicase activity. One possible way to address this question is to study the biochemical properties of specific mutants that trap, stabilise or hinder a particular stoichiometry.
A range of MthMCM complexes have been described, ranging from double hexamers and heptamers (24,28,29) to single hexamers (25,30) and heptamers (26). The WT MthMCM protein forms a labile double ring in solution, which is not stable enough to be easily visualised by electron microscopy. A recent study suggests that the likely active conformation of the MthMCM helicase, at temperature and ionic strengths comparable to those existing in M. thermautotrophicus cells, is a single ring (38). Consistent with this information, the R160A mutation hinders the formation of double ring complexes but does not interfere with helicase activity.
In a parallel study, we identified both R160 and R137 as likely to have a key role in the formation of a double ring and generated two mutants designed to disrupt the double ring interface, namely the single mutant R137A and the double mutant RR137,160AA (RRAA, Fig 1). In addition to carrying out electron microscopy studies, we complemented our investigation with analytical ultracentrifugation, and obtained a good agreement between the two techniques. Contrary to our expectations, the single mutant R137A was still able to form stable double ring complexes. EM studies showed the presence of a double heptamer, with a pronounced conformational change involving a swing-out movement of sub-domain A (Supp. Fig. 1, Fig. 3). A biochemical analysis of the mutant showed a decreased ability to bind single and double stranded DNA and a mild defect in DNA helicase activity (Fig. 4, 5).
The double mutant RRAA visualised by electron microscopy appears as a single heptamer, with a similar conformational change in sub-domain A (Supp. Fig 1, Fig. 3). Our sizing data are consistent with the RRAA protein forming a complex that is a single ring in solution, although this protein exhibits some heterogeneity. Hydrodynamic studies suggest that the RRAA mutant is slightly larger than the heptameric conformation observed by EM. This difference can be explained by the large conformational change of sub-domain A. A similar effect on the apparent size of the protein has been described for the P61L mutant (equivalent to the P83L bob1 mutant in MCM5) and has been correlated to a swing out movement of sub-domain A (30).
A comparison between the behaviour of the R160A and the R137A single mutants with the RRAA double mutant allows one to decouple the effects of the mutations. Mutation of R160 affects the stoichiometry of the complex without influencing its DNA binding or helicase activity (38). Introduction of a second mutation into the single ring (R137A) results in substantially reduced DNA binding (Fig. 4) and significantly reduced DNA helicase activity (Fig. 5, 6), despite this complex showing WT DNA-dependent ATP hydrolysis (Supp. Fig. 2). In parallel, the R137 mutation causes a large conformational change involving the position of sub-domain A (Fig. 3). The same conformational change has been proposed to trigger a critical step in the activation of MCM helicase activity (15,39), and has been related to the initial mode of association of MCM with dsDNA, before loading occurs (18). This mode of binding involves dsDNA wrapping around the N-terminal domain ring, sandwiched between the swung-out sub-domain A and sub-domain B, and our modelling, based on EM results, places the DNA very close to the R137 residue, providing a possible explanation for the reduced DNA binding ability of the R137A mutant.
We suggest that whereas the WT protein may contain a mixture of heptamers and hexamers, easily interconverted in the absence of substrates, the R137A mutation stabilises the heptameric form, “trapping” what is normally a more labile, transient intermediate and therefore interfering with the conformational changes that are required for the activation of the helicase activity. The R137A structure shows a clear asymmetry between the two rings, with a more evident swing-out of sub-domain A in the upper ring, associated with the formation of a cap (Fig. 3). This is reminiscent of the asymmetry observed in the presence of dsDNA, where the emergence of the cap was interpreted as an ordering of the C-terminal domains (28). Both the asymmetry and the conformational change at the AAA+/C-terminal end of the protein, upon mutation of a residue at the very bottom of the N-terminal domain, are further evidence of the long range inter- and intra-molecular cross-talk between the various domains.
While neither the R137 nor the R160 residues are conserved throughout the MCMs, these residues can potentially reveal the molecular mechanisms that govern loading of the MCM complex onto DNA in both archaea and eukaryotes. The loading process may involve a series of intermediate configurations, including a heptameric complex with a swung-out sub-domain A, ready for the initial association with dsDNA (as visualised in the R137A mutant), to a hexameric complex where duplex DNA initially associates with the outside N-terminal face of the MCM making interactions with the R137 side chain (as observed in (18)), to an active hexameric or double-hexameric MCM encircling the DNA as a processive helicase. Although R137 is not conserved in archaea, an arginine or lysine is present in this position in almost all MCM3 subunits in eukaryotes. Thus, this mutant may act as a paradigm for an intermediate in the association of the MCM complex with DNA.
Supplementary Material
Acknowledgments
We thank Dr Zvi Kelman for sharing data prior to publication, members of the Chong laboratory for comments on the manuscript, M. van Heel for use of the EM Facility, J. van den Elsen for help with molecular visualisations, M. Polonska and J. Cartwright (York TF) for assistance with large-scale E. coli cultures and protein purification and E. Bolt for providing helicase template oligos. This work was supported by a BBSRC David Phillips Research Fellowship (Refs 86/AF/14817 and BB/X510703/1) and University of York Innovation and Research Priming Funds awarded to JPJC, and an Imperial College studentship awarded to AC.
The abbreviations used are
- MCM
minichromosome maintenance
- WT
wild-type
- RRAA
RR137,160AA MthMCM mutant
REFERENCES
- 1.Blow JJ, Dutta A. Nat Rev Mol Cell Biol. 2005;6:476–486. doi: 10.1038/nrm1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Donovan S, Harwood J, Drury LS, Diffley JF. Proc Natl Acad Sci U S A. 1997;94:5611–5616. doi: 10.1073/pnas.94.11.5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dutta A, Bell SP. Annu Rev Cell Dev Biol. 1997;13:293–332. doi: 10.1146/annurev.cellbio.13.1.293. [DOI] [PubMed] [Google Scholar]
- 4.Lisziewicz J, Godany A, Agoston DV, Kuntzel H. Nucleic Acids Res. 1988;16:11507–11520. doi: 10.1093/nar/16.24.11507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stillman B. Science. 1996;274:1659–1664. doi: 10.1126/science.274.5293.1659. [DOI] [PubMed] [Google Scholar]
- 6.Ishimi Y. J Biol Chem. 1997;272:24508–24513. doi: 10.1074/jbc.272.39.24508. [DOI] [PubMed] [Google Scholar]
- 7.Moyer SE, Lewis PW, Botchan MR. Proc Natl Acad Sci U S A. 2006;103:10236–10241. doi: 10.1073/pnas.0602400103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prokhorova TA, Blow JJ. J Biol Chem. 2000;275:2491–2498. doi: 10.1074/jbc.275.4.2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tye BK. Proc Natl Acad Sci U S A. 2000;97:2399–2401. doi: 10.1073/pnas.97.6.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edgell DR, Doolittle WF. Cell. 1997;89:995–998. doi: 10.1016/s0092-8674(00)80285-8. [DOI] [PubMed] [Google Scholar]
- 11.Chong JP. Nat Struct Mol Biol. 2005;12:734–736. doi: 10.1038/nsmb0905-734. [DOI] [PubMed] [Google Scholar]
- 12.Koonin EV. Nucleic Acids Res. 1993;21:2541–2547. doi: 10.1093/nar/21.11.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jenkinson ER, Chong JP. Proc Natl Acad Sci U S A. 2006;103:7613–7618. doi: 10.1073/pnas.0509297103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McGeoch AT, Trakselis MA, Laskey RA, Bell SD. Nat Struct Mol Biol. 2005;12:756–762. doi: 10.1038/nsmb974. [DOI] [PubMed] [Google Scholar]
- 15.Fletcher RJ, Bishop BE, Leon RP, Sclafani RA, Ogata CM, Chen XS. Nat Struct Biol. 2003;10:160–167. doi: 10.1038/nsb893. [DOI] [PubMed] [Google Scholar]
- 16.Sakakibara N, Kasiviswanathan R, Melamud E, Han M, Schwarz FP, Kelman Z. Nucleic Acids Res. 2008;36:1309–1320. doi: 10.1093/nar/gkm1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kasiviswanathan R, Shin JH, Melamud E, Kelman Z. J Biol Chem. 2004;279:28358–28366. doi: 10.1074/jbc.M403202200. [DOI] [PubMed] [Google Scholar]
- 18.Costa A, van Duinen G, Medagli B, Chong J, Sakakibara N, Kelman Z, Nair SK, Patwardhan A, Onesti S. EMBO J. 2008;27:2250–2258. doi: 10.1038/emboj.2008.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chong JP, Hayashi MK, Simon MN, Xu RM, Stillman B. Proc Natl Acad Sci U S A. 2000;97:1530–1535. doi: 10.1073/pnas.030539597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelman Z, Lee JK, Hurwitz J. Proc Natl Acad Sci U S A. 1999;96:14783–14788. doi: 10.1073/pnas.96.26.14783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shechter DF, Ying CY, Gautier J. J Biol Chem. 2000;275:15049–15059. doi: 10.1074/jbc.M000398200. [DOI] [PubMed] [Google Scholar]
- 22.Carpentieri F, De Felice M, De Falco M, Rossi M, Pisani FM. J Biol Chem. 2002;277:12118–12127. doi: 10.1074/jbc.M200091200. [DOI] [PubMed] [Google Scholar]
- 23.Grainge I, Scaife S, Wigley DB. Nucleic Acids Res. 2003;31:4888–4898. doi: 10.1093/nar/gkg662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gomez-Llorente Y, Fletcher RJ, Chen XS, Carazo JM, San Martin C. J Biol Chem. 2005;280:40909–40915. doi: 10.1074/jbc.M509760200. [DOI] [PubMed] [Google Scholar]
- 25.Pape T, Meka H, Chen S, Vicentini G, van Heel M, Onesti S. EMBO Rep. 2003;4:1079–1083. doi: 10.1038/sj.embor.7400010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu X, VanLoock MS, Poplawski A, Kelman Z, Xiang T, Tye BK, Egelman EH. EMBO Rep. 2002;3:792–797. doi: 10.1093/embo-reports/kvf160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen YJ, Yu X, Kasiviswanathan R, Shin JH, Kelman Z, Egelman EH. J Mol Biol. 2005;346:389–394. doi: 10.1016/j.jmb.2004.11.076. [DOI] [PubMed] [Google Scholar]
- 28.Costa A, Pape T, van Heel M, Brick P, Patwardhan A, Onesti S. Nucleic Acids Res. 2006;34:5829–5838. doi: 10.1093/nar/gkl708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Costa A, Pape T, van Heel M, Brick P, Patwardhan A, Onesti S. J Struct Biol. 2006;156:210–219. doi: 10.1016/j.jsb.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 30.Fletcher RJ, Shen J, Gomez-Llorente Y, Martin CS, Carazo JM, Chen XS. J Biol Chem. 2005;280:42405–42410. doi: 10.1074/jbc.M509773200. [DOI] [PubMed] [Google Scholar]
- 31.Roark DE. Biophys. Chem. 1976;5:185–196. doi: 10.1016/0301-4622(76)80034-8. [DOI] [PubMed] [Google Scholar]
- 32.Castella S, Burgin D, Sanders CM. Nucleic Acids Res. 2006;34:3731–3741. doi: 10.1093/nar/gkl554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Heel M, Harauz G, Orlova EV, Schmidt R, Schatz M. J Struct Biol. 1996;116:17–24. doi: 10.1006/jsbi.1996.0004. [DOI] [PubMed] [Google Scholar]
- 34.Dube P, Tavares P, Lurz R, van Heel M. Embo J. 1993;12:1303–1309. doi: 10.1002/j.1460-2075.1993.tb05775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Costa A, Patwardhan A. J Mol Biol. 2008;378:273–283. doi: 10.1016/j.jmb.2008.02.054. [DOI] [PubMed] [Google Scholar]
- 36.Li M, Desiderio S. Physical characterisation of DNA-binding proteins in crude preparations. In: Latchman DS, editor. Transcription Factors - A practical approach. IRL Press; Oxford: 1993. [Google Scholar]
- 37.Shin JH, Reeve JN, Kelman Z. Nucleic Acids Res. 2005;33:e8. doi: 10.1093/nar/gni009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shin J-H, Heo G-Y, Kelman Z. J Biol Chem. doi: 10.1074/jbc.M806803200. (in press) [DOI] [PubMed] [Google Scholar]
- 39.Hoang ML, Leon RP, Pessoa-Brandao L, Hunt S, Raghuraman MK, Fangman WL, Brewer BJ, Sclafani RA. Mol Cell Biol. 2007;27:7594–7602. doi: 10.1128/MCB.00997-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schuck P. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.