

Summary
The Notch signaling pathway plays a central role in animal growth and patterning, and its deregulation leads to many human diseases, including cancer [1, 2]. Mutations in the tumor suppressor lethal giant discs (lgd) induce strong Notch activation and hyperplastic overgrowth of Drosophila imaginal discs [3–5]. However, the gene that encodes lgd and its function in the Notch pathway have not yet been identified. Here we report that Lgd is a novel, conserved C2 domain protein that regulates Notch receptor trafficking. Notch accumulates on early endosomes in lgd mutant cells and signals in a ligand-independent manner. This phenotype is similar to that seen when cells lose endosomal pathway components such as Erupted and Vps25 [6–9]. Interestingly, Notch activation in lgd mutant cells requires the early endosomal component Hrs, indicating that Hrs is epistatic to Lgd. Our data indicate that Lgd affects Notch trafficking between the actions of Hrs and the late endosomal component Vps25. Taken together, our data identify Lgd as a novel tumor suppressor protein that regulates Notch signaling by targeting Notch for degradation or recycling.
Results
Lgd is a novel cytoplasmic C2 domain protein
To enable molecular analysis of Lethal giant discs (Lgd) function, we identified the lgd gene by P-element recombination mapping (Fig. S1A) [10]. We found mutations in the coding region of CG4713 in all known lgd alleles, and a genomic rescue construct containing the entire genomic region of CG4713 (Fig. S1B) was sufficient to rescue the lethality of lgd mutations. Therefore CG4713 is lgd.
lgd encodes a novel 816 amino acid protein (Fig. 1A and Fig. S1C). Lgd is conserved in vertebrates, which have two homologs of Lgd that we call HsLgd1 and HsLgd2. The two human homologs are also known as CC2D1B and CC2D1A, respectively. A recent study by Basel-Vanagaite et al. indicated that mutations in HsLgd2 caused non-syndromic mental retardation [11], suggesting an important role for the Lgd homologs in vertebrates.
Figure 1. Lgd is a novel cytoplasmic C2 domain protein.
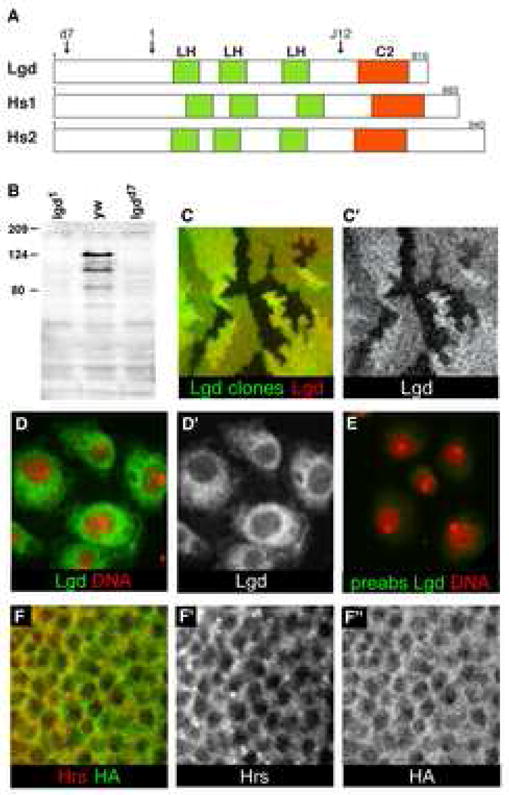
(A) Schematic representation of the protein structures of Drosophila lgd and the two human homologs. The arrow labeled ‘d7’ indicates the position at which the lgdd7 allele has a frameshift mutation: the removal of a single nucleotide results in a new translational frame and a premature stop codon after the addition of 22 new amino acids. The arrow labeled ‘1’ indicates the position at which the lgd1 allele has a frameshift mutation: two nucleotides are removed resulting in a new translational frame and a premature stop codon after the addition of 15 new amino acids. The arrow labeled ‘J12’ indicates the position at which the EMS-induced allele, lgdJ12, contains a nonsense mutation: Q626 is changed to a stop codon. Green boxes indicate the Lgd homology domains (LH), and orange boxes indicate C2 domains. (B) Western blot to detect Lgd protein in extracts from lgd1, yw, and lgdd7 third instar larvae. The lgd1 and lgdd7 alleles truncate the protein N-terminally to the epitope used to generate the Lgd antibodies. The band at approximately 120kD contained the majority of Lgd protein, and was missing in both of the lgd mutant lanes. Other specific bands of lower molecular weight were also occasionally seen. (C–C′) Lgd antibodies specifically detect Lgd protein in imaginal discs. lgd mutant clones, indicated by the lack of GFP (green), stained with α–Lgd antibodies (red in C and gray in C′). (D–E) S2 cells stained with Lgd antibodies (green in D and E and gray in D′) and Topro (red). (E) Lgd antibodies incubated with Lgd antigen prior to use for staining. The preabsorbed Lgd antibodies (green) did not display specific staining, demonstrating that the Lgd antibodies specifically detect the Lgd antigen. (F–F″) C5 Gal4 driven UAS-HA-Lgd in the wing pouch of third instar imaginal discs, detected by α-HA (green in F and gray in F″). HA did not specifically colocalize with α-Hrs (red in F and gray in F′).
The proteins in the Lgd family share a conserved C2 domain and three Lgd homology (LH) domains (Fig. 1A and Fig. S1C). The LH domain is not found in any other protein family, and its function is unknown. C2 domains are known as calcium and lipid binding domains, and they are thought to have many functions such as protein-protein interaction, membrane recruitment, protein localization, and trafficking [12, 13].
The lgdd7 and lgd1 alleles have frameshift mutations, resulting in premature stop codons in the N-terminal half of the protein, such that both alleles have all of the conserved domains of the protein deleted (see Fig. 1A legend). We also performed an EMS mutagenesis screen to find new alleles of lgd. We found an allele, lgdJ12, containing a point mutation that creates a stop codon immediately before the C2 domain of the protein (Fig. 1A). This allele has an equivalent phenotype to that of the lgdd7 and lgd1 alleles, signifying the importance of the C2 domain for the function of Lgd.
We generated antibodies against the Lgd protein and used them to analyze Lgd expression and subcellular localization. We found that α–Lgd specifically detects Lgd protein on western blots, in S2 cells, and in imaginal discs (Fig 1B–E). Lgd was present endogenously in S2 and KC cells and localized in the cytoplasm (Fig. 1D–D′). Antibodies pre-incubated with Lgd antigen did not give specific staining, indicating that the antibodies specifically recognized Lgd protein (Fig. 1E). Lgd was expressed ubiquitously in imaginal discs where it also localized in the cytoplasm (Fig. S2B′ and data not shown). We also expressed an HA-tagged version of the Lgd protein in imaginal discs, and it showed the same localization as the anti-Lgd antibodies (Fig. S2). Neither α–Lgd nor HA-tagged Lgd appeared to specifically colocalize with common organelle markers such as Hrs, an early endosomal protein (Fig. 1F–F″). All together, these data indicate a cytoplasmic localization of Lgd.
Lgd acts cell autonomously to restrict Notch activation to target cells
To further understand the function of Lgd, we focused on the phenotype in the developing wing. Notch activation along the dorsal-ventral boundary specifies margin cell fates and induces the expression of target genes such as wingless (wg) and Cut along the margin (Fig. S3D) [14–16]. Ectopic activation of Notch away from the margin induces neighboring blade tissue to grow and form extra wing tissue [17]. In lgd mutant wing discs, Notch target gene expression is expanded: the expression of E(spl) m8-LacZ (a reporter of Notch signaling [18]) extended into the entire wing pouch (Fig. S3A–C), and the expression of Cut extended into most of the wing pouch (Fig. S3D–E). The deregulation of Notch target genes eventually leads to massively overgrown imaginal discs in lgd mutants (Fig. S3A–C and [3–5]).
To determine whether Lgd was required to regulate Notch activity in the signal-sending cell or the signal-receiving cell, we generated clones of lgd mutant cells using the Flipase (FLP) - FRT system [19] and examined whether or not Notch activation was cell autonomous. We used Cut expression as a readout for Notch activation because Cut is cell-autonomously induced by high levels of Notch signaling [20, 21]. We found that only lgd mutant cells (marked by the lack of GFP expression in Fig. 2A) ectopically upregulated Notch signaling as measured by the induction of Cut expression (Fig. 2A–A′). Ectopic Cut expression did not extend into the wild-type or lgd+/− cells outside of the lgd mutant clones (Fig. 2A–A′), as we would expect if Lgd acted in signaling cells to suppress ligand function. In addition, Lgd does not act non-autonomously by repressing Notch activation on adjacent cells because mutant cells immediately adjacent to the clonal boundary expressed ectopic Cut in the region around the wing margin (Fig. 2A). We conclude that Lgd acts cell autonomously to regulate Notch signaling in the cell in which Notch is activated.
Figure 2. Lgd acts cells autonomously and restricts Notch activation in a ligand-independent manner.
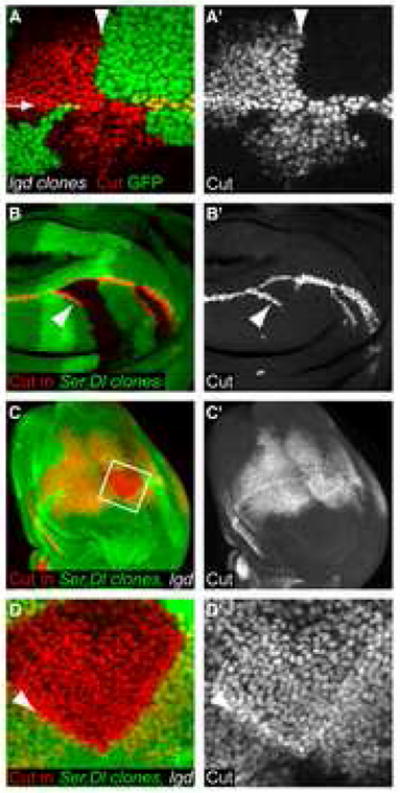
(A and A′) Third instar wing disc with lgdd10 mutant clones marked by the absence of GFP (Green). The discs were stained for Cut (red in A, gray in A′), a target of the Notch pathway. The arrow in (A) indicates the wing margin. The arrowhead in (A and A′) marks the clone boundary, showing the demarcation between Cut-expressing and non-expressing cells. Only lgd mutant cells (lacking GFP) showed Cut activation outside of the wing margin. (B–B′) Dlrev10,SerRX106 mutant clones in a wild-type wing disc. Cut (red in B and gray in B′) was ectopically expressed in some cells adjacent to the clone boundaries. (C–D′) Dlrev10,SerRX106 mutant clones in an lgd1/d7 wing imaginal disc. D and D′ show higher magnification of the region boxed in (C). The disc is stained for Cut (red in C and D, shown singly in gray in C′ and D′), and the lack of GFP expression (green in C and D) marks the clone. (D–D′) Cut expression extended throughout the clone. Also, the levels of Cut expression were higher along the boundary of the clone (arrowhead), demonstrating the presence of cis-inhibition in the absence of Lgd.
Lgd is not required for cis-inhibition
It was previously suggested that cis-inhibition is disrupted in lgd mutant cells [5]. ‘Cis-inhibition’ is the term used to describe the phenomenon where the ligands Serrate and Delta cell-autonomously suppress activation of Notch in a cell that would otherwise show activated Notch signaling [21–23]. For example, overexpression of the Notch ligand Serrate in wild-type wing disc cells caused Cut suppression in the cells that expressed high levels of Serrate [24] (See supplementary information, Fig. S3D and Fig. S3F–F′). In lgd mutant wing discs, we found that expression of Serrate could still suppress Notch activation cell-autonomously (Fig. S3G–G′). This result suggests that Lgd is not required for cis-inhibition. In addition, we found that binding between Notch and Delta was not affected by either lgd RNAi or the overexpression of Lgd (UAS-Lgd) in Alkaline-Phosphatase (AP) binding assays [25] (See supplementary information, Fig. S4).
Lgd restricts Notch activation in a ligand-independent manner
Notch activation normally requires interaction with ligands on neighboring cells [20, 26]. To determine if the presence of ligands is required for the activation of Notch in the absence of Lgd, we made clones of Dl−, Ser− double mutant cells in lgd mutant discs (Fig. 2C–D′). In contrast to the wild-type situation (Fig. 2B–B′), we found that lgd mutant cells throughout the Dl−, Ser− double mutant clones still displayed active Notch signaling (Fig. 2C–D′). Therefore the ligands were not required for Notch activation in lgd mutant cells. As the receptor and ligands are membrane bound, it is unlikely that the Notch receptor in cells in the center of the clone (~15 cell diameters away from the clone boundary) can be activated by ligands outside the clone. We conclude that Notch signaling can be activated independently of the known ligands Delta and Serrate in lgd mutant wing discs.
Mutations in lgd cause disruptions in Notch trafficking
Ligand-independent activation of Notch signaling was recently observed in cell culture studies by knocking down the endosomal ESCRT-II component Vps25 [9]. In imaginal discs, vps25 mutant cells exhibit increased accumulation of Notch on endosomes, resulting in ectopic Notch signaling (Fig. 3D–D′ and [7–9]). Similarly, we found that Notch accumulated in subcellular puncta more frequently in lgd mutant cells than in the surrounding wild-type tissue (Fig. 3A–A′). We observed this result with both Notch intracellular and extracellular domain localization (Fig. 3A and Fig. 3B). These puncta often costained with the early endosomal marker Hrs (Hepatocyte growth factor-regulated substrate) [27, 28] (Fig. 3A‴), indicating that Notch accumulates on early endosomes in lgd mutant cells. Hrs is required for maturation of endosomes into multivesicular bodies [27, 28]. Similarly to the loss of Lgd, the loss of Hrs also leads to accumulation of Notch in subcellular puncta. (Fig. 3E–E′ and [7, 29]). All together, our data indicate that Notch trafficking is altered in lgd mutant cells such that Notch accumulates in Hrs-positive early endosomes.
Figure 3. Mutations in lgd cause disruptions in Notch trafficking.

(A–A‴) An lgdd7 mutant clone in a third instar wing disc, indicated by the absence of ubiGFP (blue), stained for α-NICD (green in A and A‴, gray in A′) and α-Hrs (red in A and A‴, gray in A″). NICD and Hrs accumulated together in lgd mutant cells (arrowhead). (B–B′) An lgdd7 mutant clone in a third instar wing disc, indicated by the absence of ubiGFP (blue) stained for α-NECD (green in B, gray in B′). The arrowhead indicates one of the subcellular puncta that shows accumulation of the extracellular part of the Notch protein. (C–C′) vps25, UAS-P35 MARCM mutant clones in the third instar wing disc, indicated by the presence of UAS-GFP (blue), stained for α-Hrs (red in C, gray in C′). The arrowhead indicates one of the subcellular puncta that expressed Hrs. (D–D‴) vps25 MARCM mutant clones in a third instar wing disc, indicated by the presence of UAS-GFP (blue) stained for α-NICD (green in D and D‴, gray in D′) and α-Lgd (red in D and D‴, gray in D″). Both NICD and Lgd protein accumulate in the vps25 mutant cells. (E–E′) An hrsD28 mutant clone in a third instar wing disc, indicated by the absence of ubiGFP (blue), stained for α-NICD (green in E and E‴ and gray in E′) and α-Lgd (red in E and E‴ gray in E″). NICD staining accumulated in the mutant cells, but there was no change in Lgd protein levels or localization within the clone compared to wild-type cells.
Hrs is required for Notch activation in lgd mutant cells
In the current model of endocytosis trafficking, Hrs acts upstream of Vps25 [30]. Accordingly, Hrs protein accumulated in vps25 mutant cells (Fig. 3C–C′) [7]. We found that, like Hrs, Lgd protein also accumulated in subcellular puncta and elsewhere in vps25 mutant cells (Fig. 3D″), indicating that Lgd may also act upstream of Vps25. In hrs mutant cells, we found no change in Lgd protein levels or localization (Fig. 3E–E′). However, Hrs protein accumulated in lgd mutant cells, suggesting that Lgd could possibly act downstream of Hrs (Fig. 3A″).
If Lgd acts downstream of Hrs, then the function of Hrs may be required for the transport of Notch to the place in the endosomal pathway in which it can ectopically signal in lgd mutant cells. To test this, we compared the levels of Notch activation, as assayed by Cut induction, between hrs, lgd double mutant clones to each of the single mutant clones. hrs mutant clones did not induce ectopic Cut expression (Fig. 4B–B′). In lgd mutant clones, Cut was ectopically induced (Fig. 4A–A′). In contrast to lgd single mutant clones, hrs, lgd double mutant clones did not show ectopic Notch activation (Fig. 4C–C′). Therefore the induction of Cut in lgd mutant cells requires the function of Hrs. Taken together, our results suggest that Hrs is epistatic to Lgd.
Figure 4. Hrs is required for Notch activation in lgd mutant cells.
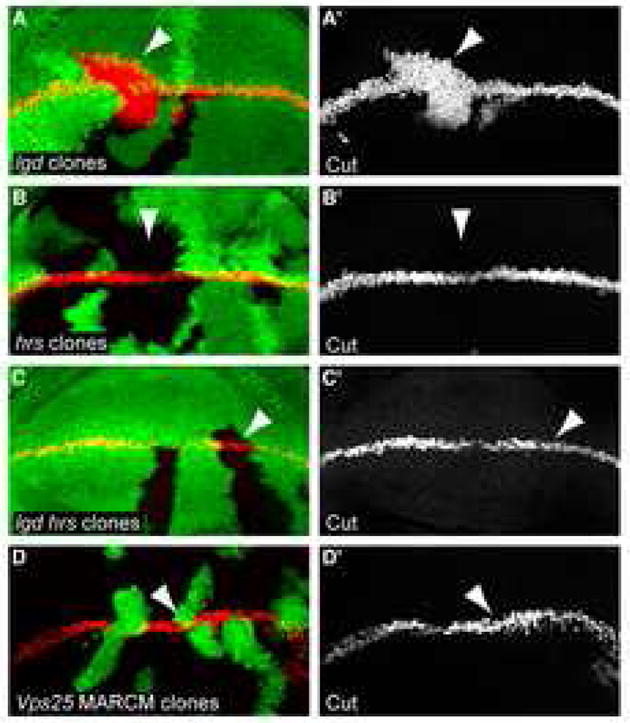
(A–A′) lgdd7 mutant clones in a third instar imaginal wing disc, indicated by the absence of ubiGFP (green), stained for Cut (red in A and gray in A′). Cut, a target gene activated by the Notch pathway, was induced in the lgd mutant tissue in the area surrounding the wing margin. The ectopic Cut induction does not appear to be autonomous in these clones only because this image and all panels in this figure are extended focus views of the whole disc, instead of single focal planes as in Fig. 1D. (B–B′) hrsD28 mutant clones in a third instar imaginal wing disc, indicated by the absence of ubiGFP (green), stained for α-Cut (red in B and gray in B′). Unlike in lgd mutant clones, Cut was not induced in the hrs mutant tissue. (C–C′) hrsD28, lgdd7 double mutant clones in a third instar imaginal wing disc, indicated by the absence of ubiGFP (green), stained for α-Cut (red in C and gray in C′). Cut was not induced in the cells lacking both Lgd and Hrs, suggesting that the hrs mutant phenotype is epistatic over the lgd mutant phenotype. (D–D′) vps25 MARCM mutant clones in the third instar wing disc, indicated by the presence of nuclear UAS-GFP (green), stained for α-Cut (red in D and gray in D′). Cut was not induced in the mutant tissue.
Discussion
In this report, we have identified Lgd as a novel C2 domain protein, and our results indicate that it acts by regulating Notch trafficking. We propose a model in which Lgd functions as a negative regulator of Notch through endosomal sorting of Notch downstream of Hrs function. The loss of Lgd resulted in the accumulation of Notch in early endosomes, and our results suggest that this triggered a signaling event that was distinct from normal activation of Notch signaling. Our data indicate that Notch can be activated in a ligand-independent manner in lgd mutant cells (Fig. 2C–D′). Additionally, cells that lack both Hrs and Lgd did not display ectopically activated Notch signaling as measured by Cut expression (Fig. 4C–C′). However, doubly mutant cells at the wing margin were still able to express margin-specific genes. Therefore Hrs is not required for normal (ligand-dependent) Notch signaling, but it is required for the ectopic activation of Cut expression found in lgd mutant cells.
lgd mutant cells display both similarities and differences compared with cells that are mutant for vps25, a known endosomal trafficking component. Both mutations induce ectopic Notch signaling resulting in tissue overgrowth, and both mutations alter Notch trafficking. However, lgd mutant cells induce higher levels of Notch signaling than do vps25 mutant cells (Cut was not notably ectopically activated in vps25 mutant cells- Fig 4D–D′), do not induce apoptosis, and can survive into adulthood. Also unlike vps25 mutants, lgd mutant cells have no significant defects in cell polarity and do not accumulate increased levels of ubiquitylated proteins (data not shown). It is thought that Vps25 is an endosomal component used to sort many different molecules, whereas Lgd might act specifically in the Notch pathway [5]. We therefore propose a model where Lgd function is required to target full length Notch for endosomal degradation or recycling. It is likely that the removal of Lgd function leaves Notch in an optimal position or modification state for γ-secretase cleavage. The molecular mechanism by which Lgd affects Notch trafficking is currently not known, as we have found no evidence of direct binding between Notch and Lgd by immunoprecipitation.
It is important to note that the subcellular location of the γ-secretase complex cleavage of Notch (S3 cleavage) remains controversial. The traditional view is that the cleavage of Notch occurs at the plasma membrane [31]. However, this view conflicts with the evidence that endocytosis is required for Notch signaling in Drosophila [32]. When protein internalization is blocked by shibire mutations, Notch signaling is eliminated [32]. A different view of the location of Notch S3 cleavage was recently developed when the γ-secretase enzyme Presenilin was shown to have a low optimal pH, suggesting that it could be active in the acidic endocytic compartments [33, 34]. It is possible that differentially processed Notch could be activated in separate cellular compartments. In accordance with the model proposed by Hori et al. [35], Notch activation in the ligand-dependent canonical pathway may occur at the plasma membrane or in endocytic vesicles, whereas Lgd-regulated activation of Notch may occur later, at Hrs-positive endosomes.
Supplementary Material
Acknowledgments
We apologize to the authors of the papers that could not be acknowledged due to space constraints. We thank N. Baker, H. Bellen, A. Bergmann, K. Choi, S. Cohen, C. Delidakis, P. R. Hiesinger, K. Irvine, H. Jafar-Nejad, Y. N. Jan, G. Mardon, A. Martinez-Arias, I. Mellman, M. Milan, D. Ready, K. Schulze, E. Seto, A. Singh, G. Struhl, P. Verstreken, M. Young, the Developmental Studies Hybridoma Bank, University of Iowa and the Bloomington Drosophila Stock Center, Indiana for fly stocks and reagents. We also thank A. Bergmann, H. M. Herz, P.R. Hiesinger, R. Johnson, R. Kopan, J. Qin, B. Su, and members of the Halder lab for disscusion. We thank Leisa McCord for assistance with graphics and Xin Wei Zhang for technical assistance. J.L.C. was supported by National Institutes of Health Training Grant 5 T32-HD07325. This work was supported in part by an NIH grant to G.H. and research grant number 1-FY05-106 from the March of Dimes birth defects foundation to G.H.
Footnotes
Competing Interests statement The authors declare that they have no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schweisguth F. Notch signaling activity. Curr Biol. 2004;14:R129–138. [PubMed] [Google Scholar]
- 2.Miele L. Notch signaling. Clin Cancer Res. 2006;12:1074–1079. doi: 10.1158/1078-0432.CCR-05-2570. [DOI] [PubMed] [Google Scholar]
- 3.Buratovich MA, Bryant PJ. Enhancement of overgrowth by gene interactions in lethal(2)giant discs imaginal discs from Drosophila melanogaster. Genetics. 1997;147:657–670. doi: 10.1093/genetics/147.2.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agrawal N, Joshi S, Kango M, Saha D, Mishra A, Sinha P. Epithelial hyperplasia of imaginal discs induced by mutations in Drosophila tumor suppressor genes: growth and pattern formation in genetic mosaics. Dev Biol. 1995;169:387–398. doi: 10.1006/dbio.1995.1155. [DOI] [PubMed] [Google Scholar]
- 5.Klein T. The tumour suppressor gene l(2)giant discs is required to restrict the activity of Notch to the dorsoventral boundary during Drosophila wing development. Dev Biol. 2003;255:313–333. doi: 10.1016/s0012-1606(02)00052-0. [DOI] [PubMed] [Google Scholar]
- 6.Moberg KH, Schelble S, Burdick SK, Hariharan IK. Mutations in erupted, the Drosophila ortholog of mammalian tumor susceptibility gene 101, elicit non-cell-autonomous overgrowth. Dev Cell. 2005;9:699–710. doi: 10.1016/j.devcel.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 7.Vaccari T, Bilder D. The Drosophila tumor suppressor vps25 prevents nonautonomous overproliferation by regulating notch trafficking. Dev Cell. 2005;9:687–698. doi: 10.1016/j.devcel.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 8.Herz HM, Chen Z, Scherr H, Lackey M, Bolduc C, Bergmann A. vps25 mosaics display non-autonomous cell survival and overgrowth, and autonomous apoptosis. Development. 2006;133:1871–1880. doi: 10.1242/dev.02356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thompson BJ, Mathieu J, Sung HH, Loeser E, Rorth P, Cohen SM. Tumor suppressor properties of the ESCRT-II complex component Vps25 in Drosophila. Dev Cell. 2005;9:711–720. doi: 10.1016/j.devcel.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 10.Zhai RG, Hiesinger PR, Koh TW, Verstreken P, Schulze KL, Cao Y, Jafar-Nejad H, Norga KK, Pan H, Bayat V, Greenbaum MP, Bellen HJ. Mapping Drosophila mutations with molecularly defined P element insertions. Proc Natl Acad Sci U S A. 2003;100:10860–10865. doi: 10.1073/pnas.1832753100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Basel-Vanagaite L, Attia R, Yahav M, Ferland RJ, Anteki L, Walsh CA, Olender T, Straussberg R, Magal N, Taub E, Drasinover V, Alkelai A, Bercovich D, Rechavi G, Simon AJ, Shohat M. The CC2D1A, a member of a new gene family with C2 domains, is involved in autosomal recessive non-syndromic mental retardation. J Med Genet. 2006;43:203–210. doi: 10.1136/jmg.2005.035709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nalefski EA, Falke JJ. The C2 domain calcium-binding motif: structural and functional diversity. Protein Sci. 1996;5:2375–2390. doi: 10.1002/pro.5560051201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ponting CP, Parker PJ. Extending the C2 domain family: C2s in PKCs delta, epsilon, eta, theta, phospholipases, GAPs, and perforin. Protein Sci. 1996;5:162–166. doi: 10.1002/pro.5560050120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blair SS. Mechanisms of compartment formation: evidence that non-proliferating cells do not play a critical role in defining the D/V lineage restriction in the developing wing of Drosophila. Development. 1993;119:339–351. doi: 10.1242/dev.119.2.339. [DOI] [PubMed] [Google Scholar]
- 15.Diaz-Benjumea FJ, Cohen SM. Serrate signals through Notch to establish a Wingless-dependent organizer at the dorsal/ventral compartment boundary of the Drosophila wing. Development. 1995;121:4215–4225. doi: 10.1242/dev.121.12.4215. [DOI] [PubMed] [Google Scholar]
- 16.Dahmann C, Basler K. Compartment boundaries: at the edge of development. Trends Genet. 1999;15:320–326. doi: 10.1016/s0168-9525(99)01774-6. [DOI] [PubMed] [Google Scholar]
- 17.Go MJ, Eastman DS, Artavanis-Tsakonas S. Cell proliferation control by Notch signaling in Drosophila development. Development. 1998;125:2031–2040. doi: 10.1242/dev.125.11.2031. [DOI] [PubMed] [Google Scholar]
- 18.Cooper MT, Tyler DM, Furriols M, Chalkiadaki A, Delidakis C, Bray S. Spatially restricted factors cooperate with notch in the regulation of Enhancer of split genes. Dev Biol. 2000;221:390–403. doi: 10.1006/dbio.2000.9691. [DOI] [PubMed] [Google Scholar]
- 19.Xu T, Rubin GM. Analysis of genetic mosaics in developing and adult Drosophila tissues. Development. 1993;117:1223–1237. doi: 10.1242/dev.117.4.1223. [DOI] [PubMed] [Google Scholar]
- 20.de Celis JF, Garcia-Bellido A, Bray SJ. Activation and function of Notch at the dorsal-ventral boundary of the wing imaginal disc. Development. 1996;122:359–369. doi: 10.1242/dev.122.1.359. [DOI] [PubMed] [Google Scholar]
- 21.Micchelli CA, Rulifson EJ, Blair SS. The function and regulation of cut expression on the wing margin of Drosophila: Notch, Wingless and a dominant negative role for Delta and Serrate. Development. 1997;124:1485–1495. doi: 10.1242/dev.124.8.1485. [DOI] [PubMed] [Google Scholar]
- 22.Jacobsen TL, Brennan K, Arias AM, Muskavitch MA. Cis-interactions between Delta and Notch modulate neurogenic signalling in Drosophila. Development. 1998;125:4531–4540. doi: 10.1242/dev.125.22.4531. [DOI] [PubMed] [Google Scholar]
- 23.Sakamoto K, Ohara O, Takagi M, Takeda S, Katsube K. Intracellular cell-autonomous association of Notch and its ligands: a novel mechanism of Notch signal modification. Dev Biol. 2002;241:313–326. doi: 10.1006/dbio.2001.0517. [DOI] [PubMed] [Google Scholar]
- 24.Klein T, Brennan K, Arias AM. An intrinsic dominant negative activity of serrate that is modulated during wing development in Drosophila. Dev Biol. 1997;189:123–134. doi: 10.1006/dbio.1997.8564. [DOI] [PubMed] [Google Scholar]
- 25.Bruckner K, Perez L, Clausen H, Cohen S. Glycosyltransferase activity of Fringe modulates Notch-Delta interactions. Nature. 2000;406:411–415. doi: 10.1038/35019075. [DOI] [PubMed] [Google Scholar]
- 26.Artavanis-Tsakonas S, Matsuno K, Fortini ME. Notch signaling. Science. 1995;268:225–232. doi: 10.1126/science.7716513. [DOI] [PubMed] [Google Scholar]
- 27.Komada M, Masaki R, Yamamoto A, Kitamura N. Hrs, a tyrosine kinase substrate with a conserved double zinc finger domain, is localized to the cytoplasmic surface of early endosomes. J Biol Chem. 1997;272:20538–20544. doi: 10.1074/jbc.272.33.20538. [DOI] [PubMed] [Google Scholar]
- 28.Lloyd TE, Atkinson R, Wu MN, Zhou Y, Pennetta G, Bellen HJ. Hrs regulates endosome membrane invagination and tyrosine kinase receptor signaling in Drosophila. Cell. 2002;108:261–269. doi: 10.1016/s0092-8674(02)00611-6. [DOI] [PubMed] [Google Scholar]
- 29.Jekely G, Rorth P. Hrs mediates downregulation of multiple signalling receptors in Drosophila. EMBO Rep. 2003;4:1163–1168. doi: 10.1038/sj.embor.7400019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Babst M. A protein’s final ESCRT. Traffic. 2005;6:2–9. doi: 10.1111/j.1600-0854.2004.00246.x. [DOI] [PubMed] [Google Scholar]
- 31.Ray WJ, Yao M, Mumm J, Schroeter EH, Saftig P, Wolfe M, Selkoe DJ, Kopan R, Goate AM. Cell surface presenilin-1 participates in the gamma-secretase-like proteolysis of Notch. J Biol Chem. 1999;274:36801–36807. doi: 10.1074/jbc.274.51.36801. [DOI] [PubMed] [Google Scholar]
- 32.Seugnet L, Simpson P, Haenlin M. Requirement for dynamin during Notch signaling in Drosophila neurogenesis. Dev Biol. 1997;192:585–598. doi: 10.1006/dbio.1997.8723. [DOI] [PubMed] [Google Scholar]
- 33.Pasternak SH, Bagshaw RD, Guiral M, Zhang S, Ackerley CA, Pak BJ, Callahan JW, Mahuran DJ. Presenilin-1, nicastrin, amyloid precursor protein, and gamma-secretase activity are co-localized in the lysosomal membrane. J Biol Chem. 2003;278:26687–26694. doi: 10.1074/jbc.m304009200. [DOI] [PubMed] [Google Scholar]
- 34.Gupta-Rossi N, Six E, LeBail O, Logeat F, Chastagner P, Olry A, Israel A, Brou C. Monoubiquitination and endocytosis direct gamma-secretase cleavage of activated Notch receptor. J Cell Biol. 2004;166:73–83. doi: 10.1083/jcb.200310098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hori K, Fostier M, Ito M, Fuwa TJ, Go MJ, Okano H, Baron M, Matsuno K. Drosophila deltex mediates suppressor of Hairless-independent and late-endosomal activation of Notch signaling. Development. 2004;131:5527–5537. doi: 10.1242/dev.01448. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.