

Abstract
Tumors initiate angiogenesis primarily by secreting VEGF-A164. The first new vessels to form are greatly enlarged, pericyte-poor sinusoids, called mother vessels (MV), that originate from preexisting venules. We postulated that the venular enlargement necessary to form MV would require a selective degradation of their basement membranes, rigid structures that resist vascular expansion. To identify the specific proteases responsible for MV formation, we induced angiogenesis in mouse tissues with an adenoviral vector expressing VEGF-A164 (Ad- VEGF-A164) or with VEGF-A-secreting TA3/St mammary tumors. We found that MV formation resulted from greatly increased activity of cathepsins (B>S>L) in venules transitioning into MV, as well as from a reciprocal decrease in the expression of several cysteine protease inhibitors (CPI), stefin A and cystatins B and C, by these same venules. Using a fluorescence probe that selectively binds cellular sites of cathepsin protease activity in vivo, we demonstrated that increased cathepsin activity was localized exclusively to perivenular cells, not to venule endothelial cells. CPI strikingly inhibited angiogenesis in the Matrigel assay, and Ad-VEGF-A164-induced angiogenesis was reduced by ∼50% in cathepsin B-null mice. Thus, VEGF-A, whether expressed by interstitial cells infected with an adenoviral vector or by tumor cells, upsets the normal cathepsin-CPI balance in nearby venules, leading to degradation of their basement membranes, an important first step in angiogenesis.
Keywords: cathepsins, cysteine protease inhibitor, VEGF, angiogenesis, mother vessels
Introduction
In order to grow beyond minimal size, tumors must induce a new vascular supply (1). They do so by overexpressing growth factors, particularly vascular endothelial growth factor/vascular permeability factor (VPF/VEGF, VEGF-A) and its 164 (mouse)/165 (human) isoform (2-4). However, unlike the angiogenesis of normal development, the new blood vessels that tumors induce are highly abnormal and differ strikingly from the microvessels of normal tissues with respect to both structure and function (2, 3, 5). The first new vessels to form in many transplantable mouse tumor models are mother vessels (MV), a blood vessel type that is also common in many autochthonous human tumors (2, 3, 6-8). MV are greatly enlarged, thin-walled, hyperpermeable, pericyte-depleted sinusoids that form from preexisting venules. The dramatic enlargement of venules leading to MV formation would seem to require proteolytic degradation of their basement membranes. Vascular basement membranes are primarily comprised of laminins and type IV collagen (9-11). They are rigid, non-compliant (non-elastic) structures and allow only an increase of ∼30% in cross-sectional area in response to increased internal pressure (12); by contrast, MV commonly have cross-sectional areas that are 4-5 times those of the venules from which they arise (2, 3, 7, 8).
The specific proteases responsible for generating MV have not been identified. Tumors are complex entities in which many different proteases participate in a wide range of simultaneous processes that include tumor, stromal, inflammatory and vascular cell proliferation and migration. Several different classes of proteases have been identified in tumors including matrix metalloproteases (MMPs) and serine and cysteine proteases (13-16). Of these, MMPs have received the most attention (15, 17, 18). However, in recent years cysteine proteases, and particularly cathepsins B, L, S and H, have been implicated in tumor cell invasion, metastasis and, more recently, in tumor angiogenesis (13, 19-26). Cathepsins are members of the papain subfamily of cysteine proteases (13); they are found in lysosomes and have traditionally been associated with intracellular functions (27, 28). More recent data indicate that cathepsins are secreted, can function extracellularly to degrade matrix proteins, and have significant roles in tumor angiogenesis (13, 20-24, 29).
Endogenous inhibitors of cathepsins, members of the cysteine protease inhibitor (CPI) family, have also been implicated in tumor progression. CPI are small, 11-13kD proteins that include stefin A and cystatins B and C (27). RIP-Tag 2 tumors grow faster in cystatin C null than in wild type mice (30), and changes in CPI have been reported in several different tumors (31-35).
The goal of the present investigation was to identify the specific proteases and protease inhibitors that participate in MV formation, as well as the cell types that make them. To avoid the complexities of the tumor environment, in which many cell types, proteases and protease inhibitors participate, we made use of an adenoviral vector that expresses VEGF-A164 (Ad-VEGF-A164); when injected into mouse tissues, Ad-VEGF-A164 induces an angiogenic response that closely mimics that induced by malignant tumors (2, 3, 7, 36). We report here that increased expression of several cathepsins (B>S>L), accompanied by a reciprocal decrease in the expression of their inhibitors, members of the CPI family, is responsible for the vascular basement membrane degradation that allows MV to form. Further, increased cathepsin and reduced CPI expression was localized selectively to venules that were transitioning into MV. We substantiated these results with a VEGF-A-expressing tumor. Together these findings implicate venule-associated cathepsins and their CPI inhibitors in the earliest stage of tumor angiogenesis. Subsequently, this degradative process was reversed as MV evolved into daughter vessels, a process accompanied by extensive new vascular basement membrane synthesis.
Materials and Methods
Animals, adenoviral vectors and tumors
Ad-VEGF-A164 and Ad-LacZ and their injection into athymic nude mice (NCI) were described previously (7). A/J mice (Jackson Laboratory) and the TA3/St tumor were described previously (37). Cathepsin B null mice on the FVB background were generously provided by Dr. Thomas Reinheckel, Freiburg, Germany (38). All studies were performed under protocols approved by the BIDMC IACUC. Microvascular density was calculated by counting six highly vascularized fields in each section at 400x. To quantify the angiogenic response, 100μl 0.5% Evans Blue dye was injected i.v. and mice were sacrificed 5 min later (8). Flank tissue images were photographed and calibrated at a standard magnification. The intensity of bluing was calculated with the IP lab program (Scanalytics), using automated segmentation of bluing intensity with a fixed threshold limit.
Histology, immunohistochemistry and confocal microscopy
Ears were fixed in 4% paraformaldehyde and 5 μm sections were immunostained (7) with antibodies specific for pan-laminins (Novus biologicals), type IV collagen (Cosmo Bio Co. LTD), stefin A (Chemicon), cystatins B and C (R&D Systems), cathepsin B (Neuromics), or CD31 (BD Biosciences). Images were obtained from 2-5 different ears or tumors at each time point.
For confocal microscopy, mice were injected i.v. with FITC-lectin (Vector Laboratories). After perfusion, ears were removed, split into dorsal and ventral halves, exposed to primary antibodies, and incubated with Texas Red-conjugated anti-rabbit immunoglobulin (Invitrogen). Images were obtained from three or more ears at each time point. For imaging cathepsin B/S/L activity, 25 nmol of GB123 was injected i.v. 8h later, FITC-Lectin was injected i.v., mice were sacrificed, and tissues were visualized in a LSM 510 Meta confocal microscope (39).
Protein extraction and Western blot analysis
Ear sites were rapidly frozen in liquid nitrogen, ground with a mortar and pestle, and lysed on ice in 1μl sample buffer/mg ear tissue: 62.5 μM Tris, 250 mM HEPES, 0.5 % NP-40, 5 % 2-mercaptoethanol, 2 % sodium dodecyl sulfate, 5 % sucrose and 0.005 % bromophenol blue. Immunoblotting was performed with a specific antibody against laminin β1 chain (CHEMICON). Experiments were repeated 4x.
Isolation of RNA, quantitative RT- PCR
Total RNA was isolated with the Qiagen RNeasy Mini Kit and reverse transcribed using TaqMan Reverse Transcription Reagents (Applied Biosystems). PCR primers were prepared as displayed on the PrimerBank website (40) (http://pga.mgh.harvard.edu/primerbank/), as follows; cystatin B (6681071a1), cystatin C (31981822a1), cathepsin B (6681079a2), cathepsin L (6753558a3), cathepsin H (7106279a3), cathepsin D (18043133a1). PCR primers for cyclophilin were forward 5′ CAGACGCCACTGTCGCTTT 3′ and reverse 5′ TGTCTTTGGAACTTTGTCTGCAA 3′. PCR primers for stefin A were designed by using Primer Express Software (Applied BioSystems) and sequences are forward 5′ CTTGAAGAGCAAACCAATGAGAAA 3′, reverse 5′ AACAAATTTTGTCCAGCAACGA 3′. Data were quantified using a statistical method that corrects for PCR efficiency for each reaction (41). The number of each gene-amplified product was normalized to the number of cyclophilin amplified product.
Cysteine cathepsin activity assays
We used the cathepsin B detection kit (Calbiochem) that measures the hydrolysis of the synthetic substrate ZArg-Arg-AMC (7-Amino-4-methylcoumarin) (42). Ear injection sites were collected with an 8 mm punch and extracted with lysis buffer. Lysates were incubated as per the manufacturer's protocol. Free AMC was measured fluorometrically at 360/465 nm Ex/Em and calculated from a standard curve. This experiment was repeated >5 times.
We also assessed cathepsin activity with the GB123 probe. Mice were injected i.v. with 25 nmol GB123. After 8 hrs, ear sites were lysed in 700 μl of lysis buffer. 75 μl of each sample was added to 25 μl sample buffer, boiled, electrophoresed on 12.5% SDS PAGE, and scanned for fluorescence at 633/670 nm Ex/Em (39)
Matrigel assays and quantification of microvessel density
Athymic nude mice were injected subcutaneously with 0.3 ml of Matrigel supplemented, as indicated, with bFGF (2.5mg/ml) and 8U heparin, 2mg/ml recombinant stefin A, cystatin C, cathepsin B (R&D Systems), or 10μM CA-047Me (Calbiochem). bFGF was used in these assays because it gives stronger and more consistent angiogenesis than VEGF-A (43). At 6 days, Matrigel plugs were harvested and processed for histology and CD31 immunohistochemistry. Mean vascular density was calculated as above, counting CD31 positive structures with lumens. This experiment was repeated 3x.
Statistics
Statistical analysis was performed using ANOVA and Dunn's multiple comparison test or unpaired t tests, as indicated.
Results
Degradation and subsequent neosynthesis of vascular basement membranes in angiogenesis
In initial studies we injected an adenoviral vector expressing VEGF-A164 (108pfu Ad-VEGF-A164) into the ears of nude mice to mimic the angiogenic response induced by many tumors (2, 3). As previously reported, the first new blood vessels to form were mother vessels (MV), greatly enlarged, thin-walled structures that arose from preexisting venules and that peaked at 5 days (7). Confocal microscopy on split ears and immunohistochemistry on tissue sections demonstrated a striking, patchy loss of both laminin and collagen IV staining in the MV induced by Ad-VEGF-A164 (Fig. 1A, S1). Similar degradation of vascular basement membrane laminin and collagen IV was observed in the MV that developed in a mouse mammary carcinoma (TA3/St) implanted s.c. in syngeneic A/J mice (Fig. 1B).
Figure 1. Degradation of vascular basement membranes in MV induced by Ad-VEGF-A164 or by TA3/St tumors.
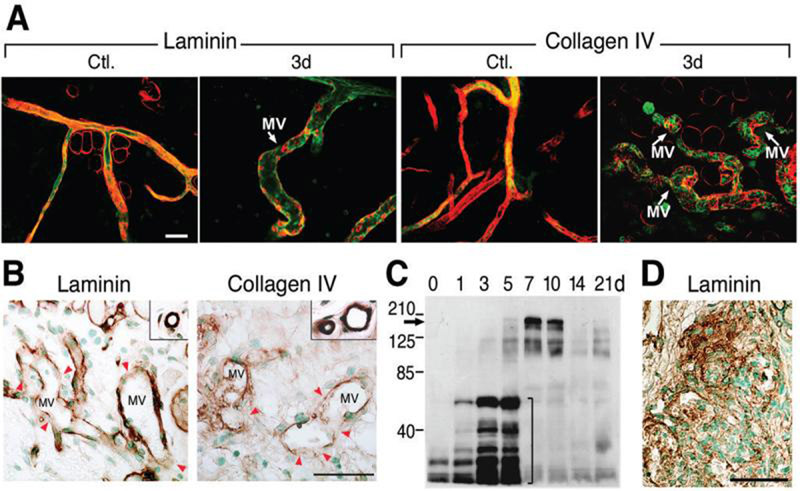
(A) Confocal microscopy of ear whole mounts from normal control (Ctl.) mice and from mice whose ears had been injected 3 days earlier with Ad-VEGF-A164. Immediately prior to sacrifice, mice were injected i.v. with FITC-lectin (green). Ears were then immunostained with antibodies against laminin or collagen IV and visualized with Texas Red conjugated secondary antibodies. Note extensive loss of red staining (laminin or collagen) in MV as compared with control venules. Scale bar, 50μm.
(B) Immunohistochemistry for laminin and collagen IV in mother vessels (MV) of TA3/St tumors harvested 4 days after implantation. Note patchily reduced MV staining (red arrowheads), compared with strong, continuous basement membrane staining of normal venules (insets). Scale bar, 50μm.
(C) Immunoblot with an antibody against laminin β1 chain performed on extracts of normal ears (time 0) and on ears harvested at 1-21 days following Ad-VEGF-A164 injection. Note increasing low molecular weight laminin β1 chain fragments (bracket) at 1-5 days. In contrast, on days 7 and 10 there is increased expression of intact laminin β1 chain (arrow), as well as high molecular weight fragments.
(D) Glomeruloid microvascular proliferations in ears 7 days after Ad-VEGF-A164 injection demonstrate extensive new laminin deposition (brown stain). Scale bar, 50μm.
Two laminin isoforms, 8 [α4β1γ1] and 10 [α5 β1γ1], are prominent components of vascular basement membranes (9-11). Because β1 chains are common to both of these isoforms, we measured β1 chains as surrogates of vascular basement membrane degradation during the course of MV formation. Western blots demonstrated progressive fragmentation of laminin β1 chain over the course of 1-5 days (Figs. 1C, loading control shown in S2), closely paralleling the course of MV formation (Figs. 1A, S1). At later times, low molecular weight laminin b1 chain fragments were no longer detected; instead, expression of intact laminin b1 chain as well as higher molecular weight complexes were significantly increased (Fig. 1C, 7 and 10 days). Consistent with this observation, formation of glomeruloid microvascular proliferations, a prominent type of daughter vessel that develops from MV after day 5 (3, 7), exhibited extensive deposits of vascular basement membrane laminin (Fig. 1D), as well as type IV collagen (not shown).
Increased cathepsin expression and activity during MV formation
To identify proteases that might be responsible for degrading venular basement membranes, we first measured matrix metalloproteases 2 and 9 because these proteases have been prominently implicated in tumor biology (15, 17, 18). However, we were unable to document a role for either in Ad-VEGF-A164-induced MV formation. As measured by RT-PCR and zymography, MMP-2 and MMP-9 expression and activity actually declined over the time course of MV formation (Fig. S3). Therefore, we performed Affymetrix gene-chip profiling and found that cathepsin B was expressed in normal ear skin and was the most upregulated (1.6-fold) protease in ears harvested at 4 days after Ad-VEGF-A164 injection as MV were approaching peak size and prominence. These findings were confirmed by quantitative RT-PCR, demonstrating that cathepsin B was highly induced (8.1-fold) after Ad-VEGF-A164 injection, reaching a peak on day 3 and then returning rapidly to baseline levels (Fig. 2A, top panel). Cathepsin B is transcribed as a preproenzyme and requires multiple posttranslational steps for activation. Of potential interest, therefore, cathepsin D, an aspartic protease that can activate procathepsin B, also increased after Ad-VEGF-A164 injection (∼5-fold increase on day 3) and with similar kinetics, though at much lower expression levels (Fig. 2A, bottom panel). Three other cysteine proteases (cathepsins S, L, and H) were also detected at low expression levels in normal mouse ears and showed different expression patterns in response to Ad-VEGF-A164 (Fig. 2A, bottom panel).
Figure 2. Cathepsin expression in developing MV.
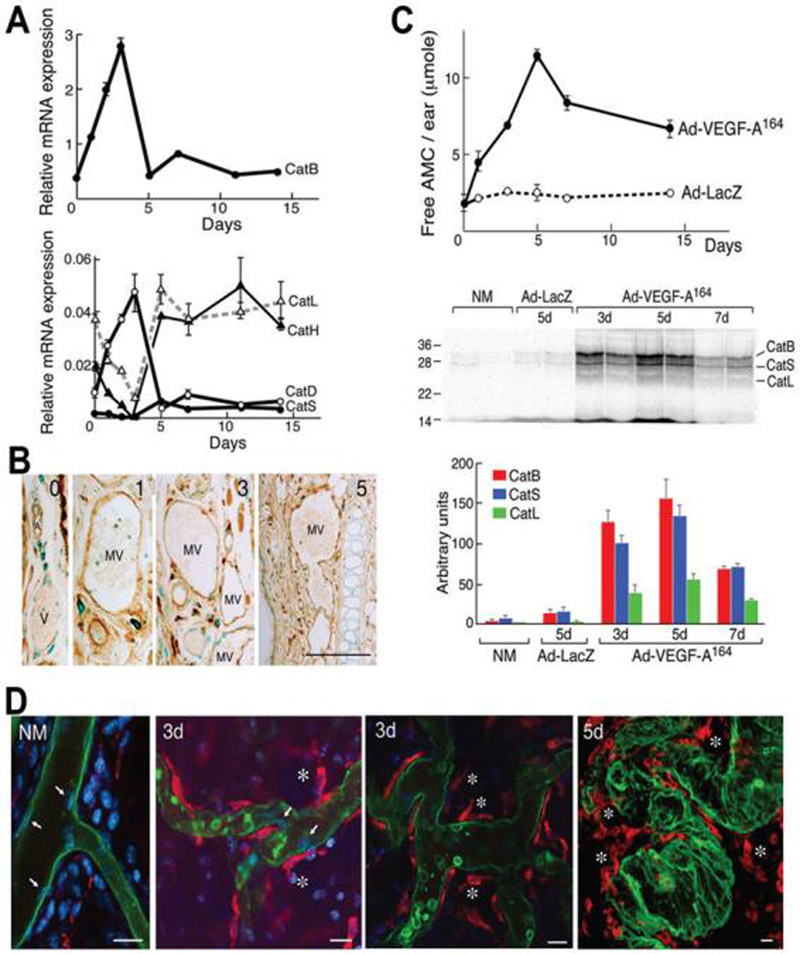
(A) Quantitative RT-PCR demonstrating expression patterns (mean ± SD) of cathepsin B, D, L, H and S mRNAs at indicated times after Ad-VEGF-A164 injection. Data are representative of nine different experiments performed on 7 different animals.
(B) Immunohistochemical localization of cathepsin B in control ears (time 0) and at 1, 3 and 5 days after Ad-VEGF-A164 injection. Cathepsin B is expressed by many different cell types in normal ears but increased expression following Ad-VEGF-A164 injection was observed only in mother vessels (MV). V, normal venule; A, arteriole. Scale bar, 50μm.
(C, top panel) Hydrolysis of Z-Arg-Arg-AMC (mean ± SD), a synthetic substrate commonly used to measure cathepsin B activity, in extracts of mouse ears that had been injected with Ad-VEGF-A164 or Ad-LacZ. Data are representative of 6 separate experiments. (C, middle panel) Fluorescence scan demonstrates cathepsin activity in extracts of mouse ears injected i.v. with GB123 at indicated times after Ad-VEGF-A164 or Ad-LacZ injection. Bands at 32 kDa, 28kDa, and 24 kDa were identified as cathepsins B, S and L, respectively. (C, bottom panel) Densitometry of pooled gel data (mean ± SD).
(D) Confocal microscopy of GB123 (red) and FITC-lectin (green) in normal mouse ears (NM) and in ears injected 3 and 5 days previously with Ad-VEGF-A164. Only rare stromal cells stain for GB123 (red) in NM ears, whereas perivascular cells, but not endothelial cells (white arrows), stain intensely in Ad-VEGF-A164-injected ears. White * indicate detaching GB123-positive perivenular cells. Blue color, DAPI staining. Scale bar, 20 μm.
We used immunohistochemistry to localize sites of cathepsin B protein expression. We found that cathepsin B was expressed strongly in many different cell types in normal ear skin, including skeletal and arterial smooth muscle cells, epidermis and some interstitial cells, but only faintly in venules. However, after Ad-VEGF-A164 injection, increased cathepsin B expression was observed selectively in venules that were evolving into MV (Fig. 2B).
To provide additional evidence that cathepsins were involved in MV formation, we incubated tissue extracts of Ad-VEGF-A164 injected ears with Z-Arg-Arg-AMC, a substrate commonly used to measure cathepsin B activity (Fig. 2C, top panel). We found a progressive increase in substrate degradation that peaked on day 5 and that remained elevated through day 14; in contrast, the low levels of protease activity present in ears injected with Ad-LacZ did not change over time.
To identify the cathepsins involved and better define the changes in their expression levels over time, we made use of GB123, a near-infrared fluorescent probe that binds to the active sites of cysteine cathepsins in vivo (39). Eight hours after injecting GB123 i.v., ears were harvested, extracted and processed for SDS-PAGE (Fig. 2C, middle panel). At 3 and 5 days after Ad-VEGF-A164 injection, and to a lesser extent at 7 days, we identified strong 32 kDa and weaker 28kDa and 24kDa bands. These molecular weights correspond to cathepsins B, S and L, respectively, as confirmed by immunoprecipitation (39). At 5 days after injecting Ad-VEGF-A164, cathepsin B activity was found to be increased ∼20-fold above control levels, whereas cathepsins S and L were increased ∼12 and ∼24-fold, respectively (Fig. 2C, bottom panel). Bands in Ad-Lac-Z injected ears were unchanged from the low levels found in uninjected control ears.
GB123 can also be used to identify intracellular sites of proteolytically active cathepsins by confocal microscopy. In normal mice, faint staining (red) was observed in the cytoplasm of occasional stromal cells, but activity was not detected in blood vessels (Fig. 2D, NM). In contrast, on days 3 and 5 after Ad-VEGF-A164 injection, strong cathepsin activity was observed in perivascular cells that were immediately enveloping or detaching from MV. In contrast, MV endothelial cells did not stain with this probe.
Reduced expression of cysteine protease inhibitors (CPIs) during MV formation
Cathepsin activity is inhibited in normal tissues by high affinity, competitive cysteine protease inhibitors (CPIs), including stefin A and cystatins B and C. We had noted in preliminary Affymetrix gene chip experiments that stefin A was downregulated ∼5-fold at 4 days after Ad-VEGF-A164 injection. We therefore measured mRNA expression levels of stefin A and of the two other CPIs at different times following Ad-VEGF-A164 injection. Quantitative RT-PCR demonstrated that cystatin B and C mRNAs were relatively abundant in normal ear skin, but fell dramatically to negligible levels over the first 3 days following Ad-VEGF-A164 injection (Fig. 3A). At day 5, however, as MV formation peaked, both rose to supra-normal levels before gradually returning to lower values. Stefin A mRNA was expressed at much lower levels in normal ears and showed a complex, fluctuating expression pattern in response to Ad-VEGF-A164 (Fig. 3B).
Figure 3. CPI expression in Ad-VEGF-A164 injected ears.
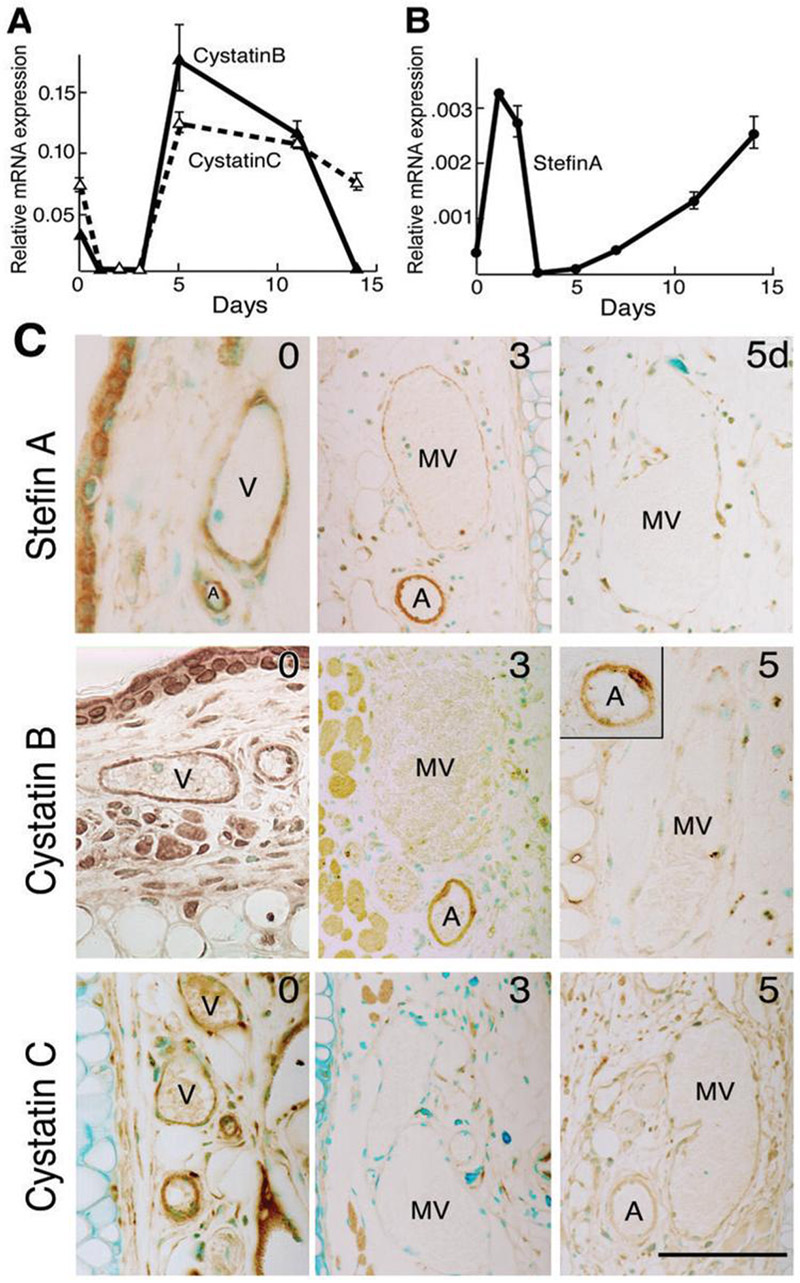
(A, B) Quantitative RT-PCR demonstrating CPI mRNA expression patterns at indicated times after Ad-VEGFA164 injection (mean ± SD). Representative data from 9 different experiments performed on 7 different animals.
(C) Immunohistochemical staining of CPIs in control ears (time 0) and at 3 and 5 days after Ad-VEGF-A164 injection. Staining for all three CPIs is greatly reduced in mother vessels (MV) compared with normal venules (V). Arteriole (A) staining for cystatin C, but not for cystatin B or stefin A, was also reduced following Ad-VEGF-A164 injection. Scale bar, 50μm.
Immunohistochemistry of normal skin demonstrated strong stefin A and cystatin B and C staining in venular and arterial endothelial cells, pericytes and arterial smooth muscle cells, as well as in epidermis, nerves, skeletal muscle and some stromal cells (Fig. 3C). However, staining of all three inhibitors was selectively lost from venules as they evolved into MV at 3 days, though by 5 days weak staining had returned in some endothelial cells. Staining for cystatin C, unlike that for stefin A and cystatin B, was also lost from arterioles during this time frame, a change that may be related to the arteriolar enlargement and arteriogenesis that Ad-VEGF-A164 induces apart from angiogenesis (44). Staining of these inhibitors did not undergo detectable changes in nonvascular cells during the course of MV formation.
Similar reciprocal vascular staining for cathepsin B and CPIs was also observed in the MV induced by TA3/St mammary carcinomas (Fig. 4).
Figure 4. Reciprocal changes in immunohistochemical CPI and cathepsin B staining in MV generated three days after s.c. injection of 106 TA3/St mammary carcinoma cells.

MV exhibit reduced CPI staining and increased cathepsin B staining as compared with control vessels (insets). Scale bar, 25μm.
Effects of cathepsin inhibitors on angiogenesis in the Matrigel assay
In order to determine whether cathepsin B had a functional role in angiogenesis in vivo, we evaluated the effects of cathepsin B inhibitors on new blood vessel formation in the Matrigel assay. As expected, bFGF induced a strong angiogenic response in Matrigel plugs and this response was strongly inhibited by stefin A (86%), cystatin C (92%) and the cathepsin inhibitor CA074Me (91%) (45, 46) (Fig. 5).
Figure 5. Effects of CPI on angiogenesis in the Matrigel assay.
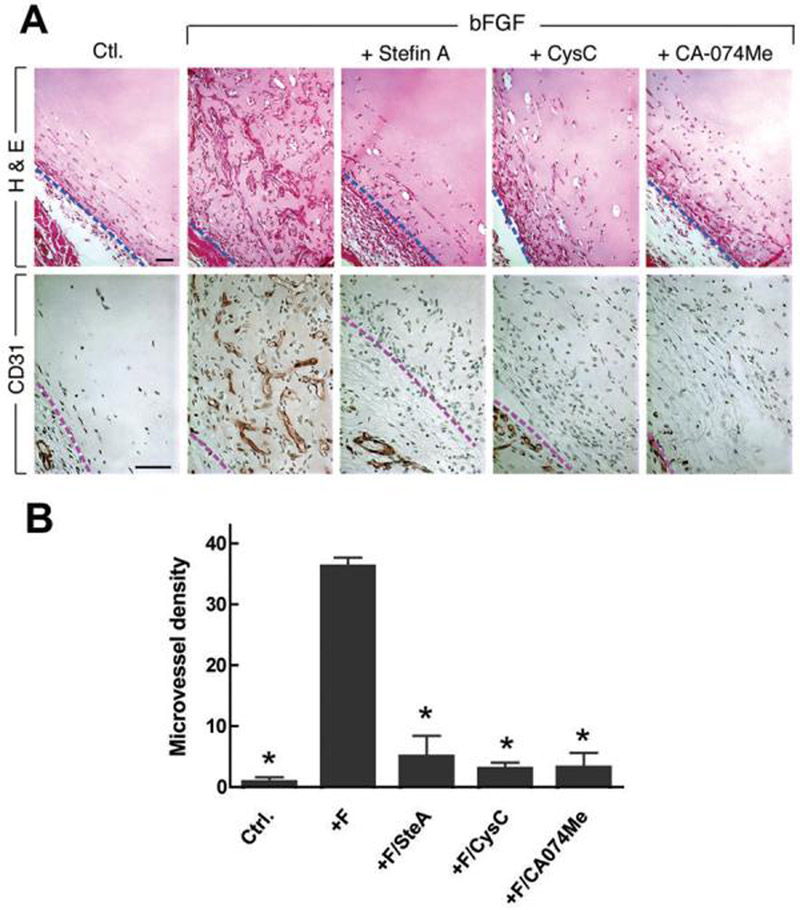
(A) Top panel is stained with H&E, bottom panel with antibodies to CD31. Dashed line indicates border between Matrigel and surrounding host tissue. Note prominent MV in Matrigels containing bFGF alone, and dearth of vessels in Ctl (no additions) and in bFGF Matrigels supplemented with stefin A, cystatin C or CA-074Me. Scale bar, 50 μm. (B) Microvascular density (mean ± SEM). *, p<0.01 all other conditions versus +F (+FGF), Dunn's multiple comparison test.
Inhibition of Ad-VEGF-A164-induced angiogenesis in cathepsin B null mice
To evaluate the importance of cathepsin B in MV formation, we evaluated the angiogenic response induced by 5×108 pfu Ad-VEGF-A164 injected into the flank skin of wild type FVB and cathepsin B null mice. As expected, Ad-VEGF-A164 induced extensive MV formation in wild type mice; by contrast, the angiogenic response in cathepsin B null mice was greatly inhibited (Fig. 6A) and mean vascular density was strikingly reduced (Fig. 6B, p<0.0001). As a further measure, we quantified Evan's blue dye accumulation in wild type and cathepsin B null mice. Evan's blue dye binds to plasma albumin and provides a measure of vascular volume as well as early leakage from hyperpermeable blood vessels. As shown in Fig. 6C, tissue bluing was reduced ∼50% in cathepsin B null mice, as compared with wild type controls.
Figure 6. Angiogenic response and vascular permeability in wild type and cathepsin B null mice 4 days after injection of Ad-VEGF-A164.
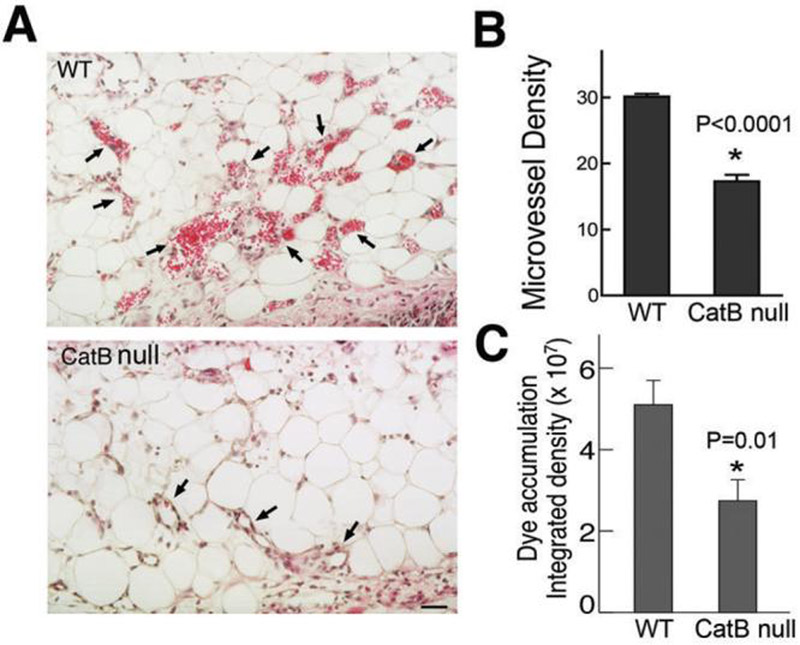
(A) Histology. Arrows identify some of the many MV present in wild type mice, whereas very few MV were found in null mice. Scale bar, 50 μm.
(B) Microvessel density (mean ± SEM) was calculated as described in Methods and analyzed statistically with an unpaired t test. *, p<0.0001.
(C) Evan's blue dye accumulation (mean ± SEM) was quantified using IPLab software. WT, n=5; CatB null, n=6. *, p=0.01, unpaired t test.
Discussion
MV, the first new blood vessels to form in response to Ad-VEGF-A164 and in many mouse and human tumors, develop from preexisting venules by a process that involves pericyte detachment and extensive enlargement of cross-sectional area (2, 3, 6-8, 36). We hypothesized that this enlargement would require proteolytic degradation of venular basement membranes, because intact basement membranes strictly limit increases in vascular diameter (12). We report here that venular basement membrane proteins are indeed degraded during the course of MV formation. Immunohistochemical staining for laminin and type IV collagen was lost (Figs. 1A, S1) and the laminin β1 chain was fragmented (Fig. 1C) over the 5 day course of MV formation. Subsequently, MV evolved into daughter glomeruloid microvascular proliferations, a vessel type that is also found in many human tumors (2). Formation of glomeruloid microvascular proliferations was accompanied by cessation of laminin degradation, increased expression of laminin β1 chain mRNA, and deposition of substantial amounts of laminin (and collagen IV, not shown) in a basement membrane distribution (Figs. 1C,D).
Matrixmetalloproteases, and especially MMPs 2 and 9, have been thought to be the most important proteases involved in tumor angiogenesis (15, 17, 18). However, we were unable to document a role for these MMPs in Ad-VEGF-A164-induced MV formation (Fig. S3). Instead, we found that MV formation was accompanied by strikingly increased expression of cathepsin B mRNA (Fig. 2A). Cathepsin B is expressed abundantly by many different cell types in normal skin. However, immunohistochemistry demonstrated that, following Ad-VEGF-A164 injection, cathepsin B expression was increased only in venules transitioning into MV (Fig. 2B). In parallel, extracts of angiogenic sites exhibited greatly increased hydrolysis of Z-Arg-Arg-AMC, a substrate commonly used to measure cathepsin B activity; this activity peaked as MV attained maximal frequency and size on day 5 (Fig. 2C). Strong cathepsin B immunostaining of MV was also observed in the MV induced by the VEGF-secreting TA3/St mammary carcinoma (Fig. 4). Taken together, increased expression of cathepsin B mRNA, protein and enzymatic activity correlated closely with each other and with MV formation.
Further evidence for the importance of cathepsins in MV formation was obtained using a recently developed fluorescent activity based probe, GB123 (39). Three major bands of increased cathepsin activity were detected at 3-7 days after Ad-VEGF-A164 injection, and these were identified as cathepsins B, S and L, respectively (Fig. 2C). All three of these cathepsins are capable of digesting basement membrane proteins (13). The GB123 probe also allowed us to identify the cells that expressed cathepsin activity by confocal microscopy. Whereas almost no staining was observed in the vasculature of normal tissues, striking activity was observed in the perivascular cells, but not in the endothelial cells, of developing MV (Fig. 2D). Thus perivascular cells, not endothelial cells, express the cathepsin activity that is responsible for venular basement membrane degradation and hence for the pericyte detachment associated with MV formation.
Additional evidence for the importance of cathepsins in angiogenesis was provided by experiments demonstrating that Ad-VEGF-A164-induced angiogenesis, as well as associated vascular permeability, were strikingly reduced in cathepsin B null mice (Fig. 6). The residual angiogenesis that developed in these mice may have resulted from the activities of cathepsins S and L, enzymes that share both complementary and compensatory activities with cathepsin B (22, 47-49). Unfortunately, doubly or triply cathepsin knockout mice are poorly viable and therefore could not be tested (47).
Ad-VEGF-A164 also induced reduced expression of CPI mRNAs and protein in venules that were transitioning into MV (Fig. 3). The MV induced by TA3/St mammary carcinomas also exhibited greatly reduced expression of stefin A and cystatin C (Fig. 4). It is likely that cystatin C has a more important role than either stefin A or cystatin B in MV formation. Cystatin C mRNA is approximately 100-fold more abundant in normal ear skin than stefin A (Fig. 3). Further, the inhibition constant (Ki) of cystatin C for cathepsin B is 0.29nM, whereas that of stefin A is 2.4nM and that of cystatin B is 19nM (50). Finally, cystatin C is a secreted protein whereas stefin A and cystatin B are thought to remain largely within cells (27). Nonetheless, both stefin A and cystatin C, as well as a non-specific cathepsin protease inhibitor, were effective in inhibiting bFGF-induced angiogenesis in the Matrigel assay (Fig. 5).
In sum, our data provide strong, mechanistic evidence that, whether induced by Ad-VEGF-A164 or by a VEGF-A-secreting tumor, the venular basement membrane degradation associated with MV formation results from a progressive increase in cathepsin activity, accompanied by a decrease in CPI expression. Further, these reciprocal changes in cathepsin and CPI expression occurred primarily in venules that were transitioning into MV and therefore seem to be independent of cathepsins or CPIs expressed by tumor or stromal cells. Together these data indicate that VEGF-A perturbs the cathepsin-CPI balance in venules and that this is a critically important step in MV formation, the earliest stage of VEGF-A-induced angiogenesis. Remaining to be determined are the signaling mechanism(s) by which VEGF-A upregulates cathepsin and downregulates CPI expression and activity in venules transitioning into MV, and the mechanisms by which the cathepsin-CPI balance is subsequently restored as MV evolve into daughter vessels. Still other questions concern the mechanisms that control cathepsin and CPI exocytosis to the pericellular space and the means by which exocytosed cathepsin B is activated. Answers to these questions are needed to determine whether the cathepsin-CPI axis can be a useful target for anti-angiogenesis therapy.
Supplementary Material
Acknowledgments
This work was supported by U.S. Public Health Service grants HL-64402, P01 CA92644 and by a contract from the National Foundation for Cancer Research (Dvorak); DK55001, DK62987, DK61688, AA13913 (Kalluri); CA118764-01, DK065298, CA45548 (Moses); NIH Roadmap Technology Center for Networks and Pathways grant U54-RR020843 (Bogyo); CA125162 and TEM U54CA122518 (Joyce).
References
- 1.Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29:15–8. doi: 10.1053/sonc.2002.37263. [DOI] [PubMed] [Google Scholar]
- 2.Dvorak H. Tumor Blood Vessels. In: Aird W, editor. Endothelial Biomedicine. Cambridge University Press; New York: 2007. pp. 1457–70. [Google Scholar]
- 3.Nagy JA, Dvorak HF, Dvorak AM. VEGF-A and the induction of pathological angiogenesis. Annu Rev Pathol Mech Dis. 2007;2:251–75. doi: 10.1146/annurev.pathol.2.010506.134925. [DOI] [PubMed] [Google Scholar]
- 4.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–76. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 5.Warren B. The vascular morphology of tumors. In: Peterson H-I, editor. Tumor blood circulation: angiogenesis, vascular morphology and blood flow of experimental and human tumors. CRC Press; Boca Raton: 1979. pp. 1–47. [Google Scholar]
- 6.Paku S, Paweletz N. First steps of tumor-related angiogenesis. Lab Invest. 1991;65:334–46. [PubMed] [Google Scholar]
- 7.Pettersson A, Nagy JA, Brown LF, et al. Heterogeneity of the angiogenic response induced in different normal adult tissues by vascular permeability factor/vascular endothelial growth factor. Lab Invest. 2000;80:99–115. doi: 10.1038/labinvest.3780013. [DOI] [PubMed] [Google Scholar]
- 8.Nagy JA, Feng D, Vasile E, et al. Permeability properties of tumor surrogate blood vessels induced by VEGF-A. Lab Invest. 2006;86:767–80. doi: 10.1038/labinvest.3700436. [DOI] [PubMed] [Google Scholar]
- 9.Hallmann R, Horn N, Selg M, Wendler O, Pausch F, Sorokin LM. Expression and function of laminins in the embryonic and mature vasculature. Physiol Rev. 2005;85:979–1000. doi: 10.1152/physrev.00014.2004. [DOI] [PubMed] [Google Scholar]
- 10.Kalluri R. Basement membranes: structure, assembly and role in tumour angiogenesis. Nat Rev Cancer. 2003;3:422–33. doi: 10.1038/nrc1094. [DOI] [PubMed] [Google Scholar]
- 11.Patarroyo M, Tryggvason K, Virtanen I. Laminin isoforms in tumor invasion, angiogenesis and metastasis. Semin Cancer Biol. 2002;12:197–207. doi: 10.1016/S1044-579X(02)00023-8. [DOI] [PubMed] [Google Scholar]
- 12.Swayne GT, Smaje LH, Bergel DH. Distensibility of single capillaries and venules in the rat and frog mesentery. Int J Microcirc Clin Exp. 1989;8:25–42. [PubMed] [Google Scholar]
- 13.Mohamed MM, Sloane BF. Cysteine cathepsins: multifunctional enzymes in cancer. Nat Rev Cancer. 2006;6:764–75. doi: 10.1038/nrc1949. [DOI] [PubMed] [Google Scholar]
- 14.van Hinsbergh VW, Engelse MA, Quax PH. Pericellular proteases in angiogenesis and vasculogenesis. Arterioscler Thromb Vasc Biol. 2006;26:716–28. doi: 10.1161/01.ATV.0000209518.58252.17. [DOI] [PubMed] [Google Scholar]
- 15.Bergers G, Brekken R, McMahon G, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2:737–44. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pepper MS. Extracellular proteolysis and angiogenesis. Thromb Haemost. 2001;86:346–55. [PubMed] [Google Scholar]
- 17.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–74. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 18.Fang J, Shing Y, Wiederschain D, et al. Matrix metalloproteinase-2 is required for the switch to the angiogenic phenotype in a tumor model. Proc Natl Acad Sci U S A. 2000;97:3884–9. doi: 10.1073/pnas.97.8.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sloane BF, Yan S, Podgorski I, et al. Cathepsin B and tumor proteolysis: contribution of the tumor microenvironment. Semin Cancer Biol. 2005;15:149–57. doi: 10.1016/j.semcancer.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 20.Premzl A, Turk V, Kos J. Intracellular proteolytic activity of cathepsin B is associated with capillary-like tube formation by endothelial cells in vitro. J Cell Biochem. 2006;97:1230–40. doi: 10.1002/jcb.20720. [DOI] [PubMed] [Google Scholar]
- 21.Joyce JA, Baruch A, Chehade K, et al. Cathepsin cysteine proteases are effectors of invasive growth and angiogenesis during multistage tumorigenesis. Cancer Cell. 2004;5:443–53. doi: 10.1016/s1535-6108(04)00111-4. [DOI] [PubMed] [Google Scholar]
- 22.Gocheva V, Zeng W, Ke D, et al. Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes Dev. 2006;20:543–56. doi: 10.1101/gad.1407406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi GP, Sukhova GK, Kuzuya M, et al. Deficiency of the cysteine protease cathepsin S impairs microvessel growth. Circ Res. 2003;92:493–500. doi: 10.1161/01.RES.0000060485.20318.96. [DOI] [PubMed] [Google Scholar]
- 24.Hazen LG, Bleeker FE, Lauritzen B, et al. Comparative localization of cathepsin B protein and activity in colorectal cancer. J Histochem Cytochem. 2000;48:1421–30. doi: 10.1177/002215540004801012. [DOI] [PubMed] [Google Scholar]
- 25.Kruszewski WJ, Rzepko R, Wojtacki J, et al. Overexpression of cathepsin B correlates with angiogenesis in colon adenocarcinoma. Neoplasma. 2004;51:38–43. [PubMed] [Google Scholar]
- 26.Yanamandra N, Gumidyala KV, Waldron KG, et al. Blockade of cathepsin B expression in human glioblastoma cells is associated with suppression of angiogenesis. Oncogene. 2004;23:2224–30. doi: 10.1038/sj.onc.1207338. [DOI] [PubMed] [Google Scholar]
- 27.Keppler D. Towards novel anti-cancer strategies based on cystatin function. Cancer Lett. 2006;235:159–76. doi: 10.1016/j.canlet.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 28.Mort JS, Buttle DJ, Cathepsin B. Int J Biochem Cell Biol. 1997;29:715–20. doi: 10.1016/s1357-2725(96)00152-5. [DOI] [PubMed] [Google Scholar]
- 29.Buck MR, Karustis DG, Day NA, Honn KV, Sloane BF. Degradation of extracellular-matrix proteins by human cathepsin B from normal and tumour tissues. Biochem J. 1992;282:273–8. doi: 10.1042/bj2820273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang B, Sun J, Kitamoto S, et al. Cathepsin S controls angiogenesis and tumor growth via matrix-derived angiogenic factors. J Biol Chem. 2006;281:6020–9. doi: 10.1074/jbc.M509134200. [DOI] [PubMed] [Google Scholar]
- 31.Sivaparvathi M, McCutcheon I, Sawaya R, Nicolson GL, Rao JS. Expression of cysteine protease inhibitors in human gliomas and meningiomas. Clin Exp Metastasis. 1996;14:344–50. doi: 10.1007/BF00123393. [DOI] [PubMed] [Google Scholar]
- 32.Li W, Ding F, Zhang L, et al. Overexpression of stefin A in human esophageal squamous cell carcinoma cells inhibits tumor cell growth, angiogenesis, invasion, and metastasis. Clin Cancer Res. 2005;11:8753–62. doi: 10.1158/1078-0432.CCR-05-0597. [DOI] [PubMed] [Google Scholar]
- 33.Strojnik T, Lah TT, Zidanik B. Immunohistochemical staining of cathepsins B, L and stefin A in human hypophysis and pituitary adenomas. Anticancer Res. 2005;25:587–94. [PubMed] [Google Scholar]
- 34.Sinha AA, Quast BJ, Wilson MJ, et al. Prediction of pelvic lymph node metastasis by the ratio of cathepsin B to stefin A in patients with prostate carcinoma. Cancer. 2002;94:3141–9. doi: 10.1002/cncr.10604. [DOI] [PubMed] [Google Scholar]
- 35.Levicar N, Kos J, Blejec A, et al. Comparison of potential biological markers cathepsin B, cathepsin L, stefin A and stefin B with urokinase and plasminogen activator inhibitor-1 and clinicopathological data of breast carcinoma patients. Cancer Detect Prev. 2002;26:42–9. doi: 10.1016/s0361-090x(02)00015-6. [DOI] [PubMed] [Google Scholar]
- 36.Nagy J, Shih S-C, Wong W, Dvorak A, Dvorak H. The adenoviral angiogenesis/lymphangiogenesis assay. Methods in Enzymology. 2008;444:43–64. doi: 10.1016/S0076-6879(08)02803-6. [DOI] [PubMed] [Google Scholar]
- 37.Nagy JA, Morgan ES, Herzberg KT, Manseau EJ, Dvorak AM, Dvorak HF. Pathogenesis of ascites tumor growth: angiogenesis, vascular remodeling, and stroma formation in the peritoneal lining. Cancer Res. 1995;55:376–85. [PubMed] [Google Scholar]
- 38.Halangk W, Lerch MM, Brandt-Nedelev B, et al. Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J Clin Invest. 2000;106:773–81. doi: 10.1172/JCI9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blum G, von Degenfeld G, Merchant MJ, Blau HM, Bogyo M. Noninvasive optical imaging of cysteine protease activity using fluorescently quenched activity-based probes. Nat Chem Biol. 2007;3:668–77. doi: 10.1038/nchembio.2007.26. [DOI] [PubMed] [Google Scholar]
- 40.Wang X, Seed B. A PCR primer bank for quantitative gene expression analysis. Nucleic Acids Res. 2003;31:e154. doi: 10.1093/nar/gng154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu W, Saint DA. Validation of a quantitative method for real time PCR kinetics. Biochem Biophys Res Commun. 2002;294:347–53. doi: 10.1016/S0006-291X(02)00478-3. [DOI] [PubMed] [Google Scholar]
- 42.Rozhin J, Robinson D, Stevens MA, et al. Properties of a plasma membrane-associated cathepsin B-like cysteine proteinase in metastatic B16 melanoma variants. Cancer Res. 1987;47:6620–8. [PubMed] [Google Scholar]
- 43.Kano MR, Morishita Y, Iwata C, et al. VEGF-A and FGF-2 synergistically promote neoangiogenesis through enhancement of endogenous PDGF-B-PDGFRbeta signaling. J Cell Sci. 2005;118:3759–68. doi: 10.1242/jcs.02483. [DOI] [PubMed] [Google Scholar]
- 44.Nagy JA, Dvorak AM, Dvorak HF. VEGF-A(164/165) and PlGF: roles in angiogenesis and arteriogenesis. Trends Cardiovasc Med. 2003;13:169–75. doi: 10.1016/s1050-1738(03)00056-2. [DOI] [PubMed] [Google Scholar]
- 45.Bogyo M, Verhelst S, Bellingard-Dubouchaud V, Toba S, Greenbaum D. Selective targeting of lysosomal cysteine proteases with radiolabeled electrophilic substrate analogs. Chem Biol. 2000;7:27–38. doi: 10.1016/s1074-5521(00)00061-2. [DOI] [PubMed] [Google Scholar]
- 46.Hill PA, Buttle DJ, Jones SJ, et al. Inhibition of bone resorption by selective inactivators of cysteine proteinases. J Cell Biochem. 1994;56:118–30. doi: 10.1002/jcb.240560116. [DOI] [PubMed] [Google Scholar]
- 47.Sevenich L, Pennacchio LA, Peters C, Reinheckel T. Human cathepsin L rescues the neurodegeneration and lethality in cathepsin B/L double-deficient mice. Biol Chem. 2006;387:885–91. doi: 10.1515/BC.2006.112. [DOI] [PubMed] [Google Scholar]
- 48.Vasiljeva O, Papazoglou A, Kruger A, et al. Tumor cell-derived and macrophage-derived cathepsin B promotes progression and lung metastasis of mammary cancer. Cancer Res. 2006;66:5242–50. doi: 10.1158/0008-5472.CAN-05-4463. [DOI] [PubMed] [Google Scholar]
- 49.Wolters PJ, Chapman HA. Importance of lysosomal cysteine proteases in lung disease. Respir Res. 2000;1:170–7. doi: 10.1186/rr29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Calkins CC, Sameni M, Koblinski J, Sloane BF, Moin K. Differential localization of cysteine protease inhibitors and a target cysteine protease, cathepsin B, by immuno-confocal microscopy. J Histochem Cytochem. 1998;46:745–51. doi: 10.1177/002215549804600607. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.