

Abstract
The diphtheria toxin repressor (DtxR) is the best-characterized member of a family of homologous proteins that regulate iron uptake and virulence gene expression in the Gram-positive bacteria. DtxR contains two domains that are separated by a short, unstructured linker. The N-terminal domain is structurally well-defined and is responsible for Fe2+ binding, dimerization, and DNA binding. The C-terminal domain adopts a fold similar to eukaryotic Src homology 3 domains, but the functional role of the C-terminal domain in repressor activity is unknown. The solution structure of the C-terminal domain, consisting of residues N130-L226 plus a 13-residue N-terminal extension, has been determined by using NMR spectroscopy. Residues before A147 are highly mobile and adopt a random coil conformation, but residues A147-L226 form a single structured domain consisting of five β-strands and three helices arranged into a partially orthogonal, two-sheet β-barrel, similar to the structure observed in the crystalline Co2+ complex of full-length DtxR. Chemical shift perturbation studies demonstrate that a proline-rich peptide corresponding to residues R125-G139 of intact DtxR binds to the C-terminal domain in a pocket formed by residues in β-strands 2, 3, and 5, and helix 3. Binding of the proline-rich peptide by the C-terminal domain of DtxR presents an example of peptide binding by a prokaryotic Src homology 3-like protein. The results of this study, combined with previous x-ray studies of intact DtxR, provide insights into a possible biological function of the C-terminal domain in regulating repressor activity.
The diphtheria toxin repressor (DtxR) regulates the expression of several iron-sensitive genes in Corynebacterium diphtheriae, including the iron-response protein genes (irp), a heme oxygenase, and the diphtheria toxin gene (tox) (1–3). DtxR is the best-characterized member of a family of homologous proteins that regulate iron uptake and virulence gene expression in the Gram-positive bacteria, including Mycobacterium tuberculosis, Brevibacterium lactofermentum, Streptomyces lividans, and Streptomyces pilosus (4, 5). The N-terminal 136 aa of this 226-aa protein form a DNA-binding dimer, whose structure has been determined by x-ray crystallography with (6–10) and without (7, 10) divalent cations, and while bound to a DNA operator sequence (11). The structure of the C-terminal 90 aa, which is more mobile than the N-terminal domain in all crystal forms, has been mapped in the Co2+-DtxR complex (9) and shown to be structurally homologous to eukaryotic Src homology 3 (SH3) domains, although the amino acid sequence homology is low. A limited number of mutations in the C-terminal domain (12) or the linker region (13) affect repressor activity, but the structural nature of this regulation by the C-terminal domain has been obscure.
To obtain a more complete understanding of the function of the intact repressor protein, we are focusing on the C-terminal domain. From the sequential assignment of resonances in heteronuclear NMR spectra of a recombinant C-terminal domain (residues N130-L226), we have shown that this isolated domain contains five β-strands and three helices (14). Here, we present the three-dimensional (3D) structure of DtxR(130–226) determined in solution by using multidimensional NMR spectroscopy and show that it adopts an SH3-like conformation. We also present evidence that this prokaryotic SH3-like domain binds to a proline-rich segment that is located in the region linking the N- and C-terminal domains of DtxR and is conserved in all known DtxR homologues. NMR chemical shift perturbation studies demonstrate that a synthetic peptide corresponding to this internal ligand interacts with specific amino acid residues in the C-terminal domain. The demonstration of peptide binding by the C-terminal domain suggests a possible mechanism for regulating the activity of the intact repressor protein.
MATERIALS AND METHODS
Protein Expression, Purification, and Sample Preparation.
The expression vector for DtxR(130–226) was constructed by first introducing a unique BamHI restriction endonuclease site in the dtxR structural gene before N130. The portion of dtxR cDNA encoding residues N130-L226 then was excised by digestion with BamHI and HindIII, and, after purification by agarose gel electrophoresis, ligated into the BamHI and HindIII sites of the pQE30 expression vector (Qiagen, Chatsworth, CA). The final protein construct, referred to as DtxR(130–226), contains a 13-residue extension at the N terminus that includes a six-residue His tag (MRGSHHHHHHGSG) to facilitate purification. DtxR(130–226) was expressed in Escherichia coli strain HMS174 grown in M9 minimal medium containing 1 g/liter 15NH4Cl and 4 g/liter glucose or 2 g/liter 13C6-glucose to produce uniformly 15N- or 15N/13C- labeled proteins, respectively. Protein expression was induced by addition of 0.4 mM isopropyl β-d-thiogalactoside to the culture at an OD600 of ≈0.6 and grown for an additional 3 h before harvesting by centrifugation. The cell pellet was resuspended in 20 ml of lysis buffer (50 mM potassium phosphate, pH 7.5, containing 0.5 M NaCl, 8 M urea, 5 mM imidazole, and 1 mM PMSF) and lysed by French press. The clarified lysate was chromatographed over a Ni2+-chelating Sepharose Fast Flow column (Amersham Pharmacia), washed with the lysis buffer (containing no urea or PMSF), and eluted with a linear gradient of imidazole (10–600 mM). Fractions containing DtxR(130–226) (at approximately 300 mM imidazole) were pooled, dialyzed, and concentrated in a Centriprep 3 (Amicon) before exchange into phosphate buffer for NMR analysis (50 mM potassium phosphate, containing 0.4% NaN3 and 10% D2O, pH 6.5). A shorter construct of the C-terminal domain, corresponding to residues 144–226 [DtxR(144–226)], was generated from DtxR(130–226) by amplifying the cDNA encoding these residues using PCR, followed by ligation into the NdeI and BamHI sites of a pET-15b expression vector (Novagen). This construct contains a 21- residue extension at the N terminus of DtxR(144–226), including a six-residue His tag and a thrombin cleavage site. DtxR(144–226) uniformly enriched in 15N was expressed in BL21(DE3) E. coli grown in M9 minimal medium supplemented with 15NH4Cl and purified as described for DtxR(130–226).
NMR Spectroscopy.
NMR spectra were collected at 30°C on a three-channel 500 MHz Varian Unityplus instrument equipped with waveform generators and three-axis pulsed field gradient accessories. A 3D 15N-separated nuclear Overhauser effect spectroscopy (NOESY)-heteronuclear single quantum correlation (HSQC) spectrum (15) was collected on a uniformly 15N-enriched DtxR(130–226) sample by using 8,333-, 1,650-, and 6,250-Hz sweep widths, and digitized as 512, 48, and 128 complex points in the ω3 (1HN), ω2 (15N), and ω1 (1H) dimensions, respectively. Two complementary 3D 13C-separated CCH-NOESY and HCH-NOESY spectra (16) were collected on uniformly 15N,13C-labeled DtxR(130–226) in the deuterated phosphate buffer by using sweep widths of 2,999.2 and 8,798.8 Hz for 1H and 13C chemical shift dimensions, respectively. A homonuclear two-dimensional (2D) NOESY spectrum was collected on a 720-MHz Varian Unityplus spectrometer, by using excitation sculpting for solvent suppression (17). All NOESY spectra were collected with a 120-ms mixing time. The φ-dihedral angle restraints were obtained from analysis of HNHA (18) and HMQC-J (19) spectra collected on 15N-labeled DtxR(130–226). Slowly exchanging amide hydrogens were identified from a 2D 1H-15N HSQC spectrum collected 24 h after dissolution in the deuterated phosphate buffer. Heteronuclear NOEs were measured as described (20). All NMR data were processed on Silicon Graphics workstations by using nmrpipe (21) and analyzed with nmrview (22).
Structure Calculation.
Structure calculations were performed by using x-plor, version 3.843 (23). The interproton NOE peaks of 2D and 3D NOESY spectra were classified as 1.8–2.8, 1.8–3.5, 1.8–5.0, and 1.8–6.0 Å corresponding to strong, medium, weak, and very weak NOEs. Pseudoatom and proton multiplicity corrections were made as described by Fletcher et al. (24). Hydrogen bond restraints were added as 2.4–3.5 Å and 1.5–2.8 Å for N-O and H-O internuclear distances, respectively, in regions where regular secondary structure elements were identified in initial structures calculated by using only NOE restraints. The φ dihedral angle restraints were applied as −120 ± 30° for β-strands with 3JHNHα > 8 Hz and −60 ± 30° for helical regions with 3JHNHα < 5.5 Hz, respectively (25). Hydrogen bond and dihedral angle restraints were combined with NOE restraints only in the final stage of the structural refinement. A total of 100 structures were calculated, of which 67 showed no restraint violations greater than 0.5 Å and 5°. From the 67 structures, 20 structures with lowest total energy were chosen for further refinement by five additional cycles of simulated annealing by decreasing the initial temperature by 100 K in each cycle from 900 K to 500 K (26). The structures were viewed by using insightii (Molecular Simulations, Sacramento, CA), and analyzed by using molmol (27), aqua, and procheck-nmr software (28).
Peptide Binding Experiments.
A 15-residue peptide (RSPFGNPIPGLDELG; residues R125-G139 of DtxR) was synthesized by using standard solid-phase methods. The peptide showed a single peak on analytical reversed-phase HPLC and gave a mass spectrum identical to that expected. Binding experiments were performed by adding aliquots of peptide to a sample of uniformly 15N-labeled DtxR(130–226) or DtxR(144–226) in the phosphate buffer, pH 6.5 at 30°C. 2D 1H-15N HSQC spectra (29) were collected by using 1,024 and 140 complex points over 8,333.3 and 1,650 Hz spectral widths in the 1H and 15N dimensions, respectively.
RESULTS AND DISCUSSION
Structure Determination.
Chemical shift assignments for the backbone and side-chain 1H, 13C, and 15N resonances of DtxR(130–226) were obtained by using the standard suite of triple-resonance NMR experiments (14). The 1Hα (Fig. 1a), 13Cα, and 13CO chemical shift deviations (30) suggested the presence of five β-strands and three helices, which subsequently were confirmed in the final 3D structures (Fig. 2). The structure of DtxR(130–226) was determined from a total of 1,142 NMR restraints in the form of NOE-derived interproton distances, φ dihedral angles, and hydrogen bonds. Structures were calculated by using a hybrid distance geometry-simulated annealing protocol (23, 31). A summary of the structural statistics for the final set of 20 structures is presented in Table 1. These 20 structures had the lowest total energies, no distance violations greater than 0.35 Å, and dihedral angle violations less than 5o. Within this family of structures, 97.6% of residues had backbone ψ,φ angles located in the allowed regions (28) of the Ramachandran plot. The rms deviation for the backbone atoms in all β-strands superimposed on the average structure was 0.80 Å (Table 1).
Figure 1.

(a) Chemical shift deviation (CSD) between the measured Hα chemical shift (14) and the random coil Hα value (25). Clusters of positive and negative values suggest β-strands and helical structures, respectively. (b) Backbone 15N-{1H} heteronuclear NOE for each residue. NOE values for some residues were not presented because of resonance overlap. (c) The number of proton–proton NOEs per residue (No. NOE/res). (d) The backbone rms deviation per residue of the 20 structures from the average structure. The helical (open ellipses) and β-strand (solid squares) regions of DtxR(130–226) are depicted at the top.
Figure 2.

Structure of DtxR(130–226) with the unstructured residues 130–145 omitted. Stereoview of the backbone superposition of the 20 refined structures determined as described in the text. The N terminus of the structure is located at lower left.
Table 1.
Structure statistics
| NOE restraints | |
| Total | 1,086 |
| Intraresidue | 548 |
| Sequential | 263 |
| Medium range | 87 |
| Long range | 188 |
| φ dihedral angle restraints | 26 |
| Hydrogen bond restraints | 30 |
| Deviation from experimental restraints | |
| Distance restraints, Å | 0.014 ± 0.003 |
| Dihedral restraints, deg | 0.17 ± 0.09 |
| Deviation from idealized covalent geometry | |
| Bonds, Å | 0.002 ± 0.000 |
| Angles, deg | 0.48 ± 0.01 |
| Impropers, deg | 0.359 ± 0.005 |
| Backbone rms deviation, Å | |
〈SA〉 to 〈 〉 residues A147-L226 〉 residues A147-L226 |
1.45 ± 0.16 |
〈SA〉 to 〈 〉 all β-strands 〉 all β-strands |
0.80 ± 0.08 |
〈SA〉 stands for the ensemble of 20 NMR structures and 〈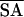 〉 is the average structure of the ensemble calculated by using x-plor. The parameter used to calculate the van der Waals (vdw) repulsion energy was 0.75 rather than 0.80 (47).
〉 is the average structure of the ensemble calculated by using x-plor. The parameter used to calculate the van der Waals (vdw) repulsion energy was 0.75 rather than 0.80 (47).
Description of NMR Structure.
The structure of DtxR(130–226) consists of a disordered N-terminal region (residues N130-A146) followed by a folded domain (residues A147-L226) (Fig. 3). The five β-strands identified in the final ensemble of structures include residues V163-Q167 (β1), V193-R198 (β2), H201-H206 (β3), K209-V211 (β4), and R222-E225 (β5). These strands are organized into a β-barrel formed by two partially orthogonal antiparallel β-sheets, with strand β2 shared by the two sheets. Sheet 1 contains strands β1, β2′ (V193-I195), and β5, while sheet 2 is formed by strands β2′′ (V196-R198), β3, and β4. Preceding β1 in the folded domain, the polypeptide chain forms two short, extended β-like structures (T150-R151 and S158-P160) that are separated by a single-turn 310 helix [residues V152-A155 (H1)]. The β-like structures of these two short segments are indicated by down-field Hα chemical shifts (Fig. 1a) and by long-range NOE contacts from residues T150-R151 to β5 and from residues S158-P160 to β2′′. Strands β1 and β2′ are connected by a long loop (residues I168-G190) containing the single α-helix [residues D177-A185 (H2)]. A short 310 helix [residues D215-A218 (H3)] is formed between strands β4 and β5, while strands β2′′-β3 and β3-β4 are connected by tight turns. Many of the hydrophobic residues in helices H1, H2, and H3 (V152, I153, A155, L182, L183, A185, and A218) showed NOE contacts with the β-barrel, forming the hydrophobic core.
Figure 3.

Stereoview of the ribbon representation of the minimized average structure. β-strands are colored in purple and helical regions in green. Orientation is the same as shown in Fig. 2. The structure was created by using molmol (27).
To obtain insight into protein chain mobility, we measured steady-state backbone 15N-{1H} heteronuclear NOE values. Heteronuclear NOEs for a limited number of residues could not be determined because of spectral overlap. Residues preceding A147 have negative heteronuclear NOEs (Fig. 1b), indicating high mobility (25). In contrast, residues A147-L226 have positive heteronuclear NOEs, indicating lower overall mobility and that these residues tumble in solution as a single folded domain. The slightly lower heteronuclear NOEs observed for residues I168-E175 suggest an increased mobility for these loop residues compared with other residues in the folded domain. The polypeptide chain mobility deduced from the heteronuclear NOE data correlated well with the number of proton–proton NOEs and the rms deviation per residue (Fig. 1 c and d), indicating that the limited number of interproton NOEs and low structural precision of the linker and the loop regions in the final family of structures reflect the internal motions of the polypeptide chains.
The C-terminal domain of DtxR adopts a similar fold in the crystal (9) and in solution, with a 2.6-Å rms deviation obtained when superimposing the Cα atoms of the two structures (residues P148-R198 and H201-L226). The largest difference between the two structures was found in residues I168-G190, consistent with their location in a long loop and their increased mobility in solution. Residues G141-A147, which were not traced in previous x-ray structures, were also highly mobile in solution and were poorly defined by the NMR data.
The C-Terminal Domain of DtxR Binds a Proline-Rich Peptide.
During purification and characterization of DtxR(130–226), it was observed that highly purified protein ran as a series of bands in nondenaturing polyacrylamide gels that correlated in mass to multiples of the monomeric protein molecular weight (Fig. 4). As seen in Fig. 4, the monomeric and trimeric forms were predominant, with lower amounts of dimer and higher aggregates observed. The formation of oligomers was not altered upon incubation with EDTA or by addition of 10 mM Ni2+, suggesting that the oligomerization was not induced by residues of the His tag binding to metal ions leached during purification. In contrast, a single molecular weight band corresponding to monomeric DtxR(130–226) was observed in denaturing PAGE gels (not shown). As noted previously (9), the structure of residues P160-L226 is homologous to eukaryotic SH3 domains. SH3 domains bind peptides with the consensus sequence PpXP, where P is a strictly conserved proline, p is generally a proline, and X is a hydrophobic residue (32–37). Following the His tag and additional residues associated with the cloning sites (see Materials and Methods), the DtxR(130–226) sequence begins as NPIPGL. We reasoned that the oligomers may result from DtxR(130–226) binding this proline-containing segment. To test this hypothesis, DtxR(144–226), in which this internal ligand is removed, was created. NMR spectra of DtxR(144–226) showed that the protein adopted the same fold as DtxR(130–226). However, in contrast to DtxR(130–226), DtxR(144–226) migrated as a single band corresponding to monomer molecular weight in nondenaturing polyacrylamide gels (Fig. 4).
Figure 4.

Nondenaturing polyacrylamide gels indicating the degree of oligomerization of DtxR(130–226) (lane A) and DtxR(144–226) (lane B). The reduced electrophoretic mobility of DtxR(144-226) monomer compared to DtxR(133-226) monomer reflects the additional residues at the N terminus of DtxR(144-226) arising from the expression construct (see Materials and Methods).
The possible binding interaction between the SH3-like C-terminal domain of DtxR and the internal proline-rich sequence was further investigated by using a synthetic peptide having the sequence RSPFGNPIPGLDELG, which corresponds to residues R125-G139 of full-length DtxR. Aliquots of this peptide were added to DtxR(130–226), and 2D HSQC spectra were collected. Because chemical shifts are extremely sensitive reporters of the local magnetic environment, ligand binding generally changes the chemical shifts of backbone and side-chain resonances. This approach is sensitive to weak binding (into the millimolar range; ref. 38) and has been used previously to demonstrate binding between proline-rich peptides and eukaryotic SH3 domains (36). When a stoichiometric amount of this peptide was added to DtxR(130–226), a limited number of protein 1H and/or 15N resonances exhibited line broadening or resonance frequency changes in 2D HSQC spectra (residues V174, I187, E192, L204, H206, D215, D216, L217, H219, and T220), but no additional resonances appeared. At approximately a 5:1 peptide/protein molar ratio, additional residues in strands β2, β3, β5, and helix H3 were shifted in an HSQC spectrum (Fig. 5). The chemical shift perturbation data demonstrate weak binding of the peptide by the SH3-like domain, in fast exchange on the NMR time scale. The perturbed residues generate a putative peptide-binding surface located between the long loop and the β-barrel (Fig. 6). The presence of an internal partial ligand that competes with the external peptide complicates a quantitative analysis of the binding affinity for the peptide. By using the existing NMR data we estimate an apparent dissociation constant in the 100 μM-1 mM range, which is slightly higher than that obtained for eukaryotic SH3 domains binding optimized peptide ligands (32–37). DtxR(144–226) also binds the R125-G139 peptide, with the same residues being shifted upon binding.
Figure 5.

Plot of 15N and 1HN chemical shift changes (in ppm) induced in DtxR(130–226) upon addition of the peptide as a function of residue number. The solid and cross-hatched bars indicate the magnitude of 1HN and 15H chemical shift changes, respectively. Only changes that are larger than twice the digital resolution in the given chemical shift dimension are indicated.
Figure 6.

Stereoview of the Cα trace of the SH3-like domain of DtxR showing the residues implicated in peptide binding. Residues that shifted upon addition of the peptide are depicted as red balls.
Except for NOEs observed between side-chain protons of A146 and I187 that were consistent with oligomer formation, no NOEs were observed between residues at the N terminus and the folded domain of DtxR(130–226), although some residues at the N terminus of DtxR(130–226) shifted after addition of the R125-G139 peptide. These intermolecular NOEs disappeared upon dilution of the DtxR(130–226) sample. The absence of NOEs from the tail to the folded domain of DtxR(130–226) may be attributed to the high flexibility of the N terminus in the monomeric species (Fig. 1b) and to the variety and low concentration of oligomeric species in solution.
A Proposed Functional Role for Peptide Binding.
A working model for transcriptional regulation by DtxR is that micromolar concentrations of Fe2+ or other divalent metals trigger the formation of the metal-bound dimeric state, which then binds to the tox and irp operators (39–42). In the absence of divalent metal ligand, DtxR is thought to exist as an inactive, monomeric apo-protein that is incapable of binding DNA. Residues R125-G139 make numerous contacts with the three helices that constitute the dimerization interface in the N-terminal domain, thereby contributing to the stabilization of the dimeric form of DtxR (9). In the current work, we found that residues R125-G139 also can interact with the C-terminal domain of DtxR. If residues R125-G139 were to dissociate from the N-terminal domain, the dimeric structure might be destabilized and dissociate into monomers. We propose that the C-terminal domain binds residues R125-G139 in the monomeric state, thereby altering the monomer-dimer equilibrium and effectively stabilizing the monomeric, inactive form. Our data is consistent with either an inter- or intramolecular binding. This model for the regulation of dimer formation by the SH3-like C-terminal domain is consistent with the weakly cooperative activation of DtxR by metal ions (4) and with the existing C-terminal domain mutants that alter repressor activity (12, 13).
Eukaryotic SH3 domains in Hck (43), Src (44), and Itk (45) regulate tyrosine kinase activities in signal transduction cascades by weak binding to an internal proline-containing peptide whose sequence differs from the high-affinity peptide sequences that activate the kinase. Here, we have postulated that binding to an internal proline-containing region by the SH3-like domain of this prokaryotic protein has significance in regulating the repressor activity of intact DtxR. According to our model, the C-terminal domain plays no direct role in the structure or function of the dimeric form of the repressor and must be flexibly linked to the N-terminal domain. This intrinsic flexibility may explain the low averaged electron density found for this domain in the existing crystal structures (6–11). Residues L120-L226 were not traced in a crystal structure of DtxR(C102D) complexed with a 33-bp DNA sequence (11), so the structure of the proline-containing region and the C-terminal domain in this state of the repressor is uncertain.
The N-terminal domain of DtxR shows strong homology with the other members of Gram-positive toxin gene repressor proteins. A recent crystal structure of the DtxR homologue from M. tuberculosis, IdeR, shows the proteins are structurally homologous as well (46). Similarly, the sequence homology of the C-terminal domains in the family of DtxR homologues suggests that they will adopt SH3-like folds (Fig. 7). Residues S126-G139 are highly conserved in all known DtxR homologues, therefore it is likely that the regulatory mechanism proposed here for DtxR is applicable to the entire family of virulence-gene repressor proteins in the Gram-positive bacteria. This regulatory model for the DtxR C-terminal domain also suggests novel routes to the development of nonmetal-ion activators of repressor activity. For example, ligands that compete with the proline-rich region for binding to the SH3-like C-terminal domain may activate repressor function and decrease toxin expression. Such molecules might serve to attenuate the virulence of the pathogens in vivo.
Figure 7.

Sequence comparison of repressor proteins homologous to DtxR found in other Gram-positive bacteria. The strongly conserved N-terminal domain is boxed; conserved residues corresponding to S126-G139 are highlighted in white text with black background; conserved or semiconserved residues in the C-terminal domain are indicated by a star. Sequences were obtained from the Swiss-Prot Data Bank (accession nos. M80338, U14190, U14191, Z50049, Z50048, and L35906).
Acknowledgments
We thank Dr. Lewis Kay (University of Toronto) for the Varian pulse sequences and for some of the data processing scheme and Gulsah Sanli (Florida State University) for assistance with the coupling constant analysis. This work was supported by U.S. Public Health Service research grants from the National Institute of General Medicine (GM54035, T.M.L.), and the National Cancer Institute (CA 47439, D.L.D.C.).
ABBREVIATIONS
- SH3
Src homology 3
- DtxR
diphtheria toxin repressor
- 3D
three-dimensional
- NOE(SY)
nuclear Overhauser effect (spectroscopy)
- HSQC
heteronuclear single quantum correlation
- 2D
two-dimensional
Note Added in Proof
A recent study by Goranson-Siekierke et al. (48) has demonstrated that single alanine substitutions for residues R80, S126, and N130 caused severely decreased DtxR activity. Crystallographic analyses of dimeric metal complexes of the native protein show that these residues coordinate an oxyanion, which has been identified as a possible co-corepressor (9, 49). In dilute solutions, the dimeric form of the protein is stabilized by low concentrations of Fe2+ or other divalent transition metal cations, but dimerization also is favored in the absence of the metal ions at high protein concentration under crystallizing conditions. High-resolution analyses of crystals of the metal-free DtxR (10) show a dimeric structure very similar to the metal-bound form, in which the segment including the conserved sequence S126-G139 (Fig. 7) is folded in an ordered conformation contacting the helices of the N-terminal domain involved in dimer formation; these polar interactions among the residues R80, S126, and N130 together with water and/or anion evidently contribute to the stability of the dimer interface. Our results demonstrate that the proline-rich segment, including residues S126 and N130, binds to the isolated C-terminal SH3-like domain of DtxR in a manner similar to the peptide binding by eukaryotic SH3 domains (43−45). According to our model for the regulation of the DtxR activity, binding of the proline-rich segment to the C-terminal SH3-like domain should stabilize the inactive monomeric form of the repressor. Because replacement of the polar residues R80, S126, and N130 with alanines will weaken the interaction between the S126-L138 segment and the N-terminal dimerization domain, we interpret the recent results reported by Goranson-Siekierke et al. (48) to indicate that destabilization of the proline-rich segment in the N-terminal domain of the dimer consequently should favor binding of this segment to the C-terminal SH3-like domain in the inactive monomer, even in the presence of activating metal ions. Thus, the sequence S126-G139 may function as an internal molecular switch, either associated with the N-terminal domain, thereby contributing to the stability of the active, metal-bound dimeric form of the repressor, or alternatively bound to the C-terminal domain, favoring the inactive monomeric form.
Footnotes
Data deposition: Chemical shift assignments reported in this paper have been deposited in the BioMagResBank, Madison, WI (accession no. 4183). The structure coordinates reported in this paper have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1bym).
References
- 1.Tao X, Schiering N, Zeng H-Y, Ringe D, Murphy J R. Mol Microbiol. 1994;14:191–197. doi: 10.1111/j.1365-2958.1994.tb01280.x. [DOI] [PubMed] [Google Scholar]
- 2.Pappenheimer A M. Annu Rev Biochem. 1977;46:69–94. doi: 10.1146/annurev.bi.46.070177.000441. [DOI] [PubMed] [Google Scholar]
- 3.Schmitt M P. Infect Immun. 1997;65:4634–4641. doi: 10.1128/iai.65.11.4634-4641.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oguiza J A, Tao X, Marcos A T, Martin J F, Murphy J R. J Bacteriol. 1995;177:465–467. doi: 10.1128/jb.177.2.465-467.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmitt M P, Predich M, Doukhan L, Smith I, Holmes R K. Infect Immun. 1995;63:4284–4289. doi: 10.1128/iai.63.11.4284-4289.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qiu X, Verlinde C L M J, Zhang S, Schmitt M P, Holmes R K, Hol W G J. Structure (London) 1995;3:87–100. doi: 10.1016/s0969-2126(01)00137-x. [DOI] [PubMed] [Google Scholar]
- 7.Schiering N, Tao X, Zeng H-Y, Murphy J R, Petsko G A, Ringe D. Proc Natl Acad Sci USA. 1995;92:9843–9850. doi: 10.1073/pnas.92.21.9843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding X, Zeng H, Schiering N, Ringe D, Murphy J R. Nat Struct Biol. 1996;3:382–387. doi: 10.1038/nsb0496-382. [DOI] [PubMed] [Google Scholar]
- 9.Qiu X, Pohl E, Holmes R K, Hol W G J. Biochemistry. 1996;35:12292–12302. doi: 10.1021/bi960861d. [DOI] [PubMed] [Google Scholar]
- 10.Pohl E, Holmes R K, Hol W G J. J Biol Chem. 1998;273:22420–22427. doi: 10.1074/jbc.273.35.22420. [DOI] [PubMed] [Google Scholar]
- 11.White A, Ding X, vanderSpek J C, Murphy J R, Ringe D. Nature (London) 1998;394:502–506. doi: 10.1038/28893. [DOI] [PubMed] [Google Scholar]
- 12.Sun L, vanderSpek J C, Murphy J R. Proc Natl Acad Sci USA. 1998;95:14985–14990. doi: 10.1073/pnas.95.25.14985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmitt M P, Holmes R K. Mol Microbiol. 1993;9:173–181. doi: 10.1111/j.1365-2958.1993.tb01679.x. [DOI] [PubMed] [Google Scholar]
- 14.Twigg P D, Wylie G P, Wang G, Murphy J R, Caspar D L D, Logan T M. J Biomol NMR. 1999;13:197–198. doi: 10.1023/a:1008391728428. [DOI] [PubMed] [Google Scholar]
- 15.Zhang O, Kay L E, Olivier J P, Forman-Kay J D. J Biomol NMR. 1994;4:845–858. doi: 10.1007/BF00398413. [DOI] [PubMed] [Google Scholar]
- 16.Vuister G, Clore G M, Gronenborn A M, Powers R, Garret D S, Tschudin R, Bax A. J Magn Reson. 1993;101:210–213. [Google Scholar]
- 17.Callihan D, West J, Kumar S, Schweitzer B I, Logan T M. J Magn Reson. 1996;112:82–85. doi: 10.1006/jmrb.1996.0114. [DOI] [PubMed] [Google Scholar]
- 18.Kuboniwa H, Grzesiek S, Delagio F, Bax A. J Biomol NMR. 1994;4:871–878. doi: 10.1007/BF00398416. [DOI] [PubMed] [Google Scholar]
- 19.Kay L E, Bax A. J Magn Reson. 1990;86:110–126. [Google Scholar]
- 20.Farrow N A, Muhandiram R, Singer A U, Pascal S M, Kay C M, Gish G, Shoelson S E, Pawson T, Forman-Kay J D, Kay L E. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 21.Delaglio F, Grzesiek S, Vuister G W, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 22.Johnson B A, Blevins R A. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 23.Brünger A T. x-plor: A System for X-Ray Crystallography and NMR. New Haven: Yale Univ. Press; 1992. , Version 3.1. [Google Scholar]
- 24.Fletcher C M, Jones D N M, Diamond R, Neuhaus D. J Biomol NMR. 1996;8:292–310. doi: 10.1007/BF00410328. [DOI] [PubMed] [Google Scholar]
- 25.Wüthrich K. NMR of Proteins and Nucleic Acids. New York: Wiley; 1986. [Google Scholar]
- 26.Briercheck D M, Wood T C, Allison T J, Richardson J P, Rule G S. Nat Struct Biol. 1998;5:393–399. doi: 10.1038/nsb0598-393. [DOI] [PubMed] [Google Scholar]
- 27.Koradi R, Billeter M, Wüthrich K. J Mol Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 28.Laskowski R A, Rullmann J A C, MacArthur M W, Kaptein R, Thornton J M. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 29.Kay L E, Keifer P, Saarinen T. J Am Chem Soc. 1992;114:10663–10665. [Google Scholar]
- 30.Wishart D S, Sykes B D. J Biomol NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- 31.Nilges M, Clore G M, Gronenborn A M. FEBS Lett. 1988;229:317–324. doi: 10.1016/0014-5793(88)81148-7. [DOI] [PubMed] [Google Scholar]
- 32.Feng S, Chen J K, Yu H, Simon J A, Schreiber S L. Science. 1994;266:1241–1247. doi: 10.1126/science.7526465. [DOI] [PubMed] [Google Scholar]
- 33.Lim W A, Richards F M, Fox R O. Nature (London) 1994;372:375–379. doi: 10.1038/372375a0. [DOI] [PubMed] [Google Scholar]
- 34.Saraste M, Musacchio A. Nat Struct Biol. 1994;1:835–837. doi: 10.1038/nsb1294-835. [DOI] [PubMed] [Google Scholar]
- 35.Viguera A R, Arrondo J L R, Musacchio A, Saraste M, Serrano L. Biochemstry. 1994;33:10925–10933. doi: 10.1021/bi00202a011. [DOI] [PubMed] [Google Scholar]
- 36.Wittekind M, Mapelli C, Farmer B T II, Suen K-L, Goldfarb V, Tsao J, Lavoie T, Barbacid M, Meyers C A, Mueller L. Biochemistry. 1994;33:13531–13539. doi: 10.1021/bi00250a004. [DOI] [PubMed] [Google Scholar]
- 37.Yu H, Chen J K, Feng S, Dalgarno D C, Brauer A W, Schreiber S L. Cell. 1994;76:933–945. doi: 10.1016/0092-8674(94)90367-0. [DOI] [PubMed] [Google Scholar]
- 38.Shuker S B, Hajduk P J, Meadows R P, Fesik S W. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 39.Tao X, Boyd J, Murphy J R. Proc Natl Acad Sci USA. 1992;89:5897–5901. doi: 10.1073/pnas.89.13.5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tao X, Zeng H Y, Murphy J R. Proc Natl Acad Sci USA. 1995;92:6803–6807. doi: 10.1073/pnas.92.15.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Z, Schmitt M P, Holmes R K. Infect Immun. 1994;62:1600–1608. doi: 10.1128/iai.62.5.1600-1608.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tao X, Murphy J R. Proc Natl Acad Sci USA. 1994;91:9646–9650. doi: 10.1073/pnas.91.20.9646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sicheri F, Maorefi I, Kuriyan J. Nature (London) 1997;385:602–609. doi: 10.1038/385602a0. [DOI] [PubMed] [Google Scholar]
- 44.Xu W, Harrison S C, Eck M L. Nature (London) 1997;385:595–602. doi: 10.1038/385595a0. [DOI] [PubMed] [Google Scholar]
- 45.Andreotti A H, Bunnell S C, Feng S, Berg L J, Screiber S L. Nature (London) 1997;385:93–97. doi: 10.1038/385093a0. [DOI] [PubMed] [Google Scholar]
- 46.Pohl E, Holmes R K, Hol W G J. J Mol Biol. 1999;285:1145–1156. doi: 10.1006/jmbi.1998.2339. [DOI] [PubMed] [Google Scholar]
- 47.Johnson P E, Joshi M D, Tomme P, Kilburn D G, McIntosh L P. Biochemistry. 1996;35:14381–14394. doi: 10.1021/bi961612s. [DOI] [PubMed] [Google Scholar]
- 48.Goranson-Siekierke J, Pohl E, Hol W G J, Holmes R K. Infect Immun. 1999;67:1806–1811. doi: 10.1128/iai.67.4.1806-1811.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pohl E, Qiu W, Must L M, Holmes R K, Hol W G J. Protein Sci. 1997;6:1114–1118. doi: 10.1002/pro.5560060519. [DOI] [PMC free article] [PubMed] [Google Scholar]