

Abstract
Incoherent quasi-elastic neutron scattering (QENS) has been used to measure the dynamics of water molecules in solutions of a model protein backbone, N-acetyl-glycine-methylamide (NAGMA), as a function of concentration, for comparison with results for water dynamics in aqueous solutions of the N-acetyl-leucine-methylamide (NALMA) hydrophobic peptide at comparable concentrations. From the analysis of the elastic incoherent structure factor, we find significant fractions of elastic intensity at high and low concentrations for both solutes, which corresponds to a greater population of protons with rotational time scales outside the experimental resolution (>13 ps). The higher-concentration solutions show a component of the elastic fraction that we propose is due to water motions that are strongly coupled to the solute motions, while for lowconcentration solutions an additional component is activated due to dynamic coupling between inner and outer hydration layers. An important difference between the solute types at the highest concentration studied is found from stretched exponential fits to their experimental intermediate scattering functions, showing more pronounced anomalous diffusion signatures for NALMA, including a smaller stretched exponent β and a longer structural relaxation time τ than those found for NAGMA. The more normal water diffusion exhibited near the hydrophilic NAGMA provides experimental support for an explanation of the origin of the anomalous diffusion behavior of NALMA as arising from frustrated interactions between water molecules when a chemical interface is formed upon addition of a hydrophobic side chain, inducing spatial heterogeneity in the hydration dynamics in the two types of regions of the NALMA peptide. We place our QENS measurements on model biological solutes in the context of other spectroscopic techniques and provide both confirming as well as complementary dynamic information that attempts to give a unifying molecular view of hydration dynamics signatures near peptides and proteins.
Introduction
Hydration layers surrounding a biological molecule show transport (and structural) signatures that differ appreciably from those of bulk water,1-19 differences whose larger implications for biological function3,6,11,17,20-24 and analogies to glass formers8,11,21,25-38 are an active area of exploration. Many experimental techniques have been used to measure the dynamics of hydration water such as magnetic resonance dispersion (MRD),18,39-41 dielectric relaxation (DR),42-47 NMR,48-51 and incoherent quasi-elastic neutron scattering (QENS) and inelastic neutron scattering2-4,6,20,21,27,32,37,38,52-57 as well as time-resolved fluorescence spectroscopy.58-65 There is some disagreement as to whether the large range of time scales measured by these techniques, from tens of picoseconds to hundreds of nanoseconds, is actually directly attributable to the hydration layer nearest the protein or peptide surface or to outer hydration layers or even coupling of the hydration dynamics to different components of the solute motion.18,47,66,67 Although there is agreement that the protein surface hydration layer dynamics are very heterogeneous, there is little information as to which components of the protein surface chemistry contribute to this heterogeneity. At present, a molecular interpretation is needed that would specify the chemical features of the protein surface, the distinct hydration layers ranging from protein surface waters to outer hydration layers to bulk liquid as well as the dynamics of the biological solute, to explain the large dynamic time scale range that is observed.
Our own recent work has sought to dissect the full complexity of heterogeneous protein surfaces and their different hydration layer dynamics through the study of model peptide systems as a function of concentration using X-ray and neutron diffraction studies, QENS experiments, and molecular dynamics simulations.37,38,68-71 In this study, we report incoherent QENS measurements at room temperature of water dynamics near a model protein hydrophilic backbone N-acetyl-glycine-methylamide (NAGMA), at 1.0 and 3.0 M concentrations, and compare them with our previous studies on aqueous solutions of a hydrophobic peptide, N-acetyl-leucine-methylamide (NALMA), at comparable concentrations of 0.5, 1.0, and 2.0 M.37,38 According to our structural X-ray scattering experiments and simulations, these solutions organize into monodispersed to very small clusters of amino acids, with the high-concentration solutions exhibiting only a single hydration layer shared between solutes (on average), while the low-concentration solutions comprise on the order of 2-3 hydration layers per solute.68-71 These model systems better enable us to detect and characterize translational and rotational motions for different hydration layers as well as the time scales of motion for the peptide solute itself37,38 and to do so while contrasting the influence of the amino acid chemistry on the hydration dynamics. Our first goal is to use the more precise definition of molecular hydration layers near the peptide model systems as a function of concentration as a way of unifying the distinctly different time scale resolutions and interpretations, probed by different spectroscopic techniques such as MRD, DR, time-resolved fluorescence spectroscopy, and QENS measurements on aqueous protein solutions.
MRD can directly probe the orientational relaxation dynamics of 17O-labeled water molecules and is a single-molecule dynamics spectroscopy.18,39-41 The resulting spectral density function is fit to a Lorentzian form to determine an average residence time and mean rotational correlation time. In MRD experiments on proteins, two populations of hydration water motions have been detected: very long-lived internal waters that reside in protein pockets on the order of a microsecond and those that interact with the protein surface with time scales on the order of 10-100 ps.18 The comparison of the hydration dynamics near protein surfaces to those near small organic solutes shows that the systems are largely similar, with water orientational relaxation times near proteins increasing by a factor of 2-3 at most. Halle has suggested that a comparison of MRD experiments to other techniques can be made through the rankindependent rotational retardation factor ⟨τhyd⟩τbulk, where ⟨τhyd⟩ and τbulk are the mean rotational correlation times of hydration water and bulk water, respectively; for MRD, this factor ranges from ~1.0 to 2.5 for small organic solutes and up to ~5.5 for hydration water near proteins at room temperature.18
Dielectric relaxation measures the collective response of dipolar reorientation to an oscillating electric field.44,45 The resulting dielectric dispersion profile measured for dilute protein solutions at room temperature shows two strong signatures: a β-dispersion corresponding to long time scale protein tumbling (~30 ns) and a γ-dispersion, which corresponds to the orientational relaxation time due to bulk water (~8.0 ps).42-47 Two weaker δ-relaxations are present and are attributed to a bimodality in the orientational response of water in the first hydration layer at the protein surface, with relaxation times on the order of ~20-60 ps and ~1-10 ns.42-45,72 The δ-dispersions measured by DR have been argued to arise from dynamic exchange of bound and free waters at the protein surface,45,72 and the two time scales give rotational retardation factors of ⟨τhyd⟩/τbulk ≈ 5 and 102-103, the latter which is in significant disagreement with MRD.18 Attempts to explain the discrepancy between these techniques have given rise to a number of experimental studies and theoretical analysis.18,46,47,63,66,73
Recently, time-resolved fluorescence spectroscopy, which measures a collective environmental response after electronic excitation of a fluorophore located near the protein surface, has been used to study the hydration water dynamics. Several solvation dynamics studies have been carried out using either an endogenous tryptophan residue in the protein as a probe or an extrinsic probe covalently attached to the protein.58,60,63-65 The shift in the fluorophore's emission frequency with time, known as time-dependent fluorescence Stokes shift (TDFSS), is measured and then interpreted in terms of linear response theory that equates the TDFSS to a time correlation function of fluctuations of solvation energy from its equilibrium value. Whether the probe is situated in the protein or on the protein surface, the technique measures both protein and aqueous solvent dynamics that must be further resolved into distinct protein and hydration components based on analysis of molecular dynamics simulations.19 Recent time-resolved measures of tryptophan fluorescence for two proteins generate a profile with two distinct time scales, one corresponding to bulk water dynamics of ~1 ps and a longer time scale of 16 ps (monellin) and 38 ps (subtilisin) attributed to the hydration layer dynamics,63,65 resulting in a rotational retardation factor of ⟨τhyd⟩/τbulk ≈ 10-40, which again is in significant disagreement with MRD.18
Fundamentally, the large difference in the measured ⟨τhyd⟩/τbulk between these techniques indicates that dielectric relaxation and time-resolved fluorescence spectroscopy characterizes water hydrogen-bonded network dynamics that are more strongly perturbed by the presence of the protein, while MRD observes a weaker perturbation in the water dynamics that suggests that the hydrogen-bonded network is minimally disrupted with the introduction of a biological interface. This work reports on a new analysis of incoherent QENS data taken on a well-defined model system for hydration layers near different side-chain chemistries, which adds further insight into this ongoing debate about the molecular view of hydration dynamics.
Incoherent QENS measures single-particle dynamics that probe both translational and rotational motions on the picosecond time scale.74-76 The QENS measurements of water orientational motion includes τrot, which is interpreted as largeamplitude librations related to the lifetime of hydrogen bonds,77 and a parameter of the jump diffusion model,78 the residence time τ0, which is interpreted as the time scale necessary to overcome caging, by rotational excitation, by surrounding water molecules to execute translational diffusion.79 In this paper, we define QENS estimates of the rotational retardation factor for the rotational motion, ⟨τrot⟩/τbulkrot, and a pseudorotational retardation factor with the residential time, ⟨τ0⟩/τbulk0, and show that these ranges are ~1.0-2.2 and ~1.0-3.5, respectively, with the largest values for each factor arising from the first hydration layer and near the more hydrophobic solute, which agrees with a fundamental description of the water dynamics as not being grossly perturbed by the solute at room temperature.
While the longer reorientational time scales observed from DR, MRD, and NMR are not directly observed in the shorter time scale QENS experiments, we propose an analysis of the elastic incoherent structure factor (EISF) determined from the incoherent QENS experiment2,15,80 as an indirect signature of possible longer reorientational relaxation times (greater than ~13 ps based on the resolution of these current experiments). Furthermore, the concentration dependence and different amino acid chemistries are manifested in different values of the elastic component and provide a complementary view of protein hydration water dynamics measured by other spectroscopic techniques. We determine that this longer reoreintational time scale involves both tight coupling of water dynamics with the solute dynamics as well as dynamic coupling of water molecules between inner and outer hydration layers. We suggest that this longer time scale shares a molecular origin with the shorter time scale δ-relaxations measured by DR and possibly the longer time scale component of TDFSS measured by time-resolved fluorescence spectroscopy.63,65
Our second goal is to contrast the peptide chemistries and their influence on hydration dynamics to clarify the role of the spatial heterogeneity of dynamics near protein surfaces.3,5,8,11,12,24-26,34,36-38 We report stretched exponential fits to the experimental intermediate scattering functions (obtained from the Fourier transform in the frequency plane of the measured incoherent dynamic structure factor) for both the hydrophobic and hydrophilic peptides at all concentrations. An important difference between the solute chemistries at the highest concentration studied is a more pronounced anomalous translational diffusion signature for NALMA,38 including a smaller stretched exponent β and a longer structural relaxation time τ than those found for NAGMA. The more normal diffusion exhibited in the hydration dynamics near the model hydrophilic backbone provides experimental support for an explanation of the origin of the anomalous diffusion behavior of NALMA as arising from frustrated interactions between water molecules when a chemical interface is formed upon addition of the hydrophobic side chain,38 thereby inducing spatial heterogeneity in the hydration dynamics in the two types of regions of the NALMA peptide.
Materials and Methods
Completely deuterated N-acetyl(d3)-glycine(d2)-methylamide-(d3) (NAGMA, MW) = 138 kDa) was purchased from CDN Isotopes, Canada. The 3.0 M solution was obtained by dissolution of the completely deuterated amino acid powder in pure H2O (18 H2O/solute), and the 1 M (55 H2O/solute) lowconcentration sample was obtained by diluting the 3.0 M solution. To remove small aggregates, the samples were centrifuged (10 min at 10 000g) before measurement, given a 0.5% error in the reported concentration.
The QENS experiments were carried out at the National Institutes for Standards and Technology Center for Neutron Research, using the time-of-flight disk chopper spectrometer (DCS)81 operating at a 7.5-Å wavelength, with an energy resolution of 35 μeV at full width at half-maximum (fwhm) and a wave vector range of 0.15 Å-1 < Q < 1.57 Å-1. The Gly(D)/H2O samples were contained in the 0.1-mm-thick annular space between two concentric thin-walled aluminum cylinders of radius 10 mm and height 100 mm. Each data collection run lasted ~12 h. All measurements were performed at room temperature. Spectra were corrected for scattering by the sample container, and a standard vanadium sample was used to determine relative detector efficiencies and the energy resolution function. The data were corrected and analyzed using the DAVE software (http://www.ncnr.nist.gov/dave/).
Experimental Analysis
The quasi-elastic neutron scattering experiment measures the double differential incoherent scattering cross section
| (1) |
where σinc is the total incoherent scattering cross section per scatterer, N is the number of scatterers, ki and ks are the wave vectors of the incident and scattered neutrons, Q is the momentum transfer, ω is the frequency transfer, and Sinc(Q,ω) is the incoherent dynamic structure factor. The analysis involves fitting the incoherent dynamic structure factor to a sum of Lorentzian contributions convoluted with the instrumental resolution. On the basis of the fits, we further interpret the data using the following analytical models traditionally applied to liquids.74,75
We assume that Sinc(Q,ω) can be expressed as a convolution of three different kinds of proton motion74,75
| (2) |
where the exponential term is the Debye-Waller factor, which represents the reduction in intensity due to molecular vibrations, ⟨u2⟩ being a mean-square displacement. The second and third terms are the translational and rotational incoherent dynamic structure factors, respectively. The translational scattering function is written as
| (3) |
where Γtrans is the half-width at half-maximum of a Lorentzian function. We have found that the translational Lorentzian is best fit to a random jump diffusion model, which considers the mean residence time τ0 for one site in a given network before jumping to another site.78
| (4) |
where the mean jump diffusion length L is defined in this model as and Dtrans is the translational diffusion coefficient between two sites.
Water rotational relaxation is described using the Sears model for hindered rotational diffusion on the surface of a sphere.82 The rotational incoherent dynamic structure factor is
| (5) |
where jl is a spherical Bessel function of order l, a is the radius of the sphere on which the motion of water protons occurs, and Drot is the rotational diffusion coefficient. For l = 1, which dominates the second term of eq 5, the half-width at half-maximum is Γrot = 2Drot, which corresponds to a characteristic rotational time of τrot =1/6Drot.
The first term in eq 5 corresponds to the form factor of the restricted volume explored by the hydrogen atoms, by hindered rotation, and is known as the elastic incoherent structure factor (EISF). In the Sears model, the EISF corresponds to a spherical form factor. Convoluting eq 3 with eq 5 and excluding terms with l > 1, eq 2 becomes
| (6) |
To the extent that terms with l > 1 can be neglected, the experimental EISF is Itrans(Q)/[Itrans(Q) + Irot+trans(Q)], where Itrans(Q) and Irot+trans(Q) are the experimental integrated intensities of the first and second terms, respectively, in eq 6.15
It is possible to estimate from the experimental EISF the fraction of hydration atoms that are rotationally immobile, i.e., hydrogen motions that are faster or slower than the experimental resolution. Therefore, the dynamics can be characterized by two populations,80 a fraction p of protons with correlation times that are very different with respect to the energy resolution, which in this experiment corresponds to rotational motion outside an experimental window of 1.0-13.5 ps, while the dynamics of the (1 - p) protons gives the total quasi-elastic signal. The EISF can therefore be written as
| (7) |
and in the limit that Q → ∞ we can determine the value of p as the constant elastic contribution. It is intuitive that the immobile fraction value p changes as a function of resolution, which has also been discussed in ref 80.
To study the distribution of the translational relaxation time, we also analyzed the experimental intermediate scattering function, FH(Q,t) for both solutes at all concentrations. The Fourier transform of Sinc(Q,ω) has been generated using the DAVE fast Fourier transform utility, and to avoid contributions from the fast dynamics component (previously characterized as large-amplitude librational movements), we normalized FH-(Q,t) to unity at t = 5 ps.
Experimental Results
Translational and Librational Dynamics
To characterize the hydration water dynamics near a primitive protein backbone model, the scattering profile of the completely deuterated NAGMA solute in H2O has been measured at room temperature for both 1.0 and 3.0 M concentrations. The 1.0 and 3.0 M NAGMA concentrations were chosen to match the 0.5-2.0 M NALMA concentrations,37,38 using the weight percentage approach. Molecular dynamics simulations to be reported elsewhere83 also confirm that for each solute the low concentrations have a hydration level of 2-3 hydration layers, while the highest solute concentrations correspond to one hydration layer shared between solutes. The high solute concentrations are unique in measuring a single hydration layer at the peptide surface, while the low concentrations allow us to distinguish, based on the high-concentration results, the outer-layer hydration dynamics. Given the small atomic fraction of solute molecules and the relatively small scattering cross sections of the deuterated solute atoms as compared with that of normal hydrogen, we shall ignore incoherent quasi-elastic scattering from the solute.
The fits to the NAGMA data all required two Lorentzians and a flat background. Figure 1 shows two examples of fits, with relative residuals, of the incoherent dynamic structure factor measured at 3.0 M NAGMA concentration, at Q = 0.84 Å-1 and Q = 1.25Å-1. The dash-dot line is the background component that takes into account all movements that are too fast to be observed within the chosen energy window, e.g., low-energy vibrational modes. The narrow Lorentzian function (dashed line) is indicative of translational motion and, based on the dependence of Γ(Q) with Q2, is best described with a jump diffusion model. The broad Lorentzian component (dotted line), which describes short time scale movement, is identified as water rotational motion.37,38 The solid line is the sum of the three fit components.
Figure 1.

Incoherent structure factor spectrum for 3.0 M deuterated NAGMA in H2O at 25 °C, measured at 35 μeV (open symbols) for (a) Q ≈ 1.25 Å-1 and (b) Q ≈ 0.84 Å-1. The solid line is the total fit component resulting from the convolution of the two Lorentzian functions and the flat background. The Lorentzian fits to the spectra (dashed lines) show good separation of widths and intensities and are typical of the qualities of the fits for all spectra measured in this study. The residuals show that the quality of fit is good in the energy range of the experiment.
Figure 2 plots the line width at half-maximum due to translation Γtrans(Q) versus Q2, while Figure 3 plots the line width at half-maximum due to rotational motion Γrot versus Q2, of the water hydration dynamics for NAGMA at the two different concentrations, respectively. We present on the same plots the translational and rotational contributions of the NALMA hydration dynamics at room temperature for 0.5, 1.0, and 2.0 M solutions. Since the NAGMA rotational component has been characterized here using the 35 μeV resolution, while the NALMA rotational data were extended by an additional experiment at 81 μeV as described in refs 37 and 38, there is a difference in the Q range measured between the two solutes. Although the rotational part of the NAGMA dynamics would be better described by an additional low-resolution experiment to characterize a wider Q range, we consider the presented data satisfactory for the general interpretation of the dynamics that we present.
Figure 2.

Half-width at half-maximum of the Lorentzian function, Γtrans(Q), plotted vs Q2 corresponding to the translational motions of protons for NAGMA and NALMA at different solute concentrations: (a) 3.0 M deuterated NAGMA and 1.0 and 2.0 M deuterated NALMA in H2O and (b) 1.0 M deuterated NAGMA and 0.5 M deuterated NALMA in H2O.
Figure 3.

Half-width at half-maximum of the Lorentzian function, Γrot, plotted vs Q2 corresponding to the rotational motions of protons for NAGMA and NALMA at different solute concentrations: (a) 3.0 M deuterated NAGMA and 1.0 and 2.0 M deuterated NALMA in H2O and (b) 1.0 M deuterated NAGMA and 0.5 M deuterated NALMA in H2O.
We analyze the new NAGMA hydration data reported in Figures 2 and 3 with the analytical model fits described in the Methods section. We report in Table 1 the hydration water translational diffusion coefficient Dtrans and the residence time τ0, obtained from the jump diffusion model, and the rotational relaxation time τrot from the Sears model as a function of concentration. For comparative purposes, we also report in Table 1 the corresponding values analyzed as a function of NALMA concentration37,38 as well as a few reported bulk water values.77,79 These measures of hydration water dynamics, Dtrans, τ0, and τrot, are averages over a population of proton motions whose time scales are directly resolvable in the experimental resolution of this QENS study.
TABLE 1.
Experimental Values for Hydration Water Dynamics at Room Temperature for NAGMA and NALMA as a Function of Concentration
| transport property | bulk water77,79 | 1.0 M NAGMA | 3.0 M NAGMA | 0.5 M NALMA | 1.0 M NALMA | 2.0 M NALMA |
|---|---|---|---|---|---|---|
| Dtrans (10-5 cm2/s) (JDa) | 2.3 | 1.65 | 1.10 | 1.65 | 1.26 | 0.75 |
| Dtrans (10-5 cm2/s) (ISFb) | 1.61 | 1.10 | 1.25 | 0.67 | ||
| τo (ps) (JDa) | 1.1 | 0.9 | 2.4 | 0.9 | 1.9 | 3.6 |
| τrot (ps) (HRc) | 1.0 | 1.0 | 2.0 | 1.0 | 1.0 | 2.2 |
| p value (EISFd) | 0.66 | 0.43 | 0.54 | 0.38 | 0.33 | |
| a value (Å) (EISF) | 2.1 | 1.8 | 1.9 | 1.8 | 1.9 |
Translational diffusion coefficient, Dtrans, and the residence time,τ0, of water based on the jump diffusion model.
Translational diffusion coefficient, Dtrans, based on the analysis from the experimental intermediate scattering function.
Rotational time scale for water, τrot, based on fit to Sears hindered rotation model.
Immobile protons.
In Figure 2a, Γtrans(Q) for the high-concentration solute data exhibits a pronounced plateau at high values of Q and a slope measured at the smallest values of Q (<1.0) to give a residential time and translational diffusion coefficient value for 3.0 M NAGMA that are substantially suppressed, approaching values more typical of a supercooled water translational diffusion coefficient and corresponding long residential time, as was seen in our earlier NALMA study. Nonetheless, as shown in Figure 2a and Table 1, the transport values from the jump diffusion model, Dtrans and τ0, are significantly faster for 3.0 M NAGMA than those measured for 2.0 M NALMA, while based in Figure 3a the rotational time scales for 3.0 M NAGMA are found to be comparable to 2.0 M NALMA. The difference in translational dynamics that the NALMA and NAGMA solutes invoke in their surrounding hydration layer stems from their different chemistries, which we examine more closely below in our analysis of the intermediate scattering function.
In Figure 2b, we show the translational hydration water dynamics comparisons between 1.0 M NAGMA and 0.5 M NALMA, the lowest concentrations measured. We attribute to this concentration an inclusion of outer-layer water dynamics in addition to the first-hydration-layer dynamics measured at the highest concentrations. Although the lower-concentration hydration water exhibits faster translational dynamics than those of the higher concentration, the translational dynamics do not fully recover to room temperature bulklike values for either solute. From Figures 2b and 3b and analysis reported in Table 1, it is evident that the translational and rotational time scales for the low-concentration NAGMA are equal to those measured at the corresponding low-concentration NALMA. For these resolvable proton populations, the outer-layer hydration dynamics for translation and libration are apparently not affected by the particular “flavor” of the solute. This is consistent with our previous work on the lower-concentration NALMA solutions, which showed that the outer-hydration-layer translational dynamics are largely independent of the first-hydration-layer dynamics. Thus, the equivalence between the translational and the rotational dynamics of low-concentration NALMA and NAGMA solutions is consistent with the molecular hypothesis of an outer-sphere hydration layer that is only affected by an excluded volume effect in the water hydrogen-bonding network.
Together, the jump diffusion and Sears models have been used to interpret hydrogen-bond lifetimes and caging effects in water dynamics that are related to different reorientational processes of water molecules in the neat liquid77,79 or in solution.84-86 The rotational component corresponds to large-amplitude librational motions that can be related to the average hydrogen-bond lifetime,77 while the residence time of the jump diffusion model can be interpreted as a measure of the time necessary for a water molecule to break from its hydrogen-bonded neighbors by rotational excitation.79 At high solute concentrations or lower temperatures, these two time scales tend to diverge since the residence time measures a caging effect requiring cooperative motion among more than one hydrogen-bonding partner to execute diffusion.87 Therefore, we can define a QENS rotational retardation factor and a pseudorotational retardation factor . For either NAGMA or NALMA at room temperature, we find that , while is ~1.0-3.5. The rotational retardation factor for the average single-hydrogen-bond dynamics is in good agreement with what is observed by MRD for small-peptide systems,18,41 and the upper bound of 2.0 is found for the high-concentration data and therefore most directly originates from the first hydration layer. The pseudorotational retardation factor for the residence time τ0 is found to range significantly beyond the upper bound of ~2.5 of the MRD measurement for small solutes. This is most certainly due to the implicit collective caging effect of the surrounding water molecules on the motion of a central water molecule that exaggerates this upper bound relative to the single-particle dynamics measured by MRD. Furthermore, the upper bound of occurs in the first hydration layer of the more hydrophobic solute.
Elastic Incoherent Structure Factor
In Figure 4, we present the EISF of hydration water, arising from rotational motions, for different concentrations of NALMA and NAGMA at room temperature. Although the EISF formally gives information about the geometry of the rotations, interpretations based on eq 7 imply that it indirectly measures hydration water dynamics over a population of proton motions whose time scales are not directly resolvable in the experimental resolution of this QENS study (corresponding to time scales >13 ps). In Table 1, we report the values of p and a for the NALMA and NAGMA solutions as a function of concentration as inferred by adjusting eq 7 to the experimental EISF. We have reevaluated the rotational EISF for all of the concentrations of NALMA solutions at room temperature using only the high-resolution data (in ref 37 we used the low-resolution data) to be consistent with the resolution used in this work for NAGMA. Therefore, the values reported in this work for the NALMA data are different from those published in ref 37 because of the different resolution.
Figure 4.

(a) EISF of hydration water plotted vs Q for 1.0 and 3.0 M NAGMA and 0.5, 1.0, and 2.0 M NALMA concentrations. (b) EISF of hydration water for 2.0 M NALMA and 1.0 M NAGMA concentrations with the fit to eq 7 of the text. The solid lines are the fits, with the fraction of immobile protons p and the radius a given in Table 1.
In the comparison between solutes, we note that both show similar trends in the elastic fraction between the high- and the low-concentration solutions (Figure 4). For Q > 0.5 Å-1, the lowest concentration NAGMA data show a higher fraction of the elastic intensity (~65%) with respect to the more concentrated NAGMA solutions (~40%). These population differences are ~50% and ~30% for the low- and high-concentration NALMA solutions, respectively. In other words, the less concentrated solutions have a greater percentage of protons with a correlation time longer than the resolution window of 13 ps. Correspondingly, the first hydration layer as measured by the high-concentration solutions can resolve a majority of protons to reside within the experimental resolution window. Thus, moving from a single hydration layer to ~2-3 hydration layers gives rise to a change in the population of protons executing short versus longer time scale reorientational motions.
Most DR experiments on hydration water near the protein surface have resolved a bimodality in the (collective) orientational response, with relaxation times on the order of 20-60 ps and 1-10 ns (δ-relaxation).42-45 The shorter time scale δ-dispersion (δ3 in the ribonuclease A study47) was assigned initially to hydration water dynamics by experiment42-44 and given a molecular origin as dynamic exchange between bound and free water at the protein surface.45,63,72 Recent DR experiments on relatively concentrated solutions of ribonuclease A,47 which observes three δ-relaxation modes, attribute the 40 ps component (δ3 mode) unambiguously to the hydration water dynamics, while the assignment of the longer time scale ~1-10 ns mode (and/or an additional ~500 ps mode) remains controversial. However, recent MD simulations and DR experiments give evidence that the longer time scale δ-dispersion is related to protein-water cross interactions.46,66 Thus, there seems to be consensus that the ~40 ps time scale measured by DR is a consistent signature of hydration water dynamics near biological solutes. Furthermore, through the use of a simple continuum model based on the relation between the single-particle orientational relaxation and DR,88,89 recent molecular dynamics simulations of a micellar solution show that the DR signal at ~40 ps correlates quite well with the ~20 ps component observed in the single-particle orientational dynamics of hydration water.73 Recent time-resolved fluorescence spectroscopy found a slower solvation time scale of ~20-40 ps (depending on protein and position of the tryptophan fluorophore) in the TDFSS profile, and it is believed to reflect the slow hydration water dynamics.63,65 In fact, a recent MD simulation study of the protein HP-36 further resolves an intermediate component of ~9-18 ps, likely to arise from “quasi-bound” water, in addition to a slow component of ~48-84 ps corresponding to waters that are likely hydrogen-bonded to the protein surface with much longer residence times.19
The higher-concentration solutions (which have on average only one hydration layer shared between solutes) show an ~30-40% population of protons that reside outside the experimental resolution. A plausible explanation is that this elastic fraction is due to water motions that are strongly coupled to the solute motions, since the single-hydration-layer dynamics that are measured are by design tightly integrated with the peptide surface. However, for low-concentration solutions, which show an ~50-65% elastic fraction, it is likely that in addition to the component arising from this coupled motion (possibly less dominant than that for high concentrations) there is an activation of an additional population due to the dynamic coupling between inner and outer hydration layers.
Together, our high- and low-concentration data from the QENS analysis of the EISF suggest a bimodality in the reorientational response. We find that the high-concentration data is consistent with peptide-driven motions of the first hydration layer, while the low-concentration data introduces a new reorientational motion that is solely due to hydration water dynamics, involving dynamic coupling between inner and outer hydration layers and consistent with the shorter time scale δ-dispersion.42-45,47 Together, these results suggest that the faster component of the δ-relaxation could be made to disappear for proteins under severe hydration conditions.47 Our QENS measurements and interpretation may also be consistent with those observed in the TDFSS profile (if relaxation due to the protein environment can be unambiguously removed).
Intermediate Scattering Function Analysis
At the relatively high solute concentrations examined here, the time scales for local librational motion are shorter than measured residence time scales because waters are caged by local neighbors, requiring a cooperative motion to execute diffusion.87 This caging effect is crudely captured by the jump diffusion model through its average residence time scale parameter τ0 that represents deviations from normal Brownian diffusion.79 To study the distribution of the translational relaxation time from the perspective of a more complex structural relaxation point of view, we analyzed the experimental intermediate scattering function, FH(Q,t) for both solutes at all concentrations.
The Fourier transform of S(Q,ω) has been generated using the DAVE fast Fourier transform utility, and to avoid contributions from the fast dynamics component (previously characterized as large-amplitude librational movements), we normalized FH(Q,t) to unity at t = 5 ps. In Figure 5, we present the FH(Q,t) for 1.0 and 3.0 M NAGMA concentrations as a function of Q. We fit the long time decay of FH(Q,t) to a stretched exponential form
| (8) |
where deviations from β = 1 are signatures of a pronounced slowdown in dynamic processes with a characteristic relaxation time τ, which is believed to be related to spatial heterogeneity in the dynamics.90-93 In fact, the origin of the nonexponential form is thought to arise from anomalous diffusion that is controlled by a local structural relaxation or “cage effect” as discussed from the MCT theory of supercooled liquids.90,94 The dependence of 1/τ versus Q2 is then proportional to the water translational diffusion coefficient in the limit of Q → 0 and numerically evaluated from the slope for Q < 1.0 Å-1. As shown in Table 1, we find good agreement between the diffusion coefficient inferred from the stretched exponential analysis and the jump diffusion model.
Figure 5.

Self-intermediate scattering function from experiment at Q ≈ 0.44 Å-1 and Q ≈ 1.54 Å-1 (symbols) and the stretched exponential fit (line): (a) 3.0 M NAGMA and (b) 1.0 M NAGMA.
Figure 6 reports the fitted stretched exponents β as a function of Q for 1.0 and 3.0 M NAGMA and 0.5 and 2.0 M NALMA from the QENS experiments. The qualitative behavior for both solutes is a β exponent that is relatively flat, as also seen in previous experiments95 in contrast with simulation results that always exhibit a β exponent with a stronger Q dependence.38,90 However, the difference between the solutes is manifest most directly in the highest concentration and therefore in the first hydration shell, where the β for NALMA deviates significantly from 1, reaching values as low as 0.75 for some Q values, while the corresponding β exponent for NAGMA shows relatively smaller deviations from 1. We also see larger structural relaxation times, τ, for high-concentration NALMA relative to NAGMA. Together, the β exponent and τ signatures imply that the first-hydration-layer water dynamics in the NALMA solution exhibit a wider distribution of time scales that is dominated by the long time tail than the water dynamics for the NAGMA solutions. This difference between solutes is also evident when the first-hydration-layer dynamics are analyzed by the jump diffusion model, in which there is evidence of a strong caging effect in the translational motion for NALMA.
Figure 6.
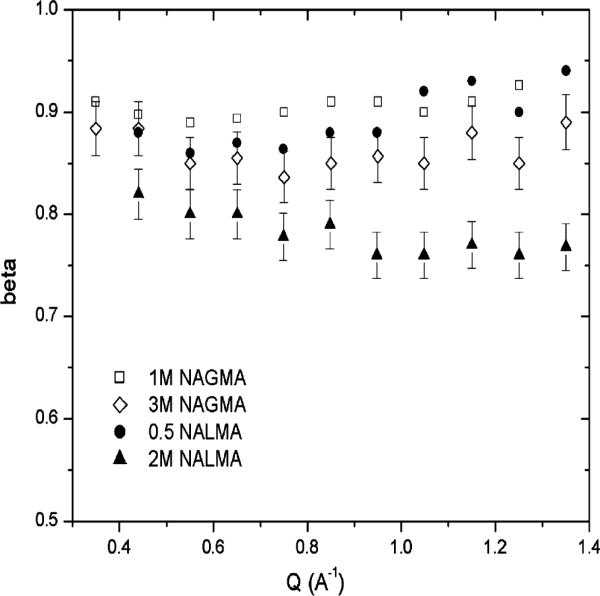
Stretched exponential parameter β, from the fit to experiment at different Q values for NAGMA and NALMA as a function of concentration. We show the error bar in regard to the fit for the highest concentration used for NAGMA and NALMA and similar error bars were found for the other concentrations as well.
We believe that the anomalous dynamics of the first hydration layer for NALMA, compared to the more normal water diffusion near the NAGMA backbone, are due to spatial heterogeneity in the water dynamics. This spatial heterogeneity in the water dynamics is in turn due to the heterogeneity of the NALMA chemistry when a chemical interface is formed upon addition of a hydrophobic side chain to the hydrophilic backbone. As we reported in ref 37 for simulations of NALMA hydration dynamics under ambient conditions, which have been observed in other simulation studies on model peptides,96 the average residence times for labeled water near the hydrophobic side chain are shorter, and the corresponding orientational correlation function is faster, while the waters near the hydrophilic backbone are dramatically slower by these two measures. Furthermore, that separation of time scales in the two regions becomes more exaggerated as the temperature is lowered.97 This experimental study shows that the loss of distinct heterogeneity in the chemistry when going from NALMA to NAGMA and the accompanying loss of frustrated interactions with water result in a hydration dynamics signature (a smaller τ and a β exponent closer to 1) that looks more like normal diffusion.
Discussion and Conclusions
In this paper, we have reported new incoherent QENS experiments on NAGMA as a model peptide backbone and contrast the hydration dynamics with those measured near the model NALMA hydrophobic peptide at similar concentrations. For dynamic averages generated over the resolvable proton motions, the 3.0 M (high) concentration NAGMA data shows a translational diffusion coefficient, residential time, and rotational diffusion constant that are suppressed with respect to bulk water but which are significantly faster than those measured for the hydrophobic NALMA peptide at a comparable high concentration. At lower solute concentrations that correspond to at least one additional hydration layer, the translational and rotational dynamics of water near the two peptides are equivalent, suggesting that the perturbation from bulk water dynamics for outer hydration layers arises from changes in the water hydrogen-bonding network due to the excluded volume effect.
However, our analysis of the EISF from incoherent QENS shows that a significant fraction of the hydration water population is slowed by at least a factor of 10 with respect to the bulk water. We find significant fractions of elastic intensity at high and low concentrations for both solutes, which correspond to a greater population of protons with rotational time scales outside the experimental resolution (>13 ps). The higher-concentration solutions show an ~30-40% elastic fraction that we attribute to water motions that are strongly coupled to the solute motions, while for low-concentration solutions, which show slow populations of ~50-65%, an additional component is activated due to dynamic coupling between inner and outer hydration layers. Some of the slow dynamics are due to water-solute couplings, perhaps manifested as conformational transitions of the peptide, similar to coupling arising from motions of side chains on the protein surface.47 As hydration layers are added and thus dynamic coupling of water molecules between hydration layers becomes possible, the activation of new reorientational responses of the water molecules takes place, consistent with the determination of a larger elastic fraction measured by QENS for the lower-concentration solutions.
Our QENS measurements and interpretation may also be consistent with the 20-40 ps component measured in the TDFSS profile using time-resolved fluorescence spectroscopy. Recent phenomenological theories45,72 involving a dynamic exchange between protein-bound water and outer hydration layers or even bulk water that have been used to explain both the shorter time scale δ-dispersion from DR as well as the longer time scale solvation dynamics from the TDFSS profile63,65 would give credence to an agreement between QENS and time-resolved fluorescence spectroscopy. We plan an extensive molecular dynamics analysis of these systems to evaluate the dielectric response and time-resolved fluorescence signatures and the molecular origin of inner- and outer-hydration-layer coupling as postulated here.
The rotational retardation factors measured by QENS on short time scales are in quantitative agreement with those measured by MRD.18 However, we conclude that the longer rotational motions measured by incoherent QENS, recent DR experiments for the fast δ-dispersion, time-resolved fluorescence spectroscopy, and MRD converge on a molecular view of hydration water dynamics, at room temperature, that is in fact perturbed by factors of 2-50 with the biological solute present but not by orders of magnitude. (Hence, the long time scale δ-dispersion from DR remains controversial but seems unlikely due to hydration water dynamics46,66). Furthermore, the range in rotational retardation factors due to hydration depends on whether we are comparing peptides versus proteins or comparing different proteins, whether we have a collective measure of dynamics (DR and TDFSS) versus a single-particle probe (MRD and QENS), whether the experiment yields a mean correlation time (DR, MRD, and certain analysis of QENS) versus a more detailed population analysis that is possible when measuring a time correlation function (TDFSS, the intermediate scattering function, and a more advanced analysis of the QENS data), and inevitable ambiguities in interpretation of each of these experiments due to intrinsic limitations of the observable that is measured. To the extent possible when these differences can be accounted for (for example, the fact that collective motions are intrinsically slower than measurements of single-particle dynamics, different proteins, etc.), then the differences in retardation factors between the different spectroscopies become even smaller, and all give a consistent molecular view of a hydrogen-bonded water network near a biological interface that exhibits a slowdown of dynamics in the vicinal water layers.
An important difference between the hydrophilic versus hydrophobic amino acid types at the highest concentration studied was found in a stretched exponential fit to their experimental intermediate scattering function, which showed a more pronounced anomalous diffusion signature for NALMA, including a smaller stretched exponent β and a longer structural relaxation time τ than those found for NAGMA. Together, the β exponent and τ signatures imply that the first-hydration-layer water dynamics in the NALMA solution exhibit a wider distribution of time scales than the water dynamics for the NAGMA solutions. This wider distribution in the NALMA dynamics is composed of long time scales in the first hydration layer due to a strong caging effect in the translational motion as well as short time scales (based on simulation studies) in which we find that the average residence time for labeled waters near the hydrophobic side chain is much shorter and the decay of the corresponding orientational correlation function is much faster, in contrast to the hydrophilic backbone.37,97 This is to be contrasted with the case when water solvates a purely hydrophobic or hydrophilic solute of the same size, where it is seen that the water residence times are always long-lived near the homogeneous but different solute chemistries.98 Returning to the population differences in the elastic fraction from the EISF, we note that the hydrophobic amino acid has the smaller elastic fraction by ~10-15% compared to the hydrophilic case at both concentrations. We speculate that this smaller fraction for NALMA is a result of faster and resolvable water molecules in the hydration layers due to instability of the water network in the hydrophobic region of the peptide.
The more normal diffusion exhibited in the hydration dynamics near the hydrophilic backbone model provides experimental support for the origin of the anomalous diffusion behavior for NALMA as arising from frustrated interactions between water molecules when a chemical interface is formed upon addition of a hydrophobic side chain, which induces spatial heterogeneity in the hydration dynamics in the two types of regions of the NALMA peptide. It provides a better molecular understanding that it is at the interface of chemically distinct domains on the protein surface that is the origin of the heterogeneous dynamics of the hydration water. An important point is that the resulting faster dynamics near hydrophobic regions of a chemically heterogeneous protein surface37,96 contradict the view of clathrate water structure that undergoes slower dynamic transitions for rearrangements of the hydrogen-bonding network, near a heterogeneous protein surface.
Acknowledgment
We gratefully acknowledge the support of the National Institutes of Health under Agreement No. GM65239-01. This work utilized facilities supported in part by the National Science Foundation under Agreement No. DMR-0086210. We acknowledge the support of the National Institute of Standards and Technology, U.S. Department of Commerce, in providing the neutron research facilities used in this work. D.R. thanks José Teixeira (LLB, France) and J.M Zanotti (LLB, France) for constructive discussions. The authors are also very grateful to Robert Dimeo (NIST) for his help with the DAVE software. Certain commercial materials are identified in this paper to foster understanding. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology nor does it imply that the materials or equipment identified are necessarily the best available for the purpose.
References and Notes
- (1).Rupley JA, Careri G. Adv. Protein Chem. 1991;41:37. doi: 10.1016/s0065-3233(08)60197-7. [DOI] [PubMed] [Google Scholar]
- (2).Bellissentfunel MC, Teixeira J, Bradley KF, Chen SH. J. Phys. I. 1992;2:995. [Google Scholar]
- (3).Bellissentfunel MC, Zanotti JM, Chen SH. Faraday Discuss. 1996;103:281. [Google Scholar]
- (4).Settles M, Doster W. Faraday Discuss. 1996;103:269. [Google Scholar]
- (5).Cheng YK, Rossky PJ. Nature. 1998;392:696. doi: 10.1038/33653. [DOI] [PubMed] [Google Scholar]
- (6).Bellissent-Funel MC. J. Mol. Liq. 2000;84:39. [Google Scholar]
- (7).Makarov VA, Andrews BK, Smith PE, Pettitt BM. Biophys. J. 2000;79:2966. doi: 10.1016/S0006-3495(00)76533-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Tarek M, Tobias DJ. Biophys. J. 2000;79:3244. doi: 10.1016/S0006-3495(00)76557-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Xu H, Berne BJ. J. Phys. Chem. B. 2001;105:11929. [Google Scholar]
- (10).Balasubramanian S, Pal S, Bagchi B. Phys. Rev. Lett. 2002;89:115505. doi: 10.1103/PhysRevLett.89.115505. [DOI] [PubMed] [Google Scholar]
- (11).Bizzarri AR, Cannistraro S. J. Phys. Chem. B. 2002;106:6617. [Google Scholar]
- (12).Marchi M, Sterpone F, Ceccarelli M. J. Am. Chem. Soc. 2002;124:6787. doi: 10.1021/ja025905m. [DOI] [PubMed] [Google Scholar]
- (13).Balasubramanian S, Bagchi B. J. Phys. Chem. B. 2002;106:3668. [Google Scholar]
- (14).Pal S, Balasubramanian S, Bagchi B. J. Chem. Phys. 2002;117:2852. doi: 10.1063/1.1635803. [DOI] [PubMed] [Google Scholar]
- (15).Russo D, Baglioni P, Peroni E, Teixeira J. Chem. Phys. 2003;292:235. [Google Scholar]
- (16).Pal S, Balasubramanian S, Bagchi B. Phys. Rev. E. 2003;67:1502. doi: 10.1103/PhysRevE.67.061502. [DOI] [PubMed] [Google Scholar]
- (17).Daniel RM, Dunn RV, Finney JL, Smith JC. Annu. Rev. Biophys. Biomol. Struct. 2003;32:69. doi: 10.1146/annurev.biophys.32.110601.142445. [DOI] [PubMed] [Google Scholar]
- (18).Halle B. Philos. Trans. R. Soc. London, Ser. B. 2004;359:1207. doi: 10.1098/rstb.2004.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Bandyopadhyay S, Chakraborty S, Balasubramanian S, Bagchi B. J. Am. Chem. Soc. 2005;127:4071. doi: 10.1021/ja042847r. [DOI] [PubMed] [Google Scholar]
- (20).Dellerue S, Bellissent-Funel MC. Chem. Phys. 2000;258:315. [Google Scholar]
- (21).Dellerue S, Petrescu AJ, Smith JC, Bellissent-Funel MC. Biophys. J. 2001;81:1666. doi: 10.1016/S0006-3495(01)75820-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Careri G, Peyrard M. Cell. Mol. Biol. 2001;47:745. [PubMed] [Google Scholar]
- (23).Pizzitutti F, Bruni F. Phys. Rev. E. 2001;64:2905. doi: 10.1103/PhysRevE.64.052905. [DOI] [PubMed] [Google Scholar]
- (24).Bruni F, Pagnotta SE. Phys. Chem. Chem. Phys. 2004;6:1912. [Google Scholar]
- (25).Green JL, Fan J, Angell CA. J. Phys. Chem. 1994;98:13780. [Google Scholar]
- (26).Angell CA. Science. 1995;267:1924. doi: 10.1126/science.267.5206.1924. [DOI] [PubMed] [Google Scholar]
- (27).Fitter J. Biophys. J. 1999;76:1034. doi: 10.1016/S0006-3495(99)77268-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Vitkup D, Ringe D, Petsko GA, Karplus M. Nat. Struct. Biol. 2000;7:34. doi: 10.1038/71231. [DOI] [PubMed] [Google Scholar]
- (29).Bizzarri AR, Paciaroni A, Cannistraro S. Phys. Rev. E. 2000;62:3991. doi: 10.1103/physreve.62.3991. [DOI] [PubMed] [Google Scholar]
- (30).Tarek M, Martyna GJ, Tobias DJ. J. Am. Chem. Soc. 2000;122:10450. [Google Scholar]
- (31).Tarek M, Tobias DJ. Phys. Rev. Lett. 2002;8813:8101. doi: 10.1103/PhysRevLett.88.138101. [DOI] [PubMed] [Google Scholar]
- (32).Orecchini A, Paciaroni A, Bizzarri AR, Cannistraro S. J. Phys. Chem. B. 2001;105:12150. [Google Scholar]
- (33).Teeter MM, Yamano A, Stec B, Mohanty U. Proc. Natl. Acad. Sci. U.S.A. 2001;98:11242. doi: 10.1073/pnas.201404398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Bellissent-Funel MC. J. Mol. Liq. 2002;96-7:287. [Google Scholar]
- (35).Ringe D, Petsko GA. Biophys. Chem. 2003;105:667. doi: 10.1016/s0301-4622(03)00096-6. [DOI] [PubMed] [Google Scholar]
- (36).Tournier AL, Xu JC, Smith JC. Biophys. J. 2003;85:1871. doi: 10.1016/S0006-3495(03)74614-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Russo D, Hura G, Head-Gordon T. Biophys. J. 2004;86:1852. doi: 10.1016/S0006-3495(04)74252-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Russo D, Murarka RK, Hura G, Verschell ER, Copley JRD, Head-Gordon T. J. Phys. Chem. B. 2004;108:19885. [Google Scholar]
- (39).Denisov VP, Halle B. Faraday Discuss. 1996;103:227. doi: 10.1039/fd9960300227. [DOI] [PubMed] [Google Scholar]
- (40).Halle B, Denisov VP. Methods Enzymol. 2001;338:178. doi: 10.1016/s0076-6879(02)38220-x. [DOI] [PubMed] [Google Scholar]
- (41).Modig K, Liepinsh E, Otting G, Halle B. J. Am. Chem. Soc. 2004;126:102. doi: 10.1021/ja038325d. [DOI] [PubMed] [Google Scholar]
- (42).Grant EH, Sheppard RJ, South PG. Dielectric Behavior of Biological Molecules in Solutions. Clarendon; Oxford, U. K.: 1978. [Google Scholar]
- (43).Dachwitz E, Parak F, Stockhauser M. Ber. Bunsen-Ges. Phys. Chem. 1989;93:1454. [Google Scholar]
- (44).Pethig R. Annu. Rev. Phys. Chem. 1992;43:177. doi: 10.1146/annurev.pc.43.100192.001141. [DOI] [PubMed] [Google Scholar]
- (45).Nandi N, Bhattacharyya K, Bagchi B. Chem. Rev. 2000;100:2013. doi: 10.1021/cr980127v. [DOI] [PubMed] [Google Scholar]
- (46).Knocks A, Weingartner H. J. Phys. Chem. B. 2001;105:3635. [Google Scholar]
- (47).Oleinikova A, Sasisanker P, Weingartner H. J. Phys. Chem. B. 2004;108:8467. [Google Scholar]
- (48).Otting G. Prog. Nucl. Magn. Reson. Spectrosc. 1997;31:259. doi: 10.1016/j.pnmrs.2016.11.001. [DOI] [PubMed] [Google Scholar]
- (49).Venu K, Svensson LA, Halle B. Biophys. J. 1999;77:1074. doi: 10.1016/s0006-3495(99)76957-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Zanotti JM, Bellissent-Funel MC, Parello J. Biophys. J. 1999;76:2390. doi: 10.1016/S0006-3495(99)77395-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Almasy L, Banki P, Bellissent-Funel MC, Bokor M, Cser L, Jancso G, Tompa K, Zanotti JM. Appl. Phys. A: Mater. Sci. Process. 2002;74:S516. [Google Scholar]
- (52).Wanderlingh U, Giordano R, Teixeira J. J. Mol. Struct. 1993;296:271. [Google Scholar]
- (53).Diehl M, Doster W, Petry W, Schober H. Biophys. J. 1997;73:2726. doi: 10.1016/S0006-3495(97)78301-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Gabel F, Bicout D, Lehnert U, Tehei M, Weik M, Zaccai G. Q. Rev. Biophys. 2002;35:327. doi: 10.1017/s0033583502003840. [DOI] [PubMed] [Google Scholar]
- (55).>Merzel F, Smith JC. Proc. Natl. Acad. Sci. U.S.A. 2002;99:5378. doi: 10.1073/pnas.082335099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Mattos C. Trends Biochem. Sci. 2002;27:203. doi: 10.1016/s0968-0004(02)02067-4. [DOI] [PubMed] [Google Scholar]
- (57).Harpham MR, Ladanyi BM, Levinger NE, Herwig KW. J. Chem. Phys. 2004;121:7855. doi: 10.1063/1.1792592. [DOI] [PubMed] [Google Scholar]
- (58).Jordanides XJ, Lang S, Fleming GR. J. Phys. Chem. B. 1999;103:7995. [Google Scholar]
- (59).Bhattacharyya K, Bagchi B. J. Phys. Chem. A. 2000;104:10603. [Google Scholar]
- (60).Pal SK, Mandal D, Sen S, Bhattacharyya K. J. Phys. Chem. B. 2001;105:1438. [Google Scholar]
- (61).Shirota H, Castner EWJ. J. Am. Chem. Soc. 2001;123:12877. doi: 10.1021/ja010290z. [DOI] [PubMed] [Google Scholar]
- (62).Frauchiger L, Shirota H, Ulrich KE, Castner EWJ. J. Phys. Chem. B. 2002;106:7463. [Google Scholar]
- (63).Pal SK, Peon J, Bagchi B, Zewail AH. J. Phys. Chem. B. 2002;106:12376. [Google Scholar]
- (64).Bhattacharyya K. Acc. Chem. Res. 2003;36:95. doi: 10.1021/ar020067m. [DOI] [PubMed] [Google Scholar]
- (65).Pal SK, Zewail AH. Chem. Rev. 2004;104:2099. doi: 10.1021/cr020689l. [DOI] [PubMed] [Google Scholar]
- (66).Boresch S, Steinhauser O. J. Phys. Chem. B. 2000;104:8743. [Google Scholar]
- (67).Boresch S, Willensdorfer M, Steinhauser O. J. Chem. Phys. 2004;120:3333. doi: 10.1063/1.1640996. [DOI] [PubMed] [Google Scholar]
- (68).Head-Gordon T, Sorenson JM, Pertsemlidis A, Glaeser RM. Biophys. J. 1997;73:2106. doi: 10.1016/S0006-3495(97)78241-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Hura G, Sorenson JM, Glaeser RM, Head-Gordon T. Perspect. Drug Discovery Des. 1999;17:97. [Google Scholar]
- (70).Pertsemlidis A, Soper AK, Sorenson JM, Head-Gordon T. Proc. Natl. Acad. Sci. U.S.A. 1999;96:481. doi: 10.1073/pnas.96.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Sorenson JM, Hura G, Soper AK, Pertsemlidis A, Head-Gordon T. J. Phys. Chem. B. 1999;103:5413. [Google Scholar]
- (72).Nandi N, Bagchi B. J. Phys. Chem. B. 1997;101:10954. [Google Scholar]; Nandi N, Bagchi B. J. Phys. Chem. A. 1998;102:8217. [Google Scholar]
- (73).Pal S, Balasubramanian S, Bagchi B. J. Chem. Phys. 2004;120:1912. doi: 10.1063/1.1635803. [DOI] [PubMed] [Google Scholar]
- (74).Bee M. Quasi-Elastic Neutron Scattering [Google Scholar]
- (75).Bee M. J. Phys. IV. 2000;10:1. [Google Scholar]
- (76).Bee M. Chem. Phys. 2003;292:121. [Google Scholar]
- (77).Teixeira J, Bellissent-Funel MC, Chen SH, Dianoux AJ. Phys. Rev. A. 1985;31:1913. doi: 10.1103/physreva.31.1913. [DOI] [PubMed] [Google Scholar]
- (78).Egelstaff PA. An Introduction to the Liquid State. Clarendon; Oxford, U. K.: 1992. [Google Scholar]
- (79).Bellissent-Funel MC, Teixeira J. J. Mol. Struct. 1991;250:213. [Google Scholar]
- (80).Zanotti JM, Parello J, Bellissent-Funel MC. Appl. Phys. A: Mater. Sci. Process. 2002;74:S1277. [Google Scholar]
- (81).Copley JRD, Cook JC. Chem. Phys. 2003;292:477. [Google Scholar]
- (82).Sears VF. Can. J. Phys. 1966;44:1299. [Google Scholar]
- (83).Murarka RK, Russo D, Batchelor J, Head-Gordon T. 2005. Unpublished work.
- (84).Cabral JT, Luzar A, Teixeira J, Bellissent-Funel MC. J. Chem. Phys. 2000;113:8736. [Google Scholar]
- (85).Smith LJ, Price DL, Chowdhuri Z, Brady JW, Saboungi ML. J. Chem. Phys. 2004;120:3527. doi: 10.1063/1.1648302. [DOI] [PubMed] [Google Scholar]
- (86).Calandrini V, Deriu A, Onori G, Lechner RE, Pieper J. J. Chem. Phys. 2004;120:4759. doi: 10.1063/1.1645782. [DOI] [PubMed] [Google Scholar]
- (87).Stanley HE, Teixeira J. J. Chem. Phys. 1980;73:3404. [Google Scholar]
- (88).Powles JG. J. Chem. Phys. 1953;21:633. [Google Scholar]
- (89).Bagchi B, Chandra A. Adv. Chem. Phys. 1991;80:1. [Google Scholar]
- (90).Sciortino F, Gallo P, Tartaglia P, Chen SH. Phys. Rev. E. 1996;54:6331. doi: 10.1103/physreve.54.6331. [DOI] [PubMed] [Google Scholar]
- (91).Ediger MD, Angell CA, Nagel SR. J. Phys. Chem. 1996;100:13200. [Google Scholar]
- (92).Ediger MD. Annu. Rev. Phys. Chem. 2000;51:99. doi: 10.1146/annurev.physchem.51.1.99. [DOI] [PubMed] [Google Scholar]
- (93).Debenedetti PG, Stillinger FH. Nature. 2001;410:259. doi: 10.1038/35065704. [DOI] [PubMed] [Google Scholar]
- (94).Gotze W, Sjogren L. Rep. Prog. Phys. 1992;55:241. [Google Scholar]
- (95).Bellissent-Funel MC, Longeville S, Zanotti JM, Chen SH. Phys. Rev. Lett. 2000;85:3644. doi: 10.1103/PhysRevLett.85.3644. [DOI] [PubMed] [Google Scholar]
- (96).Beck DAC, Alonso DOV, Daggett V. Biophys. Chem. 2003;100:221. doi: 10.1016/s0301-4622(02)00283-1. [DOI] [PubMed] [Google Scholar]
- (97).Head-Gordon T. Unpublished results.
- (98).Rasaiah JC, Lynden-Bell RM. Philos. Trans. R. Soc. London, Ser. A. 2001;359:1545. [Google Scholar]