

Abstract
N-acetyl aspartyl glutamate (NAAG) is an endogenous agonist at the metabotropic glutamate receptor 3 (mGluR3,GRM3) receptor and antagonist at the N-methyl D-aspartate (NMDA) receptor, both receptors important to the pathophysiology of schizophrenia. Glutamate carboxypeptidase II (GCPII), an enzyme that metabolizes NAAG, is also implicated in this illness. In this study, we conducted in situ hybridization experiments to examine expression of mGluR3 and GCPII transcripts along the rostrocaudal axis of the human postmortem hippocampus. We hypothesized that we would find changes in mGluR3 and/or GCPII in the AH but not posterior hippocampus (PH) in schizophrenia. We compared mRNA levels of these genes in the dentate gyrus (DG) and cornu ammonis (CA)1 and CA3 of AH and PH in 20 matched pairs of control and schizophrenia cases. In controls, mGluR3 is highly expressed in the DG and at lower levels in CA1 and CA3 while GCP II is expressed at similar levels in these regions. Group comparisons show a significant reduction of GCPII mRNA level in the AH in schizophrenia. Post hoc analyses reveal this difference is localized to the CA1 region. In addition, we find a significant positive correlation between GCPII and mGluR3 mRNA in the CA3 of the control AH (r=0.66,p=0.008) which is not present in schizophrenia (r=0.096,p=0.76). This may reflect a disrupted functional interaction between NAAG and mGluR3 in CA3 in schizophrenia. These data suggest that NAAG-mediated signaling is disrupted in the AH in schizophrenia and localize the defect to the CA1 and CA3 regions.
Keywords: glutamate, psychosis, post mortem, in situ, GRM3, mGluR3
1. Introduction
NAAG is a peptide neurotransmitter found in high concentrations in the mammalian brain (Coyle 1997; Neale et al 2000). It is concentrated in synaptic vesicles, released upon depolarization in a calcium-dependent manner and metabolized by a membrane-bound peptidase, GCP II (Neale et al 2000), to N-acetyl aspartate (NAA) and glutamate (Berger et al 1999; Slusher et al 1999; Stauch et al 1989; Zhong et al 2006). NAAG is an agonist at the mGluR3 (GRM3) receptor with potency similar to that of glutamate (Wroblewska et al 1998; Wroblewska et al 1997) and may also function as a weak antagonist at the NMDA receptor depending on concentration (Sekiguchi et al 1989; Trombley and Westbrook 1990; Westbrook et al 1986), NMDA receptor subunit composition (Hess et al 1999.) and cell type (Losi et al 2004; Fricker et al, 2009).
Altered expression of the NAAG-related genes, GCP II and mGluR3, are seen in the dorsolateral prefrontal cortex and hippocampus in schizophrenia (Ghose et al 2009; Ghose et al 2004; Guilarte et al 2008; Tsai et al 1995). In the hippocampus, the data are mixed with reports of a reduction in GCP II activity (Tsai et al 1995), reduced binding of 125I DCIT (GCP II radioligand) only in the entorhinal cortex (Guilarte et al 2008) and an increase in GCP II mRNA in a CA3 subregion (Ghose et al 2004). One reason for this apparent discrepancy could be precise anatomical regions of the hippocampus that were examined. The AH and PH are innervated by distinct afferent pathways (Lavenex and Amaral 2000; Suzuki and Amaral 1994a; Witter et al 2000) with the visual association/inferior temporal cortex projecting to the AH (through the perirhinal cortex) and the posterior visual association/posterior parietal cortex projecting to the PH (through the parahippocampal cortex) roughly mirroring the topography of the neocortex (Ansausti and Amaral 2004; Eichenbaum 2000; Lavenex and Amaral 2000; Moser and Moser 1998; Sweatt 2003; Witter et al 2000) (Ansausti and Amaral 2004; Medoff et al 2001; Suzuki and Amaral 1994a; Suzuki and Amaral 1994b; Sweatt 2003; Sweatt 2004; Tranel et al 1988). There is mounting evidence to suggest that the AH may be specifically involved in schizophrenia (Pegues et al 2003; Shenton et al 1992; Suddath et al 1990; Szeszko et al 2003; Tamminga et al 1992; Weinberger et al 1992b). In vivo brain imaging data have reported an increase in baseline perfusion and a decrease in activation, both of which correlate with severity of psychosis in the AH, but not PH, in schizophrenia (Medoff et al 2001; Tamminga et al 2003).
To further evaluate the NAAG system in the human hippocampus, we conducted experiments examining the distribution of GCPII and mGluR3 along the rostrocaudal axis in normal controls. Next, we compared gene expression levels between normal controls and cases of schizophrenia, and examined GCPII - mGluR3 correlations as a measure of coupling between NAAG and mGluR3. We hypothesized that we would find abnormal expression of GCP II and/or mGluR3 in regions of the AH.
2. Materials and methods
2.1.1. Characteristics of subjects
Human brain specimens were obtained from the Maryland Brain Collection (Baltimore, MD). Tissue was collected after obtaining consent from the next of kin. Consent was also obtained to collect medical records and conduct a telephone interview with a family member. All clinical information obtained for each case was reviewed by two research psychiatrists; diagnoses were made using DSM IV criteria. Blood toxicology screens for drugs of abuse, alcohol and prescription drugs including psychotropics were conducted on each case. The collection of human brain cases was approved by the Institutional Review Board of the University of Maryland and use of this tissue at the University of Texas Southwestern Medical Center was approved by the UT Southwestern IRB. Twenty cases were included in this study (Table 1). The two diagnostic groups were matched as closely as possible for race, sex, age, brain pH, postmortem interval (PMI) and RIN (RNA integrity number).
Table 1. a Demographics.
| CONTROLS | SCHIZOPHRENIA | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Case no. | Sex | Age | PMI | RIN | pH | Case no. | Sex | Age | PMI | RIN | pH |
| C1 | F | 31 | 21 | 7.8 | 6.66 | S1 | M | 45 | 6 | 7.2 | 6.63 |
| C2 | M | 48 | 12 | 7.1 | 6.56 | S2 | F | 60 | 18 | n | n |
| C3 | M | 43 | 20 | 7.9 | 6.45 | S3 | M | 47 | 21 | 7.7 | 6.48 |
| C4 | F | 67 | 12 | 7.8 | 6.52 | S4 | M | 23 | 20 | 7.7 | 6.96 |
| C5 | M | 46 | 23 | 8.2 | 6.31 | S5 | F | 50 | 11 | 7.6 | 6.91 |
| C6 | M | 55 | 10 | 7.8 | 6.87 | S6 | M | 22 | 10 | 6.9 | 6.56 |
| C7 | M | 52 | 15 | 7.9 | 6.32 | S7 | M | 77 | 17 | 7.3 | 6.84 |
| C8 | F | 24 | 23 | 8.4 | 6.84 | S8 | M | 62 | 12 | 4.9 | 6.84 |
| C9 | M | 42 | 13 | 8.3 | 6.6 | S9 | M | 59 | 13 | 9.1 | 6.92 |
| C10 | M | 62 | 13 | 5.8 | 6.47 | S10 | M | 53 | 5 | 9.4 | 6.85 |
| C11 | M | 51 | 15 | 7.3 | 6.31 | S11 | M | 47 | 20 | 8.6 | 6.95 |
| C12 | M | 55 | 17 | 9.6 | 6.99 | S12 | M | 38 | 6 | 9.5 | 6.57 |
| C13 | M | 47 | 14 | 9.2 | 7 | S13 | M | 24 | 23 | 8.4 | 6.65 |
| C14 | M | 35 | 16 | 8.7 | 6.72 | S14 | M | 33 | 12 | 7.4 | 6.42 |
| C15 | F | 29 | 18 | n | 6.35 | S15 | M | 31 | 14 | 8.7 | 6.81 |
| C16 | M | 50 | 5 | n | 6.59 | S16 | F | 40 | 11 | n | 6.62 |
| C17 | M | 44 | 25 | 6.9 | 6.63 | S17 | M | 28 | 34 | 7.7 | 6.81 |
| C18 | M | 35 | 11 | 8.2 | 6.71 | S18 | M | 37 | 14 | 8 | 6.86 |
| C19 | F | 72 | 20 | 7.9 | 6.81 | S19 | F | 77 | 17 | 7.3 | 6.77 |
| C20 | M | 47 | 10 | 7.7 | 6.98 | S20 | M | 41 | 19 | 7.2 | 6.57 |
| average | 5F/15M | 46.86 | 15.48 | 7.63 | 6.6 | average | 4F/16M | 44.7 | 15.24 | 7.52 | 6.71 |
| S.D. | 2 | 5.12 | 1.51 | 0.29 | S.D. | 16.37 | 6.67 | 1.64 | 0.21 | ||
Demographics on cases in the cohort used. PMI = =post mortem interval, RIN = RNA Integrity Number, The avaerge and standard deviations (S.D.) of demographic variables for each group are shown.
2.1.2. Tissue Preparation
At the time of brain dissection, the medial temporal lobe was removed in its entirety and frozen at -80 °C. A subsequent dissection was performed on the frozen tissue, whereby the hippocampus was divided into five equal blocks along the rostrocaudal axis from the anterior to the posterior end of the structure. For these experiments, anterior (blocks 1 or 2) and posterior (blocks 4 or 5) matched blocks were used. Tissue blocks were kept frozen at -80 °C until they were cryostat sectioned in the coronal plane at 14 μm at -20° C, thaw-mounted onto gelatin-coated subbed microscope slides, then dried and stored at -80°C. Every 75th section was stained for Nissl substance with thionin.
2.1.3. pH and RIN Determination
This was done as previously described (Stan et al, 2006). About 150 mg of cerebellar tissue was homogenized in 5ml ddH2O, centrifuged for 3 min at 8000g at 4 °C and pH of the supernatant measured in duplicate (Thermo-Electron Corporation). RIN determination was performed by isolating total RNA using Trizol (Invitrogen) followed by analysis with an Agilent 2100 Bioanalyzer.
2.2 In situ Hybridization
The riboprobe for GCPII was prepared as previously described (Ghose et al 2004). Antisense and sense templates were linearized using HindIII and ApaI, respectively. The mGluR3 cDNA (Loftstrand Labs, Gaithersburg, MD) is a 545 base pair fragment amplified from total human RNA and inserted into the pCRII vector. Antisense and sense templates were linearized using XbaI and BamHI respectively. 35S-UTP-labeled riboprobes were synthesized using an in vitro transcription kit (Riboprobe Systems, Promega, Madison, WI) with SP6 polymerase and T7 polymerase to generate the antisense and sense riboprobes, respectively. Two 14 μm sections per region per case were used in each in situ hybridization experiment. Tissue sections were fixed, acetylated, delipidated and dehydrated, then hybridized with 35S-UTP-labeled riboprobes as previously described (Ghose et al 2008; Ghose et al 2004). Following the in situ procedure, slides were apposed to Kodak autoradiographic film (Biomax) for 3 weeks (GCPII) or 5 weeks (mGluR3) along with 14C standards (American Radiolabeled Chemicals, Inc., St Louis, MO). Films were developed using Kodak D-19, digitized and their optical densities read in anatomical regions of interest using quantitative densitometry with the MCID densitometer and image system (Imaging Research Inc. St. Catharine's, Ontario, Canada).
3. Statistical Analysis
The demographic variables pH, PMI and age were compared between cohorts using unpaired t-tests. Correlation between GCPII and mGluR3 mRNA levels with the pH, RIN PMI and age were run with a Spearman Rank Order correlation. The effect of diagnosis on mRNA levels in specific regions (DG, CA1, CA3) was analyzed using a mixed model ANOVA with diagnosis as the between group factor and brain area as the within group factor. Separate analyses were conducted for GCP II and mGluR3 in the AH or PH. Significant findings were further analyzed using post hoc t-tests. If any demographic variable was found to influence target gene mRNA levels, an ANCOVA covarying for the variable was conducted. In all analyses, alpha < 0.05 was considered significant.
4. Results
4.1. Anatomic distribution of transcripts in the normal human hippocampal formation
4.1.1. GCPII mRNA
GCP II mRNA was detected in all sections examined from control and schizophrenia cases. As previously reported (Ghose et al 2004), GCPII mRNA hybridization signal was detected at high levels in the dentate gyrus and Ammon's horn of the hippocampus. Within each of these regions, robust expression was seen in the polymorphic and molecular layers of CA1 – 3 and in other white matter regions (fig 1a,c). Expression within the granule cell layer and pyramidal neuronal layer was minimal. Levels of GCPII expression in the DG, CA1 and CA3 subfields were similar in the AH and PH (table 2). There were no significant correlations between GCP II mRNA with age (r = 0.029 to 0.06), PMI (r = -0.069 to -0.18) or RIN (r = 0.13 to 0.21) in the DG, CA1 or CA3 regions except for RIN in the DG (r = 0.038, p = 0.004).
Figure 1. Representative autoradiographs.
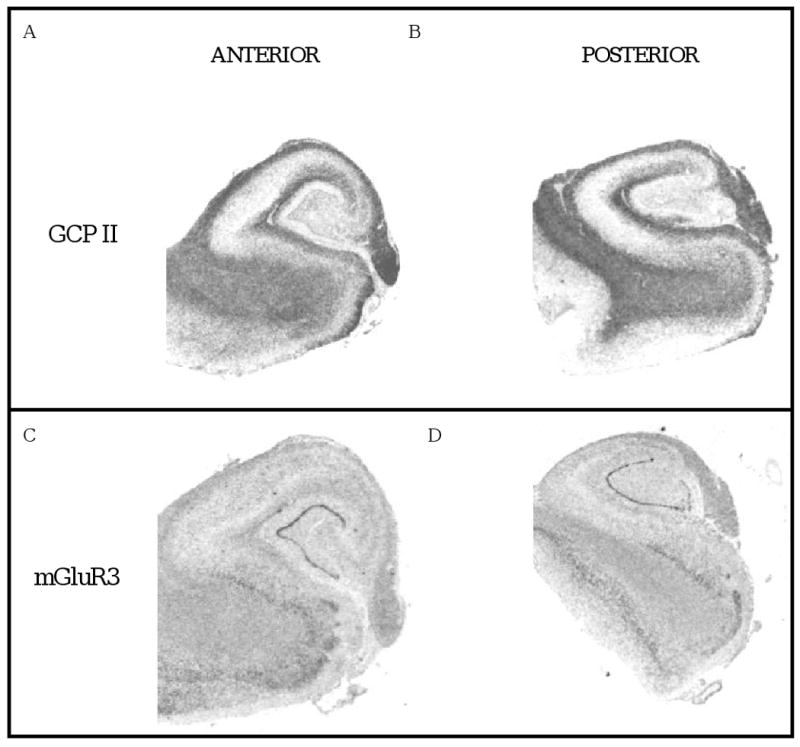
Images of tissue sections from the anterior (A, C) and posterior (B, D) hippocampus following in situ hybridization with cDNA probes for GCP II (A, B) and mGluR3 (C, D).
Table 2.
| ANTERIOR HIPPOCAMPUS | POSTERIOR HIPPOCAMPUS | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| control | schizophrenia | control | schizophrenia | ||||||||||
| mean | S.D. | mean | S.D. | p value | mean | S.D. | mean | S.D. | p value | ||||
| GCP II | DG | 19.46 | 5.89 | 18.61 | 4.22 | 0.65 | DG | 21.86 | 2.79 | 21.26 | 7.21 | 0.81 | |
| CA3 | 24.76 | 6.94 | 22.14 | 5.69 | 0.26 | CA3 | 25.70 | 2.36 | 26.01 | 7.26 | 0.92 | ||
| CA1 | 22.41 | 6.94 | 17.87 | 4.83 | 0.04 | CA1 | 23.23 | 2.49 | 21.81 | 4.92 | 0.43 | ||
| mGluR3 | DG | 22.486 | 5.90 | 22.822 | 8.15 | 0.896 | DG | 26.994 | 9.23 | 23.185 | 6.10 | 0.193 | |
| CA3 | 12.142 | 1.80 | 11.809 | 2.21 | 0.649 | CA3 | 12.829 | 3.23 | 12.408 | 2.01 | 0.671 | ||
| CA1 | 12.741 | 2.43 | 12.064 | 2.38 | 0.448 | CA1 | 14.060 | 4.32 | 15.564 | 9.74 | 0.589 | ||
GCP II and mGluR3 mRNA levels in the DG (dentate gyrus), CA3 and CA1 of the anterior and posterior hippocampus of 20 controls and cases of schizophrenia. Data shown are mean and standard deviation (S.D.).
4.1.2. mGluR3 mRNA
mGluR3 mRNA hybridization signal was observed throughout the hippocampus with the strongest signal in dentate gyrus. Lower levels were seen in the Ammon's horn with message observed in the polymorphic, pyramidal and molecular layers. Relatively high levels are seen in white matter regions consistent with the known localization of mGluR3 on glia. Expression of mRNA levels in respective subfields of AH and PH were similar (table 2). There were no significant correlations between mGluR3 mRNA with age (r = -0.23 to 0.021), PMI (r = -0.022 to 0.049) or RIN (r = -0.04 to 0.13) in the DG, CA1 or CA3 regions.
4.2. Regional comparison between schizophrenia and normal cases
4.2.1. GCPII
In the AH, we did not find a main effect of diagnosis (F = 2.35, df 1,29, p = 0.14) but did find an effect of region (F = 14.8, df 2,58, p <0.001) and a significant interaction between diagnosis and region (F= 3.59, df2,58, p = 0.034). Post hoc analyses reveal that the decreased expression of GCP II in schizophrenia is localized to CA1 (t = 2.14, df,1,31, p = 0.04) but is not apparent in DG (t = 0.45, df,1,29, p = 0.45) or CA3 (t = 1.16, df,1,30, p = 0.26) (table 2). Analysis of GCPII within subfield layers of CA1 (table 3) shows this decrease is present in the polymorphic layer (t = -2.12, df 1, 29, p = 0.04) and pyramidal layer (t = -2.3, df 1,31, p = 0.027) and shows a strong trend in the molecular layer (t = -2.04, df 1, 20, p = 0.054). Since we found a correlation between GCP II and RIN in the DG, we conducted an ANCOVA with RIN as a covariate, but this did not affect the results for the DG (F = 0.15, df 2,27, p = 0.7). Comparison of GCP II mRNA expression in the PH did not reveal any diagnosis-region interactions (F = 0.22, df2,38, p = 0.8).
Table 3.
GCP II in CA1 layers of the AH
| control | schizophrenia | ||||
|---|---|---|---|---|---|
| mean | S.D. | mean | S.D. | p value | |
| CA1 poly | 37.57 | 1.58 | 29.12 | 1.36 | 0.027 |
| CA1 pyr | 9.05 | 13.22 | 7.85 | 7.38 | 0.042 |
| CA1 mol | 24.58 | 6.74 | 19.58 | 4.56 | 0.054 |
GCP II mRNA levels in layers of CA1 – the polymorphic (poly), pyramidal (pyr) and molecular (mol) layers of the anterior hippocampus in 20 controls and cases of schizophrenia. Data shown are mean and standard deviation (S.D.).
4.2.2. mGluR3
There were no significant group differences in mGluR3 mRNA expression in AH (F = 0.02, df 2,58 p = 0.98) or PH (F = 1.33, df2,64, p = 0.27) between controls and cases of schizophrenia. Exploratory analyses of mGluR3 levels in the DG, CA1 or CA3 did not reveal any difference between the groups.
4.3. Correlations between molecular markers
We examined the correlations between these two related genes within hippocampal subfields in AH and PH. A significant positive correlation is seen between GCPII and mGluR3 in the CA3 region of the AH in controls (r = 0.66, p = 0.008) but not cases of schizophrenia (r = -0.096, p = 0.76). There were no correlations between GCP II and mGluR3 in the DG or CA1 in control or schizophrenia cases (r = 0.14 to 0.3; p = 0.18 to 0.64). In the PH, there were no significant correlations between GCP II and mGluR3 in any of the regions analyzed (DG, CA1, CA3) for either diagnostic group (r = - 0.26 to 0.45; p =0.19 to 0.87).
5. Discussion
In the present study, we examined the normal distribution pattern of mGluR3 and GCP II along the rostrocaudal axis of the human hippocampus and compared expression of these genes in cases of schizophrenia to normal controls. We found differences in these NAAG system genes in the AH, but not PH, in schizophrenia. Specifically, in CA1, GCP II was significantly reduced, while in CA3, the strong significant association between GCP II and mGluR3 was lost in schizophrenia.
5.1. Gene expression along the rostrocaudal axis of the control hippocampus
The expression of mGluR3 mRNA has not been previously examined by in situ hybridization in the human hippocampus. We found a pattern of mGluR3 mRNA expression in the human hippocampus that is similar to that described for the rodent hippocampus (Fotuhi et al 1994; Lyon et al 2008). Expression of mGluR3 was highest in the DG compared to CA1 or CA3 with signal detected throughout the hippocampus, consistent with the known expression of mGluR3 in both neurons and glia (Ghose S. 2008; Ohishi et al 1993; Tanabe et al 1993; Testa et al 1994). As previously reported, GCP II expression was reflected by its known localization to astrocytes (Ghose et al 2004); highest expression was found in the polymorphic and molecular layers. The pattern of expression for each gene was similar in the AH and PH.
5.2. Differences in Schizophrenia
There are two interesting findings from the diagnostic group comparisons. First, we report that GCP II is significantly decreased in the CA1 region of the AH in schizophrenia. This decrease is seen in the CA1 polymorphic and pyramidal layers although a strong trend (p = 0.054) is also seen in the molecular layer. This reduction in GCP II mRNA expression the enzyme that hydrolyzes NAAG, would result in an increase in NAAG in CA1 in schizophrenia. Since NAAG is a potent agonist at mGluR3 and antagonist at NMDA receptors, elevated levels of NAAG could increase mGluR3-mediated responses and diminish those that are NMDA-mediated. Electrophysiologically, NAAG has been shown to preferentially influence NMDA receptors in rodent CA1 pyramidal neurons (Bergeron et al 2005; Bergeron et al 2007) in a mGluR3-independent manner. Increasing concentrations of NAAG, either by exogenous application or by inhibiting GCP II, resulted in significant reductions in the NMDA receptor component of evoked excitatory postsynaptic currents. Blocking mGluR3 receptors did not influence these effects. These data suggest that the predominant action of elevated NAAG in CA1 in schizophrenia would be to reduce neuronal NMDA-dependent currents resulting in a hypoglutamatergic state (figure 2). Glial mGluR3 receptors could, however, still be activated by NAAG. Activation of the astrocytic mGluR3 receptor plays a role in modulating brain microcirculation (Baslow et al 2005) such that higher NAAG levels would increase local blood flow, and presumably magnetic resonance measures of perfusion of regional cerebral blood flow and volume (Baslow et al 2005). While this has not yet been replicated, these data suggest that the increase in NAAG levels could result in a hypoglutamatergic state associated with an increase in local regional blood flow or perfusion. This is reminiscent of the increased blood flow seen in the AH in medication-free human volunteers with schizophrenia (Tamminga et al 2003).
Figure 2. Model proposing a mechanism of hypoglutamatergia and increased blood flow in the anterior hippocampus in schizophrenia.
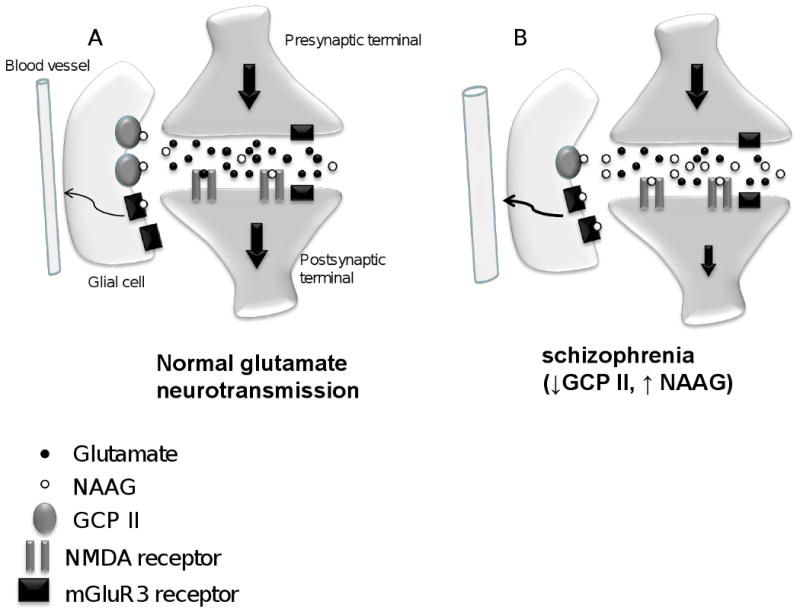
(A) In the control CA1 pyramidal neuron, NAAG is released into the synapse where it can bind to NMDA or mGluR3 receptors. The action of NAAG is terminated by hydrolysis by GCP II located on astrocytes. NAAG preferentially binds to NMDA receptors on pyramidal neurons (Bergeron et al, 2005, 2007). It could also activate mGluR3 receptors on glia to modulate local blood flow. (B) In schizophrenia, lower levels of GCP II in CA1 would lead to less metabolism of NAAG resulting in higher concentrations of this neurotransmitter in the synaptic cleft. This increase in NAAG levels would block post synaptic NMDA receptors resulting in a hypoglutamatergic state. Higher NAAG levels could also lead to increased activation of glial mGluR3 receptors to increase blood flow.
A second interesting observation of this study is the striking positive correlation between GCP II and mGluR3 in the CA3 of the AH in controls but not cases of schizophrenia. Animal electrophysiology studies demonstrate that GCP II inhibition at the mossy fiber – CA3 synapse modulates glutamate release (Sanabria et al 2004). It is postulated that NAAG released from CA3 neurons activates presynaptic mGluR3 receptors to inhibit glutamate neurotransmission (Sanabria et al 2004). The strong positive correlation in control cases suggests a tight modulation between NAAG and mGluR3 in the CA3 region of controls. This correlation is lost in cases of schizophrenia. Although we do not find differences in mGluR3 mRNA transcript levels between control and schizophrenia cases, the differences in gene expression correlations raises the possibility that NAAG-mGluR3 neurotransmission is impaired. In the control CA3, we find a tight positive correlation between GCP II and mGluR3 suggesting an inverse relationship between NAAG and mGluR3, i.e. as NAAG levels increase, mGluR3 decreases and vice versa. This may reflect a functional coupling between this neurotransmitter and its receptor. Since NAAG is known to modulate glutamate release via presynaptic mGluR3 receptors, the coupling between NAAG and mGluR3 may serve to maintain a homeostatic balance in synaptic glutamate levels. In the schizophrenia CA3, this correlation was lost. mGluR3 levels did not vary with changes in GCP II suggesting that increases/decreases in NAAG are not associated with compensatory changes in mGluR3. We speculate that this reflects a defect in the ability of the NAAG-mGluR3 system to modulate glutamate in the CA3 synapse. This disruption of glutamate neurotransmission would then impact information flow through the Schaffer collateral to CA1.
The trisynaptic pathway of the hippocampus consists of a glutamate-mediated excitatory feed forward system from the entorhinal cortex to the DG, the mossy fibers from DG to CA3 and the Schaffer collaterals from CA3 to CA1. Our data suggests that alterations in the NAAG system exist in the CA3 and CA1. These deficits could disrupt the one way flow of information through the hippocampus. Further, these data suggest that there may not be one mechanism of dysfunction but that multiple deficits could contribute to the overall hypoglutamatergic state in schizophrenia.
5.3. Previous studies
As mentioned earlier, previous studies examining GCP II in the hippocampus in schizophrenia have not been consistent (Ghose et al 2004; Guilarte et al 2008; Tsai et al 1995). Our findings in this report support the initial study by Tsai et al (Tsai et al 1995) reporting a reduction of GCP II in hippocampus in schizophrenia (Tsai et al 1995). In this study, we augmented the information in that report by specifying that this reduction is localized to the AH, in particular to the CA1 region. Although Guilarte et al. (Guilarte et al 2008) reported significant decreases in 125I DCIT binding only in the entorhinal cortex, they did find 31% lower binding, although non-significant, to GCP II in CA1 in schizophrenia. In our previous study, we reported an increase in GCP II transcript in the CA3 polymorphic region of the mid body of the hippocampus (Ghose et al 2004). The reason for this is not entirely clear but there are several factors to consider. First, the influence of antipsychotic treatment on GCP II expression in the post mortem schizophrenic brain is a potential confound. Although chronic antipsychotic treatment does not influence GCP II protein levels (Flores and Coyle 2003), it is possible that subtle differences in drug exposure might account for the differences between studies. Further, tissue quality characteristics differ from cohort to cohort and this may be a contributory factor. Finally, differences between studies may reflect heterogeneity in the etiology of schizophrenia with only a subgroup of cases with schizophrenia showing alterations in GCP II expression.
5.4. Summary
We find that schizophrenia-associated molecular alterations in the NAAG system occur predominantly in the AH while the PH is relatively unaffected. The findings could explain the observation of increased blood flow in the AH associated with a hypoglutamatergic state in schizophrenia. Further, these data lend support to the notion of AH dysfunction in schizophrenia (Pegues et al 2003; Suddath et al 1990; Szeszko et al 2003; Tamminga et al 1992; Tamminga et al 2000; Weinberger et al 1992a) suggesting that neural circuits associated with the AH are specifically disrupted in schizophrenia.
Acknowledgments
We wish to thank the next of kin of the brain tissue donors who made this study possible, the Baltimore County Medical Examiners Office for assistance with procurement of tissue and to Robert Conley, M.D. who did case diagnosis.
Role of Funding Source: This project was supported by the following grants: NARSAD Research Fund (Domenici Investigator to SG), National Institutes of Mental Health (NIMH; MH6223602 to CT, MH60744 to RR) and National Center for Research Resources (NCRR; UL1RR024982 to Milton Packer, MD). NARSAD, NIMH and NCRR had no further role in the study design; in the collection, analysis and interpretation of the data; in the writing of the report; and in the decision to submit the paper for publication.
Footnotes
Conflict of interest: None of the authors report any potential conflicts of interests.
Contributors: Subroto Ghose: conceptualized and designed the study, conducted experiments and statistical analyses, wrote first draft of the manuscript
Ronald Chin: conducted data analysis
Analysa Gallegos: conducted experiments
Rosalinda Roberts: directed the MBC, dissected tissue blocks and provided cohort of matched tissue sets
Joseph Coyle: construction of GCPII cDNA clone
Carol Tamminga: conceptualized and designed the study
All authors contributed to the final manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ansausti R, Amaral D. Hippocampal Formation in The Human Nervous System. San Diego, CA: Elsevier; 2004. [Google Scholar]
- Baslow MH, Dyakin VV, Nowak KL, Hungund BL, Guilfoyle DN. 2-PMPA, a NAAG peptidase inhibitor, attenuates magnetic resonance BOLD signals in brain of anesthetized mice: evidence of a link between neuron NAAG release and hyperemia. J Mol Neurosci. 2005;26:1–15. doi: 10.1385/JMN:26:1:001. [DOI] [PubMed] [Google Scholar]
- Berger UV, Luthi-Carter R, Passani LA, Elkabes S, Black I, Konradi C, et al. Glutamate carboxypeptidase II is expressed by astrocytes in the adult rat nervous system. The Journal of comparative neurology. 1999;415:52–64. doi: 10.1002/(sici)1096-9861(19991206)415:1<52::aid-cne4>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Bergeron R, Coyle JT, Tsai G, Greene RW. NAAG reduces NMDA receptor current in CA1 hippocampal pyramidal neurons of acute slices and dissociated neurons. Neuropsychopharmacology. 2005;30:7–16. doi: 10.1038/sj.npp.1300559. [DOI] [PubMed] [Google Scholar]
- Bergeron R, Imamura Y, Frangioni JV, Greene RW, Coyle JT. Endogenous N-acetylaspartylglutamate reduced NMDA receptor-dependent current neurotransmission in the CA1 area of the hippocampus. Journal of neurochemistry. 2007;100:346–357. doi: 10.1111/j.1471-4159.2006.04253.x. [DOI] [PubMed] [Google Scholar]
- Coyle JT. The nagging question of the function of N-acetylaspartylglutamate. Neurobiol Dis. 1997;4:231–238. doi: 10.1006/nbdi.1997.0153. [DOI] [PubMed] [Google Scholar]
- Eichenbaum H. A cortical-hippocampal system for declarative memory. Nat Rev Neurosci. 2000;1:41–50. doi: 10.1038/35036213. [DOI] [PubMed] [Google Scholar]
- Flores C, Coyle JT. Regulation of glutamate carboxypeptidase II function in corticolimbic regions of rat brain by phencyclidine, haloperidol, and clozapine. Neuropsychopharmacology. 2003;28:1227–1234. doi: 10.1038/sj.npp.1300129. [DOI] [PubMed] [Google Scholar]
- Fotuhi M, Standaert DG, Testa CM, Penney JB, Jr, Young AB. Differential expression of metabotropic glutamate receptors in the hippocampus and entorhinal cortex of the rat. Brain research. 1994;21:283–292. doi: 10.1016/0169-328x(94)90259-3. [DOI] [PubMed] [Google Scholar]
- Frickler AC, Selina Mok MH, de la Flor R, Shah AJ, Woolley M, Dawson LA, Kew JN. Effects of N-acetylaspartylglutamate (NAAG) at group II mGluRs and NMDAR. Neuropharmacology. 2009 Mar 12; doi: 10.1016/j.neuropharm.2009.03.002. Epub. [DOI] [PubMed] [Google Scholar]
- Ghose S, Crook JM, Bartus CL, Sherman TG, Herman MM, Hyde TM, et al. Metabotropic glutamate receptor 2 and 3 gene expression in the human prefrontal cortex and mesencephalon in schizophrenia. Int J Neurosci. 2008;118:1609–1627. doi: 10.1080/00207450802330702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghose S, Gleason K, Potts B, Lewis-Amezcua K, Tamminga C. Differential expression of metabotropic glutamate receptor 2 and 3 in schizophrenia: a mechanism for antipsychotic drug action? 2009 doi: 10.1176/appi.ajp.2009.08091445. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghose S, Weickert CS, Colvin SM, Coyle JT, Herman MM, Hyde TM, et al. Glutamate carboxypeptidase II gene expression in the human frontal and temporal lobe in schizophrenia. Neuropsychopharmacology. 2004;29:117–125. doi: 10.1038/sj.npp.1300304. [DOI] [PubMed] [Google Scholar]
- Ghose S, C J, Bartus C, Sherman T, Herman M, Hyde T, Kleinman J, Akil M. Metabotropic glutamate receptor 2 and 3 gene expression in the human prefrontal cortex and mesencephalon in schizophrenia. International Journal of Neuroscience. 2008 doi: 10.1080/00207450802330702. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilarte TR, Hammoud DA, McGlothan JL, Caffo BS, Foss CA, Kozikowski AP, et al. Dysregulation of glutamate carboxypeptidase II in psychiatric disease. Schizophrenia research. 2008;99:324–332. doi: 10.1016/j.schres.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess S, Pasieczny R, Rao S, Jachec C, Varney M, Johnson E. Activity of N-cetylaspartylglutamate at human recombinant glutamate receptors. 29th Annual Meeting, Society for Neuroscience; Miami Beach, FL. 1999. p. 975. [Google Scholar]
- Lavenex P, Amaral DG. Hippocampal-neocortical interaction: a hierarchy of associativity. Hippocampus. 2000;10:420–430. doi: 10.1002/1098-1063(2000)10:4<420::AID-HIPO8>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Losi G, Vicini S, Neale J. NAAG fails to antagonize synaptic and extrasynaptic NMDA receptors in cerebellar granule neurons. Neuropharmacology. 2004;46:490–496. doi: 10.1016/j.neuropharm.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Lyon L, Kew JN, Corti C, Harrison PJ, Burnet PW. Altered hippocampal expression of glutamate receptors and transporters in GRM2 and GRM3 knockout mice. Synapse (New York, NY) 2008;62:842–850. doi: 10.1002/syn.20553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medoff DR, Holcomb HH, Lahti AC, Tamminga CA. Probing the human hippocampus using rCBF: contrasts in schizophrenia. Hippocampus. 2001;11:543–550. doi: 10.1002/hipo.1070. [DOI] [PubMed] [Google Scholar]
- Moser MB, Moser EI. Functional differentiation in the hippocampus. Hippocampus. 1998;8:608–619. doi: 10.1002/(SICI)1098-1063(1998)8:6<608::AID-HIPO3>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Neale JH, Bzdega T, Wroblewska B. N-Acetylaspartylglutamate: the most abundant peptide neurotransmitter in the mammalian central nervous system. Journal of neurochemistry. 2000;75:443–452. doi: 10.1046/j.1471-4159.2000.0750443.x. [DOI] [PubMed] [Google Scholar]
- Ohishi H, Shigemoto R, Nakanishi S, Mizuno N. Distribution of the mRNA for a metabotropic glutamate receptor (mGluR3) in the rat brain: an in situ hybridization study. The Journal of comparative neurology. 1993;335:252–266. doi: 10.1002/cne.903350209. [DOI] [PubMed] [Google Scholar]
- Pegues MP, Rogers LJ, Amend D, Vinogradov S, Deicken RF. Anterior hippocampal volume reduction in male patients with schizophrenia. Schizophrenia research. 2003;60:105–115. doi: 10.1016/s0920-9964(02)00288-8. [DOI] [PubMed] [Google Scholar]
- Sanabria ER, Wozniak KM, Slusher BS, Keller A. GCP II (NAALADase) inhibition suppresses mossy fiber-CA3 synaptic neurotransmission by a presynaptic mechanism. J Neurophysiol. 2004;91:182–193. doi: 10.1152/jn.00465.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi M, Okamoto K, Sakai Y. Low-concentration N-acetylaspartylglutamate suppresses the climbing fiber response of Purkinje cells in guinea pig cerebellar slices and the responses to excitatory amino acids of Xenopus laevis oocytes injected with cerebellar mRNA. Brain research. 1989;482:87–96. doi: 10.1016/0006-8993(89)90545-3. [DOI] [PubMed] [Google Scholar]
- Shenton ME, Kikinis R, Jolesz FA, Pollak SD, LeMay M, Wible CG, et al. Abnormalities of the left temporal lobe and thought disorder in schizophrenia. A quantitative magnetic resonance imaging study. The New England journal of medicine. 1992;327:604–612. doi: 10.1056/NEJM199208273270905. [DOI] [PubMed] [Google Scholar]
- Slusher BS, Vornov JJ, Thomas AG, Hurn PD, Harukuni I, Bhardwaj A, et al. Selective inhibition of NAALADase, which converts NAAG to glutamate, reduces ischemic brain injury. Nat Med. 1999;5:1396–1402. doi: 10.1038/70971. [DOI] [PubMed] [Google Scholar]
- Stauch BL, Robinson MB, Forloni G, Tsai G, Coyle JT. The effects of N-acetylated alpha-linked acidic dipeptidase (NAALADase) inhibitors on [3H]NAAG catabolism in vivo. Neurosci Lett. 1989;100:295–300. doi: 10.1016/0304-3940(89)90702-7. [DOI] [PubMed] [Google Scholar]
- Suddath RL, Christison GW, Torrey EF, Casanova MF, Weinberger DR. Anatomical abnormalities in the brains of monozygotic twins discordant for schizophrenia. The New England journal of medicine. 1990;322:789–794. doi: 10.1056/NEJM199003223221201. [DOI] [PubMed] [Google Scholar]
- Suzuki WA, Amaral DG. Perirhinal and parahippocampal cortices of the macaque monkey: cortical afferents. J Comp Neurol. 1994a;350:497–533. doi: 10.1002/cne.903500402. [DOI] [PubMed] [Google Scholar]
- Suzuki WA, Amaral DG. Topographic organization of the reciprocal connections between the monkey entorhinal cortex and the perirhinal and parahippocampal cortices. J Neurosci. 1994b;14:1856–1877. doi: 10.1523/JNEUROSCI.14-03-01856.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweatt J. Mechanisms of Memory 2003 [Google Scholar]
- Sweatt JD. Hippocampal function in cognition. Psychopharmacology (Berl) 2004;174:99–110. doi: 10.1007/s00213-004-1795-9. [DOI] [PubMed] [Google Scholar]
- Szeszko PR, Goldberg E, Gunduz-Bruce H, Ashtari M, Robinson D, Malhotra AK, et al. Smaller anterior hippocampal formation volume in antipsychotic-naive patients with first-episode schizophrenia. The American journal of psychiatry. 2003;160:2190–2197. doi: 10.1176/appi.ajp.160.12.2190. [DOI] [PubMed] [Google Scholar]
- Tamminga CA, Lahti AC, Medoff DR, Gao XM, Holcomb HH. Evaluating glutamatergic transmission in schizophrenia. Annals of the New York Academy of Sciences. 2003;1003:113–118. doi: 10.1196/annals.1300.062. [DOI] [PubMed] [Google Scholar]
- Tamminga CA, Thaker GK, Buchanan R, Kirkpatrick B, Alphs LD, Chase TN, et al. Limbic system abnormalities identified in schizophrenia using positron emission tomography with fluorodeoxyglucose and neocortical alterations with deficit syndrome. Archives of general psychiatry. 1992;49:522–530. doi: 10.1001/archpsyc.1992.01820070016003. [DOI] [PubMed] [Google Scholar]
- Tamminga CA, Vogel M, Gao X, Lahti AC, Holcomb HH. The limbic cortex in schizophrenia: focus on the anterior cingulate. Brain Res Brain Res Rev. 2000;31:364–370. doi: 10.1016/s0165-0173(99)00053-3. [DOI] [PubMed] [Google Scholar]
- Tanabe Y, Nomura A, Masu M, Shigemoto R, Mizuno N, Nakanishi S. Signal transduction, pharmacological properties, and expression patterns of two rat metabotropic glutamate receptors, mGluR3 and mGluR4. J Neurosci. 1993;13:1372–1378. doi: 10.1523/JNEUROSCI.13-04-01372.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa CM, Standaert DG, Young AB, Penney JB., Jr Metabotropic glutamate receptor mRNA expression in the basal ganglia of the rat. J Neurosci. 1994;14:3005–3018. doi: 10.1523/JNEUROSCI.14-05-03005.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tranel D, Brady DR, Van Hoesen GW, Damasio AR. Parahippocampal projections to posterior auditory association cortex (area Tpt) in Old-World monkeys. Exp Brain Res. 1988;70:406–416. doi: 10.1007/BF00248365. [DOI] [PubMed] [Google Scholar]
- Trombley PQ, Westbrook GL. Excitatory synaptic transmission in cultures of rat olfactory bulb. J Neurophysiol. 1990;64:598–606. doi: 10.1152/jn.1990.64.2.598. [DOI] [PubMed] [Google Scholar]
- Tsai G, Passani LA, Slusher BS, Carter R, Baer L, Kleinman JE, et al. Abnormal excitatory neurotransmitter metabolism in schizophrenic brains. Archives of general psychiatry. 1995;52:829–836. doi: 10.1001/archpsyc.1995.03950220039008. [DOI] [PubMed] [Google Scholar]
- Weinberger DR, Berman KF, Suddath R, Torrey EF. Evidence of dysfunction of a prefrontal-limbic network in schizophrenia: a magnetic resonance imaging and regional cerebral blood flow study of discordant monozygotic twins. Am J Psychiatry. 1992a;149:890–897. doi: 10.1176/ajp.149.7.890. [DOI] [PubMed] [Google Scholar]
- Weinberger DR, Berman KF, Torrey EF. Correlations between abnormal hippocampal morphology and prefrontal physiology in schizophrenia. Clin Neuropharmacol. 1992b;15(Pt A) 1:393A–394A. doi: 10.1097/00002826-199201001-00205. [DOI] [PubMed] [Google Scholar]
- Westbrook GL, Mayer ML, Namboodiri MA, Neale JH. High concentrations of N-acetylaspartylglutamate (NAAG) selectively activate NMDA receptors on mouse spinal cord neurons in cell culture. J Neurosci. 1986;6:3385–3392. doi: 10.1523/JNEUROSCI.06-11-03385.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witter MP, Wouterlood FG, Naber PA, Van Haeften T. Anatomical organization of the parahippocampal-hippocampal network. Annals of the New York Academy of Sciences. 2000;911:1–24. doi: 10.1111/j.1749-6632.2000.tb06716.x. [DOI] [PubMed] [Google Scholar]
- Wroblewska B, Santi MR, Neale JH. N-acetylaspartylglutamate activates cyclic AMP-coupled metabotropic glutamate receptors in cerebellar astrocytes. Glia. 1998;24:172–179. doi: 10.1002/(sici)1098-1136(199810)24:2<172::aid-glia2>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Wroblewska B, Wroblewski JT, Pshenichkin S, Surin A, Sullivan SE, Neale JH. N-acetylaspartylglutamate selectively activates mGluR3 receptors in transfected cells. Journal of neurochemistry. 1997;69:174–181. doi: 10.1046/j.1471-4159.1997.69010174.x. [DOI] [PubMed] [Google Scholar]
- Zhong C, Zhao X, Van KC, Bzdega T, Smyth A, Zhou J, et al. NAAG peptidase inhibitor increases dialysate NAAG and reduces glutamate, aspartate and GABA levels in the dorsal hippocampus following fluid percussion injury in the rat. Journal of neurochemistry. 2006;97:1015–1025. doi: 10.1111/j.1471-4159.2006.03786.x. [DOI] [PubMed] [Google Scholar]