

Abstract
Altering the specificity of an enzyme requires precise positioning of side-chain functional groups that interact with the modified groups of the new substrate. This requires not only sequence changes that introduce the new functional groups but also sequence changes that remodel the structure of the protein backbone so that the functional groups are properly positioned. We describe a computational design method for introducing specific enzyme–substrate interactions by directed remodeling of loops near the active site. Benchmark tests on 8 native protein–ligand complexes show that the method can recover native loop lengths and, often, native loop conformations. We then use the method to redesign a critical loop in human guanine deaminase such that a key side-chain interaction is made with the substrate ammelide. The redesigned enzyme is 100-fold more active on ammelide and 2.5e4-fold less active on guanine than wild-type enzyme: The net change in specificity is 2.5e6-fold. The structure of the designed protein was confirmed by X-ray crystallographic analysis: The remodeled loop adopts a conformation that is within 1-Å Cα RMSD of the computational model.
Keywords: computational protein design, loop modeling
Computational protein design methodology has been used to optimize properties such as protein stability (1, 2) and to introduce functions such as binding of small molecules (3), proteins (4), and nucleic acids (5), as well as enzymatic activity (6, 7). In most of these studies, the implicit assumption that the structure of the polypeptide backbone would remain largely fixed despite mutations of amino acid side chains was made for the sake of computational tractability.
Explicit remodeling of the polypeptide backbone makes possible further optimization of these and other structural or functional properties. The set of combinations of protein sequences and structures is considerably larger when backbone flexibility is allowed and is likely to contain conformations that optimize a desired property to a greater degree than the original scaffold. This is illustrated by the backbone shifts that accompany functional divergence in natural protein evolution. Previous studies have achieved functional changes by backbone alteration, but relied on grafting methods that are restricted to sequences of known structure and function (8–10), which may be suboptimal with respect to the desired property.
De novo protein structure prediction methods are well suited for sampling novel backbone conformations (11). These methods have recently been extended to focus sampling on conformations that satisfy specific positional constraints (12, 13). Computational design algorithms that iterate between sequence design and backbone optimization using structure-prediction methods have been used to design previously unobserved protein fold and loop conformations (2, 14), but have not yet been applied to achieving functional changes such as alteration of an enzyme's substrate specificity.
We have developed a computational design algorithm that uses constrained backbone sampling to remodel a flexible loop subject to functional constraints. This constrained loop-remodeling protocol allows the identification of novel substitutions, insertions, and deletions that alter backbone structure so that specified functional groups satisfy precise positional constraints. This protocol can be used for the redesign of enzyme specificity by positioning side-chain groups in favorable interactions with a new substrate. In this article we first describe the algorithm, then validate the method computationally by using a benchmark set of loop structures in native enzymes. Finally, we apply the method to redesigning the specificity of human guanine deaminase (hGDA) and characterize the activity and structure of the designed enzyme experimentally.
Results and Discussion
Computational Methodology.
We developed a constrained loop-remodeling method for enzyme specificity alteration. A model of the enzyme in complex with the hypothetical transition state structure for a new substrate is created. New “anchor” residues are placed in the model in positions at which their side-chain functional groups make ideal interactions with the transition state. Backbone conformations capable of hosting these residues in the appropriate locations are identified by using techniques for modeling backbone flexibility from de novo protein structure prediction. Subsequently, sequence optimization is carried out to stabilize the novel backbone configurations, and thus stabilize the reaction transition state via interactions with the anchor residues. De novo loop modeling methods (15) are used to corroborate that designed sequences computationally fold to the desired structure when the constraint between the substrate and side chain is removed. The final designs differ from the scaffold structure in length, conformation, and sequence.
Native Structure Recapitulation.
To evaluate the method, we first benchmarked its performance in native structure recapitulation experiments. Eight protein–ligand complexes in which an anchor residue makes a strong interaction with the ligand were chosen from the Protein Data Bank (PDB). A 7-residue window centered on the anchor residue was excised from a model of the complex. The PDB ID codes, anchor residues, and excised regions are given in supporting information (SI) Table S1. To fill the excised region, loops of 9 different lengths were generated by using the protocol described in Materials and Methods. The native loop has 3 residues before and 3 residues after the anchor residue; this was replaced by loops having 2, 3, or 4 residues before and after the anchor residue. The rigid-body interaction observed in the native complex was used to position the terminal side-chain moiety of the anchor residue. No other knowledge of the native configuration of the anchor side chain and the excised backbone was used during the course of the protocol. The native sequence of the loop was also not used, except for the identity of the anchor residue. For each loop length, 100 models were generated.
The energy of the 5 lowest-energy structures for each complex and loop length are shown in Fig. 1A. In 6 of 8 cases, the lowest-energy structure corresponds to the native loop length. This indicates that the energy of structures generated with this protocol can be used to discriminate between native and nonnative loop lengths. Furthermore, low-energy structures of the correct loop length are often quite close to the native in conformation (Table S1 and Fig. S1). The lowest-energy structures for 2 cases are shown in Fig. S2. Inadequate sampling of near-native structures contributed to the failure of correct loop length discrimination in the case of PDB ID code 2IO2, because none of the structures generated for any length were as low in energy as the crystal structure conformation. Therefore, sampling a larger number of structures would likely improve performance in loop-length discrimination.
Fig. 1.

Benchmarking and application of loop design methodology. (A) Eight protein/ligand complexes were chosen from the PDB to benchmark the loop design protocol. A 7-residue window centered on an anchor residue was excised from a model of the complex. To assess our ability to recapitulate native loop lengths with this protocol, the excised region was filled by using loops of the native length (3,3; orange) and several nonnative lengths (blue). Loop lengths indicate the number of residues inserted in the excised region before and after the anchor residue. The total loop length also includes the anchor residue. The energies of the 5 lowest-energy structures generated for each loop length are shown in box-and-whiskers form. In 6 of 8 cases, the native length structures are lower in energy than the nonnative length structures. Thus, native loop lengths can be identified with this protocol. (B) The sequence requirements for hGDA specificity alteration were determined by comparing energies of several different loop lengths and anchor identities. The lowest-energy structures (orange) had loop length 3,4, which was 2 residues shorter than the native loop, and anchor identity asparagine. The lowest-energy glutamine design had loop length 2,4 and had a higher energy than the lowest-energy asparagine design. The lowest energy loop overall (orange, asparagine) was selected for experimental characterization.
Redesign of hGDA Specificity.
Encouraged by the promising results on the benchmark, we sought to use our method to redesign enzyme specificity. We applied the method to altering the specificity of human guanine deaminase (hGDA) with the long-term goal of introducing cytosine deaminase activity into a human protein scaffold. A designed cytosine deaminase with a sequence close to that of a human protein would solve an important problem in suicide gene therapy (16–18) by providing prodrug-activating ability (19–22) while retaining low immunogenicity, as described in the SI Text. We consider hGDA to be the best starting point for such an effort based on the complement of deaminases that exist in the human genome (23, 24). A comparison of the active sites of hGDA (25) and the distantly related bacterial cytosine deaminase (26) (bCD, Fig. 2) suggests that key interactions with cytosine may be introduced into hGDA by loop modeling with design while preserving the residues directly involved in catalysis. In this article, we sought to redesign guanine deaminase for activity toward the compound ammelide, which is a structural intermediate between guanine and cytosine (Fig. 3).
Fig. 2.

Active-site structures. (A) The active site of human guanine deaminase (hGDA) with product, xanthine (PDB ID code 2UZ9). Arginine 213 and phenylalanine 214 are visible at the bottom of the image. (B) The active site of bacterial cytosine deaminase (bCD) with transition state analog, di-hydropyrimidine (PDB ID code 1K70). Glutamine 156 is visible at the bottom of the image. Proton shuttling residues, colored yellow, are conserved and are responsible for positioning and transferring protons from a bound water molecule to the substrate. Metal-binding histidines, colored orange, are responsible for binding a metal (zinc in hGDA, iron in bCD), which lowers the pKa of the bound water molecule. The transition state of the reaction is the tetrahedral intermediate formed after attack of the bound water molecule and before leaving group departure.
Fig. 3.
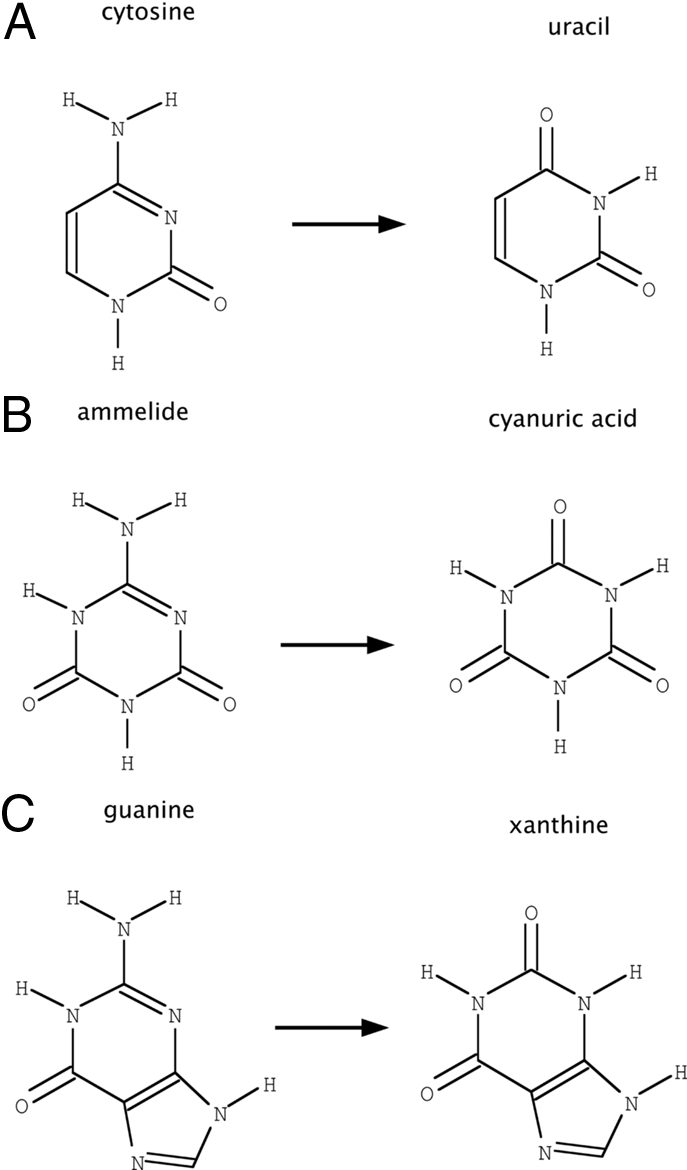
Deaminase substrates. (A) The reaction performed by wild-type human guanine deaminase (hGDA). (B) The reaction under study in this article. (C) The reaction performed by prodrug-activating cytosine deaminases, such as bacterial cytosine deaminase (bCD). Ammelide is a structural intermediate between guanine and cytosine and thus provides a stepping stone for specificity alteration of hGDA. Each reaction consumes 1 molecule of H2O and releases 1 molecule of NH3.
The application of our method to this system consisted of superimposing ammelide onto the 6-membered ring of guanine in the transition state for deamination in a model derived from the crystal structure of hGDA (25). Based on the structure of bacterial cytosine deaminase (26), in which an interaction between cytosine N1/O2 and a glutamine residue is observed (Fig. 2B), a starting model for design was created with a glutamine or asparagine residue positioned such that its amide group made analogous hydrogen bonds with ammelide. The segment between residues 211 and 220 was then remodeled to generate configurations capable of hosting this new glutamine or asparagine residue. Loops of 16 different lengths (2, 3, 4, or 5 residues, before and after the anchor residue) were built by using the protocol described above. For each loop length, 200 models were generated. The energy of the 5 lowest-energy structures is shown in Fig. 1B.
The optimal loop identified by this protocol involved sequence changes only to residues 213–218, used an asparagine to bind the substrate, and was 2 residues shorter than the wild-type loop (Fig. 4). Several sequences compatible with this conformation were identified. Based on de novo loop modeling and visual inspection, the sequence of the loop chosen for experimental verification was GNGV, which was significantly different from the wild-type loop sequence, RFSLSC.
Fig. 4.

Structure of design models. The backbone configuration of several designs are superimposed on wild-type hGDA, highlighting the differences that allow binding to the new reaction transition state. The design structures differ in length, conformation, and sequence. Two residues have been deleted, and 4 mutations have been made, including the introduction of an asparagine that directly interacts with the substrate.
Biochemical Characterization.
A gene, hGDA-des, with the substitutions and deletions required for the designed GNGV loop, was created and assayed for ammelide deaminase activity. Additional point mutants were created to establish whether each key feature of the designed enzyme was necessary for activity (Fig. 5A). First, a catalytic residue in the active site was mutated to alanine in both the wild-type enzyme (wt-E243A) and in the designed enzyme (des-E241A). Second, the asparagine anchor residue in the designed enzyme was mutated to both alanine (des-N214A) and to glutamine (des-N214Q). Third, individual residues of the wild-type loop were mutated to amide-containing amino acids without the designed 2-residue deletion (wt-R213NQ, wt-F214NQ).
Fig. 5.

Kinetic characterization. (A) Product formation over time at 20 μM enzyme, 500 μM substrate reveals that hGDA-des is highly active relative to wild type (100-fold), and asparagine or glutamine mutants without the remodeled backbone (>6-fold, Table S3). Mutations of the designed asparagine to alanine or glutamine demonstrates significantly reduced activity. (B) Velocity vs. substrate concentration at enzyme concentration of 10 μM. The estimated Michaelis–Menten parameters of hGDA-des are: kcat = 2.2 (2.1–2.4) e−4 s−1, Km = 1,300 (1,200–1,500) μM. Estimating kcat/Km from the 4 lowest substrate concentrations gives 0.15 (0.14–0.15) s−1 M−1 (95% confidence intervals in parentheses).
Negligible ammelide deaminase activity was measured for wild-type hGDA or either catalytic-residue mutant (Fig. 5A). The activity measured for the rationally designed hGDA-des was >6-fold higher than any other mutant, and 100-fold higher than the wild type or catalytic residue mutants (Table S3).
The relative activity of these mutants demonstrates that several aspects of the design were necessary for the alteration of activity. First, the negligible level of activity of the catalytic residue mutants demonstrates that the overall catalytic mechanism is preserved in the design, and that there are no contaminating activities. Second, the higher level of activity in the designed enzyme relative to the alanine or glutamine mutant of the designed enzyme shows that the correct positioning of the designed asparagine amide group was necessary for activity. Third, the higher level of activity in the designed enzyme relative to the amide-containing mutants that lacked a deletion suggests that the correct positioning of the designed asparagine was due to the alteration of backbone structure by the deletion.
Michaelis–Menten parameters for hGDA-des were determined as described, giving kcat = 2.2 (2.1–2.4) e−4 s−1, Km = 1,300 (1,200–1,500) μM (Fig. 5B). However, because saturation was not achieved due to limitations in the solubility of the substrate, these parameters are less meaningful individually than estimates of kcat/Km derived from the same data: kcat/Km = 0.15 (0.14–0.15) s−1 M−1 (95% confidence intervals are given in parentheses). This value is several orders of magnitude lower than that of wild-type hGDA for guanine. The nominal value of Km for ammelide is comparable with that of AtzC, a bacterial enzyme involved in atrazine degradation (27). Residual guanine deaminase activity in hGDA-des was <6e−4 s−1 at 90 μM guanine (Fig. S3), making the specificity switch (vad/vg)des × (vg/vad)wt > 2.5e6 (for velocities determined at 500 μM ammelide and 90 μM guanine).
Crystal Structure.
The X-ray crystal structure of hGDA-des was determined at 2.4-Å resolution (Table S4). Phases were determined by molecular replacement with a truncated model of wild-type hGDA. Residues 210–220, which span the redesigned loop, were omitted in the molecular replacement search model, as were several other regions of the structure. Clear unbiased density corresponding to the backbone of the designed region was observed immediately after molecular replacement (Fig. 6A). The asymmetric unit contained 2 individual chains, which were refined independently. Although ammelide was included in the crystallization solution, no density was observed in the location expected for bound substrate. Other features described below also suggest that the observed conformation represents the unbound state of the enzyme.
Fig. 6.

Superimposition of computational model and crystal structure. In both A and B, the computational model of hGDA-des is shown in yellow, whereas both chains of the final refined structure are shown in cyan. The backbone of the computational model is within 1-Å Cα RMSD of the crystal structure. (A) Unbiased electron density using phases derived from molecular replacement is shown at the 1σ level in mesh. The backbone of the designed loop is clearly visible in the density after molecular replacement. The side chain of designed residue N214 was not clearly visible in the electron density at this stage or after refinement. The side chain of designed residue V216 is clearly visible in the expected location. (B) No electron density was observed in the location expected for bound substrate, ammelide, or product, cyanuric acid, but for comparison, the modeled location is shown in magenta. The wild-type structure is also shown in slate blue.
The computational model of the designed loop was very similar to that observed in the crystal structure (Fig. 6 and S4). The Cα RMSD between the computational model and the A chain of the structure (B chain in parentheses) for designed residues 213–216 was 0.99 (1.0) Å. Furthermore, the φ–ψ angles of the backbone were in the same region of the Ramachandran plot for each residue in the designed loop (Fig. S5). However, whereas the backbone was clearly observed, density for the side chain of designed residue N214 beyond Cβ was not observed after molecular replacement, and after refinement was only visible for chain B. Thus, the residue designed to interact with substrate does not appear to be preordered in any single conformation beyond the Cβ atom. The side chain of designed residue V216 packs in the region predicted in the computational model.
To assess whether the designed loop is able to access a conformation compatible with catalysis, we compared the crystal structure with an isoenergetic ensemble of models generated at the corroborative loop-modeling stage (Fig. S6). This ensemble contains a configuration that is compatible with optimal transition state binding, and that differs from the crystal by only 0.48-Å Cα RMSD, suggesting that transition state binding is energetically accessible from the observed conformation of the designed loop.
The overall Cα RMSD between the computational model and the A chain (B chain in parentheses) of the crystal structure was 0.98 (0.96) Å. However, 2 segments, which contain no changes from the wild-type sequence, differ structurally between the computational model and crystal structure. The Cα RMSDs for those segments are: residues 90–109, 2.7 (2.6) Å; residues 412–422, 4.6 (4.5) Å. When these segments as well as the designed loop are excluded from the calculation, the overall Cα RMSD between the computational model and the crystal structure is 0.38 (0.39) Å.
The deviation of these 2 nondesigned segments of the structure from the conformation observed for wild-type hGDA is likely due to intrinsic flexibility of the wild-type enzyme associated with substrate binding, and not due to structural disruption by the designed loop. First, no substrate was observed in the structure, and crystallization with higher concentrations of substrate was not attempted because of substrate solubility limitations. In contrast, wild-type hGDA was cocrystallized with bound product (25). This difference suggests that the structure of hGDA-des may represent an apo conformation of the enzyme distinct from the bound conformation previously observed. Furthermore, the conformation of residues 90–109 described here resembles that of residues 67–96 in the apo structure of bCD (PDB ID code 1K6W), whereas the conformation observed for the corresponding residues in wild-type hGDA resemble the bound structure of bCD (PDB ID code 1K70). These residues of bCD form an active site lid, with an open state allowing substrate binding and product release and a closed state that excludes solvent during catalysis (28). Because hGDA-des residues 412–422 are adjacent to residues 90–109 in the opposite unit of the dimeric structure, their motion is likely necessary to accommodate this structural transition.
Other aspects of the active site are preserved between the wild type and designed structure. Importantly, the catalytic residues all adopt the same configuration in both structures, and density corresponding to the catalytic zinc and water is clearly seen.
B factors for the designed loop are not significantly different from for other exposed regions of the structure (Fig. S4). The segment with the highest average B factor is residues 242–265. This region, adjacent to the designed loop, may have been destabilized by the alteration of packing caused by deletion of 2 residues in the designed loop. Because this region is close in sequence to catalytic residues H238 and E241, and packs against the active-site lid in the bound conformation, destabilization of the conformation observed in the bound structure may be responsible for the low level of activity. Conversely, stabilization of that conformation may be a productive goal for future designs aimed at increasing activity.
Conclusions
We have developed a method for anchored loop redesign and validated the method through both computational and experimental tests. The computational results show that our protocol can accurately recapitulate features such as loop length in native structures. The relative activities measured in the experimental test, redesigning hGDA for ammelide deamination, corroborate the predictions of the method, and underscore the necessity of modeling backbone flexibility. The X-ray crystal structure shows that the designed loop adopts a conformation within 1-Å Cα RMSD of the computational model.
Although we increased activity by 2 orders of magnitude, the absolute activity toward ammelide (kcat/Km = 0.15 s−1 M−1) is still 7 orders of magnitude lower than the activity of wild-type hGDA for guanine (kcat = 17.4 s−1, Km = 9.5 μM, kcat/Km = 1.8e6 s−1 M−1) (29). Achieving levels of activity comparable with natural enzymes may require either filling the active-site cavities caused by the shorter loop and smaller substrate or positioning N214 more precisely to optimally orient the substrate for attack of the metal-bound water (30). More precisely stabilizing the optimal binding conformation of a designed loop may require flexible backbone design of the surrounding regions as well. Addressing these issues and increasing the activity of the designed enzyme to the level of wild-type hGDA for guanine are key challenges for future work.
In summary, hGDA-des offers an improved starting point for further computational design and directed evolution approaches to creating a nonimmunogenic cytosine deaminase. The accuracy of the benchmarking experiments and the close correspondence of the computational design with experimental results suggest our methodology should be broadly useful for redesign of specificity.
Materials and Methods
Full materials and methods are provided in SI Text.
Computational Method.
Our general method was implemented as an extension to the Rosetta suite of molecular modeling programs (11, 31). First, a model of a complex between a scaffold enzyme and a new reaction transition state is created, based on homology modeling or quantum chemical calculations, for use as input. Second, protein side-chain functional groups important for specific interactions between the enzyme and the reaction transition state are optimally positioned in the model, analogous to the “inverse rotamers” described in a previous study (32). Third, the polypeptide backbone of the scaffold protein is remodeled by using several techniques from structure prediction (13, 33, 34) (Fig. S7), so that it is able to host the newly introduced side chains in the optimal position. Fourth, standard computational design protocols (35) are used to identify sequences that maximally stabilize the generated backbone structures and thus optimize transition state binding. De novo loop-modeling methods (15) are used to corroborate that designed sequences computationally fold to the desired structure when the constraint between substrate and side chain is removed. Some sequences are observed to fold to configurations that do not satisfy the substrate–side chain constraint, and are thus not further evaluated. This method is for applications in which a single key interaction is to be introduced through large-scale loop remodeling. A complementary method for introducing one of a large library of equally acceptable interactions through finer-scale backbone alterations has also been developed in our group (44). The former method is tailored for enzyme-small molecule interactions, the latter, protein–DNA interactions. The methods differ algorithmically and in the amount of compute time allocated to searching for solutions that contain any single interaction.
Ammelide Deamination Assay.
cDNA for hGDA was obtained (Origene Technologies) and cloned between the NdeI and XhoI sites of pET-29b. hGDA-des and related controls were made by overlap assembly PCR (36) and Kunkel mutagenesis (37, 38) of the same vector. Proteins were expressed in an autoinduction medium (39) and purified by using NiNTA His*Bind resin (Qiagen). Reaction rates were measured in PBS (pH 7.5) at 25 °C, by using 20 μM enzyme and 50–750 μM ammelide (Sigma–Aldrich). Product formation was measured by HPLC by using an isocratic elution of 22% 5 mM sodium phosphate (pH 6.0) and 78% acetonitrile over a Zorbax NH2 Analytical Column (Agilent) at 1 mL/min (40). Absorption at 210 nm was monitored. Ammelide eluted between 12.7 and 13.0 min, whereas cyanuric acid eluted between 5.7 and 6.0 min.
Crystallography.
hGDA-des was cloned into pET-15b, expressed as above, and purified by using Talon resin (Clontech). After thrombin removal of the hexahistadine tag, size-exclusion chromatography was performed by using a Superdex 200 26/60 column (GE Healthcare). Purified protein at 10 mg/mL was dialyzed into 25 mM Hepes (pH 7.5), 100 mM sodium chloride, 2% glycerol, 1 mM TCEP, and 200 μM ammelide and crystallized by vapor-phase equilibration in the hanging-drop geometry, against a mother liquor of 100 mM Mes (pH 6.5), 200 mM sodium chloride, and 1.8 M ammonium sulfate. Crystals of space group I212121 produced diffraction data to 2.4-Å resolution at beamline 5.0.1 at the Advanced Light Source, Lawrence Berkeley Laboratory, Berkeley, CA at wavelength 0.9774. After processing and scaling with HKL-2000 (41), phases were determined by molecular replacement with PHASER using a truncated model of wild-type hGDA. Model building was performed by using the CCP4 suite of programs (42, 43), excluding a random 5% of the data for cross-validation. Statistics are provided in Table S4, and coordinates were deposited into the RCSB Protein Data Bank (accession no. 3E0L).
Supplementary Material
Acknowledgments.
We acknowledge the assistance of Betty Shen and Ryo Takeuchi in crystallographic data collection and of Meg Holmes in structure determination. Data were collected at beamline 5.0.1 at the Advanced Light Source (Lawrence Berkeley Laboratory, Berkeley, CA).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 3E0L).
This article contains supporting information online at www.pnas.org/cgi/content/full/0811070106/DCSupplemental.
References
- 1.Dahiyat BI, Mayo SL. De novo protein design: Fully automated sequence selection. Science. 1997;278:82–87. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 2.Kuhlman B, et al. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302:1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 3.Looger LL, et al. Computational design of receptor and sensor proteins with novel functions. Nature. 2003;423:185–190. doi: 10.1038/nature01556. [DOI] [PubMed] [Google Scholar]
- 4.Kortemme T, et al. Computational redesign of protein–protein interaction specificity. Nat Struct Mol Biol. 2004;11:371–379. doi: 10.1038/nsmb749. [DOI] [PubMed] [Google Scholar]
- 5.Ashworth J, et al. Computational redesign of endonuclease DNA binding and cleavage specificity. Nature. 2006;441:656–659. doi: 10.1038/nature04818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang L, et al. De novo computational design of retro-aldol enzymes. Science. 2008;319:1387–1391. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rothlisberger D, et al. Kemp elimination catalysts by computational enzyme design. Nature. 2008;453:190–195. doi: 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- 8.Riechmann L, et al. Reshaping human antibodies for therapy. Nature. 1988;332:323–327. doi: 10.1038/332323a0. [DOI] [PubMed] [Google Scholar]
- 9.Hedstrom L, et al. Converting trypsin to chymotrypsin: The role of surface loops. Science. 1992;255:1249–1253. doi: 10.1126/science.1546324. [DOI] [PubMed] [Google Scholar]
- 10.Bender GM, et al. De novo design of a single-chain diphenylporphyrin metalloprotein. J Am Chem Soc. 2007;129:10732–10740. doi: 10.1021/ja071199j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rohl CA, et al. Protein structure prediction using Rosetta. Methods Enzymol. 2004;383:66–93. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- 12.Mazur AK, Abagyan RA. New methodology for computer-aided modelling of biomolecular structure and dynamics. 1. Non-cyclic structures. J Biomol Struct Dyn. 1989;6:815–832. doi: 10.1080/07391102.1989.10507739. [DOI] [PubMed] [Google Scholar]
- 13.Bradley P, Baker D. Improved beta-protein structure prediction by multilevel optimization of nonlocal strand pairings and local backbone conformation. Proteins. 2006;65:922–929. doi: 10.1002/prot.21133. [DOI] [PubMed] [Google Scholar]
- 14.Hu X, et al. High-resolution design of a protein loop. Proc Natl Acad Sci USA. 2007;104:17668–17673. doi: 10.1073/pnas.0707977104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rohl CA, et al. Modeling structurally variable regions in homologous proteins with Rosetta. Proteins. 2004;55:656–677. doi: 10.1002/prot.10629. [DOI] [PubMed] [Google Scholar]
- 16.Bordignon C, et al. Transfer of the HSV-tk gene into donor peripheral blood lymphocytes for in vivo modulation of donor anti-tumor immunity after allogeneic bone marrow transplantation. Hum Gene Ther. 1995;6:813–819. doi: 10.1089/hum.1995.6.6-813. [DOI] [PubMed] [Google Scholar]
- 17.Verzeletti S, et al. Herpes simplex virus thymidine kinase gene transfer for controlled graft-versus-host disease and graft-versus-leukemia: Clinical follow-up and improved new vectors. Hum Gene Ther. 1998;9:2243–2251. doi: 10.1089/hum.1998.9.15-2243. [DOI] [PubMed] [Google Scholar]
- 18.Sprangers B, et al. Can graft-versus-leukemia reactivity be dissociated from graft-versus-host disease? Front Biosci. 2007;12:4568–4594. doi: 10.2741/2411. [DOI] [PubMed] [Google Scholar]
- 19.Vermes A, et al. Flucytosine: A review of its pharmacology, clinical indications, pharmacokinetics, toxicity and drug interactions. J Antimicrob Chemother. 2000;46:171–179. doi: 10.1093/jac/46.2.171. [DOI] [PubMed] [Google Scholar]
- 20.Longley DB, et al. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 21.Rooseboom M, et al. Enzyme-catalyzed activation of anticancer prodrugs. Pharmacol Rev. 2004;56:53–102. doi: 10.1124/pr.56.1.3. [DOI] [PubMed] [Google Scholar]
- 22.Loffler M, et al. Pyrimidine pathways in health and disease. Trends Mol Med. 2005;11:430–437. doi: 10.1016/j.molmed.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 23.Zielke CL, Suelter CH. In: The Enzymes. 3rd Ed. Boyer PD, editor. London: Academic; 1971. [Google Scholar]
- 24.Barthelmes J, et al. BRENDA, AMENDA and FRENDA: The enzyme information system in 2007. Nucleic Acids Res. 2007;35:D511–D514. doi: 10.1093/nar/gkl972. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moche M, et al. Human guanine deaminase (guad) in complex with zinc and its product xanthine. 2007 doi: 10.2210/pdb2uz9/2uz9. [DOI] [Google Scholar]
- 26.Ireton GC, et al. The structure of Escherichia coli cytosine deaminase. J Mol Biol. 2002;315:687–697. doi: 10.1006/jmbi.2001.5277. [DOI] [PubMed] [Google Scholar]
- 27.Shapir N, et al. Purification, substrate range, and metal center of AtzC: The N-isopropylammelide aminohydrolase involved in bacterial atrazine metabolism. J Bacteriol. 2002;184:5376–5384. doi: 10.1128/JB.184.19.5376-5384.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ireton GC, et al. The 1.14 A crystal structure of yeast cytosine deaminase: Evolution of nucleotide salvage enzymes and implications for genetic chemotherapy. Structure (London) 2003;11:961–972. doi: 10.1016/s0969-2126(03)00153-9. [DOI] [PubMed] [Google Scholar]
- 29.Yuan G, et al. Cloning and characterization of human guanine deaminase. Purification and partial amino acid sequence of the mouse protein. J Biol Chem. 1999;274:8175–8180. doi: 10.1074/jbc.274.12.8175. [DOI] [PubMed] [Google Scholar]
- 30.Wolfenden R. Are there limits to enzyme-inhibitor binding discrimination? Inferences from the behavior of nucleoside deaminases. Pharmacol Ther. 1993;60:235–244. doi: 10.1016/0163-7258(93)90008-2. [DOI] [PubMed] [Google Scholar]
- 31.Das R, Baker D. Macromolecular modeling with Rosetta. Annu Rev Biochem. 2008;77:363–382. doi: 10.1146/annurev.biochem.77.062906.171838. [DOI] [PubMed] [Google Scholar]
- 32.Zanghellini A, et al. New algorithms and an in silico benchmark for computational enzyme design. Protein Sci. 2006;15:2785–2794. doi: 10.1110/ps.062353106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simons KT, et al. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and Bayesian scoring functions. J Mol Biol. 1997;268:209–225. doi: 10.1006/jmbi.1997.0959. [DOI] [PubMed] [Google Scholar]
- 34.Canutescu AA, Dunbrack RL., Jr Cyclic coordinate descent: A robotics algorithm for protein loop closure. Protein Sci. 2003;12:963–972. doi: 10.1110/ps.0242703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuhlman B, Baker D. Native protein sequences are close to optimal for their structures. Proc Natl Acad Sci USA. 2000;97:10383–10388. doi: 10.1073/pnas.97.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sambrook J, Russell DW. Molecular Cloning. A Laboratory Manual. 3rd Ed. Cold Spring Harbor, NY: Cold Spring Harbor Lab Press; 2001. pp. 13.36–13.39. [Google Scholar]
- 37.Kunkel TA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci USA. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kunkel TA, et al. Efficient site-directed mutagenesis using uracil-containing DNA. Methods Enzymol. 1991;204:125–139. doi: 10.1016/0076-6879(91)04008-c. [DOI] [PubMed] [Google Scholar]
- 39.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 40.Sugita T, et al. Determination of melamine and three hydrolytic products by liquid chromatography. Bull Environ Contam Toxicol. 1990;44:567–571. doi: 10.1007/BF01700877. [DOI] [PubMed] [Google Scholar]
- 41.Otwinowski Z, Minor W, editors. Methods in Enzymology. New York: Academic; 1997. [DOI] [PubMed] [Google Scholar]
- 42.Collaborative Computational Project, Number 4. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 43.Potterton E, et al. A graphical user interface to the CCP4 program suite. Acta Crystallogr D. 2003;59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- 44.Havranek JJ, Baker D. Motif-directed flexible backbone design of functional interactions. Protein Sci. 2009 doi: 10.1002/pro.142. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.