

Abstract
We identified the first enzymes that use mycothiol and mycoredoxin in a thiol/disulfide redox cascade. The enzymes are two arsenate reductases from Corynebacterium glutamicum (Cg_ArsC1 and Cg_ArsC2), which play a key role in the defense against arsenate. In vivo knockouts showed that the genes for Cg_ArsC1 and Cg_ArsC2 and those of the enzymes of the mycothiol biosynthesis pathway confer arsenate resistance. With steady-state kinetics, arsenite analysis, and theoretical reactivity analysis, we unraveled the catalytic mechanism for the reduction of arsenate to arsenite in C. glutamicum. The active site thiolate in Cg_ArsCs facilitates adduct formation between arsenate and mycothiol. Mycoredoxin, a redox enzyme for which the function was never shown before, reduces the thiol-arseno bond and forms arsenite and a mycothiol-mycoredoxin mixed disulfide. A second molecule of mycothiol recycles mycoredoxin and forms mycothione that, in its turn, is reduced by the NADPH-dependent mycothione reductase. Cg_ArsCs show a low specificity constant of ∼5 m-1 s-1, typically for a thiol/disulfide cascade with nucleophiles on three different molecules. With the in vitro reconstitution of this novel electron transfer pathway, we have paved the way for the study of redox mechanisms in actinobacteria.
The frequent abundance of arsenic in the environment has guided the evolution of enzymes for the reduction of arsenate (As(V))4 (1). Arsenate reductases (ArsCs) are unusual among well studied enzyme classes, because there is not a single family of evolutionarily related sequences. The structural folds and mechanisms that they are using are fundamentally different and arose independently during evolution (2). Arsenate reductases are small cytoplasmic redox enzymes that reduce arsenate to arsenite (As(III)) by the sequential involvement of three different thiolate nucleophiles that function as a redox cascade. As such, arsenate reductases from different organisms often work together with the thiol/disulfide mechanism in the cell.
The major and most ubiquitous system for protection against oxidative stress and to maintain the intracellular thiol homeostasis is the thioredoxin system that is composed of Trx (thioredoxin) and TrxR (thioredoxin reductase) (3). In addition to the thioredoxin system, most living organisms contain low molecular weight thiol compounds that serve as a buffer to avert disulfide stress. In eukaryotes and Gram-negative bacteria, the redox level is maintained by redox cycling of glutathione (GSH) with Grx (glutaredoxin) and glutathione reductase (4). Gram-positive bacteria, like Staphylococcus aureus, produce no glutathione, but millimolar quantities of reduced coenzyme A is the predominant thiol, which is kept reduced with a NADPH-dependent flavoenzyme, coenzyme A disulfide reductase (5). Also actinobacteria, like Corynebacterium glutamicum, produce no GSH, but instead they contain millimolar concentrations of MSH (mycothiol; chemically 1D-myo-inosityl-2-[N-acetyl-l-cysteinyl] amido-2-deoxy-α-d-glucopyranoside), a pseudodisaccharide containing a cysteine moiety as a reactive thiol (6). Its oxidized form is mycothione (MSSM). In actinobacteria, MTR (mycothione reductase) is the NADPH-dependent flavoenzyme that reduces MSSM in order to maintain the intracellular redox homeostasis to allow the proper functioning of a variety of biological functions (7).
Arsenate reductases are part of a defense mechanism of the cell against toxic arsenate. Their genes are most of the time found in an operon together with arsenite sensing and efflux genes (8). Based on the mechanism used to reduce arsenate to arsenite, two distinct ArsC classes can be defined. The first one is the thioredoxin-coupled ArsC class represented by S. aureus pI258 ArsC and Bacillus subtilis ArsC (9–11). Both enzymes use the structural fold of low molecular weight tyrosine phosphatase and need Trx to start a second catalytic cycle (12–14). The second class is the GSH/glutaredoxin-coupled class represented by Escherichia coli plasmid R773 ArsC (15, 16), the eukaryotic Acr2p reductase from Saccharomyces cerevisiae (17), and ArsC from Leishmania major (18). In this second class, two different structural folds are found; E. coli R773 ArsC partially resembles glutaredoxin (19), whereas the eukaryotic ArsCs have a rhodanese fold like the Cdc25a cell cycle control phosphatase (20). Notably, all arsenate reductases have a thiolate nucleophile at the N-terminal end of an α-helix. The active site of the ArsCs with a phosphatase-like scaffold is conserved (root mean square deviation of 0.54 Å) with a catalytically important Arg on position Cys+6.
In C. glutamicum, there are four arsC genes located on different places in the chromosome (21): one orphan arsC gene (arsC4) and three arsC genes (arsC1-arsC1′ and arsC2) present in two ars operons. We show here that two of the encoded proteins, Cg_ArsC1 and Cg_ArsC2 (with 66% sequence identity) are members of a new third class, the mycothiol- and mycoredoxin-dependent arsenate reductases. Both the genes of arsC1 and arsC2, together with the genes for the enzymes of the mycothiol biosynthesis pathway are involved in arsenate resistance in C. glutamicum. We have reconstituted in vitro a novel electron transfer network containing, next to Cg_ArsC1 or Cg_ArsC2, mycothiol, mycoredoxin, and mycothione reductase. As such, the mechanism for the reduction of arsenate by C. glutamicum could be unraveled.
EXPERIMENTAL PROCEDURES
Knockouts Involved in Mycothiol Biosynthesis—MshB, MshC, and MshD mutant strains from C. glutamicum were described previously (22). Mutant strains C. glutamicum MshA and Mtr were kindly supplied by Dr. Kalinowski (Bielefel, Germany). In all of these mutants, the msh/mtr structural genes were removed by the site-specific gene deletion system based on the plasmid pK18 mobsacB (23); recombinant plasmids containing the up and down regions of the msh/mtr genes were mobilized to the recipient strain C. glutamicum RES167 and integrated into a specific site of the chromosome, allowing for marker-free deletion of the target genes when antibiotic pressure and sucrose was adequate.
As(V) and As(III) Resistance Assays—Single colonies of the C. glutamicum strains were inoculated into fresh MMC or TSB and grown for 16 h at 30 °C in aerobic conditions. Midexponential phase cells were diluted 100-fold into fresh, prewarmed low phosphate MMC or TSB containing, respectively, the indicated concentrations of As(V) or As(III) in the form of sodium arsenate or sodium arsenite. Cells were grown at 30 °C in aerobic conditions for 48 h. Growth was monitored (A600) after 12, 24, 36, and 48 h. Cell cultures were diluted to be in the linear OD range, and the obtained A600 values were multiplied with the respective dilution factors.
Arsenate Reductase Activity Assay—Cg_ArsCs were injected on a Superdex75HR column (GE Healthcare) equilibrated in 50 mm Hepes, pH 8.0, 150 mm NaCl to obtain a pure monomeric sample. Mrx1_wt (mycoredoxin), MTR, and MSH were purified as described in the supplemental material. NADPH (Sigma) was dissolved in water to a stock concentration of 10 mm and stored at 4 °C. Arsenate (Na2HAsO4·7H2O; Sigma) was freshly dissolved in water at a concentration of 2 m for making the dilution series.
The final assay mixture was prepared by diluting all components, except Cg_ArsC1 (or Cg_ArsC2) and its substrate arsenate, in the assay buffer solution to obtain the final concentration of 10 μm Mrx1_wt, 3 μm MTR, 0.47 mm MSH, and 250 μm NADPH (component mixture) taking into account the subsequent addition of arsenate and the respective Cg_ArsC enzyme.
The component mixture and different dilution series of substrate were mixed and incubated for 20 min at 37 °C in a 96-well plate (PolySorb, Nunc, Denmark) (180 μl/well) in a SPECTRAmax 340PC (Molecular Devices, Sunnyvale, CA). The assay was started by the addition of enzyme (200 nm) at 20 μl/well to obtain a final volume of 200 μl/well. As a background control for the assay, buffer solution was added.
The arsenate reduction coupled to NADPH oxidation (Δε340 = 6220 m-1 cm-1) was measured by following the decrease in absorption at 340 nm. The path length was measured after each run for each well with the PathCheck Sensor of the system and was used for kcat calculations. Initial rates were calculated with SPECTRAmaxPro (Molecular Devices). Kinetic plots were made with Prism version 4.0 using the Michaelis-Menten expression to calculate Vmax and kcat values. Adding 100 mm K2SO4 and 100 mm sodium phosphate to the assay buffer solution in the component mixture tested the influence of oxyanions on the assay. For the Trx/TrxR pathway, Trx (trxA), Mrx2, and TrxR (trxB) were used at the same concentrations as Mrx1_wt and MTR.
Inhibition and Activation of the Mrx1/MSH Pathway by Arsenite and Arsenate, Respectively—Arsenate and arsenite (each at 1 and 50 mm) were incubated for 20 min at 37 °C with 470 μm MSH, 3 μm MTR, 250 μm NADPH in 50 mm Hepes, pH 8.0. The reaction was started with the addition of 50 mm oxidized Mrx1 and monitored at 340 nm as a function of time. Oxidized Mrx1 was prepared by the addition of a 10-fold molar excess of diamide, followed by size exclusion chromatography on Superdex75 HR in 50 mm Hepes, pH 8.0, 150 mm NaCl.
Arsenite Analysis—All components (or an experimentally designed selection) were mixed in 20 mm Tris/HCl, pH 6.5, to obtain 250 μm NADPH, 3 μm MTR, 10 μm Mrx1, 0.47 mm MSH, 200 nm Cg_ArsCs, and 100 mm As(V) (varying concentrations) incubated for different times at 37 °C. A pH of 6.5 guarantees most arsenate in its dianionic form and arsenite as As(OH)3. The reaction was stopped by removing the proteins on a solid phase extraction cartridge (Waters Oasis HLB). The excess of arsenate was removed on Dowex 21K/XLT anion exchange resin (Supelco) pretreated with NaCl in Tris/HCl, pH 6.5, and thoroughly washed with water. The flow-through fraction was 0.2-μm filtered, argon-flushed, and injected on a Hamilton PRP-X100 anion exchange column (250 × 4.1 mm) operated in 20 mm KH2PO4/K2HPO4, pH 6.0, at 1 ml/min. The high pressure liquid chromatography effluent was mixed with 1.5 m HCl and 2.5% NaBH4, 2% NaOH at 1 ml/min to form gaseous arsine (AsH3). The arsines were analyzed and quantified using an atomic fluorescence spectrometer (Excalibur, PS Analytical, Orpington, UK) calibrated with a standard of arsenite and arsenate.
Softness Difference Calculation—The ArsC-arseno adduct is
modeled as
 and
and
 . The
geometries of MSH, GSH,
. The
geometries of MSH, GSH, 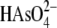 ,
,
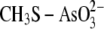 and
and
 were
optimized at the B3LYP/6-31+G** level. Subsequent single point
calculations were performed in a polarized continuum
(24) aqueous solvent model at
the B3LYP/6–31+G** level. All calculations were performed
with the Gaussian package
(25).
were
optimized at the B3LYP/6-31+G** level. Subsequent single point
calculations were performed in a polarized continuum
(24) aqueous solvent model at
the B3LYP/6–31+G** level. All calculations were performed
with the Gaussian package
(25).
The preferred reactivity between the attacking nucleophilic sulfur atom of
MSH and GSH and the accepting electrophilic arsenate or sulfur atom of
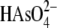 ,
,
 and
and
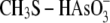 can be
extracted from the difference in the local softness between the reacting
partners (hard and soft acids and bases principle)
(26). The smaller this
difference, the higher the reactivity.
can be
extracted from the difference in the local softness between the reacting
partners (hard and soft acids and bases principle)
(26). The smaller this
difference, the higher the reactivity.
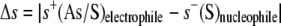 |
(Eq. 1) |
The local softness was calculated as described (27).
RESULTS
Cg_ArsC1 and Cg_ArsC2 Are Involved in the Defense against Arsenate—Resistance analyses of C. glutamicum strains showed a high tolerance of As(V) (28). With single ArsC mutant strains (C. glutamicum ArsC1 or ArsC2), the same resistance levels were obtained as for wild type (Fig. 1A). The double mutant (ArsC1-C2), however, was very sensitive to arsenate and showed resistance levels, which were 20–30 times lower. This double disruption mutant strain was further used as a host for homologous complementation analyses (Fig. 1B). Bifunctional plasmids containing the constitutively expressed arsC1, arsC2, arsC1′, and arsC4 genes from C. glutamicum (pECars derivatives; Table S1) were transferred to this double knock-out strain. Only strains containing either the arsC1 or the arsC2 gene were able to complement the arsenate reductase activity. When the single cysteine in the arsC1 and the arsC2 genes was mutated to a serine, the complementation was lost (Fig. 1B). As such, the single cysteine in Cg_ ArsCs is involved in the reaction mechanism.
FIGURE 1.

The arsC1 and arsC2 genes and the genes of the MSH pathway confer As(V) resistance. In all panels, the cell density (A600 nm) of the culture as a function of increasing concentration of arsenate or arsenite is shown. A, resistance to arsenate of C. glutamicum strains: RES167 (wild type), mutant ArsC1 (arsC1-), mutant ArsC2 (arsC2-), and mutant ArsC1-C2 (arsC1- arsC2-). B, resistance to arsenate of strain ArsC1-C2 after homologous complementation analyses using the genes arsC1 (+pECarsC1), arsC2 (+pECarsC2), arsC1′ (+pECarsC1′), and arsC4 (+pECarsC4). Also, mutant copies of the arsC1 and arsC2 genes with the Cys codon replaced by Ser were used for the complementation analyses (+pECarsC1* and +pECarsC2*). C and D, resistance to arsenate (C) or arsenite (D)of C. glutamicum wild type (RES167) and mutant strains involved in mycothiol biosynthesis or its regeneration (msh/mtr). See Table S1 for details.
We also checked the effect of the absence of Cg_ArsC1 and Cg_ArsC2 on As(III) resistance. Removing arsC1, arsC2, or both genes had no effect on the resistance against As(III), and resistance levels equivalent to those for wild type were obtained (data not shown). As such, the arsC1 and arsC2 genes confer only As(V) resistance.
To test whether these four arsC genes could rescue arsenate reductase activity across different species, we did a heterologous complementation analysis. Cloning of the four respective arsC genes from C. glutamicum (arsC1, -C2, -C1′, and -C4) in a WC3110 E. coli mutant lacking arsenate reductase activity (21, 29) was successful. However, none of the arsC genes increased the survival of the WC3110 strain in arsenate (data not shown). This might indicate that the genes for arsenate reductases in C. glutamicum have evolved to become host-specific enzymes, which depend on the proper cellular environment. It suggests that next to Cg_ArsC1 or Cg_ArsC2, there are C. glutamicum-specific molecules involved in the enzymatic reduction mechanism of arsenate.
Mycothiol Is Involved in Arsenate Defense—Since C. glutamicum produces no glutathione, unlike E. coli, but instead millimolar concentrations of mycothiol, we tested the influence of removing the genes of the biosynthesis pathway of MSH on the arsenate and arsenite resistance of C. glutamicum. In Mycobacterium species, the genes mshA, mshB, mshC, and mshD were found to encode for the enzymes that sequentially catalyze the formation of MSH starting from UDP-N-acetyl-glucosamine and inositol-P (30). Orthologs of these msh genes have been located in the genome data of C. glutamicum (31, 32).
With mutants from C. glutamicum (mshA, mshB, mshC, mshD, and mtr), the arsenate and arsenite resistance was tested. In all mutants, clear differences in resistance to arsenate were observed (Fig. 1C). Some of the msh/mtr mutants have a similar susceptibility to As(V) as observed for the ArsC1-C2 double mutant strain, suggesting a possible relation between MSH and the arsenate reductase activities. For arsenite, this was not the case, and only a slight decrease of the resistance level as compared with wild type was observed (Fig. 1D).
Next to the mycothiol redox system, C. glutamicum has a transcriptional thioredoxin unit consisting of trxB-trxA (thioredoxin reductase and thioredoxin; NCgl2984-NCgl2985) (32). In the case of S. aureus plasmid pI258 ArsC, TrxR and Trx are the redox enzymes responsible for electron transport from NADPH (12, 33). By the construction of trx gene disruption mutants, we checked the possible involvement of trxB and trxA in As(V) resistance in C. glutamicum. Unfortunately, the inactivation of the trx genes seems to be lethal, because no transconjugants were obtained after many attempts, suggesting their importance and essential role for C. glutamicum. Their involvement in the defense mechanism against arsenate could not be ruled out in vivo.
Electron Transfer Pathways in Vitro Reconstructed—In C. glutamicum, we located two hypothetical mycoredoxins: Mrx1 (NCgl0808) and Mrx2 (NCgl2445). The corresponding mrx genes were identified by homology with the E. coli glutaredoxin genes (grx) (16). Mrx2 shows 76% sequence identity (NCBI BLAST program) (34) with NrdH-redoxin from Corynebacterium ammoniagenes for which the structure shows a domain-swapped dimer (35). Mycoredoxin1 (Mrx1) is characterized with the same active site sequence motif as Grx (i.e. CPYC). Grx is a redox enzyme with a high specificity for the tripeptide GSH (36). Based on this knowledge, the in vitro electron transfer functionality of two pathways was tested, the Trx/TrxR pathway and the MSH/Mrx1 pathway (Fig. 2, A and B). Whether Mrx2 could accept electrons from MTR or TrxR was also tested. For this kinetic study, we used only enzymes of C. glutamicum, because it is well known that although the active site cysteines of redox enzymes are essential for protein reduction, the participation of the integral structure in the target recognition process modulates its efficiency in doing so (37). All enzymes of both pathways were recombinantly overexpressed in E. coli and purified to homogeneity (see supplemental material).
FIGURE 2.

Cg_ArsC1/2 thiol/disulfide exchange is linked to the MSH/Mrx1 pathway. A, oxidized Trx (trxA) and Mrx2 as substrates for 0.2 μm TrxR (trxB) were analyzed in progress curves, and the consumption of 500 μm NADPH is monitored at A340 nm. B, different concentrations of oxidized Mrx1 were analyzed in the presence of 10 μm (open symbols) and 100 μm MSH (solid symbols), and the consumption of 500 μm NADPH by 2 μm MTR was measured at 340 nm. C, progress curves with 200 nm Cg_ArsC1 or Cg_ArsC2, 0.47 mm MSH, 10 μm Mrx1, 3 μm MTR, 250 μm NADPH and progress curves with 200 nm Cg_ArsC1 or Cg_ArsC2, 3 μm Trx, 3 μm TrxR, and 500 μm NADPH in the presence of 100 mm As(V) are shown. ox, oxidized; red, reduced.
The redox enzymes Trx, Mrx1, and Mrx2 were oxidized with a 10-fold molar excess of diamide, which was subsequently removed on a size exclusion column. By monitoring the consumption of NADPH as a function of time in progress curves, the electron transfer chains for all three enzymes were tested in both pathways (Fig. 2). Mrx1 was found to be specific for the MSH/Mrx1 pathway (Fig. 2B), whereas the function of Mrx2 and Trx is restricted to accept electrons from TrxR (Fig. 2A). We have reconstituted two thiol/disulfide redox pathways from C. glutamicum and shown for the first time the functionality of two hypothetical mycoredoxins: Mrx1 and Mrx2.
Cg_ArsC1 and Cg_ArsC2 Receive Electrons from the Mycothiol/Mycoredoxin 1 Pathway—The following objective was to investigate whether Cg_ArsCs reduce arsenate to arsenite with electrons coming from the Trx/TrxR-pathway or from the MSH/Mrx1 pathway. Both Cg_ArsC1 and Cg_ArsC2 were recombinantly overexpressed in E. coli (see supplemental material). With progress curves using varying concentrations of enzymes, arsenate, and NADPH, we found that both Cg_ArsC1 and Cg_ArsC2 are only connected with the MSH/Mrx1 pathway (Fig. 2C). No electron transfer was observed with the Trx/TrxR-pathway. Remarkably, the electron transfer was only possible with a MSH concentration of at least 0.1 mm (Fig. 2B). For the in vivo situation, the cellular concentration of MSH was published to be in the millimolar range (6), which might explain this observation.
The Cg_ArsCs Kinetics Are Slow—In order to correctly interpret the kinetic parameters generated in an enzymatic assay with several components, it is of uppermost importance that the enzymes stay active during the course of the enzymatic assay (14). Especially for enzymes that contain oxidation sensitive cysteines and in assays where products are formed that could react with cysteines (like arsenite), extra attention is needed (14). We showed that millimolar concentrations of As(III) are inhibiting, whereas As(V) is activating the MSH/Mrx1 pathway (Fig. 3A). To ensure that product inhibition by As(III) is negligible, initial velocities were measured.
FIGURE 3.

The Cg_ArsC kinetics are slow. A, several concentrations of arsenite and arsenate are tested in the presence of 0.47 mm MSH, 10 μm Mrx1, and 250 μm NADPH. B, progress curves in the presence of 3 μm MTR and 250 μm NADPH and in which all possible combinations of As(V) (100 mm), Cg_ArsC1 (200 nm), MSH (0.47 mm), and Mrx1 (10 μm) were tested, are shown. C and D, Michaelis-Menten curves with 200 nm Cg_ArsC1 (C) and Cg_ArsC2 (D), 10 μm Mrx1, 3 μm MTR, 0.47 mm MSH, and 250 μm NADPH in the presence of varying As(V) concentrations in 50 mm Hepes, pH 8, are shown.
In the assay, the concentration of MSH, Mrx1, and MTR have to be high enough so that their action is not rate-limiting. Otherwise, the progress curves will show a lag phase, and the use of initial rates for calculating kinetic plots will lead to false cooperativity. We varied the concentrations of Cg_ArsC, MSH, Mrx1, and MTR with a constant concentration of 250 μm NADPH. After concentration optimization, the various components in the coupled enzyme assay necessary to yield Michaelis-Menten kinetics (Hill factor of 1) were found to be 200 nm Cg_ArsC, 0.47 mm MSH, 10 μm Mrx1, 3 μm MTR, and 250 μm NADPH. Since arsenate is already inducing nonenzymatic background electron transfer in the MSH/Mrx1 pathway (Fig. 3B), the reaction was started with the addition of Cg_ArsC. Removing the components one by one and testing different combinations resulted in lower initial velocities. As such, all components contribute to the electron transfer pathway.
Thiol/disulfide exchange reactions are pH-dependent. We tested the pH dependence of the reaction by comparing the initial rates in the presence of 10 mm arsenate at various pH values (6.5, 7.0, 7.5, and 8.0). Increasing pH resulted in increasing initial velocities, with the highest value at pH 8.0.
Finally, we measured the kinetic parameters of Cg_ArsC1 (Fig. 3C) and Cg_ArsC2 (Fig. 3D) under the optimized conditions in the presence and absence of the oxyanions phosphate and sulfate (Table 1). Phosphate and sulfate have been shown to stabilize pI258 ArsC from S. aureus and increase its Km and kcat values (14). For Cg_ArsCs, the stabilizing effect is only marginal (for the Selwyn test, see Fig. S2). The Km drops with a factor of 2, and the effect on the kcat is insignificant. All together, Cg_ ArsCs have slow kinetics (kcat of 32 and 17 min-1; see Table 1) with a specificity constant of ∼5 m-1 s-1.
TABLE 1.
Kinetic parameters of Cg_ArsC1 and Cg_ArsC2 under different conditions
| Cg_ArsC | Buffer solution | Km | kcat | kcat/Km | H |
|---|---|---|---|---|---|
| mm | min–1 | m–1 s–1 | |||
| Cg_ArsC1 | 50 mm Hepes, pH 8 | 142 ± 19 | 32 ± 2.3 | 3.8 | 1 |
| 50 mm Hepes, pH 8, 100 mm phosphate | 130 ± 7 | 36 ± 1.3 | 4.6 | 1 | |
| 50 mm Hepes, pH 8, 100 mm sulfate | 46 ± 3 | 14 ± 0.6 | 5.1 | 2.8 | |
| Cg_ArsC2 | 50 mm Hepes, pH 8 | 82 ± 13 | 17 ± 1.2 | 3.4 | 1 |
| 50 mm Hepes, pH 8, 100 mm phosphate | 36 ± 8 | 10 ± 1.1 | 4.6 | 1 | |
| 50 mm Hepes, pH 8, 100 mm sulfate | 38 ± 5 | 7 ± 0.6 | 3 | 1.8 |
Cg_ArsC and Mycoredoxin 1 Show Strict Specificity to Mycothiol, and Mycoredoxin 1 Functions as a Monothiol Oxidoreductase—Mycoredoxin 1 (Mrx1) seems to function in C. glutamicum as a glutaredoxin. Therefore, we tested whether electron transfer is also possible when mycothiol is replaced by glutathione. We analyzed NADPH consumption at 340 nm in the presence of MTR, Mrx1, GSH, Cg_ArsC, and arsenate (Fig. 4A). Progress curves showed no electron transfer in the presence of GSH. The coupled redox cascade reaction is strictly linked to MSH.
FIGURE 4.

For the reduction of arsenate with Cg_ArsC, mycothiol and the N-terminal cysteine of Mrx1 are essential for thiol/disulfide exchange. A, MSH cannot be replaced by GSH. Progress curves are shown for 0.47 mm MSH with and without 200 nm Cg_ArsC1 and for 0.47 mm GSH with and without 200 nm Cg_ArsC1. B, the N-terminal cysteine of Mrx1 is essential. Active site mutants of Mrx1 were compared in progress curves. The concentrations of As(V), Mrx1, MTR, and NADPH are the same as in Fig. 3B.
Glutaredoxins are functioning either with one or two cysteines in the active site (38, 39). In the case of a monothiol, first a mixed disulfide between glutaredoxin and glutathione is formed. By a subsequent thiol-disulfide exchange with reduced glutathione, the enzyme is regenerated. In the case of a dithiol mechanism, a mixed disulfide between the enzyme and the protein substrate is formed. This intermediate is released by a nucleophilic attack by the second cysteine residue in the active site of Grx.
To check whether Mrx1 is using one or two active site cysteines in the reaction mechanism related to Cg_ArsCs, we mutated the first, the second, and both cysteines of the CXXC motif to alanines. Their functionality was tested in progress curves and compared with wild type Mrx1 (Fig. 4B). Mutant Mrx1 CXXA is almost as functional as wild type. Its initial velocity dropped less than 10% (Figs. 3B and 4B). On the other hand, electron transfer was drastically reduced to background levels when Mrx1 AXXC or Mrx1 AXXA was present. As such, Mrx1 is functioning as a monothiol mixed disulfide reductase with an essential N-terminal nucleophilic cysteine.
Cg_ArsC1 Catalyzes the MSH/Mrx1-dependent Reduction of Arsenate to Arsenite—In a comparative study, we analyzed the catalyzed versus the noncatalyzed formation of arsenite as a function of time using the optimized assay conditions (see above) (Fig. 5A). Cg_ArsC1 is clearly decreasing the activation energy toward product formation during the reaction. Nevertheless, noncatalyzed As(III) formation is also observed, and this phenomenon is even more striking during an overnight experiment at 37 °C (Fig. 5C). Cg_ArsCs incubated with arsenate do not produce As(III). At least MSH is needed, but the reaction is more efficient in the presence of MSH and Mrx1. Under the latter conditions, the importance of the N-terminal nucleophilic cysteine of Mrx1 is confirmed.
FIGURE 5.

Cg_ArsC1 catalyzes the MSH/Mrx1-dependent reduction of arsenate to arsenite. A, in 20 mm Tris/HCl, pH 6.5, as buffer solution, 100 mm As(V), 0.47 mm MSH, 10 μm Mrx1, 3 μm MTR, and 250 μm NADPH were incubated without (gray) and with 200 nm Cg_ArsC1 (black) at 37 °C for 30, 60, and 120 min. Relative percentages of the produced As(III) are shown. B, relative percentages of the produced As(III) after 2 h of incubation with varying sample compositions are shown. The same concentrations as in A were used, except when indicated. ArsC2 was used at 200 nm. C, same as in B but after 16 h of incubation.
Both Cg_ArsC1 and Cg_ArsC2 produce about the same amount of As(III) in a 2-h incubation experiment at 37 °C (Fig. 5B). Increasing the concentration of wild type Mrx1 from 10 to 465 μm (equivalent to the concentration of MSH in the reaction) in the absence of MTR and NADPH increases the level of As(III) (Fig. 5B). To produce arsenite, MTR and NADPH are not explicitly needed, and with a reaction mixture of Cg_ArsC, arsenate, MSH, and Mrx1, similar high levels of As(III) are obtained. As such, the functional role of MTR and NADPH is most probably only to recycle the formed mycothione or the mixed disulfide between mycothiol and mycoredoxin.
Cg_ArsC Forms an Arseno Adduct to Facilitate As(V)-SM Formation—To scrutinize the reaction, MSH was tested as neutral and thiolate entity (pKa = 8.3 (40)) for its nucleophilic attack toward arsenate and toward a Cg_ArsC arseno-thiol adduct. We address the question whether MSH is performing a nucleophilic attack toward the sulfur or toward the arsenic atom in the arseno-thiol adduct.
With a pKa2 of 6.9, the majority of arsenate
is present as 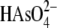 at pH 8. The
protonation state of the Cg_ArsC arseno-thiol adduct is not known but will be
mono- or dianionic; as such, both
at pH 8. The
protonation state of the Cg_ArsC arseno-thiol adduct is not known but will be
mono- or dianionic; as such, both
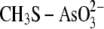 and
and
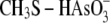 are
considered as simplified models for the enzyme arseno-thiol adduct
(Table 2). The reactivity of
MSH toward
are
considered as simplified models for the enzyme arseno-thiol adduct
(Table 2). The reactivity of
MSH toward 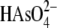 is compared with the
reactivity toward
is compared with the
reactivity toward
 and
and
 . We
found that according to the principle of minimal softness (hard and soft acids
and bases principle), the reactivity to the arsenic atom is larger (lower
difference in softness) than to the sulfur atom in both
. We
found that according to the principle of minimal softness (hard and soft acids
and bases principle), the reactivity to the arsenic atom is larger (lower
difference in softness) than to the sulfur atom in both
 and
and
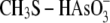 (Table 2). We also observed
that the reactivity of MSH toward the thiol-arseno adduct is higher than
toward arsenate. These data suggest that Cg_ArsCs form an arseno adduct that
facilitates As(V)-SM formation. Similar results were obtained with GSH
(Table 2).
(Table 2). We also observed
that the reactivity of MSH toward the thiol-arseno adduct is higher than
toward arsenate. These data suggest that Cg_ArsCs form an arseno adduct that
facilitates As(V)-SM formation. Similar results were obtained with GSH
(Table 2).
TABLE 2.
Softness difference calculation
|
Electrophiles
|
Nucleophiles
|
||
|---|---|---|---|
| Sulfur atom of GSH Δs | Sulfur atom of MSH Δs | Sulfur atom of MS– Δs | |
| a.u.a | a.u. | a.u. | |
Arsenic in 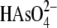
|
1.47 | 1.37 | 1.15 |
Arsenic in
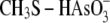
|
0.002 | 0.09 | 2.62 |
Sulfur in

|
1.19 | 1.28 | 3.81 |
Arsenic in
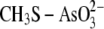
|
0.87 | 0.78 | 1.74 |
Sulfur in
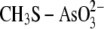
|
1.35 | 1.44 | 3.96 |
a.u., atomic units
DISCUSSION
Most of the cellular arsenate resistance is associated with the presence of cellular arsenate reductases (ArsC/Acr2p). In C. glutamicum, we have found a completely new enzymatic mechanism for the reduction of arsenate in which the electron transfer is coupled to mycothiol (MSH) and mycoredoxin1 (Mrx1) (Fig. 6). No link with the Trx/TrxR-pathway was observed.
FIGURE 6.

The thiol/disulfide-linked reaction mechanism of Cg_ArsC. A, the gas phase-optimized (hf/6–31G level) mycothiol-arseno structure is shown. MSH is shown in a stick representation, and the sulfur (S), arsenic (As) and the As(V)-oxygens are shown in ball representations. The optimized S–As interatomic distance is 2.5 Å. B, Cg_ArsC-catalyzed mycothiol-arseno adduct formation. Mrx1 reduces the thiol-arseno bond and forms As(III) and a mixed mycothiol-mycoredoxin disulfide (Mrx1-S-SM). A second molecule of MSH recycles Mrx1 and forms MSSM that is reduced by the NADPH-dependent MTR.
With MSH biosynthesis pathway mutants (22), we showed a clear link between the production of mycothiol and the level of arsenate resistance. Newton and Fahey (41) showed that MshA and MshC activities are essential for MSH production, whereas MshB and MshD mutants are still producing trace amounts of MSH in Mycobacterium. The arsenate resistance levels observed for C. glutamicum mutants (MshA, MshB, MshC, and MshD) are consistent with their findings. MshA and MshC mutants of C. glutamicum displayed equivalent resistance levels as observed for the double disruption ArsC1-C2 mutant, whereas in the MshB and MshD mutants, the resistance to As(V) was not so dramatically decreased (Fig. 1C). In a C. glutamicum Mtr mutant strain with the MSSM reduction blocked, a constitutive production of MSH maintains the arsenate resistance at a basal level of 3 mm.
Although As(III) is a stronger oxidant and more toxic than As(V), the resistance levels to As(III) for the mutants of the MSH biosynthetic pathway did not decrease to the same extent as observed for As(V) (Fig. 1D). In these mutants, the slight decrease in resistance at higher As(III) concentration might be associated with the lack of the redox buffer capacity in the absence of MSH. In E. coli, a similar gradual decrease of the resistance to As(III) was observed in the absence of GSH (42).
We showed that for the arsenate reduction mechanism, Cg_ArsC1 and Cg_ArsC2 depend not only on the presence of MSH but also on the presence of the N-terminal cysteine in both Cg_ArsC and Mrx1. As such, the reaction proceeds via a thiol/disulfide exchange mechanism. This is also confirmed by the necessity of the presence of MTR and NADPH to reduce MSSM or the MSH-Mrx1 mixed disulfide. In the absence of MTR and NADPH, no electrons are consumed, and the reaction cannot be followed spectrophotometrically at 340 nm. Adding As(V), MTR, and NADPH to MSH and Mrx1, in the absence of Cg_ArsCs, resulted already in a background level reaction (Fig. 3B). Arsenate reacts with MSH and forms arsenite (Fig. 5). For C. glutamicum, the reaction of arsenate with millimolar concentrations of MSH in the cell might even be the first line of defense against arsenate entering the cell via the phosphate uptake system (2). Arsenite induces the ars operon, since the Cg_ArsR repressor can only be released from the ars operator/promoter region with As(III) and not with As(V) (43). A slightly different model has been suggested for Desulfovibrio desulfuricans (44); here, the constitutively expressed orphan arsC gene is producing arsenite to induce the ars operon.
In the absence of Cg_ArsCs, MSH is capable of reducing As(V) to As(III). This reaction strongly suggests the formation of a MS-As(V) adduct (Fig. 6A) that will be reduced by MSH to generate As(III) (Fig. 5) and MSSM. MSH in its thiolate form seems to be the preferred nucleophile for this reaction (Table 2). MSSM will be further reduced by MTR with the consumption of NADPH. The reduction of MS-As(V) is more efficient in the presence of Mrx1 and causes the background level in the kinetic and the arsenite assays. Mrx1 uses its N-terminal nucleophilic cysteine to attack the MS-As(V) adduct with the formation of an Mrx1-S-SM complex. Subsequently, Mrx1-S-SM enters the MSH/MTR-pathway (Fig. 6B). Unfortunately, we do not have experimental evidence for As(V)-SM adduct and the Mrx1-S-SM mixed disulfide complex formation, because these transiently occurring forms are instantly reduced by MSH and/or Mrx1.
It looks as if Cg_ArsCs are not necessary to reduce As(V) to As(III), although its catalytic role was clearly shown in a short time range, where the electron transfer and the production of arsenite is increased in the presence of Cg_ArsCs (Fig. 5A). After the formation of an arseno-sulfur complex in Cg_ArsC, the nucleophilic attack with mycothiol shows regioselectivity for the arsenic atom. As such, an As-SM arseno mycothiol complex is formed (Fig. 6A). We also showed the involvement of Mrx1. Based on all our analyses, a reaction as shown in Fig. 6 will most likely occur: (i) the active site thiolate in Cg_ArsC facilitates adduct formation between arsenate and mycothiol; (ii) Mrx1 reduces the thiol-arseno bond and forms arsenite and a mixed disulfide between mycothiol and Mrx1; (iii) a second molecule of MSH recycles Mrx1 and forms MSSM; (iv) MSSM in its turn is reduced by the NADPH-dependent MTR. At a first glance, the mechanism with the formation arseno-glutathione intermediate looks similar to what has been proposed for E. coli R773 ArsC, which is coupled to GSH and Grx (19, 45). However, in the mechanism suggested for E. coli, a monohydroxy positively charged arsenite intermediate is formed on R773 ArsC, and arsenite is only released from the enzyme after hydroxylation. In C. glutamicum, the mechanism is different; the active site thiolate in Cg_ArsC lowers the energy barrier to facilitate adduct formation between arsenate and mycothiol. Here, arsenite is released after a nucleophilic attack of Mrx1.
Cg_ArsC1 and Cg_ArsC2 are very slow arsenate reductases (Table 1). We compared the kinetic constants of both Cg_ArsCs with other small cytoplasmic arsenate reductases (Table 3). Low specificity constants are also observed for the GSH/Grx-coupled arsenate reductases. Common is that arsenate reductases are using the sequential involvement of three different thiolate nucleophiles that function as a redox cascade. In pI258 ArsC from S. aureus (11), with all three thiolate nucleophiles in a single molecule, a higher catalytic efficiency is obtained; the specificity constant is several orders of magnitude higher. All arsenate reductases produce arsenite with relatively small kcat values. Low kcat values might be explained by the fact that arsenite is more toxic compared with the substrate arsenate (46). To guarantee an immediate efflux of the reactive arsenite, the Acr3p efflux pump (28, 47) has to work in concert with the Cg_ArsCs.
TABLE 3.
Kinetic parameters of arsenate reductases linked with different thiol nucleophiles
| ArsCsa | Nucleophiles | Km | kcat | kcat/Km | H | References |
|---|---|---|---|---|---|---|
| mm | min–1 | m–1 s–1 | ||||
| Cg_ArsC1 | MSH/Mrx | 142 ± 19 | 32 ± 2.3 | 3.8 | 1 | This work |
| Cg_ArsC2 | MSH/Mrx | 82 ± 13 | 17 ± 1.2 | 3.4 | 1 | This work |
| Sc_Acr2p | GSH/Grx | 35 | 6 | 2.8 | 2.7 | Mukhopadhyay et al. (29) |
| Ec_ArsC R773 | GSH/Grx | 15 | 32 | 35 | 1 | Gladysheva et al. (15) |
| Os_Acr2 | GSH/Grx | 12 | 20 | 27 | 1 | Duan et al. (55) |
| Lm_Acr2 | GSH/Grx | 10 | 5.5 × 10–3 | 10–2 | 1 | Zhou et al. (18) |
| Sa_ArsC pI258 | Trx | 68 × 10–3 | 215 | 5.2 × 104 | 1 | Messens et al. (14) |
Cg, C. glutamicum; Sc, S. cerevisiae; Ec, E. coli; Os, Oryza sativa; Lm, L. major; Sa, S. aureus
GSH and MSH are low molecular weight redox buffer components for which evolution might have selected a similar active site in glutaredoxin and mycoredoxin. We showed that Cg_ArsC and Mrx1 are not functioning with glutathione, but they have a strict specificity for mycothiol in the reaction coupled to MTR and NADPH.
Since its discovery in 1993 (6, 48), MSH has been shown to be involved in many processes. Next to its role as a storage form of cysteine in Mycobacterium smegmatis (49) and its detoxification role for alkylating agents (50), there are only two enzymes described that depend on MSH for functioning: formaldehyde dehydrogenase MscR (51), later identified as nitrosomycothiol reductase, with a role in the protection against oxidative stress (52), and maleylpyruvate isomerase (22, 53). We present the first enzymes, arsenate reductase 1 and 2, from C. glutamicum, which depend for their function on mycothiol as well as on mycoredoxin with a clear link to the mycothione reductase pathway. The postulated role for transferring electrons from mycothiol to disulfide substrates has only recently been suggested for an hypothetical mycoredoxin from Streptomyces coelicolor (54). With the thiol/disulfide reaction path used by Cg_ArsC, we have proven that the postulated electron transfer mechanism is a reality.
Supplementary Material
Acknowledgments
We thank John Blanchard, Gerald Newton, and Bob Fahey for sending mycothiol control samples; Jörn Kalinowski for sending the Msh-Mtr mutants; Tomas G. Villa for high scale C. glutamicum culture; Georges Laus and Guy Vandenbussche for mass spectrometry analysis; and Elke Brosens and Khadija Wahni for technical assistance.
This work was supported by the Vlaams Instituut voor Biotechnologie (VIB), the Fonds voor Wetenschappelijk Onderzoek Vlaanderen (FWO), the Onderzoeksraad of the Vrije Universiteit Brussel, and Science and Technology Ministry from Spain Grants BIO2008-00519 and BIO2005-02723.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1 and S2 and Figs. S1 and S2.
Footnotes
The abbreviations used are: As(V), arsenate; As(III), arsenite; MSSM, mycothione.
References
- 1.Messens, J., and Silver, S. (2006) J. Mol. Biol. 362 1-17 [DOI] [PubMed] [Google Scholar]
- 2.Mukhopadhyay, R., Rosen, B. P., Phung, L. T., and Silver, S. (2002) FEMS Microbiol. Rev. 26 311-325 [DOI] [PubMed] [Google Scholar]
- 3.Arner, E. S. J., and Holmgren, A. (2000) Eur. J. Biochem. 267 6102-6109 [DOI] [PubMed] [Google Scholar]
- 4.Fahey, R. C., Brown, W. C., Adams, W. B., and Worsham, M. B. (1978) J. Bacteriol. 133 1126-1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.delCardayre, S. B., and Davies, J. E. (1998) J. Biol. Chem. 273 5752-5757 [DOI] [PubMed] [Google Scholar]
- 6.Newton, G. L., Arnold, K., Price, M. S., Sherrill, C., Delcardayre, S. B., Aharonowitz, Y., Cohen, G., Davies, J., Fahey, R. C., and Davis, C. (1996) J. Bacteriol. 178 1990-1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rawat, M., and Av-Gay, Y. (2007) FEMS Microbiol. Rev. 31 278-292 [DOI] [PubMed] [Google Scholar]
- 8.Silver, S. (1996) Gene (Amst.) 179 9-19 [DOI] [PubMed] [Google Scholar]
- 9.Zegers, I., Martins, J. C., Willem, R., Wyns, L., and Messens, J. (2001) Nat. Struct. Biol. 8 843-847 [DOI] [PubMed] [Google Scholar]
- 10.Bennett, M. S., Guan, Z., Laurberg, M., and Su, X. D. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 13577-13582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Messens, J., Martins, J. C., Van Belle, K., Brosens, E., Desmyter, A., De Gieter, M., Wieruszeski, J. M., Willem, R., Wyns, L., and Zegers, I. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 8506-8511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ji, G., Garber, E. A., Armes, L. G., Chen, C. M., Fuchs, J. A., and Silver, S. (1994) Biochemistry 33 7294-7299 [DOI] [PubMed] [Google Scholar]
- 13.Ji, G., and Silver, S. (1992) Proc. Natl. Acad. Sci. U. S. A. 89 9474-9478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Messens, J., Martins, J. C., Brosens, E., Van Belle, K., Jacobs, D. M., Willem, R., and Wyns, L. (2002) J. Biol. Inorg. Chem. 7 146-156 [DOI] [PubMed] [Google Scholar]
- 15.Gladysheva, T. B., Oden, K. L., and Rosen, B. P. (1994) Biochemistry 33 7288-7293 [DOI] [PubMed] [Google Scholar]
- 16.Shi, J., Vlamis-Gardikas, A., Aslund, F., Holmgren, A., and Rosen, B. P. (1999) J. Biol. Chem. 274 36039-36042 [DOI] [PubMed] [Google Scholar]
- 17.Mukhopadhyay, R., and Rosen, B. P. (1998) FEMS Microbiol. Lett. 168 127-136 [DOI] [PubMed] [Google Scholar]
- 18.Zhou, Y., Messier, N., Ouellette, M., Rosen, B. P., and Mukhopadhyay, R. (2004) J. Biol. Chem. 279 37445-37451 [DOI] [PubMed] [Google Scholar]
- 19.Martin, P., DeMel, S., Shi, J., Gladysheva, T., Gatti, D. L., Rosen, B. P., and Edwards, B. F. (2001) Structure 9 1071-1081 [DOI] [PubMed] [Google Scholar]
- 20.Mukhopadhyay, R., Bisacchi, D., Zhou, Y., Armirotti, A., and Bordo, D. (2009) J. Mol. Biol. 386 1229-1239 [DOI] [PubMed] [Google Scholar]
- 21.Mateos, L. M., Ordóñez, E., Letek, M., and Gil, J. A. (2006) Int. Microbiol. 9 207-215 [PubMed] [Google Scholar]
- 22.Feng, J., Che, Y., Milse, J., Yin, Y. J., Liu, L., Ruckert, C., Shen, X. H., Qi, S. W., Kalinowski, J., and Liu, S. J. (2006) J. Biol. Chem. 281 10778-10785 [DOI] [PubMed] [Google Scholar]
- 23.Schäfer, A., Tauch, A., Jager, W., Kalinowski, J., Thierbach, G., and Pühler, A. (1994) Gene (Amst.) 145 69-73 [DOI] [PubMed] [Google Scholar]
- 24.Mennucci, B., and Tomasi, J. (1997) Chem. Phys. 106 5151-5158 [Google Scholar]
- 25.Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuceria, G. E., Robb, M. A., Cheeseman, J. R., Montgomery, J. A., Vreven, J., T., K. N. K., Burant, J. C., Millam, J. M., Iyengar, S. S., Tomasi, J., Barone, V., Mennucci, B., Cossi, M., Scalmani, G., Rega, N., Petersson, G. A., Nakatsuji, H., Hada, M, Ehara, M., Toyota, k., Fukuda, R., Hasegawa, J., Ishida, M., nakajima, T., Honda, Y., Kitao, O., Nakai, H., Klene, M., Li, X., Knox, J. E., Hratchian, H. P., Cross, J. B., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R. E., Yazyev, O., Austin, A. J., Cammi, R., Pomelli, C., Ochterski, J. W., Ayala, P. Y., Morokuma, K., Voth, G. A., Salvador, P., Dannenberg, J. J., Zakrzewski, V. G., Dapprich, S., Daniels, A. D., Strain, M. C., Farkas, O., Malick, D. K., Rabuck, A. D., Raghavachari, K., Foresman, J. B., Ortiz, J. V., Cui, Q., Baboul, A. G., Clifford, S., Cioslowski, J., Stefanov, B. B., Liu, G., Liashenko, A., Piskorz, P., Komaromi, I., Fox, D. J., Keith, T., Al-Laham, M. A., Peng, C. Y., Nanayakkara, A., Challacombe, M., Gill, P. M. W., Johnson, B., Chen, W., Wong, M. W., Gonzalez, C., and Pople, J. A. (2003) Gaussian 03, Revision A.1, Gaussian, Inc., Pittsburgh, PA
- 26.Pearson, R. G. (1997) Chemical Hardness, Wiley-VCH, Weinheim, Germany
- 27.Roos, G., Loverix, S., Brosens, E., Van Belle, K., Wyns, L., Geerlings, P., and Messens, J. (2006) ChemBioChem 7 981-989 [DOI] [PubMed] [Google Scholar]
- 28.Ordóñez, E., Letek, M., Valbuena, N., Gil, J. A., and Mateos, L. M. (2005) Appl. Environ. Microbiol. 71 6206-6215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mukhopadhyay, R., Shi, J., and Rosen, B. P. (2000) J. Biol. Chem. 275 21149-21157 [DOI] [PubMed] [Google Scholar]
- 30.Newton, G. L., Buchmeier, N., and Fahey, R. C. (2008) Microbiol. Mol. Biol. Rev. 72 471-494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kalinowski, J., Bathe, B., Bartels, D., Bischoff, N., Bott, M., Burkovski, A., Dusch, N., Eggeling, L., Eikmanns, B. J., Gaigalat, L., Goesmann, A., Hartmann, M., Huthmacher, K., Kramer, R., Linke, B., McHardy, A. C., Meyer, F., Mockel, B., Pfefferle, W., Puhler, A., Rey, D. A., Ruckert, C., Rupp, O., Sahm, H., Wendisch, V. F., Wiegrabe, I., and Tauch, A. (2003) J. Biotechnol. 104 5-25 [DOI] [PubMed] [Google Scholar]
- 32.Ikeda, M., and Nakagawa, S. (2003) Appl. Microbiol. Biotechnol. 62 99-109 [DOI] [PubMed] [Google Scholar]
- 33.Messens, J., Hayburn, G., Desmyter, A., Laus, G., and Wyns, L. (1999) Biochemistry 38 16857-16865 [DOI] [PubMed] [Google Scholar]
- 34.Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D. J. (1997) Nucleic Acids Res. 25 3389-3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stehr, M., and Lindqvist, Y. (2004) Proteins 55 613-619 [DOI] [PubMed] [Google Scholar]
- 36.Holmgren, A. (1976) Proc. Natl. Acad. Sci. U. S. A. 73 2275-2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bunik, V., Raddatz, G., Lemaire, S., Meyer, Y., Jacquot, J. P., and Bisswanger, H. (1999) Protein Sci. 8 65-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gravina, S. A., and Mieyal, J. J. (1993) Biochemistry 32 3368-3376 [DOI] [PubMed] [Google Scholar]
- 39.Yang, Y., Jao, S., Nanduri, S., Starke, D. W., Mieyal, J. J., and Qin, J. (1998) Biochemistry 37 17145-17156 [DOI] [PubMed] [Google Scholar]
- 40.Spies, H. S., and Steenkamp, D. J. (1994) Eur. J. Biochem. 224 203-213 [DOI] [PubMed] [Google Scholar]
- 41.Newton, G. L., and Fahey, R. C. (2002) Arch. Microbiol. 178 388-394 [DOI] [PubMed] [Google Scholar]
- 42.Latinwo, L. M., Donald, C., Ikediobi, C., and Silver, S. (1998) Biochem. Biophys. Res. Commun. 242 67-70 [DOI] [PubMed] [Google Scholar]
- 43.Ordóñez, E., Thiyagarajan, S., Cook, J. D., Stemmler, T. L., Gil, J. A., Mateos, L. M., and Rosen, B. P. (2008) J. Biol. Chem. 283 25706-25714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li, X., and Krumholz, L. R. (2007) J. Bacteriol. 189 3705-3711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DeMel, S., Shi, J., Martin, P., Rosen, B. P., and Edwards, B. F. (2004) Protein Sci. 13 2330-2340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Healy, S. M., Wildfang, E., Zakharyan, R. A., and Aposhian, H. V. (1999) Biol. Trace Elem. Res. 68 249-266 [DOI] [PubMed] [Google Scholar]
- 47.Achour, A. R., Bauda, P., and Billard, P. (2007) Res. Microbiol. 158 128-137 [DOI] [PubMed] [Google Scholar]
- 48.Newton, G. L., Fahey, R. C., Cohen, G., and Aharonowitz, Y. (1993) J. Bacteriol. 175 2734-2742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bzymek, K. P., Newton, G. L., Ta, P., and Fahey, R. C. (2007) J. Bacteriol. 189 6796-6805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Newton, G. L., Av-Gay, Y., and Fahey, R. C. (2000) Biochemistry 39 10739-10746 [DOI] [PubMed] [Google Scholar]
- 51.Misset-Smits, M., van Ophem, P. W., Sakuda, S., and Duine, J. A. (1997) FEBS Lett. 409 221-222 [DOI] [PubMed] [Google Scholar]
- 52.Vogt, R. N., Steenkamp, D. J., Zheng, R., and Blanchard, J. S. (2003) Biochem. J. 374 657-666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang, R., Yin, Y. J., Wang, F., Li, M., Feng, J., Zhang, H. M., Zhang, J. P., Liu, S. J., and Chang, W. R. (2007) J. Biol. Chem. 282 16288-16294 [DOI] [PubMed] [Google Scholar]
- 54.den Hengst, C. D., and Buttner, M. J. (2008) Biochim. Biophys. Acta 1780 1201-1216 [DOI] [PubMed] [Google Scholar]
- 55.Duan, G. L., Zhou, Y., Tong, Y. P., Mukhopadhyay, R., Rosen, B. P., and Zhu, Y. G. (2007) New Phytol. 174 311-321 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.