

Abstract
Ectopic delivery of HOXB4 elicits the expansion of engrafting hematopoietic stem cells (HSCs). We hypothesized that inhibition of tumor necrosis factor-α (TNF-α) signaling may be central to the self-renewal signature of HOXB4. Because HSCs derived from Fanconi anemia (FA) knockout mice are hypersensitive to TNF-α, we studied Fancc−/− HSCs to determine the physiologic effects of HOXB4 on TNF-α sensitivity and the relationship of these effects to the engraftment defect of FA HSCs. Overexpression of HOXB4 reversed the in vitro hypersensitivity to TNF-α of Fancc−/− HSCs and progenitors (P) and partially rescued the engraftment defect of these cells. Coexpression of HOXB4 and the correcting FA-C protein resulted in full correction compared with wild-type (WT) HSCs. Ectopic expression of HOXB4 resulted in a reduction in both apoptosis and reactive oxygen species in Fancc−/− but not WT HSC/P. HOXB4 overexpression was also associated with a significant reduction in surface expression of TNF-α receptors on Fancc−/− HSC/P. Finally, enhanced engraftment was seen even when HOXB4 was expressed in a time-limited fashion during in vivo reconstitution. Thus, the HOXB4 engraftment signature may be related to its effects on TNF-α signaling, and this pathway may be a molecular target for timed pharmacologic manipulation of HSC during reconstitution.
Introduction
Hematopoietic stem cell (HSC) division is a tightly regulated process that can result either in the production of committed daughter cells destined to produce mature blood cells or in self-renewal, which results in the generation of at least one daughter cell with the same HSC properties as the parent cell.1 The lifelong maintenance of hematopoiesis relies on balancing these commitment and self-renewal divisions to meet the demands of steady-state homoeostasis versus the response to stress. Further elucidation of the molecular program(s) that lead to HSC self-renewal could have significant impact on the development of cell and gene therapy using HSC targets, as well as deepening our understanding of HSC biology in normal and diseased states.
The homeobox transcription factor HOXB4 remains one of the few known cellular factors that can independently facilitate HSC expansion when expressed ectopically. Retroviral delivery of HOXB4 has been shown to mediate the in vitro and in vivo expansion of murine long-term reconstituting HSC.2–4 This effect of HOXB4 on HSC self-renewal appears to be conserved across several species, including human HSC as assayed by long-term bone marrow (BM) culture and murine xenotransplant assays, although the magnitude of HSC expansion appears to be greatest in the mouse.5–8 Thus, ectopic delivery of HOXB4 apparently skews HSC toward self-renewal divisions and can therefore be used as a tool to dissect the molecular mechanisms critical for HSC expansion.
Using an inducible system to deliver HOXB4 in murine BM enriched for primitive HSC/progenitor (P) cells and genomic transcription profiling, we have recently shown that multiple components of the tumor necrosis factor-α (TNF-α) signaling pathway are suppressed as a consequence of direct transcriptional regulation of TNF-α effectors by HOXB4.9 We thus sought to use a model system that presents a phenotype of HSC hypersensitivity to TNF-α to determine at the mechanistic level the relevance of this observation toward the documented effect of HOXB4 on HSC self-renewal.
Fanconi anemia (FA) is a rare inherited BM failure syndrome characterized on a molecular level by an inability to sense and repair damage by DNA cross-linking agents, such as mitomycin C or diepoxybutane.10,11 FA patients cluster into at least 13 different complementation groups (A-C, D1, D2, E-G, I, J, L-N) with 13 corresponding genes having been cloned to date.12 These proteins are thought to act in an epistatic pathway, which is involved in the cellular response to DNA damage and stress. Hence, inactivation of any of these proteins will disrupt the FA signaling pathway.13 Despite intrapatient variations in disease phenotype, which may be related to complementation group or specific mutations, hematopoietic failure is the almost universal outcome for FA patients14 and FA patients also have a significantly increased risk of myeloid leukemia over their lifetime. Although most genes of the FA pathway have been identified and cloned for some time, the exact mechanism of progressive HSC loss is not understood. However, in addition to increased susceptibility to DNA cross-linking agents, FA hematopoietic cells are also hypersensitive to the action of proinflammatory cytokines, such as interferon-γ (IFNγ) and TNF-α.15,16 Thus, it has been postulated that TNF-α hypersensitivity may comprise the selective pressure that drives HSC failure and the evolution of malignant clones.15,17–19
Several murine models of FA have been developed using gene-targeting methods, with the Fancc−/− mouse being the most extensively characterized with regards to hematopoietic phenotype.20 Fancc−/− BM cells display the characteristic cellular FA phenotype of hypersensitivity to mitomycin C and diepoxybutane, although progressive BM failure is not seen in either this or any of the other FA mouse models.20 Of import, Fancc−/− BM cells demonstrate hypersensitivity to TNF-α, resulting in depletion of HSC/P in vitro.15,17 Fancc−/− BM also demonstrates a profound engraftment defect compared with wild-type (WT) cells when assayed by competitive murine BM repopulation assay21 and are readily depleted by in vitro culture.22 We therefore chose to study the effect of ectopic HOXB4 expression on Fancc−/− HSC/P to further dissect at the mechanistic level the role of TNF-α signaling in the effect of HOXB4 on HSC self-renewal.
Methods
Additional details of methods are available on the Blood website; see the Supplemental Materials link at the top of the online article).
Mice
All animals were maintained in a specific pathogen-free environment, and all experiments were approved by the Institutional Animal Care and Use Committee of Cincinnati Children's Research Foundation. C57B6J (referred to as C57B6 hereafter) mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Fancc−/− mice have been previously described20 and were backcrossed with C57B6 mice for 10 generations.
Generation of retroviral vectors
The SF-FAC-IG retroviral vector used in this study has been described previously (S11FCIEG).23 The SF-IG, SF-IV, SF-HOXB4-IG, SF-HOXB4-IV, and SF-HOXB4-ERT-IG retroviral vectors were based on the SF91 backbone.24 SF-HOXB4-IG and SF-HOXB4-IV contain the human HOXB4 cDNA followed by the encephalomyocarditis virus internal ribosome entry site (IRES) and either enhanced green fluorescent protein (eGFP) or Venus, respectively. SF-IG and SF-IV contain only the IRES-eGFP or IRES-Venus cassettes, respectively. A mutant form of the murine estrogen receptor ligand-binding domain (kindly provided by Dr Tariq Enver, Weatherall Institute of Molecular Medicine, Oxford, United Kingdom)25 was fused to the C-terminus of the HOXB4 cDNA and then subcloned into SF-IG upstream of the IRES-eGFP cassette to yield SF-HOXB4-ERT-IG.
Viral supernatants were generated as previously described26 and were concentrated by ultracentrifugation.27
Isolation and transduction of murine LSK cells
Low-density mononuclear BM cells were depleted of lineage-positive (lin+) cells as described in Document S1. After antibody staining, lin−, Sca-1+, c-Kit+ (LSK) cells were isolated by flow sorting (FACSVantage, BD Biosciences, San Jose, CA).
LSK cells were prestimulated for 48 hours, after which retroviral transduction was performed in nontissue culture–coated plates (BD Biosciences) precoated with fibronectin CH296 fragment (Takara, Otsu, Japan) at 4 μg/cm2. LSK cells were seeded with concentrated retroviral supernatant at a multiplicity of infection (MOI) of 4. After 10- to 12-hour exposure to retroviral vector particles, further concentrated viral supernatant was added to the cells, corresponding to a further MOI of 4. Thirty-six hours later, cells were harvested from the FN CH296-coated plates and live GFP+/Venus+ cells were isolated by flow sorting (FACSVantage). Postsort analysis revealed a purity of more than 97% transduced cells. Transduction frequencies for LSK cells with individual vectors were in the range of 10% to 38%, whereas the transduction frequencies of double-transduced cells were in the range of 5% to 12%. Isolation and transduction of BM from mice pretreated with 150 mg/kg 5-fluorouracil were performed as described.28 Unless otherwise stated, In vitro expansion of HOXB4 transduced BM was performed in Iscove modified Dulbecco medium supplemented with 10% (vol/vol) fetal calf serum, along with 20 ng/mL murine interleukin-3, 100 ng/mL interleukin-6 (both from PeproTech, Rocky Hill, NJ), and 100 ng/mL recombinant rat stem cell factor (rrSCF).
Competitive repopulation assays
The indicated number of transduced and sorted LSK cells were injected into the tail vein of lethally irradiated recipient mice along with 106 freshly isolated whole BM competitor cells. After red cell lysis (Pharm Lyse, BD Biosciences), peripheral blood and BM cells were assessed for fluorescence (FACSCanto, BD Biosciences). For in vivo activation of the HOXB4-ERT fusion construct, tamoxifen citrate was administered via drinking water to mice starting 1 week before transplantation at a concentration of 100 μg/mL.
Colony-forming assays
Colony-forming assays were performed essentially as described.26 Plates were additionally supplemented with murine TNF-α or murine IFN-γ (both PeproTech).
Immunophenotypic analysis of TNF-α receptor expression levels
Sorted transduced LSK cells were expanded for 7 days in Iscove modified Dulbecco medium supplemented with 10% (vol/vol) fetal calf serum, along with 100 ng/mL rrSCF, 100 ng/mL megakaryocyte growth and development factor, and 100 ng/mL granulocyte colony-stimulating factor, at 37°C, 5% CO2. Cells were then stained with antibodies directed against lineage-specific markers, c-Kit, Sca-1, and either Alexa Fluor 647–conjugated hamster anti–mouse CD120a (55R-286) or Alexa Fluor 647–conjugated hamster anti–mouse CD120b (TR75-89; both Serotec, Raleigh, NC), as described in Document S1.
Flow analysis of apoptosis and reactive oxygen species levels
Transduced LSK cells were expanded for 2 days as described in “Immunophenotypic analysis of TNF-α receptor expression levels.” For reactive oxygen species (ROS) analysis, BM was labeled with CM-H2DCFDA (Invitrogen, Carlsbad, CA) for 15 minutes at 37°C according to the manufacturer's instructions, before staining for immunophenotypic markers. For analysis of apoptosis, stained BM cells were subsequently incubated with fluorescein isothiocyanate–conjugated annexin V (BD Biosciences) per the manufacturer's instructions.
In vitro treatment of transduced BM with TNF-α
LSK cells were prestimulated and transduced as described in “Isolation and transduction of murine LSK cells,” with the exception that the described media was substituted with StemSpan serum-free expansion medium (StemCell Technologies, Vancouver, BC) supplemented with 100 ng/mL rrSCF, 100 ng/mL megakaryocyte growth and development factor, and 100 ng/mL granulocyte colony-stimulating factor. At 36 hours after transduction, TNF-α was added to the media at the stated concentration. After a further 24-hour incubation at 37°C, 5% CO2, BM was stained for lineage-specific markers c-Kit and Sca-1 as described previously and analyzed by flow cytometry (FACSCanto).
Analysis of G2/M arrest and FANCD2 mono-ubiquitination in human FA lymphoblast cell lines
The derivation of Epstein-Barr virus–transformed lymphoblast cell lines (LCLs) from FA patient samples and details of their subsequent culture conditions have been previously described.23 LCLs were transduced with RD114 pseudotyped retroviral vector essentially as described,23 after which eGFP+ cells were isolated by flow sorting (FACSVantage). Analysis of cell-cycle arrest in response to treatment with 0.5 μg/mL melphalan (Sigma-Aldrich, St Louis, MO) was performed as described.23 Analysis of FANCD2 mono-ubiquitination in response to treatment with hydroxyurea (Sigma-Aldrich) was performed as described.29
Phosphospecific immunoblots
Lysate derived from transduced 32D cells treated with 20 ng/mL recombinant murine TNF-α or vehicle control was probed by immunoblot using antibodies specific for nuclear factor-κB (NF-κB) p65 phosphorylated at the Ser536 residue (rabbit anti–phospho-NF-κB p65, Ser536, clone 93H1, Cell Signaling Technology, Danvers, MA), SAPK/JNK phosphorylated at Thr183/Tyr185 (mouse anti–phospho-SAPK/JNK, Thr183/Tyr185, clone G9, Cell Signaling Technology), total NF-κB p65 (rabbit polyclonal anti–NF-κB p65, Cell Signaling Technology), total SAPK/JNK (rabbit polyclonal anti-SAPK/JNK, Cell Signaling Technology), or β-actin (mouse anti–β-actin, clone AC-15, Sigma-Aldrich).
Results
Generation of bicistronic retroviral vectors for the cotransduction of primary murine BM cells
The γ-retroviral vectors used in this study are depicted in Figure 1A. The control SF-IG vector, which expresses eGFP alone, and SF-FAC-IG (previous nomenclature S11FCIEG), which coexpresses the human FANCC protein and eGFP, have been previously described.23,26 SF-IG was modified so that eGFP was replaced with the Venus cDNA yielding SF-IV. The SF-HOXB4-IG vector comprises the SF91 backbone24 into which an expression cassette consisting of (5′ to 3′) the human HOXB4 cDNA, the EMCV IRES, and eGFP has been subcloned. SF-HOXB4-IG transduction was associated with the preservation of colony-forming activity in transduced BM after prolonged in vitro culture (Figure 1B), thus demonstrating the expression of functionally relevant levels of HOXB4 in primary cells. Initial studies focused on the effect of HOXB4 expression on cells deficient in the FA pathway in vitro. SF-HOXB4-IG vector transduction was associated with a significant in vitro expansion of both WT C57B6 and Fancc−/− cells (Figure 1C), suggesting that HOXB4 may still promote self-renewal despite a compromised Fanconi pathway.
Figure 1.

Retrovirus vectors and in vitro expansion of transduced Fancc−/− BM cells. (A) Retroviral vectors used in this study. SF indicates enhancer/promoter from the spleen focus forming virus long terminal repeat; FANCC, cDNA encoding the human FA complementation group C protein; HOXB4, cDNA encoding the human homeobox cluster B paralog group 4 protein; IRES, the internal ribososome entry site from encephalomyocarditis virus; eGFP, enhanced green fluorescent protein; Venus, modified version of yellow fluorescent protein; Pre, truncated version of the woodchuck hepatitis virus posttranscriptional regulatory element, which lacks any X protein coding sequence; Ψ, packaging signal. (B) Preservation of colony forming activity after in vitro culture. C57B6 LSK BM cells were transduced twice with either SF-HOXB4-IG or SF-IG at an MOI of 6 and cultured for an additional 14 days. The initial transduction frequency was 48.2% plus or minus 0.6% for SF-HOXB4-IG versus 55.1% plus or minus 1.5% for SF-IG–transduced LSK cells based on fluorescence at day 2 after transduction. BM was then plated out in methylcellulose, as described in “Colony-forming assays” and colonies were scored 7 days later. Data represent the mean of 3 independent experiments plus or minus SEM. □ represents SF-IG; ■, SF-HOXB4-IG. **P < .01 compared with SF-IG–transduced group by Student t test. (C) In vitro expansion of BM cells transduced with SF-HOXB4-IG. LSK BM cells from either C57B6 or Fancc−/− mice were transduced with either SF-HOXB4-IG or SF-IG and were then cultured for an additional 14 days. The percentage of eGFP+ cells in culture was determined at days 4, 9, and 14 after transduction. Data represent the mean of 5 or 6 independent experiments plus or minus SEM. □ represents C57B6 BM; Δ, Fancc−/− BM; —, SF-IG;  , SF-HOXB4-IG. **P < .01 compared with corresponding SF-IG transduced group by Student t test.
, SF-HOXB4-IG. **P < .01 compared with corresponding SF-IG transduced group by Student t test.
To facilitate the detection and isolation of primary murine BM cells that have been cotransduced with 2 different retroviral vectors, the SF-HOXB4-IG vector was modified so that eGFP was replaced with Venus. Analysis of peripheral blood from mice engrafted with BM transduced with eGFP alone, Venus alone, or both eGFP and Venus showed distinct populations of fluorescent cells (Figure S2).
HOXB4 overexpression renders Fancc−/− HSC/P resistant to TNF-α in vitro
HOXB4 overexpression suppresses the transcription of mediators of TNF-α signaling in HSC/P cells.9 Because FA cells have been shown to have increased sensitivity to the effects of proinflammatory cytokines including TNF-α, we sought to determine whether HOXB4 might affect the physiologic responses of FA cells to TNF-α. Fancc−/− and WT LSK cells were transduced with retroviral vectors (Figure 1A) and sorted, and eGFP+ cells were plated in methylcellulose in the presence of increasing doses of TNF-α. As previously demonstrated,21 control-transduced Fancc−/− progenitor cells were found to be profoundly sensitive to concentrations of TNF-α that had little effect on control-transduced WT cells (Figure 2A; P < .003 for Fancc−/− vs WT cells at both 1 ng/mL and 10 ng/mL TNF-α). Restoration of the FA pathway by transduction with SF-FAC-IG partially reversed this hypersensitivity to TNF-α, resulting in a more than 2-fold increase in colony formation compared with SF-IG transduced cells at 10 ng/mL TNF-α (P < .003). However, overexpression of HOXB4 completely abrogated the inhibitory effects of TNF-α treatment on Fancc−/− colony formation, resulting in more than 2-fold and more than 4-fold higher progenitor colony numbers compared with SF-IG–transduced Fancc−/− cells at 1 ng/mL and 10 ng/mL TNF-α, respectively. Thus, the growth of SF-HOXB4-IG–transduced Fancc−/− progenitors at 1 or 10 ng/mL TNF-α was not significantly different from either nontreated cells or from WT BM transduced with SF-IG (Figure 2A). Surprisingly, retroviral-mediated expression of HOXB4 afforded better protection of Fancc−/− BM progenitors to TNF-α than expression of the complementary FA cDNA, resulting in a significant approximately 2-fold higher colony formation at 10 ng/mL TNF-α (P < .003). Conversely, overexpression of HOXB4 did not protect Fancc−/− progenitor cells against the inhibitory effects of the proinflammatory cytokine IFN-γ, thus demonstrating that the HOXB4-mediated rescue of TNF-α treatment is agonist specific (Figure S3). In summary, ectopic delivery of HOXB4 profoundly antagonizes the inhibitory action of TNF-α in Fancc−/− BM. The correction of hypersensitivity of Fancc−/− BM toward TNF-α via HOXB4 overexpression in the absence of the FA complementation occurs despite the presence of a compromised FA core complex pathway, suggesting that TNF-α responsiveness is not a direct or necessary consequence of defective DNA repair via the FA core complex. Of note, other authors have previously demonstrated that certain mutant versions of FANCC result in a cellular phenotype of a defective stress response uncoupled from hypersensitivity to DNA crosslinking agents.30
Figure 2.

Ectopic HOXB4 protects HSC/P from the inhibitory effects of TNF-α. (A) Survival of transduced progenitor cells with increasing concentrations of TNF-α. Murine LSK cells were transduced with the indicated retroviral vectors, and sorted transduced cells were then plated in methylcellulose supplemented with 1 ng/mL, 10 ng/mL, or no (NTX) TNF-α. The frequency of surviving day 7 colonies, expressed as a percentage of nontreated controls, is shown. Data represent the least squares mean estimates plus or minus SEM from mixed-effects model analysis of 3 or 4 independent experiments. □ represents Fancc−/− + SF-IG;  , Fancc−/− + SF-FAC-IG; ■, Fancc−/− + SF-HOXB4-IG;
, Fancc−/− + SF-FAC-IG; ■, Fancc−/− + SF-HOXB4-IG;  , WT + SF-IG. *Significant compared with Fancc−/− + SF-IG group or as indicated by bars after multiple comparison adjustment (P < .003) by Bonferroni method. NS indicates not significant. (B,C) Immunophenotypic analysis of percentage HSC/P in culture after TNF-α treatment. LSK BM from C57B6 or Fancc−/− mice were transduced with either SF-IV or SF-HOXB4-IV. At 36 hours after transduction, cells were treated with the indicated dose of TNF-α as described in “Analysis of G2/M arrest and FAN CD2 mono-ubiquitination in human FA lymphoblast cell lines” and a further 24 hours later BM was stained with antibodies directed against lineage markers, c-Kit and Sca-1. The percentage of BM cells corresponding to either (B) lin− c-Kit+ or (C) lin−, c-Kit+, Sca1+ is shown. Data represent the least squares mean estimates plus or minus SEM from mixed-effects model analysis of 5 or 6 independent experiments. □ represents Fancc−/− + SF-IV; ■, Fancc−/− + SF-HOXB4-IV; , WT + SF-IV; ▩, WT + SF-HOXB4-IV. *Significant compared with nontreated control after multiple comparison adjustment (P < .05) by Tukey method.
, WT + SF-IG. *Significant compared with Fancc−/− + SF-IG group or as indicated by bars after multiple comparison adjustment (P < .003) by Bonferroni method. NS indicates not significant. (B,C) Immunophenotypic analysis of percentage HSC/P in culture after TNF-α treatment. LSK BM from C57B6 or Fancc−/− mice were transduced with either SF-IV or SF-HOXB4-IV. At 36 hours after transduction, cells were treated with the indicated dose of TNF-α as described in “Analysis of G2/M arrest and FAN CD2 mono-ubiquitination in human FA lymphoblast cell lines” and a further 24 hours later BM was stained with antibodies directed against lineage markers, c-Kit and Sca-1. The percentage of BM cells corresponding to either (B) lin− c-Kit+ or (C) lin−, c-Kit+, Sca1+ is shown. Data represent the least squares mean estimates plus or minus SEM from mixed-effects model analysis of 5 or 6 independent experiments. □ represents Fancc−/− + SF-IV; ■, Fancc−/− + SF-HOXB4-IV; , WT + SF-IV; ▩, WT + SF-HOXB4-IV. *Significant compared with nontreated control after multiple comparison adjustment (P < .05) by Tukey method.
The effect of TNF-α treatment in vitro on the prevalence of different immunophenotypically defined immature BM populations was next analyzed. LSK BM cells were isolated from WT or Fancc−/− mice and were then transduced with either the SF-IV control vector or the SF-HOXB4-IV vector. At 36 hours after transduction, BM was treated with increasing concentrations of TNF-α and stained with antibodies directed against lineage markers, c-Kit and Sca-1, after another 24 hours of culture (Figure 2B,C). In agreement with the colony-forming assays described in this section, Fancc−/− BM transduced with SF-IV demonstrated a significant reduction in the progenitor-enriched lin−, c-Kit+ (LK) fraction on treatment with TNF-α (Figure 2B). In contrast, Fancc−/− BM transduced with SF-HOXB4-IV only showed a reduction in the LK fraction at the highest dose of TNF-α. Thus, at each dose of TNF-α, there was a similar frequency of LK cells in the Fancc−/− group transduced with SF-HOXB4-IV and the WT group transduced with SF-IV. The frequency of LK cells was not altered on TNF-α treatment of SF-HOXB4-IV–transduced WT cells, demonstrating that ectopic HOXB4 also protects WT progenitor cells against the deleterious action of TNF-α treatment. Compared with transduced WT BM, there was a reduction in the frequency of more immature Fancc−/− HSC-enriched LSK cells before TNF-α treatment (Figure 2C). Nonetheless, HOXB4 overexpression completely protected Fancc−/− LSK cells against depletion by TNF-α treatment compared with control-transduced Fancc−/− BM. The immature HSC-enriched LSK fraction also showed a significant depletion on treatment of SF-IV control-transduced WT cells (Figure 2C). In summary, whereas primitive Fancc−/− LSK appear to be depleted during the prestimulation/transduction protocol relative to WT cells, overexpression of HOXB4 protects both WT and Fancc−/− HSC/P against the deleterious effects of TNF-α treatment in vitro.
HOXB4 overexpression rescues the Fancc−/− HSC engraftment defect
Because HOXB4 overexpression had a dramatic effect on TNF-α sensitivity in Fancc−/− HSC/P in vitro, we hypothesized that a potential mechanism for the reported effects of HOXB4 on HSC engraftment may involve response to this pathway in vivo. Given that (1) long-term Fancc−/− hematopoiesis has been shown to be sensitive to treatment with low doses of TNF-α in vivo and (2) Fancc−/− cells have a profound engraftment defect, we next sought to determine whether defective engraftment of Fancc−/− HSC could be corrected by ectopic HOXB4 expression. LSK cells were isolated from Fancc−/− mice and transduced separately with SF-IG, SF-FAC-IG, or SF-HOXB4-IV. Transduced cells expressing eGFP+ or Venus+ were isolated by flow sorting. In separate experiments, Fancc−/− LSKs were cotransduced with SF-FAC-IG and SF-HOXB4-IV, and cells expressing both eGFP and Venus were sorted. As a control, LSK cells derived from WT mice were transduced with SF-IG, and eGFP+ cells were isolated by flow sorting. A total of 2 × 104 cells per mouse transduced sorted BM was injected intravenously into lethally irradiated recipient mice along with 1 × 106 freshly isolated WT BM competitor cells (Figure 3A). The percentage of gene-marked peripheral blood leukocytes was assessed at 4, 13, and 26 weeks after transplantation.
Figure 3.

Effect of HOXB4 expression on engraftment, survival, and ROS. (A) Schematic representation of competitive repopulation assay used to assess relative engraftment potential of gene-modified HSC populations. (B) Competitive repopulation of transduced BM as measured by the percentage fluorescent-positive cells in peripheral blood. At the indicated time points after transplantation, the contribution of gene modified cells to the production of peripheral blood leukocytes was analyzed by flow analysis. Percentages: □ represents Fancc−/− + SF-IG; , Fancc−/− + SF-FAC-IG; ■, Fancc−/− + SF-HOXB4-IV; ▨, Fancc−/− + SF-HOXB4-IV + SF-FAC-IG; , WT + SF-IG–transduced cells in the peripheral blood. Data represent the mean plus or minus SEM of 2 independent experiments incorporating 5 to 14 mice per experimental group. *P < .05, **P < .01 compared with Fancc−/− + SF-IG group (Wilcoxon rank sum test). NS indicates not significant. (C,D) Apoptosis and ROS levels in transduced BM. LSK BM from C57B6 or Fancc−/− mice were transduced with either SF-IV or SF-HOXB4-IV. At 2 days after transduction, BM was stained with antibodies directed against lineage markers, c-Kit and Sca-1, in addition to staining with either (C) annexin V or (D) H2DCFDA. Data represent the least squares mean estimates plus or minus SEM from mixed-effects model analysis of 5 or 6 independent experiments. □ represents Fancc−/− + SF-IV; ■, Fancc−/− + SF-HOXB4-IV; , WT + SF-IV; and ▩, WT + SF-HOXB4-IV. *Significant after multiple comparison adjustment (P < .05) by Tukey method.
Uncorrected SF-IG–transduced Fancc−/− HSC demonstrated a significant and severe engraftment defect compared with SF-IG–transduced WT cells. The engraftment defect represented a 3-, 50-, and 80-fold lower engraftment rate at 4, 13, and 26 weeks after transplantation, respectively, indicating a severely compromised primitive repopulating long-term-HSC compartment (Figure 3B). Compared with uncorrected Fancc−/− cells, Fancc−/− LSK cells transduced with SF-FAC-IG resulted in a modest but consistent increase in engraftment (7.3% ± 4.5% vs 0.9% ± 0.8% at 13 weeks after engraftment and 7.8% ± 6.4% vs 0.5% ± 0.2% at 26 weeks after engraftment; SF-FAC-IG vs SF-IG, respectively). Thus, despite the expression of the FANCC cDNA, the engraftment of SF-FANC-IG–transduced Fancc−/− cells was more than 5-fold lower than the engraftment of SF-IG–transduced WT cells at all time points (P < .01, Wilcoxon rank sum test). These data are consistent with previous reports of engraftment defects in Fancc−/− BM cells.21,22,31 Overexpression of HOXB4 led to a significant increase in long-term engraftment of gene-marked Fancc−/− BM cells compared with both SF-IG– and SF-FAC-IG–transduced cells (Figure 3B). This correlated to approximately 36- and 2-fold higher engraftment, respectively, compared with SF-IG– and SF-FAC-IG–transduced Fancc−/− cells at 26 weeks after transplantation. The competitive engraftment advantage conferred by ectopic HOXB4 expression in Fancc−/− cells was also evident using lineage-depleted BM as opposed to LSK cells for transduction (Figure S4). Despite the enhanced reconstitution of Fancc−/− cells resulting from overexpression of HOXB4, transduction with SF-HOXB4-IV did not fully restore engraftment to the level of WT cells (P < .01 and P < .05 vs SF-IG transduced WT cells at 13 and 26 weeks after transplantation, respectively).
In contrast, simultaneous coexpression of HOXB4 and FANCC led to a complete restoration to WT levels in the engraftment of gene-marked Fancc−/− BM at 13 and 26 weeks after transplantation (Figure 3B). Fancc−/− LSK coexpressing HOXB4 and FANCC also demonstrated significantly enhanced engraftment compared with BM transduced with SF-HOXB4-IV at 4 weeks and 26 weeks after transplantation (Figure 3B), with the relative magnitude of engraftment suggesting a synergistic mechanism. These data demonstrate that ectopic delivery of HOXB4 facilitates the engraftment of Fancc−/− long-term-HSC, despite the absence of an intact FA core complex. Restoration of HSC engraftment to WT levels required overexpression of both HOXB4 and the complementary FA cDNA. As previously described,7,32 we observed a perturbation of T-cell differentiation from BM expressing ectopic HOXB4 (Figure S5).
Increased apoptosis resulting from exposure to inhibitory cytokines has been proposed to be a mechanism of HSC/P loss in Fancc−/− BM.15–17,33 To address the mechanistic basis of enhanced engraftment of HOXB4 overexpressing HSC/P, the level of apoptosis in transduced Fancc−/− BM, and WT LSK BM at 2 days after transduction was analyzed by annexin V staining. Although there was no difference between the percentage of annexin V–positive control- and HOXB4-transduced WT LSK cells, control-transduced Fancc−/− LSK demonstrated an almost 2-fold higher rate of apoptosis compared with HOXB4-transduced Fancc−/− LSK (Figure 3C). Because ROS have been proposed to be downstream mediators of the inhibitory action of TNF-α on FA BM and have also been shown to negatively regulate self-renewal in WT HSCs,34–36 we also examined the level of ROS generated in transduced Fancc−/− and WT cells. Fancc−/− and WT LSK BM cells were transduced with either SF-IV or SF-HOXB4-IV, and LSK cells were stained with the fluorescent ROS reporter molecule CM-H2DCFDA at 2 days after transduction. Control-transduced Fancc−/− cells were found to have more than 30% higher ROS levels than all other groups (Figure 3D), whereas Fancc−/− LSK cells transduced with SF-HOXB4-IV had ROS levels similar to those observed in WT LSK cells. Hence, the enhanced engraftment observed after transgenic expression of HOXB4 in Fancc−/− LSK cells correlates with a pronounced reduction in both apoptosis and the level of ROS compared with control-transduced Fancc−/− cells.
HOXB4 overexpression has no effect on the response to DNA damage via FA core complex function
We next sought to determine whether HOXB4 overexpression had any impact on the function of the FA core complex. Human Epstein-Barr virus–transformed LCLs (HSC536-FAC and HSC2911-FAC) derived from FA complementation group C (FA-C) patients were transduced with the retroviral vectors described in Figure 1A, and then treated with the DNA-damaging agent melphalan.23 As expected, transduction of FA-C LCL with the SF-FAC-IG vector resulting in FANCC expression led to a reversal of the melphalan-induced G2/M arrest observed in FA-C cells (Figure 4A; Figure S6). Transduction with the SF-HOXB4-IG vector and expression of HOXB4 did not reverse this melphalan-induced G2/M arrest. Furthermore, SF-HOXB4-IG transduction of LCLs failed to correct the hydroxyurea-induced defect in FANCD2 mono-ubiquitination characteristic of FA cell lines13 (Figure 4B). Taken together, these data demonstrate that ectopic expression of HOXB4 does not directly impact on FA core complex function in response to DNA damage with respect to 2 measures of this function, G2/M arrest and FANCD2 mono-ubiquitination.
Figure 4.

Characterization of the FA cellular phenotype in transduced hematopoietic cells. (A) G2/M arrest of transduced LCLs after treatment with melphalan. The human FANCC−/− lymphoblast cell line HSC536-FAC was transduced with the indicated retroviral vectors. The transduced cells were then treated with melphalan as described in “Methods analysis of G2/M arrest and FANCD2 monoubiquitination in human LCLs.” The proportion of eGFP+ cells in G2/M after melphalan treatment is indicated. NTX indicates nontreated. ▩ represents G0/G1; ▨, S; and ■, G2/M. (B) FANCD2 mono-ubiquitination in transduced LCLs after treatment with hydroxyurea. HSC526-FAC cells were transduced with the indicated retroviral vectors and were then either subject to treatment with hydroxyurea (HU) before cell lysis (+) or were directly lysed without treatment (−). Immunoblot was then performed to visualize the relative abundance of long-form mono-ubiquitinated FANCD2 (L) and short-form nonubiquitinated FANCD2 (S).
Expression of receptors for TNF-α is down-regulated in Fancc−/− LSK cells expressing ectopic HOXB4
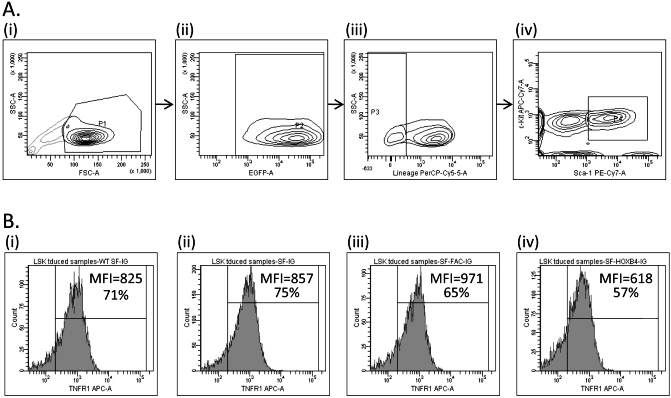
Having demonstrated that HOXB4 overexpression (1) was able to mediate enhanced engraftment of Fancc−/− HSC, (2) abolished the hypersensitivity of HSC/P to TNF-α, and (3) did not restore the cellular response to DNA damage via the FA core complex, the mechanism(s) by which HOXB4 enhanced engraftment and effected resistance to TNF-α were further examined. Fancc−/− and WT LSK cells were transduced with retroviral vectors, and eGFP+ cells were isolated by flow sorting. Cells were cultured for an additional 7 days, and LSK cells were then examined for the expression of the TNF-α receptors, TNFR1 (CD120a/p55) and TNFR2 (CD120b/p75) by flow analysis (Figure S7). Fancc−/− cells transduced with SF-HOXB4-IG had a reduced frequency of TNFR1+ and TNFR2+ LSK cells compared with all other experimental groups (Figure 5A; Figure S7). The level of expression of TNFR1 was also significantly reduced, as determined by the MFI of TNFR1 (Figure 5B; Figure S7). There was also a trend toward reduced MFI of TNFR2, although this difference did not reach significance in these experiments (Figure 5B). Ectopic HOXB4 was also found to decrease expression of both TNFR1 and TNFR2 in transduced WT LSK BM (Figure S8). These data show that expression of HOXB4 significantly down-modulates the expression of TNFR1 and, to a lesser extent, TNFR2, and suggest that the modulation of expression may have physiologic effects on HSC engraftment and reconstitution.
Figure 5.

Determination of TNF-α receptor expression levels in transduced LSK BM by flow. Transduced murine BM cells were isolated by flow sort and were cultured for 7 days. Cells were then stained with antibodies directed against lineage markers, c-Kit, Sca-1, and either TNFR1 or TNFR2. (A) The mean percentage of TNFR1+ or TNFR2+ LSK cells and (B) the mean fluorescent intensity of staining for TNFR1 or TNFR2 within different retroviral transduced populations. Data represent the least squares mean estimates plus or minus SEM from mixed-effects model analysis of 3 or 4 independent experiments. □ represents Fancc−/− + SF-IG; , Fancc−/− + SF-FAC-IG; ■, Fancc−/− + SF-HOXB4-IG; , WT + SF-IG. *Significant after multiple comparison adjustment (P < .05) by Tukey method. (C,D) 32D cells were transduced with either SF-IV or SF-HOXB4-IV, and Venus+ cells were isolated by flow sorting 2 days after transduction and expanded for 4 days in vitro. Expanded cells were harvested and treated either with 20 ng/mL TNF-α (+) or with an equivalent volume of PBS (−). After 5 minutes of incubation at 37°C, cells were harvested and lysed. The lysate from 2 × 105 cells was probed by immunoblot using an antibody specific for (C) NF-κB p65 phosphorylated at the Ser536 residue or (D) SAPK/JNK phosphorylated at the Thr183/Tyr185 residues. Membranes were then sequentially stripped of antibody and reprobed with antibodies against either total NF-κB p65 and then β-actin; or total SAPK/JNK and then β-actin.
TNF-α signaling is compromised in 32D cells, which overexpress HOXB4
Because ectopic expression of HOXB4 also protects against TNF-α–induced apoptosis in the immortalized myeloid progenitor 32D cell line, we chose to further examine the biochemical basis for the inhibition of TNF-α signaling using this model (Figure S9). We examined the phosphorylation of both NF-κB p65 at Ser536 and SAPK/JNK at Thr183/Tyr185 in 32D cells expressing the HOXB4 transgene after TNF-α treatment. Whereas control-transduced 32D cells induced phoshorylation of the NF-κB p65 subunit on treatment with TNF-α, HOXB4-expressing 32D cells had no detectable phosphorylation of NF-κB p65 either in the presence or absence of stimulation by TNF-α (Figure 5C). In addition, there was a pronounced reduction in phosphorylation of SAPK/JNK at both baseline and after TNF-α treatment in HOXB4-transduced 32D cells compared with control-transduced cells (Figure 5D).
Taken together, these data demonstrate that ectopic expression of HOXB4 results in attenuation of TNF-α signaling pathways after ligand binding and elucidate further the mechanism through which HOXB4 may elicit a physiologic effect on HSC expansion and engraftment.
Engraftment of transduced WT BM is enhanced when HOXB4 is induced after transplantation
Because TNF-α and other proinflammatory proteins are elevated after pretransplantation irradiation conditioning of recipient mice,37 the physiologic relevance of decreased TNF-α receptor expression was tested in a murine transplantation model. To regulate HOXB4 expression in a time-dependent fashion, the HOXB4 cDNA was fused to a mutated version of the ligand-binding domain from murine estrogen receptor-α (ERT) to effect the inducible activation of HOXB4 on treatment with 4-hydroxytamoxifen.9 The chimeric HOXB4-ERT cDNA was then inserted into the SF-IG vector to yield SF-HOXB4-ERT-IG (Figure 6A). To test the effect of inducible activation of HOXB4 on HSC engraftment, a competitive repopulation assay (Figure 6B) was performed. Low-density mononuclear BM cells were isolated from WT mice and were transduced with SF-HOXB4-ERT-IG and eGFP+ cells were isolated by flow sorting. A total of 1 × 105/mouse transduced BM cells were then injected into lethally irradiated recipient mice along with 1.1 × 106 freshly isolated competitor cells. Recipient mice either were given tamoxifen citrate via drinking water starting 1 week before transplantation or remained untreated. The percentage of eGFP+ leukocytes in the peripheral blood was analyzed at various time points after transplantation.
Figure 6.

Inducible activation of HOXB4 in vivo. (A) Retroviral vector used in this study. ERT indicates mutated ligand binding domain of murine estrogen receptor-α. (B) Schematic representation of competitive repopulation assay used to assess relative engraftment potential of gene-modified HSC populations in the presence or absence of active HOXB4 as induced by tamoxifen citrate treatment. (C) Competitive repopulation of transduced BM as measured by the percentage of fluorescent-positive cells in peripheral blood. At the indicated time points after transplantation, the contribution of gene modified cells to the production of peripheral blood leukocytes was analyzed by flow analysis. The mean percentage of WT + SF-HOXB4-ERT-IG–transduced cells in the peripheral blood of transplanted recipients is shown plus or minus SEM. ■ represents recipients of 100 μg/mL tamoxifen citrate in drinking water; and □, nonrecipients of tamoxifen citrate. **P < .01, Student t test; n = 5 mice per group.
In recipient mice that did not receive tamoxifen citrate, SF-HOXB4-ERT-IG–transduced cells achieved a stable level of chimerism approximately equal to their input percentage (Figure 6C). However, in tamoxifen citrate-treated mice, the percentage of SF-HOXB4-ERT-IG leukocytes in the periphery was more than 5-fold higher than in untreated mice at weeks 4, 6, and 13 after transplantationation. Thus, a significant selective engraftment advantage can be conferred to transduced murine BM cells by activation of HOXB4 exclusively during in vivo reconstitution.
Discussion
Strategies that facilitate the expansion and enhanced engraftment of repopulating HSCs have long been sought to potentiate therapeutic uses of blood cells. Because HOXB4 has been well characterized as one of the few proteins that, as a single agent, can elicit such properties in ex vivo manipulated HSCs, we sought to further explore at a mechanistic level its role in HSC expansion, to identify potential new therapeutic targets, and to further our understanding of HSC biology.
We have previously defined the TNF-α signaling pathway as a prominent direct transcriptional target of HOXB4 in HSC/P.9 As TNF-α has been shown to regulate cell fate decisions in primitive hematopoietic cells38–40 and, along with other proinflammatory cytokines, has been implicated in the pathophysiology of FA HSC depletion,15–19 we reasoned that the physiologic relevance of HOXB4 effects on this pathway could be best studied in FA HSC. Here, we demonstrate that HOXB4 overexpression completely abrogates the inhibitory effects of TNF-α on Fancc−/− HSC/P in vitro. Furthermore, we also show that ectopic HOXB4 expression partially rescues the engraftment defect of Fancc−/− HSCs. We are also the first to demonstrate that ectopic HOXB4 attenuates apoptosis and ROS production in HSC/P in vitro. Together with concomitant complementation of the relevant FA pathway component, overexpression of HOXB4 restores the function of the HSC compartment to WT levels. This observation has important translational implications because prior depletion of the HSC pool resulting from the FA defect would be predicted to result in quantitative defects in engraftment compared with WT HSCs, even with effective complementation of the FA gene by gene transfer. Indeed, deficiencies of target CD34+ cells in FA patients have recently been noted in an early-phase human gene therapy trial.41 This observation probably also reflects the loss of Fancc−/− HSC/P during the ex vivo manipulation that is required to achieve transduction with a γ-retroviral vector and highlights the need to further optimize transduction conditions to minimize ex vivo manipulations, for example, by using lentiviral vectors that facilitate shortened transduction protocols.22,42
The studies presented here also demonstrate that reversal of Fancc−/− hypersensitivity toward TNF-α by HOXB4 does not require direct correction of the FA core complex pathway. HOXB4 overexpression results in decreased expression of the TNF-α receptors in cell populations enriched for HSC/P. We also report that HOXB4 modulates the response of the NF-κB and JNK pathways to stimulation by TNF-α. This correlates with recent data from Saadatzadeh et al, who showed that JNK mediates apoptosis in Fancc−/− BM progenitor cells after exposure to TNF-α.43 These data suggest that the pharmacologic targeting of either the interaction between TNF-α and its receptors, or signal transduction pathways downstream of TNF-α receptor binding, could be used as a strategy to enhance the engraftment of both Fancc−/− and WT HSCs. Pharmacologic agents to inhibit the TNF pathway, including etanercept and infliximab, are already in clinical use for the treatment of inflammatory diseases, such as rheumatoid arthritis and Crohn disease,44 and the potential use of these agents to delay BM failure in FA has been evaluated in preclinical models18 and is currently being tested in a clinical trial.
Constitutive overexpression of HOXB4 may be undesirable because it has previously been shown to perturb differentiation of both murine and human HSC/P in a dose-specific manner.32,45,46 We also observed inhibition of T-cell differentiation in the context of HOXB4 overexpression in Fancc−/− BM. In addition, although there is no evidence in mice of leukemia induced by overexpression of HOXB4, leukemic transformation in association with insertional proto-oncogene up-regulation after retroviral delivery of HOXB4 has been recently seen in a large-animal BM transplantation setting.47 Therefore, safely harnessing the dramatic HSC phenotype conveyed by HOXB4 may rely on strategies that facilitate the transient delivery of HOXB4 or that target the self-renewal pathways regulated by HOXB4. Indeed, the in vitro delivery of recombinant HOXB4 protein has been demonstrated to elicit a modest pro-engraftment effect on HSC.6,48 However, this approach did not achieve the level of potency observed when HOXB4 is overexpressed via a retroviral vector, probably the result of dosage effects relating to uptake and short half-life of recombinant protein. Taken together, our data and previous studies suggest that overexpression of HOXB4 may be critical during the in vivo reconstitution of conditioned recipients, when HSCs are exposed to elevated TNF-α levels generated in response to radiation-induced tissue damage.37 Indeed, as shown here, the inducible activation of HOXB4 only after infusion of cells and early engraftment results in significantly higher engraftment of HSCs. Therefore, to approach the levels of enhanced engraftment that have been documented for retroviral delivery of HOXB4, it is probable that HOXB4 signaling pathways should be targeted in reconstituting HSCs in vivo. Administration of an anti–TNF-α blockade to BM transplantation recipients may comprise a strategy in which a critical component of HOXB4 signaling can be modulated in vivo.
The characterization of the TNF-α signaling axis as a downstream effector of HOXB4 in HSC/P may explain in part the limited success of transferring this approach to the expansion and engraftment of HSC to humans. In the context of xenograft studies using human CD34+ cells transplanted into immune-compromised mice, human cells have a decreased sensitivity to the inhibitory effects of murine TNF-α, and this may blunt the positive effect of HOXB4 overexpression on engraftment.49–51 Furthermore, the nonmyeloablative conditioning regimens used in mouse xenotransplants may result in reduced production of proinflammatory cytokines, such as TNF-α. In the context of the findings described here, it would seem reasonable to hypothesize that the effect of HOXB4 overexpression may be less significant when transduced cells are transplanted into an environment where the selective pressure mediated by high levels of TNF-α is reduced via reduced intensity conditioning. The data presented here shed new light on a possible mechanism through which HOXB4 may elicit enhanced engraftment of HSCs and suggest that future work should focus on the role of TNF-α signaling downstream of HOXB4 to facilitate improved engraftment of human CD34 cells.
Supplementary Material
Acknowledgments
The authors thank Jose Cancelas, Elke Grassman, Victoria Summey, Chad Harris, Qishen Pang, Paul Andreassen, Ruhikanta Meetei, and Lorna Woolford for technical support and advice, and Megan Smith and Eva Meunier for administrative support.
This manuscript is dedicated to the memory of Leslie J. Fairbairn.
This work was supported by the National Blood Foundation (M.D.M.), Cancer Research United Kingdom (L.J.F.), Deutsche Krebshilfe (B.S.), Deutsche Forschungsgemeinschaft program grants SFB566 and REBIRTH (C.B.), and National Institutes of Health HL081499 (D.A.W.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: M.D.M., B.S., C.B., L.J.F., and D.A.W. performed experimental design and interpretation of data; M.D.M., J.B., A.M.A., and M.K. performed experiments; M.-O.K., D.L., and M.J. performed statistical analysis; and M.D.M. and D.A.W. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Leslie J. Fairbairn died on July 26, 2005.
Correspondence: David A. Williams, Children's Hospital Boston, 300 Longwood Ave, Karp Family Research Laboratories, Boston, MA 02115; e-mail: DAWilliams@childrens.harvard.edu.
References
- 1.May G, Enver T. The lineage commitment and self-renewal of blood stem cells. In: Zon LI, editor. Hematopoiesis a Developmental Approach. Oxford, United Kingdom: Oxford University Press; 2001. pp. 61–81. [Google Scholar]
- 2.Sauvageau G, Thorsteinsdottir U, Eaves CJ, et al. Overexpression of HOXB4 in hematopoietic cells causes the selective expansion of more primitive populations in vitro and in vivo. Genes Dev. 1995;9:1753–1765. doi: 10.1101/gad.9.14.1753. [DOI] [PubMed] [Google Scholar]
- 3.Antonchuk J, Sauvageau G, Humphries RK. HOXB4-induced expansion of adult hematopoietic stem cells ex vivo. Cell. 2002;109:39–45. doi: 10.1016/s0092-8674(02)00697-9. [DOI] [PubMed] [Google Scholar]
- 4.Thorsteinsdottir U, Sauvageau G, Humphries RK. Enhanced in vivo regenerative potential of HOXB4-transduced hematopoietic stem cells with regulation of their pool size. Blood. 1999;94:2605–2612. [PubMed] [Google Scholar]
- 5.Buske C, Feuring-Buske M, Abramovich C, et al. Deregulated expression of HOXB4 enhances the primitive growth activity of human hematopoietic cells. Blood. 2002;100:862–868. doi: 10.1182/blood-2002-01-0220. [DOI] [PubMed] [Google Scholar]
- 6.Amsellem S, Pflumio F, Bardinet D, et al. Ex vivo expansion of human hematopoietic stem cells by direct delivery of the HOXB4 homeoprotein. Nat Med. 2003;9:1423–1427. doi: 10.1038/nm953. [DOI] [PubMed] [Google Scholar]
- 7.Schiedlmeier B, Klump H, Will E, et al. High-level ectopic HOXB4 expression confers a profound in vivo competitive growth advantage on human cord blood CD34+ cells, but impairs lymphomyeloid differentiation. Blood. 2003;101:1759–1768. doi: 10.1182/blood-2002-03-0767. [DOI] [PubMed] [Google Scholar]
- 8.Zhang XB, Beard BC, Beebe K, Storer B, Humphries RK, Kiem HP. Differential effects of HOXB4 on nonhuman primate short- and long-term repopulating cells. PLoS Med. 2006;3:e173. doi: 10.1371/journal.pmed.0030173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schiedlmeier B, Santos AC, Ribeiro A, et al. HOXB4's road map to stem cell expansion. Proc Natl Acad Sci U S A. 2007;104:16952–16957. doi: 10.1073/pnas.0703082104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cervenka J, Arthur D, Yasis C. Mitomycin C test for diagnostic differentiation of idiopathic aplastic anemia and Fanconi anemia. Pediatrics. 1981;67:119–127. [PubMed] [Google Scholar]
- 11.Latt SA, Stetten G, Juergens LA, Buchanan GR, Gerald PS. Induction by alkylating agents of sister chromatid exchanges and chromatid breaks in Fanconi's anemia. Proc Natl Acad Sci U S A. 1975;72:4066–4070. doi: 10.1073/pnas.72.10.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet. 2007;8:735–748. doi: 10.1038/nrg2159. [DOI] [PubMed] [Google Scholar]
- 13.Garcia-Higuera I, Taniguchi T, Ganesan S, et al. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- 14.Kutler DI, Singh B, Satagopan J, et al. A 20-year perspective on the International Fanconi Anemia Registry (IFAR). Blood. 2003;101:1249–1256. doi: 10.1182/blood-2002-07-2170. [DOI] [PubMed] [Google Scholar]
- 15.Haneline LS, Broxmeyer HE, Cooper S, et al. Multiple inhibitory cytokines induce deregulated progenitor growth and apoptosis in hematopoietic cells from Fac−/− mice. Blood. 1998;91:4092–4098. [PubMed] [Google Scholar]
- 16.Rathbun RK, Faulkner GR, Ostroski MH, et al. Inactivation of the Fanconi anemia group C gene augments interferon-gamma-induced apoptotic responses in hematopoietic cells. Blood. 1997;90:974–985. [PubMed] [Google Scholar]
- 17.Otsuki T, Nagakura S, Wang J, Bloom M, Grompe M, Liu JM. Tumor necrosis factor-alpha and CD95 ligation suppress erythropoiesis in Fanconi anemia C gene knockout mice. J Cell Physiol. 1999;179:79–86. doi: 10.1002/(SICI)1097-4652(199904)179:1<79::AID-JCP10>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 18.Dufour C, Corcione A, Svahn J, et al. TNF-alpha and IFN-gamma are overexpressed in the bone marrow of Fanconi anemia patients and TNF-alpha suppresses erythropoiesis in vitro. Blood. 2003;102:2053–2059. doi: 10.1182/blood-2003-01-0114. [DOI] [PubMed] [Google Scholar]
- 19.Li J, Sejas DP, Zhang X, et al. TNF-alpha induces leukemic clonal evolution ex vivo in Fanconi anemia group C murine stem cells. J Clin Invest. 2007;117:3283–3295. doi: 10.1172/JCI31772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen M, Tomkins DJ, Auerbach W, et al. Inactivation of Fac in mice produces inducible chromosomal instability and reduced fertility reminiscent of Fanconi anaemia. Nat Genet. 1996;12:448–451. doi: 10.1038/ng0496-448. [DOI] [PubMed] [Google Scholar]
- 21.Haneline LS, Gobbett TA, Ramani R, et al. Loss of FancC function results in decreased hematopoietic stem cell repopulating ability. Blood. 1999;94:1–8. [PubMed] [Google Scholar]
- 22.Li X, Le Beau MM, Ciccone S, et al. Ex vivo culture of Fancc−/− stem/progenitor cells predisposes cells to undergo apoptosis, and surviving stem/progenitor cells display cytogenetic abnormalities and an increased risk of malignancy. Blood. 2005;105:3465–3471. doi: 10.1182/blood-2004-06-2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chandra S, Levran O, Jurickova I, et al. A rapid method for retrovirus-mediated identification of complementation groups in Fanconi anemia patients. Mol Ther. 2005;12:976–984. doi: 10.1016/j.ymthe.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 24.Hildinger M, Abel KL, Ostertag W, Baum C. Design of 5′ untranslated sequences in retroviral vectors developed for medical use. J Virol. 1999;73:4083–4089. doi: 10.1128/jvi.73.5.4083-4089.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heyworth C, Gale K, Dexter M, May G, Enver T. A GATA-2/estrogen receptor chimera functions as a ligand-dependent negative regulator of self-renewal. Genes Dev. 1999;13:1847–1860. doi: 10.1101/gad.13.14.1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Milsom MD, Jerabek-Willemsen M, Harris CE, et al. Reciprocal relationship between O6-methylguanine-DNA methyltransferase P140K expression level and chemoprotection of hematopoietic stem cells. Cancer Res. 2008;68:6171–6180. doi: 10.1158/0008-5472.CAN-08-0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schambach A, Galla M, Modlich U, et al. Lentiviral vectors pseudotyped with murine ecotropic envelope: increased biosafety and convenience in preclinical research. Exp Hematol. 2006;34:588–592. doi: 10.1016/j.exphem.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 28.Milsom MD, Woolford LB, Margison GP, Humphries RK, Fairbairn LJ. Enhanced in vivo selection of bone marrow cells by retroviral-mediated coexpression of mutant O6-methylguanine-DNA-methytransferase and HOXB4. Mol Ther. 2004;10:862–873. doi: 10.1016/j.ymthe.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 29.Meetei AR, Medhurst AL, Ling C, et al. A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat Genet. 2005;37:958–963. doi: 10.1038/ng1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pang Q, Christianson TA, Keeble W, et al. The Fanconi anemia complementation group C gene product: structural evidence of multifunctionality. Blood. 2001;98:1392–1401. doi: 10.1182/blood.v98.5.1392. [DOI] [PubMed] [Google Scholar]
- 31.Haneline LS, Li X, Ciccone SL, et al. Retroviral-mediated expression of recombinant Fancc enhances the repopulating ability of Fancc−/− hematopoietic stem cells and decreases the risk of clonal evolution. Blood. 2003;101:1299–1307. doi: 10.1182/blood-2002-08-2404. [DOI] [PubMed] [Google Scholar]
- 32.Pilat S, Carotta S, Schiedlmeier B, et al. HOXB4 enforces equivalent fates of ES-cell-derived and adult hematopoietic cells. Proc Natl Acad Sci U S A. 2005;102:12101–12106. doi: 10.1073/pnas.0505624102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pang Q, Keeble W, Christianson TA, Faulkner GR, Bagby GC. FANCC interacts with Hsp70 to protect hematopoietic cells from IFN-gamma/TNF-alpha-mediated cytotoxicity. EMBO J. 2001;20:4478–4489. doi: 10.1093/emboj/20.16.4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sejas DP, Rani R, Qiu Y, et al. Inflammatory reactive oxygen species-mediated hemopoietic suppression in Fancc-deficient mice. J Immunol. 2007;178:5277–5287. doi: 10.4049/jimmunol.178.8.5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ito K, Hirao A, Arai F, et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med. 2006;12:446–451. doi: 10.1038/nm1388. [DOI] [PubMed] [Google Scholar]
- 36.Miyamoto K, Araki KY, Naka K, et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell. 2007;1:101–112. doi: 10.1016/j.stem.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 37.Mori M, Desaintes C. Gene expression in response to ionizing radiation: an overview of molecular features in hematopoietic cells. J Biol Regul Homeost Agents. 2004;18:363–371. [PubMed] [Google Scholar]
- 38.Bryder D, Ramsfjell V, Dybedal I, et al. Self-renewal of multipotent long-term repopulating hematopoietic stem cells is negatively regulated by Fas and tumor necrosis factor receptor activation. J Exp Med. 2001;194:941–952. doi: 10.1084/jem.194.7.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dybedal I, Bryder D, Fossum A, Rusten LS, Jacobsen SE. Tumor necrosis factor (TNF)-mediated activation of the p55 TNF receptor negatively regulates maintenance of cycling reconstituting human hematopoietic stem cells. Blood. 2001;98:1782–1791. doi: 10.1182/blood.v98.6.1782. [DOI] [PubMed] [Google Scholar]
- 40.Drutskaya MS, Ortiz M, Liepinsh DJ, Kuprash DV, Nedospasov SA, Keller JR. Inhibitory effects of tumor necrosis factor on hematopoiesis seen in vitro are translated to increased numbers of both committed and multipotent progenitors in TNF-deficient mice. Exp Hematol. 2005;33:1348–1356. doi: 10.1016/j.exphem.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 41.Kelly PF, Radtke S, Kalle C, et al. Stem cell collection and gene transfer in fanconi anemia. Mol Ther. 2007;15:211–219. doi: 10.1038/sj.mt.6300033. [DOI] [PubMed] [Google Scholar]
- 42.Muller LU, Milsom MD, Kim MO, Schambach A, Schuesler T, Williams DA. Rapid lentiviral transduction preserves the engraftment potential of Fanca(−/−) hematopoietic stem cells. Mol Ther. 2008;16:1154–1160. doi: 10.1038/mt.2008.67. [DOI] [PubMed] [Google Scholar]
- 43.Saadatzadeh MR, Bijangi-Vishehsaraei K, Kapur R, Haneline LS. Distinct roles of stress-activated protein kinases in Fanconi anemia type C deficient hematopoiesis. Blood. 2009;113:2655–2660. doi: 10.1182/blood-2008-09-181420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin J, Ziring D, Desai S, et al. TNFalpha blockade in human diseases: an overview of efficacy and safety. Clin Immunol. 2008;126:13–30. doi: 10.1016/j.clim.2007.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Milsom MD, Duxbury R, Gagen D, et al. Overexpression of HOXB4 confers a myelo-erythroid differentiation delay in vitro. Leukemia. 2005;19:148–153. doi: 10.1038/sj.leu.2403554. [DOI] [PubMed] [Google Scholar]
- 46.Brun AC, Fan X, Bjornsson JM, Humphries RK, Karlsson S. Enforced adenoviral vector-mediated expression of HOXB4 in human umbilical cord blood CD34+ cells promotes myeloid differentiation but not proliferation. Mol Ther. 2003;8:618–628. doi: 10.1016/s1525-0016(03)00237-5. [DOI] [PubMed] [Google Scholar]
- 47.Zhang XB, Beard BC, Trobridge GD, et al. High incidence of leukemia in large animals after stem cell gene therapy with a HOXB4-expressing retroviral vector. J Clin Invest. 2008;118:1502–1510. doi: 10.1172/JCI34371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krosl J, Austin P, Beslu N, Kroon E, Humphries RK, Sauvageau G. In vitro expansion of hematopoietic stem cells by recombinant TAT-HOXB4 protein. Nat Med. 2003;9:1428–1432. doi: 10.1038/nm951. [DOI] [PubMed] [Google Scholar]
- 49.Fransen L, Ruysschaert MR, Van der Heyden J, Fiers W. Recombinant tumor necrosis factor: species specificity for a variety of human and murine transformed cell lines. Cell Immunol. 1986;100:260–267. doi: 10.1016/0008-8749(86)90025-0. [DOI] [PubMed] [Google Scholar]
- 50.Kramer SM, Aggarwal BB, Eessalu TE, et al. Characterization of the in vitro and in vivo species preference of human and murine tumor necrosis factor-alpha. Cancer Res. 1988;48:920–925. [PubMed] [Google Scholar]
- 51.Smith RA, Kirstein M, Fiers W, Baglioni C. Species specificity of human and murine tumor necrosis factor: a comparative study of tumor necrosis factor receptors. J Biol Chem. 1986;261:14871–14874. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}