

Abstract
A proliferation-inducing ligand (APRIL), as well as its receptors transmembrane activator and calcium-modulating cyclophilin ligand (CAML) interactor (TACI) and B-cell maturation antigen (BCMA), has been shown to be important in B-cell biology, and overexpression of APRIL in mice results in development of lymphoma. Limited data are available on APRIL-specific signaling responses, but knockout models suggest that signaling through TACI is critical to B-cell homeostasis. To better understand the mechanism by which APRIL exerts its effects and how it may contribute to lymphomagenesis, we sought to characterize the outcome of APRIL-TACI interactions. In support of murine studies, we find that APRIL induces proliferation of human patient follicular lymphoma (FL) B cells in a TACI-dependent manner. This study also shows that APRIL is expressed within the tumor microenvironment and that, upon engagement with TACI, APRIL mediates activation of the phosphatidylinositol 3-kinase (PI3K) pathway. Activation of PI3K via APRIL results in phosphorylation of Akt and mammalian target of rapamycin (mTOR) and the mTOR-specific substrates p70S6 kinase and 4E-binding protein 1 in a TACI-dependent manner. APRIL-mediated signaling also results in phosphorylation of Rb and up-regulation of cyclin D1. These studies are the first to characterize APRIL-TACI–specific signaling and suggest a role for this ligand-receptor pair in FL B-cell growth.
Introduction
There is accumulating evidence that implicates the tumor necrosis factor (TNF) superfamily members B-cell–activating factor (BAFF; TNFSF13B), also called B lymphocyte stimulator,1,2 and a proliferation-inducing ligand (APRIL; TNFSF13),3 as well as their receptors, as critical factors for the growth and survival of both normal and malignant B cells.4 In vivo studies of APRIL have shown that administration of recombinant soluble APRIL to mice results in increased spleen weight and an increased proportion of B cells in the spleen,5 and transgenic overexpression of APRIL in mice results in B-cell hyperplasia and lymphoma, suggesting a significant role for this protein in malignant B-cell biology.6 Whereas the studies performed to date highlight a role for APRIL in murine models, the significance of this protein on human B-cell biology, normal or malignant, is poorly understood, although initial work suggests that, similar to BAFF, APRIL supports lymphoma B-cell survival.7,8 The downstream signaling cascades activated upon APRIL binding that may be involved in tumor formation are also yet to be characterized.
Two receptors, B-cell maturation antigen (BCMA; TNFRSF17)9 and transmembrane activator and calcium-modulating cyclophilin ligand (CAML) interactor (TACI; TNFRSF13B),10 have been identified as receptors for BAFF and APRIL. APRIL does not bind to BAFF-receptor (BAFF-R; TNFRSF13C), which is the primary receptor for BAFF on mature B cells.11,12 The significance of BCMA in B-cell function remains to be fully elucidated, but there is growing evidence that this receptor is important in normal and malignant plasma cell biology.13 An important role for TACI in B-cell homeostasis has been implicated from TACI-deficient mice that were found to have an accumulation of B cells, splenomegaly, and increased immunoglobulin production.13–16 More recently, 2 groups have recently identified mutations in TACI as an important factor in common variable immunodeficiency and immunoglobulin A deficiency, both of which predispose to development of lymphoma.17,18 TACI has also been shown to regulate B-cell differentiation and class switch recombination.19 In addition to TACI and BCMA, several reports have demonstrated that APRIL binds with low affinity to heparan sulfate proteoglycans (HSPGs) on plasma cells.20,21 The biologic significance of APRIL binding to HSPGs is not yet clear, but a recent study has shown that HSPG is essential for some APRIL-mediated responses,22 and HSPGs have also been shown to directly bind to and activate TACI.23
In previous work, we have shown that TACI is the predominant APRIL receptor on non-Hodgkin lymphoma (NHL) B cells.24 The specific mechanisms that control TACI expression as well as the biologic impact of ligand engagement remain to be fully defined. The study of signal transduction through APRIL-TACI interactions has proven to be challenging due to the complexity of the BAFF/APRIL and TACI/BCMA/BAFF-R receptor-ligand system, and signals from TACI have only been partially described in overexpression models.25 Because both APRIL and TACI have been shown to influence B-cell homeostasis and disease pathogenesis, we sought to better characterize their role in NHL B-cell growth and survival and to characterize APRIL-TACI–mediated signal transduction. In this study, we show for the first time that APRIL, through TACI, promotes proliferation of follicular lymphoma (FL) B cells and cyclin D1 expression through phosphatidylinositol 3-kinase (PI3K)–regulated mammalian target of rapamycin (mTOR) activation.
Methods
Reagents
Recombinant and biotinylated APRIL were provided by ZymoGenetics (Seattle, WA). Briefly, APRIL was generated by overlap polymerase chain reaction of human APRIL with oligonucleotides forming a FLAG-ZymoZipper 12.6 sequence at the amino-terminal side of the heparin-binding region and TNF domain. FLAG-APRIL was transfected into suspension, protein-free, media-adapted Chinese hamster ovary DXB11 cells, and purified using sequential affinity chromatographic techniques.26 Biotinylated anti-BCMA (BAF193), phycoerythrin (PE)–conjugated anti-TACI (FAB1741P), and PE-conjugated anti–Syndecan 1 (FAB2780P) were from R&D Systems (Minneapolis, MN) and were used for fluorescence-activated cell sorter (FACS) analysis. Wortmannin and LY294002 were from Calbiochem (La Jolla, CA). Rapamycin, heparin, and heparinase were from Sigma-Aldrich (St Louis, MO). Antibodies used for Western blot analysis were as follows: phospho-specific antibodies for Akt (9271), p85 (4228), p70S6 kinase (9205), S6 (2211), mTOR (2971), 4E-binding protein 1 (4E-BP1; 2855), and pRb (9307) were from Cell Signaling Technology (Beverly, MA). Total antibodies for Akt (9272), p85 (4292), p70S6 (9202), S6 (2217), 4EBP1 (9452), mTOR (2972), Rb (9309), cyclin D1 (2922), c-Myc (9402), and p27 (2552) were also from Cell Signaling Technology. Antibodies for APRIL-ED2 (2415) and TACI (2395) were from ProSci (Poway, CA). Anti-BCMA (Sc-73 732) was from Santa Cruz Biotechnology (Santa Cruz, CA).
Lymphoma samples and cell lines
Specimens were obtained according to a protocol approved by the institutional review board of Mayo Clinic (Rochester, MN). Lymph node biopsy cells, tonsil cells, and peripheral blood (PB) mononuclear cells were isolated by Ficoll-Hypaque, as previously described,27 from normal donors or FL patients after written informed consent was obtained in accordance with the Declaration of Helsinki. A total of 18 (FL1-FL18) FL specimens were used in the study. B cells were isolated using CD19 microbeads (Miltenyi Biotec, Auburn, CA) from specimens in which the percentage of neoplastic B cells (CD19+, monotypic κ or λ total CD19+) was greater than 96%. The DOHH2, Karpas-422, lymphoma cell lines (LCL), and the bone marrow-derived stromal cell line HS.528 were obtained from the Deutsch Sammlung von Mikroorganismen und Zellkulturen (Braunschweig, Germany) and ATCC (Manassas, VA). The SUDHL-6 (DHL-6) line was a gift from Dr Margaret A. Shipp (Dana-Farber Cancer Institute, Boston, MA).
Flow cytometric analysis
Cells were incubated with PE-conjugated anti-TACI or biotin-conjugated anti-BCMA, followed by streptavidin-PE. APRIL binding was assessed with 2 μg/mL biotinylated APRIL or isotype control, followed by streptavidin-PE. For the heparin experiments, FL cells were pretreated with 20 μg/mL heparin before addition of APRIL. Cells were analyzed by flow cytometry using a FACSCalibur instrument (BD Biosciences, San Jose, CA). Analysis was performed using FlowJo software (TreeStar, Ashland, OR).
Proliferation assay
Cells were cultured in 96-well round-bottom microtiter plates (Costar, Cambridge, MA) at a density of 5 × 104 cells/well with 500 ng/mL APRIL alone or in the presence of 5 μg/mL heparin for 48 hours, and proliferation was assessed by thymidine incorporation, as previously described.29 Statistical analyses were performed using the Student t test.
Immunohistochemistry
Formalin-fixed paraffin-embedded tissue blocks were obtained from Mayo Clinic Tissue Registry. Slides were dried and deparaffinized, and endogenous peroxidase activity was quenched by incubation of sections in 50%/50% solution of 3% hydrogen peroxide and absolute methanol. Antigen retrieval was performed using 1 mM EDTA (ethylenediaminetetraacetic acid), pH 8.0, in steamer for 30 minutes. The slides were placed on a DakoCytomation autostainer with the following autostainer activities: anti-APRIL (APRIL-ED2, ProSci (2415), anti-CD20 (N1502; Dako North America, Carpinteria, CA), anti-CD3 (A0452; DakoCytomation), or anti-CD68 (M0876; Dako North America); MACH horseradish peroxidase (HRP)–polymer kit (Biocare Medical, Walnut Creek, CA); and substrate. The slides were removed from autostainer, rinsed, and counterstained with hematoxylin. Slides were independently reviewed by a hematopathologist.
APRIL enzyme-linked immunosorbent assay
Enzyme-linked immunosorbent assay (ELISA) plates (Nunc Maxisorp; Nalge Nunc International, Rochester, NY) were coated with 2 μg/mL anti-APRIL monoclonal antibody (ZymoGenetics; E9779), and APRIL was detected with biotinylated anti-APRIL (ZymoGenetics; E9618), followed by streptavidin-HRP and tetramethylbenzidine substrate. APRIL levels were calculated from a standard curve generated with recombinant human APRIL (ZymoGenetics) in 20% horse sera.
Western blot analysis
Cells were stimulated with 500 ng/mL APRIL at 37°C for time indicated and lysed with radioimmunoprecipitation assay buffer supplemented with 1 mM phenylmethylsulfonyl fluoride and protease inhibitors (Roche Diagnostic, Indianapolis, IN). Total protein was resolved on 10% to 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membrane. Membranes were blocked and probed with antibodies, as indicated, and subsequently incubated with peroxidase-conjugated secondary antibody. Blots were developed using the Pierce Super Signal (Pierce, Rockford, IL). Densitometric analysis was performed using AlphaImager Software (Alpha Innotech, San Leandro, CA). Integrated density values of phopsphorylated proteins were normalized to total protein for each sample. Unstimulated cells were given a value of 1.0, and the level of phosphorylation in all other samples was normalized to this value.
Small-interfering RNA transfection
A total of 5 μL of 20 μM control or TACI or BCMA-specific small-interfering RNA (siRNA) was complexed with 6 μL of HiPerfFect (QIAGEN, Valencia, CA) in 0.2 mL of OptiMEM for 5 minutes and used for transfection of LCL. After the addition of the lipid-siRNA complexes to the cells, the cultures were incubated for 72 hours. The Akt siRNA kit (Cell Signaling Technology) was used according to manufacturer's instruction, and cultures were incubated for 48 hours.
Cell cycle analysis
Karpas cells were stimulated with APRIL for 24 hours, washed with phosphate-buffered saline, and fixed in ice-cold 70% ethanol overnight at −20°C. Cells were then treated with RNase (50 μg/mL) and subsequently stained with propidium iodide (PI; 100 μg/mL) for 30 minutes at 37°C. Cell cycle was analyzed by flow cytometry using FlowJo software.
Coculture experiments
Irradiated HS.5 cells (104) were cultured in 24-well plates for 4 hours before addition of 105 serum-starved Karpas cells. Karpas and HS.5 cells were cocultured for 48 hours with or without control Fc (5 μg/mL; ZymoGenetics) or TACI-Fc (5 μg/mL; ZymoGenetics). Proliferation was assessed, as described above. For signaling experiments, cocultured cells were harvested and lysed, and Western blot analysis was performed, as described above.
Results
Expression of TACI and BCMA and APRIL binding on FL B cells
We have previously shown that FL B cells, which are derived from germinal center follicular B cells and typically overexpress Bcl-2 driven by the 14:18 chromosomal translocation, express TACI.24 To confirm these data and to determine the APRIL-binding profile, we first assessed the surface expression of the APRIL receptors TACI and BCMA on malignant B cells from patients with FL and the Karpas and DOHH2 LCL, which also carry the 14:18 translocation. CD19+ B cells were isolated from FL patient specimens and were screened for receptor expression: 9 of 9 patients expressed TACI (mean Δ mean fluorescence intensity (MFI) = 8.9, range = 1.8-21.8), and 7 of 9 patients expressed BCMA (mean ΔMFI = 2.9, range = 0.8-6.9), although at a lower level than TACI (Figure 1A). The Karpas cell line expressed TACI, but did not express BCMA, whereas the DOHH2 cell line was found to express little to no TACI and a low level of BCMA (Figure 1A). Similar to our previous studies,29 we found that normal CD19+ B cells express TACI and have minimal expression of BCMA (Figure 1A).
Figure 1.

FL B cells express receptors for APRIL and exhibit APRIL binding. (A) Karpas, DOHH2, FL patient specimens (n = 9), and CD19+ B cells from healthy donors (n = 3) were stained with PE-conjugated anti-TACI (thick line), anti-BCMA (dashed line), or isotype control (solid gray line), and analyzed by flow cytometry. A representative patient histogram is shown. (B) Karpas, DOHH2, FL patient specimens (n = 5), and CD19+ B cells from healthy donors (n = 3) were left untreated or pretreated with heparin and then incubated with biotinylated APRIL and analyzed by flow cytometry: isotype-control (solid gray), APRIL alone (thick line), or APRIL plus heparin (dashed line).
We next examined the ability of FL B cells, the LCL, and normal CD19+ B cells to bind soluble biotinylated APRIL (Figure 1B). B cells isolated from 5 of 5 FL specimens as well as the LCL were found to consistently bind biotinylated APRIL (FL, mean ΔMFI = 3.48, range 1.4-6.1). Similar results were seen in 3 of 3 normal B-cell specimens (mean ΔMFI = 6.6, range = 5.3-8.8). Because APRIL has recently been shown to bind to the surface of tumor cells in a TACI/BCMA-independent manner via HSPGs,20 we next wanted to determine whether APRIL binding to FL B cells was mediated by TACI, BCMA, HSPG, or a combination of these receptors. We therefore used heparin as a competitive inhibitor of APRIL-HSPG interactions30 to determine APRIL-binding specificity (Figure 1B). Expression of the HSPG Syndecan 1 was detected on the LCL, normal B cells, and FL B cells (Figure S1, available on the Blood website; see the Supplemental Materials link at the top of the online article). In the presence of heparin, we observed complete inhibition of APRIL binding to DOHH2 cells, which express little to no TACI or BCMA (Figure 1B). In contrast, minimal inhibition of APRIL binding was seen in the presence of heparin on Karpas cells, FL patient cells, or normal B cells, all of which express TACI and/or BCMA (Figure 1B). Taken together with Figure 1A, these results suggest that TACI is the predominant APRIL receptor on FL cells, FL cells can bind to soluble APRIL, and APRIL binding to CD19+ FL cells is HSPG independent.
APRIL promotes FL B-cell growth
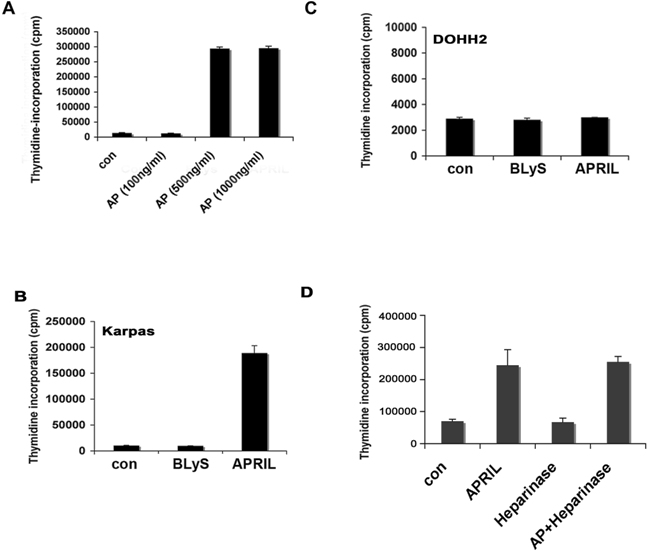
APRIL has been reported to have both prosurvival and growth-enhancing properties, and we next wanted to characterize the biologic impact of APRIL on FL cells. We first tested the ability of recombinant human APRIL (100-1000 ng/mL) to promote proliferation of the LCL and found that compared with the nil control, it significantly enhanced the growth of the Karpas cells (Figures 2A, S2A). BAFF, which has little proliferative capacity, was also tested and was found to have minimal effect on cell proliferation (Figure S2B). The DOHH2 cell line was unresponsive to APRIL (Figure S2C), and this is most likely due to its lack of TACI or BCMA expression. To confirm that the effect of APRIL on cell proliferation was independent of HSPG, we also performed the proliferation assay in the presence of heparin or heparinase and found that APRIL-induced proliferation of Karpas cells was unaffected (Figures 2A and S2D). Heparin or heparinase alone had no effect on cell proliferation. Similar results were seen in CD19+ B cells from FL tissue (data not shown), although the overall level of proliferation was low.
Figure 2.

APRIL stimulation promotes the proliferation of FL B cells. (A) Karpas cells were cultured alone or with APRIL, heparin, or both for 48 hours, and proliferation was assessed. Bar graph shows mean plus or minus SDs (n = 3). (B) Karpas cells were treated with APRIL for 48 hours, and cell-cycle analysis was performed. Results are represented as fold induction (APRIL compared with the nil control) of cells in G1, S, and G2 phase (n = 3) mean (± SDs). (C) Control and TACI siRNA-transfected (100 nM) Karpas cells were treated with APRIL for 48 hours, and proliferation was assessed. (D) Control and TACI siRNA–transfected (100 nM) Karpas cells were stained with PE-conjugated anti-TACI. (E) Expression of TACI in control siRNA (100 nM) and TACI siRNA–transfected (50 nM and 100 nM) Karpas cells was analyzed by Western blot using a TACI-specific antibody, as described in “Methods.” For panel A, *P < .05, APRIL compared with nil control.
To determine whether the effect of APRIL on FL cell proliferation was due in part to its ability to promote survival, we also measured FL cell viability by annexin/PI staining and found that APRIL did not significantly alter survival (data not shown), suggesting that the increase seen in cell proliferation is due to cell cycle progression. We then confirmed the ability of APRIL to promote entry into the cell cycle by performing cell cycle analysis on APRIL-stimulated Karpas cells. In these experiments, APRIL was able to increase the proportion of cells in S phase nearly 2-fold in comparison with untreated cells (n = 3, 1.8-fold increase, P = .08; Figure 2B).
We next wanted to determine whether APRIL-mediated cell proliferation was dependent on TACI. Karpas cells were transfected with a control or TACI-specific siRNA30 to inhibit TACI expression, and then tested for their ability to respond to APRIL in a proliferation assay (Figure 2C). To confirm loss of TACI protein expression, FACS and Western blot analysis was performed, and nearly a complete loss of TACI expression was observed 72 hours posttransfection (Figure 2D,E). Karpas cells expressing the TACI siRNA were unable to respond to APRIL, whereas the control siRNA transfected remained APRIL responsive (Figure 2C). Taken together, these data suggest that APRIL induces cell growth of FL B cells and that APRIL-mediated proliferation is HSPG independent and TACI dependent.
Expression of APRIL in FL tumor microenvironment
APRIL is expressed by many cell types, dendritic cells, macrophages, neutrophils, and tumor cells,3,31–33 many of which are found in the FL tumor microenvironment. To determine whether APRIL is expressed in FL specimens, we first used an immunohistochemistry approach to evaluate the expression of APRIL and found that APRIL was detected in all FL specimens studied (10 of 10). In a representative specimen, APRIL immunoreactivity is shown to be localized within the CD20-positive neoplastic follicles (Figure 3A). The majority of the cells within the follicles are CD20-positive B cells, with scattered, admixed CD3-positive T cells and rare CD68-positive macrophages. Because APRIL is a secreted protein, we next looked for its expression by ELISA in cell culture supernatants from 5 unsorted FL specimens and found that all the samples tested secreted soluble APRIL (Figure 3B). Expression of APRIL by FL cells was similar to that seen in normal tonsil tissue. We then examined APRIL expression by Western blot in the LCL and CD19+ FL cells and found that APRIL was expressed in 3 of 4 primary FL specimens. PB mononuclear cells were used as negative control, and the human HS.5 stromal cell line was used as a positive control (Figure 3C).
Figure 3.

Expression of APRIL in the FL tumor microenvironment. (A) Expression of APRIL, CD20, CD3, and CD68 was determined by immunohistochemistry in FL (n = 10). A representative case of FL, grade 1, is shown. APRIL immunoreactivity is seen within neoplastic follicles ([ ], top left panel) using an APRIL-specific antibody, but not an isotype control. Higher magnification (top right panel) shows the staining surrounding abnormal small lymphocytes within the neoplastic follicles. (B) Cells from normal tonsilar tissue (Ton) or FL patient specimens (n = 5) were cultured for 7 days, and supernatants were collected and assessed for APRIL by ELISA. (C) Expression of APRIL by LCL, PB mononuclear cells, CD19+ FL cells, and HS.5 stromal cells was determined by Western blot using APRIL-specific antibody. (D) Karpas cells were cultured alone or cocultured with irradiated HS.5 cells in the presence of TACI-Fc (5 μg/mL) or control-Fc (5 μg/mL), and growth was assessed by thymidine incorporation. Mean (± SDs) from 6 determinations obtained in 2 separate experiments are shown.
], top left panel) using an APRIL-specific antibody, but not an isotype control. Higher magnification (top right panel) shows the staining surrounding abnormal small lymphocytes within the neoplastic follicles. (B) Cells from normal tonsilar tissue (Ton) or FL patient specimens (n = 5) were cultured for 7 days, and supernatants were collected and assessed for APRIL by ELISA. (C) Expression of APRIL by LCL, PB mononuclear cells, CD19+ FL cells, and HS.5 stromal cells was determined by Western blot using APRIL-specific antibody. (D) Karpas cells were cultured alone or cocultured with irradiated HS.5 cells in the presence of TACI-Fc (5 μg/mL) or control-Fc (5 μg/mL), and growth was assessed by thymidine incorporation. Mean (± SDs) from 6 determinations obtained in 2 separate experiments are shown.
To better understand the significance of APRIL in the context of stromal cell support, we examined the ability of the HS.5 cells, which express APRIL, to support FL cell growth. Karpas cells were cultured alone or with irradiated HS.5 cells in the presence of TACI-Fc, which blocks TACI-APRIL interactions, or a control-Fc protein, and assayed for proliferation. Coculture of Karpas cells with stromal cells triggered Karpas cell growth, which was inhibited by TACI-Fc (Figure 3D). The control-Fc had no effect compared with the nil control. Because TACI-Fc also blocks BAFF, it is possible that the results seen in this experiment are due in part to inhibition of BAFF binding. However, BAFF alone has little effect on cell proliferation (Figure S2B), suggesting that the effects seen by TACI-Fc are APRIL mediated, although BAFF may contribute to cell survival and indirectly affect cell proliferation.29 Taken together, these data suggest that APRIL is present in the FL tumor microenvironment and that APRIL expressed by stromal cells promotes FL growth. In addition, we find that the FL tumor cells themselves may be a source of APRIL.
APRIL-TACI interactions stimulate the PI3K/Akt pathway in FL B cells
Signals from TACI have been partially described in overexpression models, and similar to other TNF receptors (TNFRs), it recruits TNFR-associated factors (TRAFs), which in turn lead to activation of nuclear factor (NF)–κB.25 In our preliminary studies, we examined whether APRIL could promote activation of Iκ-Bα and we observed a weak and transient response (data not shown). We therefore set out to determine whether additional signaling cascades were activated by APRIL-TACI interactions. Because BAFF has recently been shown to activate Akt,34 we first examined the effect of APRIL on PI3K pathway. When we stimulated Karpas cells with APRIL, we found that APRIL stimulation led to phosphorylation of the regulatory subunit of PI3K, p85. Phosphorylation of p85 could be first detected after 1 hour of APRIL stimulation and persisted for up to 5 hours after exposure to APRIL (Figure 4A). Because Akt is a substrate for PI3K, we next examined the ability of APRIL to induce phosphorylation of Akt. Similar to what we had observed with p85, APRIL stimulation led to phosphorylation of Akt in FL cells, whereas expression of total p85 and total Akt remained unchanged throughout the 5-hour period (Figure 4A). As a control, we also examined the ability of APRIL to activate Akt phosphorylation in the DOHH2 cells, which express little to no cell surface TACI (Figure 1). We found that APRIL stimulation had no effect on Akt phosphorylation in DOHH2 cells, with the exception of a minimal amount visible at the 3-hour time point (Figure 4A). Pretreatment of Karpas cells with the PI3K-specific inhibitors wortmannin and Ly294002 inhibited APRIL-induced Akt activity, suggesting that Akt phosphorylation in response to APRIL is PI3K dependent (Figure 4B).
Figure 4.

APRIL stimulates PI3K and Akt activation. (A) Karpas cells were stimulated for the indicated times with APRIL, and phosphorylation of p85 and Akt (Ser473) was analyzed by Western blot. (B) Karpas cells were treated with 100 nM or 500 nM wortmamnin or 10 μM LY294002, and then stimulated with APRIL for 180 minutes and were analyzed for Akt phosphorylation. (C) Karpas cells transfected with control or TACI siRNA (100 nM) were stimulated with APRIL for indicated times and analyzed for Akt phosphorylation. (D) DHL-6 cells were transfected with control, TACI, or BCMA siRNA (100 nM) for 72 hours and subsequently induced with APRIL for the indicated times and analyzed for Akt phosphorylation. (E) Karpas cells were stimulated with APRIL or APRIL and heparin for the indicated time and analyzed for Akt phosphorylation. (F) CD19+ FL B cells were treated with APRIL for the indicated time and analyzed for p85 and Akt phosphorylation. Data from a representative patient are shown (n = 3). Densitometric values are listed below each blot.
To determine whether APRIL-mediated Akt phosphorylation is TACI dependent, Karpas cells were transfected with a control, or TACI siRNA before exposure to APRIL. Control cells increased p-Akt in response to APRIL, whereas p-Akt remained unchanged in the TACI siRNA-transfected cells (Figure 4C). To determine the involvement of BCMA in APRIL-induced Akt signaling, we performed siRNA experiments in a NHL line that expresses both TACI and BCMA. DHL-6 cells were transfected with control, TACI, or BCMA siRNA before exposure to APRIL. Control and BCMA siRNA-transfected cells increased p-Akt in response to APRIL, whereas p-Akt remained unchanged in the TACI siRNA-transfected cells (Figure 4D). Inhibition of TACI and BCMA expression in DHL-6 cells was confirmed by FACS and Western blot analysis (Figure S3A,B). In addition, heparin treatment did not influence APRIL-induced Akt activation (Figure 4E).
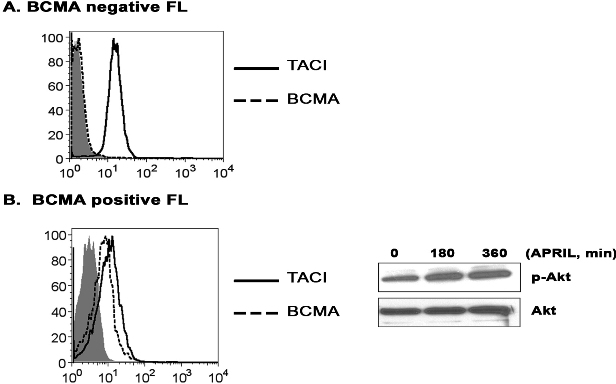
To confirm that our finding in the Karpas cell line was consistent with that seen in patient specimens, CD19+ B cells from FL tissue and examined APRIL mediated Akt phosphorylation. Similar to the Karpas cells, stimulation of patient FL cells results in phosphorylation of both p85 and Akt, with similar kinetics to that seen in the FL line (Figure 4F). APRIL-dependent phosphorylation of Akt occurred in both BCMA-negative (Figures 4F, S4A) and BCMA-positive FL cells (Figure S4B). In summary, these results suggest that APRIL induces phosphorylation of p85 and Akt in FL cells and that these effects are TACI dependent.
APRIL-TACI–mediated activation of mTOR in FL B cells
A major downstream effector of Akt is the mTOR, a highly conserved serine/threonine kinase and a major regulator of cell growth and proliferation.35 Therefore, we next evaluated the effect of APRIL on mTOR activity in Karpas by measuring the phosphorylation profile of mTOR itself, as well as its known substrates p70S6 kinase and 4E-BP1, and the p70S6 kinase substrate S6, using antibodies directed against mTOR-specific phosphorylation sites for each protein. We found that stimulation of Karpas led to phosphorylation of mTOR and 4E-BP1, p70S6 kinase, and S6, with the kinetics similar to APRIL-induced PI3K/Akt pathway (Figure 5A).
Figure 5.

APRIL induces phosphorylation of mTOR and is substrates. (A) Karpas cells were stimulated for the indicated times with APRIL and analyzed for phosphorylation of mTOR, 4E-BP1, p70S6 kinase, and S6. (B) Karpas cells were cultured alone or cocultured with irradiated HS.5 cells in the presence of TACI-Fc (5 μg/mL) or control-Fc (5 μg/mL) and activation of S6 and 4EBP1 by Western blot. Data from a representative experiment are shown (n = 3). (C) Karpas cells were treated with the indicated doses of rapamycin and then stimulated with APRIL for 180 minutes, and were analyzed for mTOR phosphorylation. (D) Karpas cells were transfected with control or Akt siRNA, and then were treated with APRIL for indicated times and analyzed for 4E-BP1 phosphorylation. Expression of Akt and β-actin is shown as controls. (E) Karpas cells were treated with 100 nM or 500 nM wortmamnin or 10 μM LY294002, and then stimulated with APRIL for 180 minutes, and were analyzed for 4E-BP1 phosphorylation. (F) CD19+ FL B cells were treated with APRIL for the indicated time and analyzed for mTOR, 4E-BP1, p70S6 kinase, and S6 phosphorylation. Data from a representative patient are shown (n = 3). (G) DHL-6 cells were transfected with control, TACI, or BCMA siRNA (100 nM) for 72 hours, and subsequently induced with APRIL for the indicated times and analyzed for S6 phosphorylation. Densitometric values are listed below each blot.
We then examined the ability of stromal cell–derived APRIL to induce activation of the mTOR pathway by measuring phosphorylation of 4E-BP1 and S6 after coculture of Karpas cells with irradiated HS5 stromal cells. This coculture resulted in an increase in S6 and 4EBP1 phosphorylation in the Karpas cells, which could be inhibited by TACI-Fc (Figure 5B). Addition of the control-Fc protein to the coculture had no effect on S6 and 4EBP1 phosphorylation. Together, these results suggest that stromal cell–derived APRIL triggers activation of the mTOR pathway.
Pretreatment of Karpas cells with the mTOR-specific inhibitor rapamycin inhibited APRIL-induced mTOR phosphorylation in a dose-dependent manner (Figure 5C), and we next determined whether APRIL-induced regulation of the mTOR pathway was Akt dependent using both a siRNA approach and pharmacologic inhibitors. Karpas cells were transfected with a control or Akt-specific siRNA before exposure to APRIL. Control siRNA-transfected cells increased mTOR pathway activation (p4E-BP1) in response to APRIL in a dose-dependent manner (Figure 5D). Treatment of APRIL-stimulated cells with PI3K inhibitors also prevented increased phosphorylation of 4E-BP1 (Figure 5E).
We next isolated CD19+ B cells from FL tissue and examined APRIL-mediated mTOR pathway activation. Similar to the Karpas cells, stimulation of patient FL cells results in phosphorylation of mTOR and its targets 4E-BP1, p70S6 kinase, and S6, with similar kinetics to that seen in the FL line (Figure 5F). Together these results suggest that APRIL induces activation of mTOR and its substrates in FL cells, and that APRIL-induced mTOR activity is directly regulated through the PI3K/Akt pathway.
To determine the involvement of BCMA in APRIL-induced mTOR signaling, we performed TACI and BCMA siRNA experiments, as described above. We found that control and BCMA siRNA-transfected cells increased phosphorylation of S6 in response to APRIL, whereas phosphorylation of Akt remained unchanged in the TACI siRNA-transfected cells (Figure 5G). In summary, these results suggest that APRIL induces activation of the mTOR pathway in FL cells and that these effects are TACI dependent.
APRIL-TACI–mediated regulation of cell-cycle proteins
We next investigated the effects of APRIL on the status of cell-cycle regulatory proteins active in the G1-S phase. Stimulation of Karpas cells with APRIL resulted in up-regulation of cyclin D1 expression in a time-dependent manner, whereas little change was observed in p27 or c-Myc expression. We also examined the effect of APRIL on phosphorylation of Rb and found that the level of phosphorylated Rb was increased after stimulation with APRIL (Figure 6A).
Figure 6.

APRIL induces expression of cyclin D1 and phosphorylation of Rb. (A) Karpas cells were stimulated for the indicated times with APRIL and analyzed for expression of cyclin D1, p27, c-myc, and phosphorylation of Rb. (B) Karpas cells transfected with control or TACI siRNA were treated with APRIL for indicated times and analyzed for cyclin D1 expression. (C) Karpas cells were treated with the indicated doses of rapamycin, and then stimulated with APRIL for 180 minutes, and analyzed for cyclin D1 expression and Rb phosphorylation. (D) CD19+ FL B cells were treated with APRIL for the indicated times and analyzed for expression of cyclin D1, p27, c-myc, and phosphorylation of Rb. Data from a representative patient are shown (n = 3). Densitometric values are listed below each blot.
To determine whether APRIL-mediated cyclin D1 expression was both TACI and mTOR dependent, Karpas cells were transfected with a control or TACI-specific siRNA before exposure to APRIL. Control siRNA-transfected cells up-regulated cyclin D1 in response to APRIL in a time-dependent manner, whereas cyclin D1 remained unchanged in the TACI siRNA-transfected cells (Figure 6B). In addition, APRIL-stimulated Karpas cells were treated with the mTOR inhibitor rapamycin and, as expected, we found that mTOR inhibition prevented APRIL-induced cyclin D1 expression and phosphorylation of Rb (Figure 6C).
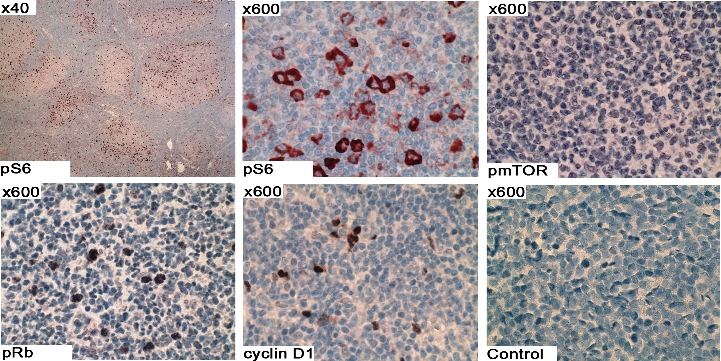
Up-regulation of cell-cycle regulatory proteins by APRIL was next examined in CD19+ patient FL cells (Figure 6D). Similar to the Karpas cells, stimulation of patient FL cells results in up-regulation of cyclin D1 and Rb phosphorylation with similar kinetics to that seen in Karpas cells. The level of c-Myc and p27 remained unchanged in response to APRIL. Taken together, these observations suggest that APRIL-TACI interactions mediate expression of cyclin D1 and phosphorylation of Rb in FL B cells. To further confirm activation of the mTOR pathway in FL patient tissue specimens (n = 10) were stained for p-S6, p-mTOR, pRb, and cyclin D1, and all were found to be expressed (Figure S5).
APRIL-mediated proliferation is PI3K/Akt dependent
We next wanted to determine whether APRIL-mediated proliferation was PI3K and Akt dependent. Pretreatment of Karpas cells with the PI3K-specific inhibitors wortmannin or Ly294002 completely inhibited APRIL-mediated FL cell proliferation (Figure 7A). In addition, we also found that transfection of Karpas cells with Akt-specific siRNA inhibited APRIL-mediated cell proliferation compared with the scramble siRNA control (Figure 7B). Pretreatment of Karpas cells with the mTOR-specific inhibitor rapamycin partially inhibited APRIL-induced proliferation of FL B cells (Figure 7C). These data suggest APRIL-induced cell proliferation is dependent on both the PI3K and Akt pathways.
Figure 7.

APRIL-TACI–mediated proliferation of FL cells is PI3K and mTOR dependent. (A) Karpas cells were cultured alone or with 500 nM wortmanin or 10 μM Ly294002 with or without the addition of APRIL for 48 hours, and proliferation was assessed. Data representative of 3 experiments are shown. (B) Control and Akt siRNA-transfected Karpas cells were left untreated or treated with APRIL for 48 hours, and proliferation was assessed. (C) Karpas cells were cultured alone or with APRIL or with 20 nM rapamycin or both for 48 hours, and proliferation was assessed. (D) Schematic diagram for APRIL-induced signaling through TACI in FL B cells. Malignant cells from FL express TACI, which delivers a proliferative signal upon engagement with APRIL. APRIL-TACI interactions activate the PI3K, Akt, and mTOR pathway, followed by phosphorylation of p70S6 kinase, 4E-BP1, and S6. Activation of mTOR results in up-regulation of cyclin D1 expression and Rb phosphorylation. The hypothetical TRAF proteins involved in APRIL-TACI–mediated PI3K and NF-κB activation are indicated by dashed lines.
Discussion
In light of recent studies suggesting a role for APRIL in lymphoma development,6 we wanted to better characterize the specific biologic process and signaling mechanisms by which this occurs. Our work now provides strong evidence that APRIL, through its interaction with TACI, promotes the growth of FL B cells and cyclin D1 up-regulation through PI3K/Akt-induced mTOR activation. Our studies clearly indicate that APRIL is present in specimens from patients with FL, and that cells from FL specimens can secrete soluble APRIL. The specific mechanisms that control APRIL expression or the cell type responsible for its secretion in the FL microenvironment are not known, and our data would now suggest that both the malignant B cells as well as stromal cells can express APRIL. However, it is also possible that the APRIL detected in purified FL B cells was prebound to the cells, as has been observed previously for BAFF.36 Tumor-infiltrating neutrophils have been previously reported to be a source of APRIL in high-grade NHL, but not in FL or other low-grade lymphomas.32 In keeping with these findings, we reviewed our cases and noted the absence of a significant neutrophilic infiltrate in all cases, suggesting that other cell types are a source for APRIL in FL.
Accumulating experimental evidence supports the notion that APRIL stimulates the growth of human and murine tumor cell lines in vitro and in vivo.37 Our finding that APRIL promotes proliferation of FL B cells is consistent with these data, and we also provide evidence that APRIL-induced proliferation of FL B cells is TACI dependent and proteoglycan independent. These results do not explain the phenotype of TACI knockout mice, which exhibit increased B-cell numbers and splenomegaly,13–16 leading some to hypothesize that TACI is a negative regulator of B-cell proliferation.15 One possible explanation for this disparity is that BAFF-R may play a compensatory role for TACI in the TACI-deficient B cells, thereby enhancing a positive survival signal through BAFF-BAFF-R. In addition, TACI, as well as BAFF-R and BCMA, may activate unique signaling cascades depending on their individual or additive expression levels. Furthermore, expression of TACI is dependent on the maturation stage of the B cell, expressed weakly by immature transitional type B cells, and up-regulated in marginal zone B cells and activated B cells.13 It is therefore possible that lack of TACI expression prevents ordered B-cell development and differentiation, resulting in expansion of certain B-cell subsets.38
Originally, TACI was described as a CAML-associated protein, and upon overexpression it was found to activate the transcription factors nuclear factor of activated T cells, AP-1, and NF-κB.39 TACI has since been found to activate c-Jun NH2-terminal kinase, and analysis of its cytoplasmic tail in a yeast 2-hybrid system revealed TRAF2, TRAF5, and TRAF6 binding.25 Beyond this, little is known about endogenous TACI-dependent signaling, specifically in response to APRIL. Our data now suggest that stimulation of TACI with APRIL results in activation of PI3K. The precise mechanism by which TACI activates PI3K is still a matter of further investigation, but it is likely to be through its association with TRAF proteins (Figure 7D). TRAF6 in particular, which binds to the TACI cytoplasmic tail, has recently been shown to be important in both CD40- and TNF-mediated PI3K activation.40–42
Although we cannot fully exclude a potential role for BCMA in APRIL-mediated signaling in FL patient specimens, inhibition of TACI expression significantly reduced both APRIL-induced proliferation and PI3K activation, whereas inhibition of BCMA had little effect, suggesting that TACI is the primary APRIL receptor responsible for these effects. Alignment of TACI and BCMA sequences reveals significant homology between the 2 receptors,43 but they do differ in their TRAF-binding profile, suggesting that their cytoplasmic tails may transmit unique signals. BCMA has been reported to associate with TRAF1, TRAF2, and TRAF3 and activate NF-κB, Elk-1, and c-Jun N-terminal kinase (JNK).44 Based on these findings, we hypothesize that TACI can specifically activate the PI3K pathway, whereas BCMA regulates activation of the p38 and JNK pathways.
One of the principal downstream targets of PI3K is Akt,45,46 which regulates cell survival, the cell cycle, and protein synthesis.47 Akt phosphorylation was detected upon stimulation of FL cells with APRIL, and it was found to occur in a TACI- and PI3K-dependent manner. Phosphorylation of Akt occurs on 2 key residues, Thr308 and Ser473. PI3K and 3-phosphoinositide dependent protein kinase (PKD1) mediate phosphorylation on Thr308, resulting in activation of mTOR through phosphorylation of tuberous sclerosis protein (TSC)1/TSC2.48–50 The mTOR complex further phosphorylates Akt on Ser473, resulting in its full activation.51 Our data indicate that APRIL induces phosphorylation of Akt on Ser473, suggesting a role for mTOR, and phophorylation of mTOR indeed occurs upon stimulation of FL cells with APRIL. Similar studies have described a role for mTOR in BAFF-mediated survival of mouse B cells, suggesting that BAFF and APRIL may activate common pathways.34
mTOR can form a complex with one of 2 distinct proteins: TOR complex 1 (mTORC1) consists of mTOR and its binding partner Raptor, whereas TOR complex 2 (mTORC2) consists of mTOR and its binding partner Rictor.52 These complexes differ in both substrate specificity and rapamycin sensitivity.52–54 Upon mTOR activation, the TORC1 complex facilitates cell-cycle progression from G1 into S phase by associating with and phosphorylating 2 proteins: p70S6 kinase and eukaryotic initiation factor 4E-BP1.55–57 p70S6K phosphorylates the S6 protein of the 40S ribosomal subunit and is involved in translational control of 5′ oligopyrimidine tract mRNAs,58 whereas 4E-BP1 inhibits the mRNA cap-binding protein eukaryotic initiation factor 4E, thereby promoting translation of genes involved in cell-cycle progression and ribosomal biogenesis.59–61 Both p70S6 kinase and 4E-BP1 are important in growth factor-mediated cell proliferation, and consistent with this, our data now suggest that APRIL mediates phosphorylation of 4E-BP1, p70S6 kinase, and its substrate S6 in FL B cells. The specificity of the substrates (4E-BP1 and p70S6 kinase) as well as the ability of rapamycin to inhibit APRIL-mediated mTOR phosphorylation suggest that APRIL signals through the mTORC1 complex; however, it does not rule out a potential role for mTORC2.
One of the downstream gene targets of the PI3K/Akt/mTORC1 pathway is cyclin D1, which, when expressed, results in its association with CDK4 and CDK5, leading to phosphorylation of Rb and subsequent progression of the cell into S phase.62,63 Our studies now suggest that APRIL, via TACI, up-regulates cyclin D1 expression and Rb phosphorylation in a mTOR-dependent manner. The ability of TACI to modulate cyclin D1 expression is further supported by studies in multiple myeloma, in which it was found that TACIhigh myeloma cells had increased expression of c-MAF, cyclin-D1, and integrin β7 and that stimulation of TACIhigh cells with BAFF results in up-regulation of c-maf, cyclin-D1, and β7 expression.64,65 The ability of APRIL to regulate cyclin D1 and Rb may have significant impact on FL tumor cell development and growth, and may be one mechanism by which APRIL overexpression results in development of lymphoma.6
The role of the mTOR pathway in cancer has been studied extensively over the past few years, and it has been found that the PI3K/Akt/mTOR pathway is constitutively active in many tumor types.57,66 Whereas our studies specifically highlight a role for APRIL-TACI–mediated PI3K activation in FL, this ligand-receptor pair may also be involved in activation of the PI3K pathway in normal B cells as well as other types of malignant B cells. Mantle cell lymphoma lines have been shown to constitutively express phospho-p70S6 kinase, and clinical targeting of this pathway with temsirolimus, an inhibitor of mTOR, resulted in substantial antitumor activity (38% overall response rate) in relapsed mantle cell lymphoma (MCL).67 Our data now suggest that APRIL-TACI interactions may be one mechanism by which the PI3K/Akt pathway is activated in lymphoma, and it may also be a mechanism by which tumor cells overcome mTOR-targeted therapies. Therefore, combined therapies that target both the PI3K/mTOR pathway and APRIL may have clinical potential in the treatment of lymphoma.
Supplementary Material
Acknowledgments
We thank Gail Bishop and Richard Bram for critical reading of the manuscript.
This work was supported in part by National Institutes of Health (NIH, Bethesda, MD) grant CA097274, ZymoGenetics, and Merck Serono International (an affiliate of Merck KGaA, Darmstadt, Germany). A.J.N. is a Lymphoma Research Foundation Follicular Lymphoma Research Grantee.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contibution: M.G. and A.J.N. designed and performed research, analyzed and interpreted data, and drafted the paper; S.R.D. provided reagents and edited the paper; S.C.Z. performed research; A.L.F. reviewed the pathology and immunohistochemistry; and T.E.W., S.M.A., and J.R.C. collected data, provided patient specimens, and edited the manuscript.
Conflict-of-interest disclosure: S.R.D. is an employee of ZymoGenetics. The remaining authors declare no competing financial interests.
Correspondence: Dr Anne J. Novak, Division of Hematology, Mayo Clinic, 200 First St SW, Rochester, MN 55905; e-mail: novak.anne@mayo.edu.
References
- 1.Schneider P, MacKay F, Steiner V, et al. BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. J Exp Med. 1999;189:1747–1756. doi: 10.1084/jem.189.11.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moore PA, Belvedere O, Orr A, et al. BLyS: member of the tumor necrosis factor family and B lymphocyte stimulator. Science. 1999;285:260–263. doi: 10.1126/science.285.5425.260. [DOI] [PubMed] [Google Scholar]
- 3.Hahne M, Kataoka T, Schroter M, et al. APRIL, a new ligand of the tumor necrosis factor family, stimulates tumor cell growth. J Exp Med. 1998;188:1185–1190. doi: 10.1084/jem.188.6.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bossen C, Schneider P. BAFF, APRIL and their receptors: structure, function and signaling. Semin Immunol. 2006;18:263–275. doi: 10.1016/j.smim.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 5.Yu G, Boone T, Delaney J, et al. APRIL and TALL-I and receptors BCMA and TACI: system for regulating humoral immunity. Nat Immunol. 2000;1:252–256. doi: 10.1038/79802. [DOI] [PubMed] [Google Scholar]
- 6.Planelles L, Carvalho-Pinto CE, Hardenberg G, et al. APRIL promotes B-1 cell-associated neoplasm. Cancer Cells. 2004;6:399–408. doi: 10.1016/j.ccr.2004.08.033. [DOI] [PubMed] [Google Scholar]
- 7.He B, Chadburn A, Jou E, et al. Lymphoma B cells evade apoptosis through the TNF family members BAFF/BLyS and APRIL. J Immunol. 2004;172:3268–3279. doi: 10.4049/jimmunol.172.5.3268. [DOI] [PubMed] [Google Scholar]
- 8.Endo T, Nishio M, Enzler T, et al. BAFF and APRIL support chronic lymphocytic leukemia B-cell survival through activation of the canonical NF-κB pathway. Blood. 2007;109:703–710. doi: 10.1182/blood-2007-04-081786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Madry C, Laabi Y, Callebaut I, et al. The characterization of murine BCMA gene defines it as a new member of the tumor necrosis factor receptor superfamily. Int Immunol. 1998;10:1693–1702. doi: 10.1093/intimm/10.11.1693. [DOI] [PubMed] [Google Scholar]
- 10.von Bulow GU, Russell H, Copeland NG, et al. Molecular cloning and functional characterization of murine transmembrane activator and CAML interactor (TACI) with chromosomal localization in human and mouse. Mamm Genome. 2000;11:628–632. doi: 10.1007/s003350010125. [DOI] [PubMed] [Google Scholar]
- 11.Thompson JS, Bixler SA, Qian F, et al. BAFF-R, a newly identified TNF receptor that specifically interacts with BAFF. Science. 2001;293:2108–2111. doi: 10.1126/science.1061965. [DOI] [PubMed] [Google Scholar]
- 12.Yan M, Brady JR, Chan B, et al. Identification of a novel receptor for B lymphocyte stimulator that is mutated in a mouse strain with severe B cell deficiency. Curr Biol. 2001;11:1547–1552. doi: 10.1016/s0960-9822(01)00481-x. [DOI] [PubMed] [Google Scholar]
- 13.Ng LG, Sutherland AP, Newton R, et al. B cell-activating factor belonging to the TNF family (BAFF)-R is the principal BAFF receptor facilitating BAFF costimulation of circulating T and B cells. J Immunol. 2004;173:807–817. doi: 10.4049/jimmunol.173.2.807. [DOI] [PubMed] [Google Scholar]
- 14.von Bulow GU, van Deursen JM, Bram RJ. Regulation of the T-independent humoral response by TACI. Immunity. 2001;14:573–582. doi: 10.1016/s1074-7613(01)00130-3. [DOI] [PubMed] [Google Scholar]
- 15.Seshasayee D, Valdez P, Yan M, et al. Loss of TACI causes fatal lymphoproliferation and autoimmunity, establishing TACI as an inhibitory BLyS receptor. Immunity. 2003;18:279–288. doi: 10.1016/s1074-7613(03)00025-6. [DOI] [PubMed] [Google Scholar]
- 16.Yan M, Wang H, Chan B, et al. Activation and accumulation of B cells in TACI-deficient mice. Nat Immunol. 2001;2:638–643. doi: 10.1038/89790. [DOI] [PubMed] [Google Scholar]
- 17.Castigli E, Wilson SA, Garibyan L, et al. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat Genet. 2005;37:829–834. doi: 10.1038/ng1601. [DOI] [PubMed] [Google Scholar]
- 18.Salzer U, Chapel HM, Webster AD, et al. Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat Genet. 2005;37:820–828. doi: 10.1038/ng1600. [DOI] [PubMed] [Google Scholar]
- 19.Sakurai D, Kanno Y, Hase H, et al. TACI attenuates antibody production costimulated by BAFF-R and CD40. Eur J Immunol. 2007;37:110–118. doi: 10.1002/eji.200636623. [DOI] [PubMed] [Google Scholar]
- 20.Ingold K, Zumsteg A, Tardivel A, et al. Identification of proteoglycans as the APRIL-specific binding partners. J Exp Med. 2005;201:1375–1383. doi: 10.1084/jem.20042309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hendriks J, Planelles L, de Jong-Odding J, et al. Heparan sulfate proteoglycan binding promotes APRIL-induced tumor cell proliferation. Cell Death Differ. 2005;12:637–648. doi: 10.1038/sj.cdd.4401647. [DOI] [PubMed] [Google Scholar]
- 22.Sakurai D, Hase H, Kanno Y, et al. TACI regulates IgA production by APRIL in collaboration with HSPG. Blood. 2007;109:2961–2967. doi: 10.1182/blood-2006-08-041772. [DOI] [PubMed] [Google Scholar]
- 23.Bischof D, Elsawa SF, Mantchev G, et al. Selective activation of TACI by syndecan-2. Blood. 2006;107:3235–3242. doi: 10.1182/blood-2005-01-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Novak AJ, Grote DM, Stenson M, et al. Expression of BLyS and its receptors in B-cell non-Hodgkin lymphoma: correlation with disease activity and patient outcome. Blood. 2004;104:2247–2253. doi: 10.1182/blood-2004-02-0762. [DOI] [PubMed] [Google Scholar]
- 25.Xia XZ, Treanor J, Senaldi G, et al. TACI is a TRAF-interacting receptor for TALL-1, a tumor necrosis factor family member involved in B cell regulation. J Exp Med. 2000;192:137–143. doi: 10.1084/jem.192.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fox BA, Moore MD. Trimerizing polypeptides. WO 2007/030803 A2. US Priority Date, September 9, 2005. [Google Scholar]
- 27.Novak AJ, Bram RJ, Kay NE, et al. Aberrant expression of B-lymphocyte stimulator by B chronic lymphocytic leukemia cells: a mechanism for survival. Blood. 2002;100:2973–2979. doi: 10.1182/blood-2002-02-0558. [DOI] [PubMed] [Google Scholar]
- 28.Roecklein BA, Torok-Storb B. Functionally distinct human marrow stromal cell lines immortalized by transduction with the human papilloma virus E6/E7 genes. Blood. 1995;85:997–1005. [PubMed] [Google Scholar]
- 29.Novak AJ, Darce JR, Arendt BK, et al. Expression of BCMA, TACI, and BAFF-R in multiple myeloma: a mechanism for growth and survival. Blood. 2004;103:689–694. doi: 10.1182/blood-2003-06-2043. [DOI] [PubMed] [Google Scholar]
- 30.Chiu A, Xu W, He B, et al. Hodgkin lymphoma cells express TACI and BCMA receptors and generate survival and proliferation signals in response to BAFF and APRIL. Blood. 2007;109:729–739. doi: 10.1182/blood-2006-04-015958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huard B, Arlettaz L, Ambrose C, et al. BAFF production by antigen-presenting cells provides T cell co-stimulation. Int Immunol. 2004;16:467–475. doi: 10.1093/intimm/dxh043. [DOI] [PubMed] [Google Scholar]
- 32.Schwaller J, Schneider P, Mhawech-Fauceglia P, et al. Neutrophil-derived APRIL concentrated in tumor lesions by proteoglycans correlates with human B cell lymphoma aggressiveness. Blood. 2006;109:331–338. doi: 10.1182/blood-2006-02-001800. [DOI] [PubMed] [Google Scholar]
- 33.Dillon SR, Gross JA, Ansell SM, et al. An APRIL to remember: novel TNF ligands as therapeutic targets. Nat Rev Drug Discov. 2006;5:235–246. doi: 10.1038/nrd1982. [DOI] [PubMed] [Google Scholar]
- 34.Woodland RT, Fox CJ, Schmidt MR, et al. Multiple signaling pathways promote B lymphocyte stimulator dependent B-cell growth and survival. Blood. 2008;111:750–760. doi: 10.1182/blood-2007-03-077222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103:253–262. doi: 10.1016/s0092-8674(00)00117-3. [DOI] [PubMed] [Google Scholar]
- 36.Jelinek DF, Darce JR. Human B lymphocyte malignancies: exploitation of BLyS and APRIL and their receptors. Curr Dir Autoimmun. 2005;8:266–288. doi: 10.1159/000082107. [DOI] [PubMed] [Google Scholar]
- 37.Rennert P, Schneider P, Cachero TG, et al. A soluble form of B cell maturation antigen, a receptor for the tumor necrosis factor family member APRIL, inhibits tumor cell growth. J Exp Med. 2000;192:1677–1684. doi: 10.1084/jem.192.11.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salzer U, Jennings S, Grimbacher B. To switch or not to switch–the opposing roles of TACI in terminal B cell differentiation. Eur J Immunol. 2007;37:17–20. doi: 10.1002/eji.200636914. [DOI] [PubMed] [Google Scholar]
- 39.von Bulow GU, Bram RJ. NF-AT activation induced by a CAML-interacting member of the tumor necrosis factor receptor superfamily. Science. 1997;278:138–141. doi: 10.1126/science.278.5335.138. [DOI] [PubMed] [Google Scholar]
- 40.Benson RJ, Hostager BS, Bishop GA. Rapid CD40-mediated rescue from CD95-induced apoptosis requires TNFR-associated factor-6 and PI3K. Eur J Immunol. 2006;36:2535–2543. doi: 10.1002/eji.200535483. [DOI] [PubMed] [Google Scholar]
- 41.Deregibus MC, Buttiglieri S, Russo S, et al. CD40-dependent activation of phosphatidylinositol 3-kinase/Akt pathway mediates endothelial cell survival and in vitro angiogenesis. J Biol Chem. 2003;278:18008–18014. doi: 10.1074/jbc.M300711200. [DOI] [PubMed] [Google Scholar]
- 42.Burow ME, Weldon CB, Melnik LI, et al. PI3-K/AKT regulation of NF-κB signaling events in suppression of TNF-induced apoptosis. Biochem Biophys Res Commun. 2000;271:342–345. doi: 10.1006/bbrc.2000.2626. [DOI] [PubMed] [Google Scholar]
- 43.Gross JA, Johnston J, Mudri S, et al. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature. 2000;404:995–999. doi: 10.1038/35010115. [DOI] [PubMed] [Google Scholar]
- 44.Hatzoglou A, Roussel J, Bourgeade MF, et al. TNF receptor family member BCMA (B cell maturation) associates with TNF receptor-associated factor (TRAF) 1, TRAF2, and TRAF3 and activates NF-κB, elk-1, c-Jun N-terminal kinase, and p38 mitogen-activated protein kinase. J Immunol. 2000;165:1322–1330. doi: 10.4049/jimmunol.165.3.1322. [DOI] [PubMed] [Google Scholar]
- 45.Franke TF, Kaplan DR, Cantley LC, et al. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 1997;275:665–668. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- 46.Klippel A, Kavanaugh WM, Pot D, et al. A specific product of phosphatidylinositol 3-kinase directly activates the protein kinase Akt through its pleckstrin homology domain. Mol Cell Biol. 1997;17:338–344. doi: 10.1128/mcb.17.1.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 48.Inoki K, Li Y, Zhu T, et al. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 49.Manning BD, Tee AR, Logsdon MN, et al. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10:151–162. doi: 10.1016/s1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- 50.Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol. 2002;4:658–665. doi: 10.1038/ncb840. [DOI] [PubMed] [Google Scholar]
- 51.Sarbassov DD, Guertin DA, Ali SM, et al. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 52.Bhaskar PT, Hay N. The two TORCs and Akt. Dev Cell. 2007;12:487–502. doi: 10.1016/j.devcel.2007.03.020. [DOI] [PubMed] [Google Scholar]
- 53.Jacinto E, Loewith R, Schmidt A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 54.Sarbassov DD, Ali SM, Kim DH, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 55.Nojima H, Tokunaga C, Eguchi S, et al. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J Biol Chem. 2003;278:15461–15464. doi: 10.1074/jbc.C200665200. [DOI] [PubMed] [Google Scholar]
- 56.Schalm SS, Fingar DC, Sabatini DM, et al. TOS motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Curr Biol. 2003;13:797–806. doi: 10.1016/s0960-9822(03)00329-4. [DOI] [PubMed] [Google Scholar]
- 57.Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–734. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- 58.Pullen N, Thomas G. The modular phosphorylation and activation of p70s6k. FEBS Lett. 1997;410:78–82. doi: 10.1016/s0014-5793(97)00323-2. [DOI] [PubMed] [Google Scholar]
- 59.Lin TA, Kong X, Haystead TA, et al. PHAS-I as a link between mitogen-activated protein kinase and translation initiation. Science. 1994;266:653–656. doi: 10.1126/science.7939721. [DOI] [PubMed] [Google Scholar]
- 60.Pause A, Belsham GJ, Gingras AC, et al. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature. 1994;371:762–767. doi: 10.1038/371762a0. [DOI] [PubMed] [Google Scholar]
- 61.Peterson RT, Schreiber SL. Translation control: connecting mitogens and the ribosome. Curr Biol. 1998;8:R248–R250. doi: 10.1016/s0960-9822(98)70152-6. [DOI] [PubMed] [Google Scholar]
- 62.Ewen ME, Sluss HK, Sherr CJ, et al. Functional interactions of the retinoblastoma protein with mammalian D-type cyclins. Cell. 1993;73:487–497. doi: 10.1016/0092-8674(93)90136-e. [DOI] [PubMed] [Google Scholar]
- 63.Hunter T, Pines J. Cyclins and cancer, II: cyclin D and CDK inhibitors come of age. Cell. 1994;79:573–582. doi: 10.1016/0092-8674(94)90543-6. [DOI] [PubMed] [Google Scholar]
- 64.Moreaux J, Cremer FW, Reme T, et al. The level of TACI gene expression in myeloma cells is associated with a signature of microenvironment dependence versus a plasmablastic signature. Blood. 2005;106:1021–1030. doi: 10.1182/blood-2004-11-4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moreaux J, Hose D, Jourdan M, et al. TACI expression is associated with a mature bone marrow plasma cell signature and C-MAF overexpression in human myeloma cell lines. Haematologica. 2007;92:803–811. doi: 10.3324/haematol.10574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cantley LC. The role of phosphoinositide 3-kinase in human disease. Harvey Lect. 2004;100:103–122. [PubMed] [Google Scholar]
- 67.Witzig TE, Geyer SM, Ghobrial I, et al. Phase II trial of single-agent temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol. 2005;23:5347–5356. doi: 10.1200/JCO.2005.13.466. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}