

Abstract
Plasmodium falciparum invasion into human erythrocytes relies on the interaction between multiple parasite ligands and their respective erythrocyte receptors. The sialic acid-independent invasion pathway is dependent on the expression of P. falciparum reticulocyte binding protein-like homologue 4 (PfRh4), as disruption of the gene abolishes the ability of parasites to switch to this pathway. We show that PfRh4 is present as an invasion ligand in culture supernatants as a 160-kDa proteolytic fragment. We confirm that PfRh4 binds to the surfaces of erythrocytes through recognition of an erythrocyte receptor that is neuraminidase resistant but trypsin and chymotrypsin sensitive. Serum antibodies from malaria-exposed individuals show reactivity against the binding domain of PfRh4. Purified immunoglobulin G raised in rabbits against the binding domain of PfRh4 blocked the binding of native PfRh4 to the surfaces of erythrocytes and inhibited erythrocyte invasion of parasites using sialic acid-independent invasion pathways and grown in neuraminidase-treated erythrocytes. Our results suggest PfRh4 is a potential vaccine candidate.
During the asexual stage of the Plasmodium falciparum life cycle, the parasite undergoes rapid replication within the erythrocytes of the human host, resulting in the clinical manifestations seen in malaria infections. The merozoite forms of P. falciparum invade erythrocytes through a multistep process that involves initial contact with the erythrocyte, apical reorientation of the merozoite, and the formation of a tight junction, which moves progressively toward the posterior end of the parasite until host cell membrane fusion is completed (for a review, see reference 9). These steps in invasion are dependent on specific interactions between multiple parasite invasion ligands and their respective host erythrocyte receptors (8, 20). Although P. falciparum has a very restricted host cell range, it has developed the ability to invade human erythrocytes using multiple parasite ligand-erythrocyte receptor interactions that have become known as alternative invasion pathways (11, 12, 14, 21, 31).
Broadly speaking, there are two major invasion pathways in P. falciparum, one that is dependent on sialic acid (SA) residues, termed the SA-dependent pathway, and one that is independent of SA, called the SA-independent pathway (23, 30, 37, 38). SA moieties on the surfaces of erythrocytes can be removed by treatment with the enzyme neuraminidase. Parasite strains that invade neuraminidase-treated erythrocytes efficiently are called SA-independent strains, whereas strains that invade inefficiently into the treated erythrocytes are called SA-dependent strains. To date, two gene families encoding invasion ligands have been identified as major players in these invasion pathways: those encoding the erythrocyte binding antigens (EBAs) (EBA-175; EBA-181, also known as JESEBL; and EBA-140, also known as BAEBL) (2, 19, 26-28, 44, 48) and those encoding the P. falciparum reticulocyte binding protein-like homologues (PfRhs) (PfRh1, PfRh2a, PfRh2b, PfRh3, PfRh4, and PfRh5) (4, 14, 22, 24, 40, 41, 43, 46, 51). All members of these families are expressed and functional, except the EBA-165 (also known as PAEBL) and PfRh3 genes, which appear to be pseudogenes (47, 52). Previous studies have shown that the EBAs and PfRh1 are involved in the SA-dependent pathway, whereas PfRh2b and PfRh4 are important in the SA-independent pathway (14, 16, 18, 26, 27, 41, 42, 46, 50). Host receptors have been identified only for EBA-175 and EBA-140, which bind to glycophorin A and C, respectively (26, 27, 29, 44). Both EBA-181 and PfRh1 have been shown to bind SA on the erythrocyte surface, although the identities of these receptors are unknown (15, 19, 41). EBA181 has also been reported to bind band 4.1 (25).
Changes in the expression and activation of some PfRhs enable the parasite to utilize alternate invasion pathways, and clinical P. falciparum isolates show diversity in invasion phenotypes and expression of EBA and PfRh proteins (5, 6, 10, 14, 16, 35, 46). For instance, W2mef parasites primarily invade via an SA-dependent pathway, using EBA-175 as a key invasion ligand (16, 46). In this strain, there is no detectable expression of PfRh4. Through a targeted knockout of EBA-175 or selection of W2mef for invasion of neuraminidase-treated erythrocytes, this strain has the ability to switch to an SA-independent invasion pathway (13, 42). The switch in invasion pathway is concurrent with an increase in PfRh4 protein expression (16, 46). PfRh4 is essential in the SA-independent pathway, as disruption of the gene in W2mef results in the inability of the strain to switch invasion pathways to allow invasion into neuraminidase-treated erythrocytes (46). The activation of PfRh4 in response to the loss of EBA-175 function suggests that the PfRh and EBA families overlap with respect to their functions in invasion (14, 46). Recent studies demonstrated that PfRh4 binds to the surfaces of erythrocytes (17). By varying the levels of expression of these invasion ligands, the parasite is able to switch receptor usage from SA-dependent to SA-independent pathways, providing a mechanism for the parasite to evade the host immune system (14, 39, 46). The ability to use different receptor-ligand interactions for invasion may also enable the parasites to adapt to different physiological conditions in different hosts.
EBAs and PfRhs are located at the merozoite apical tip to allow recognition of and binding to their erythrocyte receptor (1, 14, 46). For successful parasite entry into the erythrocyte, the tight junction formed between these transmembrane parasite ligands and their receptors must be released. It is thought that this release occurs through the cleavage of invasion ligands by rhomboid proteases (3, 34, 54). Subsequently, these proteolytic fragments are shed into the bloodstream, resulting in parasite ligands being exposed to the human immune system. Therefore, although these ligands are crucial in the invasion process, it is also highly likely that EBAs and PfRhs are targets of inhibitory antibodies of the human immune system (39). Inhibitory antibodies are thought to be an important component of acquired protective immunity through their ability to block invasion by a parasite and its subsequent rapid replication within erythrocytes. In support of the importance of inhibitory antibodies, previous studies have shown that rabbit antibodies against EBA-175, EBA-140, PfRh2b, and PfRh1 inhibit parasite invasion in vitro (14, 15, 27, 41, 45). Differential inhibition by human antibodies of P. falciparum lines that vary in their use of specific EBA and PfRh proteins pointed to these ligand families as major targets of inhibitory antibodies (39).
Although PfRh4 has an important role in the SA-independent pathway, antibodies generated against PfRh4 domains have shown no inhibition of merozoite invasion (17). A recombinant 30-kDa protein in a conserved region of PfRh4 (rRh430) has been shown to bind to erythrocytes in a neuraminidase-resistant, chymotrypsin- and trypsin-sensitive manner (17). Addition of rRh430 itself or anti-rRh430 antibodies into an erythrocyte binding assay resulted in the inhibition of native PfRh4 erythrocyte binding; however, these anti-rRh430 antibodies did not inhibit parasite invasion. In immunoblots, these antibodies detected PfRh4 as a 250-kDa protein in saponin-lysed schizont pellets, a protein size not consistent with other published reports (24, 46). Furthermore, a processed form of PfRh4 was not detected in culture supernatants using these antibodies, though others have suggested that PfRh4 is proteolytically cleaved and released into the culture supernatant by rhomboid proteases during the invasion process (3).
In light of the importance of PfRh4 in parasite invasion, we examined the binding of PfRh4 to erythrocytes, evidence that PfRh4 is proteolytically processed, and the role of antibodies against PfRh4 in inhibition of erythrocyte invasion. Our work shows that recombinant PfRh4 reacts with sera from malaria-exposed individuals and that antibodies to it inhibit parasite invasion. This suggests that PfRh4, a major invasion ligand for the SA-independent pathway, is exposed to the human immune system and provides a target of inhibitory antibodies and is therefore a potential vaccine candidate.
MATERIALS AND METHODS
Parasite culture and material.
P. falciparum asexual stages were maintained in human O+ erythrocytes and synchronized by standard methods (49). 3D7 is a cloned line derived from NF54 supplied by David Walliker, Edinburgh University. W2mef is a cloned line derived from the Indochina III/CDC strain. W2mefΔ175 and W2mefΔRh4 are cloned lines containing a disrupted EBA-175 or PfRh4 gene as previously described (13, 46). HB3 is a cloned line from South America (53).
Culture supernatants enriched in parasite invasion ligands were obtained by treating synchronized parasite cultures at 5% parasitemia with trypsin (1.0 mg/ml) and neuraminidase (25 mU/ml). These enzyme treatments on the erythrocytes effectively prevent reinvasion of the erythrocytes after schizont rupture. The supernatants were harvested approximately 48 h after enzyme treatment or when it was apparent there was an absence of reinvasion and were frozen for storage at −80°C. Total proteins from schizont stage parasites were obtained by synchronization and by saponin lysis of infected erythrocytes.
Recombinant fusion cloning and purification.
A codon-optimized version of PfRh4 containing the DNA sequence for amino acids (aa) 28 to 766 was synthesized and cloned into pUC19 (Codon Devices Inc.). From this clone, the region for Rh4.9 was digested from pUC19 using BamHI and XhoI and was subsequently cloned in frame into compatible sites in pET-45b(+), which contains an amino-terminal six-His tag. The fusion protein was expressed in BL21(DE3) (Novagen) bacterial cells and purified over an Ni-nitrilotriacetic acid column (Qiagen) under native conditions. The soluble protein expressed from Rh4.9 used in all assays underwent a second step purification in which Ni resin-purified six-His-Rh4 was concentrated and further purified on a Superdex 200 gel filtration column (10/300 GL or Hiload 16/60; Amersham Pharmacia Biotech). The protein was eluted from the columns as a monomer.
The six-His-tagged recombinant proteins Rh4.10, Rh4.11, Rh4.12, and Rh4.13 were generated in the following way. Their respective PfRh4 fragments were amplified from the codon-optimized version of PfRh4 mentioned above using the following primers: for Rh4.10, 5′-CGCGGATCCCAGCAAAGAAAAGA and 5′-GCGACTCGAGTTATTAAAAATGAGAACGCAGATCCG; for Rh4.11, 5′-CGCGGATCCCATCGACAGTGAAAACGAGAAGC and 5′-GCGCTCGAGTTATTAAATCTCGTTCAGCTTATTCAGGA; for Rh4.12, 5′-CGCGGATCCCAAGAACGAGTTTCTGAATAAATTCAT and 5′-GCGAGACTCGAGTTATTAGATATTTTGCAT; and for Rh4.13, 5′-CGCGGATCCCATCAATAACGACGATAACTTTATTGAAT and 5′-GCGCTCGAGTTATTATTTGAACAGATTGATTTTCGTTTG. These oligonucleotides also contain the BamHI and XhoI restriction sites used for subsequent cloning. The cloning and purification of these fusion proteins were as described for Rh4.9.
Erythrocyte binding and inhibition assay.
Erythrocyte binding assays were performed in the following manner. Culture supernatant (250 μl) was mixed with 50 μl of packed erythrocytes for more than 30 min at room temperature. The erythrocytes and parasite proteins were centrifuged at 12,000 rpm for 30 s through 400 μl of silicone oil (dibutyl phthalate; Sigma) to remove unbound culture supernatant material. The erythrocytes and bound proteins were washed twice with 500 μl of phosphate-buffered saline (PBS). Proteins bound to the erythrocytes were eluted by incubation with 10 μl of 1.5 M NaCl for 15 min at room temperature and then centrifuged for 30 s at 12,000 rpm, and the eluate was removed from the erythrocytes. An equal volume of 2× reducing sample buffer was added to the eluted proteins. The eluted proteins were separated on sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis and identified by immunoblotting.
Uninfected washed erythrocytes were modified by the addition of neuraminidase (66.7 mU/ml), low trypsin (0.1 mg/ml), high trypsin (1.5 mg/ml), and chymotrypsin (1.5 mg/ml) separately for 1 hour at 37°C. Soybean trypsin inhibitor was added to the enzyme-treated erythrocytes at 1.5 mg/ml. The treated erythrocytes were subsequently washed and added to the binding assay as described above.
For the binding inhibition assay, purified anti-Rh4 immunoglobulin G (IgG) or normal rabbit serum IgG was incubated with 250 μl of culture supernatant for 1 h at room temperature before the addition of the packed erythrocytes. For the antibody titration, anti-PfRh4 IgG or IgG from normal rabbit serum was added to final concentrations of 0, 0.0008, 0.0016, 0.003, 0.006, 0.012, 0.024, 0.05, 0.1, 0.2, 0.4, 0.8, and 1.5 mg/ml (amount of IgG/250 μl of invasion supernatant). The rest of the binding assay was performed as described above.
Immunoblotting and antibodies.
Proteins were separated on either 3 to 8% Tris-acetate for proteins larger than 75 kDa or 4 to 12% N,N-methylenebisacrylamide-Tris SDS-polyacrylamide gel electrophoresis gels for smaller proteins (Invitrogen). Western blotting onto nitrocellulose (0.45 mM; Schleicher and Schuell) was performed according to standard protocols, and the blots were processed with an enhanced chemiluminescence system (Amersham).
Anti-Rh4 antibodies were raised in rabbits against purified six-His-tagged Rh4.9 protein. The anti-PfRh4 mouse monoclonal antibody was raised against purified Rh4.R2-glutathione S-transferase protein (46). The other antibody used in immunodetection was rabbit anti-EBA-175, as described previously (42).
ELISA.
Enzyme-linked immunosorbent assays (ELISA) were performed as described previously (39). Ninety-six-well flat-bottom plates (Maxisorp; Nunc) were coated with recombinant fusion protein at a concentration of 1 μg/ml in human tonicity PBS overnight at 4°C. The plates were incubated with 10% skim milk/0.05% Tween 20 for 2 h at 37°C to block nonspecific binding. After the plates were washed, serum samples (1:500) were applied in 5% skim milk/0.05% Tween 20. The plates were incubated for 1 h at room temperature before the serum was removed by washing. Secondary antibody (horseradish peroxidase-conjugated goat anti-human; Chemicon) was used at 1:5,000 in 5% skim milk/0.05% Tween 20. The plates were incubated for 1 h at room temperature. Azino-bis-3-ethylbenthiazoline-6-sulfonic acid (liquid substrate; Sigma-Aldrich) was used to detect horseradish peroxidase activity. The reaction was stopped with 1% SDS, and the optical density (OD) was measured at 405 nm. All washes were done in 1× human tonicity PBS/0.05% Tween 20. Samples were all tested in duplicate. The OD from wells incubated with PBS instead of serum was considered the background and was deducted from the ODs of all samples.
Human serum samples used in ELISA were collected from malaria-exposed adult residents of the Madang area, Papua New Guinea, after informed consent. Negative control sera were obtained from unexposed Melbourne blood donors. Ethics approval was obtained from the Medical Research Advisory Committee, Papua New Guinea, and the Human Research Ethics Committee of the Walter and Eliza Hall Institute, Australia.
Invasion inhibition assay.
Invasion inhibition assays were performed as described previously (39). Neuraminidase (66.7 mU/ml)-treated or normal erythrocytes at 1% hematocrit in culture medium were inoculated with late trophozoite stage parasites to give a parasitemia of 0.2% and a hematocrit of 1% in a volume of 50 μl. The parasites were cultured in 96-well round-bottom microtiter plates (Becton Dickinson, NJ). Antibodies used for the assay were purified using protein G affinity columns. The antibodies were added to a final concentration of 2 mg/ml during the setup of the assay, prior to reinvasion. For the antibody titration invasion inhibition assay, anti-PfRh4 IgG or IgG from normal rabbit serum was added to final concentrations of 0, 0.05, 0.1, 0.22, 0.45, 0.9, 1.5, and 2.0 mg/ml (amount of IgG/55-μl final culture volume). After incubation with antibodies for two cycles of parasite growth, the parasitemia of each well was determined by flow cytometry of ethidium bromide (Bio-Rad, Hercules, CA)-stained trophozoite stage parasites using a FACSCalibur with a plate reader (Becton Dickinson, NJ). For each well, 40,000 cells or more were counted. Growth was expressed as a percentage of parasitemia for the mean of two or more PBS, rabbit prebleed, or nonimmune IgG wells, as appropriate. Two independent assays were performed, each in duplicate.
RESULTS
PfRh4 is expressed in the culture supernatant and binds the surfaces of erythrocytes.
Previous studies showed that PfRh4 is expressed during the late stages of the parasite life cycle and is located at the apical tips of merozoites, suggesting that it may function as an invasion ligand (24, 46). Many molecules of invasion ligands are present within culture supernatants, and it is thought that the proteolytic processing of these molecules from the parasite surface upon entry into erythrocytes may be an integral part of successful parasite invasion (for a review, see reference 7). To further understand the role of PfRh4 as an invasion ligand, we analyzed its expression using an anti-PfRh4 mouse monoclonal antibody in supernatants from the 3D7, HB3, W2mefΔRh4, and W2mefΔ175 strains. We detected the presence of a single band at 160 kDa in supernatants from 3D7, HB3, and W2mefΔ175 (isolates that express PfRh4), but not in supernatant from the W2mefΔRh4 isolate, which lacks PfRh4 (Fig. 1A, right). This same antibody detected the expected doublet band at 190 kDa and 180 kDa in a saponin-treated schizont pellet in the 3D7, HB3, and W2mefΔ175 strains and an absence of the doublet in the W2mefΔRh4 strain (Fig. 1A, left) (46). Three other anti-Rh4 antibodies raised against distinct regions of PfRh4 showed similar results in schizont pellets and culture supernatants, confirming the specificity of the generated reagents (Fig. 1B).
FIG. 1.
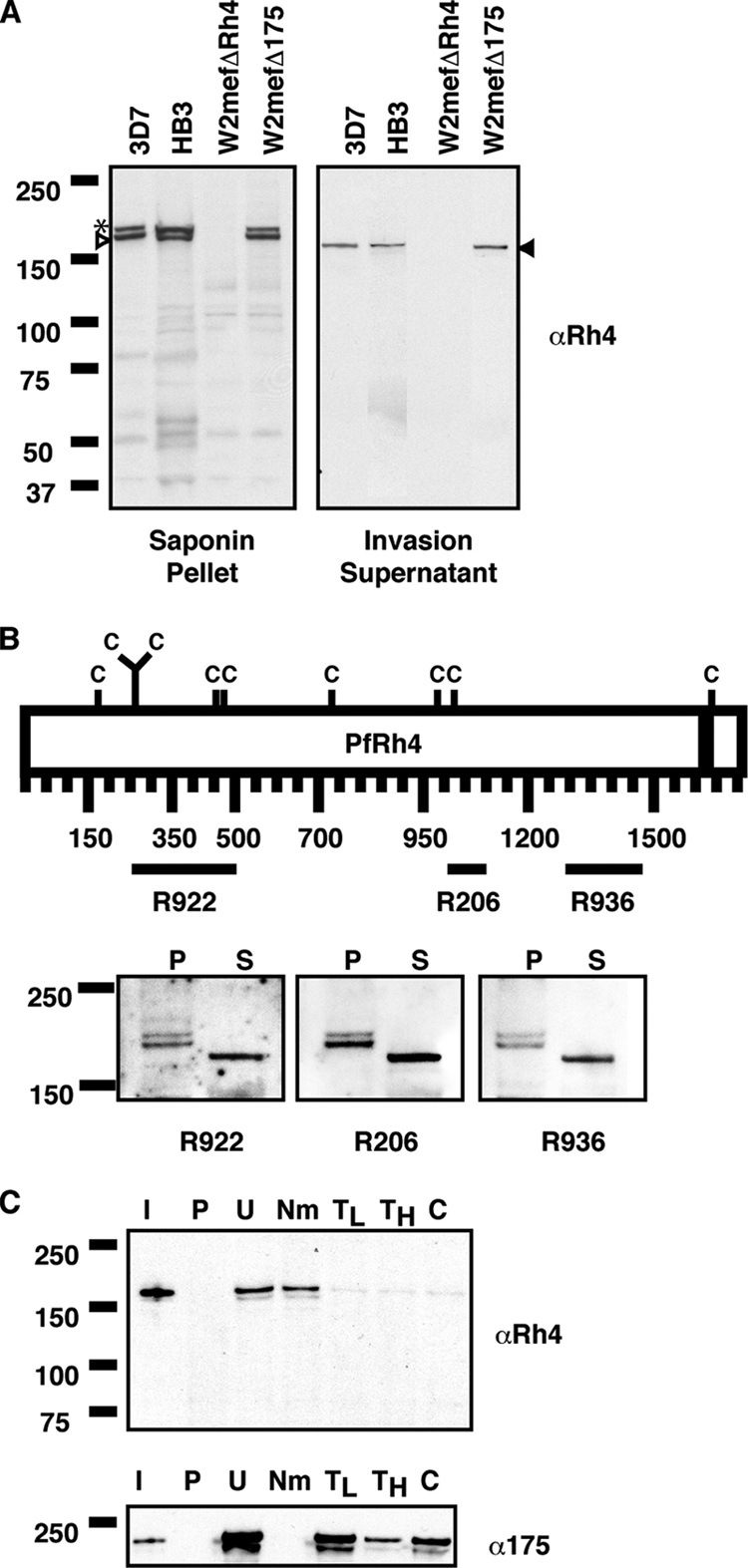
PfRh4 is expressed as a 160-kDa fragment in the invasion supernatant and binds to the surfaces of erythrocytes in an enzyme-dependent manner. (A) Western blots of saponin-treated schizont pellets (left) and invasion supernatants (right) were probed with an anti-Rh4 (αRh4) antibody. 3D7, HB3, and W2mefΔ175 express PfRh4, which is absent from W2mefΔRh4. The asterisk, white arrowhead, and black arrowhead highlight bands running at 190 kDa, 180 kDa, and 160 kDa, respectively. (B) (Top) Schematic representation of the various domains of PfRh4 against which rabbit polyclonal antibodies were raised. The black bar above each antibody name (R922, R206, and R936) highlights the region of the fusion protein used. C denotes cysteine residues, and the black bar within the schematic represents the transmembrane domain of PfRh4. (Bottom) Western blots of saponin-treated schizont pellets (lanes P) and culture supernatants (lanes S) were probed with three separate anti-Rh4 antibodies. (C) Immunodetection of parasite proteins with anti-RH4 and anti-EBA-175 antibodies after binding and elution from untreated and enzyme-treated erythrocytes. Lanes: I, input lane; P, proteins eluted from PBS control; U, untreated erythrocytes; N, neuraminidase; TL, low trypsin; TH, high trypsin; and C, chymotrypsin-treated erythrocytes. Low trypsin and high tryspin are trypsin treatments with 0.1 and 1.5 mg/ml of enzyme, respectively. Molecular masses are indicated on the left (in kDa) for all panels.
If PfRh4 functions as an invasion ligand, it would likely have the capability of binding to the surfaces of erythrocytes. To determine if PfRh4 binds to the surfaces of erythrocytes, we performed an erythrocyte binding assay. Briefly 3D7 invasion supernatants were incubated with human erythrocytes. The erythrocytes and parasite proteins were passed through oil, the bound proteins were eluted under high-salt conditions, and the eluate was analyzed by immunoblotting. Incubation of 3D7 invasion supernatants with untreated erythrocytes confirmed that PfRh4 binds erythrocytes (Fig. 1C). The specificity of binding was further determined by modifying the surfaces of the erythrocytes with neuraminidase, low-trypsin (0.1 mg/ml), high-trypsin (1.5 mg/ml), and chymotrypsin enzyme treatments. Treatment with neuraminidase, which removes SA moieties from the cell surface, did not perturb the binding of PfRh4. However, binding of PfRh4 was abolished when erythrocytes were treated with trypsin and chymotrypsin, indicating that the receptor for PfRh4 is neuraminidase resistant, trypsin sensitive, and chymotrypsin sensitive (Fig. 1C, top). The same binding eluates were probed with an anti-EBA-175 antibody (Fig. 1C, bottom). This showed that EBA-175 bound to untreated erythrocytes but not to neuraminidase-treated erythrocytes serving as controls for the specificity of the enzyme treatments and any nonspecific carryover of invasion ligands into the binding eluates. These results confirmed that PfRh4 released into the supernatant of P. falciparum cultures was able to bind to human erythrocytes.
PfRh4 binds to the erythrocyte surface through its N-terminal region.
The Rh family of proteins consists of several high-molecular-mass proteins, with PfRh4 itself being a 205-kDa protein (46). To narrow the binding domain of PfRh4, we expressed an 88-kDa region of PfRh4 (aa 28 to 766) tagged with an amino-terminal six-His tag (RH4.9) (Fig. 2A). This recombinant protein was expressed in Escherichia coli and purified from the soluble lysate using an Ni-nitrilotriacetic acid column with a second-step purification on a gel filtration column. The identity of the purified protein was confirmed by mass spectroscopy analysis (data not shown).
FIG. 2.
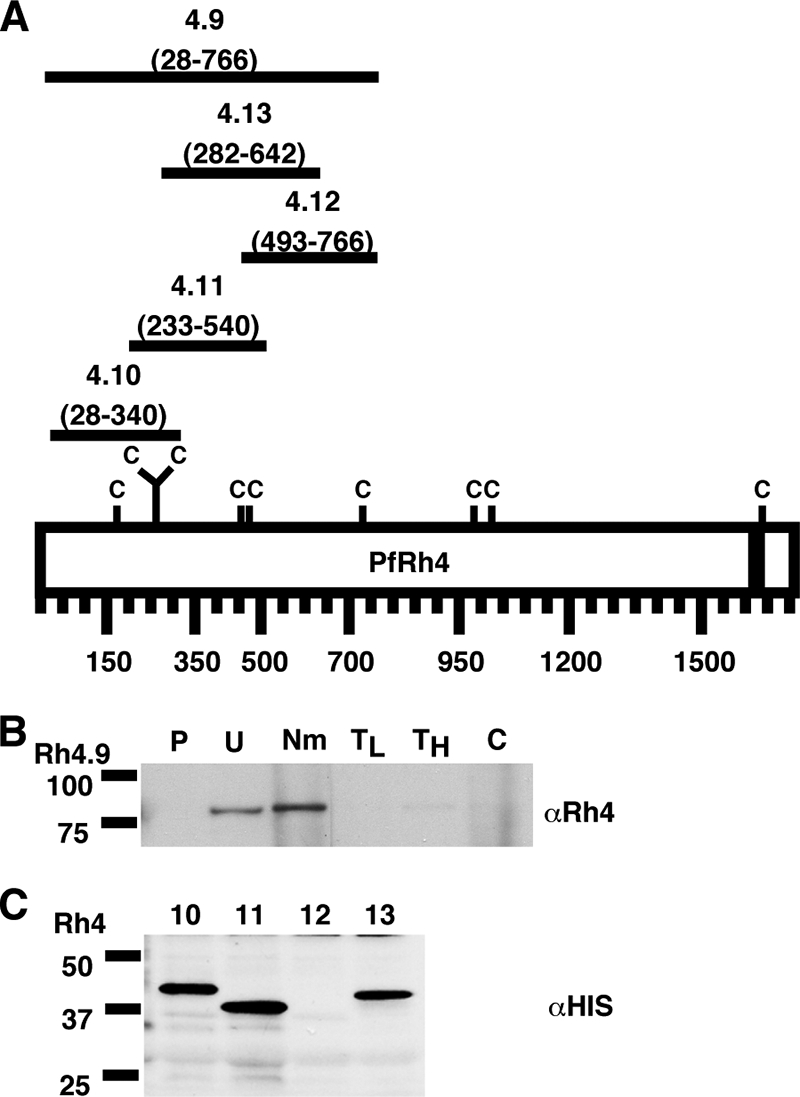
PfRh4 binds to the erythrocyte surface through its N-terminal region. (A) Schematic representation of the various six-His-tagged PfRh4 recombinant proteins. The C denotes cysteine residues, and the black bar represents the transmembrane domain of PfRh4. The number below each fusion protein indicates the amino acid sequence that it encompasses. (B) rRh4.9 binds erythrocytes in a manner similar to that of native PfRh4. Immunodetection of the recombinant fusion protein with anti-Rh4 (αRh4) antibodies after binding and elution from untreated and enzyme-treated erythrocytes is shown. Lanes: P, proteins eluted from PBS control; U, untreated erythrocytes; Nm, neuraminidase; TL, low trypsin; TH, high trypsin; and C, chymotrypsin-treated erythrocytes. Low trypsin and high tryspin refer to trypsin treatments with 0.1 and 1.5 mg/ml of enzyme, respectively. Molecular masses are indicated on the left (in kDa). (C) Minimal binding domain of PfRh4. Binding of six-His-tagged recombinant PfRh4 proteins (Rh4.10, Rh4.11, Rh4.12, and Rh4.13) to untreated erythrocytes was detected using mouse monoclonal anti-His5 (αHis) antibodies. Molecular masses are indicated on the left (in kDa).
When Rh4.9 was incubated with untreated erythrocytes in an erythrocyte binding assay, we found that it bound to the surfaces of the erythrocytes (Fig. 2B). Furthermore, Rh4.9 bound to erythrocytes treated with neuraminidase, but not to erythrocytes treated with trypsin and chymotrypsin (Fig. 2B). These binding characteristics are identical to the enzyme specificity seen with native PfRh4 binding, showing that this 88-kDa region of PfRh4 is sufficient for binding to erythrocytes and recognition of the PfRh4 erythrocyte receptor.
To further delineate the binding region of PfRh4, we expressed three overlapping recombinant proteins spanning Rh4.9: Rh4.10 (aa 28 to 340), Rh4.11 (aa 233 to 540), and Rh4.12 (aa 493 to 700) (Fig. 2A). As shown in Fig. 2C, only Rh4.10 and Rh4.11 bound to the surfaces of the erythrocytes, suggesting that the most C-terminal region of Rh4.9 is not required for binding. In addition, we expressed a region of PfRh4 that contains homology to Plasmodium vivax reticulocyte binding protein 1 (PvRBP1) as Rh4.13 (aa 282 to 642) and showed that this recombinant protein also bound erythrocytes (Fig. 2C). This result is consistent with previous results that defined a smaller recombinant fusion within this region that had erythrocyte binding capabilities (17). Combining all these analyses, we propose that the minimal region required for PfRh4 binding to its erythrocyte receptor is between aa 282 and 340.
Reactivity of recombinant rRh4 with human immune sera.
To determine whether antibodies against PfRh4 were elicited during a natural infection with P. falciparum, the rRh4.9 protein, which contains the erythrocyte binding domain, was tested for reactivity with sera collected from malaria-exposed adults from the Madang area in Papua New Guinea. By ELISA, we measured the levels of antibodies to recombinant protein in sera from 13 Madang adults with various degrees of past exposure to P. falciparum (Fig. 3). Substantial levels of IgG binding were detected in most samples (9/13 samples had ODs of >0.5), and all 13 samples were positive for IgG to Rh4.9 (positive was defined as an OD greater than the mean of Melbourne control samples M1 to M7 [OD = 0.156] plus 3 standard deviations).
FIG. 3.

Reactivities of human antibodies to rRh4.9. The reactivity of human IgG to rRh4.9 was measured by ELISA using purified rRh4.9 protein. All samples were tested in duplicate and adjusted for background reactivity. The error bars represent the ranges of two duplicates. The samples tested were from malaria-exposed residents of Madang, Papua New Guinea (numbered samples), and nonexposed Melbourne residents (M1 to M7). OD 405, OD at 405 nm.
Antibodies to the PfRh4 binding domain inhibit binding to the surfaces of erythrocytes.
We wanted to determine if antibodies raised to the binding region of PfRh4 have the ability to inhibit native PfRh4 erythrocyte binding capabilities. To this end, rabbit polyclonal antisera were raised against recombinant protein Rh4.9. Purified IgG raised against PfRh4 was incubated with Western blots of parasite proteins isolated from saponin-treated schizonts, as well as parasite proteins released into the culture supernatant. As expected the anti-PfRh4 antibodies detected the doublet bands in saponin-treated schizont pellets and a singlet 160-kDa band within culture supernatants (data not shown).
For the erythrocyte binding antibody inhibition assay, we preincubated IgG-purified anti-PfRh4 antibodies in various final concentrations (0 to 1.5 mg/ml) with 3D7 culture supernatants before proceeding with the standard erythrocyte binding assay. In Fig. 4A (left), we show that native PfRh4 binding to the surfaces of erythrocytes was blocked by the addition of anti-PfRh4 antibodies. As increasing amounts of anti-PfRh4 antibodies were added, the inhibition of PfRh4 binding to erythrocytes was also enhanced. Complete inhibition of binding was attained when a concentration of more than 0.05 mg/ml of antibody was used (Fig. 4A, right). The same binding eluates were probed with anti-EBA-175 antibodies, which showed that EBA-175 binding to erythrocytes was not perturbed, evidence that the inhibition is specific to PfRh4 (Fig. 4B). In addition, similar concentrations of IgG purified from preimmunization rabbit serum did not cause any inhibition of PfRh4 erythrocyte binding (Fig. 4C).
FIG. 4.

Antibodies raised to the Rh4.9 binding domain inhibit PfRh4 binding to the surfaces of erythrocytes. (A) Anti-Rh4 (αRh4) IgG at final concentrations of 0.0008 to 0.024 mg/ml (left) and 0 to 1.5 mg/ml (right) was incubated with 250 μl of 3D7 invasion supernatants prior to the erythrocyte binding assay. Immunodetection of parasite proteins with anti-Rh4 antibodies after binding and elution from untreated erythrocytes is shown. (B) Anti-Rh4 IgG at final concentrations of 0.0008 to 0.024 mg/ml (left) and 0 to 1.5 mg/ml (right) was incubated with 250 μl of 3D7 invasion supernatants prior to the erythrocyte binding assay. Immunodetection of parasite proteins with anti-EBA-175 antibodies after binding and elution from untreated erythrocytes is shown. (C) Nonimmune IgG from normal rabbit serum (NRS) at final concentrations of 0.0008 to 0.024 mg/ml (left) and 0 to 1.5 mg/ml (right) was incubated with 250 μl of 3D7 invasion supernatants prior to the erythrocyte binding assay. Immunodetection of parasite proteins with anti-Rh4 antibodies after binding and elution from untreated erythrocytes is shown. The concentrations mentioned above refer to the total IgG purified using protein G columns.
Antibodies to the PfRh4 binding domain inhibit parasite invasion.
Since anti-PfRh4 antibodies block native PfRh4 binding to erythrocytes, we sought to determine if these same antibodies could inhibit parasite invasion in vitro. We analyzed the PfRh4 rabbit antibody effects on invasion by W2mef and W2mefΔRh4, which have an SA-dependent invasion phenotype, and 3D7 and W2mefΔ175, which invade using SA-independent interactions. Upon incubation of anti-PfRh4 IgG-purified antibodies with the parasite strains and untreated erythrocytes, only 3D7 showed modest inhibition of parasite invasion (23%) (Fig. 5A). As the parasites utilize several different receptor-ligand interactions, we treated the erythrocytes with neuraminidase prior to the invasion assay to force the parasites to use SA-independent interactions. As a result, invasion by the SA-dependent parasites W2mef and W2mefΔRh4 was abolished due to the removal of SA moieties, and therefore, these strains were removed from further analyses. However for 3D7 and W2mefΔ175, parasite invasion was further inhibited by anti-PfRh4 antibodies when erythrocytes were treated with neuraminidase (Fig. 5B). Inhibition of parasite invasion increased with further bleeds, with third bleeds exhibiting up to 78% inhibition in 3D7 and 49% inhibition in W2mefΔ175, showing that increased inhibition may be correlated with increased immune response to PfRh4. These data are in contrast with previous data, which showed that anti-PfRh4 antibodies raised against aa 328 to 588 in both rats and rabbits did not inhibit parasite invasion in vitro (17).
FIG. 5.

Strain-specific invasion inhibition of parasite growth using PfRh4 antibodies. (A) Parasite growth for the 3D7, W2mef, W2mefΔ175, and W2mefΔRh4 parasite lines grown in the presence of purified IgG from rabbit prebleed serum (NRS) or anti-Rh4 purified IgG antibodies (αRh4) in normal erythrocytes. (B) Growth of the 3D7 and W2mefΔ175 lines in neuraminidase-treated erythrocytes in the presence of purified IgG from rabbit preimmune serum, purified IgG from second-bleed serum, and purified IgG from third-bleed serum from rabbits immunized with PfRh4. (C) Growth of the 3D7 parasite line in the presence of a dilution series for purified nonspecific IgG from rabbit prebleed serum (NRS) and purified anti-Rh4 IgG antibodies (αRh4) in neuraminidase-treated erythrocytes shows that Rh4 antibody inhibition of parasite growth is concentration dependent. The final concentrations of IgG antibodies ranged from 0 to 2 mg/ml in each invasion assay. For all panels, parasite growth was measured as a percentage of the mean parasite growth for four wells, with nonspecific IgG from rabbit prebleed serum control added in each experiment. The error bars represent the standard error of the mean for duplicate wells in two independent experiments. The concentrations mentioned above refer to the total IgG purified using protein G columns.
To determine what effect the antibody concentration has on invasion inhibition, we titrated out the amount of purified IgG from 2 to 0 mg/ml for each invasion assay. As seen in Fig. 5C, addition of more anti-PfRh4 antibodies resulted in an increase in invasion inhibition in 3D7 grown in neuraminidase-treated erythrocytes (black circles). As a control, similar amounts of IgG-purified rabbit prebleed sera were added to the assay, with no effect on 3D7 parasite invasion into erythrocytes evident (Fig. 5C).
DISCUSSION
PfRh proteins play key roles in merozoite invasion through their interaction with specific host erythrocyte receptors. In particular, PfRh4 appears to be essential to the SA-independent pathway, as disruption of the gene results in the inability of W2mef parasites to switch to this invasion pathway (46). For this study, we sought to examine the role of anti-PfRh4 antibodies in inhibition of host ligand interactions and parasite invasion in vitro as a framework for how inhibitory antibodies against PfRh4 may be effective in inhibiting parasite growth in the human host. We definitively showed the protein profile of PfRh4 in schizont pellets and invasion supernatants in multiple strains as a means to validate the specificity of our anti-PfRh4 antibodies. We defined the minimal binding domain of PfRh4 that bound to erythrocytes through the recognition of a receptor that is neuraminidase resistant but trypsin and chymotrypsin sensitive. Our results also showed that serum antibodies from malaria-exposed individuals reacted with the binding domain of PfRh4. Rabbit antibodies raised against the binding domain of PfRh4 blocked the interaction between PfRh4 and the erythrocyte receptor and inhibited SA-independent parasite invasion of neuraminidase-treated erythrocytes using 3D7 and W2mefΔ175.
We followed PfRh4 during schizont development and subsequent merozoite invasion of the host erythrocyte. In PlasmoDB, the Plasmodium Genome Resource (www.plasmodb.org), PfRh4 is annotated as a 205-kDa protein. In saponin-lysed schizont pellets, PfRh4 is detected as a doublet of 190 and 180 kDa (Fig. 1A) (46). Cleavage of the PfRh4 signal peptide at aa 27 yields a peptide of approximately 3.3 kDa. Therefore, the doublet bands must undergo further processing at the amino terminus that may be important for the function of PfRh4. Upon invasion into erythrocytes, PfRh4 is further proteolytically cleaved and shed into the culture medium, and it migrates as a single band at 160 kDa (Fig. 1A). Proteolytic cleavage of PfRh4 occurs at the C-terminal end, as a 3′ C-terminal six-His tag on native PfRh4 is not detectable in culture supernatants but is present in schizont pellets (T. Triglia, W.-H. Tham, A. N. Hodder, and A. F. Cowman, unpublished data). This C-terminal processing is thought to be mediated by the rhomboid protease PfROM4 and may be crucial for the successful completion of the invasion process (3).
Previous reports on the identification of PfRh4 showed that it migrates as a protein of >200 kDa in both saponin-lysed schizont pellets and culture supernatants (16, 17, 24). In contrast, multiple lines of evidence in this study suggest that PfRh4 migrates as a 180/190-kDa doublet in saponin-lysed schizont pellets and as a 160-kDa band in culture supernatants. We believe that our analyses of the migration of PfRh4 are definitive for the following reasons. First, we observed the same protein profile in the schizont pellet and the culture supernatant in three independent strains, 3D7, HB3, and W2mefΔ175. This was expected, given that the protein sequences of PfRh4 in these three strains are almost identical and yield the same predicted molecular weight for PfRh4 (Fig. 1A). Second, we used three separate antibodies raised to distinct regions of PfRh4, and they all showed the same profile of PfRh4 harvested from saponin pellets and culture supernatants (Fig. 1B). Third, our antibodies did not detect the presence of any specific PfRh4 bands in W2mefΔRh4, a strain that lacks expression of this protein (Fig. 1A). As antibodies raised against PfRh proteins can display major cross-reactivity in immunoblots, we believe that these are crucial controls in interpreting the specific reactivities of antibodies to PfRh4. All of our different anti-PfRh4 rabbit sera recognized PfRh4 in all strains so far tested, including 3D7, HB3, 7G8, W2mefΔ175, T994R, and FCR3, so the protein is immunologically cross-reactive across all strains (46). Furthermore, the discrepancies observed between our study and previous publications (17) are not a result of strain differences, as in some cases, genetically identical strains were used in the analyses.
Parasite invasion can be broadly categorized into two separate pathways: an SA-dependent pathway and an SA-independent pathway. Of the known invasion ligands, EBA-175, EBA-181, EBA-140, and PfRh1 are involved in mediating invasion via the SA-dependent pathway (13, 27, 42, 50). On the other hand, PfRh4 and PfRh2b are important in mediating invasion in the SA-independent pathway (14, 46). Our invasion assays were performed using W2mef and W2mefΔRh4, which invade via SA-independent interactions, and W2mefΔ175 and 3D7, which can invade using SA-independent interactions. It was not surprising that antibodies against PfRh4 did not affect the growth of W2mef or W2mefΔRh4, since PfRh4 was not expressed by these parasites. Anti-PfRh4 antibodies produced only modest inhibition of 3D7 grown in untreated erythrocytes. This could be due to the fact that EBAs and PfRh1 are still expressed and functional and may allow the parasite to overcome antibodies that block the function of PfRh4. The antibody inhibition assays showed that PfRh4 contributes to approximately a quarter of the invasion events when the SA pathway is intact (Fig. 5A). To completely remove the dependence on SA moieties in invasion, we treated the erythrocyte surfaces with neuraminidase. Upon treatment, anti-PfRh4 antibodies inhibited invasion by 78% in the 3D7 strain, suggesting that PfRh4 plays a major role in the SA-independent pathway in this strain. Anti-PfRh4 antibodies also did not affect growth in W2mefΔ175 grown in untreated erythrocytes (Fig. 5A). Upon neuraminidase treatment, anti-PfRh4 antibodies blocked approximately half the invasion events (Fig. 5B). In contrast, previous results showed that anti-PfRh4 antibodies had no effect on parasite invasion (17). These antibodies were unable to recognize the processed form of PfRh4 and showed a different banding pattern than in our analyses of PfRh4 (17).
There are circumstances in human populations under which receptor availability may be limited as a result of selection against P. falciparum infection. For example, Melanesian populations express a mutant form of glycophorin C, which is the identified host receptor for EBA-140 (26, 27, 29). This mutation effectively eliminates binding of EBA-140 to glycophorin C, thereby inactivating the contribution of this invasion pathway (27). Receptor availability may also be limited through the acquisition of inhibitory antibodies to parasite ligands involved in merozoite invasion that would block utilization of particular invasion pathways, thus limiting the pathways (14). Previous studies showed that inhibitory antibodies to both SA-dependent and SA-independent invasion pathways are acquired by exposed humans and that antibodies to the EBAs and PfRh proteins are acquired (39). Antibodies from malaria-exposed individuals that showed greater inhibition of W2mef than of W2mefΔ175 or W2mef selected on neuraminidase-treated erythrocytes may point to inhibitory antibodies acquired against ligands in the SA-dependent pathway (EBA-175, EBA-140, EBA-181, and PfRh1). In the cases where utilization of the SA-dependent pathway is limited, upregulation of PfRh4 protein expression to enable a switch to the SA-independent pathway may be crucial for parasite survival (46). Importantly, certain field isolates have high levels of PfRh4 expression, suggesting that they may be utilizing the SA-independent invasion pathway (33).
One possible mechanism by which anti-PfRh4 antibodies block invasion is through binding to a region of PfRh4 involved in interaction with its host erythrocyte receptor, thus blocking functional contact. In support of this hypothesis, we showed that anti-PfRh4 antibodies blocked binding of native PfRh4 to erythrocytes (Fig. 4A). This may be similar to the mechanism by which anti-F2/EBA-175 antibodies inhibit parasite invasion. The binding region of EBA-175 to glycophorin A involves a cysteine-rich region consisting of two domains called F1 and F2, and antibodies to the F2 domain of EBA-175 can partially inhibit invasion of P. falciparum merozoites into human erythrocytes (32, 36). From the binding and invasion inhibition assays, we observed that the inhibition was dependent on the antibody concentration, where more antibodies added to the assays resulted in enhanced inhibition (Fig. 4A and 5C). At concentrations of anti-PfRh4 antibody that completely abolished PfRh4 erythrocyte binding (>0.05 mg/ml) (Fig. 4A, right), we observed inhibition of invasion in 3D7 grown in neuraminidase-treated erythrocytes (Fig. 5C), suggesting that these two phenomena are correlated.
We observed that sera from immune individuals in Madang Province, Papua New Guinea, contained antibodies that react strongly against the binding domain of PfRh4 (Fig. 3). A previous study using recombinant PfRh4 that lies outside the binding domain showed that the level of antibodies to this domain is associated with increasing age of infected individuals (39). This finding suggests that the acquired antibodies to PfRh4 may be consistent with increasing exposure and the acquisition of immunity. Though it remains to be tested, it is highly likely that the PfRh4 binding domain provides a target of inhibitory antibodies and that these acquired antibodies may be correlated with increased exposure in the human host.
It is clear that EBAs and PfRhs are important parasite ligands in the invasion process. These ligands are located at the merozoite apical tip, where they are able to recognize and bind to their respective host erythrocyte receptors. For the merozoite to successfully invade, these invasion ligands are proteolytically cleaved and shed into the surrounding medium. Recent studies have shown that the use of alternate erythrocyte invasion pathways by P. falciparum alters the efficacy of human invasion-inhibitory antibodies and that antibodies to ligands of both SA-dependent and SA-independent invasion are acquired (39). This finding suggests that the parasite evades the human immune response through the switching of invasion pathways and the utilization of invasion ligands (46). Therefore, any strategy for vaccine development must target both sets of parasite ligands involved in these two independent invasion pathways. Encouragingly, rabbit antibodies against EBA-175, EBA-140, PfRh1, PfRh2b, and PfRh4 have been shown to inhibit parasite invasion in vitro, suggesting that a multivalent vaccine targeting different EBA and PfRh proteins to cover the spectrum of invasion phenotypes may be achievable.
Acknowledgments
We thank Tony Triglia for antibodies and the Red Cross Blood Service (Melbourne, Australia) for the supply of erythrocytes and serum. We thank the Monoclonal Facility at the Walter and Eliza Hall Institute for generation of monoclonal antibodies.
Infrastructure was supported by the Victoria State Government OIS and NHMRC IRIISS (no. 361646) grants. A.F.C. is a Howard Hughes International Scholar and an Australia Fellow from the National Health and Medical Research Council (NHMRC), Australia. J.G.B. is supported by an NHMRC Career Development Award. This work was supported by the NHMRC.
Editor: W. A. Petri, Jr.
Footnotes
Published ahead of print on 23 March 2009.
REFERENCES
- 1.Adams, J. H., D. E. Hudson, M. Torii, G. E. Ward, T. E. Wellems, M. Aikawa, and L. H. Miller. 1990. The duffy receptor family of Plasmodium knowlesi is located within the micronemes of invasive malaria merozoites. Cell 63141-153. [DOI] [PubMed] [Google Scholar]
- 2.Adams, J. H., B. K. L. Sim, S. A. Dolan, X. Fang, D. C. Kaslow, and L. H. Miller. 1992. A family of erythrocyte binding proteins of malaria parasites. Proc. Natl. Acad. Sci. USA 897085-7089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baker, R. P., R. Wijetilaka, and S. Urban. 2006. Two Plasmodium rhomboid proteases preferentially cleave different adhesins implicated in all invasive stages of malaria. PLoS Pathog. 2e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baum, J., L. Chen, J. Healer, S. Lopaticki, M. Boyle, T. Triglia, F. Ehlgen, S. A. Ralph, J. G. Beeson, and A. F. Cowman. 2009. Reticulocyte-binding protein homologue 5: an essential adhesin involved in invasion of human erythrocytes by Plasmodium falciparum. Int. J. Parasitol. 39371-380. [DOI] [PubMed] [Google Scholar]
- 5.Baum, J., A. G. Maier, R. T. Good, K. M. Simpson, and A. F. Cowman. 2005. Invasion by P. falciparum merozoites suggests a hierarchy of molecular interactions. PLoS Pathog. 1e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baum, J., M. Pinder, and D. J. Conway. 2003. Erythrocyte invasion phenotypes of Plasmodium falciparum in The Gambia. Infect. Immun. 711856-1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blackman, M. J. 2004. Proteases in host cell invasion by the malaria parasite. Cell Microbiol. 6893-903. [DOI] [PubMed] [Google Scholar]
- 8.Butcher, G. A., G. H. Mitchell, and S. Cohen. 1973. Mechanism of host specificity in malarial infection. Nature 24440-41. (Letter.) [DOI] [PubMed] [Google Scholar]
- 9.Cowman, A. F., and B. S. Crabb. 2006. Invasion of red blood cells by malaria parasites. Cell 124755-766. [DOI] [PubMed] [Google Scholar]
- 10.Deans, A. M., S. Nery, D. J. Conway, O. Kai, K. Marsh, and J. A. Rowe. 2007. Invasion pathways and malaria severity in Kenyan Plasmodium falciparum clinical isolates. Infect. Immun. 753014-3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dolan, S. A., L. H. Miller, and T. E. Wellems. 1990. Evidence for a switching mechanism in the invasion of erythrocytes by Plasmodium falciparum. J. Clin. Investig. 86618-624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dolan, S. A., J. L. Proctor, D. W. Alling, Y. Okubo, T. E. Wellems, and L. H. Miller. 1994. Glycophorin B as an EBA-175 independent Plasmodium falciparum receptor of human erythrocytes. Mol. Biochem. Parasitol. 6455-63. [DOI] [PubMed] [Google Scholar]
- 13.Duraisingh, M. T., A. G. Maier, T. Triglia, and A. F. Cowman. 2003. Erythrocyte-binding antigen 175 mediates invasion in Plasmodium falciparum utilizing sialic acid-dependent and -independent pathways. Proc. Natl. Acad. Sci. USA 1004796-4801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duraisingh, M. T., T. Triglia, S. A. Ralph, J. C. Rayner, J. W. Barnwell, G. I. McFadden, and A. F. Cowman. 2003. Phenotypic variation of Plasmodium falciparum merozoite proteins directs receptor targeting for invasion of human erythrocytes. EMBO J. 221047-1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao, X., K. P. Yeo, S. S. Aw, C. Kuss, J. K. Iyer, S. Genesan, R. Rajamanonmani, J. Lescar, Z. Bozdech, and P. R. Preiser. 2008. Antibodies targeting the PfRH1 binding domain inhibit invasion of Plasmodium falciparum merozoites. PLoS Pathog. 4e1000104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gaur, D., T. Furuya, J. Mu, L. B. Jiang, X. Z. Su, and L. H. Miller. 2006. Upregulation of expression of the reticulocyte homology gene 4 in the Plasmodium falciparum clone Dd2 is associated with a switch in the erythrocyte invasion pathway. Mol. Biochem. Parasitol. 145205-215. [DOI] [PubMed] [Google Scholar]
- 17.Gaur, D., S. Singh, S. Singh, L. Jiang, A. Diouf, and L. H. Miller. 2007. Recombinant Plasmodium falciparum reticulocyte homology protein 4 binds to erythrocytes and blocks invasion. Proc. Natl. Acad. Sci. USA 10417789-17794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilberger, T. W., J. K. Thompson, M. B. Reed, R. T. Good, and A. F. Cowman. 2003. The cytoplasmic domain of the Plasmodium falciparum ligand EBA-175 is essential for invasion but not protein trafficking. J. Cell Biol. 162317-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilberger, T. W., J. K. Thompson, T. Triglia, R. T. Good, M. T. Duraisingh, and A. F. Cowman. 2003. A novel erythrocyte binding antigen-175 paralogue from Plasmodium falciparum defines a new trypsin-resistant receptor on human erythrocytes. J. Biol. Chem. 27814480-14486. [DOI] [PubMed] [Google Scholar]
- 20.Gratzer, W. B., and A. R. Dluzewski. 1993. The red blood cell and malaria parasite invasion. Semin. Hematol. 30232-247. [PubMed] [Google Scholar]
- 21.Hadley, T. J., F. W. Klotz, G. Pasvol, J. D. Haynes, and M. H. McGinniss. 1987. Falciparum malaria parasites invade erythrocytes that lack glycophorin A and B (MkMk). Strain differences indicate receptor heterogeneity and two pathways for invasion. J. Clin. Investig. 801190-1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hayton, K., D. Gaur, A. Liu, J. Takahashi, B. Henschen, S. Singh, L. Lambert, T. Furuya, R. Bouttenot, M. Doll, F. Nawaz, J. Mu, L. Jiang, L. H. Miller, and T. E. Wellems. 2008. Erythrocyte binding protein PfRH5 polymorphisms determine species-specific pathways of Plasmodium falciparum invasion. Cell Host Microbe 440-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Howard, R. J., J. D. Haynes, M. H. McGinniss, and L. H. Miller. 1982. Studies on the role of red blood cell glycoproteins as receptors for invasion by Plasmodium falciparum merozoites. Mol. Biochem. Parasitol. 6303-315. [DOI] [PubMed] [Google Scholar]
- 24.Kaneko, O., J. Mu, T. Tsuboi, X. Su, and M. Torii. 2002. Gene structure and expression of a Plasmodium falciparum 220-kDa protein homologous to the Plasmodium vivax reticulocyte binding proteins. Mol. Biochem. Parasitol. 121275-278. [DOI] [PubMed] [Google Scholar]
- 25.Lanzillotti, R., and T. L. Coetzer. 2006. The 10 kDa domain of human erythrocyte protein 4.1 binds the Plasmodium falciparum EBA-181 protein. Malaria J. 5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lobo, C. A., M. Rodriguez, M. Reid, and S. Lustigman. 2003. Glycophorin C is the receptor for the Plasmodium falciparum erythrocyte binding ligand PfEBP-2 (baebl). Blood 66. [DOI] [PubMed] [Google Scholar]
- 27.Maier, A. G., M. T. Duraisingh, J. C. Reeder, S. S. Patel, J. W. Kazura, P. A. Zimmerman, and A. F. Cowman. 2003. Plasmodium falciparum erythrocyte invasion through glycophorin C and selection for Gerbich negativity in human populations. Nat. Med. 987-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mayer, D. C., O. Kaneko, D. E. Hudson-Taylor, M. E. Reid, and L. H. Miller. 2001. Characterization of a Plasmodium falciparum erythrocyte-binding protein paralogous to EBA-175. Proc. Natl. Acad. Sci. USA 985222-5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mayer, D. C., J. B. Mu, X. Feng, X. Z. Su, and L. H. Miller. 2002. Polymorphism in a Plasmodium falciparum erythrocyte-binding ligand changes its receptor specificity. J. Exp. Med. 1961523-1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller, L. H., F. M. McAuliffe, and S. J. Mason. 1977. Erythrocyte receptors for malaria merozoites. Am. J. Trop. Med. Hyg. 26204-208. [DOI] [PubMed] [Google Scholar]
- 31.Mitchell, G. H., T. J. Hadley, M. H. McGinniss, F. W. Klotz, and L. H. Miller. 1986. Invasion of erythrocytes by Plasmodium falciparum malaria parasites: evidence for receptor heterogeneity and two receptors. Blood 671519-1521. [PubMed] [Google Scholar]
- 32.Narum, D. L., D. Haynes, S. Fuhrmann, K. Moch, H. Liang, S. L. Hoffman, and B. K. L. Sim. 2000. Antibodies against the Plasmodium falciparum receptor binding domain of EBA-175 block invasion pathways that do not involve sialic acids. Infect. Immun. 681964-1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nery, S., A. M. Deans, M. Mosobo, K. Marsh, J. A. Rowe, and D. J. Conway. 2006. Expression of Plasmodium falciparum genes involved in erythrocyte invasion varies among isolates cultured directly from patients. Mol. Biochem. Parasitol. 149208-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O'Donnell, R. A., F. Hackett, S. A. Howell, M. Treeck, N. Struck, Z. Krnajski, C. Withers-Martinez, T. W. Gilberger, and M. J. Blackman. 2006. Intramembrane proteolysis mediates shedding of a key adhesin during erythrocyte invasion by the malaria parasite. J. Cell Biol. 1741023-1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okoyeh, J. N., C. R. Pillai, and C. E. Chitnis. 1999. Plasmodium falciparum field isolates commonly use erythrocyte invasion pathways that are independent of sialic acid residues of glycophorin A. Infect. Immun. 675784-5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pandey, K., S. Singh, P. Pattnaik, C. Pillai, U. Pillai, A. Lynn, S. Jain, and C. Chitnis. 2002. Bacterially expressed and refolded receptor binding domain of Plasmodium falciparum EBA-175 elicits invasion inhibitory antibodies. Mol. Biochem. Parasitol. 12323. [DOI] [PubMed] [Google Scholar]
- 37.Pasvol, G. 1984. Receptors on red cells for Plasmodium falciparum and their interaction with merozoites. Phil. Trans. R. Soc. Lond. B 307189-200. [DOI] [PubMed] [Google Scholar]
- 38.Pasvol, G., M. Jungery, D. J. Weatherall, S. F. Parsons, D. J. Anstee, and M. J. A. Tanner. 1982. Glycophorin as a possible receptor for Plasmodium falciparum. Lancet 2947-950. [DOI] [PubMed] [Google Scholar]
- 39.Persson, K. E., F. J. McCallum, L. Reiling, N. A. Lister, J. Stubbs, A. F. Cowman, K. Marsh, and J. G. Beeson. 2008. Variation in use of erythrocyte invasion pathways by Plasmodium falciparum mediates evasion of human inhibitory antibodies. J. Clin. Investig. 118342-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rayner, J. C., M. R. Galinski, P. Ingravallo, and J. W. Barnwell. 2000. Two Plasmodium falciparum genes express merozoite proteins that are related to Plasmodium vivax and Plasmodium yoelii adhesive proteins involved in host cell selection and invasion. Proc. Natl. Acad. Sci. USA 979648-9653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rayner, J. C., E. Vargas-Serrato, C. S. Huber, M. R. Galinski, and J. W. Barnwell. 2001. A Plasmodium falciparum homologue of Plasmodium vivax reticulocyte binding protein (PvRBP1) defines a trypsin-resistant erythrocyte invasion pathway. J. Exp. Med. 1941571-1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reed, M. B., S. R. Caruana, A. H. Batchelor, J. K. Thompson, B. S. Crabb, and A. F. Cowman. 2000. Targeted disruption of an erythrocyte binding antigen in Plasmodium falciparum is associated with a switch toward a sialic acid independent pathway of invasion. Proc. Natl. Acad. Sci. USA 977509-7514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodriguez, M., S. Lustigman, E. Montero, Y. Oksov, and C. A. Lobo. 2008. PfRH5: a novel reticulocyte-binding family homolog of Plasmodium falciparum that binds to the erythrocyte, and an investigation of its receptor. PLoS ONE 3e3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sim, B. K. L., C. E. Chitnis, K. Wasniowska, T. J. Hadley, and L. H. Miller. 1994. Receptor and ligand domains for invasion of erythrocytes by Plasmodium falciparum. Science 2641941-1944. [DOI] [PubMed] [Google Scholar]
- 45.Sim, B. K. L., P. A. Orlandi, J. D. Haynes, F. W. Klotz, J. M. Carter, D. Camus, M. E. Zegans, and J. D. Chulay. 1990. Primary structure of the 175K Plasmodium falciparum erythrocyte binding antigen and identification of a peptide which elicits antibodies that inhibit malaria merozoite invasion. J. Cell Biol. 1111877-1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stubbs, J., K. M. Simpson, T. Triglia, D. Plouffe, C. J. Tonkin, M. T. Duraisingh, A. G. Maier, E. A. Winzeler, and A. F. Cowman. 2005. Molecular mechanism for switching of P. falciparum invasion pathways into human erythrocytes. Science 3091384-1387. [DOI] [PubMed] [Google Scholar]
- 47.Taylor, H. M., T. Triglia, J. Thompson, M. Sajid, R. Fowler, M. E. Wickham, A. F. Cowman, and A. A. Holder. 2001. Plasmodium falciparum homologue of the genes for Plasmodium vivax and Plasmodium yoelii adhesive proteins, which is transcribed but not translated. Infect. Immun. 693635-3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thompson, J. K., T. Triglia, M. B. Reed, and A. F. Cowman. 2001. A novel ligand from Plasmodium falciparum that binds to a sialic acid-containing receptor on the surface of human erythrocytes. Mol. Microbiol. 4147-58. [DOI] [PubMed] [Google Scholar]
- 49.Trager, W., and J. B. Jensen. 1976. Human malaria parasites in continuous culture. Science 193673-675. [DOI] [PubMed] [Google Scholar]
- 50.Triglia, T., M. T. Duraisingh, R. T. Good, and A. F. Cowman. 2005. Reticulocyte-binding protein homologue 1 is required for sialic acid-dependent invasion into human erythrocytes by Plasmodium falciparum. Mol. Microbiol. 55162-174. [DOI] [PubMed] [Google Scholar]
- 51.Triglia, T., J. Thompson, S. R. Caruana, M. Delorenzi, T. Speed, and A. F. Cowman. 2001. Identification of proteins from Plasmodium falciparum that are homologous to reticulocyte binding proteins in Plasmodium vivax. Infect. Immun. 691084-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Triglia, T., J. K. Thompson, and A. F. Cowman. 2001. An EBA175 homologue which is transcribed but not translated in erythrocytic stages of Plasmodium falciparum. Mol. Biochem. Parasitol. 11655-63. [DOI] [PubMed] [Google Scholar]
- 53.Walliker, D., I. A. Quakyi, T. E. Wellems, T. F. McCutchan, A. Szarfman, W. T. London, L. M. B. Corcoran, T. R. Burkot, and R. Carter. 1987. Genetic analysis of the human malaria parasite Plasmodium falciparum. Science 2361661-1666. [DOI] [PubMed] [Google Scholar]
- 54.Yeoh, S., R. A. O'Donnell, K. Koussis, A. R. Dluzewski, K. H. Ansell, S. A. Osborne, F. Hackett, C. Withers-Martinez, G. H. Mitchell, L. H. Bannister, J. S. Bryans, C. A. Kettleborough, and M. J. Blackman. 2007. Subcellular discharge of a serine protease mediates release of invasive malaria parasites from host erythrocytes. Cell 1311072-1083. [DOI] [PubMed] [Google Scholar]