

Abstract
A small group of ecotropic murine retroviruses cause a spongiform neurodegenerative disease manifested by tremor, paralysis, and wasting. The neurovirulence of these viruses has long been known to be determined by the sequence of the viral envelope protein, although the nature of the neurotoxicity remains to be clarified. Studies on the neurovirulent viruses FrCasNC and Moloney murine leukemia virus ts1 indicate that the nascent envelope protein misfolds, is retained in the endoplasmic reticulum (ER), and induces an unfolded protein response. In the present study we constructed a series of viruses with chimeric envelope genes containing segments from virulent and avirulent retroviruses. Each of the viruses studied was highly neuroinvasive but differed in the severity of the neurological disease they induced. Only viruses that contained the receptor-binding domain (RBD) of the neurovirulent virus induced neurological disease. Likewise, only viruses containing the RBD of the neurovirulent virus exhibited increased binding of the ER chaperone BiP to the envelope precursor protein and induced the unfolded protein response. Thus, the RBD determined both neurovirulence and folding instability. Among viruses carrying the neurovirulent RBD, the severity of the disease was increased when envelope sequences from the neurovirulent virus outside the RBD were also present. Interestingly, these sequences appeared to further increase the degree of folding instability (BiP binding) of the viral envelope protein. These results provide strong support for the hypothesis that this spongiform neurodegenerative disease represents a virus-induced protein folding disorder.
The accumulation of misfolded proteins in the endoplasmic reticulum (ER) causes ER stress and activates a series of responses known collectively as the unfolded protein response (UPR) (33, 43). The UPR consists of three parallel signal transduction pathways activated by transmembrane proteins projecting into the ER lumen which act as sensors of misfolded proteins. The three sensors—Ire1α, ATF6, and PERK—are maintained in an inactive state by the binding of the major ER chaperone BiP to their luminal domains. The accumulation of misfolded proteins in the ER lumen causes BiP to be displaced from the sensor molecules and bind to exposed hydrophobic patches on protein-folding intermediates. This leads to activation of the sensors and initiation of the downstream signaling pathways. Ire1α an endoribonuclease, triggers the upregulation of the protein degradative machinery leading to the removal of terminally misfolded proteins. ATF6 is an ER resident transcription factor that upregulates the expression of genes encoding ER chaperones, including BiP, which improve the folding efficiency of the ER. PERK, an ER resident kinase, phosphorylates the translation initiation factor eukaryotic translation initiation factor 2α, leading to global downregulation of protein translation and consequently a decrease in the load of client proteins entering the ER. These are adaptive responses that contribute to the reestablishment of a state of protein folding homeostasis. If homeostasis is not achieved, ER linked cell death pathways are activated (29, 44).
Over the last few years, it has become clear that ER stress is associated with a variety of human degenerative diseases. These include disorders as diverse as diabetes, retinitis pigmentosa, brittle bone disease (osteogenesis imperfecta), and a variety of hereditary leukodystrophies (45), as well as Parkinson's and Alzheimer's disease (37). In some of these diseases, ER stress is activated by the accumulation of a mutant protein. In other cases, the ER stress is caused by dysfunction of one of the elements of the UPR, limiting its capacity to respond to demands on the folding machinery. The UPR is also activated by some viruses, including hantavirus, flaviviruses, paramyxoviruses, and Borna virus (17, 29), as well as prion agents (20), although a role for ER stress in disease pathogenesis remains speculative. A possible role for ER stress in the pathogenesis of Borna virus-induced neurodegeneration has been reported (53). However, it is not known whether the insult to the ER is due to misfolding of a viral protein.
The evidence is stronger for a role of ER stress in the pathogenesis of a neurodegenerative disease caused by a small group of murine retroviruses. Derivatives of CasBrE (13), a virus originally isolated from feral mice, and Moloney murine leukemia virus ts1 (MoMLVts1) (54), a temperature-sensitive strain of MoMLV, cause a noninflammatory spongiform encephalopathy resembling histopathologically the prion diseases. The neurovirulence of these viruses is determined by the sequence of the envelope protein (7, 39, 49, 55), although the nature of the toxicity has remained elusive. We have studied the role of the envelope protein in disease pathogenesis using two coisogenic viruses, FrCasNC (9) and F43 (2). FrCasNC contains the envelope gene and 3′pol sequences from the neurovirulent virus CasBrE and causes a rapidly progressive and fatal neurological disease characterized by tremor, paralysis, and wasting. F43 carries the envelope gene and 3′ pol sequences of a nonneurovirulent virus, FMLV57 (38), and is apathogenic. Both viruses are highly neuroinvasive and infect the same spectrum of cells in the brain (2). Transcriptional analysis of brain tissue from infected mice revealed that only the neurovirulent virus induced the UPR (8). Activation of the UPR was found to be a consequence of the folding instability of the FrCasNC envelope protein, which was retained in the ER and degraded by the ubiquitin-proteasome system (9). In addition, histopathologic studies revealed that one of the downstream target genes of the UPR, CHOP, is highly expressed in cells in the brain exhibiting signs of cytopathology (4). In the present study, we further explored the association between protein misfolding and neuropathology. A domain swapping strategy was used to construct a series of highly neuroinvasive viruses carrying F43/FrCasNC sequences. These viruses differed in the severity of the neurological disease they induced. Neuropathogenicity was precipitated only by those viruses carrying the receptor-binding domain (RBD) of the envelope protein from FrCasNC. Of particular interest was the observation that the severity of the neurological disease was directly related to the degree of folding instability of the envelope protein in the ER.
MATERIALS AND METHODS
Construction of chimeric viral genomes.
The cloning strategy used in the present study was similar to that reported previously (2, 9). The genomes of the avirulent viruses FB29 (accession no. NC_001362), FMuLV57 (accession no. X02794) and the neurovirulent virus CasBrE (accession no. X57540) share a common NdeI site in 3′ pol and a ClaI site in 3′ env (see Fig. 1). The parental viruses in the present study, F43 (avirulent) and FrCasNC (neurovirulent), were constructed previously by introduction of the NdeI-ClaI fragments of FMuLV57 and CasBrE, respectively, into the genome of FB29.
FIG. 1.

The neurovirulence of FrCasNC is determined by the sequence of the viral envelope gene, with no contribution by the sequence of 3′ end of the polymerase gene. (A) Schematic diagram of the complete viral genomes bounded by viral long-terminal repeats (LTR) at each end. The names of the viral genes are shown above the diagram of F43. Restriction endonuclease sites used to introduce segments of the CasBrE genome (▪) into that of F43 (□) are shown above the diagram of FrCasNC. A magnified view of the region of FrCasAC around the AvrII site is shown below the diagram of FrCasAC. The AvrII site used to construct FrCasAC is located within the signal sequence of the envelope protein just 3′ of the 3′ end of the polymerase coding sequence. Thus, the virus FrCasAC contains sequences from CasBrE only within the viral envelope coding sequence. (B) Tempo of clinical neurological disease caused by intraperitoneal inoculation of the respective viruses as depicted in two graphs. The upper graph represents early signs of neurological disease (tremor and abnormal abduction reflex of the hind limbs), and the lower graph advanced disease (paralysis and wasting). Whereas F43 was nonpathogenic (n > 60 mice), mice inoculated with FrCasNC (n = 30 mice) or FrCasAC (n = 34 mice) developed neurological disease with indistinguishable kinetics. (C) Results showing that both FrCasNC and FrCasAC induced the UPR marked by the upregulation of BiP and CHOP mRNA. NIH 3T3 cells were infected for 48 h with each virus. Quantitative real-time reverse transcription-PCR was carried out on triplicate cultures as described previously (8). The data were normalized first with β-actin mRNA and are expressed as a fold increase relative to the level of mRNA in mock-infected cells. The graphs represent the cumulative results of three experiments (i.e., nine infections per group). The data were analyzed by one-way ANOVA, and each group was compared to mock-infected cells (*, P < 0.001; NS, P > 0.05).
For the viruses FC424, FC70, and CF70 (see Fig. 2), a one-step ligation was carried out using the restriction sites BstEII near the 3′ end of the coding sequence for the surface glycoprotein (SU) and the ApaI site near the 5′ end of SU coding sequence, both of which are shared by FMuLV57 and CasBrE sequences. For construction of the virus FrCasAC, we used an AvrII site within the sequence encoding the SU signal peptide of CasBrE (nucleotide position 5818). This site is not present in FMuLV57, but was introduced by using PCR-based mutagenesis (QuikChange; Stratagene) with the forward primer 5′- TAAGGGGCCAGGAGGTCCCGCG-3′, the reverse primer 5′- AGCAGAGCGCAGATACCAAATAC-3′, and the FMuLV57 sequence as a template.
FIG. 2.

Neurovirulence is determined by the RBD of CasBrE. Schematic diagrams are shown of the envelope precursor polyproteins (pr85env) of chimeric viruses that exhibited neuroinvasiveness comparable to that of FrCasNC. The boundaries of the surface glycoprotein (SU) and the transmembrane protein (TM) are shown at the top. The brackets denote the boundaries of the domains of the SU protein. The RBD and C-terminal domain (C) are linked by a PRR hinge. Subdomains are shown as gray bars and include the variable regions of SU (VRA, VRC, and VRB) and the fusion peptide (FP) and membrane-spanning domain (MSD) of the TM protein. The numbers just above the diagram of FrCasNC represent the percent difference in the amino acid sequence between FrCasNC and F43 for each restriction enzyme fragment. The dotted lines show the restriction sites used to construct these chimeric envelope genes. Viral RNA was determined at 17 to 20 dpi by quantitative reverse transcription-PCR on RNA extracted from brainstems of mice inoculated intraperitoneally with each virus. Each dot represents a mouse. The distribution of spongiform lesions is shown as dark gray areas in diagrams of parasaggital sections of brains (under the column heading “Spongiosis”). The names of each affected region is shown for the brain of FrCasNC and include brainstem (BS), deep cerebellar nuclei (Cblm), anterior and posterior colliculi (Col), thalamus (Thal), and deep neocortex (NCtx). Histopathology was assessed on no fewer than three mice per group at 17 to 20 dpi.
For viruses CF223, FC223, and CFC a BamHI site at position 6548 of the FMuLV57 genome, near the 3′ end of the coding sequence for the receptor binding domain was introduced into the sequence of CasBrE by using PCR-based mutagenesis (QuikChange). The forward primer was 5′- CCTTTTAGCCGGATCCCAAAAGTG 3′, the reverse primer was 5′- CACTTTTGGGATCCGGCTAAAAG-3′, and the CasBrE sequence was used as a template.
Virus stocks were prepared by transfection of Mus dunni cells with linearized plasmids as described previously (41), and titers of the stocks were determined by a focal immunoassay (5) on NIH 3T3 cells. Titers are expressed as focus-forming units/ml and were as follows: F43 2 × 107, FrCasNC 6 × 106, FrCasAC 4 × 106, FC424 3 × 106, FC223 1 × 107, FC70 4 × 105, CF70 2 × 107, CF223 8 × 106, and CFC 6 × 105. The nomenclature of these viruses is based on the number of the amino acid of the FMuLV57 envelope protein, starting with the first amino acid of the mature protein (i.e., after the signal sequence), as used by Fass et al. (11). Thus, FC223 contains FMuLV57 sequences up to amino acid residue 223, the rest of the envelope protein up to the ClaI site is from CasBrE. Likewise, CF223 contains CasBrE sequences up to amino acid 223, the rest of the envelope protein up to the ClaI site is from FMuLV57. For simplicity, we chose not to use this numbering system for the virus CFC (see Fig. 2) since the name would have been too long.
Mice and virus inoculations.
Inbred Rocky Mountain White mice were bred and raised at the Rocky Mountain Laboratories (RML) and were handled according to policies of the RML Animal Care and Use Committee. Mice were inoculated with virus stocks as described previously (41). Mice were inoculated intraperitoneally on postnatal day 1 with 30 μl of virus stock containing 1.2 × 104 to 6 × 105 focus-forming units.
Evaluation of neurological disease.
Mice were followed for typical clinical signs of neurological disease as described previously (6). The earliest signs include an abnormal abduction reflex of the hind limbs when the mouse is elevated by the tail and a fine intention tremor involving all four limbs. As the disease progresses, mice develop muscle wasting and ruffled fur, followed by paralysis of hind limbs and finally forelimbs. Mice were euthanized at the first signs of paralysis. Spongiform neurodegeneration (also called spongiosis) was evaluated by histopathology as described previously (6). Brains were removed and fixed in phosphate-buffered 3.7% formaldehyde and embedded in paraffin, and the sections were stained with hematoxylin-eosin.
Quantification of RNA.
RNA was extracted and purified from NIH 3T3 cells as described previously (8). Extraction of RNA from brainstem was as described previously (8) except that the homogenization of the tissue was performed with the aid of a FastPrep FP120 instrument (MP Biomedical, Inc.). RNA preparations were treated with DNase (Ambion) to remove traces of DNA contamination. Quantification of mRNA was carried out by using TaqMan technology on an HT7900 PCR System (Applied Biosystems, Inc.) with the primer-probe sets for BiP, CHOP, and viral RNA described previously (8). In vitro studies were carried out in triplicate on individual RNA samples and, for each experiment, independent infections were repeated in triplicate. Analyses of brainstem RNA was carried out in triplicate on at least five mice per group except for CF223 in which three mice were examined.
Immunoprecipitation and Western blot analysis.
Viral envelope and BiP proteins were detected with goat anti-gp70 (SU protein) and mouse monoclonal anti-KDEL (SPA-827; Stressgen), respectively (9). NIH 3T3 were infected with viruses as described previously (9). At 48 to 96 h postinfection, cells were lysed on ice for 10 min with 0.5% NP-40, 50 mM Tris (pH 7.5), 150 mM NaCl, 0.5% deoxycholate, a 1/100 dilution of protease inhibitor cocktail (Sigma P8340), and 10 U of apyrase (Sigma catalog no. A86132)/ml. Nuclei were cleared by centrifugation at 10,000 rpm for 1 min on a Tomy MTX150 microcentrifuge. Immunoprecipitation was carried out with 2 μl of neat antibody overnight at 4°C. Immunoprecipitates were captured with protein A-agarose (Pierce catalog no. 20333) at 4°C for 1.5 h. After the agarose was washed, Laemmli sample buffer (with 2-mercaptoethanol) was added, and the samples boiled 5 min. Samples were separated on preformed NuPAGE Novex 12% Bis-Tris polyacrylamide gels (Invitrogen). Proteins were transferred to Immobilon-FL polyvinylidene difluoride membranes (Millipore catalog no. IPFL20200), incubated in blocking buffer (Licor Biosciences catalog no. 927-40000) at least 1 h at 4°C, and then incubated with primary antibodies for 1 h at room temperature. Blots were washed with phosphate-buffered saline (PBS) containing 0.1% Tween 20 (PBST), four times for 5 min each, and incubated with donkey anti-goat immunoglobulin (IRDye 800CW; Licor Biosciences catalog no. 926-32214) or goat anti-mouse immunoglobulin (IRDye 680; Licor Biosciences catalog no. 926-32220) for 1 h at room temperature. After being washed in PBST, the blots were rinsed in PBS without Tween 20 and scanned on a Odyssey infrared imager model 9120 (Licor Biosciences). Scans were analyzed using Odyssey application software v2.1. Images were exported as jpg files into Photoshop v7.0 and converted to gray scale.
Statistical analysis.
Both RNA and protein quantification data were expressed as mean ± the standard error of the mean. The data were analyzed by using one way analysis of variance (ANOVA) with a Tukey's multiple-comparison post-test. In the case of protein quantification (see Fig. 6C), we used repeat measures ANOVA.
FIG. 6.
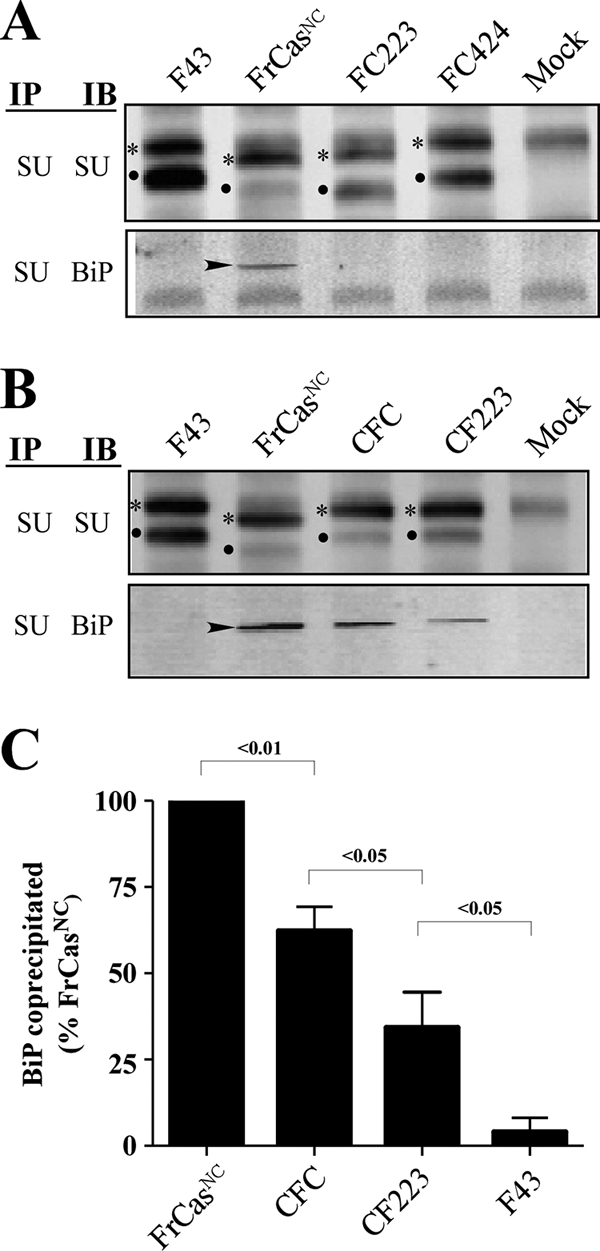
Both folding instability and ER retention were determined by the RBD of FrCasNC. NIH 3T3 cells were infected with F43, FrCasNC, and the chimeric viruses shown in Fig. 4. (A) Western blots of lysates from cells infected with viruses containing the RBD of F43 (FrCasNC served as a positive control). (B) Western blots of lysates from cells infected with viruses containing the RBD of FrCasNC (F43 served as a negative control). Lysates were immunoprecipitated with anti-SU antiserum and immunoblotted with either anti-SU or anti-BiP, as described in the legend to Fig. 5. The two deletions in PRR of the CasBrE envelope protein accounts for the differences in size of the envelope proteins of these viruses. Thus, viruses containing the PRR of F43 (FC424, CF223, and CFC) have envelope proteins of comparable molecular size to F43. The virus FC223 contains the PRR of CasBrE, and its envelope proteins are comparable in size to those of FrCasNC. The relative ratios of pr85env (asterisks) and SU protein (dots) suggest that, like the envelope protein of FrCasNC, the envelope proteins of CFC and CF223 were retained in the ER. In addition, like FrCasNC, BiP (arrowheads) was coprecipitated with the envelope proteins of CFC and CF223, both of which contain the RBD of FrCasNC (see Fig. 4). For viruses that exhibited detectable BiP binding, the relative levels of BiP coprecipitated with anti-SU is shown in panel C. Pixel density was normalized to total pr85env in each lysate and expressed as a percentage of BiP coprecipitated in lysates of FrCasNC-infected cells. The results are cumulative data from three independent experiments. The numbers above each bracket represents the P values calculated by using one-way ANOVA (repeat measures test).
RESULTS
The neurovirulence of FrCasNC is determined by the envelope gene.
The neurovirulence of the virus CasBrE has been mapped to the envelope gene (39). Since FrCasNC and F43 differ not only in the sequence of their envelope genes but also in the 3′ end of the pol gene (Fig. 1A), it was important to rule out a contribution of the polymerase gene sequences in determining neurovirulence. The coding sequence of the pol gene is in a different reading frame and overlaps that of the 5′ env gene. We therefore constructed the chimeric virus FrCasAC, which carries CasBrE sequences beginning 3′ of the pol termination codon, within the signal sequence of the envelope protein (Fig. 1A). Like FrCasNC, FrCasAC was highly neurovirulent, causing the first signs of clinical disease (abnormal hind limb abduction reflex and tremor) beginning at 14 days postinoculation (dpi), followed by signs of advanced disease (hind limb paralysis and wasting) beginning at 16 to 17 dpi (Fig. 1B). Furthermore, FrCasAC, like FrCasNC, also induced the upregulation of mRNA of the ER chaperone BiP and the bZIP transcription factor CHOP (Fig. 1C) after infection of NIH 3T3 cells in vitro, indicating that both viruses activated the UPR. Thus, both the neurovirulence and the ER stress induced by FrCasNC were determined specifically by its envelope protein.
Neurovirulence of the chimeric viruses.
Retroviral envelope proteins are synthesized in the ER as a precursor polyprotein which is cleaved in the Golgi by a furin-like protease, generating the surface glycoprotein (SU), which interacts with the viral receptor during viral entry, and the transmembrane protein (TM), which mediates membrane fusion (47). The SU protein is divided into two domains (Fig. 2), the receptor-binding domain (RBD) and the C-terminal domain joined by a proline-rich hinge region (PRR). A series of viruses was constructed carrying chimeric envelope genes in which these SU domains and the TM sequences were derived from either F43 or FrCasNC. The genetic background of all of these viruses was the same as that of FrCasNC and F43, being derived from the Friend MLV strain FB29 (46). Each of the chimeric viruses chosen for further study was highly neuroinvasive, as indicated by the high levels of viral RNA in the brainstems 17 to 20 days after intraperitoneal inoculation (Fig. 2, viral RNA). However, the occurrence of spongiform lesions differed. The histopathology caused by FrCasNC and related viruses has been described in detail in previous reports (6). Lesions are seen in the brainstem, deep cerebellar nuclei, anterior and posterior colliculi, thalamus, and deep layers of the cerebral neocortex (Fig. 2). Only viruses carrying the RBD from CasBrE induced spongiform histopathology (Fig. 2). The extent and distribution of these lesions, however, also appeared to be dependent on the presence of CasBrE sequences in other parts of the envelope gene (Fig. 2). Thus, CF223 (carrying the RBD from CasBrE) exhibited spongiform lesions exclusively localized to the brainstem, whereas mice inoculated with FC70 and CFC exhibited lesions in a wide distribution similar to FrCasNC. The extent of spongiosis in each of these locations was not quantified because morphometric analysis was beyond the scope of the present study.
Differences among these neurovirulent viruses were further revealed when these mice were monitored for signs of clinical disease (Fig. 3). All viruses containing the RBD from CasBrE caused an abnormal hind-limb abduction reflex and tremor (i.e., signs of mild disease) (Fig. 3A). Only viruses also containing CasBrE sequences outside the RBD (FrCasNC, FC70, and CFC) progressed to develop paralysis and wasting (Fig. 3B). However, the development of paralysis/wasting for FC70 and CFC was slower than that of FrCasNC. Indeed, only 25% of the mice inoculated with CFC progressed to paralytic disease within the 100 days of observation. Thus, while RBD sequences from CasBrE appeared to be necessary for induction of spongiform lesions per se, they were not sufficient for expression of the highly neurovirulent phenotype exhibited by FrCasNC. Inclusion of F43 sequences in any of the domains of the CasBrE envelope protein blunted the neurovirulence of the virus.
FIG. 3.

Tempo of the neurological disease induced by FrCasNC, FC70, CFC, and CF223. Mice were inoculated intraperitoneally on postnatal day 1 and observed for signs of neurological disease. As was done in Fig. 1B, clinical disease here was divided into the mild neurological signs (A) and severe late signs (B). Mice inoculated with FrCasNC (n = 38), FC70 (n = 32), and CFC (n = 34) all exhibited rapid onset of mild neurological signs, whereas these signs were delayed in mice inoculated with CF223 (n = 18). The tempo of paralysis and wasting, however, brought out in more striking terms the differences in neurovirulence of these viruses. Only FrCasNC induced rapidly progressive paralysis. Paralysis was seen in all of the FC70-inoculated mice but was delayed compared to FrCasNC. Only 25% of the mice inoculated with CFC exhibited paralysis and wasting. None of the mice inoculated with CF223 developed paralysis over the 100-day observation period.
Activation of the UPR is determined by the sequence of the RBD.
In previous studies comparing the viruses F43 and FrCasNC, it was found that the neurovirulence of FrCasNC was associated with the activation of the UPR (4, 8, 9). It was therefore of interest to determine whether the induction of UPR correlated with the induction of disease. NIH 3T3 cells were infected with a subset of the chimeric viruses described above, and the levels of mRNA for two UPR target genes (gpr78/BiP and GADD153/CHOP) were measured by quantitative reverse transcription-PCR. BiP, a downstream target gene of the UPR sensor ATF6, is a major ER chaperone, the upregulation of which is considered to be one of the more specific markers of UPR signaling. CHOP, a downstream target gene of the UPR sensor PERK, is a bZiP transcription factor and is associated with both adaptive and proapoptotic signaling (34). Upregulation of CHOP, however, can be induced by cellular stresses other than ER stress (52) and therefore is not as specific for UPR signaling as is BiP. The results (Fig. 4) clearly indicated that both BiP and CHOP were robustly upregulated by viruses carrying CasBrE sequences in the RBD.
FIG. 4.

Viruses containing the CasBrE RBD induced ER stress in NIH 3T3 cells. The occurrence of ER stress was detected by the upregulation of two UPR target genes, BiP and CHOP. RNA was extracted 48 h after infection and BiP and CHOP mRNA quantified by real-time reverse transcription-PCR. The data were normalized as in Fig. 1C.
Envelope sequences that determine protein folding instability.
Since the induction of UPR by FrCasNC was found to be a consequence of the misfolding and ER retention of its envelope protein (9), it was of interest to determine which regions of the envelope protein were involved in destabilizing this protein. We first carried out coprecipitation studies on FrCasNC and F43-infected cells using antisera specific for viral SU and the ER chaperone BiP (Fig. 5). BiP binds in an ATP-dependent fashion to exposed hydrophobic patches, preventing aggregation during the process of protein folding (14). The level of BiP binding at steady state can thus be used as a measure of the level of protein misfolding (19). Although cell lysates of F43 and FrCasNC contained comparable amounts of pr85env (Fig. 5, IP-SU, IB-SU), there was a striking difference in the levels of SU protein (Fig. 5) being barely detectable in the FrCasNC lysate. This is consistent with the ER retention observed previously, since cleavage of the pr85env polyprotein into SU and TM components occurs in the Golgi. Similar levels of the envelope precursor protein in these cell lysates is a consequence of the increased rate of degradation of the FrCasNC pr85env relative to that of F43 (9). The increased association of BiP with the envelope precursor of FrCasNC was revealed by the coprecipitation of BiP with anti-SU (Fig. 5, IP-SU, IB-BiP), as well as by the coprecipitation of pr85env with anti-BiP (Fig. 5, IP-BiP, IB-SU). Finally, the level of BiP protein in the lysate of FrCasNC-infected cells was elevated compared to F43 and mock-infected cells (Fig. 5, IP-BiP, IB-BiP), a finding consistent with the upregulation of BiP mRNA (Fig. 4).
FIG. 5.
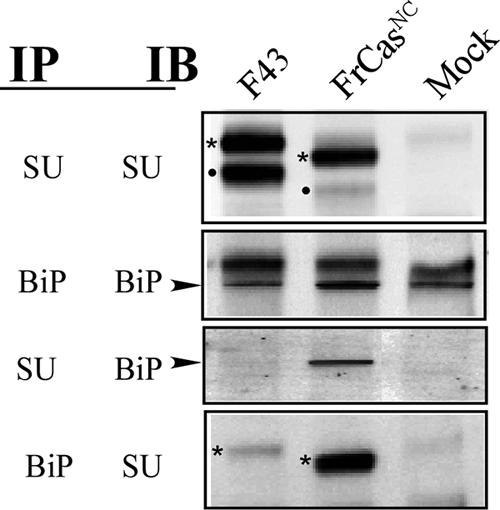
Folding stability of the envelope proteins of FrCasNC and F43 were assessed by the extent of binding of the ER chaperone BiP. NIH 3T3 cells were infected with F43 or FrCasNC, and the cells were lysed 48 h later. Lysates were immunoprecipitated (IP) with anti-SU or anti-BiP (anti-KDEL). Immunoprecipitates were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunoblotted (IB) with either anti-SU or anti-BiP antibodies. The envelope protein is synthesized in the ER as a polyprotein pr85env (asterisks), which after transit to the Golgi compartment is cleaved by a cellular protease into SU (dot) and TM (not shown) components (see top Fig. 2). The smaller molecular sizes of the pr85env and SU proteins of FrCasNC compared to those of F43 are due to two deletions of 4 and 7 amino acid residues in the PRR (see Fig. 2) of CasBrE. The envelope protein of FrCasNC but not F43 is retained in the ER, which accounts for the difference in the ratio of pr85env and SU proteins observed in F43 and FrCasNC-infected cells (IP-SU/IB-SU). The increased binding of BiP (arrowheads) to the pr85env protein of FrCasNC relative to that of F43 (IP-SU/IB-BiP and IP-BiP/IB-SU) is consistent with the folding instability of the FrCasNC envelope protein reported previously (9).
Analysis of lysates of cells infected with the chimeric viruses indicated that the increased BiP binding to the envelope precursor protein was also determined by the sequence of the RBD. Only viruses containing the CasBrE RBD exhibited noticeable coprecipitation of BiP with the anti-SU antiserum (Fig. 6A and B). Furthermore, it was apparent that the relative level of BiP binding increased when sequences outside the RBD were also derived from CasBrE (compare CF223, CFC, and FrCasNC in Fig. 6B). For viruses that exhibited increased BiP binding, the relative levels of BiP coprecipitated with anti-SU, when normalized to the relative levels of pr85env (Fig. 6C), appeared to correlate with the level of neurovirulence (i.e., FrCasNC > CFC > CF223) (Fig. 2 and 3). These results suggested a direct relationship between the degree of protein misfolding in the ER and the severity of the neurological disease.
DISCUSSION
From the histopathologic (Fig. 2) and clinical studies (Fig. 3) it was apparent that retroviral neurovirulence was dependent on the presence of RBD sequences from CasBrE. This observation is consistent with mapping studies that have been carried out on other neurovirulent murine retroviruses (35, 36, 39, 48). However, the presence of the RBD from CasBrE was not sufficient to generate a virus with neurovirulence comparable to that of FrCasNC. Thus, the virus CF223, which contained the RBD from CasBrE but the rest of the envelope gene from F43, induced spongiform lesions more restricted in their distribution, and clinical disease less severe, than that caused by FrCasNC. Addition of CasBrE sequences outside the RBD (i.e., FC70 and CFC) resulted in viruses that induced spongiform lesions not only in the brainstem but also in the subcortex and deep cerebral cortex, a lesion distribution quite comparable to that caused by FrCasNC. However, neither of these viruses induced clinical neurological disease of comparable severity and tempo to that of FrCasNC (Fig. 3). Thus, while the presence of the CasBrE RBD was necessary for the induction of spongiform lesions and clinical disease, the addition of F43 sequences in any of the domains of the envelope protein attenuated the intensity of the disease. It should be noted, however, that the addition of sequences from F43 did not blunt the neuroinvasiveness of the respective viruses. Indeed, for all of the viruses shown in Fig. 2, irrespective of the their relative neurovirulence, the levels of viral RNA in the brainstem were comparable. This observation is consistent with previous studies, suggesting that neurovirulence and neuroinvasiveness are separable phenotypes (1, 2).
The main goal of the present study was to ascertain whether the envelope sequences that determined neurovirulence also determined the induction of ER stress and protein misfolding. We found that only viruses that contained the CasBrE RBD induced ER stress in NIH 3T3 cells. In addition, like neurovirulence and ER stress, the CasBrE RBD appeared to render the envelope precursor protein (pr85env) unstable, as measured by the coprecipitation of BiP. Finally, the relative levels of BiP binding (i.e., FrCasNC > CFC > CF223) appeared to correlate with the relative neurovirulence of the respective viruses (Fig. 2 and 3). Thus, although the CasBrE RBD was primarily responsible for the instability of the envelope protein, CasBrE sequences in other parts of the envelope protein further contributed to protein misfolding. Cumulatively, these results provide strong evidence in support of the notion that protein folding instability is causally linked to neurovirulence.
The role of the RBD in the folding instability of pr85env is currently unclear. One obvious explanation is that the CasBrE RBD is inherently unstable. This could be a consequence of the amino acid sequence or perhaps to the presence of a unique N-linked glycosylation site in the CasBrE RBD (32). On the other hand, it is also possible that it is the binding of the viral receptor, CAT1, to the RBD that triggers folding instability in other parts of the envelope precursor protein. Interaction between the envelope protein and CAT1 appears to occur in the ER (12, 18, 27), although the relevance of this interaction in the virus life cycle is not known. A great deal more is known about the interaction between the RBD and CAT1 at the cell surface during viral entry into cells. Receptor binding appears to trigger an interaction between the RBD and the C-terminal domain (3), leading to disulfide bond rearrangements in the C-terminal domain of SU (51) and exposure of the fusion peptide in the TM protein. Thus, it is conceivable that the viral receptor could also influence folding of the nascent viral envelope protein in the ER.
While the correlation between protein folding instability and neurovirulence is strong, the precise role of protein misfolding in the pathogenesis of the neurological disease induced by these viruses remains unresolved by these studies. Activation of the three parallel signal transduction pathways of the UPR (driven by Ire1α, ATF6, and PERK) decrease the load of client proteins on the ER folding machinery and increase the folding capacity of the ER, all of which represents prosurvival responses. Concomitantly, however, the UPR also activates proapoptotic signaling (29, 43, 44). The activation of cell death pathways by neurovirulent murine retroviruses has been studied primarily using another neurovirulent murine retrovirus, MoMLVts1. Like FrCasNC, the envelope protein of MoMLVts1 misfolds in the ER and activates the UPR (26). In addition, MoMLVts1-infected cells exhibit activation of ER-associated caspase 12 (26), JNK (24), and BAX (25), in addition to the proapoptotic transcription factor CHOP (26). What is not clear, however, is whether these potentially cytotoxic responses are actually involved in the demise of the target cells of these viruses in the brain. For instance, although CHOP protein is upregulated specifically in the cells in the brain exhibiting signs of cytopathology (4, 26), the neurovirulence of MoMLVts1 (4) and FrCasNC (unpublished data) is undiminished in CHOP−/− mice. Nevertheless, the upregulation of CHOP may represent a surrogate marker for some other effector molecules that may be activated by the same upstream pathway that also upregulates CHOP. The upregulation of CHOP occurs downstream of the activation of the ER kinase PERK, and it has been suggested that persistent activation of this arm of the UPR in chronic ER stress is associated with cell death (28), although the precise mechanisms are not yet understood.
On the other hand, both MoMLVts1 (42) and FrCasNC (unpublished data)-infected cells exhibit signs of oxidative stress associated with the accumulation of reactive oxygen species (ROS), and treatment of mice with the antioxidant monosodium α-luminol renders them partially resistant to the neurovirulence of MoMLVts1 (21). Although the source of the ROS has not been identified, it has been estimated that up to 25% of the total ROS generated by the cell is a consequence of oxidative protein folding in the ER (50). The formation of disulfide bonds results in the transport of electrons from client proteins through protein disulfide isomerase to ERO1 and ultimately to molecular oxygen. It would be expected, therefore, that misfolding of retroviral envelope proteins, which contain multiple disulfide bonds (11 disulfides in the case of FrCasNC), would place a high demand on the redox balance of the cell. Thus, it is conceivable that while the cytotoxicity of these viruses is driven by the accumulation of misfolded envelope protein, it is oxidative stress rather than the UPR that ultimately activates the cell death pathways (42).
One of the more puzzling aspects of this disease is the cellular specificity of the toxicity. These viruses infect many different cell types in the brain, including microvascular endothelial cells, a variety of glial cells, and even certain populations of neurons (16, 31). Nevertheless, virus-induced cytopathology is observed predominantly in a subpopulation of infected oligodendrocytes, which appear to represent cells in an early stage of differentiation, and it is only these cells that exhibit signs of ER stress (4). The cell type specificity of the ER stress is supported by a recent microarray study that failed to detect signs of ER stress in infected microglial cells (10), a cell type in the brain that is heavily infected by FrCasNC (31). This implies that in infected microglial cells and by extension in endothelial cells and neurons, ER homeostasis is either not upset by FrCasNC or the cells are able to rapidly adapt to the influx of misfolded proteins. Oligodendrocytes, on the other hand, appear to be particularly vulnerable to the loss of homeostasis in the secretory pathway (15, 30, 40). This is thought to be a consequence of the large burden on the ER imposed by the myelin-forming function of these cells.
It is unclear whether the clinical disease induced by the neurovirulent retroviruses is a consequence of the death of oligodendrocytes or due to a bystander effect on neurons. Certainly, there is little evidence of demyelination in this disease. Furthermore, the observation that transgenic mice overexpressing BCL2 specifically in neurons are partially resistant to neurovirulence of CasBrE (22) suggests that the clinical manifestations of this disease are due to the loss of trophic support and secondary dysfunction or death of neurons, which have been shown not to be infected by these viruses (23).
Although the neurovirulent murine retroviruses FrCasNC and MoMLVts1 induce ER stress and activate the UPR, the role of the protein misfolding in the pathogenesis of the neurodegenerative disease caused by these viruses has remained tenuous. The results of the present study suggest that there exists a direct relationship between the severity of the neurological disease and the degree of protein misfolding, strengthening the notion that this disease represents a protein folding disorder caused by a virus.
Acknowledgments
This research was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
We thank Cynthia Martellero of the Laboratory of Persistent Viral Diseases (LPVD), RML, for technical assistance and Gary Hettrick of the Visual Medical Arts Department, RML, for assembling the figures. We also are grateful to Bruce Chesebro and Kim Hasenkrug of LPVD, RML, and Sonja Best of Laboratory of Virology, RML, for reading and critiquing the manuscript.
Footnotes
Published ahead of print on 1 April 2009.
REFERENCES
- 1.Askovic, S., C. Favara, F. J. McAtee, and J. L. Portis. 2001. Increased expression of MIP-1 alpha and MIP-1 beta mRNAs in the brain correlates spatially and temporally with the spongiform neurodegeneration induced by a murine oncornavirus. J. Virol. 752665-2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Askovic, S., F. J. McAtee, C. Favara, and J. L. Portis. 2000. Brain infection by neuroinvasive but avirulent murine oncornaviruses. J. Virol. 74465-473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barnett, A. L., R. A. Davey, and J. M. Cunningham. 2001. Modular organization of the Friend murine leukemia virus envelope protein underlies the mechanism of infection. Proc. Natl. Acad. Sci. USA 984113-4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clase, A. C., D. E. Dimcheff, C. Favara, D. Dorward, F. J. McAtee, L. E. Parrie, D. Ron, and J. L. Portis. 2006. Oligodendrocytes are a major target of the toxicity of spongiogenic murine retroviruses. Am. J. Pathol. 1691026-1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Czub, M., S. Czub, F. J. McAtee, and J. L. Portis. 1991. Age-dependent resistance to murine retrovirus-induced spongiform neurodegeneration results from central nervous system-specific restriction of virus replication. J. Virol. 652539-2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Czub, S., W. P. Lynch, M. Czub, and J. L. Portis. 1994. Kinetic analysis of spongiform neurodegenerative disease induced by a highly virulent murine retrovirus. Lab. Investig. 70711-723. [PubMed] [Google Scholar]
- 7.DesGroseillers, L., M. Barrette, and P. Jolicoeur. 1984. Physical mapping of the paralysis-inducing determinant of a wild mouse ecotropic neurotropic virus. J. Virol. 52356-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dimcheff, D. E., S. Askovic, A. H. Baker, C. Johnson-Fowler, and J. L. Portis. 2003. Endoplasmic reticulum stress is a determinant of retrovirus-induced spongiform neurodegeneration. J. Virol. 7712617-12629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dimcheff, D. E., M. A. Faasse, F. J. McAtee, and J. L. Portis. 2004. Endoplasmic reticulum (ER) stress induced by a neurovirulent mouse retrovirus is associated with prolonged BiP binding and retention of a viral protein in the ER. J. Biol. Chem. 27933782-33790. [DOI] [PubMed] [Google Scholar]
- 10.Dimcheff, D. E., L. G. Volkert, Y. Li, A. L. DeLucia, and W. P. Lynch. 2006. Gene expression profiling of microglia infected by a highly neurovirulent murine leukemia virus: implications for neuropathogenesis. Retrovirology 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fass, D., R. A. Davey, C. A. Hamson, P. S. Kim, J. M. Cunningham, and J. M. Berger. 1997. Structure of a murine leukemia virus receptor-binding glycoprotein at 2.0 angstrom resolution. Science 2771662-1666. [DOI] [PubMed] [Google Scholar]
- 12.Fujisawa, R., and M. Masuda. 2007. Ecotropic murine leukemia virus envelope protein affects interaction of cationic amino acid transporter 1 with clathrin adaptor protein complexes, leading to receptor downregulation. Virology 368342-350. [DOI] [PubMed] [Google Scholar]
- 13.Gardner, M. B., B. E. Henderson, J. E. Officer, R. W. Rongey, J. C. Parker, C. Oliver, J. D. Estes, and R. J. Huebner. 1973. A spontaneous lower motor neuron disease apparently caused by indigenous type-C RNA virus in wild mice. J. Natl. Cancer Inst. 511243-1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gething, M. J. 1999. Role and regulation of the ER chaperone BiP. Semin. Cell Dev. Biol. 10465-472. [DOI] [PubMed] [Google Scholar]
- 15.Gow, A., and R. A. Lazzarini. 1996. A cellular mechanism governing the severity of Pelizaeus-Merzbacher disease. Nat. Genet. 13422-428. [DOI] [PubMed] [Google Scholar]
- 16.Gravel, C., D. G. Kay, and P. Jolicoeur. 1993. Identification of the infected target cell type in spongiform myeloencephalopathy induced by the neurotropic Cas-Br-E murine leukemia virus. J. Virol. 676648-6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He, B. 2006. Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ. 13393-403. [DOI] [PubMed] [Google Scholar]
- 18.Heard, J. M., and O. Danos. 1991. An amino-terminal fragment of the Friend murine leukemia virus envelope glycoprotein binds the ecotropic receptor. J. Virol. 654026-4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hellman, R., M. Vanhove, A. Lejeune, F. J. Stevens, and L. M. Hendershot. 1999. The in vivo association of BiP with newly synthesized proteins is dependent on the rate and stability of folding and not simply on the presence of sequences that can bind to BiP. J. Cell Biol. 14421-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hetz, C., M. Russelakis-Carneiro, K. Maundrell, J. Castilla, and C. Soto. 2003. Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J. 225435-5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang, Y., V. L. Scofield, M. Yan, W. Qiang, N. Liu, A. J. Reid, W. S. Lynn, and P. K. Wong. 2006. Retrovirus-induced oxidative stress with neuroimmunodegeneration is suppressed by antioxidant treatment with a refined monosodium alpha-luminol (Galavit). J. Virol. 804557-4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jolicoeur, P., C. Hu, T. W. Mak, J. C. Martinou, and D. G. Kay. 2003. Protection against murine leukemia virus-induced spongiform myeloencephalopathy in mice overexpressing Bcl-2 but not in mice deficient for interleukin-6, inducible nitric oxide synthetase, ICE, Fas, Fas ligand, or TNF-R1 genes. J. Virol. 7713161-13170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kay, D. G., C. Gravel, Y. Robitaille, and P. Jolicoeur. 1991. Retrovirus-induced spongiform myeloencephalopathy in mice: regional distribution of infected target cells and neuronal loss occurring in the absence of viral expression in neurons. Proc. Natl. Acad. Sci. USA 881281-1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim, H. T., W. Qiang, N. Liu, V. L. Scofield, P. K. Wong, and G. Stoica. 2005. Up-regulation of astrocyte cyclooxygenase-2, CCAAT/enhancer-binding protein-homology protein, glucose-related protein 78, eukaryotic initiation factor 2 alpha, and c-Jun N-terminal kinase by a neurovirulent murine retrovirus. J. Neurovirol. 11166-179. [DOI] [PubMed] [Google Scholar]
- 25.Kim, H. T., S. Tasca, W. Qiang, P. K. Wong, and G. Stoica. 2002. Induction of p53 accumulation by Moloney murine leukemia virus-ts1 infection in astrocytes via activation of extracellular signal-regulated kinases 1/2. Lab. Investig. 82693-702. [DOI] [PubMed] [Google Scholar]
- 26.Kim, H. T., K. Waters, G. Stoica, W. Qiang, N. Liu, V. L. Scofield, and P. K. Wong. 2004. Activation of endoplasmic reticulum stress signaling pathway is associated with neuronal degeneration in MoMuLV-ts1-induced spongiform encephalomyelopathy. Lab. Investig. 84816-827. [DOI] [PubMed] [Google Scholar]
- 27.Kim, J. W., and J. M. Cunningham. 1993. N-linked glycosylation of the receptor for murine ecotropic retroviruses is altered in virus-infected cells. J. Biol. Chem. 26816316-16320. [PubMed] [Google Scholar]
- 28.Lin, J. H., H. Li, D. Yasumura, H. R. Cohen, C. Zhang, B. Panning, K. M. Shokat, M. M. Lavail, and P. Walter. 2007. IRE1 signaling affects cell fate during the unfolded protein response. Science 318944-949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin, J. H., P. Walter, and T. S. Yen. 2008. Endoplasmic reticulum stress in disease pathogenesis. Annu. Rev. Pathol. 3399-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin, W., H. P. Harding, D. Ron, and B. Popko. 2005. Endoplasmic reticulum stress modulates the response of myelinating oligodendrocytes to the immune cytokine interferon-gamma. J. Cell Biol. 169603-612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lynch, W. P., S. Czub, F. J. McAtee, S. F. Hayes, and J. L. Portis. 1991. Murine retrovirus-induced spongiform encephalopathy: productive infection of microglia and cerebellar neurons in accelerated CNS disease. Neuron 7365-379. [DOI] [PubMed] [Google Scholar]
- 32.Lynch, W. P., and A. H. Sharpe. 2000. Differential glycosylation of the Cas-Br-E Env protein is associated with retrovirus-induced spongiform neurodegeneration. J. Virol. 741558-1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malhotra, J. D., and R. J. Kaufman. 2007. The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 18716-731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marciniak, S. J., C. Y. Yun, S. Oyadomari, I. Novoa, Y. Zhang, R. Jungreis, K. Nagata, H. P. Harding, and D. Ron. 2004. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 183066-3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Masuda, M., P. M. Hoffman, and S. K. Ruscetti. 1993. Viral determinants that control the neuropathogenicity of PVC-211 murine leukemia virus in vivo determine brain capillary endothelial cell tropism of the virus in vitro. J. Virol. 674580-4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murphy, S. L., M. J. Honczarenko, N. V. Dugger, P. M. Hoffman, and G. N. Gaulton. 2004. Disparate regions of envelope protein regulate syncytium formation versus spongiform encephalopathy in neurological disease induced by murine leukemia virus TR. J. Virol. 788392-8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakamura, T., and S. A. Lipton. 2008. Emerging roles of S nitrosylation in protein misfolding and neurodegenerative diseases. Antioxid. Redox Signal. 1087-101. [DOI] [PubMed] [Google Scholar]
- 38.Oliff, A. I., G. L. Hager, E. H. Chang, E. M. Scolnick, H. W. Chan, and D. R. Lowy. 1980. Transfection of molecularly cloned Friend murine leukemia virus DNA yields a highly leukemogenic helper-independent type C virus. J. Virol. 33475-486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paquette, Y., Z. Hanna, P. Savard, R. Brousseau, Y. Robitaille, and P. Jolicoeur. 1989. Retrovirus-induced murine motor neuron disease: mapping the determinant of spongiform degeneration within the envelope gene. Proc. Natl. Acad. Sci. USA 863896-3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pennuto, M., E. Tinelli, M. Malaguti, C. U. Del, M. D'Antonio, D. Ron, A. Quattrini, M. L. Feltri, and L. Wrabetz. 2008. Ablation of the UPR-mediator CHOP restores motor function and reduces demyelination in Charcot-Marie-Tooth 1B mice. Neuron 57393-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Portis, J. L., S. Czub, C. F. Garon, and F. J. McAtee. 1990. Neurodegenerative disease induced by the wild mouse ecotropic retrovirus is markedly accelerated by long terminal repeat and gag-pol sequences from nondefective Friend murine leukemia virus. J. Virol. 641648-1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qiang, W., J. M. Cahill, J. Liu, X. Kuang, N. Liu, V. L. Scofield, J. R. Voorhees, A. J. Reid, M. Yan, W. S. Lynn, and P. K. Wong. 2004. Activation of transcription factor Nrf-2 and its downstream targets in response to Moloney murine leukemia virus ts1-induced thiol depletion and oxidative stress in astrocytes. J. Virol. 7811926-11938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ron, D., and P. Walter. 2007. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell. Biol. 8519-529. [DOI] [PubMed] [Google Scholar]
- 44.Rutkowski, D. T., and R. J. Kaufman. 2007. That which does not kill me makes me stronger: adapting to chronic ER stress. Trends Biochem. Sci. 32469-476. [DOI] [PubMed] [Google Scholar]
- 45.Schroder, M., and R. J. Kaufman. 2005. The mammalian unfolded protein response. Annu. Rev. Biochem. 74739-789. [DOI] [PubMed] [Google Scholar]
- 46.Sitbon, M., H. Ellerbrok, F. Pozo, J. Nishio, S. F. Hayes, L. H. Evans, and B. Chesebro. 1990. Sequences in the U5-gag-pol region influence early and late pathogenic effects of Friend and Moloney murine leukemia viruses. J. Virol. 642135-2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Swanstrom, R., and J. W. Wills. 1997. Synthesis, assembly, and processing of viral proteins, p. 263-334. In J. M. Coffin, S. H. Hughes, and H. E. Varmus (ed.), Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [PubMed]
- 48.Szurek, P. F., P. H. Yuen, J. K. Ball, and P. K. Wong. 1990. A Val-25-to-Ile substitution in the envelope precursor polyprotein, gPr80env, is responsible for the temperature sensitivity, inefficient processing of gPr80env, and neurovirulence of ts1, a mutant of Moloney murine leukemia virus TB. J. Virol. 64467-475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Szurek, P. F., P. H. Yuen, R. Jerzy, and P. K. Y. Wong. 1988. Identification of point mutations in the envelope gene of Moloney murine leukemia virus TB temperature-sensitive paralytogenic mutant ts1: molecular determinants for neurovirulence. J. Virol. 62357-360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tu, B. P., and J. S. Weissman. 2004. Oxidative protein folding in eukaryotes: mechanisms and consequences. J. Cell Biol. 164341-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wallin, M., M. Ekstrom, and H. Garoff. 2004. Isomerization of the intersubunit disulphide-bond in Env controls retrovirus fusion. EMBO J. 2354-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wek, R. C., H. Y. Jiang, and T. G. Anthony. 2006. Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 347-11. [DOI] [PubMed] [Google Scholar]
- 53.Williams, B. L., and W. I. Lipkin. 2006. Endoplasmic reticulum stress and neurodegeneration in rats neonatally infected with Borna disease virus. J. Virol. 808613-8626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wong, P. K. Y., M. M. Soong, R. MacLeod, G. E. Gallick, and P. H. Yuen. 1983. A group of temperature-sensitive mutants of Moloney leukemia virus which is defective in cleavage of env precursor polypeptide in infected cells also induces hind-limb paralysis in newborn CFW/D mice. Virology 125513-518. [DOI] [PubMed] [Google Scholar]
- 55.Yuen, P. H., E. Tzeng, C. Knupp, and P. K. Wong. 1986. The neurovirulent determinants of ts1, a paralytogenic mutant of Moloney murine leukemia virus TB, are localized in at least two functionally distinct regions of the genome. J. Virol. 5959-65. [DOI] [PMC free article] [PubMed] [Google Scholar]