

Abstract
Background
The circulating hormone hepcidin plays a central role in iron homeostasis. Our goal was to establish an ex vivo iron-sensing model and to characterize the molecular mechanisms linking iron to hepcidin.
Design and Methods
Murine hepatocytes were isolated by the collagenase method, either from wild type or HFE knockout mice, and cultured 42 h without serum before treatments.
Results
After 42 h of serum-free culture, hepcidin gene expression was undetectable in the hepatocytes. Hepcidin gene expression could, however, be re-activated by an additional 24 h of incubation with 10% serum. Interestingly, addition of 30 μM holotransferrin consistently increased serum-dependent hepcidin levels 3- to 5-fold. The effects of serum and serum+holotransferrin were direct, transcriptional, independent of de novo protein synthesis and required the presence of bone morphogenetic protein. Transferrin receptor-2 activation by its ligand holotransferrin led to extracellular signal regulated kinase (ERK)/mitogen activated protein kinase pathway stimulation and the ERK specific inhibitor U0-126 blunted holotransferrin-mediated induction of hepcidin. ERK activation by holotransferrin provoked increased levels of phospho-Smad1/5/8 highlighting cross-talk between the bone morphogenetic protein/hemojuvelin and ERK1/2 pathways. Finally, we demonstrated, using hepatocytes isolated from Hfe−/− mice, that HFE was not critical for the hepcidin response to holotransferrin but important for basal hepcidin expression.
Conclusions
We demonstrate that hepatocytes are liver iron-sensor cells and that transferrin receptor-2, by signaling through the ERK1/2 pathway, and bone morphogenetic protein/hemojuvelin, by signaling through the Smad pathways, coordinately regulate the iron-sensing machinery linking holotransferrin to hepcidin.
Keywords: hepcidin, iron, hepatocyte, ERK1/2, BMP/HJV, HFE, TfR2
Introduction
Circulating plasma iron reflects dietary iron absorption by the enterocytes of the duodenum and release of iron from the macrophages of the reticuloendothelial system. Iron export from these cells is regulated by the liver-derived peptide hepcidin. Following its synthesis and secretion from the liver, circulating hepcidin reduces iron export by binding to the iron exporter ferroportin1 present on the surface of enterocytes and macrophages leading to its degradation and subsequent cellular iron retention.1 As befits an iron-regulatory hormone, hepcidin synthesis is under the control of body iron status. Although hepcidin synthesis has clearly been shown to be increased by iron treatment to limit further iron absorption,2–4 the setting of the iron-dependent molecular circuitry regulating hepcidin is still incompletely understood. Emerging evidence from studies of inherited defects does however, suggest that hepcidin expression in the liver is influenced by hemojuvelin (HJV), transferrin receptor 2 [TfR2, a recently described liver-specific homolog of transferrin receptor-1 (TfR1)] and HFE. Indeed, mutations of the genes coding for these three liver-enriched membrane proteins are associated with human hereditary hemochromatosis, an iron overload disease characterized by increased iron absorption, increased reticuloendothelial cell iron release, elevated serum iron levels and increased tissue iron deposition.5 The common pathogenic mechanism responsible for these iron disorders, as demonstrated in humans and mice, is hepcidin deficiency.5
The signal transduction pathway linking HJV to hepcidin has recently been elucidated. HJV acts as a co-receptor for bone morphogenetic protein (BMP) signaling, ultimately activating receptor Smad/Smad4 complexes to increase hepcidin transcription.6–8 BMP, like other transforming growth factor superfamily ligands, induce apposition of type I and type II receptors to cause phosphorylation of type I receptors. Activated BMP type I receptors phosphorylate the BMP-responsive Smad proteins 1/5/8, leading to the interaction of these proteins with the common mediator Smad4, subsequent nuclear translocation of the complexes, and finally, transcriptional regulation of BMP-responsive genes. BMP-2, 4, 5, 6, 7 and 9 were all shown to induce hepcidin gene expression robustly in hepatoma cells8,9 and BMP2 injection was demonstrated in vivo to increase hepcidin expression and decrease serum iron levels.8 The final effector Smad4 is believed to be important for maintaining basal hepcidin expression by keeping the hepcidin promoter in an active state. Accordingly, hepcidin gene expression was markedly decreased in Smad4 liver-specific knock-out mice, which are unresponsive to both iron and inter-leukin-6, another well-known inducer of hepcidin gene expression.7 Importantly, in contrast to cellular HJV, which positively regulates hepcidin gene expression, a soluble hemojuvelin (sHJV) form, present in the serum, was shown to suppress hepcidin gene expression in liver cells8,10 by a yet uncharacterized mechanism.
In contrast to HJV, not much is known concerning HFE and TfR2 signaling pathways, except that these proteins are not required for hepcidin’s response to BMP.9 TfR2 has emerged as a strong candidate likely to be part of the regulatory system at the membrane of the hepatocyte sensing diferric iron. This assumption is based mainly on the following. TfR2 affinity for holotransferrin is low (25-fold lower than that of TfR1); its tissue distribution is limited with prominent expression in the liver; and, the biological function of TfR2, in contrast to that of TfR1, is most likely not related to iron uptake. Indeed, human mutations in TfR2 are associated with liver iron overload, a clinical observation which is not consistent with a function of TfR2 in iron uptake.11
HFE is an atypical major histocompatibility class I like molecule that was shown to form protein complexes with both TfR1 and TfR2.12,13 It was recently proposed that, as serum iron saturation increases, HFE is dislodged from its overlapping binding site on TfR1 by iron-transferrin. HFE would then be able to interact with TfR2 and to signal, by unknown mechanisms, the upregulation of hepcidin.14
So far, in vitro studies in hepatoma cell lines have failed to reproduce increased hepcidin synthesis in response to iron, hampering the study of integrated mechanisms for iron-responsive hepcidin regulation. Here, we investigated the capacity of murine hepatocytes in primary culture to respond to holotransferrin and asked how TfR2 and HFE might link holotransferrin, the physiological form of circulating serum iron, to hepcidin.
Design and Methods
Animals
Female C57BL/6 mice (Charles River) were cared for in accordance with the principles of the “European convention for the protection of laboratory animals” and the Animal Welfare Committee of the University Descartes-Paris 5 (Paris, France). Animals were given free access to tap water and a standard laboratory mouse chow diet (AO3, iron content 280 mg/kg, UAR, France). The HFE knockout mice were kindly provided by the laboratory of Nancy Andrews. Sex and age-matched wild type and HFE knock-out mice on a 129/SvEvTac background were used in this study.15
Primary culture of hepatocytes
Hepatocytes were isolated from 2-4-month old C57Bl/6 female mice or from HFE+/+ and HFE−/− female mice on a 129Tc strain, by a modification of the collagenase method.16 Hepatocytes were seeded at a density of 2×106 cells in 60-mm Petri dishes and cultured at 37°C (5% CO2) with medium containing 10% serum for 4 h. After cell attachment, the medium was replaced by fresh M199 medium without serum for 42 h. Unless indicated for the kinetic studies, hepatocytes were incubated for an additional 24 h with complete fresh medium containing either no serum (control cells), 10% fetal calf serum, 30 μM human holotransferrin (Sigma), 30 μM apotransferrin (Sigma), 10% serum+30 μM human holotransferrin, or 10% serum+30 μM bovine holotransferrin. Treatments were as follow: actinomycin D and cycloheximide (Sigma) were added for 24 h at 1 μg/mL and 50 μg/mL, respectively. Monoclonal anti-human BMP2/4 antibody and recombinant mouse noggin (R&D systems) were used for 24 h at 20 μg/mL and 1 μg/mL, respectively. Epidermal growth factor (EGF) treatment consisted of 50 ng/mL (Sigma) for 15 min. Finally, the inhibitor U0-126 (Cell Signaling) was added 1 h before serum and serum+holotransferin treatments, and used at 10 μM for 24 h.
RNA isolation and analysis
Total RNA was isolated using the Qiagen RNAeasy kit according to the manufacturer’s instructions (Qiagen). Northern blot analysis was carried out as previously described.17 Real-time quantification of transcripts was performed in a LightCycler 1.5 instrument (Roche) using fast start DNA master SYBR® Green 1 (Roche). The sequences of the primers were: hepc1, forward 5’-CCTATCTCCATCAACAGATG-3’ and reverse 5’-TGCAACAGATACCACACTG-3’ ; β actin forward 5’-AGGCCCAGAGCAAGAGAGG-3’ and reverse 5’-TACATGGCTGGGGTGTTGAA-3’, BMP2 forward 5’-GTTTGGCCTGAAGCAGAGAC-3’ and reverse 5’-AAGTTCCTCCACGGCTTCTT-3’, BMP4 forward 5’-GAGGGATCTTTACCGGCTCC-3’ and reverse 5’-GTTGAAGAGGAAACGAAAAGCAG-3’, BMP6 forward 5’-ATGGCAGGACTGGATCATTGC-3’ and reverse 5’-CCATCACAGTAGTTGGCAGCGT-3’.
Protein analysis
Cells were harvested and total protein extracts were prepared as described elsewhere18 in 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 10% (vol/vol) glycerol, 1 mM dithiothreitol, 50 mM NaF, 5 mM sodium pyrophosphate, 1 mM Na3VO4, 0.5 mM phenylmethyl-sulfonyl fluoride, and 1% NP40. For western blot analysis, membranes were incubated with primary antibody: rabbit anti-TfR2 (Alpha Diagnostic), rabbit anti-p44/42 MAP kinase (Cell Signaling), rabbit anti-active MAPKpAb (Promega), rabbit anti-phospho-Smad1/5/8 (Cell Signaling), rabbit anti-Smad4 (Cell Signaling) and mouse anti-α-tubulin (Sigma, France). The antigen-antibody complexes were detected using horseradish peroxidase-labeled rabbit anti-IgG or mouse anti-IgG and a SuperSignal West Pico chemiluminescence kit detection system (Pierce).
Statistics
Results are expressed as mean ± standard deviation. Student’s t test was used for estimation of statistical significance.
Results
To investigate the molecular mechanisms involved in the induction of hepcidin synthesis by holotransferrin, we first used human and murine hepatoma cell lines. In these transformed cells, we were unable, using different conditions, to show any activation of hepcidin expression by holotransferrin (data not shown). Furthermore, we confirmed that non-transferrin bound iron led to a strong inhibition of hepcidin expression (data not shown) as previously reported.19,20 We thus decided to explore hepcidin gene regulation in a more physiologically relevant model using mouse hepatocytes in primary culture.
Hepcidin regulation by serum and holotransferrin in hepatocytes in primary culture
Mouse hepatocytes isolated by collagenase treatment were cultured for 42 h in serum-free medium. Fresh medium containing either 10% serum, 30 μM human holotransferrin, or both, was then added for an additional 24 h. We found that, compared to hepcidin levels in total liver, hepcidin mRNA levels dropped dramatically in isolated hepatocytes cultured in the absence of serum, with levels being undetectable after 42 h of culture (Figure 1). Addition of 30 μM holotransferrin alone had no effect on hepcidin expression, while addition of serum dramatically increased hepcidin expression. Interestingly, addition of 30 μM holotransferrin significantly and reproducibly increased the serum-dependent hepcidin expression. Quantification by real-time PCR (Online Supplementary Figure S1) showed a 4-fold increase of hepcidin gene expression by holotransferrin relative to hepcidin levels in the presence of serum alone, demonstrating that isolated hepatocytes are capable of sensing iron and of responding, in the presence of serum, by increasing hepcidin gene expression. The specificity of the response to iron-bound transferrin was further demonstrated by the fact that apo-transferrin was unable to induce hepcidin gene expression in this system (Online Supplementary Figure S1). Interestingly, Kawabata et al. reported that human holotransferrin was able to bind both human TfR1 and TfR2, while, in contrast, bovine holotransferrin recognized only human TfR2.21 We thus tested the effect of bovine holotransferrin and demonstrated the same efficiency of bovine and human holotransferrin in inducing hepcidin expression (Online Supplementary Figure S1). Assuming that, as in human cells, bovine holotransferrin did not recognize mouse TfR1 (mouse and human TfR1 share 95% similarity in the C-ter region binding holotransferrin), but specifically mouse TfR2, this result favors the hypothesis that TfR2 might preferentially be the receptor activated by holotransferrin for signaling to hepcidin.
Figure 1.
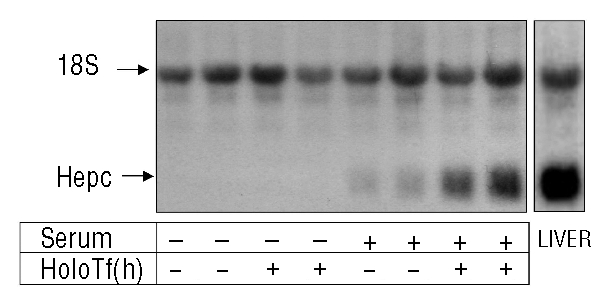
Hepcidin mRNA levels in hepatocytes in primary culture treated with serum and serum plus human holotransferrin [(holoTf (h)] Hepcidin mRNA levels in primary hepatocytes was determined by northern blot analysis. Hepatocytes were treated, after a 42 h period of serum-free culture, with either 0% serum (control cells), 10% serum, 30 μM human holotransferrin, or both, for 24 h. Total liver RNA (20 μg) was subjected to electrophoresis, blotted, and hybridized with hepcidin and 18S-labeled probes. The experiment was performed at least three times and a representative result is shown.
The effects of serum and serum+holotransferrin on hepcidin gene expression were further investigated. The hepcidin response to serum was rapid, occurring at 2 h after the addition of serum into the medium, being maximal at 6 h and then reaching a plateau (Online Supplementary Figure S2A). The specific response of hepcidin to serum+holotransferrin was delayed and significantly different to that of the response to serum only after 12 h of culture (Online Supplementary Figure S2A). De novo synthesis of protein was not required for the effects of serum and serum+holotransferrin since hepcidin response was maintained in conditions of serum+cycloheximide, and serum+holotransferrin+cycloheximide (Online Supplementary Figure S2B). Finally, the addition of actinomycin D, an inhibitor of transcription, when added to the medium in the presence of the serum, inhibited the serum-induced effect on hepcidin expression indicating that the upregulation of the hepcidin gene is transcriptionally dependent (Online Supplementary Figure S2B). Similarly, when actinomycin D was added 8 h after the addition of serum in the medium, the effect of holotransferrin-induced hepcidin expression was blunted, suggesting a transcriptional effect of holotransferrin on hepcidin gene expression (Online Supplementary Figure S2C).
Role of bone morphogenetic protein on hepcidin regulation by serum and holotransferrin in hepatocytes in primary culture
Since BMP signaling is important for hepcidin expression,6,8,9 we looked at the role of BMP in the effects of serum and serum+holotransferrin in inducing hepcidin gene expression. First, we confirmed that in our cell culture system, the hepatocytes were sensitive to BMP and showed a 500–1000 fold increase of hepcidin with 24 h treatment of 42 h serum-starved hepatocytes with 20 ng/mL BMP2 (data not shown). We then tested the effects of noggin, a soluble BMP inhibitor that binds to BMP ligands and blocks the binding epitope for BMP receptors,22 in the presence of serum or serum+holotransferrin. As shown in Figure 2, noggin inhibited the hepcidin response to serum by 50% and completely blunted the response to holotransferrin, decreasing the level of hepcidin to that with serum in the presence of noggin. Similar results were obtained using neutralizing antibodies against BMP2/BMP4 (Figure 2). Since hepatocytes produce BMP, we asked whether serum or serum+holotransferrin conditions were capable of inducing BMP gene expression. In the time-course experiment shown in Figure 2B, endogenous BMP2 mRNA levels were not affected by the presence of serum or serum+holotransferrin in the medium. The same results were obtained for BMP4 and BMP6, i.e., no significant increase of mRNA levels was induced by serum and holotransferrin treatment (data not shown). Altogether, these results are in agreement with our previous data suggesting that de novo protein synthesis is not required for the effects of serum and holotransferrin on hepcidin expression, thus strongly favoring the hypothesis that the effect of serum and holotransferrin are due, partly, to the presence of BMP2/4 contained in the serum.
Figure 2.
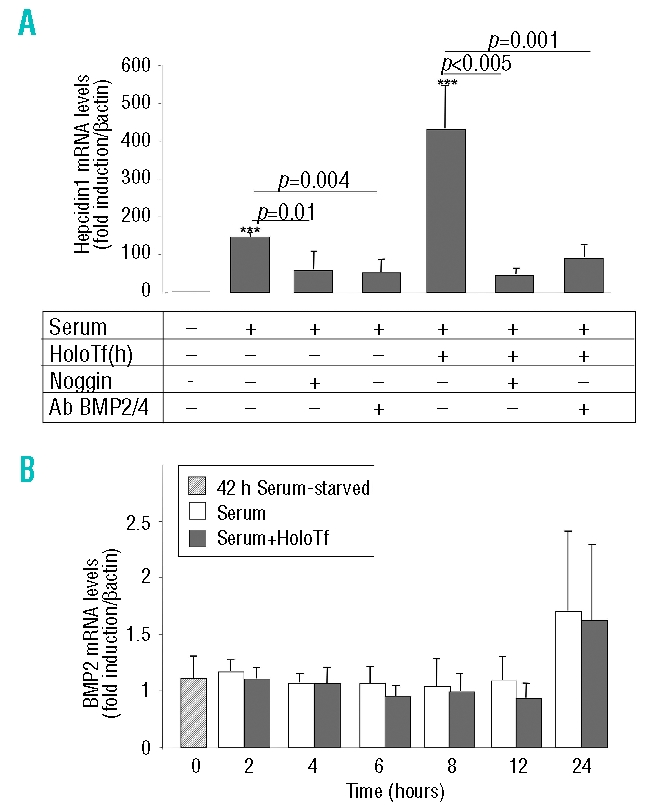
Effects of noggin and neutralizing BMP2/4 antibodies on hepcidin response to serum and serum+holotransferrin and kinetic study of BMP2 mRNA levels. Relative changes in hepcidin mRNA levels were quantified by real-time quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) performed on cDNA synthesized from 2 μg total RNA (β-actin normalized, arbitrary units) (A) Hepatocytes were treated, after a 42 h period of serum-free culture, with either 0% serum (control cells), 10% serum, or 10% serum + 30 μM human holotransferrin [holoTf(h)], in the presence of either 1 μg/mL noggin or 20 μg/mL BMP2/4 antibodies (Ab BMP2/4), for 24 h. Results are expressed relative to hepatocytes cultured for 66 h without serum. Mean±SD for three independent samples. The experiment was performed three times and a representative result is shown. Statistical analysis was performed using Student’s t test (unpaired, two-tailed). ***p<0.001 as compared to untreated hepatocytes. (B) Hepatocytes were treated, after a 42 h period of serum-free culture, with either 0% serum (control cells), 10% serum or 10% serum in the presence of 30 μM human holoTf for 2, 4, 6, 8, 12 or 24 h. Relative changes in BMP2 mRNA levels were quantified by real-time qRT-PCR performed on cDNA synthesized from 2 μg total RNA (β-actin normalized, arbitrary units). Results are expressed relative to hepatocytes cultured for 66 h without serum. Mean±SD for three independent samples. The experiment was performed three times and a representative result is shown. Statistical analysis was performed using Student’s t test (unpaired, two tailed).
Transferrin receptor-2 activation leads to the stimulation of the ERK1/2 pathway and hepcidin response to holotransferrin
The next question we addressed was the role of TfR2 in hepcidin expression after activation by its ligand, holotransferrin. The recent observation that specific stimulation of TfR2, and not of TfR1, by holotransferrin, led to the activation of the ERK1/2 cascade in K562 cells23 prompted us to ask whether holotransferrin was able to activate this pathway in isolated hepatocytes. That this was indeed the case is shown in Figure 3 by the rapid and transient phosphorylation of ERK1/2 after short-term exposure of 42 h serum-starved hepatocytes to either holotransferrin alone (Figure 3A, 30 μM holotransferrin for the indicated time), serum alone (Figure 3B, 10% serum for the indicated time), or both (Figure 3C). Noteworthy, in these latter conditions, the activation of the ERK1/2 pathway was enhanced and sustained for a longer period of time. To determine whether ERK1/2 activation in hepatocytes was involved in hepcidin induction by serum and serum+holotransferrin, hepatocytes were cultured in the presence of the specific ERK1/2 inhibitor, U0-126. The efficacy of the inhibitor on P-ERK1/2 is illustrated in the western blot in Figure 3C. Figure 3D shows that, in the presence of U0-126, the effect of hepcidin activation by serum tended to decrease but without statistical significance. In contrast, the effect of holotransferrin was completely inhibited in the presence of the inhibitor, suggesting that the ERK1/2 pathway might convey transcriptional information from holotransferrin to hepcidin.
Figure 3.
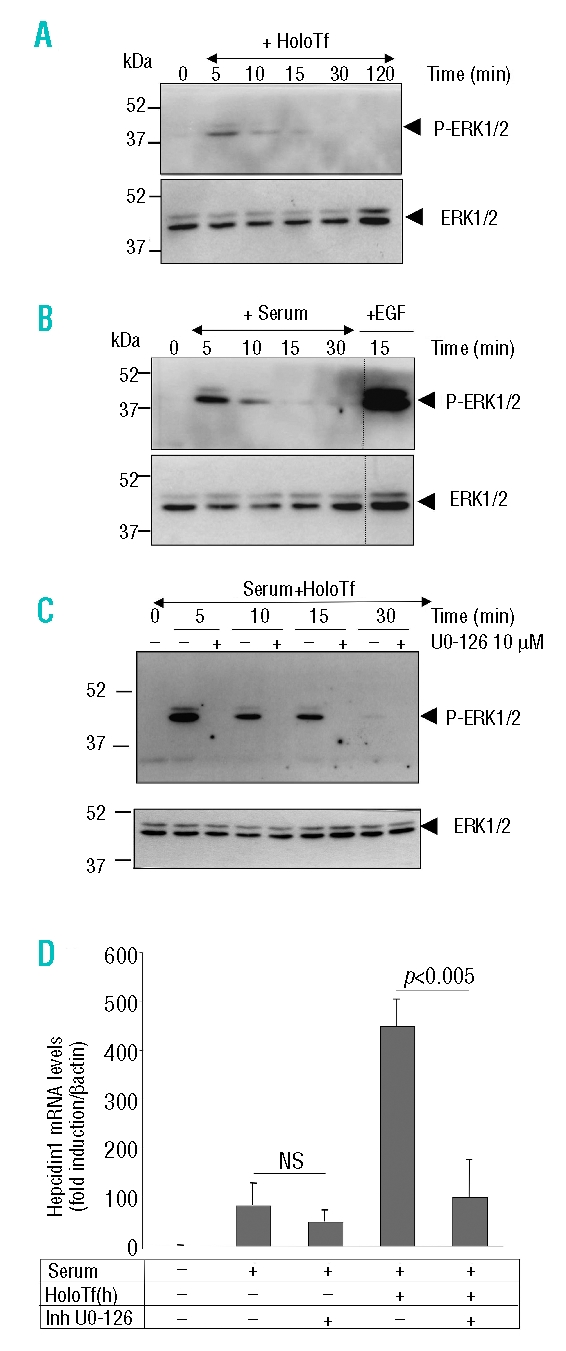
Effect of holotransferrin treatment on ERK1/2 activation and of a specific ERK1/2 inhibitor on hepcidin mRNA induction by holotransferrin. (A) After a 42 h period of serum-free culture, hepatocytes were treated either with 30 μM holotransferrin (holoTf) alone (A), 10% serum alone (B), or both (C) for 5, 10, 15, 30, or 120 min. Activation of the ERK1/2 pathway was assessed by immunoblotting 80 μg hepatocyte extracts with the phospho-ERK1/2 antibody. The relative amount of ERK1/2 was ascertained by immunoblot analysis using ERK1/2 antibody. Hepatocytes were treated for 15 min with 50 ng/mL EGF as a positive control for the activation of the ERK1/2 pathway. Molecular weight markers (kDa) are indicated on the left. (D) Relative changes in hepcidin mRNA levels as quantified by real-time qRT-PCR performed on cDNA synthesized from 2 μg total RNA (β-actin normalized, arbitrary units). Hepatocytes were treated for 24 h, after a 42 h period of serum-free culture, with either 0% serum (control cells) or 10% serum with or without 30 μM holoTf, in the presence or absence of 10 μM U0-126 inhibitor added 1 h prior treatment. Results are expressed relative to hepatocytes cultured for 66 h without serum. Mean±SD for four independent samples. The experiment was performed at least three times and a representative result is shown Statistical analysis was performed using Student’s t test (unpaired, two tailed). NS: non significant.
ERK activation by holotransferrin increased phospho Smad 1/5/8
Since BMP signaling occurs through phosphorylation of Smad1/5/8, we next investigated the effects of serum or serum+holotransferrin on hepatic P-Smad1/5/8 (Figure 4). The presence of serum led to the activation of P-Smad1/5/8 beginning 30 min after the treatment with maximum activation reached at 120 min. Holotransferrin alone was inefficient in activating this pathway throughout the kinetics. Interestingly, in the serum+holotransferrin condition, the amount of P-Smad1/5/8 was increased at both 15 and 30 min (comparing lanes 3 to 1, and 7 to 5, respectively) suggesting that the presence of holotransferrin could enhance phosphorylation of Smad1/5/8. This observation lead to the hypothesis that holotransferrin-induced ERK activation could be responsible for the increase of Smad1/5/8 phosphorylation. When serum+holotransferrin treated cells were incubated in the presence of the ERK inhibitor, the activation of Smad1/5/8 phosphorylation was indeed blunted at 15 and 30 min (comparing lanes 4 to 3, and 8 to 7, respectively) strongly suggesting that short-term activation of ERK is required for increased levels of P-Smad1/5/8. At 120 min, when P-ERK had returned to basal levels, the amount of P-Smad1/5/8 was found to be similar in the absence or presence of the ERK inhibitor, suggesting that an additional event is necessary beyond Smad1/5/8 phosphorylation for full hepcidin gene activation.
Figure 4.
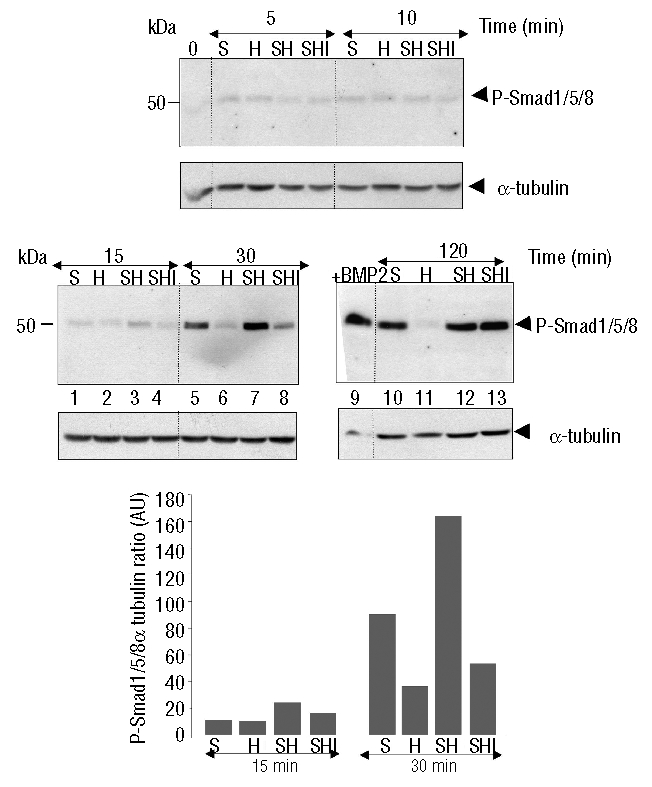
Effect of holotransferrin (holoTf) treatment on Smad1/5/8 activation. After a 42 h period of serum-free culture, hepatocytes were treated with either 10% serum alone (S), 30 μM holotransferrin alone (H), serum + holoTf (SH), or serum + holoTf in the presence of 10 μM U0-126 (SHI) for 5, 10, 15, 30, or 120 min. Activation of the Smad pathway was assessed by immunoblotting 120 μg of hepatocyte extracts with the phospho-Smad1/5/8 antibody. Lane 9 is a positive control with hepatocytes treated for 24 h with 20 ng/mL BMP2. Molecular weight markers (kDa) are indicated on the left. Blot densitometry of two independent experiments is shown.
HFE is not required for the hepcidin response to holotransferrin
HFE has recently been described as being a key component of the liver-centered homeostatic regulatory mechanism to signal for hepcidin production.13,14,24 To determine to what extent HFE is pivotal for hepcidin regulation by serum and holotransferrin in culture, similar experiments were performed with hepatocytes isolated from Hfe knockout (Hfe−/−) mice (Figure 5). As in hepatocytes from the C57Bl/6 strain, in the Hfe+/+ control hepatocytes isolated from the 129Tc strain, hepcidin expression was induced by serum and hepcidin mRNA levels were further increased about 4-fold in the presence of holotransferrin. Interestingly, in Hfe−/− hepatocytes, although the effect of serum was reduced by 50% as compared to that in Hfe+/+ hepatocytes, the fold induction of hepcidin by holotransferrin was similar to that observed in Hfe+/+ hepatocytes. Activation of the ERK and HJV/BMP pathways were found to be identical in Hfe+/+ and Hfe−/− hepatocytes in the presence of serum, holotransferrin and serum+holotransferrin (data not shown).
Figure 5.
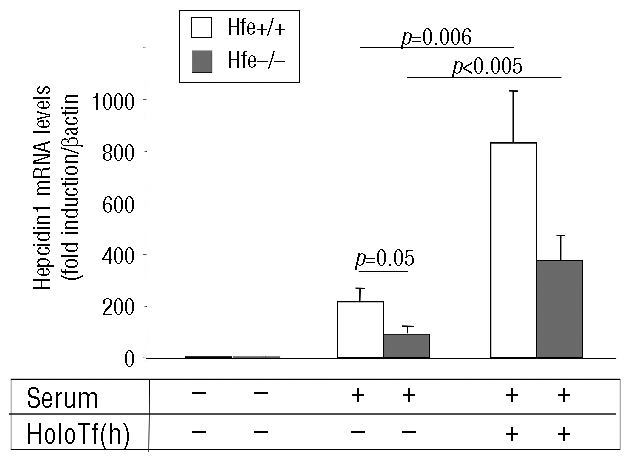
Effects of HFE deficiency on hepcidin response to serum and serum+holotransferrin. Relative changes in hepcidin mRNA levels were quantified by real-time qRT-PCR performed on cDNA synthesized from 2 μg total RNA (β-actin normalized, arbitrary units). (A) Hepatocytes isolated from either Hfe+/+ mice or Hfe−/−mice were treated, after a 42 h period of serum-free culture, with either 0% serum (control cells), 10% serum or 10% serum in the presence of 30 μM human holotransferrin [holoTf (h)] for 24 h. Results are expressed relative to hepatocytes cultured for 66 h without serum. Mean±SD for three independent samples. The experiment was performed twice and a representative result is shown. Statistical analysis was performed using Student’s t test (unpaired, two tailed).
Discussion
In this work, we developed culture conditions allowing transcriptional activation of hepcidin by serum and holotransferrin in mouse hepatocytes in primary culture. The effect of serum was robust (100- to 200-fold depending on the batch of serum), rapid (and sustained at least over 24 h), direct (no requirement for de novo protein synthesis) and partly due to the presence of BMP. The fact that BMP expression was similar in starved or serum-treated hepatocytes (Figure 2B) suggests that, in our model system, the endogenous levels of BMP were not sufficient to induce hepcidin levels in the absence of serum, and that the level of BMP activity necessary to induce hepcidin expression in serum conditions is likely due to BMP present in the serum. Knowing the important role of BMP during mammalian fetal development, one may predict that fetal bovine serum is highly enriched in BMP. However, an increase in BMP maturation or secretion induced by serum could not be completely ruled out. To test a possible effect of serum on the proteolytic activation of BMP precursors by furin,25 furin mRNA levels were measured and found to be unchanged regardless of the cell culture conditions (data not shown). In hepatoma cells, the contribution of endogenous BMP ligands to basal hepcidin expression was recently investigated using short interfering RNA.8 It was shown that endogenous BMP 2, 4 and 6 ligands all contribute to basal hepcidin expression. The authors hypothesized that these endogenously expressed BMP ligands could be inactivated after interaction with sHJV, thereby explaining the mechanism by which sHJV inhibits hepcidin expression in these cells.6,8 Interestingly, Kautz et al. recently suggested that, among the various BMP, BMP6 could have the preponderant role in the activation of the Smad signaling pathway in vivo. They showed that liver expression of BMP6 was transcriptionally regulated by iron, i.e. induced by iron overload and repressed by iron deficiency, suggesting its critical role in the maintenance of systemic iron homeostasis.26
The remaining hepcidin transcriptional activity measured in the presence of noggin suggests that there are other stimulatory effectors present in the serum, with the inflammatory cytokines (such as interleukin-6 and inter-leukin-1β) being obvious candidates.27,28 Finally, in our system, ERK activation seemed not to be required for serum-dependent hepcidin activation since the presence of U0-126 did not significantly alter hepcidin expression. In addition, activation of the ERK pathway by EGF alone was not sufficient to activate hepcidin expression in starved hepatocytes (not shown).
We then tested the effect of 30 μM holotransferrin in our culture system. This physiological concentration (holotransferrin levels in healthy individuals range from approximately 9–18 μM) was chosen since it was previously estimated to be the level at which hepatocytes could sense holotransferrin to regulate hepcidin gene expression.29 Holotransferrin alone was unable to activate hepcidin gene expression. In contrast, we demonstrated that, in the presence of serum, holotransferrin was able to reproducibly and significantly induce a 3- to 5-fold activation of hepcidin gene expression, suggesting the synergistic activation of hepcidin by two cooperating pathways. This result contrasts with recent data from Lin et al. showing that primary hepatocytes cultured for 48 h before treatment with 30 μM holotransferrin no longer responded by increasing hepcidin mRNA.29 The main difference is that, in our study, hepatocytes were previously serum-starved before serum+holotransferrin treatment. This could suggest the presence of long-term dominant negative regulators present in the serum or activated by the serum. It is noteworthy that the increased effect of holotransferrin on hepcidin was not due to contamination of the transferrin preparations by endotoxin (a potent inducer of hepcidin synthesis) as indicated by the absence of inducing effect of holotransferrin by itself. Furthermore, the effect of holotransferin was not observed with the iron-free ligand apotransferrin or with non-transferrin bound iron.
It was recently shown that TfR2, unlike TfR1, was present in lipid rafts and that binding of holotransferrin to TfR2 can activate the ERK signaling pathway in K562 cells.11 We present here the first evidence that the same activation is operating in hepatocytes and that ERK activation is necessary for holotransferrin-induced hepcidin gene expression. Interestingly, we also showed that holotransferrin regulates hepcidin mRNA concentrations through a HJV/BMP dependent pathway since the effect of holotransferrin was lost in the presence of noggin and BMP2/4 antibodies. This result confirms the recent observation of Lin et al., who reported that holotransferrin was an inducer of hepcidin gene expression in freshly isolated murine hepatocytes.29 We thus sought to investigate whether the ERK and HJV signaling pathways were dependent on each other. Smad4 is an interesting putative target of ERK since it was reported that ERK can phosphorylate Smad4 in vitro and that this phosphorylation could lead to enhanced Smad4 nuclear accumulation and, as a consequence, increased Smad4 transcriptional activity.30 To determine whether holotransferrin-induced ERK1/2 triggered Smad4 to translocate to the nucleus of the treated cells, nuclear extracts were prepared at different times after serum, holotransferrin or serum+holotransferrin treatments and assessed by western blot analysis (Online Supplementary Figure S3). However, there was no detectable change in the amount of Smad4 in either the nucleus, or the cytosol, regardless of the culture conditions, suggesting that Smad4 is most likely necessary but not sufficient to sense holotransferrin. We then measured the amount of P-Smad1/5/8 after hepatocyte stimulation by serum+holotransferrin and found that ERK inhibition led to a strong decrease in the amount of P-Smad1/5/8, possibly responsible for the decrease of hepcidin gene expression observed in the presence of the ERK inhibitor (Figure 3). Whether Smad1/5/8 are direct targets of ERK is unknown, although the kinetics of the pathway activation (very rapid for ERK and delayed for Smad1/5/8) does not favor this hypothesis. Importantly, regulation of Smad activity by kinase pathways has already been widely documented.31 Increasing evidence suggests that the amount of phosphorylation of Smad1/5/8 is directly related to iron-dependent expression of hepcidin in vivo. In mice, iron dextran injection32 as well as an iron-rich diet26 were shown to increase liver P-Smad1/5/8. Conversely, sHJV injected into mice was shown to induce decreased serum iron and decreased liver P-Smad1/5/8.8 The regions of the hepcidin promoter necessary for the trancriptional response of the gene to iron have not yet been characterized. Truksa et al. have, however, recently reported that they might be distally located, up to 1.6 kb from the start site of hepcidin gene transcription.33 Our hepatocytes in primary culture will constitute an invaluable model for studies aimed at determining the iron-responsive regions of the hepcidin gene.
The iron-sensitive mechanism for up-regulating hepcidin expression in response to iron overload in vivo involves TfR2 and HJV/Smad pathways. After iron injection, hepcidin mRNA levels remained unchanged in TfR2,34 HJV35 and liver-specific Smad4 knock-out mice.7 In patients with hemochromatosis related to TfR236 or HJV37 mutations, urinary hepcidin levels were found to be decreased or barely detectable, respectively. The role of HFE in iron sensing has remained more controversial. In both Hfe knockout mice and patients with HFE-related hemochromatosis, most data suggest that functional HFE is required to maintain basal hepcidin mRNA levels. However, according to some authors, this level could be increased above basal levels in response to altered iron status,38–40 suggesting that HFE deficiency is not impairing iron sensing. In contrast, in a study by Piperno et al., hepcidin response to oral iron was shown to be blunted in patients with HFE mutations.41 Here, we demonstrated that Hfe deficiency in isolated hepatocytes is associated with decreased basal levels of hepcidin but an unaltered response to iron, suggesting that the HFE-related phenotype is more likely to be related to absolute reduced levels of hepcidin rather than to a problem of iron sensing.
In conclusion, we show that murine hepatocytes in primary culture provide a suitable model for iron sensing and that holotransferrin, in cooperation with circulating serum factors, induces hepcidin expression through both BMP/HJV and ERK1/2 kinase pathways. We further confirm that HFE is necessary for the establishment of basal hepcidin levels but dispensable for hepcidin regulation by the iron-sensing machinery.
Acknowledgments
we warmly thank Germain Margall-Ducos for technical advice concerning liver perfusion, Nancy Andrews for kindly providing the Hfe knockout mice and all the team for critical discussions and reading the manuscript
Footnotes
Authorship and Disclosures
GR and J-CD: designed and performed the research, analyzed the data and contributed to writing the paper. SV: designed the research, analyzed the data and wrote the paper. The authors declare that they have no conflicts of interest.
The online version of this article contains a supplementary appendix.
Funding: this work was supported by funds from the ANR and EU contract (LSHM-CT-2006-037296).
References
- 1.Ganz T. Hepcidin and its role in regulating systemic iron metabolism. Hematology Am Soc Hematol Educ Program. 2006:29–35. doi: 10.1182/asheducation-2006.1.29. [DOI] [PubMed] [Google Scholar]
- 2.Pigeon C, Ilyin G, Courselaud B, Leroyer P, Turlin B, Brissot P, et al. A new mouse liver specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem. 2001;276:7811–9. doi: 10.1074/jbc.M008923200. [DOI] [PubMed] [Google Scholar]
- 3.Nicolas G, Chauvet C, Viatte L, Danan JL, Bigard X, Devaux I, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest. 2002;110:1037–44. doi: 10.1172/JCI15686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004;113:1271–6. doi: 10.1172/JCI20945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vaulont S, Lou DQ, Viatte L, Kahn A. Of mice and men: the iron age. J Clin Invest. 2005;115:2079–82. doi: 10.1172/JCI25642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Babitt JL, Huang FW, Wrighting DM, Xia Y, Sidis Y, Samad TA, et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat Genet. 2006;38:531–9. doi: 10.1038/ng1777. [DOI] [PubMed] [Google Scholar]
- 7.Wang RH, Li C, Xu X, Zheng Y, Xiao C, Zerfas P, et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2005;2:399–409. doi: 10.1016/j.cmet.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 8.Babitt JL, Huang FW, Xia Y, Sidis Y, Andrews NC, Lin HY. Modulation of bone morphogenetic protein signaling in vivo regulates systemic iron balance. J Clin Invest. 2007;117:1933–9. doi: 10.1172/JCI31342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Truksa J, Peng H, Lee P, Beutler E. Bone morphogenetic proteins 2, 4, and 9 stimulate murine hepcidin 1 expression independently of Hfe, transferrin receptor 2 (Tfr2), and IL-6. Proc Natl Acad Sci USA. 2006;103:10289–93. doi: 10.1073/pnas.0603124103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin L, Goldberg YP, Ganz T. Competitive regulation of hepcidin mRNA by soluble and cell-associated hemojuvelin. Blood. 2005;106:2884–9. doi: 10.1182/blood-2005-05-1845. [DOI] [PubMed] [Google Scholar]
- 11.Calzolari A, Oliviero I, Testa U. Transferrin receptor 2 is emerging as a major player in the control of iron metabolism. CEJB. 2007;2:34–55. [Google Scholar]
- 12.Feder JN, Penny DM, Irrinki A, Lee VK, Lebron JA, Watson N, et al. The hemochromatosis gene product complexes with the transferrin receptor and lowers its affinity for ligand binding. Proc Natl Acad Sci USA. 1998;95:1472–7. doi: 10.1073/pnas.95.4.1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goswami T, Andrews NC. Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J Biol Chem. 2006;281:28494–8. doi: 10.1074/jbc.C600197200. [DOI] [PubMed] [Google Scholar]
- 14.Schmidt PJ, Toran PT, Giannetti AM, Bjorkman PJ, Andrews NC. The transferrin receptor modulates hfe-dependent regulation of hepcidin expression. Cell Metab. 2008;7:205–14. doi: 10.1016/j.cmet.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levy JE, Montross LK, Cohen DE, Fleming MD, Andrews NC. The C282Y mutation causing hereditary hemochromatosis does not produce a null allele. Blood. 1999;94:9–11. [PubMed] [Google Scholar]
- 16.Berry MN, Friend DS. High-yield preparation of isolated rat liver parenchymal cells: a biochemical and fine structural study. J Cell Biol. 1969;43:506–20. doi: 10.1083/jcb.43.3.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nicolas G, Bennoun M, Devaux I, Beaumont C, Grandchamp B, Kahn A, et al. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci USA. 2001;98:8780–5. doi: 10.1073/pnas.151179498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramey G, Faye A, Durel B, Viollet B, Vaulont S. Iron overload in Hepc1(−/−) mice is not impairing glucose homeostasis. FEBS Lett. 2007;581:1053–7. doi: 10.1016/j.febslet.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 19.Nemeth E, Valore EV, Territo M, Schiller G, Lichtenstein A, Ganz T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood. 2003;101:2461–3. doi: 10.1182/blood-2002-10-3235. [DOI] [PubMed] [Google Scholar]
- 20.Gehrke SG, Kulaksiz H, Herrmann T, Riedel HD, Bents K, Veltkamp C, et al. Expression of hepcidin in hereditary hemochromatosis: evidence for a regulation in response to the serum transferrin saturation and to non-transferrin-bound iron. Blood. 2003;102:371–6. doi: 10.1182/blood-2002-11-3610. [DOI] [PubMed] [Google Scholar]
- 21.Kawabata H, Tong X, Kawanami T, Wano Y, Hirose Y, Sugai S, et al. Analyses for binding of the transferrin family of proteins to the transferrin receptor 2. Br J Haematol. 2004;127:464–73. doi: 10.1111/j.1365-2141.2004.05224.x. [DOI] [PubMed] [Google Scholar]
- 22.Groppe J, Greenwald J, Wiater E, Rodriguez-Leon J, Economides AN, Kwiatkowski W, et al. Structural basis of BMP signalling inhibition by the cystine knot protein Noggin. Nature. 2002;420:636–42. doi: 10.1038/nature01245. [DOI] [PubMed] [Google Scholar]
- 23.Calzolari A, Raggi C, Deaglio S, Sposi NM, Stafsnes M, Fecchi K, et al. TfR2 localizes in lipid raft domains and is released in exosomes to activate signal transduction along the MAPK pathway. J Cell Sci. 2006;119:4486–98. doi: 10.1242/jcs.03228. [DOI] [PubMed] [Google Scholar]
- 24.Vujic Spasic M, Kiss J, Herrmann T, Galy B, Martinache S, Stolte J, et al. Hfe acts in hepatocytes to prevent hemochromatosis. Cell Metab. 2008;7:173–8. doi: 10.1016/j.cmet.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 25.Cui Y, Jean F, Thomas G, Christian JL. BMP-4 is proteolytically activated by furin and/or PC6 during vertebrate embryonic development. Embo J. 1998;17:4735–43. doi: 10.1093/emboj/17.16.4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kautz L, Meynard D, Monnier A, Darnaud V, Bouvet R, Wang RH, et al. Iron regulates phosphorylation of Smad1/5/8 and gene expression of Bmp6, Smad7, Id1, and Atoh8 in the mouse liver. Blood. 2008;112:1503–9. doi: 10.1182/blood-2008-03-143354. [DOI] [PubMed] [Google Scholar]
- 27.Andrews NC. Anemia of inflammation: the cytokine-hepcidin link. J Clin Invest. 2004;113:1251–3. doi: 10.1172/JCI21441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee P, Peng H, Gelbart T, Wang L, Beutler E. Regulation of hepcidin transcription by interleukin-1 and interleukin-6. Proc Natl Acad Sci USA. 2005;102:1906–10. doi: 10.1073/pnas.0409808102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin L, Valore EV, Nemeth E, Goodnough JB, Gabayan V, Ganz T. Iron-transferrin regulates hepcidin synthesis in primary hepatocyte culture through hemojuvelin and BMP2/4. Blood. 2007;110:2182–9. doi: 10.1182/blood-2007-04-087593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roelen BA, Cohen OS, Raychowdhury MK, Chadee DN, Zhang Y, Kyriakis JM, et al. Phosphorylation of threonine 276 in Smad4 is involved in transforming growth factor-beta-induced nuclear accumulation. Am J Physiol Cell Physiol. 2003;285:C823–30. doi: 10.1152/ajpcell.00053.2003. [DOI] [PubMed] [Google Scholar]
- 31.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 32.Yu PB, Hong CC, Sachidanandan C, Babitt JL, Deng DY, Hoyng SA, et al. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat Chem Biol. 2008;4:33–41. doi: 10.1038/nchembio.2007.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Truksa J, Lee P, Peng H, Flanagan J, Beutler E. The distal location of the iron responsive region of the hepcidin promoter. Blood. 2007;110:3436–7. doi: 10.1182/blood-2007-05-091108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawabata H, Fleming RE, Gui D, Moon SY, Saitoh T, O’Kelly J, et al. Expression of hepcidin is down-regulated in TfR2 mutant mice manifesting a phenotype of hereditary hemochromatosis. Blood. 2005;105:376–81. doi: 10.1182/blood-2004-04-1416. [DOI] [PubMed] [Google Scholar]
- 35.Niederkofler V, Rishard S, Arber S. Hemojuvelin is essential for dietary iron-sensing and its mutation leads to severe iron overload. J Clin Invest. 2005;115:2180–6. doi: 10.1172/JCI25683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nemeth E, Roetto A, Garozzo G, Ganz T, Camaschella C. Hepcidin is decreased in TFR2-hemochromatosis. Blood. 2004;105:4103–5. doi: 10.1182/blood-2004-08-3042. [DOI] [PubMed] [Google Scholar]
- 37.Papanikolaou G, Samuels ME, Ludwig EH, MacDonald ML, Franchini PL, Dube MP, et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat Genet. 2004;36:77–82. doi: 10.1038/ng1274. [DOI] [PubMed] [Google Scholar]
- 38.Bridle KR, Frazer DM, Wilkins SJ, Dixon JL, Purdie DM, Crawford DH, et al. Disrupted hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homeostasis. Lancet. 2003;361:669–73. doi: 10.1016/S0140-6736(03)12602-5. [DOI] [PubMed] [Google Scholar]
- 39.Constante M, Jiang W, Wang D, Raymond VA, Bilodeau M, Santos MM. Distinct requirements for Hfe in basal and induced hepcidin levels in iron overload and inflammation. Am J Physiol Gastrointest Liver Physiol. 2006;291:G229–37. doi: 10.1152/ajpgi.00092.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gehrke SG, Herrmann T, Kulaksiz H, Merle U, Bents K, Kaiser I, et al. Iron stores modulate hepatic hepcidin expression by an HFE-independent pathway. Digestion. 2005;72:25–32. doi: 10.1159/000087400. [DOI] [PubMed] [Google Scholar]
- 41.Piperno A, Girelli D, Nemeth E, Trombini P, Bozzini C, Poggiali E, et al. Blunted hepcidin response to oral iron challenge in HFE-related hemochromatosis. Blood. 2007;110:4096–100. doi: 10.1182/blood-2007-06-096503. [DOI] [PubMed] [Google Scholar]