

Abstract
Pyrrole-imidazole polyamides are DNA binding molecules that are programmable for a large repertoire of DNA sequences. Typical syntheses of this class of heterocyclic oligomers rely on solid-phase methods. Solid-phase methodologies offer rapid assembly on a micromole scale sufficient for biophysical characterizations and cell culture studies. In order to produce gram-scale quantities necessary for efficacy studies in animals, polyamides must be readily synthesized in solution. An 8-ring hairpin polyamide 1, which targets the DNA sequence 5'-WGWWCW-3', was chosen for our synthesis studies as this oligomer exhibits androgen receptor antagonism in cell culture models of prostate cancer. A convergent solution-phase synthesis of 1 from a small set of commercially available building blocks is presented which highlights principles for preparing gram quantities of pyrrole-imidazole oligomers with minimal chromatography.
Introduction
Pyrrole-imidazole polyamides are a class of small molecules that bind the minor groove of DNA sequence specifically.1,2 Encoded by side-by-side arrangements of N-methylpyrrole (Py) and N-methylimidazole (Im) carboxamide monomers, Im/Py pairs distinguish G•C from C•G base pairs, whereas Py/Py pairs are degenerate for T•A and A•T.3 Hairpin Py-Im polyamides have been shown to bind a broad repertoire of DNA sequences,4 permeate cell membranes and traffic to the nucleus,5 access chromatin,6 and disrupt protein-DNA interfaces.2 Hairpin polyamide inhibition of transcription factor-DNA binding of HIF-1α,7 androgen receptor (AR),8 and AP-19 has been exploited for controlling expression of medically relevant genes such as VEGF, PSA, TGF-β1 and LOX-1 in cell culture experiments.
An underpinning to transition polyamide studies from cell culture to small animal disease models is the ability to synthesize Py-Im polyamides on gram-scale. Over the years advances in polyamide solid-phase synthesis have been reported, including Boc- and Fmoc-based approaches from our laboratories and others.10 Solid-phase methodologies offer many advantages for milligram-scale polyamide syntheses, including rapid and reliable amino acid couplings and facile purifications owing to immobilization of the polyamide oligomer on a solid support. However, these techniques intrinsically limit the scale of synthesis. Conversely, efficient gram-scale solution-phase methods for polyamide synthesis that avoid arduous chromatographic purifications and employ commercially available Py-Im amino acid building blocks as reagents are less well developed. Remarkably, solution-phase synthesis of hairpin polyamides was the standard in our laboratory prior to the development of solid-phase methodologies,11a and many variations on this theme have been published.11 However, laborious chromatographic purifications and modest reaction yields are commonplace. Therefore, we sought to develop a general solution-phase polyamide synthesis method that would allow access to gram quantities of material in high yield with minimal chromatography.
We report a proof-of-principle study demonstrating that hairpin Py-Im polyamides can be synthesized in solution from a small set of building blocks on large scale with minimal use of chromatography. This method involves Boc-protected dimers, trimers, and tetramers of heterocycles suitable for convergent syntheses. By exploiting differences in the physical solubility properties of starting materials versus products, a solution-phase synthesis of an 8-ring hairpin Py-Im polyamide 1 (Figure 1) has been achieved. Notably, our synthesis permits core polyamide 2 (Figure 2) to be prepared without a single chromatographic purification, thereby providing large quantities of 2 for subsequent modification at the C-terminus such as 1. Py-Im polyamide 1, which targets the DNA sequence 5'-WGWWCW-3', was selected for our studies because it antagonizes AR binding to androgen response elements (ARE) in gene promoters and regulates a subset of AR-driven genes, such as PSA.8 The regulation of aberrant AR-activated gene expression in prostate cancer is a promising strategy for developing novel therapeutics.8 This biological activity, coupled with our desire to conduct small animal efficacy experiments with 1, renders this polyamide an ideal candidate for scale-up and optimization studies. In addition, we discuss unifying principles for planning solution-phase polyamide syntheses of different Py-Im arrangements.
Figure 1.
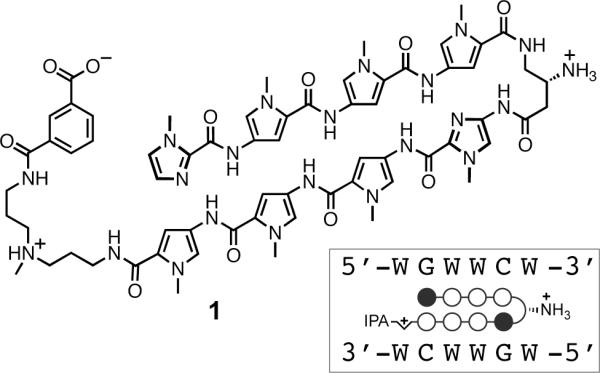
Structure of Py-Im hairpin polyamide 1 targeting the DNA sequence 5'-WGWWCW-3' and its ball and stick representation.
Figure 2.

Retrosynthetic strategy for the convergent solution-phase synthesis of polyamide 1. 7-10 are commercially available building blocks. Boc = t-butyl carbamate, Bt = benzotriazole, Cbz = benzyl carbamate.
Results and Discussion
Our retrosynthetic approach for the preparation of an 8-ring hairpin polyamide, ImPyPyPy-(R)β-H2 Nγ-ImPyPyPy-(+)-IPA (1), is shown in Figure 2. Sequential couplings of ImPyPyPy tetramer 3 to turn moiety 5, followed by ester saponification and coupling to PyPyPy trimer 4 affords polyamide 2 in a convergent manner. Advanced intermediates 3-5 were prepared from commercially available building blocks 7-10,12 which have been previously synthesized by our laboratory and others.10a,10c,13 The cornerstone of our synthesis strategy capitalizes on the disparate physical properties of starting materials versus products, which permit purification of each intermediate to be achieved by combinations of precipitation, trituration, and crystallization. Such details are described below.
The synthesis of pyrrole trimer 4 begins with pyrrole amine salt 10 (Scheme 1). Amide coupling of 10 with activated pyrrole monomer 7 affords dimer 11 in 93% yield. The utilization of a small excess of 10 relative to 7 drives the reaction to completion, and residual 10 is readily separated from 11 following precipitation in water and aqueous washing of the residual solid 11. Reaction of dimer 11 with anhydrous HCl in diethyl ether removes the carbamate protecting group and facilitates precipitation of 12 as the HCl salt during the course of the reaction. Isolation of solid 12 by filtration, followed by washing of the residual solid with excess Et2O provides the amine HCl salt in 96% yield. By exploiting the aqueous solubility of 10 versus insolubility of 11, PyPy dimer 11 is easily purified from a small excess of 10 by precipitation, whereas deprotected 12 is separable from Boc-protected 11 by virtue of the Et2O solubility of 11 versus insolubility of 12. This reaction sequence highlights our synthesis strategy: exploiting the different solubility profiles of reactants versus products for chromatography-free purifications. Accordingly, pyrrole trimer 4 was obtained from dimer 12 by coupling with 7, followed by acidic deprotection to yield 4 in 95% yield (2 steps) from dimer 12. The ImPyPyPy tetramer 3 was synthesized in two steps from trimer 4. 1-Methyl-2-trichloroacetylimidazole (6), prepared in one step from N-methylimidazole,10c,13 was allowed to react with a small excess of 4 to deliver tetramer 14 in 83% yield following precipitation in H2O, trituration with Et2O, and drying in vacuo. Saponification of 14 with aqueous NaOH in 1,4-dioxane, followed by neutralization with aqueous HCl, precipitation, and Et2O trituration, afforded tetramer 3 in 77% overall yield from trimer 4.
Scheme 1.

Preparation of 3 and 4.a
aReagents: (i) DMF, DIEA, 7, 23°C, 8 h, 93%; (ii) 2.0 M HCl in Et2O, 23°C, 18 h, 99%; (iii) DMF, DIEA, 7, 23°C, 8 h, 96%; (iv) 4.0 M HCl in 1,4-dioxane, 23°C, 18 h, 99%; (v) DMF, DIEA, 6, 23°C, 2 h, 83%; (vi) NaOH (aq), 1,4-dioxane, 42°C, 2 h, 93%.
The Im-turn fragment 5 was synthesized in two steps from the Im•HCl salt monomer10a 9 by coupling to PyBOP activated (R)-3,4-Cbz-Dbu(Boc)-OH yielding protected dimer 15 in 95% yield (Scheme 2). The utilization of a small excess of 9 drives the coupling reaction to completion and is easily separated in the aqueous wash step. Removal of the carbamate protecting group with anhydrous HCl in Et2O yielded the final synthon for our studies, imidazole-turn dimer 5, in quantitative yield following filtration and washing of the residual salt. With compound 5 in hand, the assembly of core polyamide 2 was initiated. PyBOP-mediated coupling of tetramer 3 to a small excess of water-soluble Im-turn dimer 5 yielded the advanced intermediate 16 in 97% yield. Saponification of 16 to acid 17, followed by amide coupling with an excess of water-soluble trimer 4, delivered core Py-Im hairpin polyamide 2 in 88% yield for the two steps. Consistent with the previously discussed strategy for synthesizing intermediates 3-5, the differences in solubility of reactants versus products were exploited to isolate pure material by precipitation, washing, and trituration. In most cases a low boiling-point solvent was employed in the final trituration step to facilitate efficient solvent removal in vacuo. Core polyamide 2 was synthesized without a single chromatographic purification in high overall purity, as depicted by the analytical HPLC analysis of 2 shown in Figure 3. Multi-gram quantities of 2 have been readily synthesized by this method, providing a stockpile of material for elaboration at the C-terminus into discrete polyamide conjugates, such as 1.
Scheme 2.

Preparation of 5 and assembly of core polyamide 2.a
aReagents: (i) DMF, DIEA, PyBOP, (R)-3,4-Cbz-Dbu(Boc)-OH (8), 23°C, 8 h, 95%; (ii) 4.0 M HCl in 1,4-dioxane, 23°C, 16 h, 99%; (iii) DMF, DIEA, PyBOP, 3, 23°C, 2 h, 97%; (iv) KOH (aq), MeOH, 1,4-dioxane, 42°C, 2 h, 92%; (i) DMF, DIEA, PyBOP, 4, 23°C, 10 h, 96%.
Figure 3.

Analysis of polyamide 2 purity by analytical HPLC. Wavelength shown is 310 nm.
Py-Im polyamide 1 was synthesized in solution from advanced core 2 by coupling the preassembled C-terminal tail moiety 21 with saponified core 22. This convergent approach begins by preparing Boc-protected C-terminus moiety 20 (Scheme 3). PyBOP-mediated coupling of mono-Boc-protected triamine linker 1814 with mono-benzyl-protected isophthalic acid 1915 afforded Boc-protected 20 in 98% yield. Deprotection of 20 with anhydrous CF3CO2H in dichloromethane (1:1) yielded amine 21, which was used immediately following concentration under high vacuum. Saponification of core polyamide 2 with aqueous NaOH in 1,4-dioxane at 23 °C yielded 8-ring acid 22 in 89% yield (Scheme 4). The transformation of 2 to 22 proved somewhat difficult in early studies due to formation of an unidentified side product. Avoiding reaction temperatures above 23 °C suppresses most of the byproduct formation, whereas a screen of aqueous bases commonly used for ester saponification (KOH and LiOH) failed to identify a better reagent. Coupling of residual acid 22 with freshly prepared 21 delivered the penultimate oligomer 23 in 87% yield. Unfortunately, crude oligomer 23 could not be satisfactorily purified by our standard method and required chromatography on silica gel to achieve pure material. Hundreds of milligrams of 23 have been prepared by this method in a single reaction sequence. Global deprotection of 23 by hydrogenation (Pd/C, ~ 1 atm H2) at 23 °C for 48 hours yields Py-Im polyamide 1 in 81% yield. Final product 1 can be separated from residual catalyst by solid-phase extraction and then purified by preparative reverse-phase HPLC.
Scheme 3.
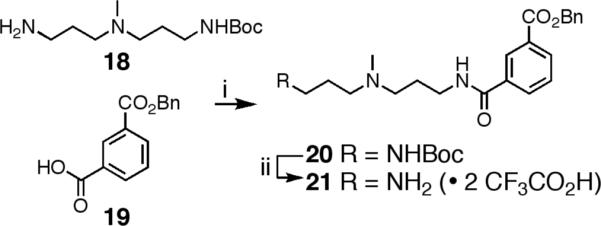
Preparation of 21.a
aReagents: (i) DMF, DIEA, PyBOP, 23 °C, 3 h, 98%; (ii) CF3CO2H, CH2Cl2, 23 °C, 20 min, concentration in vacuo and used crude.
Scheme 4.
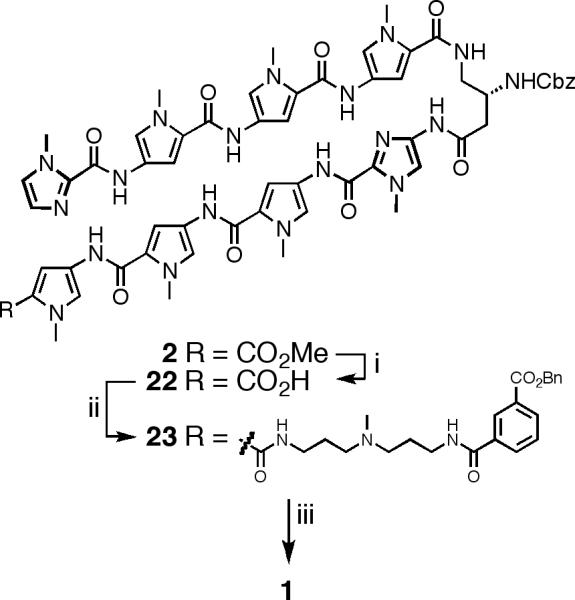
Final steps for the synthesis of Py-Im polyamide 1.a
aReagents: (i) NaOH (aq), 1,4-dioxane, 23°C, 11 h, 89%. (ii) DIEA, DMF, PyBOP, 21, 23°C, 12 h, 87%. (iii) DMF, Pd/C, H2 (1 atm), 23°C, 48 h, 81%.
With multi-milligram quantities of 1 on hand, we investigated in detail the UV properties of Py-Im polyamide 1 in a variety of laboratory and biologically relevant solvents. As shown in Figure 4 a strong solvent influence on the molar extinction coefficient of 1 is observed as the amount of organic cosolvent is increased. For example, an extinction coefficient (ε, M-1cm-1) of 26,500 was measured for 1 in distilled and deionized H2O, whereas this value doubled to 54,800 in 50% acetonitrile in aqueous CF3CO2H (0.1% v/v CF3CO2H), a widely utilized laboratory solvent for purifying and quantifying peptides. A stock solution frequently encountered for preparing biological samples, 10% DMSO in DEPC-treated H2O yielded an intermediary value of 40,900 M-1cm-1. Hence, care must be taken to consider the solvent system utilized when performing UV spectroscopy to determine polyamide concentrations.
Figure 4.

UV properties of polyamide 1 in solvent systems I-VIII. (A) UV traces of I-VIII from 250-600 nm and (B) tabular form of data. Molar extinction coefficients were calculated from the λmax for each individual system, which ranged from 313 - 317 nm. DEPC = diethylpyrocarbonate treated H2O.
Conclusion
A solution-phase synthesis of Py-Im hairpin polyamide 1 is presented, highlighting unifying principles for the preparation of related polyamides. A convergent synthesis was developed, requiring no chromatographic purifications to arrive at core 8-ring polyamide 2 on multi-gram scale. Final elaboration of the C-terminus affords AR polyamide antagonist 1 in high yield. The synthetic methodology permits gram-scale synthesis of Py-Im polyamides, a minimum next step as we transition these small molecules to animal models for biological efficacy.
Experimental Section
General
Chemicals were purchased from Sigma-Aldrich and were used without further purification. (R)-3,4-Cbz-Dbu(Boc)-OH was purchased from Senn Chemicals AG (code number 44159). Bulk grade solvents were from Fisher Scientific. Centrifugation was performed in a Beckman Coulter bench-top centrifuge (Allegra 21R) equipped with a Beckman swing-out rotor (model S4180). Analytical HPLC analysis was conducted on a Beckman Gold instrument equipped with a Phenomenex Gemini analytical column (250 × 4.6 μm, 5 μm), a diode array detector, and the mobile phase consisted of a gradient of acetonitrile (MeCN) in 0.1% (v/v) aqueous CF3CO2H. Preparative HPLC was performed on an Agilent 1200 system equipped with a solvent degasser, diode array detector, and a Phenomenex Gemini column (250 × 21.2 mm, 5 μm). A gradient of MeCN in 0.1% (v/v) aqueous CF3CO2H was utilized as the mobile phase. UV-Vis measurements were made on a Hewlett-Packard diode array spectrophotometer (Model 8452 A). NMR spectroscopy was performed on a Varian instrument operating at 499.8 MHz (for 1H) or 125.7 MHz (for 13C) at ambient temperature. All NMR analyses were performed in DMSO-d6, and chemical shifts are reported in parts per million relative to the internal solvent peak referenced to 2.49 (for 1H) or 39.5 (for 13C). High-resolution mass spectrometry (HRMS) was recorded in positiveion mode by fast-atom bombardment (FAB+) on a JOEL JMS-600H instrument or by electrospray ionization (ESI+) on a Waters Acquity UPLC-LCT Premiere XE TOF-MS system.
HCl•H2N-Py-CO2Me (10)
Prepared as previously described.10a 1H NMR: δ 10.09 (br s, 3H), 7.25 (d, J = 2.0 Hz, 1H), 6.80 (d, J = 2.2 Hz, 1H), 3.85 (s, 3H), 3.74 (s, 3H); 13C NMR: δ 160.2, 123.7, 120.8, 113.9, 111.4, 51.3, 36.6; HRMS (FAB+) calc'd for C7H11N2O2 [M+H]+ 155.0821, found 155.0847.
BocHN-PyPy-CO2Me (11)
A solution of BocHN-Py-OBt 7 (6.16 g, 17.2 mmol) and HCl•H2N-Py-CO2Me 10 (3.61 g, 19.0 mmol) in DMF (39 mL) and DIEA (6 mL, 34.4 mmol) was stirred at 23 °C for 8 h. The solution was then added to distilled H2O (500 mL) pre-acidified with aqueous HCl (1N, 300 mL, 300 mmol), yielding a precipitate that was isolated by centrifugation (~ 4500 rpm). The residual solid was again suspended in distilled H2O (80 mL) and collected by centrifugation (repeated 2X). The resultant solid, which contained a small amount of residual H2O, was frozen and lyophilized to dryness. Drying of the light-brown solid in vacuo yielded dimer 11 (6.03 g, 93%). 1H NMR: δ 9.84 (s, 1H), 9.10 (s, 1H), 7.44 (d, J = 1.7 Hz, 1H), 6.88 (m, 2H), 6.82 (s, 1H), 3.82 (s, 3H), 3.79 (s, 3H), 3.72 (s, 3H), 1.44 (s, 9H); 13C NMR: δ 160.8, 158.4, 152.8, 123.0, 122.6, 122.4, 120.7, 118.4, 117.1, 108.3, 103.8, 78.3, 50.9, 36.1, 36.0, 28.2; HRMS (FAB+) calc'd for C18H25N4O5 [M+H]+ 377.1825, found 377.1835.
HCl•H2N-PyPy-CO2Me (12)
Dimer 11 (4.0 g, 10.6 mmol) in a solution of anhydrous HCl in Et2O (2.0 M, 400 mL) was stirred at 23 °C for 18 h. The mixture was then diluted with 400 mL of anhydrous Et2O and filtered over a sintered glass funnel. The resultant solid was washed with copious amounts of anhydrous Et2O and dried in vacuo to yield dimer 12 as a tan solid (3.3 g, 99%). 1H NMR: δ 10.12 (s, 1H), 10.07 (br s, 3H), 7.46 (d, J = 2.0 Hz, 1H), 7.10 (d, J = 2.0 Hz, 1H), 7.00 (d, J = 2.0 Hz, 1H), 6.91 (d, J = 2.0 Hz, 1H), 3.87 (s, 3H), 3.83 (s, 3H), 3.72 (s, 3H); 13C NMR: δ 160.8, 157.7, 124.6, 122.6, 121.7, 120.8, 118.6, 113.1, 108.4, 107.2, 51.0, 36.6, 36.2; HRMS (FAB+) calc'd for C H13H17N4O3[M+H]+ 277.1301, found 277.1292.
BocHN-PyPyPy-CO2Me (13)
A solution of BocHN-Py-OBt 7 (3.1 g, 8.7 mmol) and dimer 12 (3.0 g, 9.59 mmol) in DMF (20 mL) and DIEA (3 mL, 17.4 mmol) was stirred at 23 °C for 8 h. The solution was then added to distilled H2O (250 mL) pre-acidified with aqueous HCl (1N, 150 mL, 150 mmol), yielding a precipitate that was isolated by centrifugation (~ 4500 rpm). The residual solid was again suspended in distilled H2O (40 mL) and collected by centrifugation (repeated 2X). The resultant solid, which contained a small amount of residual H2O, was frozen and lyophilized to dryness. Drying of the light-brown solid in vacuo yielded trimer 13 (4.17 g, 96%). 1H NMR: δ 9.91 (s, 1H), 9.86 (s, 1H), 9.09 (s, 1H), 7.46 (d, J = 2.0 Hz, 1H), 7.21 (d, J =1.7 Hz, 1H), 7.05 (d, J = 1.5 Hz, 1H), 6.89 (m, 2H), 6.83 (s, 1H), 3.83 (s, 6H), 3.80 (s, 3H), 3.73 (s, 3H), 1.45 (s, 9H); 13C NMR: δ 160.8, 158.5, 158.4, 152.8, 123.0, 122.8, 122.4, 122.30, 122.29, 120.7, 118.48, 118.47, 117.0, 108.3, 104.8, 103.8, 78.3, 50.9, 36.2, 36.05, 36.04, 28.2; HRMS (FAB+) calc'd for C24H30N6O6 [M]• + 498.2227, found 498.2233.
HCl•H2N-PyPyPy-CO2Me (4)
Dimer 13 (4.0 g, 8.02 mmol) in a solution of anhydrous HCl in 1,4-dioxane (4.0 M, 300 mL) was stirred at 23 °C for 18 h. The mixture was then diluted with 600 mL of anhydrous Et2O and filtered over a sintered glass funnel. The resultant solid was washed with copious amounts of anhydrous Et2O and dried in vacuo to yield trimer 4 as a brown-orange solid (3.45 g, 99%). 1H NMR: δ 10.16 (s, 3H), 10.13 (s, 1H), 9.97 (s, 1H), 7.46 (d, J = 2.0 Hz, 1H), 7.25 (d, J = 1.7 Hz, 1H), 7.11 (d, J = 2.0 Hz, 1H), 7.08 (d, J = 2.0 Hz, 1H), 7.02 (d, J = 2.0 Hz, 1H), 6.91 (d, J = 2.0 Hz, 1H), 3.88 (s, 3H), 3.84 (s, 3H), 3.82 (s, 3H), 3.72 (s, 3H); 13C NMR: δ 160.8, 158.4, 157.7, 124.8, 123.0, 122.6, 121.9, 121.6, 120.8, 118.7, 118.5, 113.0, 108.4, 107.2, 104.8, 51.0, 36.6, 36.2, 36.1; HRMS (FAB+) calc'd for C19H22N6O4 [M•]+ 398.1702, found 398.1685.
ImPyPyPy-CO2Me (14)
A solution of trimer 4 (1.019 g, 2.34 mmol) and 1-methyl-2-trichloroacetylimidazole (6)10c,13 (478 mg, 2.10 mmol) in DMF (4.5 mL) and DIEA (910 μL, 5.22 mmol) was stirred at 23 °C for 2 h. The solution was then added to distilled H2O (40 mL) pre-acidified with aqueous HCl (1N, 910 μL, 0.91 mmol), yielding a precipitate that was isolated by centrifugation (~ 4500 rpm). The residual solid was again suspended in distilled H2O (40 mL) and collected by centrifugation (repeated 1X). The resultant solid, which contained a small amount of residual H2O, was frozen and lyophilized to dryness. The solid was triturated with anhydrous Et2O and filtered over a sintered glass funnel. The resultant solid was washed with copious amounts of anhydrous Et2O and dried in vacuo to yield tetramer 14 as a light brown solid (886 mg, 83%). +H NMR: δ 10.68 (s, 1H), 10.00 (s, 1H), 9.94 (s, 1H), 7.48 (s, 1H), 7.47 (d, J = 2.0 Hz, 1H), 7.31 (d, J = 1.7 Hz, 1H), 7.24 (d, J = 1.7 Hz, 1H), 7.18 (s, 1H), 7.17 (d, J = 2.0 Hz, 1H), 7.08 (d, J = 2.0 Hz, 1H), 6.91 (d, J = 1.7 Hz, 1H), 4.01 (s, 3H), 3.86 (s, 3H), 3.84 (s, 3H), 3.83 (s, 3H), 3.73 (s, 3H); 13C NMR: δ 160.8, 158.5, 158.4, 155.0, 138.2, 126.4, 125.6, 123.1, 123.0, 122.5, 122.2, 121.2, 120.7, 118.8, 118.6, 118.5, 108.3, 104.9, 104.8, 50.9, 36.18, 36.17, 36.1, 35.4; HRMS (FAB+) calc'd for C24H27N8O5 [M+H]+ 507.2104, found 507.2116.
ImPyPyPy-CO2H (3)
A solution of tetramer 14 (804 mg, 1.59 mmol) in 1,4-dioxane (8 mL) and aqueous NaOH (1N, 8.0 mL, 8.00 mmol) was stirred at 42 °C for 2 h. The solution was then added to distilled H2O (40 mL) pre-acidified with aqueous HCl (1N, 8 mL, 8.00 mmol), yielding a precipitate that was isolated by centrifugation (~ 4500 rpm). The residual solid was again suspended in distilled H2O (40 mL) and collected by centrifugation (repeated 1X). The resultant solid, which contained a small amount of residual H2O, was frozen and lyophilized to dryness. The solid was triturated with anhydrous Et2O and filtered over a sintered glass funnel. The resultant solid was washed with copious amounts of anhydrous Et2O and dried in vacuo to yield tetramer 3 as a brown solid (728 mg, 93%). 1H NMR: δ 10.45 (s, 1H), 9.97 (s, 1H), 9.91 (s, 1H), 7.43 (s, 1H), 7.38 (s, 1H), 7.29 (s, 1H), 7.24 (s, 1H), 7.17 (s, 1H), 7.07 (s, 1H), 7.03 (s, 1H), 6.85 (s, 1H), 3.99 (s, 3H), 3.85 (s, 3H), 3.84 (s, 3H), 3.82 (s, 3H); 13C NMR: δ 162.0, 158.48, 158.46, 156.1, 138.8, 127.0, 126.4, 123.0, 122.7, 122.6, 122.2, 121.5, 120.3, 119.5, 118.7, 118.5, 108.4, 105.0, 104.8, 36.14, 36.07, 35.1; HRMS (FAB+) calc'd for C23H25N8O5 [M+H]+ 493.1948, found 493.1952.
BocHN-(R)β-CbzHNγ-Im-CO2Et (15)
A solution of (R)-3,4-Cbz-Dbu(Boc)-OH 8 (1.03 g, 2.93 mmol) and PyBOP (1.83 g, 3.51 mmol) in DMF (12 mL) and DIEA (1.5 mL, 8.8 mmol) was stirred at 23 °C for 22 min. The solution was then added to solid (powdered) HCl•H2N-Im-CO2Et 9 (850 mg, 3.15 mmol) and stirred at 23 °C for 8 h. The solution was then added to distilled H2O (30 mL) pre-acidified with aqueous HCl (1N, 9 mL, 9 mmol), yielding a precipitate that was isolated by centrifugation (~ 4500 rpm). The residual solid was again suspended in distilled H2O (40 mL) and collected by centrifugation (repeated 2X). The resultant solid, which contained a small amount of residual H2O, was frozen and lyophilized to dryness. Drying of the brown solid in vacuo yielded dimer 15 (1.4 g, 95%). 1H NMR: δ 10.57 (s, 1H), 7.51 (s, 1H), 7.31-7.27 (m, 5H), 7.02 (d, J = 8.5 Hz, 1H), 6.79 (m, 1H), 4.97 (s, 2H), 4.25 (q, J = 7.2 Hz, 2H), 3.93 (m, 1H), 3.89 (s, 3H), 3.01 (m, 2H), 2.44-2.35 (m, 2H), 1.35 (s, 9H), 1.27 (t, J = 7.2 Hz, 3H); 13C NMR: δ 167.6, 158.4, 155.8, 155.4, 137.4, 137.1, 130.7, 128.2, 127.6, 127.5, 114.8, 77.7, 65.1, 60.5, 48.6, 43.5, 38.1, 35.4, 28.2, 14.0; HRMS (FAB+) calc'd for C24H34 N5O7 [M+H] 504.2458, found 504.2462.
HCl•H2N-(R)β-CbzHNγ-Im-CO2Et (5)
Dimer 15 (500 mg, 0.993 mmol) in a solution of anhydrous HCl in 1,4-dioxane (4.0 M, 10 mL) and anhydrous Et2O (4 mL) was stirred at 23 °C for 16 h. The mixture was then diluted with 20 mL of anhydrous Et2O and filtered over a sintered glass funnel. The resultant solid was washed with copious amounts of anhydrous Et2O and dried in vacuo to yield dimer 5 as a white solid (432 mg, 99%). 1H NMR:δ10.75 (s, 1H), 8.05 (m, 3H), 7.52 (s, 1H), 7.38 (d, J = 8.5 Hz, 1H), 7.34-7.28 (m, 5H), 5.01 (m, 2H), 4.25 (q, J = 7.1 Hz, 2H), 4.13 (m, 1H), 3.90 (3, 3H), 2.96-2.84 (m, 2H), 2.60-2.51 (m, 2H), 1.27 (t, J = 7.1 Hz, 3H); 13C NMR:δ166.8, 158.4, 155.7, 137.2, 136.8, 130.9, 128.3, 127.8, 127.7, 114.9, 65.5, 60.6, 46.6, 42.1, 38.2, 35.4, 14.0; HRMS (FAB+) calc'd for C19H26N5O5 [M+H]+ 404.1934, found 404.1928.
ImPyPyPy-(R)β-CbzHNγ-Im-CO2Et (16)
A solution of tetramer 3 (1.28 g, 2.59 mmol) and PyBOP (1.49 g, 2.86 mmol) in DMF (8 mL) and DIEA (1.8 mL, 10.4 mmol) was stirred at 23 °C for 20 min. The solution was then treated with solid (powdered) HCl•H2N-(R)β-CbzHNγ-Im-CO2Et 5 (1.2 g, 2.73 mmol) and stirred at 23 °C for 2 h. The solution was then added to distilled H2O (30 mL) pre-acidified with aqueous HCl (1N, 20 mL, 20 mmol), yielding a precipitate that was isolated by centrifugation (~ 4500 rpm). The residual solid was again suspended in distilled H2O (40 mL) and collected by centrifugation (repeated 2X). The resultant solid, which contained a small amount of residual H2O, was frozen and lyophilized to dryness. The solid was triturated with anhydrous Et2O and filtered over a sintered glass funnel. The resultant solid was washed with copious amounts of anhydrous Et2O and dried in vacuo to yield ImPyPyPy-(R)β-CbzHNγ-Im-CO2Et 16 as a tan solid (2.2 g, 97%). 1H NMR:δ10.60 (s, 1H), 10.46 (s, 1H), 9.96 (s, 1H), 9.91 (s, 1H), 7.97 (t, J = 5.6 Hz, 1H), 7.52 (s, 1H), 7.39 (s, 1H), 7.29-7.26 (m, 6H), 7.24 (m, 1H), 7.18-7.14 (m, 3H), 7.04 (m, 2H), 6.90 (m, 1H), 4.98 (m, 2H), 4.24 (q, J = 7.1 Hz, 2H), 4.10 (m, 1H), 3.99 (s, 3H), 3.89 (s, 3H), 3.845 (s, 3H), 3.839 (s, 3H), 3.77 (s, 3H), 3.29 (m, 2H), 2.49 (m, 2H, obstructed by NMR solvent), 1.27 (t, J = 7.2 Hz, 3H); 13C NMR:δ167.7, 161.5, 158.49, 158.48, 158.45, 156.0, 155.6, 138.7, 137.4, 137.1, 130.8, 128.2, 127.7, 127.6, 126.8, 126.4, 123.0, 122.8, 122.7, 122.24, 122.17, 121.4, 118.7, 118.5, 118.0, 114.9, 105.0, 104.7, 104.4, 65.2, 60.5, 48.6, 42.1, 38.2, 36.13, 36.11, 36.0, 35.4, 35.2, 14.0; HRMS (FAB+) calc'd for C42H48N13O9 [M+H]+ 878.3698, found 878.3668.
ImPyPyPy-(R)β-CbzHNγ-Im-CO2H (17)
A solution of ImPyPyPy-(R)β-CbzHNγ-Im-CO2Et 16 (2.0 g, 2.28 mmol) dissolved in 1,4-dioxane (2 mL), MeOH (6 mL), and aqueous KOH (1N, 9.1 mL, 9.1 mmol) was stirred at 42 °C for 2 h. The solution was then acidified with aqueous HCl (1N, ~9.1 mL, ~9.1 mmol) to a pH = 4.5, yielding a precipitate that was isolated by centrifugation (~ 4500 rpm). The residual solid was again suspended in distilled H2O (10 mL) and collected by centrifugation (repeated 1X). The resultant solid, which contained a small amount of residual H2O, was frozen and lyophilized to dryness. The solid was triturated with anhydrous Et2O and filtered over a sintered glass funnel. The resultant solid was washed with copious amounts of anhydrous Et2O and dried in vacuo to yield ImPyPyPy-(R)β-CbzHNγ-Im-CO2H 17 as a tan solid (1.78 g, 92%). 1H NMR:δ10.50 (s, 1H), 10.47 (s, 1H), 9.96 (s, 1H), 9.92 (s, 1H), 7.98 (m, 1H), 7.48 (s, 1H), 7.40 (s, 1H), 7.30-7.27 (m, 6H), 7.24 (m, 1H), 7.19-7.14 (m, 3H), 7.05 (m, 2H), 6.90 (m, 1H), 4.99 (m, 2H), 4.10 (m, 1H), 3.99 (s, 3H), 3.88 (s, 3H), 3.845 (s, 3H), 3.838 (s, 3H), 3.77 (s, 3H), 3.30 (m, 2H), 2.49 (m, 2H, obstructed by NMR solvent); 13C NMR:δ167.7, 161.5, 160.0, 158.5, 155.8, 155.6, 138.6, 137.1, 131.6, 128.3, 127.7, 127.6, 126.7, 126.4, 123.0, 122.8, 122.7, 122.23, 122.16, 121.4, 118.7, 118.5, 118.0, 114.6, 105.0, 104.7, 104.4, 65.2, 48.6, 42.2, 38.2, 36.13, 36.10, 36.0, 35.4, 35.2; HRMS (FAB+) calc'd for C40H44N13O9 [M+H]+ 850.3385, found 850.3383.
ImPyPyPy-(R)β-CbzHNγ-ImPyPyPy-CO2Me (2)
A solution of ImPyPyPy-(R)β-CbzHNγ-Im-CO2H 17 (1.5 g, 1.77 mmol) and PyBOP (546 mg, 1.85 mmol) in DMF (8.8 mL) and DIEA (922 μL, 5.3 mmol) was stirred at 23 °C for 10 min. The solution was then treated with solid (powdered) HCl•H2N-PyPyPy-CO2Me 4 (806 mg, 1.85 mmol) and stirred at 23 °C for 10 h. The solution was then added to distilled H2O (35 mL) pre-acidified with aqueous HCl (1N, 5 mL, 5 mmol), yielding a precipitate that was isolated by centrifugation (~ 4500 rpm). The residual solid was again suspended in distilled H2O (40 mL) and collected by centrifugation (repeated 3X). The resultant solid, which contained a small amount of residual H2O, was frozen and lyophilized to dryness. The solid was triturated with anhydrous Et2O and filtered over a sintered glass funnel. The resultant solid was washed with copious amounts of anhydrous Et2O and dried in vacuo to yield ImPyPyPy-(R)β-CbzHNγ-ImPyPyPy-CO2Me 2 as a tan solid (2.09 g, 96%). 1H NMR:δ10.66 (s, 1H), 10.21 (s, 1H), 10.00 (s, 1H), 9.98 (s, 1H), 9.96 (s, 1H), 9.93 (s, 1H), 9.92 (s, 1H), 8.00 (m, 1H), 7.48 (s, 1H), 7.46 (d, J = 2.0 Hz, 1H), 7.45 (s, 1H), 7.31-7.27 (m, 7H), 7.23 (m, 2H), 7.20-7.14 (m, 5H), 7.06 (m, 2H), 6.92 (m, 1H), 6.90 (d, J = 2.0 Hz, 1H), 5.00 (m, 2H), 4.11 (m, 1H), 4.00 (s, 3H), 3.95 (s, 3H), 3.854 (s, 3H), 3.850 (s, 3H), 3.842 (s, 3H), 3.837 (s, 3H), 3.828 (s, 3H), 3.78 (s, 3H), 3.73 (s, 3H), 3.32 (m, 2H), 2.53 (m, 2H); 13C NMR:δ167.9, 161.6, 160.8, 158.5, 158.42, 158.38, 155.8, 155.6, 137.6, 137.1, 136.0, 134.0, 128.3, 127.7, 127.6, 126.4, 123.3, 123.1, 123.0, 122.80, 122.77, 122.5, 122.26, 122.24, 122.1, 121.2, 120.9, 120.8, 118.9, 118.70, 118.64, 118.5, 118.0, 114.1, 108.4, 104.9, 104.79, 104.76, 104.5, 65.2, 51.0, 48.8, 42.2, 38.4, 36.25, 36.21, 36.19, 36.13, 36.10, 36.0, 35.8, 35.0; HRMS (FAB+) calc'd for C59H64N19O12 [M+H]+ 1230.498, found 1230.504.
ImPyPyPy-(R)β-CbzHNγ-ImPyPyPy-CO2H (22)
A solution of polyamide 2 (500 mg, 0.406 mmol) dissolved in 1,4-dioxane (8 mL) and aqueous NaOH (1N, 8.0 mL, 8.0 mmol) was stirred at 23 °C for 11 h. The solution was then cooled to 0 °C in an ice bath and the pH adjusted to pH = 4.0 with aqueous HCl (1N, ~8 mL, 8 mmol), yielding a precipitate that was isolated by centrifugation (~ 4500 rpm). The residual solid was again suspended in distilled H2O (40 mL) and collected by centrifugation (repeated 2X). The resultant solid, which contained a small amount of residual H2O, was frozen and lyophilized to dryness. The solid was triturated with anhydrous Et2O and filtered over a sintered glass funnel. The resultant solid was washed with copious amounts of anhydrous Et2O and dried in vacuo to yield ImPyPyPy-(R)β-CbzHNγ-ImPyPyPy-CO2H 22 as a tan solid (442 mg, 89%). 1H NMR: δ 10.52 (s, 1H), 10.22 (s, 1H), 10.00 (s, 1H), 9.97 (s, 1H), 9.95 (s, 1H), 9.93 (s, 1H), 9.90 (s, 1H), 8.00 (m, 1H), 7.45 (s, 1H), 7.42 (m, 2H), 7.30-7.27 (m, 7H), 7.24 (m, 2H), 7.19-7.14 (m, 4H), 7.09 (s, 1H), 7.06 (m, 2H), 6.92 (m, 1H), 6.84 (d, J = 2.0 Hz, 1H), 5.01 (m, 2H), 4.11 (m, 1H), 3.99 (s, 3H), 3.95 (s, 3H), 3.851 (s, 3H), 3.848 (s, 3H), 3.843 (s, 3H), 3.838 (s, 3H), 3.81 (s, 3H), 3.79 (s, 3H), 3.32 (m, 2H), 2.53 (m, 2H); 13C NMR: δ 167.9, 162.0, 161.6, 158.48, 158.47, 158.44, 158.40, 155.8, 155.6, 138.6, 137.1, 136.0, 134.0, 128.3, 127.7, 127.6, 126.4, 123.06, 123.05, 122.8, 122.74, 122.70, 122.6, 122.24, 122.19, 122.15, 121.4, 121.2, 120.3, 119.5, 118.7, 118.55, 118.47, 118.0, 114.1, 108.4, 104.9, 104.86, 104.80, 104.75, 104.5, 65.2, 48.7, 42.2, 38.3, 36.2, 36.13, 36.10, 36.07, 35.97, 35.2, 34.9; HRMS (FAB+) calc'd for C58H62 N19O12[M+H] 1216.483, found 1216.487.
BocHN-(+)-BnOIPA (20)
A solution of acid 1915 (211 mg, 0.824 mmol) and PyBOP (624 mg, 1.2 mmol) in DMF (3 mL) and DIEA (211 μL, 1.2 mmol) was stirred at 23 °C for 10 min. Protected triamine 1814 (373 mg, 1.52 mmol) was then added to the solution and stirring was continued at 23 °C for 3 h. The solution was then added to distilled H2O (15 mL) and a viscous oil was isolated following centrifugation (~ 4500 rpm) and decanting of the aqueous layer. The residual oil was again washed with distilled H2O (10 mL) and collected by centrifugation (repeated 3X). Drying of the residual oil in vacuo yielded BocHN-(+)-BnOIPA 20 as a viscous amber oil (391 mg, 98%). 1H NMR: δ 8.77 (t, J = 5.5 Hz, 1H), 8.43 (app t, J = 1.5 Hz, 1H), 8.14-8.09 (m, 2H), 7.63 (app t, J = 7.8 Hz, 1H), 7.48-7.34 (m, 5H), 6.85 (t, J = 4.9 Hz, 1H), 5.38 (s, 2H), 3.30 (m, 2H), 3.00 (m, 1H), 2.94 (m, 2H), 2.72-2.51 (m, 4H), 2.38 (br s, 3H), 1.75 (m, 2H), 1.59 (m, 2H), 1.34 (s, 9H); 13C NMR: δ 165.3, 165.2, 155.6, 136.0, 135.0, 131.9, 131.7, 129.8, 129.0, 128.6, 128.2, 128.1, 127.9, 77.5, 66.4, 54.3, 54.0, 40.6, 37.7, 37.3, 28.2, 25.9, 25.5; HRMS (FAB+) calc'd for C27H38N3O5 [M+H]+ 484.2811, found 484.2793.
ImPyPyPy-(R)β-CbzHNγ-ImPyPyPy-(+)-BnOIPA (23)
A solution of ImPyPyPy-(R)β-CbzHNγ-ImPyPyPy-CO2H 22 (192 mg, 0.158 mmol) and PyBOP (99 mg, 0.189 mmol) in DMF (2 mL) and DIEA (220μ L, 1.3 mmol) was stirred at 23 °C for 30 min. In a separate vial BocHN-(+)-BnOIPA 20 (122 mg, 0.252 mmol) was deprotected by treating with a solution of CH2Cl2:CF3CO2H (1:1, 2 mL) for 20 minutes at 23 °C followed by concentration to dryness in vacuo. This residual material was then treated with the pre-activated solution of ImPyPyPy-(R)β-CbzHNγ-ImPyPyPy-CO2H 22 and allowed to stir at 23 °C for 12 h. The solution was then added to distilled H2O (30 mL) pre-acidified with aqueous HCl (1N, 2 mL, 2 mmol), yielding a precipitate that was isolated by centrifugation (~ 4500 rpm). The residual solid was again suspended in distilled H2O (40 mL) and collected by centrifugation (repeated 3X). The resultant solid, which contained a small amount of residual H2O, was frozen and lyophilized to dryness. The residual brown solid was purified by SiO2 chromatography with the mobile phase consisting of a step gradient of 49:1 CH2Cl2:MeOH to 44:5:1 CH2Cl2:MeOH:NH3 to provide ImPyPyPy-(R)β-CbzHNγ-ImPyPyPy-(+)-BnOIPA 23 as a tan solid (217 mg, 87%) after drying under high vacuum. 1H NMR: δ 10.43 (s, 1H), 10.20 (s, 1H), 9.98 (s, 1H), 9.95 (s, 1H), 9.93 (s, 1H), 9.92 (s, 1H), 9.87 (s, 1H), 8.73 (t, J = 5.5 Hz, 1H), 8.41 (app t, J = 1.6 Hz, 1H), 8.11-8.07 (m, 2H), 8.03-7.98 (m, 2H), 7.60 (app t, J = 7.7 Hz, 1H), 7.48-7.32 (m, 7H), 7.30-7.23 (m, 9H), 7.19-7.14 (m, 5H), 7.06 (d, J = 1.7 Hz, 1H), 7.05 (d, J = 1.7 Hz, 1H), 7.03 (d, J = 1.0 Hz, 1H), 6.92 (d, J = 1.2 Hz, 1H), 6.84 (d, J = 1.7 Hz, 1H), 5.37 (s, 2H), 5.00 (m, 2H), 4.11 (m, 1H), 3.99 (s, 3H), 3.95 (s, 3H), 3.85 (s, 3H), 3.84 (m, 6H), 3.83 (3, 3H), 3.79 (s, 3H), 3.77 (s, 3H), 3.30 (m, 4H), 3.18 (m, 2H), 2.53 (m, 2H), 2.35 (m, 4H), 2.16 (br s, 3H), 1.71-1.60 (m, 4H); 13C NMR: δ 167.9, 165.20, 165.16, 161.6, 161.2, 158.50, 158.46, 158.42, 156.1, 155.8, 155.6, 138.8, 137.1, 136.0, 135.2, 134.0, 131.9, 131.6, 129.7, 129.0, 128.5, 128.3, 128.2, 128.1, 128.0, 127.8, 127.7, 127.6, 127.00, 126.99, 126.3, 123.1, 123.0, 122.80, 122.77, 122.75, 122.26, 122.18, 122.14, 122.11, 121.4, 121.2, 118.7, 118.5, 118.0, 117.7, 114.1, 105.0, 104.9, 104.8, 104.5, 104.1, 66.4, 65.2, 55.1, 55.0, 48.8, 42.2, 41.6, 38.3, 37.9, 37.0, 36.2, 36.10, 36.09, 36.07, 35.96, 35.89, 35.1, 34.9, 26.9, 26.6; HRMS (ESI+) calc'd for C80H89N22O14 [M+H]+ 1581.6929, found 1581.6992.
ImPyPyPy-(R)bbb-H N2ggg-ImPyPyPy-(+)-IPA (1)
A solution of ImPyPyPy-(R)β-CbzHNγ-ImPyPyPy-(+)BnOIPA 23 (25 mg, 0.016 mmol) and Pd/C (10 wt% dry, 10 mg) in DMF (2 mL) was stirred at 23 °C for 48 h under H2 (~ 1 atm, balloon). (Note: The protecting groups are cleaved at different rates and early reaction aliquots will reveal a mono-protected compound.) The reaction was then filtered through a Sep-Pak cartridge (5g C-18 sorbent) and the Sep-Pak was washed with DMF (4 mL), aqueous MeCN (50%, 20 mL), MeCN (250 mL), and MeOH (250 mL). The filtrate was then concentrated in vacuo, purified by reverse-phase HPLC, and lyophilized to dryness. The solid was triturated with anhydrous Et2O and filtered over a sintered glass funnel. The resultant solid was washed with copious amounts of anhydrous Et2O and dried in vacuo to yield ImPyPyPy-(R)β-H2Nγ-ImPyPyPy-(+)-IPA 1 as a light tan solid (18.9 mg, 81%). 1H NMR: δ 10.61 (s, 1H), 10.46 (s, 1H), 9.95 (s, 2H), 9.94 (s, 1H), 9.92 (s, 1H), 9.89 (s, 1H), 9.46 (br s, 1H), 8.82 (t, J = 5.8 Hz, 1H), 8.42 (app t, J = 1.4 Hz, 1H), 8.20 (m, 1H), 8.15 (m, 1H), 8.08 (d, J = 1.7 Hz, 1H), 8.07 (d, J = 1.7 Hz, 1H), 7.97 (m, 3H), 7.60 (app t, J = 7.6 Hz, 1H), 7.45 (s, 1H), 7.40 (d, J = 0.7 Hz, 1H), 7.28 (d, J = 2.0 Hz, 1H), 7.26 (d, J = 1.7 Hz, 1H), 7.22 (d, J = 2.0 Hz, 1H), 7.21 (d, J = 1.7 Hz, 1H), 7.18 (d, J = 1.7 Hz, 1H), 7.16 (m, 2H), 7.15 (d, J = 1.7 Hz, 1H), 7.08 (m, 2H), 7.05 (d, J = 1.0 Hz, 1H), 7.03 (d, J = 1.7 Hz, 1H), 6.94 (d, J = 1.8 Hz, 1H), 3.99 (s, 3H), 3.96 (s, 3H), 3.85 (s, 3H), 3.844 (s, 3H), 3.842 (s, 3H), 3.83 (s, 3H), 3.82 (s, 3H), 3.79 (s, 3H), 3.69 (m, 1H), 3.44 (m, 2H), 3.35 (m, 2H), 3.24 (m, 2H), 3.18-3.12 (m, 2H), 3.11-3.04 (m, 2H), 2.76 (m, 3H), 2.50 (m, 2H, obstructed by NMR solvent), 1.95-1.91 (m, 2H), 1.90-1.83 (m, 2H); 13C NMR: δ 166.84, 166.82, 165.7, 162.1, 161.6, 158.52, 158.49, 158.40, 158.1, 157.8, 156.0, 155.7, 138.7, 135.6, 134.6, 134.2, 131.9, 131.5, 131.0, 128.7, 128.0, 126.9, 126.3, 123.1, 123.0, 122.8, 122.7, 122.5, 122.4, 122.24, 122.16, 122.10, 121.4, 121.1, 118.7, 118.5, 118.3, 118.0, 115.7, 105.0, 104.9, 104.8, 104.54, 104.52, 59.2, 53.3, 48.2, 36.5, 36.2, 36.08, 36.07, 35.99, 35.6, 35.38, 35.35, 35.1, 35.0, 24.3, 24.0; HRMS (ESI+) calc'd for C65H77N22O12 [M+H]+ 1357.6086, found 1357.6091.
Calculation of Molar Extinction Coefficients
Solid 1 (0.51 mg, 0.35 μmoles) was weighed into a tared vial on a microbalance (Sartorius Micro M4). A molecular weight of 1471.46 was utilized for 1, which assumes 1 exists as the CF3CO2H salt. The material was dissolved in distilled and deionized H2O (1000 μL), yielding a 0.35 μM stock solution. 50-fold dilutions of this stock solution were made into the appropriate solvent systems (Figure 4). Data was collected at 23 °C in a 1 cm quartz cuvette, and molar extinction coefficients are based on the λmax for each solvent system (range: 313-317 nm). The instrument was blanked on each discrete solvent system prior to data collection. Duplicate analysis of each sample yielded similar results (data not shown). A second polyamide stock solution generated by dissolving solid 1 in 50% MeCN in aqueous CF3CO2H (0.1% v/v), followed by 50-fold dilution into the individual solvent systems and data collection yielded similar results to those shown in Figure 4 (data not shown). DEPC-treated H2O (RNase & DNase free) was purchased from USB Corporation. EtOH was absolute grade from Pharmaco-AAPER and DMSO was molecular biology grade from Sigma-Aldrich. DMF was anhydrous synthesis grade from Sigma-Aldrich and acetonitrile was HPLC grade from Fisher.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (GM27681). D.M.C. is grateful for a Caltech Kanel predoctoral fellowship. D.A.H. thanks the California Tobacco-Related Disease Research Program (16FT-0055) for a postdoctoral fellowship. The National Science Foundation Chemistry Research Instrumentation and Facilities Program (CHE-0541745) is acknowledged for providing the UPLC-MS instrument.
Footnotes
Supporting Information Available: 1H and 13C NMR spectra for synthesized compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Dervan PB. Bioorg. Med. Chem. 2001;9:2215–2235. doi: 10.1016/s0968-0896(01)00262-0. [DOI] [PubMed] [Google Scholar]
- (2).Dervan PB, Edelson JA. Curr. Opin. Struct. Biol. 2003;13:284–299. doi: 10.1016/s0959-440x(03)00081-2. [DOI] [PubMed] [Google Scholar]
- (3).(a) Trauger JW, Baird EE, Dervan PB. Nature. 1996;382:559–561. doi: 10.1038/382559a0. [DOI] [PubMed] [Google Scholar]; (b) White S, Szewczyk JW, Turner JM, Baird EE, Dervan PB. Nature. 1998;391:468–470. doi: 10.1038/35106. [DOI] [PubMed] [Google Scholar]; (c) Kielkopf CL, Baird EE, Dervan PB, Rees DC. Nat. Struct. Biol. 1998;5:104–109. doi: 10.1038/nsb0298-104. [DOI] [PubMed] [Google Scholar]; (d) Kielkopf CL, White S, Szewczyk JW, Turner JM, Baird EE, Dervan PB, Rees DC. Science. 1998;282:111–115. doi: 10.1126/science.282.5386.111. [DOI] [PubMed] [Google Scholar]
- (4).Hsu CF, Phillips JW, Trauger JW, Farkas ME, Belitsky JM, Heckel A, Olenyuk BZ, Puckett JW, Wang CC, Dervan PB. Tetrahedron. 2007;63:6146–6151. doi: 10.1016/j.tet.2007.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Belitsky JM, Leslie SJ, Arora PS, Beerman TA, Dervan PB. Bioorg. Med. Chem. 2002;10:3313–3318. doi: 10.1016/s0968-0896(02)00204-3. [DOI] [PubMed] [Google Scholar]; (b) Crowley KS, Phillion DP, Woodard SS, Scheitzer BA, Singh M, Shabany H, Burnette B, Hippenmeyer P, Heitmeier M, Bashkin JK. Bioorg. Med. Chem. Lett. 2003;13:1565–1570. doi: 10.1016/s0960-894x(03)00152-5. [DOI] [PubMed] [Google Scholar]; (c) Best TP, Edelson BS, Nickols NG, Dervan PB. Proc. Natl. Acad. Sci. U. S. A. 2003;100:12063–12068. doi: 10.1073/pnas.2035074100. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Edelson BS, Best TP, Olenyuk B, Nickols NG, Doss RM, Foister S, Heckel A, Dervan PB. Nucleic Acids. Res. 2004;32:2802–2818. doi: 10.1093/nar/gkh609. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Xiao X, Yu P, Lim HS, Sikder D, Kodadek T. Angew. Chem. Int. Ed. Engl. 2007;46:2865–2868. doi: 10.1002/anie.200604485. [DOI] [PubMed] [Google Scholar]; (f) Nickols NG, Jacobs CS, Farkas ME, Dervan PB. Nucleic Acids. Res. 2007;35:363–370. doi: 10.1093/nar/gkl1042. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Dose C, Farkas ME, Chenoweth DM, Dervan PB. J. Am. Chem. Soc. 2008;130:6859–6866. doi: 10.1021/ja800888d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Hsu CF, Dervan PB. Bioorg. Med. Chem. Lett. 2008;18:5851–5855. doi: 10.1016/j.bmcl.2008.05.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Gottesfeld JM, Melander C, Suto RK, Raviol H, Luger K, Dervan PB. J. Mol. Biol. 2001;309:615–629. doi: 10.1006/jmbi.2001.4694. [DOI] [PubMed] [Google Scholar]; (b) Suto RK, Edayathumangalam RS, White CL, Melander C, Gottesfeld JM, Dervan PB, Luger K. J. Mol. Biol. 2003;326:371–380. doi: 10.1016/s0022-2836(02)01407-9. [DOI] [PubMed] [Google Scholar]; (c) Edayathumangalam RS, Weyermann P, Gottesfeld JM, Dervan PB, Luger K. Proc. Natl. Acad. Sci. U. S. A. 2004;101:6864–6869. doi: 10.1073/pnas.0401743101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Dudouet B, Burnett R, Dickinson LA, Wood MR, Melander C, Belitsky JM, Edelson B, Wurtz N, Briehn C, Dervan PB, Gottesfeld JM. Chem. Biol. 2003;10:859–867. doi: 10.1016/j.chembiol.2003.09.001. [DOI] [PubMed] [Google Scholar]
- (7).(a) Olenyuk BZ, Zhang GJ, Klco JM, Nickols NG, Kaelin WG, Dervan PB. Proc. Natl. Acad. Sci. U. S. A. 2004;101:16768–16773. doi: 10.1073/pnas.0407617101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kageyama Y, Sugiyama H, Ayame H, Iwai A, Fujii Y, Huang LE, Kizaka-Kondoh S, Hiraoka M, Kihara K. Acta Oncol. 2006;45:317–324. doi: 10.1080/02841860500486648. [DOI] [PubMed] [Google Scholar]; (c) Nickols NG, Jacobs CS, Farkas ME, Dervan PB. ACS Chem. Biol. 2007;2:561–571. doi: 10.1021/cb700110z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Nickols NG, Dervan PB. Proc. Natl. Acad. Sci. U. S. A. 2007;104:10418–10423. doi: 10.1073/pnas.0704217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Matsuda H, Fukuda N, Ueno T, Tahira Y, Ayame H, Zhang W, Bando T, Sugiyama H, Saito S, Matsumoto K, others O. J. Am. Soc. Neph. 2006;17:422–432. doi: 10.1681/ASN.2005060650. [DOI] [PubMed] [Google Scholar]; (b) Yao EH, Fukuda N, Ueno T, Matsuda H, Matsumoto K, Nagase H, Matsumoto Y, Takasaka A, Serie K, Sugiyama H, Sawamura T. Hypertension. 2008;52:86–92. doi: 10.1161/HYPERTENSIONAHA.108.112797. [DOI] [PubMed] [Google Scholar]
- (10).(a) Baird EE, Dervan PB. J. Am. Chem. Soc. 1996;118:6141–6146. [Google Scholar]; (b) Belitsky JM, Nguyen DH, Wurtz NR, Dervan PB. Bioorg. Med. Chem. 2002;10:2767–2774. doi: 10.1016/s0968-0896(02)00133-5. [DOI] [PubMed] [Google Scholar]; (c) Wurtz NR, Turner JM, Baird EE, Dervan PB. Org. Lett. 2001;3:1201–1203. doi: 10.1021/ol0156796. [DOI] [PubMed] [Google Scholar]; (d) Krutzik PO, Chamberlin AR. Bioorg. Med. Chem. Lett. 2002;12:2129–2132. doi: 10.1016/s0960-894x(02)00359-1. [DOI] [PubMed] [Google Scholar]; (e) Krutzik PO, Chamberlin AR. Methods Mol. Biol. 2002;201:77–92. doi: 10.1385/1-59259-285-6:77. [DOI] [PubMed] [Google Scholar]; (f) Ayame H, Saito T, Bando T, Fukuda N, Sugiyama H. Nucleic Acids. Res. Suppl. 2003:67–68. doi: 10.1093/nass/3.1.67. [DOI] [PubMed] [Google Scholar]; (g) Moore MJB, Cuenca F, Searcey M, Neidle S. Org. Biomol. Chem. 2006;4:3479–3488. doi: 10.1039/b607707b. [DOI] [PubMed] [Google Scholar]
- (11).(a) Mrksich M, Parks ME, Dervan PB. J. Am. Chem. Soc. 1994;116:7983–7988. [Google Scholar]; (b) Xiao J, Yuan G, Huang W, Chan AS, Lee KL. J. Org. Chem. 2000;65:5506–5513. doi: 10.1021/jo000135n. [DOI] [PubMed] [Google Scholar]; (c) Harris D, Stewart M, Sielaff A, Mulder K, Brown T, Mackay H, Lee M. Heterocycl. Commun. 2007;13:17–23. [Google Scholar]; (d) Xiao J-H, Huang W-Q, Tang F-L, Yuan G, Chan ASC, Lee KLD. Chin. J. Chem. 2000;18:603–607. [Google Scholar]; (e) Boger DL, Fink BE, Hedrick MP. J. Am. Chem. Soc. 2000;122:6382–6394. [Google Scholar]; (f) Mamidyala SK, Firestine SM. Tetrahedron Lett. 2006;47:7431–7434. [Google Scholar]
- (12).Sources for commercially available building blocks 7-10 shown in Figure 2: 7 (Fluorochem, Cat # 019570), 8 (Fluka, Cat # 17974), 9 (Bachem, Cat # F-3480), 10 (Bachem, Cat # F-3485).
- (13).Jaramillo D, Liu Q, Aldrich-Wright J, Tor Y. J. Org. Chem. 2004;69:8151–8153. doi: 10.1021/jo048686r. [DOI] [PubMed] [Google Scholar]
- (14).Design of sequence specific DNA binding molecules: bis(distamycin)phenoxazone. Dervan PB, Sluka JP. New Synth. Methodol. Funct. Interesting Compd. Elsevier; Amsterdam: 1986. Studies in Organic Chemistry 25; pp. 307–322.
- (15).Adlington RM, Baldwin JE, Becker GW, Chen B, Cheng L, Cooper SL, Hermann RB, Howe TJ, McCoull W, McNulty AM, Neubauer BL, Pritchard GJ. J. Med. Chem. 2001;44:1491–1508. doi: 10.1021/jm000145g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.