

Abstract
An affinity purification strategy was developed to characterize human proteasome complexes diversity as well as endogenous proteasome-interacting proteins (PIPs). This single step procedure, initially used for 20 S proteasome purification, was adapted to purify all existing physiological proteasome complexes associated to their various regulatory complexes and to their interacting partners. The method was applied to the purification of proteasome complexes and their PIPs from human erythrocytes but can be used to purify proteasomes from any human sample as starting material. The benefit of in vivo formaldehyde cross-linking as a stabilizer of protein-protein interactions was studied by comparing the status of purified proteasomes and the identified proteins in both protocols (with or without formaldehyde cross-linking). Subsequent proteomics analyses identified all proteasomal subunits, known regulators, and recently assigned partners. Moreover other proteins implicated at different levels of the ubiquitin-proteasome system were also identified for the first time as PIPs. One of them, the ubiquitin-specific protease USP7, also known as HAUSP, is an important player in the p53-HDM2 pathway. The specificity of the interaction was further confirmed using a complementary approach that consisted of the reverse immunoprecipitation with HAUSP as a bait. Altogether we provide a valuable tool that should contribute, through the identification of partners likely to affect proteasomal function, to a better understanding of this complex proteolytic machinery in any living human cell and/or organ/tissue and in different cell physiological states.
Proteasome-mediated and lysosomal degradations are the two main mechanisms involved in turnover of intracellular proteins. The 26 S proteasome is the proteolytic machine of the ubiquitin-proteasome pathway (UPP)1 (1, 2). In most cases, the degradation is processed by two successive steps: (i) polyubiquitination of the substrate and (ii) proteolysis of the tagged protein by the 26 S proteasome (2). The proteasome degrades abnormal and non-functional proteins generated under normal and stress conditions but also tightly regulates major cellular processes (cell cycle progression, transcription, apoptosis, DNA repair, epitope generation, etc.) by controlling the cellular pool of key regulatory proteins (3). Therefore a dysregulation of this machinery can lead to various pathologies such as neurodegenerative diseases (4) or cancers (5). The proteasome has recently been identified as a therapeutic target for cancer treatment (6).
The eukaryotic 26 S proteasome can be divided into subcomplexes, one 670-kDa 20 S core particle where proteolysis occurs and one or two axially positioned 900-kDa 19 S regulatory particles responsible for polyubiquitinated substrates recognition, ATP-dependent substrate unfolding, and ubiquitin recycling (7). The eukaryotic 20 S proteasome is a stable complex (8) composed of 28 subunits, arranged in four stacked rings with seven unique α subunits in the two outer rings and seven unique β subunits in the two inner rings (9). Six catalytic proteolytic active sites are located on the proteasome subunits β1, β2, and β5. Upon interferon-γ-induced immune response in mammals, these catalytic subunits are replaced by the immunosubunits β1i, β2i, and β5i, respectively, which induce some changes in the proteolytic activities of the complex (10).
Eukaryotic 19 S regulatory particle, also called PA700, is connected to the 20 S catalytic core through the α ring. It is composed of ∼16 electrophoretically distinct subunits with molecular masses ranging from 25 to 112 kDa (11) corresponding to at least 23 proteins at the present knowledge. The 19 S regulatory particle has a base comprising six proteasomal ATPases (Rpt1–Rpt6 in yeast), three additional non-ATPase subunits (Rpn1, Rpn2, and Rpn13), and a lid structure composed of at least 14 non-ATPase subunits and that is assumed to be connected to the base by the Rpn10 subunit. Although some subunits have been identified as key components for substrate recognition (Rpn10 and Rpt5) (12, 13), for opening the core particle gate (Rpt2) (14), and for deubiquitination (Rpn11) (15), the precise function of most subunits still remains to be elucidated. Functional characterization of 19 S subunits is difficult because the structural organization of the complex is not well defined on account of labile and dynamic interactions of several subunits (16).
In addition to PA700, the two outer α rings of the 20 S proteasome can associate to other regulatory caps, PA28αβ, PA28γ, and PA200; the main role of these regulators is to open the gate into and out of the catalytic chamber. This leads to the formation of several different subpopulations of highly dynamic proteasome complexes because one 20 S core particle can interact at its two sides with either two identical regulators or two different ones, thus forming hybrid proteasomes (17). These hybrid proteasomes have been implicated in major cellular processes such as immune surveillance, regulation of cell size and growth, apoptosis, or DNA repair, but the exact proteolytic function of each of these different complexes is mainly unresolved and constitutes today a challenge in proteasome biology (18). Moreover several transient interacting proteins of human 26 S proteasome have recently been identified, but their functional impact remains, for most of them, to be elucidated (18, 19). They are, however, of particular importance because proteasome dynamic association to various proteins is likely to regulate its stability and activity upon diverse stimuli.
Multidimensional chromatography has been used to purify the 26 S proteasome from various species to homogeneity (8, 20–24). These methods have improved our knowledge on proteasome structure but failed to catch labile interactors due to the use of high salt concentrations. Affinity purification or co-immunoprecipitation methods (25–28) as well as genome-wide two-hybrid surveys (29) have proven to be much more efficient and led to the identification of numerous additional proteasome subunits and associated proteins from yeast.
The few works dealing with the characterization of the human proteasome-interacting protein network have been published very recently and were conducted by proteomics approaches (17, 30–32). A strategy relying on the tagging of a 19 S subunit and mass spectrometric analyses of co-immunoprecipitated complexes identified Adrm1, the human orthologue of the yeast Rpn13 subunit (30, 31). A similar approach combined with a SILAC (stable isotope labeling with amino acids in cell culture) strategy enabled distinguishing proteins that transiently interact with the proteasome from stable proteasome-interacting proteins (PIPs) (32).
These strategies, involving mass spectrometry-based identification associated to biochemical approaches such as immunoprecipitation or affinity purification, have been successfully applied to the study of PIPs and support the idea that recent developments in proteomics are powerful tools for the study of protein networks (33). Protein-protein interactions inside protein complexes are difficult to maintain during the purification procedure, and the use of chemical cross-linkers can be useful for transient interactor recovery (34) and have been successfully used for 26 S proteasomes structural studies (35).
However, all the affinity-based methods described to date to purify proteasome complexes and PIPs rely on overexpression or tagging strategies. Moreover most of them are based on the use of a 19 S subunit as a bait, which implies that only proteasome containing at least one 19 S regulatory particle can be purified. Proteasome pools involving only PA28 or PA200 regulators might be associated with specific partners. Therefore, methods to efficiently purify all proteasome complexes would be of great interest.
We developed a single step affinity purification protocol to characterize all physiological proteasome complexes and their PIPs in human cells. It is based on the high affinity binding of a subunit of the 20 S core particle to a monoclonal antibody. A differential proteomics strategy with a control antibody was used to distinguish specific PIPs from nonspecific interactors. Moreover the benefit of in vivo formaldehyde cross-linking on overall proteasome complexes recovery was also assessed. This strategy led to the identification of new PIPs of the human proteasome, including the deubiquitinase HAUSP for which specific interaction was further confirmed.
EXPERIMENTAL PROCEDURES
Purification of Proteasome Complexes
Proteasome complexes were purified from human erythrocytes by affinity chromatography with the mouse IgG1 monoclonal antibody MCP21 (European Collection of Cell Cultures) directed against the human α2 subunit of 20 S proteasome. A control purification was performed using the mouse IgG1 monoclonal antibody OX8 directed against rat CD8α (a kind gift from Dr. Saoudi, INSERM U563, Toulouse, France). OX8 is a monoclonal antibody belonging to the same isotype as the MCP21 IgG1 antibody used to catch proteasomes. Eight milligrams of each antibody were immobilized separately on 1 g of CNBr-activated Sepharose (GE Healthcare).
One hundred milliliters of human blood from healthy volunteers (transfer agreement of blood products for non-therapeutic use, reference EFS-PM: 21/PNVT/TOU/IPBS01/2006-0074 and additional clauses) were mixed with 100 ml of 2% dextran T500 (GE Healthcare) in PBS to sediment the erythrocytes. The pellets were washed three times with cold PBS, frozen in liquid N2, and stored at −80 °C. The same volume of erythrocytes was prepared similarly and separately and was submitted to in vivo protein-protein cross-linking prior to freezing. The protocol was adapted from Vasilescu et al. (34): formaldehyde (1% final concentration) was added for 1 h at 4 °C, and formaldehyde action was stopped with 250 mm glycine for 15 min at 4 °C. The erythrocytes were then washed three times with cold PBS prior to freezing in liquid N2 and storage at −80 °C.
The 26 S proteasome purification was performed using a protocol adapted from Bousquet-Dubouch et al. (36). ATP and glycerol were added to buffers all along the procedure to preserve the interactions between the 20 S core particle and its regulators (8). The aliquots were thawed at 4 °C in the presence of lysis buffer (50 mm Tris-HCl, pH 8, 1 mm DTT, 1 mm EGTA, 5 mm EDTA, 5 mm MgCl2, 10% glycerol, 2 mm ATP) containing protease (Roche Applied Science) and phosphatase (1 mm sodium orthovanadate, 10 mm sodium pyrophosphate, and 20 mm sodium fluoride) inhibitors. After removal of cellular debris by centrifugation, the supernatant was split into two aliquots. Each aliquot was incubated overnight at 4 °C with rotary shaking with either the MCP21-Sepharose or the OX8-Sepharose antibody as negative control. The Sepharose beads were then washed with 50 bed volumes of equilibration buffer (20 mm Tris-HCl, 150 mm NaCl, 1 mm EDTA, 10% glycerol, 5 mm MgCl2, 2 mm ATP, pH 7.6), and proteins interacting with the beads were finally eluted by an increase of the ionic strength from 150 mm to 3 m NaCl. Two-milliliter fractions were collected and stored at 4 °C. Protein concentration was determined using the Bio-Rad Protein Assay, and proteasome content could be estimated by measuring the in vitro chymotrypsin-like (ChT-like) activity as described below. The fractions containing the most proteasome activity were then dialyzed against water and TCA/acetone-precipitated for further analysis.
The fractions obtained from the negative control experiment (containing proteins interacting with the OX8-Sepharose) were also analyzed for their protein and proteasome contents. Then they were pooled, concentrated, and simultaneously desalted on Centriprep® YM-10 centrifugal filter device (Amicon® Bioseparations, Millipore Corp., Bedford, MA) from 15 ml to approximately 500 μl. They were finally precipitated using TCA/acetone.
In Vitro Chymotrypsin-like Activity Assay
Five microliters of purified 26 S proteasome fractions were mixed with 95 μl of 50 mm Tris-HCl, pH 8 and 200 μm Suc-LLVY-AMC (BIOMOL International LP, Plymouth Meeting, MA) in 96-well black plates (Greiner Bio-One, Frickenhausen, Germany). The kinetic assays were performed at 37 °C in an FLX-800 spectrofluorometer (BIO-TEK, Winooski, VT) over 90 min with one reading every 5 min at 360 nm for excitation and 460 nm for emission. The concentration of liberated products was calculated using a standard curve for AMC. Lactacystin (BIOMOL International LP) was added at 10 μm/well when necessary to specifically inhibit the chymotrypsin-like activity of proteasome. The quantity of proteasome in the purification fractions was estimated by measuring in the same conditions the activity of 0.5 μg of commercially available 20 S proteasome or 26 S proteasome purified from human erythrocytes (BIOMOL International LP).
Immunoprecipitation Using the Anti-HAUSP Antibody
Immunoprecipitation of HAUSP and HAUSP complexes was performed using an anti-HAUSP rabbit polyclonal antibody (Merck/Calbiochem) that we cross-linked to Dynabeads® protein A (Invitrogen Dynal AS). After washing the beads as indicated by the supplier, 50 μl of beads were used to capture 12 μg of antibody, anti-HAUSP antibody or OX8 antibody as negative control, at a final antibody concentration of 0.25 μg/ml and in 0.1 m sodium phosphate buffer, pH 8. The incubation was carried out overnight at 4 °C under gentle mixing. Antibodies were cross-linked using 20 mm dimethyl pimelimidate dihydrochloride (Pierce) in 0.2 m triethanolamine, pH 8.2, for 1 h at 4 °C under gentle shaking.
Erythrocytes were prepared using the same protocol as the one used for the 26 S proteasome purification. No formaldehyde in vivo cross-linking of proteins was performed. Two 1.5-ml aliquots of frozen erythrocytes were thawed at 4 °C in lysis buffer containing protease and phosphatase inhibitors. After a 3-h centrifugation at 20,000 × g to remove cellular debris, each supernatant was incubated overnight at 4 °C with rotary shaking with either the anti-HAUSP-Dynabeads or the OX8-Dynabeads as negative control. Another tube containing 1.5 ml of lysis buffer was incubated with anti-HAUSP-Dynabeads as a second negative control in the same conditions. After washing three times with 30 bed volumes of equilibration buffer (20 mm Tris-HCl, 150 mm NaCl, 1 mm EDTA, 10% glycerol, 5 mm MgCl2, 2 mm ATP, pH 7.6), the elution was performed with 0.1 m citrate, pH 3.
Western Blots
Multiplex Detection of 26 S Proteasome in Protein Samples Purified with the MCP21 Antibody—
Proteins separated by SDS-PAGE on a 12% acrylamide gel were transferred to a nitrocellulose membrane (Hybond ECL, GE Healthcare). Blocking was performed using 2% blocking agent (ECL Advance Blocking Agent, GE Healthcare) in PBS, 0.1% Tween. Mouse monoclonal primary antibodies against 19 S proteasome subunits Rpt1 (1:5,000) and Rpn12 (1:1,000) and rabbit polyclonal antibodies against 20 S “core” subunits (α5/α7, β1, β5, β5i, and β7) (BIOMOL International LP) (1:10,000) were used for the immunoblot staining. ECL Plex CyDye-conjugated antibodies, goat α-mouse IgG-Cy3 (1:2,000) and goat α-rabbit IgG-Cy5 (1:1,250) (GE Healthcare), were used as secondary antibodies. The multiplex detection was performed using the Typhoon Trio fluorescence scanner (GE Healthcare) at 532 nm excitation and 580 nm emission for the Cy3-conjugated antibody and 633 nm excitation and 670 nm emission for the Cy5-conjugated antibody. Commercially available 20 S proteasome and 26 S proteasome purified from human erythrocytes (BIOMOL International LP) were used as positive controls.
Detection of 20 S Core Particle in Protein Samples Immunoprecipitated with the Anti-HAUSP Antibody—
After electrophoresis, protein blots were transferred to a nitrocellulose membrane (Hybond-C Super, GE Healthcare). The membrane was blocked with 1% bovine serum albumin in PBS, 0.1% Tween and incubated for 1 h with the rabbit polyclonal antibodies against 20 S core subunits (1:10,000) at room temperature. After washing three times with PBS, 0.1% Tween, the membrane was incubated at room temperature for 1 h with horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA) diluted in PBS, 0.1% Tween at 1:10,000. The detected protein signals were visualized by an enhanced chemiluminescence reaction system using the Amersham Biosciences ECL kit (GE Healthcare) and exposition to Kodak XAR film (Eastman Kodak Co.).
In-gel Digestion, Identification of Proteins by Nano-LC-ESI-LTQ-Orbitrap MS/MS Analysis, and Database Searching
After separation of proteins by SDS-PAGE on an 8 × 10-cm acrylamide gel (12%) and Coomassie Blue staining, whole gel lanes were analyzed by cutting them into several bands. Proteins were reduced and alkylated by successive incubation in solutions of 10 mm DTT in 100 mm NH4HCO3 for 35 min at 56 °C and 55 mm iodoacetamide in 100 mm NH4HCO3 for 30 min at room temperature, respectively. In-gel tryptic digestion was then performed as described previously (37) with minor modifications. Briefly after several washing steps to eliminate the stain, the gel pieces were dried under vacuum to be rehydrated in a sufficient covering volume (50–75 μl) of modified trypsin solution (20 ng/μl in 25 mm NH4HCO3; Promega, Madison, WI) for 15 min in an ice bath. Trypsin digestion was performed overnight at 37 °C under shaking. Peptides were extracted three times at 37 °C for 15 min under shaking using 50 mm NH4HCO3 once and 5% formic acid in 50% ACN twice. The peptide mixture was dried under vacuum and stored at −20 °C.
For MS analysis, tryptic peptides were resuspended with 12 μl of 5% ACN, 0.05% TFA and were submitted to nano-LC-MS/MS using an Ultimate3000 system (Dionex, Amsterdam, The Netherlands) coupled to an ESI-LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) operating in positive mode with a spray voltage of 1.4 kV. Five microliters of each sample were loaded on a C18 precolumn (300-μm inner diameter × 5 mm; Dionex) at 20 μl/min. After 5 min of desalting, the precolumn was switched on line with the analytical column (75-μm inner diameter × 15 cm; PepMap C18, Dionex) equilibrated in 95% solvent A (5% ACN, 0.2% formic acid) and 5% solvent B (80% ACN, 0.2% formic acid). Peptides were eluted using a 5–50% gradient of solvent B for 80 min at a 300 nl/min flow rate. Data were acquired with Xcalibur (LTQ-Orbitrap Software version 2.2 beta 5, Thermo Fisher Scientific). The mass spectrometer was operated in the data-dependent mode and was externally calibrated as recommended by the supplier in an automatic mode, leading to typical mass errors lower than 5 ppm. Survey MS scans were acquired in the Orbitrap on the 300–2000 m/z range with the resolution set to a value of 60,000 at m/z 400. Up to five of the most intense multiply charged ions (2+ and 3+) per scan were CID fragmented in the linear ion trap. A dynamic exclusion window was applied within 60 s. All tandem mass spectra were collected using a normalized collision energy of 35%, an isolation window of 4 m/z, and one microscan. Other instrumental parameters included maximum injection times and automatic gain control targets of 250 ms and 500,000 ions, respectively, for the FTMS and 100 ms and 10,000 ions, respectively, for LTQ MS/MS. Data were analyzed using Xcalibur software (version 2.0 SR2, Thermo Fisher Scientific), and MS/MS centroid peak lists were generated using the executable extract_msn.exe (Thermo Fisher Scientific) integrated into the Mascot Daemon software (Mascot version 2.2.1, Matrix Sciences, Boston, MA). The following parameters were set to create the peak lists: parent ions in the mass range 400–4500, no grouping of MS/MS scans, and threshold at 1,000. A peak list was created for each fraction analyzed (i.e. gel band), and individual Mascot searches were performed for each fraction.
Data were searched using Mascot server (Mascot version 2.2.01, Matrix Sciences) against Homo sapiens sequences in the Swiss-Prot TrEMBL database (68,579 sequences). This database consists of UniProtKB/Swiss-Prot Protein Knowledgebase Release 53.1 merged in house with UniProtKB/TrEMBL Protein Database Release 36.1. The mass tolerances in MS and MS/MS were set to 5 ppm and 0.8 Da, respectively, and the instrument setting was specified as “ESI Trap.” Trypsin was designated as the protease (specificity set for cleavage after Lys or Arg), and up to two missed cleavages were allowed. Oxidation of methionine, amino-terminal protein acetylation, ubiquitination of lysine (GG or LRGG), carbamidomethylation, and propionamide on cysteine were searched as variable modifications; no fixed modification was set. Protein hits were automatically validated if they satisfied one of the following criteria: identification with at least one top ranking peptide (bold and red) with a Mascot score of more than 50 (p value <0.001), at least two top ranking peptides each with a Mascot score of more than 35 (p value <0.03), or at least three top ranking peptides each with a Mascot score of more than 30 (p value <0.1) as determined by the Mascot Search program and using the automatic validation module of our in-house developed software MFPaQ (version 4.0.0) (38). Proteins identified with a single peptide as well as post-translational modifications on peptides were confirmed by manual inspection of the MS/MS spectra (provided as supplemental Data 3 and 6, respectively). Proteins identified with exactly the same set of peptides were grouped, and only one member of the protein group was reported in Tables I and II and supplemental Data 2 for greater clarity (the one that we considered as the most significant according to the functional description given in the UniProt Knowledgebase). However, detailed protein groups are shown in supplemental Data 1. Highly homologous protein hits, i.e. proteins identified with top ranking MS/MS queries also assigned to another protein hit of higher score (red, non-bold peptides), were detected by the MFPaQ software and were considered as individual hits and included in the final list only if they were additionally assigned a specific top ranking (red and bold) peptide of score higher than 33 (p value <0.05). From all the validated result files corresponding to the fractions of a one-dimensional gel lane, MFPaQ was used to generate a unique, non-redundant list of proteins by comparing proteins or protein groups (composed of all the protein sequences matching the same set of peptides) according to accession numbers and creating clusters from protein groups found in different gel slices if they have one common member. Protein list comparisons were based on the comparison of protein groups (hits matching the same set of peptides) from different lists, and the MFPaQ software assigned these protein groups as “shared” or “specific” depending on whether or not they have common members.
Table I.
Proteins specifically interacting with the MCP21 antibody: proteasome subunits and proteasome activators/inhibitors/stabilizers
Proteasome subunits listed in the table were purified as described under “Experimental Procedures” with or without formaldehyde cross-linking, separated by SDS-PAGE, and identified by nano-LC-ESI-LTQ-Orbitrap MS/MS analysis and database searching in the Swiss-Prot TrEMBL database. Nonspecific interactions could be eliminated by performing a differential analysis with proteins purified with the OX8 antibody and similarly identified. Criteria for acceptance of protein identification are described under “Experimental Procedures.” The best results in terms of protein recovery are reported. N, “normal,” i.e. without formaldehyde cross-linking; F, “formaldehyde,” i.e. with formaldehyde cross-linking.
| Namea | Accession no. | No. of unique peptides
|
Sequence coverage
|
MS/MS counting (n = 3)
|
Ref.c | |||
|---|---|---|---|---|---|---|---|---|
| N | F | N | F | Averaged ratio F/Nb | S.D. | |||
| % | ||||||||
| 20 S proteasome subunits | ||||||||
| α1 | P60900 | 29 | 32 | 83 | 84 | 1.0 | 0.0 | 46 |
| α2 | P25787 | 30 | 29 | 88 | 86 | 0.9 | 0.0 | 46 |
| α3 | P25789 | 27 | 29 | 87 | 88 | 1.2 | 0.6 | 46 |
| α4 | O14818 | 33 | 34 | 85 | 85 | 0.9 | 0.2 | 46 |
| α5 | P28066 | 18 | 26 | 75 | 80 | 0.9 | 0.1 | 46 |
| α6 | P25786 | 35 | 43 | 95 | 96 | 1.0 | 0.2 | 46 |
| α7 | P25788 | 33 | 41 | 79 | 80 | 0.9 | 0.4 | 46 |
| β1 | P28072 | 15 | 15 | 78 | 76 | 1.1 | 0.2 | 46 |
| β2 | Q99436 | 18 | 22 | 76 | 77 | 0.9 | 0.1 | 46 |
| β3 | P49720 | 17 | 22 | 62 | 64 | 1.1 | 0.1 | 46 |
| β4 | P49721 | 21 | 27 | 95 | 98 | 0.7 | 0.2 | 46 |
| β5 | P28074 | 25 | 22 | 89 | 89 | 0.9 | 0.1 | 46 |
| β6 | P20618 | 22 | 27 | 81 | 82 | 1.0 | 0.1 | 46 |
| β7 | P28070 | 12 | 12 | 57 | 59 | 1.0 | 0.1 | 46 |
| β1i | P28065 | 6 | 5 | 34 | 38 | 1.1 | 0.2 | 46 |
| β2i | P40306 | 7 | 7 | 37 | 33 | 0.6 | — | 46 |
| β5i | P28062 | 13 | 15 | 68 | 63 | 1.1 | 0.3 | 46 |
| Proteasome regulators | ||||||||
| 19 S subunits | ||||||||
| S7 (Rpt1) | P35998 | 38 | 37 | 80 | 79 | 1.2 | 0.2 | 46 |
| S4 (Rpt2) | P62191 | 29 | 36 | 58 | 72 | 1.2 | 0.2 | 46 |
| S6b (Rpt3) | P43686 | 36 | 37 | 84 | 81 | 1.5 | 0.2 | 46 |
| S10b (Rpt4) | P62333 | 28 | 42 | 74 | 77 | 1.6 | 0.3 | 46 |
| S6a (Rpt5) | P17980 | 29 | 33 | 66 | 77 | 1.6 | 0.2 | 46 |
| S8 (Rpt6) | P62195 | 32 | 32 | 74 | 76 | 1.9 | 0.2 | 46 |
| S2 (Rpn1) | Q13200 | 68 | 64 | 69 | 72 | 1.1 | 0.1 | 46 |
| S1 (Rpn2) | Q99460 | 56 | 57 | 67 | 73 | 1.1 | 0.3 | 46 |
| S3 (Rpn3) | O43242 | 33 | 50 | 58 | 73 | 1.5 | 0.1 | 46 |
| p55 (Rpn5) | O00232 | 34 | 35 | 65 | 67 | 1.6 | 0.2 | 46 |
| S9 (Rpn6) | O00231 | 34 | 39 | 75 | 80 | 1.8 | 0.3 | 46 |
| S10a (Rpn7) | Q15008 | 33 | 36 | 79 | 79 | 1.4 | 0.3 | 46 |
| S12 (Rpn8) | P51665 | 17 | 23 | 56 | 61 | 3.2 | 0.7 | 46 |
| S11 (Rpn9) | Q9UNM6 | 25 | 28 | 79 | 86 | 2.3 | 0.6 | 46 |
| S5a (Rpn10) | P55036 | 12 | 11 | 41 | 35 | 3.5 | 0.6 | 46 |
| S13 (Rpn11) | O00487 | 13 | 18 | 65 | 66 | 4.9 | 2.0 | 46 |
| S14 (Rpn12) | P48556 | 12 | 14 | 64 | 68 | 3.8 | 1.2 | 46 |
| ADRM1 (hRpn13) | Q16186 | 4 | 8 | 11 | 17 | 4.8 | — | 46 |
| PAAF1 (Rpn14) | Q9BRP4 | 0 | 6 | 0 | 17 | — | — | 46 |
| Other activators | ||||||||
| PA28α (11Sα) | Q06323 | 21 | 19 | 76 | 63 | 1.1 | 0.1 | 46 |
| PA28β (11Sβ) | Q9UL46 | 22 | 19 | 84 | 85 | 1.3 | 0.1 | 46 |
| PA200 | Q14997 | 26 | 6 | 16 | 4 | 0.2 | 0.1 | 46 |
| Inhibitors/stabilizers | ||||||||
| PI31 | Q92530 | 14 | 13 | 61 | 46 | 0.9 | 0.1 | 41 |
| ECM29 | Q5VYK3 | 0 | 5 | 0 | 4 | — | — | 42, 70 |
Several nomenclatures exist for subunits of 20 S proteasome and 19 S regulatory particle. For 20 S proteasome subunits, the nomenclature from Baumeister et al. (71) was used. For most 19 S regulatory subunits, the nomenclature usually applied in human (72) was used. For better understanding, the nomenclature applied in yeast (73) is provided in parentheses.
MS/MS counting ratios (F/N) were calculated when a significant number of MS/MS scans (>50) was performed at least for one experiment (without or with formaldehyde cross-linking). Statistical data were obtained from three independent replicates. If no standard deviation is indicated, the MS/MS threshold of 50 was reached only with one of the experiments. Dashes indicate that no value could be obtained for the reasons explained above.
Selected references reporting this protein as a proteasome subunit or a PIP by affinity purification strategies or other biochemical approaches.
Table II.
Proteins specifically interacting with the MCP21 antibody: proteins known to belong to or to be related to the UPP
Proteins listed in the table were purified as described under “Experimental Procedures,” separated by SDS-PAGE, and identified by nano-LC-ESI-LTQ-Orbitrap MS/MS analysis and database searching in the Swiss-Prot TrEMBL database. Nonspecific interactions could be eliminated by performing a differential analysis with proteins purified with the OX8 antibody and similarly identified. Criteria for acceptance of protein identification are described under “Experimental Procedures.” For proteins identified in several repeats, the best results in terms of protein recovery are reported. N, “normal,” i.e. without formaldehyde cross-linking; F, “formaldehyde,” i.e. with formaldehyde cross-linking.
| Name | Accession no.a | No. of unique peptides
|
Sequence coverage
|
MS/MS counting (n = 2)
|
Ref.c | |||
|---|---|---|---|---|---|---|---|---|
| N | F | N | F | Averaged ratio F/Nb | S.D. | |||
| % | ||||||||
| Ubiquitind | P62988 | 6 | 6 | 68 | 68 | 1.8 | 0.3 | 32 |
| Deubiquitinating enzymes | ||||||||
| USP14d | P54578 | 11 | 25 | 23 | 55 | 3.8 | 0.5 | 32 |
| UCH37d | Q9Y5K5 | 8 | 10 | 26 | 37 | 1.3 | — | 32 |
| USP7 | Q93009 | 28 | 0 | 25 | 0 | — | — | |
| E3 ligases | ||||||||
| F-box protein7e | Q5TGC3 | 14 | 10 | 25 | 19 | 0.4 | 0.1 | 25 |
| SKP1e | P63208 | 18 | 14 | 85 | 64 | 0.5 | 0.1 | 25 |
| Cullin-1e | Q13616 | 49 | 0 | 65 | 0 | — | — | 25 |
| Ring-box protein 1f | P62877 | 1 | 0 | 26 | 0 | — | — | |
| Ubiquitin-protein ligase E3Cd,f | Q15386 | 19 | 0 | 21 | 0 | — | — | 32 |
| Cullin-associated NEDD8-dissociated protein 1f | Q86VP6 | 0 | 7 | 0 | 9 | — | — | |
| Multiubiquitin chain-binding proteins | ||||||||
| hHR23Bd | P54727 | 0 | 3 | 0 | 7 | — | — | 32 |
| DDI1 homolog 2e,f | Q5TDH0 | 0 | 3 | 0 | 9.5 | — | — | 55 |
| eEF1α1d | P68104 | 0 | 5 | 0 | 16 | — | — | 32 |
| PACs | ||||||||
| PAC1 | Q6FHD3 | 8 | 9 | 31 | 37 | 1.3 | 0.4 | 52, 53 |
| PAC2 | Q969U7 | 8 | 13 | 28 | 38 | 2.3 | — | 52, 53 |
| PAC3 | A4D216 | 0 | 3 | 0 | 26 | — | — | 52, 53 |
| Chaperones | ||||||||
| HSP70 1d | P08107 | 12 | 2 | 27 | 4 | 0.5 | — | 32 |
| Heat shock cognate 71d | P11142 | 28 | 10 | 46 | 17 | 0.3 | — | 32 |
| HSP90-βd | P08238 | 0 | 5 | 0 | 14 | — | — | 32 |
| HSPA5 | Q2KHP4 | 0 | 3 | 0 | 8 | — | — | |
| Valosin-containing proteind | Q0V924 | 7 | 20 | 12 | 31 | 3.6 | 0.1 | 74 |
| TCP-1γd,f | P49368 | 2 | 0 | 4 | 0 | — | — | 21, 32 |
| TCP-1ηd,f | Q99832 | 2 | 0 | 6 | 0 | — | — | 21, 32 |
For greater clarity, the reported member of the protein group is the one that we considered as the most significant according to the functional description given in the UniProt Knowledgebase.
MS/MS counting ratios (F/N) were calculated when a significant number of MS/MS scans (>50) was performed at least for one experiment (without or with formaldehyde cross-linking). Statistical data were obtained from two independent replicates. When no averaged ratio is indicated, it means that the total MS/MS number for the protein did not reach 50. If no standard deviation is provided, the MS/MS threshold of 50 was reached only with one of the experiments. Dashes indicate that no value could be obtained for the reasons explained above.
Selected references reporting this protein as a PIP by affinity purification strategies or other biochemical approaches.
Reported human PIPs.
Reported PIPs only in other species.
Identified in one experiment only.
To evaluate false positive rates, all the initial database searches were performed using the “decoy” option of Mascot, i.e. the data were searched against a combined database containing the real specified protein sequences (target database, Swiss-Prot TrEMBL human) and the corresponding reversed protein sequences (decoy database). MFPaQ used the same criteria to validate decoy and target hits, calculated the false discovery rate (FDR = number of validated decoy hits/(number of validated target hits + number of validated decoy hits) × 100) for each gel band analyzed, and calculated the average of false discovery rate for all bands belonging to the same gel lane (i.e. to the same sample).
RESULTS
Purification of Endogenous Human Proteasome Complexes by Affinity Chromatography—
Human proteasome complexes and PIPs were purified by a single step affinity chromatography procedure using a Sepharose-bound monoclonal antibody, MCP21, showing a high affinity for the α2 subunit of human 20 S proteasome. The experimental design was adapted from an initially developed protocol to purify 20 S proteasome from various human cellular types (36, 39). Importantly ATP and glycerol, added in all buffers, enabled the weak interactions between the core particle and its regulatory complexes to be maintained. Moreover the benefit of an in vivo protein-protein interactions cross-linking step using formaldehyde as cross-linking agent was assessed. The method was optimized using human erythrocyte cells as starting material for which no tagging strategy can be used. After cell lysis, proteins were purified with either the MCP21-Sepharose antibody or the OX8-Sepharose antibody as negative control (Fig. 1). No ChT-like activity could be detected in the fractions eluted from the OX8-Sepharose column (Fig. 2). On the contrary, a peak of ChT-like activity was observed in the fractions purified with the human IgG1 MCP21 antibody. This enzymatic activity was almost completely abolished in the presence of 10 μm lactacystin and could therefore be attributed to the presence of proteasome in the purified fractions. Moreover despite a similar protein content in the two preparations (with and without formaldehyde), the ChT-like activity of the most active fraction was significantly increased in the preparation obtained after formaldehyde cross-linking (specific activity of 23 ± 6 pmol of AMC·min−1·μg−1) compared with the preparation without in vivo formaldehyde cross-linking (specific activity of 12 ± 2 pmol of AMC·min−1·μg−1). The total ChT-like activity of all of the purified fractions was calculated as the sum of the activities measured in each fraction. This activity was superior by a factor of 2.1 in the formaldehyde-treated sample compared with the non-treated sample. Such an assay requires intact 20 S proteasome complex, but the 19 S regulatory particles as well as other activators such as PA28 or PA200 are known to enhance the ChT-like proteolytic activity of 20 S proteasome. Therefore, it could be hypothesized that proteasome complexes obtained after formaldehyde treatment contain more 20 S proteasome associated to its activators.
Fig. 1.

Differential analysis strategy to identify specific human PIPs. Erythrocytes submitted or not to in vivo cross-linking were lysed, and proteins were purified by overnight incubation at 4 °C with either the MCP21-Sepharose antibody or the OX8-Sepharose antibody as negative control. After extensive washing with 20 mm Tris-HCl, 150 mm NaCl, 1 mm EDTA, 10% glycerol, 5 mm MgCl2, 2 mm ATP, pH 7.6, proteins were eluted with a saline step using the same buffer containing 3 m NaCl. Two-milliliter fractions were collected and stored at 4 °C. Protein concentration was determined using the Bio-Rad Protein Assay, and proteasome content could be estimated by measuring the in vitro chymotrypsin-like activity as described under “Experimental Procedures.” The fractions containing the most proteasome activity were then further analyzed by Western blotting and by SDS-PAGE followed by trypsin digestion and LC-MS/MS analysis. Proteins identified in each case (experiment and negative control) were subjected to a differential analysis using the MFPaQ software (38) so that a list of specific MCP21-interacting proteins could be generated. ChT-L, ChT-like.
Fig. 2.

Proteasome purification from human erythrocytes without (A) or with (B) in vivo formaldehyde cross-linking. Proteasome complexes were purified from human erythrocytes by affinity chromatography with the mouse IgG1 monoclonal antibody MCP21 coupled to Sepharose beads. A control purification was performed using the mouse IgG1 monoclonal antibody OX8 directed against rat CD8α. Proteins were cross-linked in vivo by incubation of human erythrocytes with 1% formaldehyde as indicated under “Experimental Procedures.” After incubation of 50 ml of erythrocyte lysate with either the MCP21-Sepharose or the OX8-Sepharose, proteins interacting with the beads were eluted by a saline step of 3 m NaCl. The eluted fractions were analyzed for their protein concentrations as well as for their proteasome contents. Statistical results were obtained from three independent experiments for each condition (formaldehyde-treated or not). Error bars indicate standard deviations (n = 3). ♦ and ⋄ represent protein concentrations in the fractions from the MCP21-Sepharose beads and from the OX8-Sepharose beads, respectively. ▴ and ▵ represent the in vitro ChT-like activity without and with 10 μm lactacystin, respectively, in the fractions eluted from the MCP21-Sepharose beads. No ChT-like activity could be detected in the fractions from the OX8 negative control experiment.
The most active fractions from the two preparations were further analyzed for their 19 S and 20 S content by multiplex detection of several subunits from the 19 S and 20 S particles (Fig. 3). The most active fractions (fraction 10 and fractions 8–10 from purifications with and without formaldehyde cross-linking, respectively) contain both 20 S proteasome and 19 S activators as demonstrated by the presence of a subunit of both the base (Rpt1) and the lid (Rpn12) (Fig. 3, B and C). Another interesting feature is that, as expected, formaldehyde treatment seems to stabilize the weak interactions between the 20 S proteasome and its 19 S interactors as the ratio of 19 S signal/20 S signal is higher in the most active fraction (fraction 10) obtained after purification of formaldehyde-treated erythrocytes than in its counterpart fraction in the non-treated sample (Fig. 3B). Thus, the 26 S proteasome content in the preparation obtained after purification of formaldehyde-treated erythrocytes is higher than that of the non-treated preparation. One can speculate that a higher portion of the 19 S regulators (and probably also some of the other regulators and PIPs) were lost during the purification procedure in the untreated erythrocytes sample compared with the treated sample. Moreover the elution patterns of fractions 8–10 from the non-treated erythrocytes preparation show that, for a similar 20 S subunit signal, the 19 S content is higher in the early eluted fraction (fraction 8) than in the later fractions (fractions 9 and 10) (Fig. 3C). This means that the 19 S subunits elute earlier from the MCP21-Sepharose column than the 20 S subunits, which is in accordance with the fact that the interaction between MCP21 and the 20 S proteasome is strong compared with that between the 20 S core particle and the 19 S regulatory particle. Thus, one can speculate that the other PIPs would behave similarly, and identification of PIPs from the non-treated preparation therefore can rather be performed in the early eluted fractions.
Fig. 3.

Detection of 20 S core particle and 19 S activators in the purified proteasome preparations. Proteins separated by SDS-PAGE were transferred to a nitrocellulose membrane: 2 μg of commercial 20 S and 26 S proteasome from human erythrocytes as standards (A), 20 μg of total proteins from fractions containing the maximal ChT-like activity (fractions 9 and 10 from purifications without and with formaldehyde cross-linking, respectively) (B), and 10 μg of estimated 20 S proteasome (based on the ChT-like activity measurement) from fractions 8, 9, and 10 from the purification without formaldehyde cross-linking (C). Mouse monoclonal primary antibodies against 19 S proteasome subunits Rpt1 and Rpn12 and rabbit polyclonal antibodies against 20 S core subunits were used for the immunoblot staining. ECL Plex CyDye-conjugated antibodies, goat α-mouse IgG-Cy3 and goat α-rabbit IgG-Cy5, were used as secondary antibodies. The detection was performed using the Typhoon Trio fluorescence scanner at 532 nm excitation and 580 nm emission for the Cy3-conjugated antibody and 633 nm excitation and 670 nm emission for the Cy5-conjugated antibody. Lane M, molecular mass markers.
Identification of Proteasome Complex Components by Proteomics Analysis—
To identify proteins contained in the early eluted fractions from the non-treated sample (fractions 6–9) and the formaldehyde-treated sample (fractions 7–10), proteins were fractionated by SDS-PAGE. The entire gel lanes were cut into bands for further trypsin-mediated protein digestion and nano-LC-ESI-LTQ-Orbitrap MS/MS analyses. A similar procedure was conducted for the two protein samples corresponding to the two negative control experiments (erythrocytes treated or not with formaldehyde and protein extracts incubated with the OX8-Sepharose) except that the whole eluted protein sample was loaded onto the gel in that case. As displayed on Fig. 4, the characteristic pattern of 26 S proteasome is only observed on lanes 5 and 6 corresponding to MCP21-eluted proteins from non-treated and treated erythrocytes, respectively, as compared with lane 1 (commercially available 26 S proteasome from human erythrocytes). This confirms the presence of 26 S subunits in our purified fractions. Moreover the higher 19 S/20 S ratio in the fractions purified from formaldehyde-treated erythrocytes (Fig. 4, lane 6) than that in their counterpart fractions from non-treated erythrocytes (Fig. 4, lane 5) is confirmed as the 20 S subunits (molecular mass ranging approximately from 20 to 30 kDa) can easily be identified from the 19 S subunits, which exhibit higher molecular masses.
Fig. 4.

Separation of proteasomes and proteasome-interacting proteins by SDS-PAGE. Proteins were separated by SDS-PAGE on a 8 × 10-cm acrylamide gel (12%) and detected by Coomassie Brilliant Blue staining. Whole gel lanes were analyzed. In-gel tryptic digestion of proteins was performed prior to identification by nano-LC-ESI-LTQ-Orbitrap MS/MS analysis and database searching as described under “Experimental Procedures.” Lanes 1 and 2, 2 μg of commercially available human erythrocyte 26 S and 20 S proteasomes, respectively; lanes 3 and 4, the whole protein content of eluted proteins from the OX8-Sepharose was loaded after concentration by ultrafiltration (lane 3, non formaldehyde-treated erythrocytes; lane 4, formaldehyde-treated erythrocytes); lanes 5 and 6, 40 μg of proteins from fractions 6–9 and 7–10 from purifications without and with formaldehyde cross-linking, respectively.
After nano-LC-ESI-LTQ-Orbitrap MS/MS analysis and identification of proteins using Mascot, the automatic validation module of MFPaQ was used to generate a list of proteins identified from each sample deposited in Fig. 4, lanes 3, 4, 5, and 6, as explained under “Experimental Procedures.” For the two experiments (non-treated erythrocytes and formaldehyde-treated erythrocytes), a protein list was generated for each condition (experiment and control). Proteins interacting only with the control antibody or only with the bait antibody, i.e. specific proteins after comparison of the two lists, were discarded or considered as specific interactors, respectively. Indeed it was very improbable that a nonspecific protein present in the MCP21-interacting proteins sample would not be detected and identified in the OX8 list because only 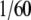 of the total proteins from the MCP21 sample was loaded onto the gel, whereas the whole protein sample, purified from the same quantity of erythrocytes cells, was analyzed in the control. A list of proteins found in common, i.e. shared proteins after comparison of the two lists, was also generated by MFPaQ as explained under “Experimental Procedures” and actually contained several 20 S proteasome subunits in both conditions (experiment and control). Therefore, these proteins in common were further submitted to a relative quantification to rescue specific MCP21-interacting proteins. The spectral counting approach, adapted from the method described by Liu et al. (40), was chosen because it does not require any labeling of proteins but is based on the counting of the total number of MS/MS spectra acquired for a protein. For each protein identified and validated using the MFPaQ automatic validation module, the number of spectral copies (given by Mascot) to identify this particular protein was used. When a protein was identified several times in consecutive gel bands, all the MS/MS spectra attributed to peptides belonging to the protein were taken into account. The relative quantity of a protein present in the two samples was estimated as the ratio of the total number of MS/MS spectra of the protein in each sample. Interestingly MCP21/OX8 MS/MS ratios obtained for 20 S proteasome subunits were the highest; they were superior to those obtained for keratins (ratios around 1), which are expected to be equally distributed between the two samples (see supplemental Data 5 for detailed information). This indicated that 20 S proteasome subunits were significantly more abundant in the MCP21-interacting protein sample. These subunits were therefore rescued as MCP21-specific interacting proteins using this approach. Their presence in the OX8-eluted protein fractions can be explained by the high abundance of 20 S proteasome in cells and knowing that the whole protein sample was analyzed in that case. All other proteins interacting both with the MCP21 and the control antibodies, including abundant globin chains and hemoglobin subunits, displayed lower MCP21/OX8 MS/MS ratios and were discarded.
of the total proteins from the MCP21 sample was loaded onto the gel, whereas the whole protein sample, purified from the same quantity of erythrocytes cells, was analyzed in the control. A list of proteins found in common, i.e. shared proteins after comparison of the two lists, was also generated by MFPaQ as explained under “Experimental Procedures” and actually contained several 20 S proteasome subunits in both conditions (experiment and control). Therefore, these proteins in common were further submitted to a relative quantification to rescue specific MCP21-interacting proteins. The spectral counting approach, adapted from the method described by Liu et al. (40), was chosen because it does not require any labeling of proteins but is based on the counting of the total number of MS/MS spectra acquired for a protein. For each protein identified and validated using the MFPaQ automatic validation module, the number of spectral copies (given by Mascot) to identify this particular protein was used. When a protein was identified several times in consecutive gel bands, all the MS/MS spectra attributed to peptides belonging to the protein were taken into account. The relative quantity of a protein present in the two samples was estimated as the ratio of the total number of MS/MS spectra of the protein in each sample. Interestingly MCP21/OX8 MS/MS ratios obtained for 20 S proteasome subunits were the highest; they were superior to those obtained for keratins (ratios around 1), which are expected to be equally distributed between the two samples (see supplemental Data 5 for detailed information). This indicated that 20 S proteasome subunits were significantly more abundant in the MCP21-interacting protein sample. These subunits were therefore rescued as MCP21-specific interacting proteins using this approach. Their presence in the OX8-eluted protein fractions can be explained by the high abundance of 20 S proteasome in cells and knowing that the whole protein sample was analyzed in that case. All other proteins interacting both with the MCP21 and the control antibodies, including abundant globin chains and hemoglobin subunits, displayed lower MCP21/OX8 MS/MS ratios and were discarded.
Eighty-six proteins specifically interacting with the MCP21-Sepharose are displayed in Tables I and II and supplemental Data 2, which include results obtained with or without in vivo formaldehyde treatment and from three different biological samples. Criteria to accept a protein identification are indicated under “Experimental Procedures,” and such criteria led to false positive rates inferior to 1% in all cases. Identification overlaps between experiments were high for proteins from the UPP (58 of 64 proteins, i.e. 91%; Tables I and II) and slightly lower (69 of 86 proteins, i.e. 80%; Tables I and II and supplemental Data 2) if all identified proteins were considered. The variability observed concerned mostly low abundance proteins identified with a low number of peptides.
As displayed in Table I, all 17 subunits of 20 S standard proteasome and immunoproteasome as well as numerous subunits of the 19 S particle were identified in both experiments (non-treated and treated) with high sequence coverages. Moreover PA28α, PA28β, and PA200 proteasome regulators were also identified with good recoveries. Our purification strategy also enabled the detection of PI31 and ECM29, already known as an inhibitor and stabilizer of 26 S proteasome, respectively (41, 42).
Twenty-three other proteins involved in the ubiquitin-proteasome degradation pathway were identified in the purified proteasome fractions from non-treated and formaldehyde-treated erythrocytes (Table II). They are ubiquitin, deubiquitinating enzymes, E3 ligases or related proteins, multiubiquitin chain-binding proteins, and proteasome-assembling chaperones as well as chaperones, such as proteins of the HSP family, valosin-containing protein, and TCP-1 subunits (Table II). This result strongly validates the strategy used to purify proteasome complexes and their interacting proteins.
Two tryptic peptides of ubiquitin, 43LIFAGK(GG)QLEDGR54 and 43LIFAGK(LRGG)QLEDGR54, which were indicative of Lys48-linked polyubiquitin chains, were identified (see supplemental Data 1 and 6). Ubiquitination sites were identified based on the sequencing of signature peptides containing a GG tag (114.1 Da) and/or an LRGG tag (383.2 Da) either on internal or on carboxyl-terminal lysine residues (43). Identification of diglycine-containing lysine residues after tryptic digestion of proteins can be biased by iodoacetamide-induced artifacts mimicking ubiquitination in MS experiment (44), but the LRGG tag confirms that Lys48 is ubiquitinated. Polyubiquitination involving lysine at residue 48 is the major signal for protein degradation by the multicatalytic proteasome complex (45).
Twenty-two other proteins identified in this work are not directly linked to the UPP (supplemental Data 2). They are proteins from the nucleotide excision repair pathway, proteins involved in the actomyosin cytoskeleton signaling, and other proteins. Seven proteins were already known as PIPs or are related to known PIPs like histones, actins, myosins, or 14-3-3 proteins (21, 32). Others constitute new putative PIPs and will need further investigation to be confirmed.
To compare our results with previously published surveys, tables (Tables I and II and supplemental Data 2) indicate selected studies reporting each identified protein as a PIP by similar strategies or other biochemical approaches in human or other species. The most recent work concerned human HEK293 cells PIP characterization by TAP strategies followed by systematic MS identification of proteins (32, 46). Venn diagrams indicating overlaps between our interaction data and those published in these surveys are shown in Fig. 5 and supplemental Data 4. Forty-eight proteins were found in common, 38 were specific to our work, and 44 were not identified by us. Among the common proteins, 36 subunits of the proteasome complexes or activator proteins could be observed as well as 11 other proteins (supplemental Data 4); some of these had already been reported as PIPs in yeast (25, 28, 47) or in other species (21) (Tables I and II). Among the 46 putative PIPs specifically identified by Huang and co-workers (32, 46), six proteins are linked to the UPP, and 38 are not. We have identified 38 new putative PIPs. Of these, 17 are directly linked to proteasome function: 10 proteins had already been reported as PIPs in other species, but seven proteins identified only by us constitute new putative PIPs using proteomics analysis (Fig. 5).
Fig. 5.

Venn diagram illustrating the overlap between human PIPs identified by us and the most recent surveys on human PIPs interaction data obtained by TAP strategies followed by systematic MS identification of proteins (32, 46).
One of these, Usp7, better known as HAUSP for herpes virus-associated ubiquitin-specific protease, was undoubtedly detected and validated in the pool of proteins identified after proteasome complex purification from non-treated erythrocytes with 28 top ranking unique peptides and 25% sequence coverage. It was not identified in proteasomes purified from formaldehyde-treated red blood cells, and no signal corresponding to tryptic peptides of this protein could be detected in the LC-MS traces obtained after digestion of protein gel bands from the SDS-PAGE-separated formaldehyde-treated samples. HAUSP belongs to the ubiquitin-specific protease (USP) subfamily of the deubiquitinating enzymes (DUBs), a well conserved family of cysteine proteases (48), but is a new reported PIP. HAUSP is a major player in the p53-MDM2 pathway (49). This protease is reported to localize to promyelocytic leukemia bodies (50), but an important fraction of this protein was also observed in the cytosol (51).
Benefit of Formaldehyde Treatment on PIP Recovery—
The benefit of formaldehyde treatment was evaluated by estimating the relative abundance of PIPs in purified samples from formaldehyde-treated and non-treated erythrocytes. The spectral counting approach was used to calculate the ratio of spectral copies of these proteins in the formaldehyde-treated sample versus in the non-treated sample (Tables I and II). For more precision, only proteins identified with a high number of spectral copies (>50; arbitrary chosen threshold) at least in one experiment were considered for the relative quantitation reported in Tables I and II. Results show that an average ratio of 1.0 ± 0.1 was obtained for 20 S subunits, whereas higher ratios (1.0–4.9) were reported for 19 S subunits. These results highlight again the benefit of formaldehyde reticulation for the stabilization of the interactions between the 20 S proteasome and its 19 S activators. Interestingly it seems that the 19 S subunits that are the most remote from the 20 S particle are more difficult to maintain without formaldehyde treatment as observed from the average ratios of the base subunits (ratio = 1.4 ± 0.3; subunits Rpt1 to -6 and Rpn1 and -2) compared with the lid subunits (ratio = 2.7 ± 1.2; subunits Rpn3 to -12). Formaldehyde cross-linking did not significantly stabilize PA28 interaction as the MS/MS ratios (1.1 and 1.3 for PA28α and PA28β, respectively) were very close to 1. This was also the case for PI31, which competes with PA28 for binding to 20 S proteasomes (41). On the contrary, it dramatically affected PA200 recovery.
As far as the other PIPs were concerned, formaldehyde had a global positive effect because it enabled important PIPs like ECM29, HSP90-β, and multiubiquitin chain-binding proteins (hHR23B, DDI1 homolog 2, and eEF1α1) to be identified or greatly increased the recovery of others like USP14 or valosin-containing protein, a PIP that had not been identified before by TAP strategies. However, for others, like HAUSP or the four identified components of the SCF complex, F-box protein7, SKP1, Cullin-1, and Ring-box protein 1, the formaldehyde treatment was undoubtedly detrimental (Tables I and II and supplemental Data 1 and 2).
Validation of HAUSP as a Newly Identified PIP—
To validate that HAUSP belongs to proteasome complexes, a reverse immunoprecipitation experiment was carried out by using a polyclonal antibody directed against HAUSP. Two negative control experiments were run in parallel as explained under “Experimental Procedures.” Immunoprecipitated proteins were analyzed by Western blotting using the rabbit polyclonal antibody directed against core subunits of 20 S proteasome (α5/α7, β1, β5, β5i, and β7). As displayed in Fig. 6, the characteristic pattern of 20 S proteasome subunits was observed in the immunoprecipitated protein sample obtained after incubation of human erythrocytes with the anti-HAUSP antibody; on the contrary, no significant signal could be detected in the control samples, indicating that 20 S proteasome specifically interacts with HAUSP. Nano-LC-ESI-LTQ-Orbitrap MS/MS analyses enabled the identification of HAUSP with a very high sequence coverage (77%) and confirmed the presence of 20 S subunits but also 19 S and PA28 regulators in the protein sample immunoprecipitated with the anti-HAUSP antibody only (results not shown).
Fig. 6.

Detection of 20 S core particle in the immunoprecipitated HAUSP complexes. HAUSP complexes were immunoprecipitated using the anti-HAUSP antibody or a control antibody as described under “Experimental Procedures,” separated by SDS-PAGE, and transferred to a nitrocellulose membrane. Rabbit polyclonal antibodies against 20 S core subunits were used for the immunoblot staining. Lane 1, 0.05 μg of commercially available human erythrocyte 20 S proteasome; lanes 2 and 4, whole protein content of eluted proteins from a 1.5-ml erythrocyte aliquot precipitated with the anti-HAUSP antibody (lane 2) or with the OX8 antibody (control) (lane 4); lane 3, protein sample eluted from the anti-HAUSP-Dynabeads after incubation with 1.5 ml of lysis buffer.
DISCUSSION
The Affinity Purification Procedure Described Is Efficient for the Capture of Physiological Proteasome Complexes and PIPs—
We report in this work a single step affinity purification strategy of endogenous proteasome complexes and PIPs from human erythrocytes based on the high affinity between a monoclonal antibody and the α2 subunit of the 20 S proteasome.
Compared with other strategies of 26 S proteasome purification using a tandem affinity purification method that decreases the number of false positive interactors (28), a single step affinity purification method can be more efficient to maintain weaker and more transient protein-protein interactions. Moreover when using careful experimental design including proper controls, true interactors can be discriminated from contaminants (33). A negative control purification was conducted, and a label-free differential quantitative method was carried out to distinguish false positives from putative PIPs. An originality of this approach compared with already published strategies to purify human 26 S proteasome is that the bait protein is the α2 subunit, a constitutive subunit of the 20 S complex present in all existing subtypes of 20 S particles. Therefore, in principle, all cellular proteasome complexes can be captured. Any other constitutive subunit of the 20 S core might have been used as a prey as far as the epitope is not hindered when the physiological complex is assembled. Another significant improvement of our approach over those described previously for human 26 S proteasome purification by affinity chromatography (30–32) is that wild-type endogenous complexes are targeted because no overexpression or tagging strategies are involved. Moreover the strategy can be used on any human sample, tissues, biopsies, cultured cells, or biological fluids.
All 20 S proteasomal subunits from the standard proteasome and from the immunoproteasome were identified with very good recoveries. All known regulatory complexes associated to the 20 S core particle, 19 S subunits, PA28αβ, and PA200 were also co-purified in high quantities except PA28γ, which is known to be exclusively nuclear in localization (18). These results therefore validate our method to efficiently purify proteasome complexes. Moreover because the prey protein is a subunit from the 20 S core particle, the approach enables evaluation of the heterogeneity of regulatory complexes associated to 20 S proteasomes. For example, the presence of premature 20 S complexes is suspected as three reported proteasome-assembling chaperones (PACs) (52, 53) were co-purified. It seems therefore that our technique might also be of interest for the study of mammalian 20 S proteasome biogenesis. Interestingly we could identify PI31 with a good sequence coverage of 61%. This well known proteasome inhibitor was not reported as a PIP using 19 S epitope tagging strategies possibly because this protein directly interacts with the 20 S proteasome (41) and may not belong to proteasome complexes involving 19 S activators.
We have identified several proteins belonging or related to the UPP that have been reported previously as human PIPs using 19 S tagging strategies (31, 32, 46, 54). This confirms the validity of our strategy to identify physiological PIPs. Other UPP-related proteins identified in this work had only been reported as PIPs in others species, such as Ddi1, a well characterized PIP in yeast (55), and some proteins of the SCF complex interacting with proteasomes in yeast (25) and in Arabidopsis thaliana (56), or had never been reported so far, such as Ring-box protein 1, PACs, HAUSP, HSPA5, and Cullin-associated NEDD8-dissociated protein 1.
Other proteins identified as putative PIPs in this work do not belong to the UPP. Histones (32) or proteins of the actomyosin cytoskeleton signaling pathway (21, 57) have already been observed in TAP purified proteasome preparations or co-immunoprecipitate with proteasomes. Subunit α of the nascent polypeptide-associated complex (NAC) has never been reported as a PIP so far. It contains a ubiquitin-associated (UBA) domain but does not associate with ubiquitin chains in yeast (58). It interacts with nascent polypeptide chains emerging from the ribosome and might link, as eEF1α does, the proteasome with the protein synthesis quality control. Moreover one of our newly identified putative PIPs, activated RNA polymerase II transcription cofactor 4 variant, confirmed the link already reported between the proteasome and RNA polymerase II (32, 59), suggesting that the proteasome plays an important role in transcription. Other proteins have not been reported so far to interact directly or indirectly with proteasomes. They could constitute either functionally active proteasome interactors or proteasome substrates as polyubiquitin chains with Lys48-ubiquitin attachment were detected by mass spectrometry in the gel migration zone corresponding to high molecular mass proteins. Importantly although proteomics quantitative strategies efficiently help in distinguishing contaminants from true complex components, not all specific interactors can be unambiguously determined (60). Moreover although the MCP21 antibody has been shown in previous studies to be highly specific (61), we cannot rule out that our more sensitive detection method may have led to the identification of non-proteasomal proteins cross-reacting with this antibody. Therefore, further studies will be required to validate the candidate interactors.
Discrepancies observed between our results and those published previously might be accounted for by the cell type or the localization specificity of some PIPs. Nuclear proteins, like PA28γ, reported by others (46) have not been identified in erythrocytes, whereas some PIPs identified in this work might be specific to erythrocyte cells. Proteasome complexes and PIPs purification from human erythrocytes, which cannot be undertaken with tagging strategies, has never been reported to our knowledge. The diversity of PIPs in such cells containing no nuclei might be lower than that in other cell types. Another explanation might be the differences in the strategies used. Tagging one 19 S subunit might be more efficient to catch labile and dynamic interactors of 19 S particle than using the 20 S proteasome as a bait especially if no cross-linker is used. On the contrary, our approach should be more appropriate to broaden the study to the overall cellular proteasome complexes. Indeed PA28 or PA200 activators might be associated with specific interactors. Our work can therefore constitute a complementary approach to already published surveys.
Native and Formaldehyde Cross-link Purifications Are Complementary to Identify PIPs—
The improvement in protein recovery and identification due to the use of formaldehyde was measured using the spectral counting approach (40), which showed that formaldehyde efficiently stabilized the association between the 20 S core particle and its regulators. Formaldehyde fulfills several criteria for efficient endogenous protein-protein interactions stabilization (34). Interestingly formaldehyde treatment did not affect the catalytic activity of the purified complex against fluorogenic peptides. Therefore, mild formaldehyde cross-linking might not affect the buried active sites of the 20 S catalytic core and does not apparently perturb the overall structural arrangement of the protease.
The benefit of the cross-linking process in terms of protein linkage stabilization seems to be the most significant for the most remote proteins from the prey protein (α2 subunit of 20 S proteasome). Indeed subunits from the 19 S lid were recovered with significantly higher spectral copies when the cross-linking process was realized contrary to subunits from the base for which the positive effect was less pronounced. Among the base subunits, recoveries of Rpn1 and Rpn2 seem to be roughly the same with and without formaldehyde, which correlates nicely with the latest published results on 19 S base subunit structural arrangements showing that these subunits are the most tightly anchored to the 20 S particle (62). Other major proteins from the UPP exhibited increased recoveries after formaldehyde treatment or were identified only in the cross-linked purified proteasome complexes. These proteins might therefore constitute transient or labile proteasome interactors. This is in accordance with previous results from Wang and Huang (32) showing that hHR23B, USP14, and ubiquitin are dynamic interactors of proteasome using the QTAX (quantitative analysis of tandem affinity-purified in vivo cross-linked (X) protein complexes) strategy.
Our results show, however, that the use of formaldehyde can be detrimental for the detection of several high molecular mass proteins, such as HAUSP (∼130 kDa), PA200 (∼210 kDa), Cullin-1 (∼90 kDa), or ubiquitin ligase E3C (∼120 kDa), or of proteins belonging to high molecular mass complexes such as the four identified proteins from the large SCF ubiquitin ligase complex. These results can be explained by the fact that covalent linkages mediated by formaldehyde treatment might be incompletely broken before SDS-PAGE separation or might induce their precipitation/insolubility.
Finally a total of 17 of 86 proteins identified were specific to the formaldehyde-treated purified sample, whereas 13 were specific to the non-treated purified one. This seems to indicate that both native and cross-linked approaches might be necessary to increase the number of identified proteins. This conclusion can be of particular importance for the development of optimized strategies for new PIP identification in other human biological samples.
HAUSP Is a Newly Identified Proteasome-associated DUB—
HAUSP, a processive DUB, was unambiguously identified as a PIP in human erythrocytes, and a reverse immunoprecipitation experiment using HAUSP as a bait confirmed this biological interaction. The question to be solved now is “what is the role of HAUSP within proteasome complexes?”
Three DUBs only, Rpn11, ubiquitin protease 14, and ubiquitin carboxyl hydrolase 37 (UCH37), have been reported to associate to proteasomes so far. They are abundant components of proteasome complexes, but they are not substrate-specific, and their main role is to deubiquitinate substrates fated to proteolysis and to regenerate ubiquitin (63).
HAUSP, on the contrary, probably has a selective set of substrates (64). Indeed its removal using RNA interference-based strategies does not lead to global changes in ubiquitination (64) even if it has major implications on cellular growth through action on the p53 pathway (65). Moreover HAUSP is reported to deubiquitinate its substrates, like p53 and MDM2 (66), so that they can escape from proteasome proteolysis.
The binding of HAUSP to the proteasome might enhance its deubiquitinating activity as it demonstrated a 300-fold increase in the in vitro activity of Ubp6, the yeast orthologue of mammalian USP14, when bound to the proteasome (27). Ubp6 interacts with the Rpn1 subunit of the 26 S proteasome through its amino-terminal ubiquitin-like domain (67). Therefore one cannot exclude that HAUSP could also interact with the 19 S regulatory complex through its recently predicted four carboxyl-terminal ubiquitin-like domains (68).
The presence of HAUSP in proteasome complexes might be a mean to regulate the activity and the efficiency of this important DUB. Indeed the probability to find its substrates might be very high in the proteasome vicinity. Therefore the inhibition of this interaction might constitute a new therapeutic strategy especially in hematopoietic tumors where mutations on the p53 gene are rare compared with solid tumors (69). Future investigations will be necessary to characterize the domains of the enzyme responsible for its presence in proteasome complexes. Identification of proteins associated to HAUSP will also represent an important step in the understanding of its role in the degradation machinery.
Finally our purification strategy represents a precious tool to identify new physiological PIPs. When associated to relative proteomics quantitation approaches, it could be used to perform differential analysis of proteasome networks in particular cellular physiological states so that the biological significance of HAUSP and other PIPs could be better understood.
Supplementary Material
Acknowledgments
We are very grateful to Dr. Monique Erard and to David Bouyssié for generous help in bioinformatics and to Marie Anceaux for contribution to the revision experiments.
Footnotes
Published, MCP Papers in Press, February 3, 2009, DOI 10.1074/mcp.M800193-MCP200
The abbreviations used are: UPP, ubiquitin-proteasome pathway; PIP, proteasome-interacting protein; MFPaQ, Mascot file parsing and quantification; HAUSP, herpes virus-associated ubiquitin-specific protease; DUB, deubiquitinating enzyme; ChT, chymotrypsin; Suc, succinyl; AMC, 7-amino-4-methylcoumarin; E3, ubiquitin-protein isopeptide ligase; HSP, heat shock protein; TAP, tandem affinity purification; PAC, proteasome-assembling chaperone; SCF, Skp-Cullin-F-box.
This work was supported, in part, by grants from the “Fondation pour la Recherche Médicale” (contrat “Grands Equipements”), the Génopole Toulouse Midi-Pyrénées (program Biologie-Santé), the Région Midi-Pyrénées, and the Agence Nationale de la Recherche.
The on-line version of this article (available at http://www.mcponline.org) contains supplemental material.
REFERENCES
- 1.Hershko, A., and Ciechanover, A. ( 1998) The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 [DOI] [PubMed] [Google Scholar]
- 2.Glickman, M. H., and Ciechanover, A. ( 2002) The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol. Rev. 82, 373–428 [DOI] [PubMed] [Google Scholar]
- 3.Ciechanover, A., Orian, A., and Schwartz, A. L. ( 2000) Ubiquitin-mediated proteolysis: biological regulation via destruction. BioEssays 22, 442–451 [DOI] [PubMed] [Google Scholar]
- 4.Goldberg, A. L. ( 2007) On prions, proteasomes, and mad cows. N. Engl. J. Med. 357, 1150–1152 [DOI] [PubMed] [Google Scholar]
- 5.Mani, A., and Gelmann, E. P. ( 2005) The ubiquitin-proteasome pathway and its role in cancer. J. Clin. Oncol. 23, 4776–4789 [DOI] [PubMed] [Google Scholar]
- 6.Adams, J. ( 2004) The development of proteasome inhibitors as anticancer drugs. Cancer Cell 5, 417–421 [DOI] [PubMed] [Google Scholar]
- 7.Glickman, M. H., Rubin, D. M., Coux, O., Wefes, I., Pfeifer, G., Cjeka, Z., Baumeister, W., Fried, V. A., and Finley, D. ( 1998) A subcomplex of the proteasome regulatory particle required for ubiquitin-conjugate degradation and related to the COP9-signalosome and eIF3. Cell 94, 615–623 [DOI] [PubMed] [Google Scholar]
- 8.Hirano, Y., Murata, S., and Tanaka, K. ( 2005) Large- and small-scale purification of mammalian 26S proteasomes. Methods Enzymol. 399, 227–240 [DOI] [PubMed] [Google Scholar]
- 9.Groll, M., Ditzel, L., Lowe, J., Stock, D., Bochtler, M., Bartunik, H. D., and Huber, R. ( 1997) Structure of 20S proteasome from yeast at 2.4 Å resolution. Nature 386, 463–471 [DOI] [PubMed] [Google Scholar]
- 10.Gaczynska, M., Rock, K. L., Spies, T., and Goldberg, A. L. ( 1994) Peptidase activities of proteasomes are differentially regulated by the major histocompatibility complex-encoded genes for LMP2 and LMP7. Proc. Natl. Acad. Sci. U. S. A. 91, 9213–9217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeMartino, G. N., Moomaw, C. R., Zagnitko, O. P., Proske, R. J., Chu-Ping, M., Afendis, S. J., Swaffield, J. C., and Slaughter, C. A. ( 1994) PA700, an ATP-dependent activator of the 20 S proteasome, is an ATPase containing multiple members of a nucleotide-binding protein family. J. Biol. Chem. 269, 20878–20884 [PubMed] [Google Scholar]
- 12.Lam, Y. A., Lawson, T. G., Velayutham, M., Zweier, J. L., and Pickart, C. M. ( 2002) A proteasomal ATPase subunit recognizes the polyubiquitin degradation signal. Nature 416, 763–767 [DOI] [PubMed] [Google Scholar]
- 13.Mayor, T., Lipford, J. R., Graumann, J., Smith, G. T., and Deshaies, R. J. ( 2005) Analysis of polyubiquitin conjugates reveals that the Rpn10 substrate receptor contributes to the turnover of multiple proteasome targets. Mol. Cell. Proteomics 4, 741–751 [DOI] [PubMed] [Google Scholar]
- 14.Kohler, A., Cascio, P., Leggett, D. S., Woo, K. M., Goldberg, A. L., and Finley, D. ( 2001) The axial channel of the proteasome core particle is gated by the Rpt2 ATPase and controls both substrate entry and product release. Mol. Cell 7, 1143–1152 [DOI] [PubMed] [Google Scholar]
- 15.Verma, R., Aravind, L., Oania, R., McDonald, W. H., Yates, J. R., III, Koonin, E. V., and Deshaies, R. J. ( 2002) Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 298, 611–615 [DOI] [PubMed] [Google Scholar]
- 16.Fu, H., Reis, N., Lee, Y., Glickman, M. H., and Vierstra, R. D. ( 2001) Subunit interaction maps for the regulatory particle of the 26S proteasome and the COP9 signalosome. EMBO J. 20, 7096–7107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shibatani, T., Carlson, E. J., Larabee, F., McCormack, A. L., Fruh, K., and Skach, W. R. ( 2006) Global organization and function of mammalian cytosolic proteasome pools: Implications for PA28 and 19S regulatory complexes. Mol. Biol. Cell 17, 4962–4971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmidt, M., Hanna, J., Elsasser, S., and Finley, D. ( 2005) Proteasome-associated proteins: regulation of a proteolytic machine. Biol. Chem. 386, 725–737 [DOI] [PubMed] [Google Scholar]
- 19.Demartino, G. N., and Gillette, T. G. ( 2007) Proteasomes: machines for all reasons. Cell 129, 659–662 [DOI] [PubMed] [Google Scholar]
- 20.Yang, P., Fu, H., Walker, J., Papa, C. M., Smalle, J., Ju, Y. M., and Vierstra, R. D. ( 2004) Purification of the Arabidopsis 26 S proteasome: biochemical and molecular analyses revealed the presence of multiple isoforms. J. Biol. Chem. 279, 6401–6413 [DOI] [PubMed] [Google Scholar]
- 21.Horiguchi, R., Dohra, H., and Tokumoto, T. ( 2006) Comparative proteome analysis of changes in the 26S proteasome during oocyte maturation in goldfish. Proteomics 6, 4195–4202 [DOI] [PubMed] [Google Scholar]
- 22.Glickman, M. H., Rubin, D. M., Fried, V. A., and Finley, D. ( 1998) The regulatory particle of the Saccharomyces cerevisiae proteasome. Mol. Cell. Biol. 18, 3149–3162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Papa, F. R., Amerik, A. Y., and Hochstrasser, M. ( 1999) Interaction of the Doa4 deubiquitinating enzyme with the yeast 26S proteasome. Mol. Biol. Cell 10, 741–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoffman, L., Pratt, G., and Rechsteiner, M. ( 1992) Multiple forms of the 20 S multicatalytic and the 26 S ubiquitin/ATP-dependent proteases from rabbit reticulocyte lysate. J. Biol. Chem. 267, 22362–22368 [PubMed] [Google Scholar]
- 25.Verma, R., Chen, S., Feldman, R., Schieltz, D., Yates, J., Dohmen, J., and Deshaies, R. J. ( 2000) Proteasomal proteomics: identification of nucleotide-sensitive proteasome-interacting proteins by mass spectrometric analysis of affinity-purified proteasomes. Mol. Biol. Cell 11, 3425–3439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Babbitt, S. E., Kiss, A., Deffenbaugh, A. E., Chang, Y. H., Bailly, E., Erdjument-Bromage, H., Tempst, P., Buranda, T., Sklar, L. A., Baumler, J., Gogol, E., and Skowyra, D. ( 2005) ATP hydrolysis-dependent disassembly of the 26S proteasome is part of the catalytic cycle. Cell 121, 553–565 [DOI] [PubMed] [Google Scholar]
- 27.Leggett, D. S., Hanna, J., Borodovsky, A., Crosas, B., Schmidt, M., Baker, R. T., Walz, T., Ploegh, H., and Finley, D. ( 2002) Multiple associated proteins regulate proteasome structure and function. Mol. Cell 10, 495–507 [DOI] [PubMed] [Google Scholar]
- 28.Guerrero, C., Tagwerker, C., Kaiser, P., and Huang, L. ( 2006) An integrated mass spectrometry-based proteomic approach: quantitative analysis of tandem affinity-purified in vivo cross-linked protein complexes (QTAX) to decipher the 26 S proteasome-interacting network. Mol. Cell. Proteomics 5, 366–378 [DOI] [PubMed] [Google Scholar]
- 29.Cagney, G., Uetz, P., and Fields, S. ( 2001) Two-hybrid analysis of the Saccharomyces cerevisiae 26S proteasome. Physiol. Genomics 7, 27–34 [DOI] [PubMed] [Google Scholar]
- 30.Hamazaki, J., Iemura, S., Natsume, T., Yashiroda, H., Tanaka, K., and Murata, S. ( 2006) A novel proteasome interacting protein recruits the deubiquitinating enzyme UCH37 to 26S proteasomes. EMBO J. 25, 4524–4536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qiu, X. B., Ouyang, S. Y., Li, C. J., Miao, S., Wang, L., and Goldberg, A. L. ( 2006) hRpn13/ADRM1/GP110 is a novel proteasome subunit that binds the deubiquitinating enzyme, UCH37. EMBO J. 25, 5742–5753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang, X., and Huang, L. ( 2008) Identifying dynamic interactors of protein complexes by quantitative mass spectrometry. Mol. Cell. Proteomics 7, 46–57 [DOI] [PubMed] [Google Scholar]
- 33.Gingras, A. C., Gstaiger, M., Raught, B., and Aebersold, R. ( 2007) Analysis of protein complexes using mass spectrometry. Nat. Rev. Mol. Cell Biol. 8, 645–654 [DOI] [PubMed] [Google Scholar]
- 34.Vasilescu, J., Guo, X., and Kast, J. ( 2004) Identification of protein-protein interactions using in vivo cross-linking and mass spectrometry. Proteomics 4, 3845–3854 [DOI] [PubMed] [Google Scholar]
- 35.Hartmann-Petersen, R., Tanaka, K., and Hendil, K. B. ( 2001) Quaternary structure of the ATPase complex of human 26S proteasomes determined by chemical cross-linking. Arch. Biochem. Biophys. 386, 89–94 [DOI] [PubMed] [Google Scholar]
- 36.Bousquet-Dubouch, M. P., Uttenweiler-Joseph, S., Ducoux-Petit, M., Matondo, M., Monsarrat, B., and Burlet-Schiltz, O. ( 2008) Purification and proteomic analysis of 20S proteasomes from human cells. Methods Mol. Biol. 432, 301–320 [DOI] [PubMed] [Google Scholar]
- 37.Wilm, M., Shevchenko, A., Houthaeve, T., Breit, S., Schweigerer, L., Fotsis, T., and Mann, M. ( 1996) Femtomole sequencing of proteins from polyacrylamide gels by nano-electrospray mass spectrometry. Nature 379, 466–469 [DOI] [PubMed] [Google Scholar]
- 38.Bouyssie, D., de Peredo, A. G., Mouton, E., Albigot, R., Roussel, L., Ortega, N., Cayrol, C., Burlet-Schiltz, O., Girard, J. P., and Monsarrat, B. ( 2007) Mascot file parsing and quantification (MFPaQ), a new software to parse, validate, and quantify proteomics data generated by ICAT and SILAC mass spectrometric analyses: application to the proteomics study of membrane proteins from primary human endothelial cells. Mol. Cell. Proteomics 6, 1621–1637 [DOI] [PubMed] [Google Scholar]
- 39.Claverol, S., Burlet-Schiltz, O., Girbal-Neuhauser, E., Gairin, J. E., and Monsarrat, B. ( 2002) Mapping and structural dissection of human 20 S proteasome using proteomic approaches. Mol. Cell. Proteomics 1, 567–578 [DOI] [PubMed] [Google Scholar]
- 40.Liu, H., Sadygov, R. G., and Yates, J. R., III ( 2004) A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 76, 4193–4201 [DOI] [PubMed] [Google Scholar]
- 41.Zaiss, D. M., Standera, S., Kloetzel, P. M., and Sijts, A. J. ( 2002) PI31 is a modulator of proteasome formation and antigen processing. Proc. Natl. Acad. Sci. U. S. A. 99, 14344–14349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kleijnen, M. F., Roelofs, J., Park, S., Hathaway, N. A., Glickman, M., King, R. W., and Finley, D. ( 2007) Stability of the proteasome can be regulated allosterically through engagement of its proteolytic active sites. Nat. Struct. Mol. Biol. 14, 1180–1188 [DOI] [PubMed] [Google Scholar]
- 43.Denis, N. J., Vasilescu, J., Lambert, J. P., Smith, J. C., and Figeys, D. ( 2007) Tryptic digestion of ubiquitin standards reveals an improved strategy for identifying ubiquitinated proteins by mass spectrometry. Proteomics 7, 868–874 [DOI] [PubMed] [Google Scholar]
- 44.Nielsen, M. L., Vermeulen, M., Bonaldi, T., Cox, J., Moroder, L., and Mann, M. ( 2008) Iodoacetamide-induced artifact mimics ubiquitination in mass spectrometry. Nat. Methods 5, 459–460 [DOI] [PubMed] [Google Scholar]
- 45.Pickart, C. M. ( 2004) Back to the future with ubiquitin. Cell 116, 181–190 [DOI] [PubMed] [Google Scholar]
- 46.Wang, X., Chen, C. F., Baker, P. R., Chen, P. L., Kaiser, P., and Huang, L. ( 2007) Mass spectrometric characterization of the affinity-purified human 26S proteasome complex. Biochemistry 46, 3553–3565 [DOI] [PubMed] [Google Scholar]
- 47.Imai, J., Maruya, M., Yashiroda, H., Yahara, I., and Tanaka, K. ( 2003) The molecular chaperone Hsp90 plays a role in the assembly and maintenance of the 26S proteasome. EMBO J. 22, 3557–3567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nijman, S. M., Luna-Vargas, M. P., Velds, A., Brummelkamp, T. R., Dirac, A. M., Sixma, T. K., and Bernards, R. ( 2005) A genomic and functional inventory of deubiquitinating enzymes. Cell 123, 773–786 [DOI] [PubMed] [Google Scholar]
- 49.Brooks, C. L., and Gu, W. ( 2006) p53 ubiquitination: Mdm2 and beyond. Mol. Cell 21, 307–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Everett, R. D., Meredith, M., Orr, A., Cross, A., Kathoria, M., and Parkinson, J. ( 1997) A novel ubiquitin-specific protease is dynamically associated with the PML nuclear domain and binds to a herpesvirus regulatory protein. EMBO J. 16, 1519–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Becker, K., Marchenko, N. D., Maurice, M., and Moll, U. M. ( 2007) Hyperubiquitylation of wild-type p53 contributes to cytoplasmic sequestration in neuroblastoma. Cell Death Differ. 14, 1350–1360 [DOI] [PubMed] [Google Scholar]
- 52.Hirano, Y., Hendil, K. B., Yashiroda, H., Iemura, S., Nagane, R., Hioki, Y., Natsume, T., Tanaka, K., and Murata, S. ( 2005) A heterodimeric complex that promotes the assembly of mammalian 20S proteasomes. Nature 437, 1381–1385 [DOI] [PubMed] [Google Scholar]
- 53.Hirano, Y., Hayashi, H., Iemura, S., Hendil, K. B., Niwa, S., Kishimoto, T., Kasahara, M., Natsume, T., Tanaka, K., and Murata, S. ( 2006) Cooperation of multiple chaperones required for the assembly of mammalian 20S proteasomes. Mol. Cell 24, 977–984 [DOI] [PubMed] [Google Scholar]
- 54.Yao, T., Song, L., Xu, W., DeMartino, G. N., Florens, L., Swanson, S. K., Washburn, M. P., Conaway, R. C., Conaway, J. W., and Cohen, R. E. ( 2006) Proteasome recruitment and activation of the Uch37 deubiquitinating enzyme by Adrm1. Nat. Cell Biol. 8, 994–1002 [DOI] [PubMed] [Google Scholar]
- 55.Kaplun, L., Tzirkin, R., Bakhrat, A., Shabek, N., Ivantsiv, Y., and Raveh, D. ( 2005) The DNA damage-inducible UbL-UbA protein Ddi1 participates in Mec1-mediated degradation of Ho endonuclease. Mol. Cell. Biol. 25, 5355–5362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Farras, R., Ferrando, A., Jasik, J., Kleinow, T., Okresz, L., Tiburcio, A., Salchert, K., del Pozo, C., Schell, J., and Koncz, C. ( 2001) SKP1-SnRK protein kinase interactions mediate proteasomal binding of a plant SCF ubiquitin ligase. EMBO J. 20, 2742–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Olink-Coux, M., Arcangeletti, C., Pinardi, F., Minisini, R., Huesca, M., Chezzi, C., and Scherrer, K. ( 1994) Cytolocation of prosome antigens on intermediate filament subnetworks of cytokeratin, vimentin and desmin type. J. Cell Sci. 107, 353–366 [PubMed] [Google Scholar]
- 58.Andersen, K. M., Semple, C. A., and Hartmann-Petersen, R. ( 2007) Characterisation of the nascent polypeptide-associated complex in fission yeast. Mol. Biol. Rep. 34, 275–281 [DOI] [PubMed] [Google Scholar]
- 59.Gillette, T. G., Gonzalez, F., Delahodde, A., Johnston, S. A., and Kodadek, T. ( 2004) Physical and functional association of RNA polymerase II and the proteasome. Proc. Natl. Acad. Sci. U. S. A. 101, 5904–5909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Trinkle-Mulcahy, L., Boulon, S., Lam, Y. W., Urcia, R., Boisvert, F. M., Vandermoere, F., Morrice, N. A., Swift, S., Rothbauer, U., Leonhardt, H., and Lamond, A. ( 2008) Identifying specific protein interaction partners using quantitative mass spectrometry and bead proteomes. J. Cell Biol. 183, 223–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hendil, K. B., Kristensen, P., and Uerkvitz, W. ( 1995) Human proteasomes analysed with monoclonal antibodies. Biochem. J. 305, 245–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rosenzweig, R., Osmulski, P. A., Gaczynska, M., and Glickman, M. H. ( 2008) The central unit within the 19S regulatory particle of the proteasome. Nat. Struct. Mol. Biol. 15, 573–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Koulich, E., Li, X., and DeMartino, G. N. ( 2008) Relative structural and functional roles of multiple deubiquitylating proteins associated with mammalian 26S proteasome. Mol. Biol. Cell 19, 1072–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kessler, B. M., Fortunati, E., Melis, M., Pals, C. E., Clevers, H., and Maurice, M. M. ( 2007) Proteome changes induced by knock-down of the deubiquitylating enzyme HAUSP/USP7. J. Proteome Res. 6, 4163–4172 [DOI] [PubMed] [Google Scholar]
- 65.Meulmeester, E., Maurice, M. M., Boutell, C., Teunisse, A. F., Ovaa, H., Abraham, T. E., Dirks, R. W., and Jochemsen, A. G. ( 2005) Loss of HAUSP-mediated deubiquitination contributes to DNA damage-induced destabilization of Hdmx and Hdm2. Mol. Cell 18, 565–576 [DOI] [PubMed] [Google Scholar]
- 66.Brooks, C. L., Li, M., Hu, M., Shi, Y., and Gu, W. ( 2007) The p53-Mdm2-HAUSP complex is involved in p53 stabilization by HAUSP. Oncogene 26, 7262–7266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hanna, J., Hathaway, N. A., Tone, Y., Crosas, B., Elsasser, S., Kirkpatrick, D. S., Leggett, D. S., Gygi, S. P., King, R. W., and Finley, D. ( 2006) Deubiquitinating enzyme Ubp6 functions noncatalytically to delay proteasomal degradation. Cell 127, 99–111 [DOI] [PubMed] [Google Scholar]
- 68.Zhu, X., Menard, R., and Sulea, T. ( 2007) High incidence of ubiquitin-like domains in human ubiquitin-specific proteases. Proteins 69, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cheon, K. W., and Baek, K. H. ( 2006) HAUSP as a therapeutic target for hematopoietic tumors. Int. J. Oncol. 28, 1209–1215 [PubMed] [Google Scholar]
- 70.Gorbea, C., Goellner, G. M., Teter, K., Holmes, R. K., and Rechsteiner, M. ( 2004) Characterization of mammalian Ecm29, a 26 S proteasome-associated protein that localizes to the nucleus and membrane vesicles. J. Biol. Chem. 279, 54849–54861 [DOI] [PubMed] [Google Scholar]
- 71.Baumeister, W., Walz, J., Zuhl, F., and Seemuller, E. ( 1998) The proteasome: paradigm of a self-compartmentalizing protease. Cell 92, 367–380 [DOI] [PubMed] [Google Scholar]
- 72.Dubiel, W., Ferrell, K., and Rechsteiner, M. ( 1995) Subunits of the regulatory complex of the 26S protease. Mol. Biol. Rep. 21, 27–34 [DOI] [PubMed] [Google Scholar]
- 73.Finley, D., Tanaka, K., Mann, C., Feldmann, H., Hochstrasser, M., Vierstra, R., Johnston, S., Hampton, R., Haber, J., McCusker, J., Silver, P., Frontali, L., Thorsness, P., Varshavsky, A., Byers, B., Madura, K., Reed, S. I., Wolf, D., Jentsch, S., Sommer, T., Baumeister, W., Goldberg, A., Fried, V., Rubin, D. M., Glickman, M. H., and Toh-e, A. ( 1998) Unified nomenclature for subunits of the Saccharomyces cerevisiae proteasome regulatory particle. Trends Biochem. Sci. 23, 244–245 [DOI] [PubMed] [Google Scholar]
- 74.Dai, R. M., Chen, E., Longo, D. L., Gorbea, C. M., and Li, C. C. ( 1998) Involvement of valosin-containing protein, an ATPase Co-purified with IκBα and 26 S proteasome, in ubiquitin-proteasome-mediated degradation of IκBα. J. Biol. Chem. 273, 3562–3573 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.