

Abstract
Most contemporary accounts of psychopathology acknowledge the importance of both biological and environmental influences on behavior. In developmental psychopathology, multiple etiological mechanisms for psychiatric disturbance are well recognized, including those operating at genetic, neurobiological, and environmental levels of analysis. However, neuroscientific principles are rarely considered in current approaches to prevention or intervention. In this article, we explain why a deeper understanding of the genetic and neural substrates of behavior is essential for the next generation of preventive interventions, and we outline 10 specific reasons why considering biological processes can improve treatment efficacy. Among these, we discuss (a) the role of biomarkers and endophenotypes in identifying those most in need of prevention; (b) implications for treatment of genetic and neural mechanisms of homotypic comorbidity, heterotypic comorbidity, and heterotypic continuity; (c) ways in which biological vulnerabilities moderate the effects of environmental experience; (d) situations in which Biology×Environment interactions account for more variance in key outcomes than main effects; and (e) sensitivity of neural systems, via epigenesis, programming, and neural plasticity, to environmental moderation across the life span. For each of the 10 reasons outlined we present an example from current literature and discuss critical implications for prevention.
Throughout much of the 20th century, psychology was portrayed as a fledgling discipline compared with other physical sciences. Less than five decades ago, the eminent philosopher Thomas Kuhn (1962) described psychology as preparadigmatic, with no established network of widely agreed upon principles or facts. Psychologists of the time were engaged in controversy over the proper approach to understanding human behavior, from unobservable unconscious motives at one end of the continuum to decontextualized operant behaviors at the other. Although the operant approach yielded some degree of prediction and control over behavior, as demonstrated by Hull’s (1943) elaboration of Thorndike’s (1911) law of effect, philosophers of science often compared psychology to physics and chemistry, which are replete with largely indisputable laws and axioms. These laws provide a level of precision in predicting future events that psychology will likely never achieve (see Beauchaine, Gatzke-Kopp, & Mead, 2007;Beauchaine, Lenzenweger, & Waller,2008).
Direct comparisons between psychology and the hard sciences are now recognized as simplistic, given the overwhelming number of causal influences affecting human behavior. Yet criticisms of psychology as a soft science and disagreement over the proper level of analysis for studying human behavior remain. This is particularly evident in clinical psychology, where debates often emerge over appropriate foci of scientific inquiry. Many have argued that clinical psychology should be first and foremost an applied discipline that develops cognitive, behavioral, and social interventions to prevent and treat maladaptive behavior (e.g., Davison, 1998). Some have even argued that focusing on genetic and neurobiological influences on behavior diverts our attention away from social and familial processes that promote psychopathology (e.g., Albee & Joffe, 2004). In contrast, others have advocated for a clinical science that examines genetic and neural mechanisms of vulnerability, assuming that understanding such mechanisms will improve our ability to reduce psychiatric morbidity and mortality (e.g., Beauchaine & Marsh, 2006; Cicchetti & Dawson, 2002; Davidson, Pizzagalli, Nitschke, & Putnam, 2002; Fishbein, 2000; Nelson et al., 2002).
Internecine squabbles over the identity of clinical psychology tend to emerge whenever the discipline reacts, often slowly, to paradigm shifts occurring in other areas of psychology. For example, behavioral principles offered by learning theorists including Hull (1943) and Skinner (1938) led to a gradual shift in clinical thinking that culminated in the 1970s, when the power of operant principles in shaping and changing human behavior was acknowledged. As a result, behaviorism supplanted psychoanalysis as the dominant clinical paradigm of the time.
However, as most readers are undoubtedly aware, the heyday of behaviorism was short lived. During the late 1970s and 1980s, a cognitive revolution swept psychology, shifting emphasis away from strict stimulus–response models of learning toward social–cognitive motivations for behavior. As a result, clinical scientists including Beck (e.g., Beck, Rush, Shaw, & Emery, 1979), Ellis (e.g., 1981), and Linehan (1993) formulated cognitive behavioral therapies for a wide range of psychiatric disorders. This change was less protracted than past paradigm shifts because cognitive models were fully compatible with the behavioral principles that preceded them. The two approaches could therefore be combined into an inclusive set of therapeutic methods. The cognitive behavioral approach that resulted remains the dominant paradigm in clinical psychology today.
By the late 1990s, yet another paradigm shift had permeated most of psychology. Basic scientists had begun using modern neuroscience techniques to probe the genetic and neural correlates of behavior. With sophisticated methods such as molecular genetics, electroencephalography (EEG), functional magnetic resonance imaging (fMRI), and positron emission tomography (PET), scientists were poised to tackle some of the most longstanding questions about learning and behavior. How do genes affect personality? Where are memories stored? How does the human brain learn? What are the brain bases of language? What neural processes give rise to emotions? How does psychopathology develop?
Despite the enormous promise of these methods, many early studies using molecular genetics and imaging technologies were crude because scientists did not yet appreciate the complexity of the systems they were observing. Naive searches for “the gene” controlling complex disorders such as depression and schizophrenia were not uncommon. Similarly, claims were made that the brain loci of mood disorders, anxiety, and emotion had been found. Although such simplistic assertions still appear in the popular press, geneticists and neuroscientists are now well aware that (a) genes do not control behaviors directly, (b) almost all behavioral traits emerge from complex interactions between multiple genes and environments, and (c) the brain bases of both personality and psychopathology are distributed across complex neural networks and are not caused by single loci, except in the most extreme cases of focal lesions. Thus, modern neuroscience is much more sophisticated than its earlier instantiations (Cicchetti & Posner, 2005; Davidson, 2003).
Biology, Neuroscience, and Prevention
The tendency of clinical psychology to respond slowly to paradigm shifts that are embraced by other areas of psychological science is clearly evident in neuroscience. Most clinical psychology programs offer minimal training in the brain bases of behavior, if they offer any such training at all, and clinical neuroscience articles are still a rarity in the Journal of Abnormal Psychology and the Journal of Consulting and Clinical Psychology, flagship journals of the profession. Perhaps of more importance, neuroscientific principles are almost completely absent from current theoretical formulations of prevention and intervention. Although exceptions to this generalization can be found in research on autism spectrum disorders (e.g., Dawson, this issue; Dawson et al., 2002), posttraumatic stress disorder (PTSD; e.g., Bryant, 2006), and borderline pathology (e.g., Schnell & Herpertz, 2007), clinical psychology as a whole has not embraced neuroscience, despite extraordinary advances in our understanding of the brain bases of motivation, emotion, and self-control. Yet dysfunction in one or more of these aspects of behavior is observed in all forms of psychopathology. Even though neuroscientific findings have resulted in rich theoretical models of major psychiatric disorders (Berridge & Robinson, 2003; Davidson et al., 2002; Gatzke-Kopp & Beauchaine, 2007a; Sagvolden, Johansen, Aase, & Russell, 2005), they are usually not considered in clinical prevention or intervention programs.
To be fair, the behavioral and cognitive paradigm shifts described above yielded principles that were much easier to implement in applied clinical settings. Nevertheless, the current under-appreciation of biological processes in prevention and intervention research is striking. Of the few studies that have included such variables, some have explored the effects of treatment on the functioning of biological systems implicated in stress reactivity and self-regulation (e.g., Fisher, Gunnar, Chamberlain, & Reid, 2000), and others have examined the moderating effects of biological variables on treatment outcome. For example, Fishbein, Hyde, Coe, and Paschall (2004) examined the responses of adolescents to a preventive intervention for drug abuse. Skin conductance was included as one measure of emotional functioning, and differentiated in part between responders and nonresponders. Similarly, Raine, Mellingen, Liu, Venables, and Mednick (2003) explored the potential moderating effect of children’s autonomic functioning on schizotypy and antisocial behavior in adolescence and adulthood following a generic preschool prevention program. Although they did not find evidence for moderation, the preschool prevention program did yield significant increases in psychophysiological orienting and arousal (as measured by skin conductance and EEG) at age 11, perhaps suggesting enhanced information processing abilities (Raine et al., 2001). Such use of biological markers in longitudinal outcome research represents a first step toward a more integrated prevention science.
In addition to exploring moderators of outcome, some researchers have included biological variables as indicators of vulnerability to psychopathology. Following arguments that etiological heterogeneity among high-risk groups should predict differential treatment responses (e.g., Beauchaine & Marsh, 2006; Cicchetti & Rogosch, 2002), researchers have sought to identify particularly vulnerable individuals based on specific genetic and neurobiological markers (see, e.g., Gottesman & Gould, 2003). Bryant (2006), for example, explored heart rate as a marker of risk for PTSD following trauma. After reviewing a number of studies, he concluded that heart rate, in combination with diagnostic status shortly after the trauma (whether or not participants met criteria for acute stress disorder), predicted the emergence of PTSD. In molecular genetics research, Young, Lawford, Nutting, and Noble (2004) conducted a series of meta-analyses examining the role of the dopamine receptor D2 (DRD2) A1 allele, concluding that it “shows promise as a marker of substance use, and of severe substance misuse in particular”(p.1288). Such findings high-light the potential role of genetic and biological characteristics in identifying individuals who are at increased risk of adverse outcomes, and who are therefore most in need of prevention.
Despite such examples, inclusion of biological processes in prevention research lags well behind recent advances in our understanding of the neurobiological substrates of psychopathology. This may result in part from anachronistic notions about the respective roles of neurobiological and environmental influences on behavior. Indeed, decisions regarding whether and how to incorporate biological variables into prevention research often revolve around the degree to which psychological outcomes are conceptualized as stemming from biological vulnerabilities versus social and environment risk factors.
A recent special issue of the Journal of Primary Prevention explored the question of whether those at risk for psychopathology would be best served by a prevention science that emphasizes biological or psychosocial factors. Joffe (2004) and others argued that emphasizing biological vulnerabilities frames psychological problems as medical illnesses, a comparison that many find unsatisfying, for a number of reasons. For example, Albee and Joffe (2004) assert that there is insufficient evidence of reliable brain abnormalities in individuals with most psychiatric disorders. With the exception of conditions such as Alzheimer disease, they argue that “the evidence for mental disorders being caused by biochemical or structural abnormalities of the brain is generally sparse and inconsistent at best” (p. 425). Similarly, Boyle (2004) stated that there is “no evidence of a causal relationship between schizophrenia diagnoses and any genetic or biochemical event or process” (p. 450).
In our view, such statements (a) oversimplify complex relations between biological vulnerabilities and environmental risk factors in producing psychopathology; (b) place too much emphasis on the main effects of biology and environment, when research in developmental psychopathology indicates that interaction effects often account for more variance in adverse outcomes (see below); and (c) are in some cases clearly inaccurate. For example, a large volume of research indicates that schizophrenia is about 80% heritable (see Rapoport, Addington, & Frangou, 2005), and is caused by genetic and neurobiological processes that give rise to compromises in both the structure and function of the brain (see, e.g., Gottesman & Gould, 2003; Reichenberg & Harvey, 2007). Although debate exists over the precise mechanisms that underlie these structural and functional compromises (e.g., Craddock, O’Donovan, & Owen, 2007; Gottesman & Gould, 2003; McClellan, Susser, & King, 2007), denying that schizophrenia has biological bases can only result from ignoring overwhelming evidence to the contrary, as discussed in later sections.
Arguments against psychopathology as medical illness are also based on the claim that psychiatric disorders do not represent distinct diagnostic entities, but instead reflect points along symptom continua. As noted by Albee and Joffe (2004) and others (see Beauchaine, 2003; Lilienfeld & Marino, 1999), decisions regarding where along such continua normative functioning ends and psychopathology begins are often based more heavily on value judgments than on knowledge about underlying biological mechanisms, clear and specific biobehavioral links, or established causal factors. However, this argument ignores the fact that many life-threatening medical conditions, such as hypertension and type II diabetes, are also expressed along symptom continua and have multiple etiological influences, both biological and environmental. Nevertheless, preventive interventions targeting those at highest biological risk for these medical conditions have saved uncounted numbers of lives, and can delay the onset of functional impairment by decades.
Related arguments against incorporating biological processes into prevention research also stem from beliefs that doing so wrongly reinforces a medical or “defect” model of psychopathology in which “all mental illnesses are caused by biological, biochemical, and/or other organic defects” (Albee & Joffe, 2004, p. 424). Yet contemporary models of psychopathology acknowledge the importance of both neurobiological and environmental influences on behavior. In developmental psychopathology, multiple etiologies for many psychiatric conditions are well recognized. Different individuals presenting with what appears to be a single psychiatric disorder can arrive at similar levels of functioning via diverse equifinal outcomes, some of which include strong biological vulnerability and less environmental risk, and others of which include less biological vulnerability and strong environmental risk (see, e.g., Beauchaine & Marsh, 2006; Cicchetti & Rogosch, 1996). This is why it is critically important to measure both classes of variables and their interactions in clinical research.
Opponents of biological approaches to prevention and intervention also argue that by emphasizing genetic and neurobiological processes, we divert attention and resources away from important psychosocial causes of maladjustment, such as stress, poverty, and family interactions: “If all ‘mental illnesses’ result from pathologies in the brain … then efforts at prevention need pay little attention to the social environment in which the affected person lives and has developed” (Albee & Joffe, 2004, p. 434). Some authors have suggested that clinical research and prevention programs should therefore focus exclusively on environmental risks, and that individuals with psychiatric disorders are using normal mechanisms to adjust to aberrant environmental inputs (Silvestri & Joffe, 2004).
Other researchers have offered compelling reasons to include genetic and other biological processes in prevention research (e.g., Agrawal & Hirsch, 2004; Ames & McBride, 2006; Fishbein, 2000; Gottlieb & Willoughby, 2006; Gunnar & Fisher, 2006). Perhaps most fundamentally, assessing biological variables advances our understanding of the etiological complexities of developing psychopathology, eventually leading to targeted interventions (Beauchaine & Marsh, 2006). For example, although early conceptualizations of schizophrenia posited single genetic loci (e.g., Meehl, 1962), extensive research on the biological substrates of the disorder has revealed complex polygenic influences that interact with environmental risk to potentiate psychiatric morbidity (e.g., Gottesman & Gould, 2003). As we outline in detail below, by specifying behavioral and biological endophenotypes that mark this genetic risk (e.g., Erlenmeyer-Kimling, Golden, & Cornblatt, 1989; Tyrka et al., 1995), prevention researchers can now identify children and adolescents who are particularly vulnerable to developing schizophrenia, and implement preventive interventions that reduce the likelihood of future psychosis and improve long-term prognosis (see, e.g., Beauchaine & Marsh, 2006; McGorry et al., 2002).
Similar interactive complexities have been identified at neurobiological levels of analysis. For example, psychophysiological research indicates that basic motivational and emotion regulatory mechanisms interact with one another to potentiate disorders of impulse control including attention-deficit/hyperactivity disorder (ADHD), conduct disorder (CD), and intentional self-injury (Beauchaine, Katkin, Strassberg, & Snarr, 2001; Crowell, Beauchaine, McCauley, Smith, Vasilev, & Stevens, 2008; Gatzke-Kopp et al., 2007; Shannon, Beauchaine, Brenner, Neuhaus, & Gatzke-Kopp, 2007). These biobehavioral vulnerabilities also interact with environmental risk factors to predict adverse outcomes (Beauchaine et al., 2007; Patterson, DeGarmo, & Knutson, 2000; Snyder, Schrepferman, & St. Peter, 1997).
Finally, biologically informed research has already yielded tremendous advances in the prevention of some psychiatric disorders. Once thought to be solely the result of inadequate parenting (Klinger, Dawson, & Renner, 2003), autism spectrum disorders are now recognized to stem from multiple genes (Schellenberg et al., 2006), yet early environmental interventions for those exhibiting endophenotypic markers of risk offer profound protective effects for many children with the disorder (Dawson, this issue; Dawson & Zanolli, 2003).
Developmental Psychopathology and Prevention
Several tenets of the developmental psychopathology perspective are relevant to this discussion. Developmental psychopathologists acknowledge that both biological vulnerabilities and environmental risk factors contribute to adjustment and maladjustment, and that apparent maladaptation can often be understood as adaptation to noxious environmental contexts (e.g., Cicchetti, 2006). This framework emphasizes interactions between individuals and their environments (Rutter & Sroufe, 2000; Sroufe & Rutter, 1984), which occur at multiple levels of analysis, including genetic, epigenetic, neurobiological, familial, and social (Cicchetti, 2007; Cicchetti & Dawson, 2002; Moffitt, Caspi, & Rutter, 2006). This is a transactional approach, as influence flows across all levels of analysis. Family environments, social conditions, and psychological processes all affect biological processes, and biological functioning and predispositions influence the ways in which an individual selects and shapes the environment (see Rutter, 2002, 2007).
In acknowledging these interactive processes, developmental psychopathologists must also recognize the probabilistic nature of predicting adverse outcomes. Psychopathology results from unique combinations of environmental risk factors, genetic vulnerabilities, and biological processes specific to each individual. The same set of vulnerabilities may be associated with various outcomes depending on a multitude of intervening risk factors (multifinality), and individuals can arrive at the same outcome via different combinations of vulnerability and risk (equifinality; Cicchetti & Rogosch, 1996). Multifinality is demonstrated in studies of maternal depression, where children are at increased risk of both depression and CD (Kopp & Beauchaine, 2007). Thus, the same risk factor operates differently for different children. Equifinality is also observed in the development of CD. Adolescents who meet criteria form a heterogeneous group, often varying in both developmental history and symptom presentation (Hinshaw & Lee, 2003; Moffitt & Caspi, 2001).
Exploring biological processes is fundamental to the developmental psychopathology framework. For the past two decades, developmental psychopathologists have emphasized that including biological variables in studies of psychopathology will improve our understanding of risk factors and predictors of later functioning, both independently and in combination with various environmental and psychosocial characteristics. The very concept of prevention implies inferred risk to an individual, which may lead to a harmful outcome, either directly or through another potentiating risk factor (e.g., early-onset CD potentiates risk for substance use disorders [SUDs]).
In targeted prevention programs, individuals are selected for treatment based on exposure to one or more risk factors that are known to promote psychopathology. In the traditional approach to prevention research, these risk exposures are usually environmental (e.g., family history, neighborhood), although individual characteristics are sometimes targeted (e.g., IQ, age, gender). Yet, biological vulnerabilities may be equally important. Recent research indicates quite clearly that an individual’s genetic constitution may confer risk directly or through interactions with adverse environments (e.g., Caspi et al., 2002; Cicchetti, 2007; Jaffee et al., 2005).
It should also be noted, however, that biological markers of vulnerability are rarely deterministic. As we discuss in more detail, neurobiological systems that are implicated in vulnerability to psychopathology are often malleable (e.g., Raine et al., 2001), especially early in life, which is sometimes overlooked by opponents of biological research. Thus, identification of biologically based vulnerabilities may provide fruitful targets for both prevention and intervention. Similarly, biological variables that moderate the relationship between various risk factors and adverse outcomes should be targets of treatment when possible.
From this discussion it should be clear that we strongly favor an approach to prevention and intervention that includes consideration and/or assessment of biological vulnerabilities, environmental risk factors, and their interactions. We state at the outset that we are not suggesting that biological variables be measured in all prevention and intervention trials, although there are many cases in which measuring appropriate biological systems will be fruitful. Rather, we suggest that the efficacy and/or efficiency of many treatment programs will be improved by considering biological mechanisms of psychopathology. In the following sections we provide 10 compelling reasons for such an inclusive approach, most of which are supported by one or more examples from existing research. Readers should note that any one of these items could be addressed in a full-length article, so our descriptions are necessarily limited in scope. Although several of these items are interrelated, points of emphasis vary enough to warrant separate sections for each.
Ten Good Reasons to Consider Biological Variables in Prevention and Intervention Research
Markers of biological vulnerability can identify those at greatest risk for psychopathology
Over four decades ago, Dawes and Meehl (1966) suggested that premorbid identification of individuals at risk for psychopathology should be a high priority because it is a necessary antecedent to prevention. Findings discussed briefly above suggest that by measuring relevant biological markers and/or endophenotypes (for discussion of the distinction between biomarkers and endophenotypes, see Gould & Gottesman, 2006), we may be able to isolate those who are at risk for future psychopathology, and develop prevention and intervention programs targeting these individuals. Blanket prevention programs that enroll children at all levels of risk are often inefficient, and can result in underestimates of intervention effects because significant behavior change is not expected among children who are not at risk for psychopathology.
Research addressing biological risk among the offspring of a parent with schizophrenia provides a particularly compelling example of using endophenotypes to identify vulnerable children premorbidly (Beauchaine & Marsh, 2006). By performing taxometric analyses on measures of sustained visual attention, neuromotor performance, and intelligence, Erlenmeyer-Kimling et al. (1989) identified a discrete group of 7- to 12-year-old children of a parent with schizophrenia who were at especially high risk of developing the disorder. Although the base rate of genetic risk for schizophrenia (schizotypy) is 5% in the general population (Blanchard, Gangestad, Brown, & Horan, 2000; Golden & Meehl, 1979; Korfine & Lenzenweger, 1995; Lenzenweger, 1999; Lenzenweger & Korfine, 1992), 47% of children with an affected parent were members of the identified schizotypy taxon, compared with the expected 4% of controls. Of more importance, 43% of the schizotypy group were either hospitalized or had received significant treatment by age 22–29. Similar results were reported by Tyrka et al. (1995), who used behavioral data derived from school reports and psychiatric interviews to identify a discrete schizotypy group of 10- to 19-year-old offspring of mothers with schizophrenia. The 48% taxon base rate was nearly identical to that reported by Erlenmeyer-Kimling et al. Moreover, 40% of taxon group members were diagnosed with a schizophrenia spectrum disorder 24–27 years later. Thus, taxometric analyses of selected behavioral and endophenotypic markers of genetic risk can identify particularly vulnerable individuals prospectively.
Implications for prevention and intervention
The importance of these findings for prevention is difficult to overstate. Blanket prevention programs for all children of parents with schizophrenia are inefficient because only 10–15% eventually develop a schizophrenia spectrum disorder (see Cornblatt, Obuchowski, Roberts, Pollack, & Erlenmeyer-Kimling, 1999). Yet in the two studies cited above, taxon group members were at nearly 50% risk. This level of vulnerability renders schizophrenia (and other) prevention programs much more pragmatic (Cornblatt, 2001). Furthermore, advances in identification of endophenotypes that mark schizophrenia liability, including impaired attention, saccadic intrusions in smooth pursuit eye tracking, and spatial working memory deficits (Cornblatt & Malhotra, 2001; Glahn et al., 2003; Lenzenweger, McLachlan, & Rubin, 2007; Reichenberg& Harvey, 2007; Ross, 2003), should facilitate premorbid identification at even younger ages. The earlier environmentally focused interventions are implemented, the more likely they are to prevent the onset of schizophrenia because accumulated environmental risk across development potentiates genetic vulnerability (see Goldsmith, Gottesman, & Lemery, 1997; Gottesman & Gould, 2003; Rutter et al., 1997).
Although these findings have not been incorporated into prevention trials to date, considerable advances in the prevention of schizophrenia have been reported. Prevention programs that include both cognitive behavioral and pharmacological components appear to be especially promising for those at risk for schizophrenia. McGorry et al. (2002) demonstrated that cognitive behavioral therapy (CBT), combined with a low dose of Risperidone (1–2 mg/day) substantially reduced the onset of first-episode psychosis in high-risk patients who had a positive family history and incipient but subthreshold symptoms. Those who took a low dose of Risperidone and participated in CBT were 95% psychosis-free 3 years later, compared with only 40% of patients who did not adhere to the Risperidone treatment and 30% of patients who received a typical needs-based intervention. These findings are important because early treatment of psychosis is associated with improved long-term prognosis (see Cornblatt, Lencz, & Kane, 2001). Similar effects of early intervention on delaying the age of onset for bipolar disorder have recently been described (Chang, Gallelli, & Howe, 2007; Miklowitz, 2007).
Heritable effects on behavior increase across the life span
Behavioral genetics studies have demonstrated increasing heritability coefficients across the life span for a wide range of psychiatric disorders (Lemery & Doelger, 2005). This generalization applies to almost all forms of psychopathology for which heritability has been assessed at different points in development, which (a) bears directly on the often repeated claim that environmental contexts are the primary “cause” of psychopathology (e.g., Albee & Joffe, 2004); (b) has received almost no attention in the clinical psychology and developmental psychopathology literature; (c) has direct implications for interpretations of the relative contributions of biological vulnerability and environmental risk for psychiatric disturbance; and (d) affects the long-term efficacy of prevention programs, which by nature target early environments to effect behavioral change.
A number of examples of rising heritability across development are noteworthy. Twin studies of depression indicate that heritability contributes minimally to symptom expression in childhood (Lemery & Doelger, 2005; Rice, Harold, & Thapar, 2002) yet increases during adolescence (Scourfield et al., 2003). By adulthood, most behavioral genetics studies yield large heritability coefficients for major depression, with nonsignificant environmental effects (Sullivan, Neale, & Kendler, 2000).
Heritability coefficients for eating disorders among females and antisocial behavior among males also increase across the life span (Hicks et al., 2007; Klump, McGue, & Iacono, 2000; Lyons et al., 1995). Furthermore, although environmental factors contribute strongly to the initiation of smoking and drinking, behavioral genetics studies indicate that both smoking maintenance and heavy drinking are accounted for primarily by heritable effects (e.g., Boomsma, Koopsman, Van Doormen, & Orlebeke, 1994; Koopsman, Slutzke, Heath, Neale, & Boomsma, 1999; Koopsman, van Doornen, & Boomsma, 1997; McGue, Iacono, Legrand, & Elkins, 2001; Viken, Kaprio, Koskenvuo, & Rose, 1999).
To explain such findings, researchers have speculated that the nature of psychiatric disorders may be qualitatively different in children than in adolescents and adults (e.g., Klein, Torpey, Bufferd, & Dyson, 2008), different genetic factors operate in childhood versus adolescence (Jacobson, Prescott, & Kendler, 2000), and observed differences in heritability may reflect diverse pathways to psychopathology (e.g., Silberg, Rutter, & Eaves, 2001; Silberg, Rutter, Neale, & Eaves, 2001). Although these and related mechanisms may be at play, developmental increases in heritability coefficients are a mathematical necessity in twin and adoption studies given individual differences in age of onset, even when the etiologies of the psychiatric disorder being assessed are quite similar across members of a population. This is illustrated in Figure 1, which depicts 10 hypothetical twin pairs, all of whom are at high genetic risk for schizophrenia. Because of differences in age of onset, concordance rates rise from childhood through adulthood. Because age of onset is dispersed across many years for most psychiatric disorders as a result of nonshared environmental effects, unmeasured stochastic effects, and allostatic load, heritability coefficients must increase across the life span.1 The only exceptions to this rule are disorders with very early and relatively invariant ages of onset, such as autism.
Figure 1.

A hypothetical distribution of individual differences in age of onset for schizophrenia across 10 twin pairs. Solid bars indicate age of onset for each individual. Concordance is determined by the proportion of the 10 twin pairs who are both afflicted. Because individual differences in age of onset are observed for almost all forms of psychopathology, concordance rates necessarily increase across the life span. Note that the final concordance rate of .80 indicates a highly heritable trait. Although dichotomous outcomes are used for simplicity of presentation, the same argument applies to continuously assessed traits.
Implications for prevention and intervention
There are several potential implications of such developmental increases in heritability for prevention. Although targeted preventions can delay and in some cases offset the emergence of highly heritable psychiatric conditions such as schizophrenia (McGorry et al., 2002), life span increases in heritability suggest that the effectiveness of many prevention programs should erode over time. This is consistent with outcome data from a wide range of prevention and intervention studies. Although significant resources have been invested in both primary and targeted prevention for a range of disorders (see, e.g., Evans et al., 2005), the long-term efficacy of many of these programs is limited at best (e.g., Fingeret, Warren, Cepeda-Benito, & Gleaves, 2006; Foxcroft, Ireland, Lister-Sharp, Lowe, & Breen, 2003; Lynam et al., 1999). Worse yet, iatrogenic effects have been demonstrated for many prevention and early intervention programs that aggregate those who are at risk for psychopathology (see Dishion, McCord, & Poulin, 1999; Lilienfeld, 2007; Petrosino, Turpin-Petrosino, & Buehler, 2003; Rhule, 2005). One possible explanation for these negative effects is that too little attention has been paid to the power of such programs to potentiate genetic risk via exposure to the high-risk behaviors of other enrollees. For example, children who are at high genetic risk for delinquency should not be aggregated with delinquent peers, as such exposure constitutes a potentiating (high risk) environment.
When we study only child and adolescent samples, as many prevention researchers do, it is easy to overlook the powerful role that heritability plays in the expression of adult psychopathology. Because heritability coefficients for childhood disorders are low, prevention researchers may conclude erroneously that environmental risk “causes” psychopathology, and that biological vulnerabilities are unimportant (e.g., Albee & Joffe, 2004; Boyle, 2004; Joffe, 2004). Even among prevention researchers who are aware of behavioral genetics data on child and adolescent psychopathology, the importance of heritability may be underestimated unless a life span approach is adopted. As the above discussion illustrates, the long-term costs of underestimating heritability may be high.
Prevention researchers should expect erosion of treatment efficacy over time, and design programs to mitigate such effects through empirically supported adjunctive treatments, including available medication management that targets neurobiological sequelae of genetic risk (see, e.g., Jensen, Hinshaw, Swanson et al., 2001;McGorry et al., 2001) and intensive and regular follow-ups (“booster sessions”) that prevent behavioral drift (e.g., Lochman, 1992).
In addition, those who are at high genetic risk for psychopathology, as determined by family history interviews, genetic assays, or positive endophenotypes (see below), should not be aggregated in prevention or intervention trials unless there is clear evidence for the effectiveness of doing so.2 Many group treatments constitute high-risk environments that can potentiate genetic risk for psychiatric morbidity and mortality.
Finally, prevention researchers should evaluate the effects of biological risk and Biology×Environment interactions in predicting long-term treatment outcomes. Such efforts will lead to advances in our understanding of complex disorders, and to refined treatments for subgroups of patients with different etiologies (Cicchetti, 2007; see the following).
Genetic vulnerabilities give rise to broad classes of homotypic disorder
Most disorders within the externalizing spectrum share a common heritable vulnerability, as do most disorders within the internalizing spectrum (Baker, Jacobson, Raine, Lozano, & Bezdjian, 2007; Kendler, Prescott, Myers, & Neale, 2003; Krueger et al., 2002; see also Krueger & Markon, 2006). These within-spectrum vulnerabilities give rise to homotypic comorbidity: the co-occurrence of multiple externalizing disorders within an individual or the co-occurrence of multiple internalizing disorders within an individual. For example, ADHD, oppositional defiant disorder (ODD), CD, antisocial personality disorder (ASPD), and SUDs often co-occur (Lewinsohn, Shankman, Gau, & Klein, 2004; Nadder, Rutter, Silberg, Maes, & Leaves, 2002). Comorbidity of internalizing disorders, including depression, dysthymia, and many anxiety disorders, is also high (Angold & Costello, 1993; Brady & Kendall, 1992; Cloninger, 1990; Donaldson, Klein, Riso, & Schwartz, 1997; Ferdinand, Dieleman, Ormel, & Verhulst, 2007).
Traditionally, it has been assumed that comorbidity reflects distinct yet co-occurring disorders with different etiologies (see Beauchaine, 2003; Kopp & Beauchaine, 2007). This interpretation is implied by the Diagnostic and Statistical Manual of Mental Disorders (American Psychiatric Association, 2000), which treats most comorbidity as a differential diagnostic issue. Yet when we assume that different syndromes within the internalizing or externalizing spectra are diagnostically distinct, our approach to science, including prevention and intervention, can become artificially fractionated. This is illustrated in research on externalizing disorders, where largely separate literatures, both basic and applied, have evolved to address different diagnostic syndromes. Researchers and clinicians alike tend to specialize in particular disorders, such as ADHD, CD, or substance use. Even though homotypic comorbidity among these disorders is extremely high (e.g., Beauchaine et al., 2001; Cohen, Chen, Crawford, Brook, & Gordon, 2007; Hinshaw, 1987), very different treatment approaches have evolved for each. Psychostimulants are the front line treatment for ADHD (MTA Cooperative Group, 1999), behavioral, and multisystemic interventions are preferred for CD (Brestan & Eyberg, 1998; Nock, 2003), and motivational techniques are often favored for SUDs, particularly among adolescent users (Masterman & Kelly, 2003; Monti et al., 1999).
With the exception of ADHD, most treatments for externalizing disorders were developed with little or no attention to biological vulnerabilities. This is understandable, given how little was known about the biological substrates of externalizing risk until quite recently. However, it is now clear that dysfunction in the mesolimbic dopamine (DA) system, including the ventral tegmental area and its projections to the nucleus accumbens, the caudate, and the putamen, is a core neural substrate of risk for all or most externalizing behaviors (Beauchaine & Neuhaus, 2008; Gatzke-Kopp & Beauchaine, 2007a, 2007b; Gatzke-Kopp et al., 2007). Studies using both PET and fMRI indicate that inadequately low levels of DA and associated neural activity in the primary reward centers of the brain predispose to sensation seeking, irritability, negative affectivity, and low motivation, which are core symptoms of externalizing psychopathology (Beauchaine et al., 2007; Durston, 2003; Laakso et al., 2003; Leyton et al., 2002; Scheres, Milham, Knutson, & Castellanos, 2007; Vaidya et al., 1998). This central DA dysfunction is a likely endophenotype of genetic risk. As noted above, additive genetic effects account for roughly 80% of the variance in vulnerability to disorders across the externalizing spectrum (Krueger et al., 2002). Parallel findings apply to internalizing disorders as well (Kendler et al., 2003).
In addition to explaining homotypic comorbidity, central DA dysfunction likely also accounts for homotypic continuity, or the sequential development of different internalizing or externalizing disorders across the life span (see, e.g., Ferdinand et al., 2007). For example, seriously delinquent adult males are likely to have traversed a developmental pathway that began with hyperactive/impulsive behaviors in toddlerhood, followed by ODD in preschool, early-onset CD in elementary school, SUDs in adolescence, and ASPD in adulthood (see Loeber & Hay, 1997; Loeber & Keenan, 1994; Lynam, 1996, 1998).
Implications for prevention and intervention
Depending on when in this developmental progression prevention or intervention is initiated, treatments are likely to be very different, having emerged from distinct intellectual and empirical traditions. As a result, many empirically supported treatments target specific diagnostic syndromes (e.g., SUDs), and are limited in their capacity to address homotypic comorbidities (e.g., ADHD, CD, delinquency; see, e.g., Conrod & Stewart, 2005). Yet comorbidity that is mischaracterized may misdirect therapeutic efforts and reduce treatment efficacy. Furthermore, comorbidity can be a critical barrier to the dissemination of evidenced-based treatments, most of which do not address co-occurring disorders (Addis, Wade, & Hatgis, 1999). Thus, a detailed understanding of biological risk for homotypic comorbidity and continuity has very tangible implications for prevention and intervention. Some of these implications are outlined below.
First, prevention and intervention programs should not focus on single disorders. Individuals who are vulnerable to one internalizing or externalizing disorder are usually vulnerable to others within the same spectrum. Homotypic comorbidity should be expected and addressed explicitly.
In addition, the effects of most prevention programs on distal outcomes will be modest at best. Treated individuals are likely to develop additional externalizing behaviors and/or disorders as they move into different developmental epochs. For example, children treated for ODD in preschool are at risk for serious conduct problems later (see Campbell, Shaw, & Gilliom, 2000). Similarly, hyperactive boys with conduct problems who undergo pharmacological and/or behavioral treatments early in childhood are not protected from delinquency in later childhood, or from criminality as adults, even when short-term outcomes are favorable (Molina et al., 2007; Satterfield et al., 2007). Programs should therefore be designed to anticipate and mitigate homotypic continuity through empirically supported adjunctive treatments and intensive regular follow-ups.
Prevention and intervention programs should also be multifaceted. In the case of externalizing disorders, in addition to being treated for specific diagnostic syndromes such as CD, enrollees should also be taught strategies for coping with impulsivity, because this broad behavioral predisposition confers vulnerability to other externalizing disorders. Similarly, those being treated for specific internalizing disorders should also be taught strategies for coping with trait anxiety (see, e.g., Conrod, Stewart, Comeau, & Maclean, 2006). In other words, prevention and intervention programs need to address risk for psychopathology across the internalizing or externalizing spectra, even for disorders that are not yet apparent but lie in homotypically continuous pathways. For example, interventions for CD should include substance use prevention modules, even when treated individuals have not yet developed SUDs.
Common genetic and neural effects influence diverse classes of heterotypic disorder
In contrast to comorbidity within the internalizing and externalizing spectra, comorbidity across domains (e.g., depression and CD) is more surprising given that symptoms overlap minimally. This has been referred to as heterotypic comorbidity because internalizing and externalizing disorders are often assumed to be of different origins, whether biological or environmental. Depression includes symptoms of depressed mood, anhedonia, and feelings of guilt or worthlessness, and is usually manifested in a withdrawn behavioral presentation. In contrast, CD is characterized by symptoms such as sensation seeking, lying, property destruction, and aggression. However, despite these different presentations, rates of comorbidity of CD and depression are far greater than expected by chance (e.g., Capaldi, 1991; Drabick, Beauchaine, Gadow, Carlson, & Bromet, 2006).
A key assumption of most research on heterotypic comorbidity is that identifying which disorder is primary will lead to more effective prevention and intervention programs. Once the primary disorder is identified and treated, secondary disorders are expected to remit. For example, some have argued that among children with CD, comorbid depression follows from the consequences brought about by oppositional, aggressive, and otherwise delinquent behaviors (Capaldi, 1991, 1992; Patterson & Capaldi, 1990; Patterson, DeBaryshe, & Ramsey, 1989). These actions elicit peer rejection, restrict access to resources, promote school failure, and result eventually in institutionalization. Any or all of these consequences may precipitate an endogenous depression that is expected to abate after successful treatment of CD.
Following this reasoning, many researchers have used family history methods to ascertain whether CD or depression is primary in comorbid cases. Yet such family history analyses suggest that neither disorder is primary, and that CD and depression are transmitted across generations separately, through complex biological and environmental mechanisms (Kopp & Beauchaine, 2007). This argues against targeting primary disorders to improve treatment efficacy, and implies that both disorders should be treated simultaneously.
Studies of overlapping biological vulnerabilities for CD and depression indicate why heterotypic comorbidity is so common. At the behavioral level, both disorders are characterized by negative affectivity, irritability, and anhedonia. At the neural level, each of these symptoms has been linked with reduced activation in DA-mediated structures involved in approach motivation, regardless of whether CD or depression is the “primary” disorder (Bogdan, & Pizzagalli, 2006; Forbes, Shaw, & Dahl, 2007; Keedwell, Andrew, Williams, Brammer, & Phillips, 2005; Nestler & Carlezon, 2006; Shankman, Klein, Tenke, & Bruder, 2007). Neuroimaging studies reveal blunted activation in mesolimbic and mesocortical brain regions during reward tasks in both CD and depression (Epstein, Hong, Kocsis, Yang, Butler, & Chusid, 2006; Forbes et al., 2006; Gatzke-Kopp & Beauchaine, 2007a; Scheres et al., 2007; Vaidya et al., 1998). Thus, these disorders appear to share a common neural deficiency that accounts for overlap in symptoms. This conclusion is consistent with results from behavioral genetics studies indicating common heritable vulnerability for depression and antisocial behavior (O’Connor, McGuire, Reiss, Hetherington, & Plomin, 1998).
This deficiency in DA-mediated reward circuitry is moderated by other biologically influenced traits to affect behavior. One such trait is behavioral inhibition (see Figure 2), which differentiates between those who present principally with CD and those who present principally with depression (Beauchaine, 2001; Beauchaine & Neuhaus, 2008). In this model, high trait anxiety potentiates depression among those with blunted reward systems, whereas low trait anxiety potentiates delinquency. Trait anxiety is modulated by an entirely different (primarily serotonergic) neural network, often referred to as the septohippocampal system (Gray & McNaughton, 2000).
Figure 2.
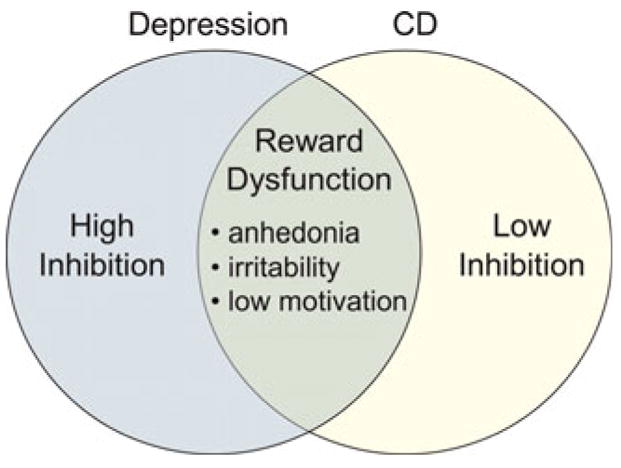
Symptom overlap in CD and depression. Both disorders are characterized by CNS reward dysfunction, leading to common symptoms. The disorders are differentiated by behavioral inhibition. [A color version of this figure can be viewed online at journals.cambridge.org/dpp]
Implications for prevention and intervention
Understanding common and unique mechanisms of internalizing and externalizing psychopathology has several implications for prevention and intervention. As noted above, treatment programs should not focus on single disorders. Individuals who are vulnerable to externalizing disorders such as CD may also be vulnerable to internalizing disorders such as depression, and vice versa. It should not be assumed that treating one disorder will eliminate the other. Although not as common as homotypic comorbidity, heterotypic comorbidity should be evaluated and addressed explicitly in prevention and intervention programs.
When possible, such programs should also be designed to capitalize on heterotypically comorbid conditions that moderate treatment response. For example, symptoms of anxiety are not uncommon among impulsive children with ADHD and CD (Angold, Costello, & Erkanli, 1999; MTA cooperative group, 1999). Compared with their nonanxious counterparts, anxious children with ADHD and conduct problems are more responsive to behavioral interventions, and to interventions that include a classroom behavior management component (Beauchaine, Webster-Stratton, & Reid, 2005; Jensen, Hinshaw, Kraemer et al., 2001). By assessing trait anxiety early in the treatment process, children can be assigned to intervention conditions from which they are most likely to benefit.
In addition, prevention and intervention programs should be designed to mitigate increased risk that is indicated by the presence or absence of heterotypically comorbid conditions (Conrod & Stewart, 2005). For example, an impulsive child who is low on trait anxiety may be especially vulnerable to developing extremely serious externalizing disorders. Psychopathy, a behavior pattern characterized by manipulation of others, superficial charm, callousness, and lack of remorse, is probably the most intractable form of externalizing conduct (Lykken, 2006). Psychopaths exhibit excessive approach behaviors that are coupled with a disturbing lack of anxiety and fear (e.g., Fowles & Dindo, 2006). Because their impulsive tendencies are not inhibited by impending consequences, callous and unemotional males with conduct problems are very resistant to current treatments (e.g., Hawes & Dadds, 2005). Thus, further refinement of intervention programs for these individuals with these traits is needed.
Individual differences in neurobiologically based traits affect treatment response
Differential treatment responses of externalizing children with and without comorbid anxiety provide one example of a neurobiologically based trait that moderates intervention effects (Beauchaine et al., 2005; Jensen, Hinshaw, Kraemer et al., 2001). Preliminary data suggest additional neurobiological moderators for other psychiatric conditions. For example, patterns of both amygdala and subgenual cingulate cortex reactivity to emotional stimuli predict treatment response for those with depression (Siegle, Carter, & Thase, 2006). Similarly, pilot data indicate that skin conductance reactivity to gambling tasks predicts prevention response to programs aimed at curbing adolescent substance use (Fishbein et al., 2004). Finally, heart rate variability appears to moderate the effects of treatment for boys with conduct problems and depression (Beauchaine, Gartner, & Hagen, 2000).
A particularly compelling example comes from the work of Conrod and colleagues (2006), who have conducted a series of studies indicating that individual differences in biologically based personality traits affect both vulnerability to SUDs and intervention responses. Impulsive and sensation-seeking individuals tend to use substances for their reward properties, whereas those high on trait anxiety tend to use for the anxiolytic effects (Conrod, Pihl, Stewart, & Dongier, 2000). These individual differences can be detected in part from physiological responses to alcohol (Conrod, Peterson, & Pihl, 1997; Conrod, Peterson, Pihl, & Mankowski, 1997; Conrod, Pihl, & Vassileva, 1998). For example, men who score high on measures of reward sensitivity and sensation seeking show larger heart rate responses to alcohol than men who score low on such measures (Brunelle et al., 2004). Among women, those with depressive symptoms of introversion and hopelessness preferentially use substances with analgesic properties (Conrod et al., 2000).
Implications for prevention and intervention
Until quite recently, most prevention and intervention programs aimed at curbing adolescent risk for SUDs were generic, with the same content applied to everyone (Conrod & Stewart, 2005). Although outcome data on drug use are sparse, such programs have very limited effects on drinking behaviors (Stewart et al., 2005). Findings outlined above specifying different motives for substance use suggest that targeted interventions might improve treatment efficacy. Preliminary research supports this conjecture. Alcohol use interventions with treatment manuals targeting specific personality risk factors (anxiety sensitivity, sensation seeking, and hopelessness) and associated coping strategies yield beneficial effects in terms of abstinence, binge drinking, and drinking quantity. These interventions also yield Personality × Treatment Condition interactions in predicting outcome, suggesting an important role for targeted interventions in future research and practice (Conrod et al., 2006). Moreover, such interventions can reduce the impact of comorbid psychological problems (depression, panic attacks, truancy) that are associated with personality risk (Castellanos & Conrod, 2006).
Although considerable research remains in the development of targeted interventions for SUDs, including additional treatment–outcome studies (Conrod & Stewart, 2005), these results suggest a clear role for evaluating high-risk traits that predispose to different motives for use. It is important that, even though impulsivity, anxiety sensitivity, and sensation-seeking have clear genetic and neurobiological substrates (see Beauchaine, 2001; Cloninger, Svrakic, & Svrakic, 1997; Corr, 2004; Gray & McNaughton, 2000; Sagvolden et al., 2005), each can be assessed reliably using self-report measures.
Finally, assessing biological factors that affect treatment response can indicate why some children do not respond to current treatment approaches, information that can be used to develop targeted interventions that are more effective (see Beauchaine et al., 2005; Gunnar & Fisher, 2006). As noted above, children, adolescents, and adults who exhibit callous unemotional traits and autonomic underarousal benefit little from current interventions for conduct problems and delinquency (see e.g., Fowles & Dindo, 2006; Hawes & Dadds, 2005; Lykken, 2006). Nevertheless, few prevention or intervention programs target such traits, a disservice to those at highest risk. Nearly a decade ago, Brestan and Eyberg (1998) implored the child psychopathology research community to ask the questions “For whom does this treatment work?” and “When is this treatment not enough?” By identifying characteristics (both biological and psychological) that predict poor treatment response, we begin to address these important questions. To date, very little research aimed at refining prevention and intervention programs to accommodate such individual differences has been conducted.
Biological vulnerabilities moderate the effects of environment on behavior
In addition to their moderating effects on treatment outcome, biologically based vulnerabilities also moderate the effects of broader environmental contexts on behavioral adjustment. For example, respiratory sinus arrhythmia (RSA), a measure of parasympathetic-linked cardiac activity that is roughly 50% heritable (Kupper et al., 2005), consistently predicts strong emotion regulation capabilities in both children and adults (see Beauchaine, 2001; Beauchaine et al., 2007), and protects children from developing psychopathology in high-risk environments. In a large study of conduct problems and depression among children and adolescents, high RSA offered partial protection from the development of conduct problems among participants with antisocial fathers (Shannon et al., 2007). In the same study, high RSA also conferred partial protection from the development of depression among participants of mothers with symptoms of melancholia.
Similarly, children with high RSA who witness marital conflict and hostility or are exposed to problem drinking by their parents are buffered from associated risk of developing both internalizing and externalizing behaviors (El-Sheikh, 2005; El-Sheikh, Harger, & Whitson, 2001; Katz & Gottman, 1995, 1997). Thus, across a wide range of environmental risks, high RSA confers partial protection from psychopathology. According to the nomenclature proposed by Luthar, Cicchetti, and Becker (2000), these are protective–reactive interactions. Similar results were obtained by Boyce et al. (2006), who reported that children’s autonomic reactivity moderated the effect of father involvement on later mental health outcomes.3
Implications for prevention and intervention
A corollary of these findings is that children with low RSA are particularly vulnerable to developing psychopathology in high-risk environments. In the study of conduct problems and depression cited above, children with low RSA were vulnerable to developing both conduct problems and depression (Shannon et al., 2007). These findings imply that assessing RSA may help to identify children who are in greatest need of prevention services given familial risk factors including parental antisocial behavior, depression, marital conflict, and problem drinking (see also Beauchaine et al., 2007).
Similar findings have been reported for other neurobiological systems. For example, Davies, Sturge-Apple, Cicchetti, and Cummings (2007) reported that low cortisol reactivity among kindergarteners in response to parental conflict marked risk for developing externalizing behaviors at 2-year follow-up. This longitudinal relation indicates some degree of prospective risk identification. Early detection of risk is extremely important given the well-documented erosion of treatment effects across development for programs aimed at curbing externalizing behaviors (Dishion & Patterson, 1992; Ruma, Burke, & Thompson, 1996). The earlier at-risk children are enrolled in prevention programs, the better. Thus, any means of identifying risk prospectively should be embraced by researchers and practitioners (Beauchaine & Marsh, 2006).
Many null findings for biological (and other) treatment moderators are likely the result of underpowered statistical tests
One argument for questioning the role of biological vulnerabilities as moderators of treatment response and other outcomes is that few such moderators have been identified to date, with several null findings reported (e.g., Coryell & Turner, 1985; Insel & Goodwin, 1983; Klein-Hessling & Lohaus, 2002; Raine et al., 2003).4 An often underappreciated reason for null findings in tests of moderation is that statistical interactions, which specify the moderation effect, have considerably less power than the main effects in a regression or analysis of variance (ANOVA) model (Aiken & West, 1991; Beauchaine & Mead, 2006; Kraemer, Wilson, Fairburn, & Agras, 2002; Whisman & McClelland, 2005). Although there are several reasons for this, three issues stand out as particularly important. First, the power to detect any effect in statistics depends on the reliability of the measures used. The more measurement error, the lower the statistical power. Unreliability is compounded when testing moderation effects because the reliability of the interaction term (α × β) equals the product of the reliabilities of the main effects. Even with high reliabilities for both the independent variables (IV) and the moderator (e.g., .85 each), the reliability of the interaction term is reduced considerably (.85×.85 =.72). As a result, achieving a conventionally acceptable power level of .80 (α = .05) to detect a medium-sized interaction effect (partial r2 = .13; see Cohen, 1988) requires a 50% increase in participants over that required to detect a medium-sized main effect (r2 = .24). Thus, many studies with adequate power to detect main effects do not have sufficient power to detect moderation. Because most researchers conduct power analyses only for main effects, frequent null findings for interaction effects should be expected.
Second, many moderating effects in psychological research are ordinal, particularly in treatment–outcome studies. In other words, the slopes of the regression lines for different treatment groups have the same sign at different levels of the moderator. Such is the case when two groups improve during treatment, but one group improves more than the other. Power to detect moderators is considerably less for ordinal interactions than for crossover interactions (see Whisman & McClelland, 2005).
Third, despite stern warnings from statisticians, many researchers dichotomize variables to test for moderation. It remains a common practice to first divide the putative moderator (often by performing a median split) and then test the correlation between the IV and the dependent variable (DV) in the resulting subgroups, a strategy advanced originally by Baron and Kenny (1986). Although it is essential to interpret a moderating effect by examining the relation between the IV and DV at different levels of the moderator, this should be done only after a significant effect is found using the continuous product term (α×β) to compute the interaction. Although there are several compelling reasons to avoid dichotomizing continuous variables (MacCallum, Zhang, Preacher, & Rucker, 2001), the point here is that doing so results in further erosion of statistical power for effects that are already underpowered given the considerations noted.
Implications for prevention and intervention
Many studies in which moderators of treatment outcome are tested do not have sufficient power to detect interaction effects. Tests of Biology×Environment (and any other) interactions are often ancillary to tests of main effects on which power calculations are based. The likely aggregate effect is a literature-wide underestimation of the importance of treatment moderators, including biological vulnerabilities. Indeed, an interaction effect that is considerably larger than a significant main effect may go undetected in the same study. This may be in part responsible for some concluding that biological moderators are irrelevant for prevention and intervention (e.g., Albee & Joffe, 2004).
Accordingly, prevention and intervention researchers who consider biologically based individual differences as moderators of treatment outcome should calculate the statistical power of interaction effects in advance to ensure that any null findings are not the result of inadequate sample size (Kraemer et al., 2002; Whisman & McClelland, 2005). As summarized above, ignoring issues of power often leads to erroneous null conclusions (Type II errors). This suggests that many null findings from tests of moderation should be revisited.
Biology×Environment interactions sometimes explain more variance in outcome than main effects
Despite the problems with power noted above, Biology × Environment interactions sometimes account for considerably more variance in key outcomes than main effects. Yet in the history of clinical science, most research has evaluated the effects of single variables (either biological or environmental) on behavior (see Porges, 2006). Questions such as “What are the genetic determinants of schizophrenia?” and “How do neighborhood influences promote delinquency?” reflect a predominant focus on main effects. Although such questions are clearly important, evaluating main effects in isolation can obscure equally important interactions between biological vulnerabilities and environmental risk factors in predicting psychopathology. This can lead researchers to conclude that one class of variables (biology or environment) is unrelated to outcome, even when such variables are critical determinants of adjustment.
For example, in our research on self-injury among adolescent girls (see Crowell et al., 2005, 2008; Crowell, Beauchaine, & Lenzenweger, 2008), we reported a significant interaction between participant’s peripheral serotonin levels and the quality of dyadic discussions with their mothers in predicting self-harm (e.g., cutting, overdoses, etc.). Adolescent girls with low levels of peripheral serotonin tended toward self-harm regardless of observed dyadic negativity with their mothers. In contrast, girls with high levels of peripheral serotonin were only at risk when dyadic interactions with their mothers were highly negative. Yet serotonin levels and dyadic negativity were unrelated to one another, and accounted for only 3 and 23% of the variance in self-harm, respectively. However, by adding a Serotonin × Dyadic Negativity interaction term into the model, we accounted for 64% of the variance in self-harm. In this case, had we measured only peripheral serotonin, we would have concluded that it is unrelated to self-harm. Similarly, had we measured only dyadic negatively, we would have vastly underestimated its importance. As illustrated in Figure 3, only by assessing the interaction between both variables were we able to explain so much variance in a critically important outcome.
Figure 3.

The main effects of (top) dyadic mother–daughter negativity and (middle) adolescent peripheral serotonin on lifetime self-harm events in a sample of 41 adolescent girls. Negativity and peripheral serotonin accounted for 23 and 3% of the variance in self-harm, respectively. In contrast, (bottom) the conjoint effects of negativity and peripheral serotonin accounted for 64% of the variance in self-injury. From “Parent–Child Interactions, Peripheral Serotonin, and Self-Inflicted Injury in Adolescents,” by S. E. Crowell, T. P. Beauchaine, E. McCauley, C. J. Smith, C. A. Vasilev, and A. L. Stevens, 2008, Journal of Consulting and Clinical Psychology. Copyright 2008 by American Psychological Association. Adapted with permission.
Fortunately, much greater appreciation for interactions between biology and environment has evolved in recent years (see Moffitt et al., 2006; Rutter, 2002, 2007). Many researchers are now evaluating the combined effects of endogenous and exogenous influences on behavior, which form the crux of both diathesis–stress and moderation models of psychopathology (e.g., Beauchaine et al., 2005; Gottesman & Gould, 2003; Kraemer, Stice, Kazdin, Offord, & Kupfer, 2001). It is now widely recognized that Gene × Environment and Neurobiology×Environment interactions are critical in the expression of diverse classes of disorder (Cicchetti, 2007), including schizophrenia (e.g., Gottesman & Gould, 2003; Lenzenweger, 2006) delinquency (e.g., Beauchaine et al., 2007; Caspi et al., 2002; Lynam et al., 2000), depression (e.g., Caspi et al., 2003) posttraumatic stress disorder (e.g., Orr et al., 2003; Stein, Jang, Taylor, Vernon, & Livesley, 2002), ADHD (e.g., Seeger, Schloss, Schmidt, Rüter-Jungfleisch, & Henn, 2004), and SUDs (e.g., Kendler et al., 2003). This research reveals that for many forms of psychopathology, neither biological vulnerabilities nor high-risk environments are sufficient in isolation to explain etiology; their combined effects must be considered (Beauchaine, Hinshaw, & Gatzke-Kopp, 2008).
Environments potentiate and mollify biological vulnerabilities through mechanisms of epigenesis, neural plasticity, and pruning
Although underappreciated until quite recently, environmental influences shape and maintain biological vulnerabilities for psychopathology in a number of ways. Adverse experiences, particularly trauma and adverse rearing conditions faced early in life, can alter gene expression, with downstream consequences for both central nervous system development and behavior. The term epigenesis refers to changes in gene expression that result from alterations in DNA structure (as opposed to sequence; Hartl & Jones, 2002). Such alterations are mediated by methylation processes that are triggered by environmental events. For example, Weaver et al. (2004) reported epigenetically transmitted genetic variation in the glucocorticoid receptor gene promoter in the hippocampi of rat pups that experienced high levels of maternal licking, grooming, and arched-back nursing compared with pups that experienced low levels of these rearing behaviors. This epigenetic maternal programming effect transmits adaptive variations in stress responding to offspring (see Meany, 2007). Rat pups reared in high-risk environments where normal maternal behaviors are compromised have more reactive hypothalamic–pituitary–adrenocortical responses, and are therefore more fearful and wary. Thus, they are better prepared for the high-risk environment that they are likely to face as they continue to develop.
Although clear epigenetic effects among humans have yet to be identified (see Rutter, 2007), mammals are particularly susceptible to environmentally mediated changes in gene expression (Hartl & Jones, 2002), and increasingly divergent patterns of DNA methylation emerge over the life spans of monozygotic twins (Fraga et al., 2005). Accordingly, several authors have noted the potential importance of epigenetic effects for research in child psychopathology (e.g., Kramer, 2005; Rutter, 2005). However, demonstrating these effects in humans is difficult because it requires random assignment of groups to different environments (e.g., impoverished vs. enriched). Nevertheless, theoretical models of antisocial behavior that include epigenetic effects have been described (Tremblay, 2005). Moreover, recent studies suggest that brain-derived neurotrophic factor, which is involved in the differentiation of DA neurons in developing mesolimbic structures, may be susceptible to paternally mediated epigenetic effects that confer risk for ADHD and other externalizing behaviors (Kent et al., 2005).
In contrast to epigenesis, neural plasticity refers to structural and functional brain changes that result from development, experience, and/or learning. Plasticity has been defined as “permanent functional transformations … in particular systems of neurons as a result of appropriate stimuli or their combination” (Konorski, 1948). Mechanisms of plasticity alter the efficiency, activation thresholds, and time course of responding within and across neural systems (see Ciccetti & Blender, 2006; Pollak, 2005). These transformations include both short-term modulations at neural synapses and long-term changes involving anatomical growth and pruning of neural connections. Pruning refers to the selective elimination of cells and synapses, and may occur as a result of programmed cell death or experience. Presumably, pruning eliminates unused and inefficient connections to improve the overall efficiency and specificity of neurotransmission.
Plasticity, programmed cell death, and pruning are critical mechanisms of neural development (see Perry, 2008). These processes are widespread prenatally, but continue postnatally, and to a lesser extent, into adulthood. Both heredity and experience impact neural plasticity and pruning, shaping the function of neural systems subserving motivation, emotion, and self-control. An individual’s genes create boundaries on developmental trajectories, functioning, and plasticity of neural systems, and provide the bases for later integration of experience (Cicchetti & Curtis, 2006; Hammock & Levitt, 2006). The impact of experiences on neural and behavioral development is influenced by the timing, duration, and intensity of stimuli, and by biological vulnerabilities, resiliencies, potentiating risk factors, and protective effects (Gunnar & Fisher, 2006; Pollak, 2005). Hammock and Levitt (2006) describe how the timing of an adverse event is a critical determinant of the brain region affected. For example, post-natal disruptions in maternal infant care in rodents result in delays of synaptic formation in the developing amygdala, cortex, and brain stem, but not in other regions that developed prenatally such as hypothalamic–brain stem connections. Such studies also document how disturbances in early maternal care create long-lasting changes in neural development that persist into the rodent homologues of human adolescence and adulthood (see Gunnar & Fisher, 2006).
The prenatal period is a critical time of development during which exposure to teratogens can result in structural and functional brain changes and persistent risk for severe psychopathology (see Fryer, Crocker, & Mattson, 2008). For example, in addition to the well-known effects of in utero ethanol exposure on children’s risk for psychiatric disturbance, maternal nicotine exposure during pregnancy is associated with risk for externalizing behaviors among offspring. It is important that significant second-hand exposure may be just as harmful as maternal smoking. In a recent study of 7- to 15-year-olds, children of mothers who did not smoke but reported regular exposure to second-hand smoke during pregnancy, either at home or in the workplace, were at similar risk for impulsivity and conduct problems compared with children whose mothers smoked (Gatzke-Kopp & Beauchaine, 2007b). In addition to inducing long-term reductions in central DA reactivity (Kane, Fu, Matta, & Sharp, 2004), nicotine mimics the effects of acetylcholine neurotransmission (Oliff & Gallardo, 1999), eliciting changes in a number of cellular processes including replication, differentiation, and sensitivity to later stimulation (Slotkin, 1998). These alterations in both dopaminergic and cholinergic neurotransmission are enduring enough to translate into problems with externalizing psychopathology well into childhood and adolescence.
As noted, neural plasticity effects are not limited to prenatal and perinatal development. Genetic predispositions toward fearfulness may interact with environmental events to alter neural circuits involved in the experience and expression of emotion, thereby amplifying and maintaining anxious behavior throughout the life span (Fox, Hane, & Pine, 2007).
Implications for prevention and intervention
In addition to these negative effects, neural plasticity and pruning may provide opportunities for resilience and positive adaptation (Cicchetti & Blender, 2006; Cicchetti & Curtis, 2006, 2007; Masten, 2007). Rodent studies of optimal maternal care document changes in gene expression in offspring associated with improved stress resilience (Kaffman & Meaney, 2007). Among humans, effective psychosocial interventions may result in adaptive changes in brain function well into adulthood. For example, CBT for adult patients with PTSD results in functional brain changes as measured by MRI (Felmingham et al., 2007). Improvement in PTSD symptoms is associated with increased activity in the anterior cingulate cortex and decreased activity in the amygdala during fear processing. Functional brain changes resulting from psychotherapy have been reported for a host of other disorders as well (e.g., Baxter et al., 1992; Goldapple et al., 2004; Paquette et al., 2003). These changes are often effected through the same neural pathways that are targeted by pharmacologic interventions (see Kumari, 2006).
Although some degree of epigenesis and neural plasticity are observed across the life span (e.g., Eriksson et al., 1998), neural adaptations are more frequent and occur more readily in childhood (see e.g., Perry, 2008), with certain exceptions for later-maturing brain regions such as the oribitofrontal and prefrontal cortices, areas that exhibit considerable plasticity into adulthood (Sowell et al., 2003). This general trend of decreasing plasticity across development underscores points outlined above concerning the urgency of initiating prevention and early intervention programs as soon as possible for those at highest risk for psychopathology (Beauchaine & Marsh, 2006). Because younger brains are more malleable, early intervention provides greater opportunities for (a) conferring protective long-term functional changes in neural systems subserving mood, motivation, and self-regulation; (b) halting the progression of emergent neural vulnerabilities; and (c) reversing existing neural vulnerabilities.
Extensive neural plasticity during prenatal, perinatal, and early childhood development also underscores the need to develop more effective prevention programs targeting young and expectant mothers and their families. Improved prenatal and early childhood care and increased public health awareness are areas where universal prevention programs are potentially most effective. Indeed, nonspecific early educational and health-promoting interventions for disadvantaged pre-schoolers confer long-term functional changes in autonomic arousal, and protect enrollees from developing both antisocial and schizotypal behaviors as adults (Raine et al., 2001; Raine et al., 2003). Expansion and rigorous evaluation of such programs should therefore be a high priority for prevention researchers.
Despite compelling arguments for the earliest interventions possible, we are not suggesting that all is lost for older children and adolescents who experience psychopathology. As noted above, the prefrontal cortex (PFC) maintains considerable plasticity into young adulthood (Sowell et al., 2003). Given the importance of the PFC in executive functioning, planning, and affect regulation, interventions should be developed that capitalize on skill acquisition in these areas. Indeed, some have suggested that attentional neural networks including the PFC may be modifiable through targeted interventions (see Rueda, Rothbart, Saccomanno, & Posner, 2007).
Early developing neural systems affect the organization and functioning of later developing neural systems
As the discussion above indicates, neural functioning is affected throughout the life span by an individual’s developmental history, including both exogenous and endogenous influences. Environmental events affect brain development beginning in utero and extending into adulthood, interacting with genetic predispositions to shape the structure and function of neural pathways subserving behavior regulation. Often these heritable and environmental influences on development are interdependent (see, e.g., Rutter, 2007). For example, heritable vulnerabilities in early-maturing neural systems can predispose individuals to behaviors that elicit and/or exacerbate environmental risk. In turn, educed environmental risk can feed back to influence continuing brain development through mechanisms of neural plasticity (see above). Over time, these evocative Biology × Environment interactions can solidify behavior patterns that were previously malleable.
One behavioral trait that often evokes self-reinforcing effects is impulsivity. Highly impulsive children present with difficult to manage behaviors, which can elicit and strengthen ineffective parenting practices that, in turn, amplify risk for progression to more serious externalizing behaviors (Beauchaine et al., 2005; Patterson et al., 1989, 2000). As noted above, deficient mesolimbic DA activity is a primary neural substrate of impulsivity and disinhibition (Gatzke-Kopp & Beauchaine, 2007a, 2007b). Deficient mesolimbic DA functioning can result from a number of causes, included heritable vulnerability, prenatal exposure to stimulants, hypoxia, and head injury (Beauchaine & Neuhaus, 2008; Gatzke-Kopp & Shannon, 2008). In the case of prenatal stimulant exposure, abnormally low mesolimbic DA activity in childhood reflects a lingering neural adaptation to earlier overactivation at the time of exposure (e.g., Kane et al., 2004). Thus, excessive mesolimbic activation from DA agonists leads to down-regulation of a key neural system of behavioral control. In turn, downregulated DA activity predisposes to sensation-seeking behaviors characteristic of conduct problems, criminality, antisocial behavior, and SUDs (Gatzke-Kopp & Beauchaine, 2007a, 2007b; see also Sagvolden et al., 2005).
Each of these conditions may further exacerbate neurological vulnerabilities, particularly through early and prolonged exposure to substances of abuse in childhood and adolescence, which induces further changes in DA functioning in mesolimbic structures (e.g., Catlow & Kirstein, 2007). In this manner, biological vulnerabilities and environmental risk factors reinforce one another, leading to worse outcomes than either factor alone. Similar evocative effects have been described for other high-risk traits, such as anxiety (Fox et al., 2007).
In addition to cumulative effects within discrete neural systems, evocative cascades that begin in childhood may compromise later developing neural networks, conferring additional vulnerability for heterotypically continuous disorders. The mesolimbic DA system, which develops very early in life, is modulated in adolescence and adulthood by the mesocortical DA system (see Halperin & Schulz, 2006; Spear, 2007), a frontal neural network that exhibits experience-dependent development into young adulthood (see above). Animal studies identify adolescence as a period of maximum DA activity in the PFC (Tunbridge et al., 2007), and as a period marked by a DA-dependent shift in the effects of stimulant drugs from dysphorigenic to euphorigenic (Andersen, Leblanc, & Lyss, 2001).
Children with already compromised yet mature mesolimbic DA systems are placed at even higher risk for psychopathology if neurodevelopment of immature frontal DA systems is also compromised by adverse events. Thus, functional deficiencies in the mesolimbic DA system that give rise to impulsive behaviors and risk for externalizing disorders may be amplified by neuroregulatory processes and evoked environments that compromise experience-dependent neuromaturation of cortical DA networks (see Beauchaine & Neuhaus, 2008).
Implications for prevention and intervention
The influence of early developing neural systems on later developing neural systems suggests the opportunity for targeted prevention programs. For example, individuals with known prenatal exposure to substances are likely to experience atypical neural development and resulting behavioral and psychological difficulties (Fryer et al., 2008), marking a clear need for early intervention. Furthermore, regardless of the source, functional deficiencies in key neural systems may confer vulnerability for maladaptive neurodevelopment in later-maturing brain regions, giving rise to additional behavioral difficulties that were not apparent at earlier ages. As already discussed, early dysregulation of the mesolimbic DA system that underlies impulsivity also confers vulnerability for later prefrontal dysfunction, which is associated with deficiencies in complex reasoning, planning, and self-regulation. Early intervention programs should provide protective environments by (a) minimizing exposure to neighborhood risk, delinquent peers, and early initiation of substance use (all of which have been linked with especially poor outcomes for impulsive children), and (b) teaching empirically supported strategies to encourage attentional control and self-regulation (see Rueda et al. 2007), which may serve to dampen or halt escalating cascades of interdependency among biological vulnerabilities and environmental risks.
It is worth reemphasizing that heterogeneous sources of vulnerability may be important moderators of prevention and intervention outcomes. A single behavioral outcome (e.g., impulsivity) may stem from multiple etiologies across individuals, despite similar behavioral presentations (equifinality). It is possible that impulsivity derived from one source (e.g., brain injury) responds differently to prevention and intervention programs than impulsivity derived from another source (e.g., heritability). For example, although both prenatal ethanol exposure and heritable DA deficiencies give rise to impulsivity, the former condition is typically associated with more widespread neurodevelopmental compromises that are more resistant to current treatment approaches (O’Malley & Nanson, 2002). For these children, new interventions may be required. Thus, an understanding of the specific vulnerabilities and insults underlying adaptive and maladaptive behaviors within individuals, and an understanding of how resulting behavioral trajectories are likely to unfold over time, are critical to the design, evaluation, and implementation of the next generation of interventions.
Conclusion
Each of the points outlined above underscores the value of adopting an explicit developmental psychopathology approach to prevention and intervention. Almost all forms of psychopathology emerge over the course of development as a result of complex interactions between biological vulnerabilities and environmental risks, neither of which can be ignored in efforts to prevent and treat debilitating psychiatric conditions. Trajectories to psychopathology typically begin very early in development, often expressed initially as heritable vulnerabilities such as impulsivity and trait anxiety. Although insufficient alone to produce severe impairment, these traits interact with high-risk environments (e.g., ineffective parenting, abuse, neglect, neighborhood crime) to potentiate psychopathology in affected individuals. Often cascades of evocative effects begin very early in life, initiating trajectories of maladjustment that become more intractable over time. Accordingly, prevention efforts should be initiated as early in development as possible, and continue throughout childhood and adolescence to prevent the emergence of heterotypically continuous disorders. At the same time, the effectiveness of prevention programs is likely to vary for children with different biological vulnerabilities, even when such vulnerabilities are expressed similarly (equifinality). Thus, children presenting with the same disorders may require different intervention approaches, as demonstrated for those with pure ADHD versus ADHD plus comorbid anxiety (Jensen, Hinshaw, Kraemer, et al., 2001).
Among the points outlined above, one theme that arises consistently is that considering biological mechanisms in prevention science will increase program efficiency. For some disorders, biological indicators of risk can already identify those most in need of prevention, information that could be used to direct limited resources more effectively. Moreover, understanding biological vulnerabilities that moderate treatment response facilitates more efficient matching of individuals to treatments. As outlined above, this strategy is already yielding important advances in substance abuse interventions for adolescents (e.g., Castellanos & Conrod, 2006; Conrod et al., 2006). Finally, knowledge of the neurobiological substrates of heterotypic continuity allows us to identify those at especially high risk of life-long impairment, which could facilitate more concentrated and targeted treatment programs.
As discussed, clinical psychology is often slow to respond to paradigm shifts that affect other areas of science much earlier. We have argued that this reluctance slows progress in prevention science, and that efforts to understand biological processes involved in the expression of psychopathology should be embraced. It is our hope that the promise of neuroscience will extend to prevention research and practice in the years ahead, and that more at risk children will benefit as a result.
Acknowledgments
Work on this article was supported by Grant MH63699 from the National Institute of Mental Health to Theodore P. Beauchaine. We thank Sheila Crowell, Penny Marsh, Hilary Mead, and Katherine Shannon for their helpful contributions.
Footnotes
This statement begs an obvious question: if nonshared environmental effects and allostatic load affect age of onset for those who are genetically predisposed to psychopathology, should not it be the interaction between heritable vulnerability (G) and environmental risk (E) that rises across the life span, rather than the main effect of heritability? In brief, the answer to this question is often “yes.” However, Heritability×Environment (G×E) interactions cannot be disentangled from pure heritability effects in behavioral genetics research unless the specific environmental variable that interacts with heritability is measured, which is often not the case. When the effects of environmental are not measured directly, G×E interactions are subsumed within the heritability effect (see, e.g., Rutter, 2007). Although full discussion of this issue is beyond the scope of this article, unmeasured effects of environment can result in inflated estimates of heritability.
As researchers have learned more about the iatrogenic effects of group interventions, some have taken steps in subsequent trials to prevent these adverse effects. The Reconnecting Youth Prevention Research Program at the University of Washington has used group formats to decrease suicide risk and drug use among high-risk adolescents by (a) carefully training instructors about the potential for iatrogenic influences and (b) setting guidelines within the group to maintain prosocial interactions (e.g., Thompson, Eggert, Randell, & Pike, 2001; Thompson, Horn, Herting, & Eggert, 1997).
Although Boyce et al. (2006) suggested that father involvement moderated the effects of autonomic activity on mental health, independent variables (IVs) and moderators are fully interchangeable in statistical tests of interaction. Thus, the mathematics are identical whether father involvement is considered the IV (predictor) and autonomic reactivity is considered the moderator or vice versa. Either way, the effect of one variable (e.g., autonomic activity) differs as a function of the other variable (father involvement) in predicting outcome.
In addition, although biological variables have rarely been used to assign participants to different treatment groups, some large scale and highly publicized interventions in which participants were matched to different treatment conditions based on key individual differences have yielded no detectable moderation effects (DiClemente et al., 2001; Project MATCH research group, 1997).
References
- Addis ME, Wade WA, Hatgis C. Barriersto dissemination of evidence-based practices: Addressing practitioners’ concerns about manual-based psychotherapies. Clinical Psychology Science and Practice. 1999;6:430–441. [Google Scholar]
- Agrawal N, Hirsch SR. Schizophrenia: Evidence for conceptualising it as a brain disease. Journal of Primary Prevention. 2004;24:437–444. [Google Scholar]
- Aiken LS, West SG. Multiple regression: Testing and interpreting interactions. Thousand Oaks, CA: Sage; 1991. [Google Scholar]
- Albee GW, Joffe JM. Mental illness is NOT “an illness like any other. Journal of Primary Prevention. 2004;24:419–436. [Google Scholar]
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 4. Washington, DC: Author; 2000. [Google Scholar]
- Ames SL, McBride C. Translating genetics, cognitive science, and other basic science research findings into applications for prevention. Evaluation and the Health Professions. 2006;29:277–301. doi: 10.1177/0163278706290407. [DOI] [PubMed] [Google Scholar]
- Andersen SL, Leblanc CJ, Lyss PJ. Maturational increases in c-fos expression in the ascending dopamine systems. Synapse. 2001;41:345–350. doi: 10.1002/syn.1091. [DOI] [PubMed] [Google Scholar]
- Angold A, Costello EJ. Depressive comorbidity in children and adolescents: Empirical, theoretical, and methodological issues. American Journal of Psychiatry. 1993;150:1779–1791. doi: 10.1176/ajp.150.12.1779. [DOI] [PubMed] [Google Scholar]
- Angold A, Costello EJ, Erkanli A. Comorbidity. Journal of Child Psychology and Psychiatry. 1999;40:57–87. [PubMed] [Google Scholar]
- Baker LA, Jacobson KC, Raine A, Lozano DI, Bezdjian S. Genetic and environmental bases of child antisocial behavior: A multi-informant twin study. Journal of Abnormal Psychology. 2007;116:219–235. doi: 10.1037/0021-843X.116.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron RM, Kenny D. The moderator–mediator variable distinction in social psychological research: Conceptual, strategic, and statistical considerations. Journal of Personality and Social Psychology. 1986;51:1173–1182. doi: 10.1037//0022-3514.51.6.1173. [DOI] [PubMed] [Google Scholar]
- Baxter LR, Schwartz JM, Bergman KS, Szuba MP, Guze BH, Mazziotta JC, et al. Caudate glucose metabolic rate changes with both drug and behavior therapy for obsessive–compulsive disorder. Archives of General Psychiatry. 1992;49:690–694. doi: 10.1001/archpsyc.1992.01820090009002. [DOI] [PubMed] [Google Scholar]
- Beauchaine TP. Vagal tone, development, and Gray’s motivational theory: Toward an integrated model of autonomic nervous system functioning in psychopathology. Development and Psychopathology. 2001;13:183–214. doi: 10.1017/s0954579401002012. [DOI] [PubMed] [Google Scholar]
- Beauchaine TP. Taxometrics and developmental psychopathology. Development and Psychopathology. 2003;15:501–527. doi: 10.1017/s0954579403000270. [DOI] [PubMed] [Google Scholar]
- Beauchaine TP, Gartner J, Hagen B. Comorbid depression and heart rate variability as predictors of aggressive and hyperactive symptom responsiveness during inpatient treatment of conduct-disordered, ADHD boys. Aggressive Behavior. 2000;26:425–441. [Google Scholar]
- Beauchaine TP, Gatzke-Kopp L, Mead HK. Polyvagal theory and developmental psychopathology: Emotion dysregulation and conduct problems from pre-school to adolescence. Biological Psychology. 2007;74:174–184. doi: 10.1016/j.biopsycho.2005.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauchaine TP, Hinshaw SP, Gatzke-Kopp L. Genetic and environmental influences on behavior. In: Beauchaine TP, Hinshaw SP, editors. Child psychopathology: Genetic, neurobiological, and environmental substrates. Hoboken, NJ: Wiley; 2008. pp. 58–91. [Google Scholar]
- Beauchaine TP, Katkin ES, Strassberg Z, Snarr J. Disinhibitory psychopathology in male adolescents: Discriminating conduct disorder from attention-deficit/hyperactivity disorder through concurrent assessment of multiple autonomic states. Journal of Abnormal Psychology. 2001;110:610–624. doi: 10.1037//0021-843x.110.4.610. [DOI] [PubMed] [Google Scholar]
- Beauchaine TP, Lenzenweger MF, Waller NG. Schizotypy, taxometrics, and disconfirming theories in soft science. Comment on Rawlings, Williams, Haslam, and Claridge. Personality and Individual Differences. 2008;44:1652–1662. [Google Scholar]
- Beauchaine TP, Marsh P. Taxometric methods: Enhancing early detection and prevention of psychopathology by identifying latent vulnerability traits. In: Cicchetti D, Cohen D, editors. Developmental psychopathology. 2. Hoboken, NJ: Wiley; 2006. pp. 931–967. [Google Scholar]
- Beauchaine TP, Mead HK. Some recommendations for testing mediating and moderating effects in treatment-outcome research. International Journal of Psychology Research. 2006;1:1. [Google Scholar]
- Beauchaine TP, Neuhaus E. Behavioral disinhibition and vulnerability to psychopathology. In: Beauchaine TP, Hinshaw SP, editors. Child psychopathology: Genetic, neurobiological, and environmental substrates. Hoboken, NJ: Wiley; 2008. pp. 129–156. [Google Scholar]
- Beauchaine TP, Webster-Stratton C, Reid MJ. Mediators, moderators, and predictors of one-year outcomes among children treated for early-onset conduct problems: A latent growth curve analysis. Journal of Consulting and Clinical Psychology. 2005;73:371–388. doi: 10.1037/0022-006X.73.3.371. [DOI] [PubMed] [Google Scholar]
- Beck AT, Rush AJ, Shaw BF, Emery G. Cognitive therapy of depression. New York: Guilford Press; 1979. [Google Scholar]
- Berridge KC, Robinson TE. Parsing reward. Trends in Neuroscience. 2003;26:507–513. doi: 10.1016/S0166-2236(03)00233-9. [DOI] [PubMed] [Google Scholar]
- Blanchard JJ, Gangestad SW, Brown SA, Horan WP. Hedonic capacity and Schizotypy revisited: A taxometric analysis of social anhedonia. Journal of Abnormal Psychology. 2000;109:87–95. doi: 10.1037//0021-843x.109.1.87. [DOI] [PubMed] [Google Scholar]
- Bogdan R, Pizzagalli DA. Acute stress reduces reward responsiveness: Implications for depression. Biological Psychiatry. 2006;60:1147–1154. doi: 10.1016/j.biopsych.2006.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boomsma DI, Koopsman JR, Van Doornen LJ, Orlebeke JF. Genetic and social influences on starting to smoke: A study of Dutch adolescent twins and their parents. Addiction. 1994;89:219–226. doi: 10.1111/j.1360-0443.1994.tb00881.x. [DOI] [PubMed] [Google Scholar]
- Boyce WT, Essex MJ, Alkon A, Goldsmith HH, Kraemer HC, Kupfer DJ. Early father involvement moderates biobehavioral susceptibility to mental health problems in middle childhood. Journal of the American Academy of Child & Adolescent Psychiatry. 2006;45:1510–1520. doi: 10.1097/01.chi.0000237706.50884.8b. [DOI] [PubMed] [Google Scholar]
- Boyle M. Preventing a non-existent illness?: Some issues in the prevention of “schizophrenia. Journal of Primary Prevention. 2004;24:445–469. [Google Scholar]
- Brady EU, Kendall PC. Comorbidity of anxiety and depression in children and adolescents. Psychological Bulletin. 1992;111:244–255. doi: 10.1037/0033-2909.111.2.244. [DOI] [PubMed] [Google Scholar]
- Brestan EV, Eyberg SM. Effective psychosocial treatments of conduct-disordered children and adolescents: 29 years, 82 studies, and 5,272 kids. Journal of Clinical Child Psychology. 1998;27:180–189. doi: 10.1207/s15374424jccp2702_5. [DOI] [PubMed] [Google Scholar]
- Brunelle C, Assaad JM, Barrett SP, Avila C, Conrod PJ, Tremblay RE, et al. Heightened heart rate response to alcohol intoxication is associated with reward-seeking personality profile. Alcoholism Clinical and Experimental Research. 2004;28:394–401. doi: 10.1097/01.alc.0000117859.23567.2e. [DOI] [PubMed] [Google Scholar]
- Bryant RA. Longitudinal psychophysiological studies of heart rate: Mediating effects and implications for treatment. Annals of the New York Academy of Sciences. 2006;1071:19–26. doi: 10.1196/annals.1364.002. [DOI] [PubMed] [Google Scholar]
- Campbell SB, Shaw DS, Gilliom M. Early externalizing behavior problems: Toddlers and pre-schoolers at risk for later maladjustment. Development and Psychopathology. 2000;12:467–488. doi: 10.1017/s0954579400003114. [DOI] [PubMed] [Google Scholar]
- Capaldi DM. Co-occurrence of conduct problems and depressive symptoms in early adolescent boys: I. Familial factors and general adjustment at Grade 6. Development and Psychopathology. 1991;3:277–300. doi: 10.1017/s0954579499001959. [DOI] [PubMed] [Google Scholar]
- Capaldi DM. Co-occurrence of conduct problems and depressive symptoms in early adolescent boys: II. A 2-year follow up at grade 8. Development and Psychopathology. 1992;4:125–144. doi: 10.1017/s0954579499001959. [DOI] [PubMed] [Google Scholar]
- Caspi A, McClay J, Moffitt T, Mill J, Martin J, Craig IW, et al. Role of genotype in the cycle of violence in maltreated children. Science. 2002;297:851–854. doi: 10.1126/science.1072290. [DOI] [PubMed] [Google Scholar]
- Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, et al. Influence of life stress on depression: Moderation by a polymorphism in the 5-HTTgene. Science. 2003;301:386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- Castellanos N, Conrod P. Brief interventions targeting personality risk factors for adolescent substance misuse reduce depression, panic and risk-taking behaviours. Journal of Mental Health. 2006;15:645–658. [Google Scholar]
- Catlow BJ, Kirstein CL. Cocaine during adolescence enhances dopamine in response to a natural reinforcer. Neurotoxicology and Teratology. 2007;29:57 –65. doi: 10.1016/j.ntt.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang K, Gallelli K, Howe M. Early identification and prevention of early-onset bipolar disorder. In: Romer D, Walker EF, editors. Adolescent psychopathology and the developing brain. Oxford: Oxford University Press; 2007. pp. 315–346. [Google Scholar]
- Cicchetti D. Development and psychopathology. In: Cicchetti D, Cohen DJ, editors. Developmental psychopathology: Vol. 1. Theory and method. 2. Hoboken, NJ: Wiley; 2006. pp. 1–23. [Google Scholar]
- Cicchetti D. Gene–environment interaction. Development and Psychopathology. 2007;19:957–1208. [PubMed] [Google Scholar]
- Cicchetti D, Blender JA. A multiple-levels-of-analysis perspective on resilience. Annals of the New York Academy of Sciences. 2006;1094:248–258. doi: 10.1196/annals.1376.029. [DOI] [PubMed] [Google Scholar]
- Cicchetti D, Curtis WJ. The developing brain and neural plasticity: Implications for normality, psychopathology, and resilience. In: Cicchetti D, Cohen DJ, editors. Developmental psychopathology: Vol. 2. Developmental neuroscience. 2. Hoboken, NJ: Wiley; 2006. pp. 1–64. [Google Scholar]
- Cicchetti D, Curtis WJ, editors. Development and Psychopathology. Vol. 19. 2007. A multilevel approach to resilience; pp. 627–955. [DOI] [PubMed] [Google Scholar]
- Cicchetti D, Dawson G, editors. Development and Psychopathology. Vol. 14. 2002. Multiple levels of analysis; pp. 417–666. [DOI] [PubMed] [Google Scholar]
- Cicchetti D, Posner MI. Cognitive and affective neuroscience and developmental psychopathology. Development and Psychopathology. 2005;17:569–575. doi: 10.1017/S0954579405050273. [DOI] [PubMed] [Google Scholar]
- Cicchetti D, Rogosch FA. Equifinality and multifinality in developmental psychpathology. Development and Psychopathology. 1996;8:597–600. [Google Scholar]
- Cicchetti D, Rogosch FA. A developmental psychopathology perspective on adolescence. Journal of Consulting and Clinical Psychology. 2002;70:6–20. doi: 10.1037//0022-006x.70.1.6. [DOI] [PubMed] [Google Scholar]
- Cloninger CR. Comorbidity of anxiety and depression. Journal of Clinical Psychopharmacology. 1990;10:43S–46S. doi: 10.1097/00004714-199006001-00009. [DOI] [PubMed] [Google Scholar]
- Cloninger CR, Svrakic NM, Svrakic DM. Role of personality self-organization in development of mental order and disorder. Development and Psychopathology. 1997;9:881–906. doi: 10.1017/s095457949700148x. [DOI] [PubMed] [Google Scholar]
- Cohen J. Statistical power analysis for the behavioral sciences. 2. New York: Academic Press; 1988. [Google Scholar]
- Cohen P, Chen H, Crawford TN, Brook J, Gordon K. Personality disorders in early adolescence and the development of later substance use disorders in the general population. Drug and Alcohol Dependence. 2007;88:S71–S84. doi: 10.1016/j.drugalcdep.2006.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrod PJ, Peterson JB, Pihl RO. Disinhibited personality and sensitivity to alcohol reinforcement: Independent correlates of drinking behavior in sons of alcoholics. Alcoholism Clinical and Experimental Research. 1997;21:1320–1332. [PubMed] [Google Scholar]
- Conrod PJ, Peterson JB, Pihl RO, Mankowski S. Biphasic effects of alcohol on heart rate are influenced by alcoholic family history and rate of alcohol ingestion. Alcoholism Clinical and Experimental Research. 1997;21:140–149. [PubMed] [Google Scholar]
- Conrod PJ, Pihl RO, Stewart SH, Dongier M. Validation of a system of classifying female substance abusers on the basis of personality and motivational risk factors for substance abuse. Psychology of Addictive Behaviors. 2000;14:243–256. doi: 10.1037//0893-164x.14.3.243. [DOI] [PubMed] [Google Scholar]
- Conrod PJ, Pihl RO, Vassileva J. Differential sensitivity to alcohol reinforcement in groups of men at risk for distinct alcoholism subtypes. Alcoholism Clinical and Experimental Research. 1998;22:585–597. doi: 10.1111/j.1530-0277.1998.tb04297.x. [DOI] [PubMed] [Google Scholar]
- Conrod PJ, Stewart SH. A critical look at dual-focused cognitive-behavioral treatments for comorbid substance use and psychiatric disorders: Strengths, limitations, and future dirsctions. Journal of Cognitive Psychotherapy. 2005;19:261–284. [Google Scholar]
- Conrod PJ, Stewart SH, Comeau N, Maclean AM. Efficacy of cognitive–behavioral interventions targeting personality risk factors for youth alcohol misuse. Journal of Clinical Child and Adolescent Psychology. 2006;35:550–563. doi: 10.1207/s15374424jccp3504_6. [DOI] [PubMed] [Google Scholar]
- Cornblatt BA. Predictors of schizophrenia and preventive intervention. In: Breier A, Tran P, editors. Current issues in the psychopharmacology of schizophrenia. Philadelphia: Lippincott Williams & Wilkins; 2001. pp. 389–406. [Google Scholar]
- Cornblatt BA, Lencz T, Kane JM. Treating the schizophrenia prodrome: Is it presently ethical? Schizophrenia Research. 2001;51:31–38. doi: 10.1016/s0920-9964(01)00236-5. [DOI] [PubMed] [Google Scholar]
- Cornblatt BA, Malhotra AK. Impaired attention as an endophenotype for molecular genetics studies of schizophrenia. American Journal of Medical Genetics. 2001;105:11–15. [PubMed] [Google Scholar]
- Cornblatt BA, Obuchowski M, Roberts S, Pollack S, Erlenmeyer-Kimling L. Cognitive and behavioral precursors of schizophrenia. Development and Psychopathology. 1999;11:487–508. doi: 10.1017/s0954579499002175. [DOI] [PubMed] [Google Scholar]
- Corr PJ. Reinforcement sensitivity theory and personality. Neuroscience and Biobehavioral Reviews. 2004;28:317–332. doi: 10.1016/j.neubiorev.2004.01.005. [DOI] [PubMed] [Google Scholar]
- Coryell W, Turner R. Outcome with desipramine therapy in subtypes of nonpsychotic major depression. Journal of Affective Disorders. 1985;9:149–154. doi: 10.1016/0165-0327(85)90094-1. [DOI] [PubMed] [Google Scholar]
- Craddock N, O’Donovan MC, Owen MJ. Phenotypic and genetic complexity of psychosis: Invited commentary on schizophrenia: A common disease caused by multiple rare alleles. British Journal of Psychiatry. 2007;190:200–203. doi: 10.1192/bjp.bp.106.033761. [DOI] [PubMed] [Google Scholar]
- Crowell SE, Beauchaine TP, Lenzenweger M. The development of borderline personality and self-injurious behavior. In: Beauchaine TP, Hinshaw S, editors. Child psychopathology: Genetic, neurobiological, and environmental substrates. Hoboken, NJ: Wiley; 2008. pp. 510–541. [Google Scholar]
- Crowell S, Beauchaine TP, McCauley E, Smith C, Stevens AL, Sylvers P. Psychological, autonomic, and serotonergic correlates of parasuicidal behavior in adolescent girls. Development and Psychopathology. 2005;17:1105–1127. doi: 10.1017/s0954579405050522. [DOI] [PubMed] [Google Scholar]
- Crowell SE, Beauchaine TP, McCauley E, Smith CJ, Vasilev CA, Stevens AL. Parent–child interactions, peripheral serotonin, and self-inflicted injury in adolescents. Journal of Consulting and Clinical Psychology. 2008;76:15–21. doi: 10.1037/0022-006X.76.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson RJ. Affective neuroscience and psychophysiology: Toward a synthesis. Psychophysiology. 2003;40:655–665. doi: 10.1111/1469-8986.00067. [DOI] [PubMed] [Google Scholar]
- Davidson RJ, Pizzagalli D, Nitschke JB, Putnam KM. Depression: Perspectives from affective neuroscience. Annual Review of Psychology. 2002;53:545–574. doi: 10.1146/annurev.psych.53.100901.135148. [DOI] [PubMed] [Google Scholar]
- Davies PT, Sturge-Apple ML, Cicchetti D, Cummings EM. The role of child adrenocortical functioning in pathways between interparental conflict and child maladjustment. Developmental Psychology. 2007;43:918–930. doi: 10.1037/0012-1649.43.4.918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davison GC. Being bolder with the Boulder model: The challenge of education and training in empirically supported treatments. Journal of Consulting and Clinical Psychology. 1998;66:163–167. doi: 10.1037//0022-006x.66.1.163. [DOI] [PubMed] [Google Scholar]
- Dawes RM, Meehl PE. Mixed group validation: A method for determining the validity of diagnostic signs without using criterion groups. Psychological Bulletin. 1966;66:63–67. doi: 10.1037/h0023584. [DOI] [PubMed] [Google Scholar]
- Dawson G. Early behavioral intervention, brain plasticity, and the prevention of autism spectrum disorder. Development and Psychopathology. 2008;20:775–803. doi: 10.1017/S0954579408000370. [DOI] [PubMed] [Google Scholar]
- Dawson G, Webb S, Schellenberg G, Aylward E, Richards T, Dager S, Friedman S. Defining the phenotype of autism: Genetic, brain, and behavioral perspectives. Development and Psychopathology. 2002;14:581–611. doi: 10.1017/s0954579402003103. [DOI] [PubMed] [Google Scholar]
- Dawson G, Zanolli K. Early intervention and brain plasticity in autism. In: Bock G, Goode J, editors. Autism: Neural basis and treatment possibilities (Novartis Foundation Symposium. Vol. 251. Chichester: Wiley; 2003. pp. 266–280. [PubMed] [Google Scholar]
- DiClemente CC, Carbonari JP, Daniels JW, Donovan D, Bellino L, Neavins T. Self-efficacy as a matching hypothesis. Causal chain analysis. In: Longabaugh RH, Wirtz PW, editors. Project MATCH: A priori matching hypotheses, results, and mediating mechanisms (NIAAA Project MATCH Monograph Series. 8. Washington, DC: US Government Printing Office; 2001. pp. 239–257. [Google Scholar]
- Dishion TJ, McCord J, Poulin F. When interventions harm: Peer groups and problem behavior. American Psychologist. 1999;54:755–764. doi: 10.1037//0003-066x.54.9.755. [DOI] [PubMed] [Google Scholar]
- Dishion TJ, Patterson GR. Age effects in parent training outcome. Behavior Therapy. 1992;23:719–729. [Google Scholar]
- Donaldson SK, Klein DN, Riso LP, Schwartz JE. Comorbidity between dysthymic and major depressive disorders: A family study analysis. Journal of Affective Disorders. 1997;42:103–111. doi: 10.1016/s0165-0327(96)00106-1. [DOI] [PubMed] [Google Scholar]
- Drabick DAG, Beauchaine TP, Gadow KD, Carlson GA, Bromet EJ. Risk factors for conduct problems and depressive symptoms in a cohort of Ukrainian children. Journal of Clinical Child and Adolescent Psychology. 2006;35:244–252. doi: 10.1207/s15374424jccp3502_8. [DOI] [PubMed] [Google Scholar]
- Durston S. A review of the biological bases of ADHD: What have we learned from imaging studies? Mental Retardation & Developmental Disabilities Reviews. 2003;9:184–195. doi: 10.1002/mrdd.10079. [DOI] [PubMed] [Google Scholar]
- Ellis A. Rational–emotive therapy and cognitive behavior therapy: Similarities and differences. Cognitive Therapy and Research. 1981;4:325–340. [Google Scholar]
- El-Sheikh M. Does poor vagal tone exacerbate child maladjustment in the context of parental problem drinking? A longitudinal examination. Journal of Abnormal Psychology. 2005;114:735–741. doi: 10.1037/0021-843X.114.4.735. [DOI] [PubMed] [Google Scholar]
- El-Sheikh M, Harger J, Whitson SM. Exposure to interparental conflict and children’s adjustment and physical health: The moderating role of vagal tone. Child Development. 2001;72:1617–1636. doi: 10.1111/1467-8624.00369. [DOI] [PubMed] [Google Scholar]
- Epstein J, Hong P, Kocsis JH, Yang Y, Butler T, Chusid J. Lack of ventral striatal response to positive stimuli in depressed versus normal subjects. American Journal of Psychiatry. 2006;163:1784–1790. doi: 10.1176/ajp.2006.163.10.1784. [DOI] [PubMed] [Google Scholar]
- Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, Peterson DA, et al. Neurogenesis in the adult human hippocampus. Nature Medicine. 1998;4:1313–1317. doi: 10.1038/3305. [DOI] [PubMed] [Google Scholar]
- Erlenmeyer-Kimling L, Golden RR, Cornblatt BA. A taxometric analysis of cognitive and neuromotor variables in children at risk for schizophrenia. Journal of Abnormal Psychology. 1989;98:203–208. doi: 10.1037//0021-843x.98.3.203. [DOI] [PubMed] [Google Scholar]
- Evans DL, Foa EB, Gur RE, Hendin H, O’Brien CP, Seligman MEP, et al. Treating and preventing adolescent mental health disorders. New York: Oxford University Press; 2005. [Google Scholar]
- Felmingham K, Kemp A, Williams L, Das P, Hughes G, Peduto A, et al. Changes in anterior cingulated and amygdala after cognitive behavior therapy of posttraumatic stress disorder. Psychological Science. 2007;18:127–129. doi: 10.1111/j.1467-9280.2007.01860.x. [DOI] [PubMed] [Google Scholar]
- Ferdinand RF, Dieleman G, Ormel J, Verhulst FC. Homotypic versus heterotypic continuity of anxiety symptoms is adolescents: Evidence for distinction between DSM-IV subtypes. Journal of Abnormal Child Psychology. 2007;35:325–333. doi: 10.1007/s10802-006-9093-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fingeret MC, Warren CS, Cepeda-Benito A, Gleaves DH. Eating disorder prevention research: A meta-analysis. Journal of Treatment and Prevention. 2006;14:191–213. doi: 10.1080/10640260600638899. [DOI] [PubMed] [Google Scholar]
- Fishbein D. The importance of neurobiological research to the prevention of psychopathology. Prevention Science. 2000;1:89–106. doi: 10.1023/a:1010090114858. [DOI] [PubMed] [Google Scholar]
- Fishbein D, Hyde C, Coe B, Paschall MJ. Neurocognitive and physiological prerequisites for prevention of adolescent drug abuse. Journal of Primary Prevention. 2004;24:471–495. [Google Scholar]
- Fisher PA, Gunnar MR, Chamberlain P, Reid JB. Preventive intervention for maltreated pre-school children: Impact on children’s behavior, neuroendocrine activity, and foster parent functioning. Journal of the American Academy of Child & Adolescent Psychiatry. 2000;39:1356–1364. doi: 10.1097/00004583-200011000-00009. [DOI] [PubMed] [Google Scholar]
- Forbes EE, May JC, Siegle GJ, Ladouceur CD, Ryan ND, Carter CS, et al. Reward-related decision-making in pediatric major depressive disorder: An fMRI study. Journal of Child Psychology and Psychiatry. 2006;47:1031–1040. doi: 10.1111/j.1469-7610.2006.01673.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes EE, Shaw DS, Dahl RE. Alterations in reward-related decision making in boys with recent and future depression. Biological Psychiatry. 2007;61:633–639. doi: 10.1016/j.biopsych.2006.05.026. [DOI] [PubMed] [Google Scholar]
- Fowles DC, Dindo L. A dual-deficit model of psychopathy. In: Patrick CJ, editor. Handbook of psychopathy. New York: Guilford Press; 2006. pp. 14–34. [Google Scholar]
- Fox NA, Hane AA, Pine DS. Plasticity for affective neurocircuitry: How the environment shapes gene expression. Current Directions in Psychological Science. 2007;16:1–5. [Google Scholar]
- Foxcroft DR, Ireland D, Lister-Sharp DJ, Lowe G, Breen R. Longer-term primary prevention for alcohol misuse in young people: A systematic review. Addiction. 2003;98:397–411. doi: 10.1046/j.1360-0443.2003.00355.x. [DOI] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer SL, Crocker NA, Mattson SN. Exposure to teratogenic agents as a risk factor for psychopathology. In: Beauchaine TP, Hinshaw SP, editors. Child psychopathology: Genetic, neurobiological, and environmental substrates. Hoboken, NJ: Wiley; 2008. pp. 180–207. [Google Scholar]
- Gatzke-Kopp LM, Beauchaine TP. Central nervous system substrates of impulsivity: Implications for the development of attention-deficit/hyperactivity disorder and conduct disorder. In: Coch D, Dawson G, Fischer K, editors. Human behavior and the developing brain: Atypical development. New York: Guilford Press; 2007a. pp. 239–263. [Google Scholar]
- Gatzke-Kopp LM, Beauchaine TP. Direct and passive prenatal nicotine exposure and the development of externalizing psychopathology. Child Psychiatry and Human Development. 2007b doi: 10.1007/s10578-007-0059-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatzke-Kopp LM, Beauchaine TP, Shannon KE, Chipman-Chacon J, Fleming AP, Crowell SE, et al. Neurological correlates of reward responding in adolescents with and without externalizing disorders. 2007 doi: 10.1037/a0014378. Manuscript submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatzke-Kopp LM, Shannon KE. Brain injury as a risk factor for psychopathology. In: Beauchaine TP, Hinshaw SP, editors. Child and adolescent psychopathology. Hoboken, NJ: Wiley; 2008. pp. 208–233. [Google Scholar]
- Glahn DC, Therman S, Manninen M, Huttunen M, Kapiro J, Lönnqvist J, et al. Spatial working memory as an endophenotype for schizophrenia. Biological Psychiatry. 2003;53:624–626. doi: 10.1016/s0006-3223(02)01641-4. [DOI] [PubMed] [Google Scholar]
- Goldapple K, Segal Z, Garson C, Lau M, Bieling P, Kennedy S, et al. Modulation of cortical-limbic pathways in major depression: Treatment-specific effects of cognitive behavior therapy. Archives of General Psychiatry. 2004;61:34–41. doi: 10.1001/archpsyc.61.1.34. [DOI] [PubMed] [Google Scholar]
- Golden RR, Meehl PE. Detection of the schizoid taxon with MMPI indicators. Journal of Abnormal Psychology. 1979;88:212–233. doi: 10.1037//0021-843x.88.3.217. [DOI] [PubMed] [Google Scholar]
- Goldsmith HH, Gottesman II, Lemery KS. Epigenetic approaches to developmental psychopathology. Development and Psychopathology. 1997;9:365–387. doi: 10.1017/s0954579497002095. [DOI] [PubMed] [Google Scholar]
- Gottesman II, Gould TD. The endophenotype concept in psychiatry: Etymology and strategic intentions. American Journal of Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- Gottlieb G, Willoughby MT. Probabilistic epigenesis of psychopathology. In: Cicchetti D, Cohen DJ, editors. Developmental psychopathology: Vol. 1. Theory and method. 2. Hoboken, NJ: Wiley; 2006. pp. 673–700. [Google Scholar]
- Gould TD, Gottesman II. Psychiatric endophenotypes and the development of valid animal models. Genes Brain and Behavior. 2006;5:113–119. doi: 10.1111/j.1601-183X.2005.00186.x. [DOI] [PubMed] [Google Scholar]
- Gray JA, McNaughton N. The Neuropsychology of anxiety: An enquiry into the functions of the septo-hippocampal system. Oxford: Oxford University Press; 2000. [Google Scholar]
- Gunnar MR, Fisher PA. Bringing basic research on early experience and stress neurobiology to bear on preventive interventions for neglected and maltreated children. Development and Psychopathology. 2006;18:651–677. [PubMed] [Google Scholar]
- Halperin JM, Schulz KP. Revisiting the role of the prefrontal cortex in the pathophysiology of attention-deficit/hyperactivity disorder. Psychological Bulletin. 2006;132:560–581. doi: 10.1037/0033-2909.132.4.560. [DOI] [PubMed] [Google Scholar]
- Hammock EAD, Levitt P. The discipline of neurobehavioral development: The emerging interface of processes that build circuits and skills. Human Development. 2006;49:294–309. [Google Scholar]
- Hartl DL, Jones EW. Essential genetics: A genomics perspective. 3. Boston: Jones and Bartlett; 2002. [Google Scholar]
- Hawes DJ, Dadds MR. The treatment of conduct problems in children with callous-unemotional traits. Journal of Consulting and Clinical Psychology. 2005;73:737–741. doi: 10.1037/0022-006X.73.4.737. [DOI] [PubMed] [Google Scholar]
- Hicks BM, Blonigen DM, Kramer MD, Krueger RF, Patrick CJ, Iacono WG, et al. Gender differences and developmental change in externalizing disorders from late adolescence to early adulthood: A longitudinal twin study. Journal of Abnormal Psychology. 2007;116:433–447. doi: 10.1037/0021-843X.116.3.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinshaw SP. On the distinction between attention deficits/hyperactivity and conduct problems/aggression in child psychopathology. Psychological Bulletin. 1987;101:443–463. [PubMed] [Google Scholar]
- Hinshaw SP, Lee SS. Conduct and oppositional defiant disorders. In: Mash EJ, Barkley RA, editors. Child psychopathology. 2. New York: Guilford Press; 2003. pp. 144–198. [Google Scholar]
- Hull CL. Principles of behavior. New York: Appleton–Century–Crofts; 1943. [Google Scholar]
- Insel TR, Goodwin FK. The dexamethasone suppression test: Promises and problems of diagnostic laboratory tests in psychiatry. Hospital and Community Psychiatry. 1983;34:1131–1138. doi: 10.1176/ps.34.12.1131. [DOI] [PubMed] [Google Scholar]
- Jacobson KC, Prescott CA, Kendler KS. Genetic and environmental influences on juvenile antisocial behavior based on two occasions. Psychological Medicine. 2000;30:1315–1325. doi: 10.1017/s0033291799002780. [DOI] [PubMed] [Google Scholar]
- Jaffee SR, Caspi A, Moffitt TE, Dodge KA, Rutter M, Taylor A, et al. Nature×nurture: Genetic vulnerabilities interact with physical maltreatment to promote conduct problems. Development and Psychopathology. 2005;17:67–84. doi: 10.1017/s0954579405050042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen PS, Hinshaw SP, Kraemer HC, Lenora N, Newcorn JH, Abikoff HB, et al. ADHD comorbidity findings from the MTA study: Comparing comorbid subgroups. Journal of the American Academy of Child & Adolescent Psychiatry. 2001;40:147–158. doi: 10.1097/00004583-200102000-00009. [DOI] [PubMed] [Google Scholar]
- Jensen P, Hinshaw SP, Swanson J, Greenhill L, Conners CK, Arnold LE, et al. Findings from the NIMH Multimodal Treatment Study of ADHD (MTA): Implications and applications for primary care providers. Journal of Developmental and Behavioral Pediatrics. 2001;22:60–73. doi: 10.1097/00004703-200102000-00008. [DOI] [PubMed] [Google Scholar]
- Joffe JM. Mental disorders: Should our emphasis be on biological or psychosocial factors? An introduction to the special issue. Journal of Primary Prevention. 2004;24:415–418. [Google Scholar]
- Kaffman A, Meaney MJ. Neurodevelopmental sequelae of postnatal maternal care in rodents: Clinical and research implications of molecular insights. Journal of Child Psychology and Psychiatry. 2007;48:224–244. doi: 10.1111/j.1469-7610.2007.01730.x. [DOI] [PubMed] [Google Scholar]
- Kane VB, Fu Y, Matta SG, Sharp BM. Gestational nicotine exposure attenuates nicotine-stimulated dopamine release in the nucleus accumbens shell of adolescent Lewis rats. Journal of Pharmacological and Experimental Therapeutics. 2004;308:521–528. doi: 10.1124/jpet.103.059899. [DOI] [PubMed] [Google Scholar]
- Katz LF, Gottman JM. Vagal tone protects children from marital conflict. Development and Psychopathology. 1995;7:83–92. [Google Scholar]
- Katz LF, Gottman JM. Buffering children from marital conflict and dissolution. Journal of Clinical Child Psychology. 1997;26:157–171. doi: 10.1207/s15374424jccp2602_4. [DOI] [PubMed] [Google Scholar]
- Keedwell PA, Andrew C, Williams SCR, Brammer MJ, Phillips ML. The neural correlates of anhedonia in major depressive disorder. Biological Psychiatry. 2005;58:843–853. doi: 10.1016/j.biopsych.2005.05.019. [DOI] [PubMed] [Google Scholar]
- Kendler KS, Prescott CA, Myers J, Neale MC. The structure of genetic and environmental risk factors for common psychiatric and substance use disorders in men and women. Archives of General Psychiatry. 2003;60:929–937. doi: 10.1001/archpsyc.60.9.929. [DOI] [PubMed] [Google Scholar]
- Kent L, Green E, Hawi Z, Kirley A, Dudbridge F, Lowe N, et al. Association of the paternally transmitted copy of common valine allele of the Val66-Met polymorphism of the brain-derived neurotrophic factor (BDNF) gene with susceptibility to ADHD. Molecular Psychiatry. 2005;10:939–943. doi: 10.1038/sj.mp.4001696. [DOI] [PubMed] [Google Scholar]
- Klein DN, Torpey DC, Bufferd SJ, Dyson MW. Depressive disorders. In: Beauchaine TP, Hinshaw SP, editors. Child and adolescent psychopathology. Hoboken, NJ: Wiley; 2008. pp. 477–509. [Google Scholar]
- Klein-Hessling J, Lohaus A. Benefits and inter-individual differences in children’s responses to extended and intensified relaxation training. Anxiety Stress and Coping. 2002;15:275–288. [Google Scholar]
- Klinger LG, Dawson G, Renner P. Autistic disorder. In: Mash EJ, Barkley RA, editors. Child psychopathology. 2. New York: Guilford Press; 2003. pp. 409–454. [Google Scholar]
- Klump KL, McGue M, Iacono WG. Differential heritability of eating attitudes and behaviors in prepubertal versus pubertal twins. International Journal of Eating Disorders. 2000;33:287–292. doi: 10.1002/eat.10151. [DOI] [PubMed] [Google Scholar]
- Konorski J. Conditioned reflexes and neuron organization. Cambridge: Cambridge University Press; 1948. [Google Scholar]
- Koopsman JR, Slutzke WS, Heath AC, Neale MC, Boomsma DI. The genetics of smoking initiation and quantity smoked in Dutch adolescent and young adult twins. Behavior Genetics. 1999;29:383–393. doi: 10.1023/a:1021618719735. [DOI] [PubMed] [Google Scholar]
- Koopsman JR, van Doornen LJ, Boomsma DI. Association between alcohol use and smoking in adolescent and young adult twins: A bivariate genetic analysis. Alcoholism: Clinical and Experimental Research. 1997;21:537–546. [PubMed] [Google Scholar]
- Kopp LM, Beauchaine TP. Patterns of psychopathology in the families of children with conduct problems, depression, and both psychiatric conditions. Journal of Abnormal Child Psychology. 2007;35:301–312. doi: 10.1007/s10802-006-9091-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korfine L, Lenzenweger MF. The taxonicity of schizotypy: A replication. Journal of Abnormal Psychology. 1995;104:26–31. doi: 10.1037//0021-843x.104.1.26. [DOI] [PubMed] [Google Scholar]
- Kraemer HC, Stice E, Kazdin A, Offord D, Kupfer D. How do risk factors work together? Mediators, moderators, and independent, overlapping, and proxy risk factors. American Journal of Psychiatry. 2001;158:848–856. doi: 10.1176/appi.ajp.158.6.848. [DOI] [PubMed] [Google Scholar]
- Kraemer HC, Wilson GT, Fairburn CG, Agras WS. Mediators and moderators of treatment effects in randomized clinical trials. Archives of General Psychiatry. 2002;59:877–883. doi: 10.1001/archpsyc.59.10.877. [DOI] [PubMed] [Google Scholar]
- Kramer DA. Commentary: Gene–environment interplay in the context of genetics, epigenetics, and gene expression. Journal of the American Academy of Child & Adolescent Psychiatry. 2005;44:19–27. doi: 10.1097/01.chi.0000145804.30112.6b. [DOI] [PubMed] [Google Scholar]
- Krueger RF, Hicks BM, Patrick CJ, Carlson SR, Iacono WG, McGue M. Etiologic connections among substance dependence, antisocial behavior, and personality: Modeling the externalizing spectrum. Journal of Abnormal Psychology. 2002;111:411–424. [PubMed] [Google Scholar]
- Krueger RF, Markon KE. Reinterpreting co-morbidity: A model-based approach to understanding and classifying psychopathology. Annual Review of Clinical Psychology. 2006;2:111–133. doi: 10.1146/annurev.clinpsy.2.022305.095213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn T. The structure of scientific revolutions. Chicago: University of Chicago Press; 1962. [Google Scholar]
- Kumari V. Do Psychotherapies produce neurobiological effects? Acta Neuropsychiatrica. 2006;18:61–70. doi: 10.1111/j.1601-5215.2006.00127.x. [DOI] [PubMed] [Google Scholar]
- Kupper N, Willemsen G, Posthuma D, De Boer D, Boomsma DI, De Geus EJC. A genetic analysis of ambulatory cardiorespiratory coupling. Psychophysiology. 2005;42:202–212. doi: 10.1111/j.1469-8986.2005.00276.x. [DOI] [PubMed] [Google Scholar]
- Laakso A, Wallius E, Kajander J, Bergman J, Eskola O, Solin O, et al. Personality traits and striatal dopamine synthesis capacity in healthy subjects. American Journal of Psychiatry. 2003;160:904–910. doi: 10.1176/appi.ajp.160.5.904. [DOI] [PubMed] [Google Scholar]
- Lemery KS, Doelger L. Genetic vulnerabilities to the development of psychopathology. In: Hankin BL, Abela JRZ, editors. Development of psychopathology: A vulnerability–stress perspective. Thousand Oaks, CA: Sage; 2005. pp. 161–198. [Google Scholar]
- Lenzenweger MF. Deeper into the schizotypy taxon: On the robust nature of maximum covariance analysis. Journal of Abnormal Psychology. 1999;108:182–187. doi: 10.1037//0021-843x.108.1.182. [DOI] [PubMed] [Google Scholar]
- Lenzenweger MF. Schizotaxia, schizotypy and schizophrenia: Paul E. Meehl’s blueprint for experimental psychopathology and the genetics of schizophrenia. Journal of Abnormal Psychology. 2006;115:195–200. doi: 10.1037/0021-843X.115.2.195. [DOI] [PubMed] [Google Scholar]
- Lenzenweger MF, Korfine L. Confirming the latent structure and base rate of schizotypy: A taxometric analysis. Journal of Abnormal Psychology. 1992;101:567–571. doi: 10.1037//0021-843x.101.3.567. [DOI] [PubMed] [Google Scholar]
- Lenzenweger MF, McLachlan G, Rubin DB. Resolving the latent structure of schizophrenia endophenotypes using expectation-maximization-based finite mixture modeling. Journal of Abnormal Psychology. 2007;116:16–29. doi: 10.1037/0021-843X.116.1.16. [DOI] [PubMed] [Google Scholar]
- Lewinsohn PM, Shankman SA, Gau JM, Klein DN. The prevalence and co-morbidity of sub-threshold psychiatric conditions. Psychological Medicine. 2004;34:613–622. doi: 10.1017/S0033291703001466. [DOI] [PubMed] [Google Scholar]
- Leyton M, Boileau I, Benkelfat C, Diksic M, Baker G, Dagher A. Amphetamine-induced increases in extracellular dopamine, drug wanting and novelty seeking: A PET/[11C]Raclopride study in healthy men. Neuropsychopharmacology. 2002;27:1027–1035. doi: 10.1016/S0893-133X(02)00366-4. [DOI] [PubMed] [Google Scholar]
- Lilienfeld SO. Psychological treatments that cause harm. Perspectives on Psychological Science. 2007;2:53–70. doi: 10.1111/j.1745-6916.2007.00029.x. [DOI] [PubMed] [Google Scholar]
- Lilienfeld SO, Marino L. Essentialism revisited: Evolutionary theory and the concept of mental disorder. Journal of Abnormal Psychology. 1999;108:400–411. doi: 10.1037//0021-843x.108.3.400. [DOI] [PubMed] [Google Scholar]
- Linehan MM. Cognitive–behavioral treatment of borderline personality disorder. New York: Guilford Press; 1993. [Google Scholar]
- Lochman JE. Cognitive–behavioral intervention with aggressive boys: Three-year follow-up and preventive effects. Journal of Consulting and Clinical Psychology. 1992;60:426–432. doi: 10.1037//0022-006x.60.3.426. [DOI] [PubMed] [Google Scholar]
- Loeber R, Hay D. Key issues in the development of aggression and violence from childhood to early adulthood. Annual Review of Psychology. 1997;48:371–410. doi: 10.1146/annurev.psych.48.1.371. [DOI] [PubMed] [Google Scholar]
- Loeber R, Keenan K. Interaction between conduct disorder and its comorbid conditions: Effects of age and gender. Clinical Psychology Review. 1994;14:497–523. [Google Scholar]
- Luthar SS, Cicchetti D, Becker B. The construct of resilience: A critical evaluation and guidelines for future work. Child Development. 2000;71:543–562. doi: 10.1111/1467-8624.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykken DT. Psychopathic personality. In: Patrick CJ, editor. Handbook of psychopathy. New York: Guilford Press; 2006. pp. 3–13. [Google Scholar]
- Lynam DR. The early identification of chronic offenders: Who is the fledgling psychopath? Psychological Bulletin. 1996;120:209–234. doi: 10.1037/0033-2909.120.2.209. [DOI] [PubMed] [Google Scholar]
- Lynam DR. Early identification of the fledgling psychopath: Locating the psychopathic child in the current nomenclature. Journal of Abnormal Psychology. 1998;107:566–575. doi: 10.1037//0021-843x.107.4.566. [DOI] [PubMed] [Google Scholar]
- Lynam DR, Caspi A, Moffitt TE, Wikström PH, Loeber R, Novak S. The interaction between impulsivity and neighborhood context on offending: The effects of impulsivity are stronger in poorer neighborhoods. Journal of Abnormal Psychology. 2000;109:563–574. doi: 10.1037//0021-843x.109.4.563. [DOI] [PubMed] [Google Scholar]
- Lynam DR, Milich R, Zimmerman R, Novak SP, Logan TK, Martin C, et al. Project DARE: No effects at 10-year follow-up. Journal of Consulting and Clinical Psychology. 1999;67:590–593. doi: 10.1037//0022-006x.67.4.590. [DOI] [PubMed] [Google Scholar]
- Lyons MJ, True WR, Eisen SA, Goldberg J, Meyer JM, Faraone SV, et al. Effects of genes and environment on antisocial traits. Archives of General Psychiatry. 1995;52:906–915. doi: 10.1001/archpsyc.1995.03950230020005. [DOI] [PubMed] [Google Scholar]
- MacCallum RC, Zhang S, Preacher KJ, Rucker DD. On the practice of dichotomization of quantitative variables. Psychological Methods. 2001;7:19–40. doi: 10.1037/1082-989x.7.1.19. [DOI] [PubMed] [Google Scholar]
- Masten AS. Competence, resilience, and development in adolescence: Clues for prevention science. In: Romer D, Walker EF, editors. Adolescent psychopathology and the developing brain. Oxford: Oxford University Press; 2007. pp. 31–52. [Google Scholar]
- Masterman PW, Kelly AB. Reaching adolescents who drink harmfully: Fitting intervention to developmental reality. Journal of Substance Abuse Treatment. 2003;24:347–355. doi: 10.1016/s0740-5472(03)00047-3. [DOI] [PubMed] [Google Scholar]
- McClellan JM, Susser E, King MC. Schizophrenia: A common disease caused by multiple rare alleles. British Journal of Psychiatry. 2007;190:194–199. doi: 10.1192/bjp.bp.106.025585. [DOI] [PubMed] [Google Scholar]
- McGorry PD, Yung AR, Phillips LJ, Yuen HP, Francey S, Cosgrave EM, et al. Randomized controlled trial of interventions designed to reduce the risk of progression to firstepisode psychosis in a clinical sample with sub-threshold symptoms. Archives of General Psychiatry. 2002;59:921–928. doi: 10.1001/archpsyc.59.10.921. [DOI] [PubMed] [Google Scholar]
- McGue M, Iacono WG, Legrand LN, Elkins I. Origins and consequences of age at first drink: II. Familial risk and heritability. Alcoholism: Clinical and Experimental Research. 2001;25:1166–1173. [PubMed] [Google Scholar]
- Meany MJ. Maternal programming of defensive responses through sustained effects on gene expression. In: Romer D, Walker EF, editors. Adolescent psychopathology and the developing brain. Oxford: Oxford University Press; 2007. pp. 148–172. [Google Scholar]
- Meehl PE. Schizotaxia, schizotypy, schizophrenia. American Psychologist. 1962;17:827–838. [Google Scholar]
- Miklowitz DJ. The role of the family in the course and treatment of bipolar disorder. Current Directions in Psychological Science. 2007;16:192–196. doi: 10.1111/j.1467-8721.2007.00502.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt T, Caspi A. Childhood predictors differentiate life-course persistent and adolescence-limited antisocial pathways among males and females. Development and Psychopathology. 2001;13:355–375. doi: 10.1017/s0954579401002097. [DOI] [PubMed] [Google Scholar]
- Moffitt T, Caspi A, Rutter M. Measured gene–environment interactions in psychopathology: Concepts, research strategies, and implications for research, intervention, and public understanding of genetics. Perspectives on Psychological Science. 2006;1:5–27. doi: 10.1111/j.1745-6916.2006.00002.x. [DOI] [PubMed] [Google Scholar]
- Molina BSG, Flory K, Hinshaw SP, Greiner AR, Arnold LE, Swanson JM, et al. Delinquent behavior and emerging substance use in the MTA at 36 months: Prevalence, course, and treatment effects. Journal of the American Academy of Child & Adolescent Psychiatry. 2007;46:1028–1040. doi: 10.1097/chi.0b013e3180686d96. [DOI] [PubMed] [Google Scholar]
- Monti PM, Colby S, Barnett NP, Spirito A, Rohsenow DJ, Myers M, et al. Brief intervention for harm reduction with alcohol-positive older adolescents in a hospital emergency department. Journal of Consulting and Clinical Psychology. 1999;67:989–994. doi: 10.1037//0022-006x.67.6.989. [DOI] [PubMed] [Google Scholar]
- MTA Cooperative Group. A 14-month randomized clinical trial of treatment strategies for attention/deficit hyperactivity disorder. Archives of General Psychiatry. 1999;56:1073–1086. doi: 10.1001/archpsyc.56.12.1073. [DOI] [PubMed] [Google Scholar]
- Nadder TS, Rutter M, Silberg JL, Maes HH, Leaves LJ. Genetic effects on the variation and covariation of attention deficit-hyperactivity disorder (ADHD) and oppositional-defiant disorder/conduct disorder (ODD/CD) symptomatologies across informant and occasion of measurement. Psychological Medicine. 2002;32:39–53. doi: 10.1017/s0033291701004792. [DOI] [PubMed] [Google Scholar]
- Nelson CA, Bloom FE, Cameron JL, Amaral D, Dahl RE, Pine D. An integrative, multidisciplinary approach to the study of brain–behavior relations in the context of typical and atypical development. Development and Psychopathology. 2002;14:499–520. doi: 10.1017/s0954579402003061. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Carlezon WA. The mesolimbic dopamine reward circuit in depression. Biological Psychiatry. 2006;59:1151–1159. doi: 10.1016/j.biopsych.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Nock MK. Progress review of the psychosocial treatment of child conduct problems. Clinical Psychology Science and Practice. 2003;10:1–28. [Google Scholar]
- O’Connor TG, McGuire S, Reiss D, Hetherington EM, Plomin R. Co-occurrence of depressive symptoms and antisocial behavior in adolescence: A common genetic liability. Journal of Abnormal Psychology. 1998;107:27–37. doi: 10.1037//0021-843x.107.1.27. [DOI] [PubMed] [Google Scholar]
- Oliff HS, Gallardo KA. The effects of nicotine on developing catecholamine systems. Frontiers in Bioscience. 1999;4:d833–d897. doi: 10.2741/oliff. [DOI] [PubMed] [Google Scholar]
- O’Malley KD, Nanson J. Clinical implications of a link between fetal alcohol spectrum disorder and attention-deficit hyperactivity disorder. Canadian Journal of Psychiatry. 2002;47:349–354. doi: 10.1177/070674370204700405. [DOI] [PubMed] [Google Scholar]
- Orr SP, Metzger LJ, Lasko NB, Macklin ML, Hu FB, Shalev AY, Pitman RK. Physiologic responses to sudden, loud tones in monozygotic twins discordant for combat exposure: Association with posttraumatic stress disorder. Archives of General Psychiatry. 2003;60:283–288. doi: 10.1001/archpsyc.60.3.283. [DOI] [PubMed] [Google Scholar]
- Paquette V, Lévesque J, Mensour B, Leroux JM, Beaudoin G, Bourgouin P, et al. Change the mind and you change the brain: Effects of cognitive–behavioral therapy on the neural correlates of spider phobia. Neuroimage. 2003;18:401–409. doi: 10.1016/s1053-8119(02)00030-7. [DOI] [PubMed] [Google Scholar]
- Patterson GR, Capaldi DM. A mediational model for boys’ depressed mood. In: Rolf J, Masten AS, Cicchetti D, Nuechterlein KH, Weintraub S, editors. Risk and protective factors in the development of psychopathology. New York: Cambridge University Press; 1990. pp. 141–163. [Google Scholar]
- Patterson GR, DeBaryshe BD, Ramsey E. A developmental perspective on antisocial behavior. American Psychologist. 1989;44:329–335. doi: 10.1037//0003-066x.44.2.329. [DOI] [PubMed] [Google Scholar]
- Patterson GR, DeGarmo DS, Knutson NM. Hyperactive and antisocial behaviors: Comorbid or two points in the same process? Development and Psychopathology. 2000;12:91–107. doi: 10.1017/s0954579400001061. [DOI] [PubMed] [Google Scholar]
- Perry BD. Child maltreatment: A neurodevelopmental perspective on the role of abuse and neglect in Psychopathology. In: Beauchaine TP, Hinshaw SP, editors. Child psychopathology: Genetic, neurobiological, and environmental substrates. Hoboken, NJ: Wiley; 2008. pp. 93–128. [Google Scholar]
- Petrosino A, Turpin-Petrosino C, Buehler J. “Scared Straight” and other juvenile awareness programs for preventing juvenile delinquency. Annals of the American Academy of Political and Social Science. 2003;589:41–62. [Google Scholar]
- Pollak SD. Early adversity and mechanisms of plasticity: Integrating effective neuroscience with developmental approaches to psychopathology. Development and Psychopathology. 2005;17:735–752. doi: 10.1017/S0954579405050352. [DOI] [PubMed] [Google Scholar]
- Porges SW. Asserting the role of biobehavioral; sciences in translational research: The behavioral neurobiology revolution. Development and Psychopathology. 2006;18:923–933. doi: 10.1017/s0954579406060457. [DOI] [PubMed] [Google Scholar]
- Project MATCH Research Group. Matching alcoholism treatments to client heterogeneity: Project MATCH posttreatment drinking outcomes. Journal of Studies on Alcohol. 1997;58:7–29. [PubMed] [Google Scholar]
- Raine A, Mellingen K, Liu J, Venables P, Mednick S. Effects of environmental enrichment at ages 3–5 years on schizotypal personality and antisocial behavior at ages 17 and 23 years. American Journal of Psychiatry. 2003;160:1627–1635. doi: 10.1176/appi.ajp.160.9.1627. [DOI] [PubMed] [Google Scholar]
- Raine A, Venables P, Dalais C, Mellingen K, Reynolds C, Mednick S. Early educational and health enrichment at age 3–5 years is associated with increased autonomic and central nervous system arousal and orienting at age 11 years: Evidence from the Mauritius Child Health Project. Psychophysiology. 2001;38:254–266. [PubMed] [Google Scholar]
- Rapoport JC, Addington AM, Frangou S. The neurodevelopmental model of schizophrenia: Update 2005. Molecular Psychiatry. 2005;10:434–449. doi: 10.1038/sj.mp.4001642. [DOI] [PubMed] [Google Scholar]
- Reichenberg A, Harvey PD. Neuropsychological impairments in schizophrenia: Integration of performance-based and brain findings. Psychological Bulletin. 2007;133:833–858. doi: 10.1037/0033-2909.133.5.833. [DOI] [PubMed] [Google Scholar]
- Rhule DM. Take care not to do harm: Harmful interventions for youth problem behavior. Professional Psychology: Research and Practice. 2005;36:618–625. [Google Scholar]
- Rice F, Harold GT, Thapar A. The genetic aetiology of childhood depression: A review. Journal of Child Psychology and Psychiatry. 2002;43:65–79. doi: 10.1111/1469-7610.00004. [DOI] [PubMed] [Google Scholar]
- Ross RG. Early expression of a pathophysiological feature of schizophrenia: Saccadic intrusions into smooth-pursuit eye movements in school-age children vulnerable to schizophrenia. Journal of the American Academy of Child & Adolescent Psychiatry. 2003;42:468–476. doi: 10.1097/01.CHI.0000046818.95464.42. [DOI] [PubMed] [Google Scholar]
- Rueda MR, Rothbart MK, Saccomanno L, Posner MI. Modifying brain networks underlying self-regulation. In: Romer D, Walker EF, editors. Adolescent psychopathology and the developing brain. Oxford: Oxford University Press; 2007. pp. 401–419. [Google Scholar]
- Ruma PR, Burke RV, Thompson RW. Group parent training: Is it effective for children of all ages? Behavior Therapy. 1996;27:159–169. [Google Scholar]
- Rutter M. The interplay of nature, nurture, and developmental influences: The challenge ahead for mental health. Archives of General Psychiatry. 2002;59:996–1000. doi: 10.1001/archpsyc.59.11.996. [DOI] [PubMed] [Google Scholar]
- Rutter M. Environmentally mediated risk for psychopathology: Research strategies and findings. Journal of the American Academy of Child & Adolescent Psychiatry. 2005;44:3–18. doi: 10.1097/01.chi.0000145374.45992.c9. [DOI] [PubMed] [Google Scholar]
- Rutter M. Gene–environment interdependence. Developmental Science. 2007;10:12–18. doi: 10.1111/j.1467-7687.2007.00557.x. [DOI] [PubMed] [Google Scholar]
- Rutter M, Dunn J, Plomin R, Simonoff E, Pickles A, Maughan B, et al. Integrating nature and nurture: Implications of person-environment correlations and interactions for developmental psychopathology. Development and Psychopathology. 1997;9:335–364. doi: 10.1017/s0954579497002083. [DOI] [PubMed] [Google Scholar]
- Rutter M, Sroufe LA. Developmental psychopathology: Concepts and challenges. Development and Psychopathology. 2000;12:265–296. doi: 10.1017/s0954579400003023. [DOI] [PubMed] [Google Scholar]
- Sagvolden T, Johansen EB, Aase H, Russell VA. A dynamic developmental theory of attention-deficit/hyperactivity disorder (ADHD) predominantly hyperactive/impulsive and combined subtypes. Behavioral and Brain Sciences. 2005;28:397–468. doi: 10.1017/S0140525X05000075. [DOI] [PubMed] [Google Scholar]
- Satterfield JH, Faller KJ, Crinella FM, Schell AM, Swanson JM, Homer LD. A 30-year prospective follow-up study of hyperactive boys with conduct problems: Adult criminality. Journal of the American Academy of Child & Adolescent Psychiatry. 2007;46:601–610. doi: 10.1097/chi.0b013e318033ff59. [DOI] [PubMed] [Google Scholar]
- Schellenberg GD, Dawson G, Sung YJ, Estes A, Munson J, Rosenthal E, et al. Evidence for multiple loci from a genome scan of autism kindreds. Molecular Psychiatry. 2006;11:1049–1060. doi: 10.1038/sj.mp.4001874. [DOI] [PubMed] [Google Scholar]
- Scheres A, Milham MP, Knutson B, Castellanos FX. Ventral striatal hyporesponsiveness during reward anticipation in attention-deficit/hyperactivity disorder. Biological Psychiatry. 2007;61:720–724. doi: 10.1016/j.biopsych.2006.04.042. [DOI] [PubMed] [Google Scholar]
- Schnell K, Herpertz SC. Effects of dialectical behavior-therapy on the neural correlates of affective hyperarousal in borderline personality disorder. Journal of Psychiatric Research. 2007;41:837–847. doi: 10.1016/j.jpsychires.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Scourfield J, Rice F, Thapar A, Harold GT, Martin N, McGuffin P. Depressive symptoms in children and adolescents: Changing aetiological influences with development. Journal of Child Psychology and Psychiatry. 2003;44:968–976. doi: 10.1111/1469-7610.00181. [DOI] [PubMed] [Google Scholar]
- Seeger G, Schloss P, Schmidt MH, Rüter-Jungfleisch A, Henn FA. Gene–environment interaction in hyperkinetic conduct disorder (HD + CD) as indicated by season of birth variations in dopamine receptor (DRD4) gene polymorphism. Neuroscience Letters. 2004;366:282–286. doi: 10.1016/j.neulet.2004.05.049. [DOI] [PubMed] [Google Scholar]
- Shankman SA, Klein DN, Tenke CE, Bruder GE. Reward sensitivity in depression: A biobehavioral study. Journal of Abnormal Psychology. 2007;116:95–104. doi: 10.1037/0021-843X.116.1.95. [DOI] [PubMed] [Google Scholar]
- Shannon KE, Beauchaine TP, Brenner SL, Neuhaus E, Gatzke-Kopp L. Familial and temperamental predictors of resilience in children at risk for conduct disorder and depression. Development and Psychopathology. 2007;19:701–727. doi: 10.1017/S0954579407000351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegle GJ, Carter CS, Thase ME. Use of fMRI to predict recovery from unipolar depression with cognitive behavior therapy. American Journal of Psychiatry. 2006;163:735–738. doi: 10.1176/ajp.2006.163.4.735. [DOI] [PubMed] [Google Scholar]
- Silberg JL, Rutter M, Eaves L. Genetic and environmental influences on the temporal association between earlier anxiety and later depression in girls. Biological Psychiatry. 2001;49:1040–1049. doi: 10.1016/s0006-3223(01)01161-1. [DOI] [PubMed] [Google Scholar]
- Silberg J, Rutter M, Neale M, Eaves L. Genetic moderation of environmental risk for depression and anxiety in girls. British Journal of Psychiatry. 2001;179:116–121. doi: 10.1192/bjp.179.2.116. [DOI] [PubMed] [Google Scholar]
- Silvestri AJ, Joffe JM. You’d have to be sick not to be crazy. Journal of Primary Prevention. 2004;24:497–511. [Google Scholar]
- Skinner BF. The behavior of organisms: An experimental analysis. New York: Appleton–Century; 1938. [Google Scholar]
- Slotkin TA. Fetal nicotine or cocaine exposure: Which one is worse? Journal of Pharmacology and Experimental Therapeutics. 1998;285:931–945. [PubMed] [Google Scholar]
- Snyder J, Schrepferman L, St Peter C. Origins of antisocial behavior: Negative reinforcement and affect dysregulation of behavior as socialization mechanisms in family interaction. Behavior Modification. 1997;21:187–215. doi: 10.1177/01454455970212004. [DOI] [PubMed] [Google Scholar]
- Sowell ER, Peterson BS, Thompson PM, Welcome SE, Henkenius AL, Toga AW. Mapping cortical change across the human lifespan. Nature Neuroscience. 2003;6:309–315. doi: 10.1038/nn1008. [DOI] [PubMed] [Google Scholar]
- Spear L. The developing brain and adolescent-typical behavior patterns: An evolutionary approach. In: Romer D, Walker EF, editors. Adolescent psychopathology and the developing brain. Oxford: Oxford University Press; 2007. pp. 9–30. [Google Scholar]
- Sroufe LA, Rutter M. The domain of developmental psychopathology. Child Development. 1984;55:17–29. [PubMed] [Google Scholar]
- Stein MB, Jang KL, Taylor S, Vernon PA, Livesley WJ. Genetic and environmental influences on trauma exposure and posttraumatic stress disorder symptoms: A twin study. American Journal of Psychiatry. 2002;159:1675–1681. doi: 10.1176/appi.ajp.159.10.1675. [DOI] [PubMed] [Google Scholar]
- Stewart SH, Conrod PJ, Marlatt GA, Comeau N, Thush C, Krank M. New developments in prevention and early intervention for alcohol abuse in youths. Alcoholism Clinical and Experimental Research. 2005;29:278–286. doi: 10.1097/01.alc.0000153547.34399.e8. [DOI] [PubMed] [Google Scholar]
- Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depresssive disorder: Review and meta-analysis. American Journal of Psychiatry. 2000;157:1552–1562. doi: 10.1176/appi.ajp.157.10.1552. [DOI] [PubMed] [Google Scholar]
- Thompson EA, Eggert LL, Randell BP, Pike KC. Evaluation of indicated suicide risk prevention approaches for potential high school dropouts. American Journal of Public Health. 2001;91:742–752. doi: 10.2105/ajph.91.5.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson EA, Horn M, Herting JR, Eggert LA. Enhancing outcomes in an indicated drug prevention program for high-risk youth. Journal of Drug Education. 1997;27:19 –41. doi: 10.2190/G6XY-U2CQ-GFQF-PNEV. [DOI] [PubMed] [Google Scholar]
- Thorndike EL. Animal intelligence. New York: Macmillan; 1911. [Google Scholar]
- Tremblay RE. Towards an epigenetic approach to experimental criminology: The 2004 Joan McCord Prize Lecture. Journal of Experimental Criminology. 2005;1:397–415. [Google Scholar]
- Tunbridge EM, Weickert CS, Kleinman JE, Herman MM, Chen J, Kolachana BS, et al. Catechol-o-methyltransferase enzyme activity and protein expression in human prefrontal cortex across the postnatal lifespan. Cerebral Cortex. 2007;17:1206–1212. doi: 10.1093/cercor/bhl032. [DOI] [PubMed] [Google Scholar]
- Tyrka AR, Cannon TD, Haslam N, Mednick SA, Schulsinger F, Schulsinger H, et al. The latent structure of schizotypy: I. Premorbid indicators of a taxon in individuals at risk for schizophrenia-spectrum disorders. Journal of Abnormal Psychology. 1995;104:173–183. doi: 10.1037//0021-843x.104.1.173. [DOI] [PubMed] [Google Scholar]
- Vaidya C, Austin G, Kirkorian G, Ridlehuber HW, Desmond JE, Glover G, et al. Selective effects of methylphenidate in attention deficit hyperactivity disorder: A functional magnetic resonance study. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:14494–14499. doi: 10.1073/pnas.95.24.14494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viken RJ, Kaprio J, Koskenvuo M, Rose RJ. Longitudinal analyses of the determinants of drinking and of drinking to intoxication in adolescent twins. Behavior Genetics. 1999;29:455–461. doi: 10.1023/a:1021631122461. [DOI] [PubMed] [Google Scholar]
- Weaver ICG, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, et al. Epigenetic programming by maternal behavior. Nature Neuroscience. 2004;7:847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- Whisman MA, McClelland GH. Designing, testing, and interpreting interactions and moderator effects in family research. Journal of Family Psychology. 2005;19:111–120. doi: 10.1037/0893-3200.19.1.111. [DOI] [PubMed] [Google Scholar]
- Young RM, Lawford BR, Nutting A, Noble EP. Advances in molecular genetics and the prevention and treatment of substance misuse: Implications of association studies of the A1allele of the D2dopamine receptor gene. Addictive Behaviors. 2004;29:1275–1294. doi: 10.1016/j.addbeh.2004.06.012. [DOI] [PubMed] [Google Scholar]