

Abstract
The α-helical coiled-coil is one of the most common oligomerization motifs found in both native and engineered proteins. To better understand the stability and dynamics of coiled-coil motifs, including those modified by fluorination, several fluorinated and non-fluorinated parallel dimeric coiled-coil protein structures were designed and modeled. We also attempt to investigate how changing the length and geometry of the important stabilizing salt bridges influences the coiled-coil protein structure. Molecular dynamics (MD) and free energy simulations with AMBER employed a particle mesh Ewald treatment of the electrostatics in explicit TIP3P solvent with balanced force field treatments. Preliminary studies with legacy force fields (ff94, ff96, ff99) show a profound instability of the coiled-coil structures in short MD simulation. Significantly better behavior is evident with the more balanced ff99SB and ff03 protein force fields. Overall, the results suggest that the coiled-coil structures can readily accommodate the larger acidic arginine or S-2,7-diaminoheptanedoic acid mutants in the salt bridge, whereas substitution of the smaller L-ornithine residue leads to rapid disruption of the coiled-coil structure on the MD simulation time scale. This structural distortion of the secondary structure allows both the formation of large hydration pockets proximal to the charged groups and within the hydrophobic core. Moreover, the increased structural fluctuations and movement lead to a decrease in the water occupancy lifetimes in the hydration pockets. In contrast, analysis of the hydration in the stable dimeric coiled coils shows high occupancy water sites along the backbone residues with no water occupancy in the hydrophobic core, although transitory water interactions with the salt bridge residues are evident. The simulations of the fluorinated coiled-coils suggest that in some cases fluorination electrostatically stabilizes the intermolecular coiled-coil salt bridges. Structural analyses also reveal different side chain rotamer preferences for leucine compared to 5,5,5,5′,5′,5′-hexafluoroleucine mutants. These observed differences in the side chain rotamer populations suggest differential changes in the side chain conformational entropy upon coiled-coil formation when the protein is fluorinated. The free energy of hydration of the isolated 5,5,5,5′,5′,5′-hexafluoroleucine amino acid is calculated to be 1.1 kcal/mol less stable than leucine; this hydrophobic penalty in the monomer may provide a driving force for coiled-coil dimer formation. Estimation of the ellipticity at 222 nm from a series of snapshots from the MD simulations with DicroCalc show distinct increases in the ellipticity when the coiled-coil is fluorinated which suggests that the helicity in the folded coiled-coils is greater when fluorinated.
Keywords: computational chemistry; free energy of hydration; 5, 5, 5, 5′, 5′, 5′-hexafluoroleucine; thermodynamic integration; rotamers
Introduction
Coiled-coils are a very common and well studied protein motif composed of two or more α-helices wound around each other1–6. They can form both homo- and hetero-multimers with two to seven helices7 and can have helix orientations either parallel or anti-parallel. Their regular structure and ease of synthesis has led to their use as the basis for stimuli-sensitive hydrogels, as epitope display scaffolds, and as components of biosensors8–15. In cells, coiled-coil motifs can act as an oligomerization domain and coiled-coils have been found in a wide variety of proteins including transcription factors16, motor proteins17, structural filaments, chaperone proteins18, tRNA synthetases19, SNARE complexes20, and cell or viral fusion proteins21. Functionally, coiled-coil motifs can act as levers, scaffolds, moving arms, and potentially as springs or nano-motors22. Potential pharmaceutical applications of coiled-coils include their use as multifunctional delivery, targeting and imaging agents13,23). Imaging takes advantage of fluorination and 19F NMR. Fluorinated amino acids can be easily incorporated into proteins both by solid state24–26 and in vivo27–29 protein synthesis, and fluorination leads to increased hydrophobicity30, increased stability31,32, and decreased drug metabolism33. Incorporation of fluorinated amino acids, such as by replacement of leucine with 5,5,5,5′,5′,5′-hexafluoroleucine (hFLeu) in the hydrophobic core of coiled-coils, leads to enhanced stability and resistance to both thermal and chemical denaturation28,29,31,34–38. Although fluorine incorporation in the hydrophobic core tends to increase protein stability, the influence of fluorine on the protein structure is not without controversy. In α-helices, the incorporation of fluorine has been reported to cause structural changes, structural distortions, lower helical propensity, and decreased ellipticity as measured in CD experiments39. This contrasts with other studies where incorporating fluorine leads to increased stability and secondary structure38. Measurements of the energetic contribution of fluorine to thermostability in α-helices vary; however, when fluorine is incorporated into the hydrophobic core it favorably contributes ~0.5–1.2 kcal/mol-residue to the stability28,36,39,40. In addition to differences in the free energetics, there are concerns related to the larger steric bulk of fluorinated compounds. Tirrell and co-workers incorporated fluorinated residues in α-helices by the introduction of 5,5,5-trifluoroleucine into the p1 region of the leucine zipper GCN434. They proposed that these fluorinated residues were isomorphic to their non-fluorinated versions34,41. Although the fluorinated amino acids maintain a shape very similar to their non-fluorinated counterparts, since the steric bulk of a trifluoromethyl group is closer in size to an isopropyl group42, and since hFLeu is approximately 37 Å3 larger than leucine (consistent with the difference in van der Waals radii for hydrogen of 1.20 Å and 1.47 Å for fluorine)37,43, fluorination leads to alterations in the core structure38.
The primary sequences of coiled-coil proteins are characterized by a highly recognizable heptad repeat denoted as abcdefg (see Figure 1). Positions a and d are typically occupied by hydrophobic residues while positions e and g are typically charged residues that form an intermolecular salt bridge44. As each turn of the α-helix results in the progression of 3.6 residues, each heptad repeat progresses slightly less than two full twists around the helix45. The coiled-coil structure is formed when the component helices come together, bury their hydrophobic regions, and form further stabilizing interactions such as salt bridges; the coiled-coil formation leads to a net-stabilization of each individual helix46. As the hydrophobic interface slowly twists around the helix, association of multiple helices at the hydrophobic domain results in a super-coil with classically defined inter-digitations of hydrophobic side chains between neighboring helices47.
Figure 1.

A heptad helical wheel and molecular graphics representation of a parallel three heptad repeat coiled-coil showing the heavy atoms and a ribbon representation of backbone secondary structure. The helical wheel shows a top-view looking down the α-helix of the two interacting helices that shows where each residue side chain is approximately located. The arrows within each circle show the residue connectivity. The crossed arrows at the center denote the interactions of the hydrophobic residues a and a′ and d and d′. The arcing arrows at the top and bottom denote the classical salt bridge interactions between g and e′ and e and g′. (The helical wheel representation shown is adapted from 12). The ribbon structure of two helices interacting as a coiled-coil is also shown including side-chain heavy atoms. The coiled-coil represented in the molecular graphics is the parallel three heptad repeat IAAL-E3/K3 dimer (PDB ID: 1U0I) 48.
As one of the simplest and most common tertiary structural motifs, coiled-coils are widely studied scaffolds for protein engineering and design5,44,48–59. Coiled-coils are also a commonly employed model systems in protein folding and stability studies60–74. Although predicting the secondary and tertiary structure of coiled-coil proteins is relatively straightforward as compared to other protein motifs, 3D structure prediction is still difficult as coiled-coil proteins display a rich tapestry of structure and motion. Specifically small changes in sequence can switch the oligomerization state, orientation, or alter the coiled-coil structure and dynamics14,55,75–84. The structure and dynamics are also influenced by extrinsic factors, such as concentration, temperature, pH, ligand binding, and dielectric8,62,85–93. Even within well-behaved dimeric structures, distinct conformational sub-states in slow exchange may be populated73,94.
Molecular Modeling and Molecular Dynamics Simulations of Coiled-coils
The computational studies of coiled-coils to-date include molecular modeling, model building, and MD simulation approaches aimed at better understanding the physical forces and chemical interactions that define the structure and dynamics of coiled-coil proteins. Initial modeling work focused on extending a set of principles and general formulas developed by Crick to describe ideal parallel coiled-coil dimer structure with knobs into holes packing of the hydrophobic core45. Later, more detailed modeling and MD simulation studies were applied to reproduce experimental structure and dynamics, coiled-coil oligomerization states, and ultimately to estimate relative energies of coiled-coil formation. The initial studies used rigid backbones and side chain packing in the hydrophobic domain of parallel coiled-coil95. As more structures emerged, and the regularity of the coiled-coil structure was confirmed, a wide arsenal of different programs and methods to build coiled-coil structures emerged. These each use various combinations of distinct means to pack the hydrophobic cores, explore the flexibility of side chains, optimize the electrostatics, and/or sample different orientations and oligomeric states60,87,96–125
Highlights of the theoretical approaches towards understanding coiled-coil structure and dynamics include:
Early simulation approaches using molecular dynamics simulation with explicit representations did remarkably well in reproducing coiled-coil structure. This includes unrestrained MD of the yeast transcriptional activator GCN4 starting from Ca atoms followed by automatic building with simulated annealing to produce a structure 1.25 Å RMSd (backbone) from the crystal structure126,127. Additionally, Monte Carlo folding simulations followed by atomistic MD on the GCN4 dimer predicted structure with an accuracy of 0.81 Å (backbone); these calculations facilitated quaternary structure predictions60,106,109. Other work performed comparative MD simulations on different coiled-coil protein sequences to qualitatively understand stability101. These early successes are notable in comparison with our recent work that shows considerable force field dependence in the simulation of the coiled-coils studied in this work (see the methods and more data in the supplementary material).
Investigations of the influence of a membrane environment on the coiled-coil structure and dynamics114,115,123,128,129, including a MD study showing formation of a well-defined coiled-coil structure in the presence of an external electrostatic field86.
Investigations of coiled-coil proteins using various free energetic approaches to better understand helical propensity99, stability122, and the influence of sequence and the environment on oligomerization84,130,131.
Detailed validation and prediction aimed at understanding the preferential oligomerization state of different coiled-coils88,113,125.
The use of targeted or force-induced MD simulations to investigate alterations in coiled-coil structure and dynamics, including calculation of a conformational exchange pathway connecting two different crystal structures of the anti-parallel coiled-coil of seryl tRNA synthetase132, transitions from the native to the putative fusogenic conformations of influenza hemagglutinin at low pH88, transitions among different GCN4 structures92, and investigation of putative conformational transitions in engineered prion peptides89.
Together, these results provide strong validation of the modern force fields and MD simulation protocols and their ability to model complex protein tertiary behavior and structure. Yet, very few simulations have investigated the effect of either fluorination or changes in the salt bridges on coiled-coil protein structure and dynamics. The only MD of fluorinated coiled-coils published to-date includes the studies by Tirrell’s group where a combination of experiment and MD simulation with an implicit solvent model were applied to understand the effect of hFLeu substitution in the GCN4 coiled-coil28.
In this paper, we present the results of structural and energetic studies of parallel dimeric coiled-coils, with and without fluorination, pursued through the use of biomolecular simulation methods. The aim of this study is to decompose contributions to the structural and energetics differences between fluorinated and non-fluorinated coiled-coils and give context to variations seen in early fluorination studies. To decompose the contributions of these differences, we have designed point mutations in the IAAL-E3/K3 coiled-coil133 to both fluorinate the peptide and to vary the length and geometry of the salt bridge domain. As seen in Table 1, the lysine residues in positions e and g of the basic monomer (labeled Monomer B in our tables) have been mutated to ornithine, arginine, and S-2,7-diaminoheptanoic acid. Fluorination mutations have also been designed at position d of both monomers to substitute 5,5,5,5′,5′,5′-hexafluoroleucine (hFLeu) for leucine. Analysis of the MD simulation runs to investigate the influence of fluorination and salt bridge deformation were pursued through calculations of circular dichroism ellipicity values to characterize helicity, studies of rotamer preferences in the hydrophobic domain, and calculations of salt bridge length. The results of energetic calculations to measure the free energy of hydration and pair wise electrostatic contributions are also discussed.
Table 1. Summary of the coiled-coil models investigated.
The sequence of each coiled-coil model is listed using single letter amino acid codes. The control sequence is listed at the top and changes in sequence are bolded and colored red. The monomer A is identical in each coiled-coil.

|
Methods
Starting geometries
The initial structures were derived from the first NMR structure model of the IAAL-E3/K3 coiled-coil dimer (PDB ID: 1U0I)134. Point mutations at positions e and g of the IAAL-E3/K3 basic monomer were made using Swiss PDB viewer135 and the AMBER LEaP program136 to vary salt bridge length and geometry.
Force fields
Simulations of 3- and 4- heptad repeat coiled-coils were validated and completed using the AMBER ff99SB modification137 of the Cornell et al. force field (ff94)138. Although not complete, validation of the force field was facilitated by visualization of the model structures and calculations of the RMSd values to relevant experimental structures. Unless otherwise mentioned, the model structures were built as straight coordinate averages of best-fit structures after equilibration; the models were created from 2 ns windows from the trajectory (which was stored at 1 ps intervals), using ptraj. Earlier attempts to simulate these and related coiled-coil protein structures using the AMBER ff94138, ff96139, and ff99140 force fields and equivalent simulation protocols resulted in significant structural distortion. The coiled-coils became bent and unraveled in short ns-scale simulations (see the supplementary material). The unwinding suggests instability of the helices comprising the coiled-coil; this was somewhat surprising as the ff94 and ff99 are known to over-stabilize α-helices141,142. Consistent with intuition and experiment, simulations of larger coiled-coil protein structures with the ff96 and ff99 force fields showed less of a dependence on the force field parameters and comparatively less movement away from the experimental structure; however, the absence of movement away from a starting structure in ns-scale simulation does not necessarily imply greater stability. In fact, well-known force field biases can be hidden in short MD simulations143 or in simulations of larger protein assemblies. As the best agreement with the experimental structures was observed with the ff99SB force field, this force field was adopted in the present studies.
Molecular dynamics simulations
All simulations of the parallel coiled-coil protein dimers and monomers were performed with the AMBER suite of programs136,144 using the ff99SB force field141. Periodic boundary conditions were applied using the particle mesh Ewald method (PME) with a less than 1 Å charge grid and cubic B-spline interpolation145. Proteins were solvated by surrounding the protein with at least a 12 Å water layer in each direction within a truncated octahedron using TIP3P waters146. This amounts to on the order of 4500–6000 waters. After the equilibration protocol was simulated, MD simulations without restraints were continued until convergence—as determined by plateaus in RMSd plots—was observed. This typically required MD sampling times on the order of ~15 – 65 ns for each coiled-coil, with the longer convergence times necessary for the structures where larger amino acid mutations were made, such as the replacement of lysine by arginine in the salt bridge. Note that these simulations are significantly longer than the < 2 ns MD sampled in simulations with ff94, ff96, and ff99 where significant structural distortions were evident (see supplementary material). All of the molecular dynamics (MD) simulations were performed with a 2 fs time step and a direct space non-bonded cutoff of 10 Å with the pair list of atomic interactions built out to 11 Å and heuristic update of the pair list triggered when any atom moved more than 0.5 Å since the previous update. During MD, bond lengths involving hydrogen atoms were constrained with SHAKE147,148 with a geometric tolerance for the constraint of 0.00001 Å during the coordinate resetting. Initial minimization was followed by heating to 300K at constant volume over a period of 10 ps using harmonic restraints of 2 kcal mol−1 Å−1 on the protein atoms. During production runs, the center of mass translational motion of the entire system was removed after the initial velocity assignments and subsequently every 5000 MD steps. Constant temperature was maintained using the weak coupling algorithm and heat bath coupling with a 2 ps time constant149. Pressure (1 atm) was maintained using isotropic position scaling with Berendsen weak coupling algorithm with a 1.0 ps pressure relaxation time149.
Residue Parameterization
The development of new force field parameters for the residues ornithine, 5,5,5,5′,5′,5′-hexafluoroleucine (hFLeu), and S-2,7-diaminoheptanoic acid (Figure 2d) were completed consistent with the original Cornell et al. force field 138 and recent ff99SB modifications 141. The ff99SB modifications to the torsional potential of the peptide backbones were necessary to improve the conformational ensemble sampled by the glycine residues and also, through modifications to the Φ/Ψ torsional potentials, to provide a better energetic balance between α-helix and β-sheet peptide geometries. The existing force field parameters, along with RESP derived charges at a consistent level of theory, have proven effective not only for modeling both peptides and nucleic acids 150,151, but also small organic molecules152,153. To build the new residues, atom types were chosen consistent with the Cornell et al. and ff99SB force fields. Torsional and angle parameters were assigned consistent with existing parameters from the ff99SB force field. No new angle or dihedral force field parameters were required beyond those already available in ff99SB. Although these residues are new, the fluorine parameters in the ff99SB force field have been previously validated, albeit with a less accurate charge model, through estimations of fluorophilicity for a series of small molecules 154 and simulation of aggregation behavior of fluoroalkanes compared to alkanes 155,156. More rigorous tests of solvation free energies for small fluorine containing molecules using the AMBER fluorine parameters and a RESP treatment of the charges suggest the applied model is appropriate for fluorine containing compounds 152,153.
Figure 2.

Shown are the structures of the various amino acid substitutions used at the e and g positions of the basic monomer, monomer B. a. L-ornithine, b. L-lysine, c. L-arginine, d. S-2,7-diaminoheptanedoic acid, and e. 5,5,5,5′,5′,5′-hexafluoro-leucine.
Di-peptide analogues of the amino acids were created by capping the N- and C-terminal ends of the amino acids with acetyl (ACE) and N-methyl (NME) groups, (respectively). These structures were then optimized using the Gaussian 98 software157 at the HF/6-31+G* level. An SCF convergence criterion of 10−8 with tight optimization was used to ensure a fully minimized structure. This minimized structure was then used to calculate a molecular electrostatic potential (MEP) on a three dimensional grid using the GAMESS quantum mechanics software package158 (again at the HF/6–31+G* level). Six distinct orientations of the structure were calculated and exported to the AMBER RESP program159 which was used to fit atom centered RESP charges to the MEP. Charge constraints were placed on the capping groups of the di-peptides such that the sum of charges of the ACE and NME groups was neutral. The parameterization was greatly facilitated by the RED II program which provides an automated method to create the MEP and fit the RESP charges160.
AMBER off libraries containing these parameters have been deposited in the AMBER Parameter Database maintained by Richard Bryce161 and are summarized in the supplementary material.
Molecular Dynamics Trajectory Analysis
Rotamer preferences and salt bridge distances were calculated using the AMBER ptraj module162. Rotamer definitions are defined in tables 3 and 4 consistent with Dunbrack and Cohen’s work163. Rotamers were calculated over a 10–12 ns window post equilibration of the trajectories with sampling every 10 ps using the dihedral function of ptraj. The dihedral measurements for the leucine and isoleucine rotamers were taken from the d and a position, respectively, of the middle heptad of both monomers. Salt bridge distances were calculated using atoms as described in Table 9. Distances were also calculated over a 10–12 ns window, sampling at 10 ps intervals, using the distance function of ptraj.
Table 3.
Standard dihedral angle definitions for leucine and isoleucine156
| Residue | Angle | Rotamer | Angle Definitions |
|---|---|---|---|
| Ile | χ1 | r1 | N-Cα-Cβ-Cγ1 |
| Ile | χ2 | r2 | Cα-Cβ-Cγ1-Cδ |
| Leu | χ1 | r1 | N-Cα-Cβ-Cγ |
| Leu | χ2 | r2 | Cα-Cβ-Cγ-Cδ1 |
Table 4.
Angle limits and conformation definitions for leucine and isoleucine156
| Conformation | χ Range |
|---|---|
| gauche+, g+ | 0 – 120 degrees |
| trans, t | 120 – 240 degrees |
| gauche−, g− | −120 – 0 degrees |
Table 9.
Salt Bridge Distance Summary
| Dimer Pair | Average Salt Bridge Distance | Participating Atoms |
|---|---|---|
| IAAL-E3K3 | 8.4 Å (stdev 2.1) | Glu13@Cδ Lys29@Nζ |
| IAA(hFLeu)-E3/K3 | 6.8 Å (stdev 1.9) | Glu13@Cδ Lys29@ζ |
| IAAL-E3O3 | 21.3 Å (stdev 1.5) | Glu13@Cδ Orn29@Nε |
| IAA(hFLeu)-E3/O3 | 15.6 Å (stdev 1.7) | Glu13@Cδ Orn29@Nε |
| IAAL-E3/R3 | 6.2 Å (stdev 2.3) | Glu13@Cδ Arg29@Nη1, Nη2 |
| IAA(hFLeu)-E3/R3 | 4.1 Å (stdev 1.0) | Glu13@Cδ Arg29@Nη1, Nη2 |
| IAAL-E3/Ĥ3 | 6.1 Å (stdev 2.7) | Glu13@Cδ DAH29@Nη |
| IAA(hFLeu)-E3/Ĥ3 | 5.4 Å (stdev 2.2) | Glu13@Cδ DAH29@Nη |
Hydration analyses of the coiled-coil structures were performed using the ptraj hbond and grid utilities. A maximum cutoff angle of 120.0° and cutoff length of 3.4 Å were used in hydrogen bond definitions. In the fluorinated coiled-coil dimer models, the carbon bonded fluorine was considered a hydrogen bond acceptor (electron donor). Measured hydrogen bonds and calculated water densities used in hydration calculations were taken during the last 1 ns (1000 frames) of MD simulation. Hydrations sites were determined using solvent distributions calculated by binning atom positions from RMS coordinate fitting over all protein atoms at 1 ps intervals into (0.5 Å)3 grids164. These grids were contoured using the volume visualization module of UCSF Chimera165.
Thermodynamic Integration Free Energy Calculations
Calculations of the relative free energy of hydration were completed using thermodynamic integration166. The use and accuracy of thermodynamic integration to calculate the free energy of hydration of amino acid residues has been shown to possibly be as accurate as sub kcal/mol measurements167. On the basis of a thermodynamic cycle, leucine (λ = 1) was perturbed in both gas (vacuum) and aqueous environments to 5,5,5,5′,5′,5′-hexafluoroleucine (λ = 0). Relative free energy of hydration values were determined by subtracting gas phase perturbation measurements from aqueous phase measurements. Aqueous simulations were performed using a particle mesh Ewald treatment of electrostatics, as previously described. These simulations were performed using the thermodynamic integration module in AMBER162. Residues were modeled as a di-peptide, using the original residue capped with an acetyl group (ACE) on the N-terminus and an N-methyl group (MNE) on the C-terminus. Minimization and equilibration of di-peptides followed procedures as described previously. The structures were allowed to relax during 6 ns of molecular dynamics simulation followed by thermodynamic integration sampling. Accurate and sufficient sampling was ensured by calculating the perturbation energy by the reverse pathway and by observation of ΔV/Δλ measurement as the simulation progressed. For our calculations, we used twelve sampling points of λ, based on Gaussian quadrature (0.00922, 0.04794, 0.11505, 0.20634, 0.31608, 0.43738, 0.56262, 0.68392, 0.79366, 0.88495, 0.95206, and 0.99078), sampled over 3 ns.
Free Energy Decomposition
In some studies presented below, the interactions of specific residues and their relative contributions to the free energy of binding were studied by performing component analysis. As described elsewhere168, each residue was split into its component atoms. Internal energies were calculated if all atoms contributed to bond, angle, or torsion angle energy terms. van der Waals interactions were calculated as one half of the pair wise energy calculations for atoms composing a single residue, as well as calculations of inter-residue contacts. The electrostatic potential of pair-wise interactions was calculated using the General Born (GB) equation, to model a given charge distribution for a solute embedded in a uniform (high) dielectric solvent. A dielectric constant of 80.0 was applied for GB based electrostatic potential calculations.
Circular Dichroism Calculations
Quantification and comparison of structural helical content was measured by calculating mean residue ellipticities representing the CD spectra of 20 individual structures spanning the final 2 ns of simulation of each coiled-coil dimer using the DichroCalc program169. A Gaussian curve type was assumed with a bandwidth at half maximum of 12.5 nm and two backbone transitions.
Results
Observation of the Simulation Results
With the exception of the “short” salt bridged, or ornithine substituted, (E3/O3) coiled-coil models, all of the molecular dynamics simulations with the ff99SB force field maintained the characteristic parallel dimeric coiled-coil structure over the course of 15–65 ns of MD simulation. In Figure 4, the root mean square deviations (RMSD) for the simulation of the IAAL-E3/K3, IAA(hFLeu)-E3/K3, IAAL-E3/O3, and IAA(hFLeu)-E3/O3 coiled-coils relative to their initial starting structures are shown. Comparison of the final IAAL-E3/K3 structure to published NMR structures134 showed a RMSD of 0.84 Å (using the backbone atoms and omitting the terminal three residues). Whereas the standard-length salt bridged E3/K3 coiled-coils are stable on the MD simulation time scale, the shorter salt bridged IAAL-E3/O3 and IAA(hFLeu)-E3/O3 coiled-coils both undergo rapid loss of secondary structure during the MD simulation. With the IAAL-E3/O3 coiled-coil, this loss of secondary structure occurs between 10 and 20 ns of simulation time and once the structure moves away, it never comes back (see Figure 4). Initially, the unraveling of the secondary structure in the terminal regions of both helices precedes exposure to solvent of the interior hydrophobic interface of the central heptad at approximately 10 ns. At 14 ns, the monomers begin to separate at the terminal regions, bending inward in opposing directions. Bending in the basic monomer persists until the folded regions resemble a horseshoe with a face perpendicular to the acidic monomer (15 ns). Final rearrangement occurs as the acidic monomer folds around the face of the horseshoe maximizing hydrophobic interactions. This is shown in greater detail in the supplemental material, Figure S6. Similar disruption of structure was seen in the hFLeu substituted E3/O3 coiled-coil.
Figure 4. Root Mean Square Deviation Plots of Selected Coiled-coil Dimers.

–Shown from left to right and top to bottom are IAAL-E3/K3, IAA(hFLeu)-E3/Ĥ3, IAAL-E3/Ĥ3, IAA(hFLeu)-E3/R3, IAAL-E3/u3, IAAL-E3/u3, IAAL-E3/O3, and IAA(hFLeu)-E3/O3. Note that the scales in each RMSD plot are different and the units are Å versus time (ps). Large fluctuations seen in IAAL-E3/O3 and IAA(hFLeu)-E3/O3 dimers reveal large conformation changes and loss of secondary structure.
The arginine (IAAL-E3/R3, IAA(hFLeu)-E3/R3) and diaminoheptanoic acid (IAAL-E3/Ĥ3, and IAA(hFLeu)-E3/Ĥ3) salt bridged coiled-coils showed similar RMSD plots compared to those observed for IAAL-E3/K3 with the exception of slower equilibration time for IAA(hFLeu)-E3/R3. We expect that the slight delay in equilibration is due to the requisite side chain reorientation and expansion in an initially constrained starting structure (all dimers were created using homology modeling to a non-fluorinated dimer). The observed remodeling of the salt bridges and coiled-coil structure are consistent with experiment14,63,170.
Hydration of Modeled Coiled-coils
Investigation of the solvation of the folded coiled-coils was enabled by analysis and visualization of the average water occupancy in grids surrounding the protein. These grids were contoured at a level corresponding to ~2–3x bulk water density and revealed specific hydration sites along the coiled-coil backbone. Clear hydration, evidenced by high occupancy water sites, was found at the distal hydrophilic face of the coiled-coil near the b and c backbone oxygen and nitrogen atoms and also near the extended f and f′ residues. Distinct hydration around the residues participating in the salt bridges (positions e/g′ or e′/g) was observed peripheral to the dimer interface, although very few high occupancy hydration sites were observed directly at the salt bridge interface. Hydrogen bond analysis shows that the side chain atoms of the salt bridge residues form multiple transitory solvent-peptide hydrogen bonds and transient salt bridges. As the salt bridge interactions are rather dynamic, this tends to inhibit the formation of long term, high occupancy water interactions at the salt bridge. For all of the coiled-coils, with the exception of the ornithine substituted coiled-coils, essentially no ordered hydration sites are evident at the helix-helix hydrophobic interface made up of residues a and d. Different hydration patterns where observed for the relatively unstable ornithine coiled-coil dimers (IAAL-E3/O3 and IAA(hFLeu)-E3/O3). The loss of secondary structure in the ornithine coiled-coils effectively allows on the order of 2–4 waters into the hydrophobic core. One of the three largest hydration pockets shows contributory interactions from six separate hydrogen bond forming atoms (Ala11@N, Glu13@OE1, OE2, Leu12@N, Ala10@N, and Ile37@O). Smaller hydration pockets can be found along backbone atoms in structured domains with hydration pockets in the unfolded regions where water interacts with multiple proximal ionic groups. In Figure 5, distinctly less order hydration is seen around the backbone atoms in the IAAL-E3/O3 and IAA(hFLeu)-E3/O3 dimers; this is likely a result of the larger fluctuations in the structure and larger RMSD values which smear the time average.
Figure 5. Hydration sites of selected coiled-coil dimers.

High occupancy water hydration sites are displayed in gray in both side view and top down views of the (a) IAAL-E3/K3 and (b) IAA(hFLeu)-E3/K3 coiled-coils. Also shown are the side views of the (c) IAAL-E3/O3 and (d) IAA(hFLeu)-E3/O3 coiled-coils. The latter figures display distinct hydration within the hydrophobic core. The contouring of the water occupancy grids is shown at 12.0 hits per (0.5 Å)3 grid from a 1 ns portion of the trajectory which is an occupancy ~2.87 times greater than bulk solvation157. The water density is superimposed on an average structure calculated over the same interval.
Helicity Measurements
Fluorinating α-helices introduces structural changes that have been reported to both increase and decrease helicity34,37. To better understand and quantify the changes in helicity as the core coiled-coil is mutated or fluorinated, CD spectra were calculated using the DichroCalc program using equivalent time samplings from the individual MD calculations. In Table 2, we present the calculated mean residue ellipticity for the simulated coiled-coil dimer pairs. In each of the coiled-coil structures, the MD simulations suggest that the fluorinated coiled-coils are more helical—as inferred from the calculated ellipicities—than their nonfluorinated counterparts. As seen in Table 2, Figure 5, and also in the supplementary material, it is clear that the ornithine substituted coiled-coils (IAAL-E3/O3 and IAA(hFLeu)-E3/O3) lose secondary and tertiary structure in the nanosecond time scales sampled in MD simulation. Individual snapshots of these dimers from the MD trajectory do show regions of α-helical secondary structure, however less than is observed with the other coiled-coil structures. Moreover, the hydrophobic interface is largely disrupted. Both effects are likely the result of steric strain induced by the shortened salt bridge.
Table 2. Calculated Mean Residue Ellipticity.
Shown are the calculated average ellipicities (degree cm2 dmol−1) at 222 nm and their standard deviations calculated from twenty representative snapshots from stable portions of the MD trajectories. The ellipicities were calculated using the DichroCalc program161.
| Coiled Coil Dimer | [θ]222 | Stdev |
|---|---|---|
| IAAL-E3/K3 | −17229.4 | 1176.1 |
| IAA(hFLeu)-E3/K3 | −17516.1 | 1154.2 |
| IAAL-E3/O3 | −6573.1 | 688.8 |
| IAA(hFLeu)-E3/O3 | −8903.8 | 1540.2 |
| IAAL-E3/R3 | −14831.7 | 1485.9 |
| IAA(hFLeu)-E3/R3 | −17100.9 | 912.2 |
| IAAL-E3/Ĥ3 | −14714.0 | 777.9 |
| IAA(hFLeu)-E3/Ĥ3 | −16546.6 | 975.8 |
For all of the other models where the coiled-coil structure was largely maintained, the measured ellipticity measurements are comparable. The standard lysine bridged coiled-coil (IAAL-E3/K3) was calculated to be the most helical of the non-fluorinated coiled-coil structures, suggesting that the lysine-glutamate salt bridge length to be most ideal for non-fluorinated coiled-coils. A similar trend is seen in the fluorinated dimers, through the degree to which salt bridge length and orientation increase ellipticity is less resolvable since the fluorinated monomers are essentially fully helical. Our results contrast somewhat with findings from Kennan et al.14 who used variable length non-natural amino acids in the e and g positions of the acid monomer (our work focused on the basic monomer). They found the optimal salt bridge length to be ~7–8 methylene units in the pHHGlu-pLys and pHGlu-pLys coiled-coil dimers (where H here represents a single methylene unit)14. However our simulations agree with their experiment in suggesting that smaller salt bridges (such as with the E3/O3 coiled-coils) are less stable. Together these differences suggest that salt bridge interactions are very subtle and contextual, consistent with previous work14,54,57,77,78,85,131,134,170.
Free energy calculations
Thermodynamic integration calculations were conducted to better understand the free energy consequences of replacing leucine with 5,5,5,5′,5′,5′-hexafluoroleucine (hFLeu). These were performed on very simple di-peptide model systems (see Figure 3) to allow better understanding of the solvation free energy costs of fluorination when the side chain is exposed. Twelve Gaussian quadrature weighted sampling points were used to continuously transform di-peptide models of leucine to hFLeu in aqueous and gas phase environments. These thermodynamic integration results show that hFLeu has a less favorable free energy of hydration by 1.1 kcal/mol (std. dev. 0.35 kcal/mol) compared to leucine. This less favorable free energy of hydration would lead to energetic penalties if the residue is exposed to an aqueous environment. If fluorinated residues are incorporated at binding domain interfaces or positions in α-helices that undergo higher level interactions (such as coiled-coils or α-helical bundles), this decreased free energy of hydration, or destabilization of the exposed side chain, would drive higher order association leading to hydrophobic burial. In coiled-coils where secondary structure in the monomers is driven by association, fluorination in the hydrophobic heptad positions leads to both increased association and helicity. Conversely, incorporation of fluorinated residues in α-helices that do not undergo higher order interactions (or solvent exposed positions in coiled-coils) would lead to structural distortions and decreased helicity in an attempt to bury hydrophobic atoms. Additional discussion of structural distortional in α-helices from incorporation of fluorinated residues can be found in related work39.
Figure 3.
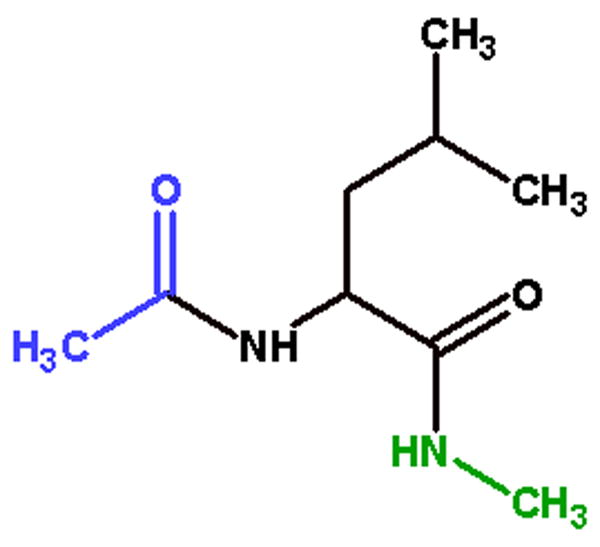
A representative capped amino acid dipeptide analog used in charge derivation and thermodynamic integration experiments. Acetyl (ACE) groups in blue are covalently bonded to the N-terminus of the amino acid, N-methyl (NME) groups in green are covalently bonded to the C-terminus of the amino acid. ACE-Leucine-NME is shown here.
Rotamer Preferences
Using the AMBER program ptraj, we analyzed the rotamers for leucine (hFLeu in the fluorinated molecules) and isoleucine in the coiled-coil dimers that we simulated. We found that incorporating fluorine leads to rotamer changes in residue side chains. Using the rotamer definitions in tables 3 and 4, we found a strong preference in hFLeu for the rotamer r1 as gauche− in the χ1 dihedral angle (−60 degrees) and r2 as trans in the χ2 dihedral angle (180 degrees).
This preference for gauche−, trans rotamers in 5,5,5,5′,5′,5′-hexafluoroleucine is likely a result of steric hindrance. Fluorine has a larger radius and is more electronegative than hydrogen. In the trans, gauche+ rotamers, significant charge repulsion and steric hindrance are likely to occur as shown schematically in the molecular graphics cartoon of Figure 6.
Figure 6. Selected leucine/hFLeu side chain rotamers.

These two side chain leucine rotamers are the most common choices (for leucine and hexafluoroleucine residues) seen in our simulations of three heptad repeat coiled-coils. Green arrows added to emphasize differences in overall length and linear persistence of the two rotamer presented here. For hFLeu, the trans, gauche+ rotamer is disfavored due to the unfavorable steric interaction of the larger trifluoromethyl groups with the backbone and unfavorable electrostatic interactions of the electronegative fluorines with oxygen.
The two rotamer pairs, (r1 = gauche−, r2 = trans) and (r1 = trans, r2 = gauche+), seem to be the dominant rotamer choices seen in leucine residues of the coiled-coils dimers modeled. The choice of the rotamer pair appears to correlate with salt bridge distance and is likely to be part of a compensatory mechanism where the side chains in the hydrophobic domain expand or compress subject to the constraints imposed by salt bridge length and fluorination. We speculate that increased salt bridge length allows an expansion of the hydrophobic side chains, while still maintaining the hydrophobic core, and that fluorination increases the steric size of fluorinated residues and leads to side chain compression in the hydrophobic domain.) The choice of gauche−, trans rotamer pairs is found in both coiled-coil dimers with normal or compressed dimerization domains. This is consistent with what is seen, with few exceptions, for the structures of coiled-coil dimers found in the RCSB Protein Data Bank (see supplementary material). When the salt bridge length is increased (for example with mutations of lysine to arginine or (S)-2,7-diaminoheptanoic acid), the rotamer preference is shifted towards expanded side chain geometries with trans, gauche+ rotamers.
Our simulation results show greater variability and less predictability among isoleucine rotamers compared to leucine rotamers. Rotamer choices for isoleucine residues are more influenced by salt bridge length than fluorination. In Table 6, we summarize rotamer pairs for both sets of monomers. A trend from (r1 = gauche−, r2 = gauche−) to (r1 = trans, r2 = gauche+) rotamers is seen as the protein-protein interaction domain is first compressed then expanded (see Table 6 and Figure 7). This pattern strongly correlates to salt bridge length with the sole exception of IAA(hFLeu)-E3/R3 which is likely influenced by the favorable electrostatic interactions with fluorine in the core (see below). Barring this exception, the choice of isoleucine rotamers can be described as a tradeoff between the total expansion, or compression, of the side chain necessary to satisfy the salt bridge while also providing the space filling bulk necessary to maintain the hydrophobic interface. In general, the fluorinated coiled-coils show a decrease in the total number of side chain rotamer states accessed by the fluorinated residues. This decrease in populated rotamer states reflects a decrease in the hydrophobic side chain conformation entropy (Sχ) in fluorinated coiled-coils171. The larger size of the fluorinated residues rigidifies the folded structures and leads to a loss of conformational entropy. Such a loss should be compensated by other terms if the net effect of fluorination is stabilizing171. In Table 8, the conformational entropy of the hydrophobic side chain rotamers (Sχ) was calculated by considering relative distributions of rotamers in fluorinated and nonfluorinated coiled-coils (as measured by percentage contribution, pi) using Equation 1. For this calculation, fluorinated and nonfluorinated dimer measurements were pooled respective to fluorine incorporation to allow for complete sampling of all potential rotamers irrespective of preferences derived from salt bridge lengths and geometries. Direct interactions of hydrophobic residues require that conformational entropy be calculated as a function of rotamer occupancy across the dimer interphase (both monomers) producing a set of 81 unique rotamers (3 conformations per rotamer, 2 rotamers per side-chain, 2 residues = 34 unique rotamer sets). From Table 8, a ten fold difference in side chain conformational entropy is seen between leucine and hexafluoroleucine while the isoleucine rotameric conformational entropies are nearly equivalent comparing fluorinated and non-fluorinated. The simulations also show that multiple rotameric sub-states are sampled over the course of the MD simulations. Time courses for the rotamer angles for leucine and isoleucine from the IAAL-E3/R3 coiled coil are shown in the supplementary material.
Table 6. Sidechain Rotamer Distributions for Isoleucine Residues in Coiled-coil Dimers.
Coiled-coil dimers are ordered from the most compressed to the most expanded dimer interface in descending order as determined by the isoleucine rotamer choices (see Figure 7). Rotamer measurements were taken from the isoleucine residues (position a) of the central heptad in both monomers. In cases where calculated rotamers are not identical for both monomers, the acid monomer isoleucine rotamer tends to be more expanded in the nonfluorinated dimers and more compressed in the fluorinated dimers.
| Dimer | Monomer1 (r1,r2) | Monomer2 (r1,r2) | Occupied (%) |
|---|---|---|---|
| IAA(hFLeu)-E3K3 | gauche−, gauche− | gauche+, trans | 24.6 |
| gauche−, trans | gauche+, trans | 75.4 | |
| IAAL-E3K3 | gauche−, gauche− | gauche+, trans | 2.9 |
| gauche−, trans | gauche−, trans | 7.8 | |
| gauche+, trans | gauche−, trans | 89.2 | |
| IAA(hFLeu)-E3/Ĥ3 | gauche−, gauche− | trans, trans | 2.1 |
| gauche−, gauche+ | trans, trans | 1.8 | |
| gauche−, trans | trans, trans | 84.9 | |
| trans, trans | trans, trans | 2.1 | |
| trans, gauche+ | gauche−, trans | 9.2 | |
| IAAL-E3/Ĥ3 | gauche−, trans | trans, trans | 22.2 |
| gauche−, trans | trans, gauche+ | 1.8 | |
| trans, trans | trans, trans | 71.8 | |
| trans, gauche+ | trans, trans | 4.2 | |
| IAA(hFLeu)-E3/R3 | gauche−, trans | trans, trans | 30.6 |
| trans, trans | gauche−, trans | 9.3 | |
| trans, trans | trans, trans | 4.1 | |
| trans, gauche+ | gauche−, gauche− | 19.2 | |
| trans, gauche+ | gauche−, trans | 36.7 | |
| IAAL-E3/R3 | gauche−, trans | trans, trans | 7.7 |
| gauche−, trans | trans, gauche+ | 4.5 | |
| trans, trans | gauche−, gauche− | 1.6 | |
| trans, trans | trans, trans | 70.8 | |
| trans, gauche+ | trans, trans | 13.3 | |
| trans, gauche+ | trans, gauche+ | 2.1 |
Figure 7. Isoleucine rotamer choices at varying coiled-coil compression levels.

Trends in isoleucine rotamer choices (on left) seen as a function of coiled-coil compression. Rotamer choices are often complex adjustments to compensate for salt bridge lengths and to optimize for interior space filling and optimizing hydrophobic interactions.
Table 8. Calculated conformational entropy for hydrophobic sidechain rotamers.
Measurements taken from pooled fluorinated and nonfluorinated coiled-coil dimers using measurement of the rotamers sampled by the central heptad repeat. Individual rotamer contributions to overall conformational entropy calculations may be found in the supplemental materials table S3.
| Leucine/hFLeu | Isoleucine | Combined | |
|---|---|---|---|
| Fluorinated | 0.12 cal/mol K | 1.41 cal/mol K | 1.53 cal/mol K |
| Nonfluorinated | 1.16 cal/mol K | 1.20 cal/mol K | 2.36 cal/mol K |
| Equation 1 |
Salt Bridges and Electrostatics
Coiled-coils are a flexible, dynamic protein-protein binding motif172. For most of the structural mutations, small conformational and rotameric changes are observed. These compensatory changes in side chain rotations and salt bridge length do have limits. The trajectories for both IAAL-E3/O3 and IAA(hFLeu)-E3/O3 dimers show a rapid loss of secondary structure during simulation. This suggests that the salt bridge formed by the glutamate-ornithine interaction was too short to span the hydrophobic interface. The resulting dimer maintained association, however much of the secondary structure and binding interface was lost or distorted. Similar instability due to shorted salt bridge lengths was seen in related work14. Table 9 displays the distance of the salt bridge for many of the coiled-coil dimers that were successfully modeled.
The relatively small standard deviation and short average distance seen in the salt bridge formed by the IAA(hFLeu)-E3/R3 coiled-coil is distinctive. Residue by residue pair-wise free energy decomposition shows a stabilization of −1.8 kcal/mol for combined van der Waals and electrostatic energetics among the Arg, Glu, and hFLeu interacting residues (see Figure 7) compared to IAAL-E3/R3. The driving force for this stabilization derives from an increased average electrostatic interaction of −2.8 kcal/mol between Arg and hFLeu pairs. This suggests that the orientation of the hFLeu residue allows for additional favorable electrostatic interactions to stabilize the positioning of the arginine residue close to the glutamate residue. This increased favorable interaction allows for a stronger and more stable glutamate – arginine salt bridge. Additionally, fluctuations in the MD simulation RMSd (after convergence) appear lower with the hFLeu substituted E3/R3 coiled-coil. Note also that the salt bridges are transient over the course of the MD simulations as shown through the time-sources of salt bridge distances in Figure S9 in the supplementary material.
Conclusion
In this study we have employed simulation methods to explore the structural and energetic differences between fluorinated and nonfluorinated coiled-coils with varying salt bridge lengths and geometries. We found that independent of salt bridge length, fluorinated coiled-coils showed a higher ellipticity than their complementary nonfluorinated pairs.
Using thermodynamic integration we were able to calculate the free energy of hydration of 5,5,5,5′,5′,5′-hexafluoroleucine (hFLeu) to be 1.1 kcal/mol less stable than leucine. We speculate that this destabilizing characteristic of fluorinated residues drives higher order association in coiled-coils to facilitate the burial of hydrophobic atoms exposed to an aqueous environment when the fluorinated atoms are found in the binding domain. This hydrophobic driving force in coiled-coils results in favorable ellipticity, suggestive of increased helicity, due to the coupling of association and secondary structure in coiled-coils173. In contrast, helicity is often reduced when incorporation of fluorinated residues occurs in isolated α-helices since the unfavorable free energy of hydration of hFLeu drives burial of the hydrophobic atoms at the expense of helicity39.
In structural studies, we found that hFLeu and leucine have different sidechain rotamer preferences. Leucine sidechain rotamers progress from gauche−, trans in compressed and normal trajectories to trans, gauche+ in expanded trajectories. 5,5,5,5′,5′,5′-hexafluoroleucine shows a strong preference for the single rotamer gauche−, trans. This difference in rotamer preferences is likely a result of both structural and electrostatic contributions. Fluorine atoms have a larger radius than hydrogen, so that the increased atomic size could lead to steric clashes between fluorine atoms and backbone oxygen atoms in the trans, gauche− rotamer state (see Figure 6). Flourine covalently attached to carbon atoms is more negative than carbon bound hydrogens. In the trans, gauche− rotamer the negatively charged fluorine atoms come in close contact to the negatively charged backbone oxygen which could result in electrostatic repulsion. Sidechain conformation entropy in fluorinated coiled-coil dimers was calculated (see Table 8) and found to be significantly different for leucine and hexalfuorleucine sidechains due to size and geometric differences in fluorinated residues. These rotamer preference differences combined with earlier studies on steric mass37,43 suggest that fluorinated residues cannot be considered isomorphic with their nonfluorinated compliments.
Observation of the average salt bridge lengths revealed that electrostatic differences in fluorinated residues may also play a role in stabilizing salt bridge interactions that reinforce coiled-coil association. Pairwise energetic decomposition of IAA(hFLeu)-E3/R3 trajectories show an increased average electrostatic interaction of −2.4 kcal/mol between Arg and hFLeu pairs compared to IAAL-E3/R3. Analysis of the coiled-coil structural interactions (see Figure 7), suggests that the orientation of the electronegative fluorine atoms of the hFLeu residue stabilizes the positioning of Arg proximal to the Glu residue.
Finally, the differences that we have shown between fluorinated and nonfluorinated coiled-coil dimers suggest that additional engineering and optimization of key residues in fluorinated dimers may lead to additional stabilization of the coiled-coil. This could prove useful for planned applications of coiled-coils as targeting agents, biosensors, and nano-manipulators. Extension of these findings to anti-parallel coiled-coils could be applicable in principle, however the results are likely to be context specific. While coiled-coil ellipticity, expansion, hydration, energetics and salt-bridge behavior will likely remain similar, anti-parallel coiled-coils differ in the residue and position-specific interactions in the hydrophobic binding domain. Trends seen in isoleucine and potentially in leucine and 5,5,5,5′,5′,5′-hexafluoroleucine rotamer preferences and behaviormay vary significantly from those reported here.
Figure 8.

Increased electrostatic interactions between Arg29, Glu13, and hFLeu33 allow the formation of an unusually strong salt bridge. Superimposed IAAL-E3/R3 and IAA(hFLeu)-E3/R3 structures are represented by secondary structure ribbon with explicit tube representation of residues Arg29 and Glu13 in blue for IAAL-E3/R3, Arg29 and Glu13 in light green for IAA(hFLeu)-E3/R3. hFLeu33 is shown in brown and red lines are drawn for emphasis. Due to averaging artifacts, this figure was generated from a single representative snapshot at equivalent stable simulation times from both IAAL-E3/K3 and IAA(hFLeu)-E3/K3 trajectories. As discussed in the text, salt bridge formation is a transitory event and the increased displacement of Arg29 from Glu13 in the IAAL-E3/R3 representation reflects salt bridge separation.
Table 5. Leucine/hFLeu sidechain rotamer distributions in coiled-coil dimers calculated from the central heptads in both monomers.
Note that occupancy calculations were measured as specific pairwise rotamer contributions of both monomer rotamer sets; calculated occupancy reflects rotamer interactions across the entire hydrophobic domain rather than within a single monomer.
| Dimer | Monomer1 rotamers (r1,r2) | Monomer2 rotamers (r1,r2) | Occupied (%) |
|---|---|---|---|
| IAAL-E3K3 | gauche−, trans | gauche−, trans | 95.7 |
| trans, gauche+ | gauche−, trans | 4.3 | |
| IAA(hFLeu)-E3/K3 | gauche−, trans | gauche−, trans | 100.0 |
| IAAL-E3/R3 | trans, gauche+ | trans, gauche+ | 48.5 |
| trans, gauche+ | gauche−, trans | 35.8 | |
| trans, gauche+ | gauche−, gauche+ | 15.8 | |
| IAA(hFLeu)-E3/R3 | gauche−, trans | gauche−, trans | 99.6 |
| IAAL-E3/Ĥ3 | trans, gauche+ | trans, gauche+ | 49.9 |
| trans, gauche+ | gauche−, trans | 43.0 | |
| trans, gauche+ | gauche−, gauche− | 4.2 | |
| gauche−, trans | gauche−, trans | 2.9 | |
| IAA(hFLeu)-E3/Ĥ3 | gauche−, trans | gauche−, trans | 90.7 |
| trans, gauche+ | gauche−, trans | 9.26 |
Table 7. Intrahelical Cα-Cα distances between Leucine/hFLeu residues as a measurement of the hydrophobic interface.
Measurements taken from snapshots spanning the final 2ns of stable simulation time between residues 9 and 30 in the central heptad.. Expansion and compression in coiled-coils can be seen to be influenced strongly by both fluorination and salt bridge length.
| Coiled Coil Dimer | Leu/hFLeu Cα-Cα Distance (Å) | Stdev. |
|---|---|---|
| IAAL-E3/K3 | 6.49 | 0.27 |
| IAA(hFLeu)-E3/K3 | 6.76 | 0.30 |
| IAAL-E3/R3 | 7.48 | 0.30 |
| IAA(hFLeu)-E3/R3 | 7.11 | 0.30 |
| IAAL-E3/Ĥ3 | 7.34 | 0.27 |
| IAA(hFLeu)-E3/Ĥ3 | 7.23 | 0.36 |
Acknowledgments
We wish to thank Seonah Kim for useful discussion of methods to calculate mean residue ellipticity. Support from the NIH R01-GM079383-01 and NIH R21-EB002880-02 and computer time from NSF LRAC MCA01S027P and the University of Utah Center for High Performance Computing (including resources provided by the NIH 1S10RR17214-01 on the Arches metacluster) is greatly acknowledged.
Footnotes
Supplementary Material
Supplementary material is available that includes full sets of RMSd plots, results from force field comparisons, full description of new parameters, plots of water occupancy, more details regarding the free energy calculations, analysis of rotamer populations, and molecular graphics of the structural distortion in the ornithine substituted coiled-coil.
References
- 1.Adamson JG, Zhou NE, Hodges RS. Structure, function and application of the coiled-coil protein folding motif. Curr Op Biotech. 1993;4:428–437. doi: 10.1016/0958-1669(93)90008-k. [DOI] [PubMed] [Google Scholar]
- 2.Lupas AN. Coiled coils: New structures and new functions. Trends Biochem Sci. 1996;21:375–382. [PubMed] [Google Scholar]
- 3.Burkhard P, Stetefeld J, Strelkov SV. Coiled coils: a highly versatile protein folding motif. Trends Cell Biol. 2001;11:82–88. doi: 10.1016/s0962-8924(00)01898-5. [DOI] [PubMed] [Google Scholar]
- 4.Mason JM, Arndt KM. Coiled coil domains: Stability, specificity, and biological implications. Chembiochem. 2004;6:170–176. doi: 10.1002/cbic.200300781. [DOI] [PubMed] [Google Scholar]
- 5.Woolfson DN. The design of coiled-coil structures and assemblies. Advances in protein chemistry. 2005;70:79–112. doi: 10.1016/S0065-3233(05)70004-8. [DOI] [PubMed] [Google Scholar]
- 6.Lupas AN, Gruber M. The structure of alpha-helical coiled coils. Advances in protein chemistry. 2005;70:37–78. doi: 10.1016/S0065-3233(05)70003-6. [DOI] [PubMed] [Google Scholar]
- 7.Liu J, Zheng Q, Deng Y, Cheng CS, Kallenbach NR, Lu M. A seven-helix coiled coil. Proc Natl Acad Sci. 2006;103:15457–15462. doi: 10.1073/pnas.0604871103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gonzalez L, Jr, Plecs JJ, Alber T. An engineered allosteric switch in leucine-zipper oligomerization. Nat Stuct Bio. 1996;3:510–514. doi: 10.1038/nsb0696-510. [DOI] [PubMed] [Google Scholar]
- 9.Chao H, Bautista DL, Litowski J, Irvin RT, Hodges RS. Use of a heterodimeric coiled-coil system for biosensor application and affinity purification. J Chromatogr B Biomed Sci Appl. 1998;715:307–329. doi: 10.1016/s0378-4347(98)00172-8. [DOI] [PubMed] [Google Scholar]
- 10.Wang C, Stewart RJ, Kopecek J. Hybrid hydrogels assembled from synthetic polymers and coiled-coil protein domains. Nature. 1999;397:417–420. doi: 10.1038/17092. [DOI] [PubMed] [Google Scholar]
- 11.Tang A, Wang C, Stewart RJ, Kopecek J. The coiled coils in the design of protein-based constructs: Hybrid hydrogels and epitope displays. J Control Release. 2001;72:57–70. doi: 10.1016/s0168-3659(01)00262-0. [DOI] [PubMed] [Google Scholar]
- 12.Litowski JR, Hodges RS. Designing heterodimeric two-stranded alpha-helical coiled-coils. Effects of hydrophobicity and alpha-helical propensity on protein folding, stability, and specificity. J Biol Chem. 2002;277:37272–37279. doi: 10.1074/jbc.M204257200. [DOI] [PubMed] [Google Scholar]
- 13.Yu YB. Coiled-coils: stability, specificity, and drug delivery potential. Advanced drug delivery reviews. 2002;54:1113–1129. doi: 10.1016/s0169-409x(02)00058-3. [DOI] [PubMed] [Google Scholar]
- 14.Ryan SJ, Kennan AJ. Variable stability heterodimeric coiled-coils from manipulation of electrostatic interface residue chain length. J Amer Chem Soc. 2007;129:10255–10260. doi: 10.1021/ja073717w. [DOI] [PubMed] [Google Scholar]
- 15.Doerr AJ, McLendon GL. Design, folding, and activities of metal-assembled coiled coil proteins. Inorg Chem. 2004;43:7916–7925. doi: 10.1021/ic0490573. [DOI] [PubMed] [Google Scholar]
- 16.Yang J, Wang H. The central coiled-coil domain and carboxyl-terminal WD-repeat domain of Arabidopsis SPA1 are responsible for mediating repression of light signaling. Plant J. 2006 doi: 10.1111/j.1365-313X.2006.02811.x. [DOI] [PubMed] [Google Scholar]
- 17.Hildebrandt ER, Gheber L, Kingsbury TJ, Hoyt MA. Homotetrameric form of CIN8P, an S. cerevisiae kinesin-5 motor, is essential for its in vivo function. J Biol Chem. 2006 doi: 10.1074/jbc.M604817200. [DOI] [PubMed] [Google Scholar]
- 18.Watanabe YH, Takano M, Yoshida M. ATP binding to nucleotide binding domain (NBD)1 of the ClpB chaperone induces motion of the long coiled-coil, stabilizes the hexamer, and activates NBD2. J Biol Chem. 2005;280(26):24562–24567. doi: 10.1074/jbc.M414623200. [DOI] [PubMed] [Google Scholar]
- 19.Fukai S, Nureki O, Sekine S, Shimada A, Vassylyev DG, Yokoyama S. Mechanism of molecular interactions for tRNA(Val) recognition by valyl-tRNA synthetase. Rna. 2003;9(1):100–111. doi: 10.1261/rna.2760703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sorensen JB, Wiederhold K, Muller EM, Milosevic I, Nagy G, de Groot BL, Grubmuller H, Fasshauer D. Sequential N- to C-terminal SNARE complex assembly drives priming and fusion of secretory vesicles. Embo J. 2006;25(5):955–966. doi: 10.1038/sj.emboj.7601003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. Crystal structure of glycoprotein B from herpes simplex virus 1. Science. 2006;313(5784):217–220. doi: 10.1126/science.1126548. [DOI] [PubMed] [Google Scholar]
- 22.Rose A, Meier I. Scaffolds, levers, rods and springs: Diverse cellular functions of long coiled-coil proteins. Cell Mol Life Sci. 2004;61:1996–2009. doi: 10.1007/s00018-004-4039-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu YB. Fluorocarbon nanoparticles as multifunctional drug delivery vehicles. J Drug Target. 2006;14:663–669. doi: 10.1080/10611860600957887. [DOI] [PubMed] [Google Scholar]
- 24.Kukhar VP, Soloshonok VA. Synthesis and properties. John Wiley & Sons, Ltd; 1995. Fluorine-containing amino acids; p. 422. [Google Scholar]
- 25.Xing X, Fichera A, Kumar K. A novel synthesis of enantiomerically pure 5,5,5,5′,5′,5′-hexafluoroleucine. Org Lett. 2001;3:1285–1286. doi: 10.1021/ol015567e. [DOI] [PubMed] [Google Scholar]
- 26.Jiang ZX, Yu YB. The synthesis of a geminally perfluoro-tert-butylated beta-amino acid and its protected forms as a potential pharmacokinetic modulator and reporter for peptide-based pharmaceuticals. J Org Chem. 2007;72:1464–1467. doi: 10.1021/jo0616308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rennert OM, Anker HS. On the incorporation of 5′,5′,5′-trifluoroleucine into proteins of E.coli. Biochem. 1963;2:471–476. doi: 10.1021/bi00903a013. [DOI] [PubMed] [Google Scholar]
- 28.Tang Y, Ghirlanda G, Vaidehi N, Kua J, Mainz DT, Goddard WA, III, DeGrado WF, Tirrell DA. Stabilization of coiled-coil peptide domains by introduction of trifluoroleucine. Biochemistry. 2001;40(9):2790–2796. doi: 10.1021/bi0022588. [DOI] [PubMed] [Google Scholar]
- 29.Wang P, Tang Y, Tirrell DA. Incorporation of trifluoroisoleucine into proteins in vivo. J Amer Chem Soc. 2003;125:6900–6906. doi: 10.1021/ja0298287. [DOI] [PubMed] [Google Scholar]
- 30.Doyon JB, Jain A. The pattern of fluorine substitution affects binding affinity in a small library of fluoroaromatic inhibitors for carbonic anhydrase. Org Lett. 1999;1(2):183–185. doi: 10.1021/ol9905250. [DOI] [PubMed] [Google Scholar]
- 31.Bilgicer B, Fichera A, Kumar K. A coiled coil with a fluorous core. Journal of the American Chemical Society. 2001;123(19):4393–4399. doi: 10.1021/ja002961j. [DOI] [PubMed] [Google Scholar]
- 32.Meng H, Kumar K. Antimicrobial activity and protease stability of peptides containing fluorinated amino acids. J Amer Chem Soc. 2007;129:15615–15622. [Google Scholar]
- 33.Lin JH, Lu AY. Role of pharmacokinetics and metabolism in drug discovery and development. Pharmacol Rev. 1997;49(4):403–449. [PubMed] [Google Scholar]
- 34.Tang Y, Ghirlanda G, Petka WA, Nakajima T, DeGrado WF, Tirrell DA. Fluorinated Coiled-Coil Proteins Prepared In Vivo Display Enhanced Thermal and Chemical Stability. Angewandte Chemie International. 2001;40(8):1494–1496. doi: 10.1002/1521-3773(20010417)40:8<1494::AID-ANIE1494>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 35.Bilgicer B, Kumar K. Synthesis and thermodynamic characterization of self-sorting coiled coils. Tetrahedron. 2002;58(20):4105–4112. [Google Scholar]
- 36.Yoder NC, Kumar K. Fluorinated amino acids in protein design and engineering. Chem Soc Rev. 2002;31(6):335–341. doi: 10.1039/b201097f. [DOI] [PubMed] [Google Scholar]
- 37.Lee K-H, Lee H-Y, Slutsky MM, Anderson JT, Marsh ENG. Fluorous effect in proteins: de novo design and characterization of a four-alpha-helix bundle protein containing hexafluoroleucine. Biochemistry. 2004;43(51):16277–16284. doi: 10.1021/bi049086p. [DOI] [PubMed] [Google Scholar]
- 38.Lee H-Y, Lee K-H, Al-Hashimi HM, Marsh ENG. Modulating protein structure with fluorous amino acids: increased stability and native-like structure conferred on a 4-helix bundle protein by hexafluoroleucine. Journal of the American Chemical Society. 2006;128(1):337–343. doi: 10.1021/ja0563410. [DOI] [PubMed] [Google Scholar]
- 39.Chiu HP, Suzuki Y, Gullickson D, Ahmad R, Kokona B, Fairman R, Cheng RP. Helix propensity of highly fluorinated amino acids. J Amer Chem Soc. 2006;128:15556–15557. doi: 10.1021/ja0640445. [DOI] [PubMed] [Google Scholar]
- 40.Bilgicer B, Xing X, Kumar K. Programmed self-sorting of coiled coils with leucine and hexafluoroleucine cores. Journal of the American Chemical Society. 2001;123(47):11815–11816. doi: 10.1021/ja016767o. [DOI] [PubMed] [Google Scholar]
- 41.Pal PP, Bae JH, Azim MK, Hess P, Friedrich R, Huber R, Moroder L, Budisa N. Structural and spectral response of Aequorea victoria green fluorescent proteins to chromophore fluorination. Biochemistry. 2005;44(10):3663–3672. doi: 10.1021/bi0484825. [DOI] [PubMed] [Google Scholar]
- 42.Nagai T, Nishioka G, Koyama M, Ando A, Miki T, Kumadaki I. Reactions of trifluoromethyl ketones. IX. Investigation of the steric effect of a trifluoromethyl group based on the stereochemistry on the dehydration of trifluoromethyl homoallyl alcohols. J Fluorine Chem. 1992;57:229–237. [Google Scholar]
- 43.Bondi A. van der Waals volumes and radii. The Journal of Physical Chemistry. 1964;68:441–451. [Google Scholar]
- 44.Mason JM, Muller KM, Arndt KM. Considerations in the design and optimization of coiled coil structures. Methods Mol Biol. 2007;352:35–70. doi: 10.1385/1-59745-187-8:35. [DOI] [PubMed] [Google Scholar]
- 45.Crick F. The packing of [alpha]-helices: simple coiled-coils. Acta Crystallographica. 1953;6:689–697. [Google Scholar]
- 46.Bosshard HR, Durr E, Hitz T, Jelesarov I. Energetics of coiled coil folding: the nature of the transition states. Biochem. 2001;40(12):3544–3552. doi: 10.1021/bi002161l. [DOI] [PubMed] [Google Scholar]
- 47.Woolfson An Introduction to Coiled Coils. 2000;2006 [Google Scholar]
- 48.Lumb KJ, Kim PS. A buried polar interaction imparts structural uniqueness in a designed heterodimeric coiled coil. Biochem. 1995;34:8642–8648. doi: 10.1021/bi00027a013. [DOI] [PubMed] [Google Scholar]
- 49.Nautiyal S, Woolfson DN, King DS, Alber T. A designed heterotrimeric coiled coil. Biochem. 1995;34:11645–11651. doi: 10.1021/bi00037a001. [DOI] [PubMed] [Google Scholar]
- 50.Nautiyal S, Alber T. Crystal structure of a designed, thermostable heterotrimeric coiled coil. Prot Sci. 1999;8:84–90. doi: 10.1110/ps.8.1.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McClain DL, Woods HL, Oakley MG. Design and characterization of a heterodimeric coiled coil that forms exclusively with an antiparallel relative helix orientation. J Amer Chem Soc. 2001;123:3151–3152. doi: 10.1021/ja004099l. [DOI] [PubMed] [Google Scholar]
- 52.van Hest JC, Tirrell DA. Protein-based materials, toward a new level of structural control. Chemical Communications. 2001;(19):1897–1904. doi: 10.1039/b105185g. [DOI] [PubMed] [Google Scholar]
- 53.Arndt KM, Pelletier JN, Muller KM, Pluckthun A, Alber T. Comparison of in vivo selection and rational design of heterodimeric coiled coils. Structure. 2002;10:1235–1248. doi: 10.1016/s0969-2126(02)00838-9. [DOI] [PubMed] [Google Scholar]
- 54.Campbell KM, Lumb KJ. Complementation of buried lysine and surface polar residues in a designed heterodimeric coiled coil. Biochemistry. 2002;41(22):7169–7175. doi: 10.1021/bi025559l. [DOI] [PubMed] [Google Scholar]
- 55.Schnarr NA, Kennan AJ. Peptide tic-tac-toe: Heterotrimeric coiled-coil specificity from steric matching of multiple hydrophobic side chains. J Amer Chem Soc. 2002;124:9779–9783. doi: 10.1021/ja0174940. [DOI] [PubMed] [Google Scholar]
- 56.Gurnon DG, Whitaker JA, Oakley MG. Design and characterization of a homodimeric antiparallel coiled coil. J Amer Chem Soc. 2003;125:7518–7519. doi: 10.1021/ja0357590. [DOI] [PubMed] [Google Scholar]
- 57.Schnarr NA, Kennan AJ. Sequential and specific exchange of multiple coiled-coil components. J Amer Chem Soc. 2003;125:13046–13051. doi: 10.1021/ja035666i. [DOI] [PubMed] [Google Scholar]
- 58.Schnarr NA, Kennan AJ. Strand orientation by steric matching: A designed antiparallel coiled-coil trimer. J Amer Chem Soc. 2004;126:14447–14451. doi: 10.1021/ja047496v. [DOI] [PubMed] [Google Scholar]
- 59.Yadav M, Redman JE, Leman LJ, Alvarez-Gutierrez JM, Zhang Y, Stout CD, Ghadiri MR. Structure-based engineering of internal cavities in coiled-coil peptides. Biochem. 2005;44:9723–9732. doi: 10.1021/bi050742a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vieth M, Kolinski A, Brooks CL, 3rd, Skolnick J. Prediction of the folding pathways and structure of the GCN4 leucine zipper. J Mol Biol. 1994;237:361–367. doi: 10.1006/jmbi.1994.1239. [DOI] [PubMed] [Google Scholar]
- 61.Lumb KJ, Carr CM, Kim PS. Subdomain folding of the coiled coil leucine zipper from the bZIP transcriptional activator GCN4. Biochem. 1994;33:7361–7367. doi: 10.1021/bi00189a042. [DOI] [PubMed] [Google Scholar]
- 62.Gonzalez L, Jr, Brown RA, Richardson D, Alber T. Crystal structures of a single coiled-coil peptide in two oligomeric states reveal the basis for structural polymorphism. Nat Stuct Bio. 1996;3:1002–1010. doi: 10.1038/nsb1296-1002. [DOI] [PubMed] [Google Scholar]
- 63.Burkhard P, Ivaninskii S, Lustig A. Improving coiled-coil stability by optimizing ionic interactions. Journal of molecular biology. 2002;318(3):901–910. doi: 10.1016/S0022-2836(02)00114-6. [DOI] [PubMed] [Google Scholar]
- 64.Dragan AI, Potekhin SA, Sivolob A, Lu M, Privalov PL. Kinetics and thermodynamics of the unfolding and refolding of the three-stranded alpha-helical coiled coil, Lpp-56. Biochem. 2004;43:14891–14900. doi: 10.1021/bi048365+. [DOI] [PubMed] [Google Scholar]
- 65.Meisner WK, Sosnick TR. Fast folding of a helical protein initiated by the collision of unstructured chains. Proc Natl Acad Sci. 2004;101:13478–13482. doi: 10.1073/pnas.0404057101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Meisner WK, Sosnick TR. Barrier-limited, microsecond folding of a stable protein measured with hydrogen exchange: Implications for downhill folding. Proc Natl Acad Sci. 2004;101:15639–15644. doi: 10.1073/pnas.0404895101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Missimer JH, Steinmetz MO, Jahnke W, Winkler FK, van Gunsteren WF, Daura X. Molecular-dynamics simulations of C- and N-terminal peptide derivatives of GCN4-p1 in aqueous solution. Chem Biodivers. 2005;2:1086–1104. doi: 10.1002/cbdv.200590078. [DOI] [PubMed] [Google Scholar]
- 68.Bjelic S, Karshikoff A, Jelesarov I. Stability and folding/unfolding kinetics of the homotrimeric coiled coil Lpp-56. Biochem. 2006;45:8931–8939. doi: 10.1021/bi0608156. [DOI] [PubMed] [Google Scholar]
- 69.Bunagan MR, Cristian L, DeGrado WF, Gai F. Truncation of a cross-linked GCN4-p1 coiled coil leads to ultrafast folding. Biochem. 2006;12:10981–10986. doi: 10.1021/bi0606142. [DOI] [PubMed] [Google Scholar]
- 70.d’Avignon DA, Bretthorst GL, Holtzer ME, Schwarz KA, Angeletti RH, Mints L, Holtzer A. Site-specific experiments on folding/unfolding of Jun coiled coils: Thermodynamic and kinetic parameters from spin inversion transfer nuclear magnetic resonance at leucine-18. Biopol. 2006;83:255–267. doi: 10.1002/bip.20555. [DOI] [PubMed] [Google Scholar]
- 71.Steinmetz MO, Jelesarov I, Matousek WM, Honnappa S, Jahnke W, Missimer JH, Frank S, Alexandrescu AT, Kammerer RA. Molecular basis of coiled-coil formation. Proc Natl Acad Sci. 2007;104(17):7062–7067. doi: 10.1073/pnas.0700321104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mason JM, Hagemann UB, Arndt KM. Improved stability of the Jun-Fos activator protein-1 coiled coil motif: A stopped-flow circular dichroism kinetic analysis. J Biol Chem. 2007;282:23015–23024. doi: 10.1074/jbc.M701828200. [DOI] [PubMed] [Google Scholar]
- 73.Nikolaev Y, Pervushin K. NMR spin state exchange spectroscopy reveals equilibrium of two distinct conformations of leucine zipper GCN4 in solution. J Amer Chem Soc. 2007;129:6461–6469. doi: 10.1021/ja0685295. [DOI] [PubMed] [Google Scholar]
- 74.Bornschlogl T, Reif M. Single-molecular dynamics of mechanical coiled-coil unzipping. Langmuir. 2007 doi: 10.1021/la7023567. [ASAP] [DOI] [PubMed] [Google Scholar]
- 75.O’Shea EK, Klemm JD, Kim PS, Alber T. X-ray structure of the GCN4 leucine zipper, a two-stranded, parallel coiled coil. Science. 1991;254(5031):539–544. doi: 10.1126/science.1948029. [DOI] [PubMed] [Google Scholar]
- 76.Harbury PB, Zhang T, Kim PS, Alber T. A switch between two-, three-, and four-stranded coiled coils in GCN4 leucine zipper mutants. Science. 1993;262(5138):1401–1407. doi: 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]
- 77.Junius FK, Mackay JP, Bubb WA, Jensen SA, Weiss AS, King GF. Nuclear magnetic resonance characterization of the Jun leucine zipper domain: unusual properties of coiled-coil interfacial polar residues. Biochem. 1995;34:6164–6174. doi: 10.1021/bi00018a020. [DOI] [PubMed] [Google Scholar]
- 78.Yu Y, Monera OD, Hodges RS, Privalov PL. Investigation of electrostatic interactions in two-stranded coiled-coils through residue shuffling. Biophys Chem. 1996;59:299–314. doi: 10.1016/0301-4622(95)00131-x. [DOI] [PubMed] [Google Scholar]
- 79.Gonzalez L, Jr, Woolfson DN, Alber T. Buried polar residues and structural specificity in the GCN4 leucine zipper. Nature structural biology. 1996;3:1011–1018. doi: 10.1038/nsb1296-1011. [DOI] [PubMed] [Google Scholar]
- 80.Caballero-Herrera A, Nilsson L. Molecular dynamics simulations of the E1/E2 transmembrane domain of the Semliki Forest virus. Biophys J. 2003;85:3646–3658. doi: 10.1016/S0006-3495(03)74782-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Danciulescu C, Nick B, Wortmann FJ. Structural stability of wild type and mutated alpha-keratin fragments: molecular dynamics and free energy calculations. Biomacromol. 2004;5:2165–2175. doi: 10.1021/bm049788u. [DOI] [PubMed] [Google Scholar]
- 82.Smith TA, Steinert PM, Parry DA. Modeling effects of mutations in coiled-coil structures: Case study using epidermolysis bullosa simplex mutations in segment 1a of K5/K14 intermediate filaments. Proteins. 2004;55:1043–1052. doi: 10.1002/prot.20089. [DOI] [PubMed] [Google Scholar]
- 83.Ambroggio XI, Kuhlman B. Computational design of a single amino acid sequence that can switch between two distinct protein folds. J Amer Chem Soc. 2006;128:1154–1161. doi: 10.1021/ja054718w. [DOI] [PubMed] [Google Scholar]
- 84.Yadav MK, Leman LJ, Price DJ, Brooks CL, 3rd, Stout CD, Ghadiri MR. Coiled coils at the edge of configurational heterogeneity. Structural analyses of parallel and antiparallel homotetrameric coiled coils reveal configurational sensitivity to a single solvent-exposed amino acid substitution. Biochem. 2006;45:4463–4473. doi: 10.1021/bi060092q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yu Y, Monera OD, Hodges RS, Privalov PL. Ion pairs significantly stabilize coiled-coils in the absence of electrolyte. J Mol Biol. 1996;255:367–372. doi: 10.1006/jmbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 86.Zhong Q, Moore PB, Newns DM, Klein ML. Molecular dynamics study of the LS3 voltage-gated ion channel. FEBS letters. 1998;427(2):267–270. doi: 10.1016/s0014-5793(98)00304-4. [DOI] [PubMed] [Google Scholar]
- 87.Gorfe AA, Ferrara P, Caflisch A, Marti DN, Bosshard HR, Jelesarov I. Calculation of protein ionization equilibria with conformational sampling: pK(a) of a model leucine zipper, GCN4 and barnase. Proteins. 2002;46:41–60. doi: 10.1002/prot.10027. [DOI] [PubMed] [Google Scholar]
- 88.Madhusoodanan M, Lazaridis T. Investigation of pathways for the low-pH conformational transition in influenza hemagglutinin. Biophysical journal. 2003;84(3):1926–1939. doi: 10.1016/S0006-3495(03)75001-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ding F, LaRocque JJ, Dokholyan NV. Direct observation of protein folding, aggregation, and a prion-like conformational conversion. J Biol Chem. 2005;280:40235–40240. doi: 10.1074/jbc.M506372200. [DOI] [PubMed] [Google Scholar]
- 90.Hawkins RJ, McLeish TC. Dynamic allostery of protein alpha helical coiled-coils. J Royal Soc Interface. 2006;3:125–138. doi: 10.1098/rsif.2005.0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Vagt T, Zschornig O, Huster D, Koksch B. Membrane binding and structure of de novo designed alpha-helical cationic coiled-coil-forming peptides. Chemphyschem. 2006;7:1361–1371. doi: 10.1002/cphc.200600010. [DOI] [PubMed] [Google Scholar]
- 92.Laughton CA, Luisi BF, Pratap JV, Calladine CR. A potential molecular switch in an alpha-helical coiled coil. Proteins. 2007;70:25–30. doi: 10.1002/prot.21596. [DOI] [PubMed] [Google Scholar]
- 93.Palaiomylitou M, Tartas A, Vlachakis D, Tzamarias D, Vlassi M. Investigating the structural stability of the Tup1-interaction domain of Ssn6: Evidence for a conformational change on the complex. Proteins. 2007;70:72–82. doi: 10.1002/prot.21489. [DOI] [PubMed] [Google Scholar]
- 94.Holtzer ME, Bretthorst GL, d’Avignon DA, Angeletti RH, Mints L, Holtzer A. Temperature dependence of the folding and unfolding kinetics of the GCN4 leucine zipper via 13C(alpha)-NMR. Biophys J. 2001;80:939–951. doi: 10.1016/s0006-3495(01)76073-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nishikawa K, Scheraga HA. Geometrical criteria for formation of coiled-coil structures of polypeptide chains. Macromolecules. 1976;9(3):395–407. doi: 10.1021/ma60051a004. [DOI] [PubMed] [Google Scholar]
- 96.Lupas A, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- 97.Tropsha A, Bowen JP, Brown FK, Kizer JS. Do interhelical side chain-backbone hydrogen bonds participate in formation of leucine zipper coiled coils? Proc Natl Acad Sci. 1991;88:9488–9492. doi: 10.1073/pnas.88.21.9488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cregut D, Liautard JP, Heitz F, Chiche L. Molecular modeling of coiled-coil alpha-tropomyosin: analysis of staggered and in register helix-helix interactions. Prot Eng. 1993;6:51–58. doi: 10.1093/protein/6.1.51. [DOI] [PubMed] [Google Scholar]
- 99.Zhang L, Hermans J. Molecular dynamics study of structure and stability of a model coiled coil. Proteins. 1993;16:384–392. doi: 10.1002/prot.340160407. [DOI] [PubMed] [Google Scholar]
- 100.Kerr ID, Sankararamakrishnan R, Smart OS, Sansom MS. Parallel helix bundles and ion channels: molecular modeling via simulated annealing and restrained molecular dynamics. Biophys J. 1994;67:1501–1515. doi: 10.1016/S0006-3495(94)80624-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rozzelle JE, Jr, Tropsha A, Erickson BW. Rational design of a three-heptad coiled-coil protein and comparison by molecular dynamics simulation with the GCN4 coiled coil: presence of interior three-center hydrogen bonds. Prot Sci. 1994;3:345–355. doi: 10.1002/pro.5560030217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Berger B, Wilson DB, Wolf E, Tonchev T, Milla M, Kim PS. Predicting coiled coils by use of pairwise residue correlations. Proc Natl Acad Sci. 1995;92:8259–8263. doi: 10.1073/pnas.92.18.8259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Harbury PB, Tidor B, Kim PS. Repacking protein cores with backbone freedom: structure prediction for coiled coils. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(18):8408–8412. doi: 10.1073/pnas.92.18.8408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Offer G, Sessions R. Computer modelling of the alpha-helical coiled coil: packing of side-chains in the inner core. Journal of molecular biology. 1995;249(5):967–987. doi: 10.1006/jmbi.1995.0352. [DOI] [PubMed] [Google Scholar]
- 105.Sankararamakrishnan R, Sansom MS. Modelling packing interactions in parallel helix bundles: pentameric bundles of nicotinic receptor M2 helices. Biochimica et biophysica acta. 1995;1239(2):122–132. doi: 10.1016/0005-2736(95)00165-y. [DOI] [PubMed] [Google Scholar]
- 106.Vieth M, Kolinski A, Brooks CL, 3rd, Skolnick J. Prediction of quaternary structure of coiled coils. Application to mutants of the GCN4 leucine zipper. J Mol Biol. 1995;251:448–467. doi: 10.1006/jmbi.1995.0447. [DOI] [PubMed] [Google Scholar]
- 107.Woolfson DN, Alber T. Predicting oligomerization states of coiled coils. Prot Sci. 1995;4:1596–1607. doi: 10.1002/pro.5560040818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hirst JD, Vieth M, Skolnick J, Brooks CL., 3rd Predicting leucine zipper structures from sequence. Prot Eng. 1996;9:657–662. doi: 10.1093/protein/9.8.657. [DOI] [PubMed] [Google Scholar]
- 109.Vieth M, Kolinski A, Skolnick J. Method for predicting the state of association of discretized protein models. Application to leucine zippers Biochem. 1996;35:955–967. doi: 10.1021/bi9520702. [DOI] [PubMed] [Google Scholar]
- 110.Garnier N, Genest D, Duneau JP, Genest M. Molecular modeling of c-erbB2 receptor dimerization: coiled-coil structure of wild and oncogenic transmembrane domains--stabilization by interhelical hydrogen bonds in the oncogenic form. Biopoly. 1997;42:157–168. doi: 10.1002/(SICI)1097-0282(199708)42:2<157::AID-BIP5>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 111.Wolf E, Kim PS, Berger B. MultiCoil: a program for predicting two- and three-stranded coiled coils. Prot Sci. 1997;6:1179–1189. doi: 10.1002/pro.5560060606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lupas A. Predicting coiled-coil regions in proteins. Curr Opin Struct Biol. 1997;7:388–393. doi: 10.1016/s0959-440x(97)80056-5. [DOI] [PubMed] [Google Scholar]
- 113.Harbury PB, Plecs JJ, Tidor B, Alber T, Kim PS. High-resolution protein design with backbone freedom. Science. 1998;282(5393):1462–1467. doi: 10.1126/science.282.5393.1462. [DOI] [PubMed] [Google Scholar]
- 114.Zhong Q, Jiang Q, Moore PB, Newns DM, Klein ML. Molecular dynamics simulation of a synthetic ion channel. Biophys J. 1998;74:3–10. doi: 10.1016/S0006-3495(98)77761-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhong Q, Husslein T, Moore PB, Newns DM, Pattnaik P, Klein ML. The M2 channel of influenza A virus: a molecular dynamics study. FEBS Lett. 1998;434:265–271. doi: 10.1016/s0014-5793(98)00988-0. [DOI] [PubMed] [Google Scholar]
- 116.Kukol A, Arkin IT. vpu transmembrane peptide structure obtained by site-specific fourier transform infrared dichroism and global molecular dynamics searching. Biophys J. 1999;77:1594–1601. doi: 10.1016/S0006-3495(99)77007-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yang PK, Tzou WS, Hwang MJ. Restraint-driven formation of alpha-helical coiled coils in molecular dynamics simulations. Biopoly. 1999;50:667–677. doi: 10.1002/(SICI)1097-0282(199911)50:6<667::AID-BIP11>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 118.Kukol A, Arkin IT. Structure of the influenza C virus CM2 protein transmembrane domain obtained by site-specific infrared dichroism and global molecular dynamics searching. J Biol Chem. 2000;275:4225–4229. doi: 10.1074/jbc.275.6.4225. [DOI] [PubMed] [Google Scholar]
- 119.Walshaw J, Woolfson DN. Open-and-shut cases in coiled-coil assembly: alpha-sheets and alpha-cylinders. Prot Sci. 2001;10:668–673. doi: 10.1110/ps.36901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Walshaw J, Woolfson DN. Socket: a program for identifying and analysing coiled-coil motifs within protein structures. J Mol Biol. 2001;307:1427–1450. doi: 10.1006/jmbi.2001.4545. [DOI] [PubMed] [Google Scholar]
- 121.Briki F, Doucet J, Etchebest C. A procedure for refining a coiled coil protein structure using x-ray fiber diffraction and modeling. Biophys J. 2002;83:1774–1783. doi: 10.1016/S0006-3495(02)73943-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Orzechoski M, Cieplak P, Piela L. Theoretical calculations of the coiled-coil stability in water in the context of its possible use as a molecular rack. J Comp Chem. 2002;23:106–110. doi: 10.1002/jcc.10020. [DOI] [PubMed] [Google Scholar]
- 123.Ash WL, Stockner T, MacCallum JL, Tieleman DP. Computer modeling of polyleucine-based coiled coil dimers in a realistic membrane environment: insight into helix-helix interactions in membrane proteins. Biochem. 2004;43:9050–9060. doi: 10.1021/bi0494572. [DOI] [PubMed] [Google Scholar]
- 124.Gruber M, Soding J, Lupas AN. Comparative analysis of coiled-coil prediction methods. J Struct Biol. 2006;155:140–145. doi: 10.1016/j.jsb.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 125.Ramos J, Lazaridis T. Energetic determinants of oligomeric state specificity in coiled coils. Journal of the American Chemical Society. 2006;128(48):15499–15510. doi: 10.1021/ja0655284. [DOI] [PubMed] [Google Scholar]
- 126.Nilges M, Brunger AT. Automated modeling of coiled coils: Application to the GCN4 dimerization domain. Prot Eng. 1991;4:649–659. doi: 10.1093/protein/4.6.649. [DOI] [PubMed] [Google Scholar]
- 127.Nilges M, Brunger AT. Successful prediction of the coiled coil geometry of the GCN4 leucine zipper domain by simulated annealing: comparison to the X-ray structure. Proteins. 1993;15:133–146. doi: 10.1002/prot.340150205. [DOI] [PubMed] [Google Scholar]
- 128.Mottamal M, Zhang J, Lazaridis T. Energetics of the native and non-native states of the glycophorin transmembrane helix dimer. Proteins. 2006;62(4):996–1009. doi: 10.1002/prot.20844. [DOI] [PubMed] [Google Scholar]
- 129.Samna Soumana O, Garnier N, Genest M. Molecular dynamics simulation approach for the prediction of transmembrane helix-helix heterodimers assembly. Eur Biophys J. 2007;36:1071–1082. doi: 10.1007/s00249-007-0195-6. [DOI] [PubMed] [Google Scholar]
- 130.Pineiro A, Villa A, Vagt T, Koksch B, Mark AE. A molecular dynamics study of the formation, stability, and oligomerization state of two designed coiled coils: Possibilities and limitations. Biophys J. 2005;89:3701–3713. doi: 10.1529/biophysj.104.055590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Missimer JH, Steinmetz MO, Baron R, Winkler FK, Kammerer RA, Daura X, van Gunsteren WF. Configurational entropy elucidates the role of salt-bridge networks in protein thermostability. Protein Sci. 2007;16(7):1349–1359. doi: 10.1110/ps.062542907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.El-Kettani MA, Smith JC. Pathways for conformational change in seryl-tRNA synthetase from Thermus thermophilus. C Royal Acad Sci III. 1996;319:161–169. [PubMed] [Google Scholar]
- 133.Litowski JR, Hodges RS. Designing heterodimeric two-stranded alpha-helical coiled-coils: the effect of chain length on protein folding, stability and specificity. J Pept Res. 2001;58(6):477–492. doi: 10.1034/j.1399-3011.2001.10972.x. [DOI] [PubMed] [Google Scholar]
- 134.Lindhout DA, Litowski JR, Mercier P, Hodges RS, Sykes BD. NMR solution structure of a highly stable de novo heterodimeric coiled-coil. Biopolymers. 2004;75(5):367–375. doi: 10.1002/bip.20150. [DOI] [PubMed] [Google Scholar]
- 135.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18(15):2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 136.Case DA, Cheatham TE, III, Darden TA, Gohlker H, Luo R, Merz KM, Jr, Onufriev AV, Simmerling C, Wang B, Woods R. The AMBER biomolecular simulation programs. J Comp Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Simmerling C, Strockbine B, Roitberg AE. All-atom structure prediction and folding simulations of a stable protein. Journal of the American Chemical Society. 2002;124(38):11258–11259. doi: 10.1021/ja0273851. [DOI] [PubMed] [Google Scholar]
- 138.Cornell WD, Cieplak P, Baylyl CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 139.Kollman PA, Dixon R, Cornell W, Fox T, Chipot C, Pohorille A. The development/application of a ‘minimalist’ organic/biochemical molecular mechanic force field using a combination of ab initio calculations and experimental data. In: Wilkinson A, PWaWFvG, editors. Computer Simulation of Biomolecular Systems. Vol. 3. Elsevier; 1997. pp. 83–96. [Google Scholar]
- 140.Wang J, Cieplak P, Kollman PA. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J Comput Chem. 2000;21:1049–1074. [Google Scholar]
- 141.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65(3):712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Roe DR, Okur A, Wickstrom L, Hornak V, Simmerling C. Secondary Structure Bias in Generalized Born Solvent Models: Comparison of Conformational Ensembles and Free Energy of Solvent Polarization from Explicit and Implicit Solvation. J Phys Chem B Condens Matter Mater Surf Interfaces Biophys. 2007;111(7):1846–1857. doi: 10.1021/jp066831u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Perez A, Marchan I, Svozil D, Sponer J, Cheatham TE, III, Laughton CA, Orozco M. Refinement of the AMBER force field for nucleic acids. Improving the description of alpha/gamma conformers. Biophys J. 2007;11:3817–3829. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE, Debolt S, Ferguson D, Seibel G, Kollman P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structure and energetic properties of molecules. Comp Phys Comm. 1995;91(1–3):1–41. [Google Scholar]
- 145.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A Smooth Particle Mesh Ewald Method. J Chem Phys. 1995;103(19):8577–8593. [Google Scholar]
- 146.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparisons of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 147.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. Journal of Computational Physics. 1977;23:327–341. [Google Scholar]
- 148.Barth E, Kuczera K, Leimkuhler B, Skeel RD. Algorithms for constrained molecular dynamics. Journal of computational chemistry. 1995;16(10):1192–1209. [Google Scholar]
- 149.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. Journal of Computational Physics. 1984;81:3684–3690. [Google Scholar]
- 150.Hobza P, Kabelac M, Sponer J, Mejzlik P, Vondrasek J. Performance of empirical potentials (AMBER, CFF95, CVFF, CHARMM, OPLS, POLTEV), semiempirical quantum chemical methods (AM1, MNDO/M, PM3), and ab initio Hartree-Fock method for interaction of DNA bases: Comparison with nonempirical beyond Hartree-Fock results. J Comp Chem. 1997;18:1136–1150. [Google Scholar]
- 151.Showalter SA, Bruschweiler R. Quantitative molecular ensemble interpretation of NMR dipolar couplings without restraints. J Amer Chem Soc. 2007;129:4158–4159. doi: 10.1021/ja070658d. [DOI] [PubMed] [Google Scholar]
- 152.Mobley DL, Dumont E, Chodera JD, Dill KA. Comparison of charge models for fixed-charge force fields: small-molecule hydration free energies in explicit solvent. J Phys Chem B. 2007;111:2242–2254. doi: 10.1021/jp0667442. [DOI] [PubMed] [Google Scholar]
- 153.Nicholls A, Mobley DL, Guthrie JP, Chodera JD, Bayly CI, Cooper MD, Pande VS. Predicting small-molecule solvation free energies: an informal blind test for computational chemistry. J Med Chem. 2008;51:769–779. doi: 10.1021/jm070549+. [DOI] [PubMed] [Google Scholar]
- 154.Daniels SM, Saunders RA, Platts JA. Prediction of fluorophilicity of organic and transition metal compounds using molecular surface areas. J Fluorine Chem. 2004;125:1291–1298. [Google Scholar]
- 155.Friedemann R, Naumann S, Brickmann J. Aggregation of alkane and fluoroalkane clusters: Molecular dynamics simulation results. Phys Chem Chem Phys. 2001;3:4195–4199. [Google Scholar]
- 156.Friedemann R, Naumann S, Brickmann J. Molecular dynamics studies on the aggregation of Y-shaped fluoroalkanes. J Mol Mod. 2002;8:266–271. doi: 10.1007/s00894-002-0092-y. [DOI] [PubMed] [Google Scholar]
- 157.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Tokoyo K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Sannenberg JJ, Zakrewski VG, Dapprich S, Daniels S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 2003. Gaussian, Inc; Pittsburgh, PA: 2003. [Google Scholar]
- 158.Granovsky AA. PC GAMESS version 7.0. 2006.
- 159.Bayly CI, Cieplak P, Cornell WD, Kollman PA. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges-the RESP model. J Phys Chem. 1993;97(40):10269–10280. [Google Scholar]
- 160.Zaffran TC, P.;, Dupradeau FY. R.E.D. II. 2005.
- 161.Bryce R. Amber Parameter Database. 2006.
- 162.Case DA, Darden TA, Cheatham ITE, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Wang B, Pearlman DA, Crowley M, Brozell S, Tsui V, Gohlke H, Mongan J, Hornak B, Cui G, Beroza P, Scafmeister C, Caldwell JW, Ross WS, Kollman PA. AMBER8. University of California; San Francisco: 2004. [Google Scholar]
- 163.Dunbrack RL, Jr, Cohen FE. Bayesian statistical analysis of protein side-chain rotamer preferences. Protein Sci. 1997;6(8):1661–1681. doi: 10.1002/pro.5560060807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Cheatham TE, Kollman PA. Molecular Dynamics Simulations Highlight the Structural Differences among DNA:DNA, RNA:RNA, and DNA:RNA Hybrid Duplexes. Journal of the American Chemical Society. 1997;119:4805–4825. [Google Scholar]
- 165.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. Journal of computational chemistry. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 166.Mezei M. The finite difference thermodynamic integration, tested on calculating the hydration free energy difference between acetone and dimethylamine in water. J Chem Phys. 1987;86(12):7084–7088. [Google Scholar]
- 167.Shirts MR, Pitera JW, Swope WC, Pande VS. Extremely Precise Free Energy Calculations of Amino Acid Side Chain Analogs: Comparison of Common Molecular Mechanics Force Fields for Proteins. J Chem Phys. 2003;119:5740. [Google Scholar]
- 168.Gohlke H, Kiel C, Case DA. Insights into protein-protein binding by binding free energy calculation and free energy decomposition for the Ras-Raf and Ras-RalGDS complexes. J Mol Biol. 2003;330:891–913. doi: 10.1016/s0022-2836(03)00610-7. [DOI] [PubMed] [Google Scholar]
- 169.Bulheller BM, Rodger A, Hirst JD. Circular and linear dichroism of proteins. Phys Chem Chem Phys. 2007;9(17):2020–2035. doi: 10.1039/b615870f. [DOI] [PubMed] [Google Scholar]
- 170.Arndt KM, Pelletier JN, Muller KM, Alber T, Michnick SW, Pluckthun A. A heterodimeric coiled-coil peptide pair selected in vivo from a designed library-versus-library ensemble. J Mol Biol. 2000;295(3):627–639. doi: 10.1006/jmbi.1999.3352. [DOI] [PubMed] [Google Scholar]
- 171.Yu YB, Lavigne P, Privalov PL, Hodges RS. The Measure of Interior Disorder in a Folded Protein and Its Contribution to Stability. Journal of the American Chemical Society. 1999;121(37):8443–8449. [Google Scholar]
- 172.Mucke N, Kreplak L, Kirmse R, Wedig T, Herrmann H, Aebi U, Langowski J. Assessing the flexibility of intermediate filaments by atomic force microscopy. Journal of molecular biology. 2004;335(5):1241–1250. doi: 10.1016/j.jmb.2003.11.038. [DOI] [PubMed] [Google Scholar]
- 173.Jelesarov I, Bosshard HR. Thermodynamic characterization of the coupled folding and association of heterodimeric coiled coils (leucine zippers) Journal of molecular biology. 1996;263(2):344–358. doi: 10.1006/jmbi.1996.0579. [DOI] [PubMed] [Google Scholar]