

Abstract
We have developed a new biosensor architecture, which is comprised of a polypeptide–peptide nucleic acid tri-block copolymer and which we have termed chimeric peptide beacons (CPB), that generates an optical output via a mechanism analogous to that employed in DNA-based molecular beacons.
Phage display and other in vitro selection techniques produce short polypeptides that tightly and specifically bind to any of a wide range of macromolecular targets. Here we demonstrate a novel means of converting such polypeptides into optical biosensors. The sensing architecture we have developed, a continuation of our peptide beacon approach,1 utilizes a chimeric block-copolymer comprised of complementary peptide nucleic acid (PNA) sequences flanking a short recognition polypeptide. In the absence of target, the flexible polypeptide allows the PNA to form a duplex, bringing a terminally attached fluorophore/quencher pair into proximity and reducing fluorescence. Target binding rigidifies the polypeptide, breaking the PNA “stem” and enhancing emission three-fold. Using this approach we report here the robust optical detection of anti-HIV antibodies at picomolar concentrations.
Molecular beacons2 (MBs), stem-loop DNA molecules that undergo a large-scale conformational change upon target binding, have proven to be of significant utility for the optical3 detection of oligonucleotides. In the absence of target, the MB’s stem-loop structure holds terminally attached quencher and fluorophore moieties in proximity, enhancing quenching and minimizing fluorescence emission. Target binding to the single-stranded loop disrupts the double-stranded stem, segregating the termini and producing a large increase in emission with a variety of different fluorophores.4 The formation of the double-stranded stem in the unbound sensor ensures efficient quenching and low background emission, rendering MBs among the more sensitive means of detecting oligonucleotides.5 The generality of the approach is limited, however, to targets that bind oligonucleotides, and while this can include proteins and small molecules (e.g., ref. 6), many analytes, including many diagnostically relevant antibodies,7 are better detected using polypeptide-based recognition elements. The development of polypeptide-based sensors analogous to MBs could thus significantly extend the scope of the beacon detection approach.
The development of polypeptide-based sensors equivalent to molecular beacons has been hampered by the fact that polypeptides generally do not form stable, “closed” structures (equivalent to the stem in an unbound MB stem-loop) that can subsequently be disrupted by target binding.8 One solution to this problem has been demonstrated in a class of optical biosensors, termed peptide beacons (PB), which exploit the observation that, while unbound polypeptides are almost invariably highly dynamic, their structure becomes fixed upon binding to a macromolecular target. This effect, in turn has been used to generate binding-induced changes in the fluorescence of terminally attached reporter groups via two different mechanisms. The first employs reporters, such as pyrene, that form weak duplexes, the disruption of which modulates emission.1a,9 The second, in contrast, relies on the contact (electron transfer-based) quenching of a fluorophore that is long-lived relative to the tens of nanoseconds time constants of intra-chain collisions.1b,10
The above PB architectures are limited to employing either those uncommon dyes that form stable dimers (and for which dimer formation alters emission) or to dyes with lifetimes that are long relative to intra-chain collisions rates, which are, unfortunately, orders of magnitude slower than the fluorescence decay rates of typical organic fluorophores. Here we have overcome these limitations by fabricating a polypeptide-based sensing platform that is more closely analogous to DNA-based MBs.‡ The new architecture, which we have termed chimeric peptide beacons (CPB) is comprised of a block co-polymer consisting of a polypeptide recognition unit flanked by two, complementary peptide nucleic acids (PNA) sequences terminally-modified with a fluorophore/contact-quencher pair. In the absence of target, the complementary PNA sequences form a stem, holding the fluorophore in proximity to the quencher and reducing emission. This stem is broken upon target binding, leading to enhanced fluorescence.
In order to test this new sensor architecture we have synthesized a CPB aimed at the detection of a specific class of anti-HIV antibodies and fabricated from a highly antigenic,11 six-residue epitope from the HIV protein p17 (Fig. 1). The epitope is contiguous and adopts a modestly extended conformation in both the native protein, and when complexed with its target antibody.12 We have added short, complementary PNA stems to this polypeptide recognition unit and terminated them with the dye bodipy and the amino acid tryptophan, which is an efficient quencher of this and dye and dyes in the ATTO series.13 Tryptophan quenches the fluorescence of these dyes, apparently via photoinduced electron transfer,14 in an effectively contact process that is disrupted by even relatively modest segregation of the dye-quencher pair.
Fig. 1.
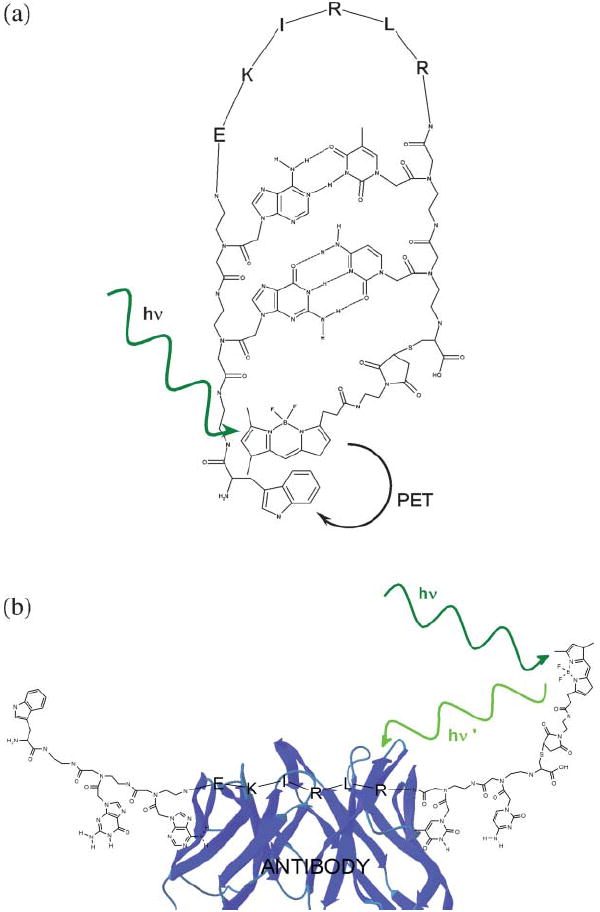
Chimeric peptide beacons are tri-block copolymers consisting of a polypeptide recognition element flanked by complementary peptide nucleic acid stems. The termini of the PNA elements, in turn, are modified with a fluorophore/contact quencher pair. Here we have employed a contiguous epitope from the HIV protein p17 as the recognition element and tryptophan and the tryptophan-quenched fluorophore, bodipy as the optical reporters. (a) In the absence of target, formation of the PNA stem ensures efficient, contact-mediated fluorescence quenching. (b) Upon target binding (here an anti-HIV antibody) the fluorophore is segregated from the quencher, enhancing fluorescence.
A CPB comprised of the p17 epitope flanked by complementary, two-base PNA sequences exhibits a three-fold increase in emission upon addition of the target antibody (Fig. 2). The signal gain of the sensor is independent of its concentration, confirming that the observed quenching is intramolecular (data not shown). The dissociation constant for the CPB–antibody complex, 4 nM (Fig. 3) is well above the ~200 pM dissociation constant previously reported for an unmodified polypeptide epitope.15 Thermal melts of the free CPB indicate that the equilibrium constant for stem formation is approximately 20 under the conditions employed (see ESI†), thus accounting for the observed reduction in affinity. Despite the reduced affinity, however, the 300% signal gain and good emissivity of the CPB sensor allow us to readily and rapidly quantify the target anti-HIV antibody at concentrations as low as 300 pM using only an inexpensive desktop fluorimeter (Fig. 3).
Fig. 2.
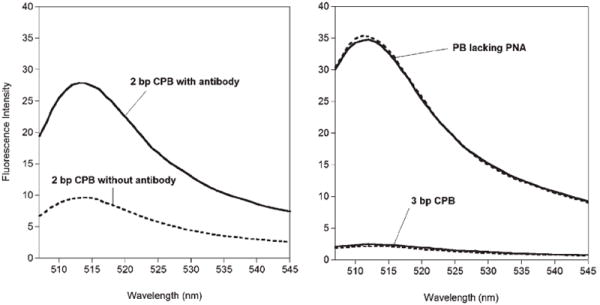
(Left) A 2bp CPB produces a strong, three-fold enhancement in fluorescence upon target binding. (Right) In contrast, a three base pair construct exhibits only a small (~15%) increase in signal and a PB construct lacking PNA base pairs does not produce any observable gain. Here the intensity of the construct in the absence of antibody is represented by a dashed line; the intensity of the CPBs in the presence of saturating (40–80 nM) antibody is represented by a solid line.
Fig. 3.
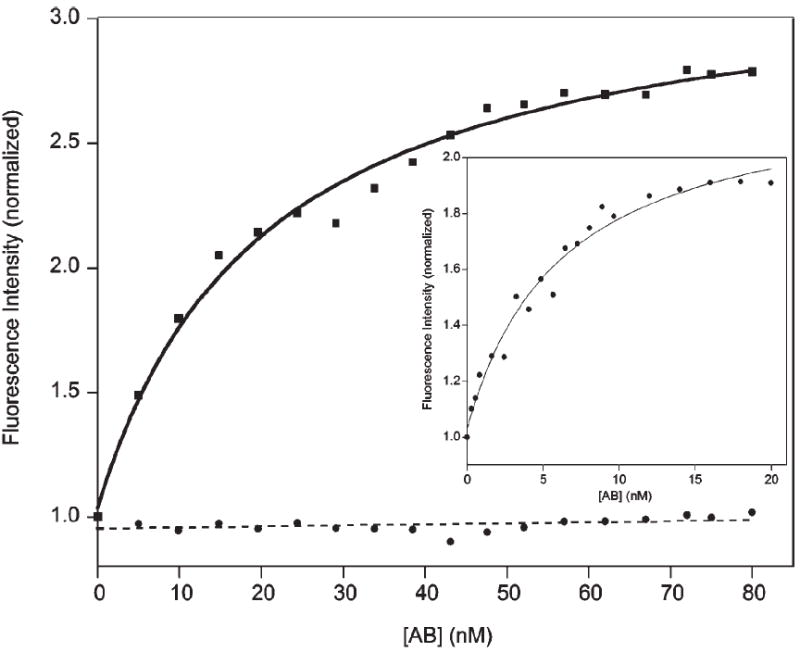
The 2bp CPB sensitively and specifically detects its target antibody at concentrations as low as a few hundred picomolar. Shown here is the normalized fluorescence response observed when the CPB (at 20 nM) is titrated with the anti-HIV-1 p17 antibody (solid line) vs. titration with a mixture of non-specific human IgG antibody isotypes (dashed line). (Inset) Using a lower concentration of the CPB (1.0 nM) we have determined that the dissociation constant of the CPB is ~4 nM.
Achieving optimal CPB performance requires optimization of the length of the PNA stem. For example, because the two ends of a six-residue, unstructured polypeptide collide on a timescale much slower than the ~2 ns lifetime of bodipy and most other organic dyes,16 a construct lacking a PNA stem is quite bright and does not exhibit any measurable increase in fluorescence upon binding to the target antibody (Fig. 2). Similarly, while a construct comprised of the epitope and a three-base-pair stem exhibits saturable binding (see ESI†), it is relatively dim and exhibits only a 15% signal increase upon saturation with the target antibody (Fig. 2 and SI). In contrast the quantum yield of the two-base-pair CPB in the complex approaches that of the construct lacking a PNA stem (Fig. 2). Taken together, these observations support the hypothesis that the poor gain of the three-base-pair CPB occurs because its longer stem remains partially intact in the complex. Of note, the optimal two-base stem length of this CPB is short relative to the 4 to 10 base-pair stems typically employed in DNA molecular beacons.17 PNA–PNA duplexes, however, are significantly more stable than the equivalent DNA duplexes, due to reduced electrostatic repulsion and improved hydrophobic interactions in the former polymer.3
Here we have demonstrated a new, label-free optical biosensor based on the binding-induced folding of a polypeptide. The approach is sensitive, with a detection limit that is competitive with absorption-based, label-free sensing approaches, such as surface plasmon resonance and quartz crystal microbalance measurements.18 The CPB approach is also operationally convenient in that it allows us to utilize any dye for which a contact quenching mechanism is available, largely irrespective of the lifetime of the dye. Moreover, the new architecture is fabricated via well developed solid phase synthesis methods, and thus can be ordered ready-to-use from commercial vendors or, if one wishes to use exogenous or proprietary fluorophores or quenchers, require only a single conjugation step after the CPB sequence is constructed. The relative ease with which CPBs are synthesized suggests they may be of more widespread utility than some of the other, previously described PB architectures (including our own1), which require significantly more cumbersome synthesis and purification.
The CPB approach also appears to be a more general platform than previous polypeptide-based optical sensing platforms19 in that CPBs neither require that binding specifically sequester a quencher or a fluorophore from the solvent,20 nor do they rely upon long-lived fluorophores or fluorophores that form stable, optically-modulated dimers.21 The only requirements of the CPB approach are that: (1) The unbound peptide is sufficiently flexible enough such that the two ends can be held together via base pairing of the PNA stem, (2) that the bound state of the peptide segregates the fluorophore/quencher pair by enough distance to disrupt the PNA stem, and (3) that base pairing and fluorophore/quencher pair does not excessively reduce the affinity of the recognition sequence. Given the elucidation of large libraries of polypeptide-based recognition elements by phage and bacterial display techniques,22 it appears that the CPB approach will be applicable to a wide range of macromolecular targets.
Acknowledgments
This work was supported by the National Institutes of Health Grant EB002046. This work made use of MRL Central Facilities supported by the MRSEC Program of the National Science Foundation under award No. DMR05-20415.
Footnotes
Electronic supplementary information (ESI) available: Thermal melt of the 2bp CPB. Saturable binding of the 3bp CPB. See DOI: 10.1039/b709776j
The chimeric beacon constructs, WgaEKIRLRtcC-BODIPY and WgagEKIRLRctcC-BODIPY (where letters in capitals represent amino acid residues, and letters in lowercase represent PNA bases), were ordered from Panagene, Inc. (Daejeon, South Korea) and used without further purification. The chimeric beacon construct that does not contain any PNA bases, sequence WGKIRLRPGC, was purchased from Biopeptide. A solution of peptide (62 μM) was made with 75 mM TRIS buffer pH 7.5. This solution was purged with argon for 30 min, and 10 eq. of TCEP was added to the solution and allowed to react for 30 min under argon. 10 eq. of Bodipy-FL maleimide in a 5.3 uM acetonitrile solution was added dropwise to the reaction over 1 h. The reaction was allowed to proceed stirred in the dark for 2 h, then stored at −20 °C. The reaction was purified using reverse phase HPLC with a Waters XTerra C18 prep scale column. The mobile phase was a gradient of 30 to 70% acetonitrile in H2O and 0.1% trifluoroacetic acid over 80 min with the product eluting at 11 min. The product in solution was lyophilized to a dry power and stored at −20 °C. The product was confirmed on an Applied Biosystems PE Sciex QSTAR-Pulsar quadrapole orthogonal time-of-flight mass spectrometer fitted with an electrospray source, m/z 1600.
Fluorescence measurements were performed on a Varian Cary Eclipse Fluorimeter. Samples were excited at 480 nm. Titrations were performed with a fixed concentration of CPB in a 1 ml quartz cuvette, in 50 mM phosphate buffer, pH 7.4 containing 200 mM NaCl and 200 mM MgCl2. The anti-HIV-1 p17 monoclonal antibody (Zeptometrix, Buffalo, New York) clone 32/1.24.89 was aliquoted and stored at −20 °C until titrations were performed. The working solutions were diluted from concentrated stocks, and allowed to stand for 30 min at 37 °C. After each addition, the solution was allowed to stand for 2 min at 20 °C, and then a measurement was acquired. The reported emission intensities represent the mean of multiple spectra acquired and averaged at 515 nm.
References
- 1.(a) Oh KJ, Cash KJ, Plaxco KW. J Am Chem Soc. 2006;128:14018–14019. doi: 10.1021/ja0651310. [DOI] [PubMed] [Google Scholar]; (b) Oh KJ, Cash KJ, Hugenberg V, Plaxco KW. Bioconjugate Chem. 2007;18:607–609. doi: 10.1021/bc060319u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tyagi S, Kramer FR. Nat Biotechnol. 1996;14:303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- 3.Kuhn H, Demidov VV, Coull JM, Fiandaca MJ, Gildea BD, Frank-Kamenetskii MD. J Am Chem Soc. 2002;124:1097–1103. doi: 10.1021/ja0041324. [DOI] [PubMed] [Google Scholar]
- 4.Tyagi S, Bratu DP, Kramer FR. Nat Biotechnol. 1998;16:49–53. doi: 10.1038/nbt0198-49. [DOI] [PubMed] [Google Scholar]
- 5.Zhao X, Tapec-Dytioco R, Wang K, Tan W. Anal Chem. 2003;75:3476–3483. doi: 10.1021/ac034330o. [DOI] [PubMed] [Google Scholar]
- 6.(a) Fang X, Sen A, Vicens M, Tan W. ChemBioChem. 2003;4:829–834. doi: 10.1002/cbic.200300615. [DOI] [PubMed] [Google Scholar]; (b) Baker BR, Lai RY, Wood MS, Doctor EH, Heeger AJ, Plaxco KW. J Am Chem Soc. 2006;128:3138–3139. doi: 10.1021/ja056957p. [DOI] [PubMed] [Google Scholar]
- 7.Winter G, Griffiths AD, Hawkins RE, Hoogenboom HR. Annu Rev Immunol. 1994;12:433–455. doi: 10.1146/annurev.iy.12.040194.002245. [DOI] [PubMed] [Google Scholar]
- 8.Pincus MR. Biopolymers. 1992;32:347–351. doi: 10.1002/bip.360320409. [DOI] [PubMed] [Google Scholar]
- 9.(a) Wei AP, Blumenthal DK, Herron JN. Anal Chem. 1994;66:1500–1506. doi: 10.1021/ac00081a023. [DOI] [PubMed] [Google Scholar]; (b) Geoghegan KF, Rosner PJ, Hoth LR. Bioconjugate Chem. 2000;11:71–77. doi: 10.1021/bc9900845. [DOI] [PubMed] [Google Scholar]
- 10.Krieger F, Fierz B, Bieri O, Drewello M, Kiefhaber T. J Mol Biol. 2003;332:265–274. doi: 10.1016/s0022-2836(03)00892-1. [DOI] [PubMed] [Google Scholar]
- 11.Papsidero LD, Sheu M, Ruscetti FW. J Virol. 1989;63:267–272. doi: 10.1128/jvi.63.1.267-272.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Massiah MA, Starich MR, Paschall C, Summers MF, Christensen AM, Sundquist WI. J Mol Biol. 1994;244:198–223. doi: 10.1006/jmbi.1994.1719. [DOI] [PubMed] [Google Scholar]
- 13.Marme N, Knemeyer J-P, Sauer M, Wolfrum J. Bioconjugate Chem. 2003;14:1133–1139. doi: 10.1021/bc0341324. [DOI] [PubMed] [Google Scholar]
- 14.Karolin J, Johansson L, Strandberg L, Ny T. J Am Chem Soc. 1994;116:7801–7806. [Google Scholar]
- 15.Ota A, Ueda S. Hybridoma. 1998;17:471–477. doi: 10.1089/hyb.1998.17.471. [DOI] [PubMed] [Google Scholar]
- 16.(a) Lapidus LJ, Eaton WA, Hofrichter J. Proc Natl Acad Sci U S A. 2000;97:7220–7225. doi: 10.1073/pnas.97.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Haugland RP. Handbook of Fluorescent Probes and Research Chemicals. 10. Molecular Probes; Eugene, OR: 2005. [Google Scholar]
- 17.Tsourkas A, Behlke MA, Rose SD, Bao G. Nucleic Acids Res. 2003;31:1319–1330. doi: 10.1093/nar/gkg212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Kößlinger C, Uttenthaler E, Drost S, Aberl F, Wolf H, Brink G, Stanglmaier A, Sackmann E. Sens Actuators, B. 1995;24:107–112. [Google Scholar]; (b) de Haard HJW, Kazermier B, Koolen MJM, Nijholt LJ, Meloen RH, van Gemen B, Hoogenboom HR, Arends J-W. Clin Diagn Lab Immunol. 1998;5:636–644. doi: 10.1128/cdli.5.5.636-644.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Kohn JE, Plaxco KW. Proc Natl Acad Sci U S A. 2005;102:10841–10845. doi: 10.1073/pnas.0503055102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ting AY, Kain KH, Klemke RL, Tsien RY. Proc Natl Acad Sci U S A. 2001;98:15003–15008. doi: 10.1073/pnas.211564598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jean JM, Clerte C, Hall KB. Protein Sci. 1999;8:2110–2120. doi: 10.1110/ps.8.10.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lotte K, Plessow R, Brockhinke A. Photochem Photobiol Sci. 2004;3:348–359. doi: 10.1039/b312436c. [DOI] [PubMed] [Google Scholar]
- 22.Bessette PH, Rice JJ, Daugherty PS. Protein Eng. 2004;17:731–739. doi: 10.1093/protein/gzh084. [DOI] [PubMed] [Google Scholar]