

Abstract
The accumulation of protein aggregates is thought to be an important component in the pathogenesis of mutant SOD1 induced disease. Mutant SOD1 aggregates appear to be cleared by proteasomes, at least in vitro, suggesting a potentially important role for proteasome degradation pathways in vivo. G93A SOD1 transgenic mice show an increase in proteasome activity and induction of immuno-proteasome subunits within spinal cord as they develop neurological symptoms. To determine what role immuno-proteasomes may have in mutant SOD1 induced disease, we crossed G93A SOD1 transgenic mice with LMP2−/− mice to obtain G93A SOD1 mice lacking the LMP2 immuno-proteasome subunit. G93A SOD1/LMP2−/− mice show significant reductions in proteasome function within spinal cord compared to G93A SOD1 mice. However, G93A SOD1/LMP2−/− mice show no change in motor function decline, or survival compared to G93A SOD1 mice. These results indicate that the loss of immuno-proteasome function in vivo does not significantly alter mutant SOD1 induced disease.
Keywords: ALS, motor neuron, spinal cord, astrocyte, microglia, aggregation
Mutations in the gene encoding Cu, Zn superoxide dismutase (SOD1) cause one form of familial amyotrophic lateral sclerosis (FALS) linked to chromosome 21q (Rosen et al., 1993). Studies using SOD1 knockout or transgenic mice expressing mutant SOD1 have established a toxic gain of function model for the abnormal SOD1 protein that may be related to protein misfolding and aggregation (Gurney et al., 1994; Ripps et al., 1995; Wong et al., 1995; Reaume et al., 1996; Kabashi and Durham, 2006). Indeed, the presence of SOD1 positive aggregates is a pathologic hallmark of disease both in transgenic mutant SOD1 mice and in patients dying from SOD1 related FALS (Shibata et al., 1996; Watanabe et al., 2001). SOD1 aggregates accumulate as the disease progresses and can be visualized as high molecular weight protein complexes (HMWPCs) on Western gels or as ubiquitin positive cellular inclusions via immuno-staining (Bruijn et al., 1997; Johnston et al., 2000; Wang et al., 2002). The finding that mutant SOD1 protein and SOD1 aggregates appear to be cleared largely by proteasomes has raised important questions concerning the nature of proteasome function in ALS (Urushitani et al., 2002; Puttaparthi et al., 2003; Di Noto et al., 2005). Early in vitro work with protein aggregates had predicted that proteasome function might be inhibited in neurodegenerative diseases, but studies using transgenic rodent models of Huntington’s disease and ALS found that instead there appeared to be induction of proteasome activity within the CNS as the animals developed neurological disease (Bence et al., 2001; Diaz-Hernandez et al., 2003; Kabashi et al., 2004; Cheroni et al., 2005; Puttaparthi and Elliott, 2005; Ahtoniemi et al., 2007). The 26S proteasome is composed of one 20S proteolytic complex and two axially positioned 19S (PA700) regulatory complexes (DeMartino and Slaughter, 1999). The 20S complex is itself composed of 2 copies each of 7 α and β type subunits, each encoded by a distinct gene. Each β ring contains three proteolytic sites that differs in its specificity, including a chymotrypsin-like site that cleaves after hydrophobic residues, a trypsin-like site that cleaves after basic residues, and a post-glutamyl peptide hydrolase (PGPH) or caspase like activity that cleaves after acidic residues particularly aspartate (Kisselev et al., 2003). Certain β subunits of the 20S core, such as LMP2 (β1i), MECL-1 (β2i), and LMP7 (β5i), are inducible and can replace the normal constitutive β1,β2 and β5 subunits, conveying differing proteolytic function to the immuno-proteasome (Peters et al., 2002). Knockout mice lacking LMP2, MECL-1 or LMP7 subunits are viable, show modest reductions in proteasome activity as well as in immunologic function but manifest no neurological deficits (Van Kaer et al., 1994; Stohwasser et al., 1996; Martin et al., 2004; Basler et al., 2006).
Several groups have analyzed the expression pattern of individual proteasome subunits within spinal cords from transgenic mutant SOD1 animals in order to better define the molecular basis for the increased proteasome activity observed with disease progression (Cheroni et al., 2005; Puttaparthi and Elliott, 2005; Ahtoniemi et al., 2007). It is the inducible and immuno-proteasome specific subunits (LMP2, MECL1 and LMP7) that are substantially upregulated in the spinal cords of mutant SOD1 transgenic animals while levels of constitutive proteasome subunits remain unchanged. This immuno-proteasome induction occurs primarily in astrocytes and microglia, rather than neurons, and is likely regulated by differential cytokine expression within the affected spinal cord (Puttaparthi and Elliott, 2005; Ahtoniemi et al., 2007). Because non-neuronal cells significantly contribute to disease progression in mutant SOD1 mice, assessing the potential role of immuno-proteasome function within astrocytes and microglia is relevant (Puttaparthi et al., 2002; Clement et al., 2003; Boillee et al., 2006). Pharmacological blockade of immuno-proteasome subunit induction using pyrrolidine dithiocarbamate, an inhibitor of nuclear transcription factor kappa-B (NF-κB) results in a reduction in lifespan for mutant SOD1 rats (Ahtoniemi et al., 2007). However, largely due to the non-specificity of drug effect, this study does not provide definitive answers to importance of immuno-proteasome function on disease course. We therefore have used a genetic approach to more specifically assess the role of immuno-proteasomes in mutant SOD1 induced disease in vivo by crossing G93A SOD1 transgenic mice with LMP2 knockout mice.
Material and Methods
Animals
Transgenic mice expressing the low copy number human G93A SOD1 mutation (B6SJL-TgNSOD1-G93A; JR2300) were used in these experiments. These mice have a mean survival of 250 days with the onset of motor dysfunction beginning at 6 months of age (Puttaparthi et al., 2002). The generation of LMP2 knockout mice has been described (Van Kaer et al., 1994). LMP2 knockout mice were originally on a mixed 129xB6 background, and subsequently backcrossed ten times to a B6 background. G93A SOD1 transgenic mice were crossed to LMP2 knock out mice. Appropriate F1 offspring were bred allowing all possible genotypes to be generated with adequate internal controls. All animal protocols were approved by our university IACRAC in compliance with NIH guidelines. Genotypes were identified by PCR on DNA isolated from tail clippings.
Western Blotting
Animals were overdosed with sodium pentobarbital (250mg/kg, i.p.). Tissues were dissected and homogenized in 20mMTris-Hcl, pH7.5, 2mM DTT, 0.1mg of leupeptin, 1mM EDTA, and 1mM EGTA. Total protein (40μg) from each sample was run on a 4–20% Tris-glycine gel (Invitrogen). The experiments were performed using the following primary antibodies: polyclonal anti-SOD1 (1:1000; Calbiochem), anti-actin (1:1000; Sigma), monoclonal anti-LMP2 (1:500; Biomol), polyclonal anti-LMP7 (1:20,000; Biomol), polyclonal anti-β5 (1:1000; Affinity BioReagents) and monoclonal anti-α7 (1:2000; Biomol). After several washes, membranes were incubated with a secondary goat anti-mouse or goat anti-rabbit antibody (1:8000; Santa Cruz Biotechnology) followed by ECL detection. Protein load was normalized by blotting with anti-actin antibody.
Immunohistochemistry
Anaesthetized mice were perfused transcardially with ice-cold PBS followed by 4% paraformaldehyde and embedded in paraffin. Spinal cords sections (6μm) were deparaffinized, hydrated and subjected to antigen retrieval at 90°C for 2 min using Target Unmasking Fluid (Pharmingen). The spinal cord sections were pretreated for 2 hours with 5% normal goat serum and 0.1% Triton X-100 and then incubated overnight at 4°C with primary antibodies. Primary antibodies used were as follows: rabbit anti-LMP7 (1:400; Biomol International); monoclonal anti-GFAP (1:2000; Sigma); monoclonal anti-NeuN (1:1000; Chemicon); isolectin B4 (1:100; Sigma); polyclonal anti-ubiquitin (1:1000; Dako). According to the manufacturer, the anti-ubiquitin antibody recognizes both mono- and polyubiquitin. Sections were washed, incubated with secondary antibodies at the following dilutions: goat anti-mouse IgG labeled with Oregon Green 488 from Molecular Probes (1:400) and goat anti-rabbit IgG labeled with Texas Red from Molecular Probes (1:400). Next, they were incubated for one minute with DAPI (1:1000) from Molecular Probes. Slides were viewed with a fluorescent microscope (Nikon Instruments Inc.) or under confocal microscopy.
Survival analysis and motor testing
The survival analysis, stride lengths and grip strengths were done with G93A SOD1/LMP2−/− dual transgenic mice, G93A SOD1/LMP2+/− transgenic mice and G93A SOD1/LMP+/+ transgenic mice. Stride lengths (cm), grip strengths (dg) and survival analysis were done as previously described (Puttaparthi et al., 2002). Because of inter-gender differences in stride length and grip strength, we selected only female animals for measurements of motor function.
Measurement of proteasome activity
For measurement of proteasome activity, 1 month and 8 month old mice were euthanized with sodium pentobarbital (250mg/kg, i.p) and perfused intracardiac with 1X PBS. Tissue was harvested immediately and placed on ice, sonicated in Buffer H (20mM Tris, 20mM NaCl, 1mM EDTA, 5mM β-mercaptoethanol, pH7.6) on ice. Lysates were cleared by centrifugation and the supernatant was used for determination of protein concentration and enzymatic activity. Proteasome activity was assessed using synthetic peptide substrates linked to the fluorimetric reporter, aminomethylcoumarin (for chymotrypsin-like activity, Suc-LLVY-AMC; for peptidylglutamyl-peptide hydrolase activity, Z-LLE-AMC; for trypsin-like activity, Z-VVR-AMC). Lysates (10μl) were assayed by addition of 200 μl of substrate. AMC hydrolyzed from the peptides was quantitated in a BioTek Synergy HT plate reader using 360 nm excitation and 460 nm emission wavelengths. Enzymatic activity was normalized for protein concentration and expressed as percent of activity in control lysates. Experiments were repeated at least three times. The specificity of these substrates for measuring proteasome activity was confirmed by adding a known specific inhibitor, lactacystin (Calbiochem), which dropped activity levels by 99%.
Results
Proteasome function and subunit expression pattern is altered in G93A SOD1 mice lacking LMP2
We first wished to determine whether the loss of the LMP2 immuno-proteasome subunit affected overall proteasome function in the spinal cord of G93A SOD1 transgenic mice. We therefore assayed the three forms of proteasome activity, chymotrypsin-like, trypsin-like and PGPH, in spinal cords from G93A SOD1 mice and G93A SOD1/LMP2−/− mice (Figure 1A & B). Both chymotrypsin-like and trypsin-like activities are significantly reduced in G93A SOD1/LMP2−/− mice compared to G93A SOD1 mice. This reduction is evident at an early 1 month time point when G93A SOD1 mice are asymptomatic, as well as in later adult life (7.5 months) when motor deficits are present. In contrast, there is no significant change in PGPH activity within spinal cords of G93A SOD1/LMP2−/− mice. We then used Western blots to assess the changes in proteasome subunit expression that accompanied the decline in measurable proteasome activity observed in G93A SOD1/LMP2−/− mice (Figure 2). As expected, LMP2 expression is absent in LMP2−/− and G93A SOD1/LMP2−/− mice (Figure 2; lanes 2, 4 and 6). There is compensatory increase in the expression of the LMP7, another immuno-proteasome subunit, in the spinal cords of mice lacking LMP2. This up-regulation of LMP7 subunit expression within the spinal cords of G93A SOD1/LMP2−/− mice is also evident by immuno-staining (data not shown). In contrast, expression levels of two constitutive proteasome subunits, α7 and β5, are unchanged in non-transgenic or G93A SOD1 mice with or without LMP2 expression. These results indicate that the loss of LMP2 subunits in G93A SOD1 mice alters specific aspects of proteasome function, chymotrypsin-like and trypsin-like activity, in spinal cord. Because constitutive proteasome subunit levels are unchanged in G93A SOD1/LMP2−/− mice, the decline in measurable proteasome function is likely related to a selective loss of immuno-proteasomes.
Fig 1.
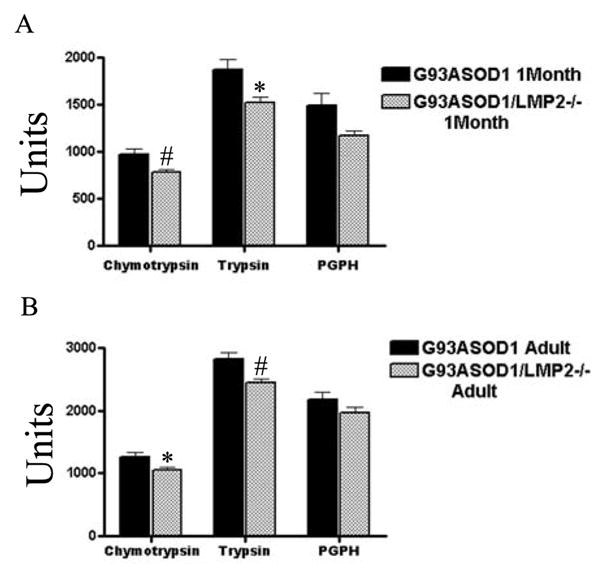
Proteasome activity is altered in spinal cords of G93A SOD1 mice lacking LMP2. Chymotrypsin-like, trypsin-like and PGPH activities were measured in the spinal cords of 1 month (A) and 7.5 month old (B) G93A SOD1 or G93A SOD1/LMP2−/− mice. * = p<.04; # = p< .02 N = 6 for each group. Units represent fluorescence activity.
Fig 2.

Proteasome subunit expression in spinal cord. Spinal cord extracts from 7.5 month WT (1), 7.5 month LMP2−/− mice (2), 1 month G93A SOD1 (3), 1 month G93A SOD1/LMP2−/− (4), 7.5 month G93A SOD1 (5) and 7.5 month G93A SOD1/LMP2−/− (6) mice probed with antibodies against immuno-proteasome subunits (LMP2 and LMP7) and constitutive proteasome subunits (α7 and β5). 40 μg of total protein loaded per lane. Actin was used as a loading control.
Survival, motor function and pathology in G93A SOD1 mice lacking LMP2
In order to determine the effect of immuno-proteasome function on G93A SOD1 induced motor neuron disease, we crossed LMP2 knockout mice with G93A SOD1 mice and longitudinally followed offspring for changes in survival and motor function. Overall, the loss of LMP2 immuno-proteasome subunit had no significant effect on survival in G93A SOD1 mice (Figure 3). The mean survival of G93A SOD1 mice was 247 ± 5 days compared to 248 ± 3 days for G93A SOD1/LMP2+/− mice or 241 ± 4 days for G93A SOD1/LMP2−/− mice. Analysis of motor function using stride length or grip strength was also performed longitudinally to discern whether loss of LMP2 subunit might alter onset or disease pattern (Figure 4A& B ). However, G93A/SOD1 LMP2−/− and G93A SOD1 mice showed neither significant difference in disease onset as measured by motor function testing or alteration in the progressive loss of motor function. Although G93A SOD1/LMP2−/− mice have reduced proteasome activity within spinal cord, there appeared to be no significant difference in the accumulation of SOD1 aggregates, either SOD1 positive HMWPCs on Western blots or as ubiquitin positive cellular inclusions on immuno-staining (Figure 5A–C). These results indicate that the loss of the LMP2 subunit and immuno-proteasome function does not significantly impact survival, disease course or aggregate accumulation in G93A SOD1 mice.
Fig 3.
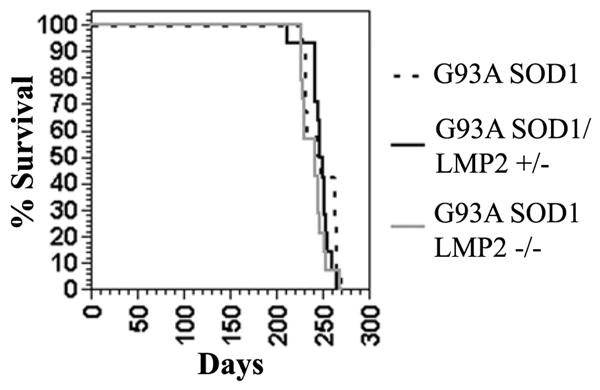
The loss of immuno-proteasome LMP2 subunit does not impact survival of G93A SOD1 mice. Survival curve for G93A SOD1, G93A SOD1/LMP2+/− and G93A SOD1/LMP2−/− mice shows no difference between the three groups. N = 14 for each group.
Fig 4.
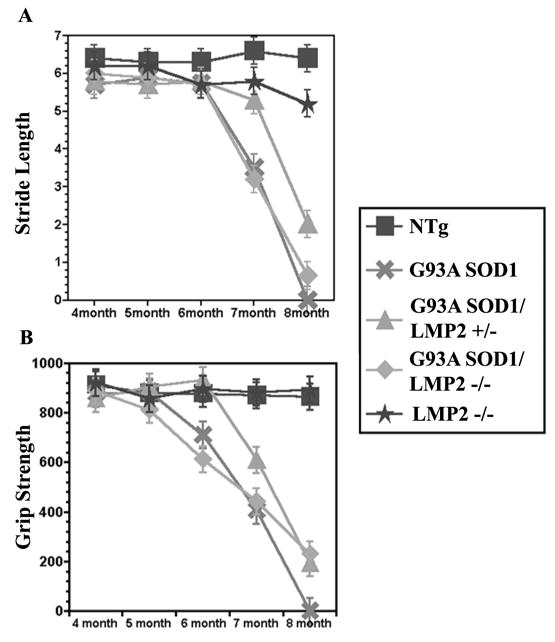
The loss of immuno-proteasome LMP2 subunit does not impact motor function loss in G93A SOD1 mice. Stride length (A) in centimeters and grip strength (B) in decigrams were measured longitudinally in G93A SOD1, G93A SOD1/LMP2+/−, and G93A SOD1/LMP2−/− female mice. There was no significant difference in onset or course of motor dysfunction among the three groups. N = 14 for each group.
Fig 5.

Aggregate pathology in G93A/LMP2−/− mouse spinal cord. Immuno-staining of spinal cord from 7.5 month old G93A SOD (A) or G93A SOD1/LMP2−/− (B) mice for ubiquitin (red) or the neuronal marker, NeuN (green). (C) Western blot of spinal cord extract from 1 month G93A SOD1 (1), 1 month G93A SOD1/LMP2−/− (2), 7.5 month G93A SOD1 (3) or 7.5 month G93A SOD1/LMP2−/− mice probed with antibody to SOD1. Bracket highlights area of high molecular weight SOD1 positive complexes. 20 μg of protein loaded per lane. (D) Confocal microscopy showing immuno-staining for LMP7 (red) and IB4 lectin (green) within a microglial cell from the spinal cord ventral horn of 7.5 old G93A SOD1/LMP2−/− mouse. Magnification = 400× (A and B); 600× for D.
Because immuno-proteasome induction in G93A SOD1 mice occurs in non-neuronal cells, we asked whether G93A SOD1/LMP2 −/− mice show changes in the astrocytosis or microgliosis that normally accompanies G93A SOD1 induced disease. Staining sections of symptomatic G93A SOD1/LMP2−/− mice for GFAP and IB4 lection reactivity, showed prominent astroglial and microglial change that was associated with LMP7 subunit induction (Figure 5D). However, the degree of increased GFAP and IB4 lectin reactivity was comparable in G93A SOD1 mice with or without LMP2 subunit expression (data not shown).
Discussion
The presence of ubiquitin positive inclusions within spinal cord is a pathological hallmark of disease in both human patients with familial ALS as well as in the mutant SOD1 transgenic mouse model. That these aggregates are central to the pathogenesis of disease forms the basis for the aggregation or protein mishandling model of ALS (Kabashi and Durham, 2006). Much experimental evidence has found that mutant SOD1 and its aggregates are cleared by proteasome mediated degradation pathways raising the possibility that alterations in proteasome function in vivo might influence the rate of aggregate accumulation and change disease course (Johnston et al., 2000; Puttaparthi et al., 2003; Di Noto et al., 2005). Several groups have found that there is induction of immuno-proteasomes in the spinal cords of mutant SOD1 transgenic mice as they develop neurological dysfunction suggesting a potential compensatory role for immuno-proteasomes as a response to the increasing accumulation of SOD1 aggregates (Cheroni et al., 2005; Puttaparthi and Elliott, 2005; Ahtoniemi et al., 2007).
In order to determine whether the immuno-proteasome does play an important role in disease, we made use of knockout mice lacking the immuno-proteasome subunit, LMP2, by crossing them with G93A SOD1 mice. Our results using these LMP2 knockout mice indicate that the loss of immuno-proteasomes does not impact motor function or survival in G93A SOD1 mice. This finding differs from the recent work which found that the addition of pharmacologic agents which in part reduce immuno-proteasome induction do affect survival in mutant SOD1 animals (Ahtoniemi et al., 2007). Although Ahtoniemi and colleagues use a mutant SOD1 transgenic rat rather than a transgenic mouse model, the more likely explanation to explain this difference is that their group employed pharmacologic agents (dithiocarbamates) which have non-specific effects beyond prevention of immune-proteasome induction. In contrast, our genetic approach using LMP2 knockout mice offers a more targeted approach aimed selectively at the immuno-proteasome. We cannot exclude that strain background of the mice used in some way did not mitigate the effect of LMP2 loss.
These results do not imply that loss of the LMP2 subunit has no physiological effect on G93A SOD1 mice. In fact, we find clear evidence to indicate that loss of the LMP2 subunit results in significant decreases in proteasome function within G93A SOD1 spinal cord for both chymotrypsin-like and tryspsin-like activities. In addition, we find clear compensatory induction of another immune-proteasome subunit, LMP7, within spinal cord. However, there is no change in the expression levels of constitutive proteasome subunit, α7 and β5, indicating little if any compensatory effort by the constitutive proteasomes. The loss of the LMP2 subunit clearly decreases immuno-proteasome function within G93A SOD1 mice, but that this loss does not significantly impact G93A SOD1 induced disease.
Although, immuno-proteasome subunits are expressed in both neuronal and non-neuronal cells, immuno-proteasome induction within G93A SOD1 spinal cord is largely limited to astrocytes and microglia (Diaz-Hernandez et al., 2003; Puttaparthi and Elliott, 2005; Ahtoniemi et al., 2007). This pattern of expression means that loss of immuno-proteasome function would affect non-neuronal cells to a greater extent than neuronal cells and might explain why we did not observe a more significant impact on disease course. Although non-neuronal cells contribute to the overall phenotype observed in G93A SOD1, it is likely that the primary site of mutant SOD1 action is within neurons (Gong et al., 2000; Puttaparthi et al., 2002; Clement et al., 2003; Boillee et al., 2006). For this reason it is possible, that alterations of constitutive proteasome function within neuronal cells might significantly impact mutant SOD1 induced disease.
Acknowledgments
This work was supported by grants from the NINDS (JLE), Muscular Dystrophy Association (JLE) and the ALS Research Fund (JLE).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahtoniemi T, Goldsteins G, Keksa-Goldsteine V, Malm T, Kanninen K, Salminen A, Koistinaho J. Pyrrolidine dithiocarbamate inhibits induction of immunoproteasome and decreases survival in a rat model of amyotrophic lateral sclerosis. Mol Pharmacol. 2007;71:30–37. doi: 10.1124/mol.106.028415. [DOI] [PubMed] [Google Scholar]
- Basler M, Moebius J, Elenich L, Groettrup M, Monaco JJ. An altered T cell repertoire in MECL-1-deficient mice. J Immunol. 2006;176:6665–6672. doi: 10.4049/jimmunol.176.11.6665. [DOI] [PubMed] [Google Scholar]
- Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- Cheroni C, Peviani M, Cascio P, Debiasi S, Monti C, Bendotti C. Accumulation of human SOD1 and ubiquitinated deposits in the spinal cord of SOD1G93A mice during motor neuron disease progression correlates with a decrease of proteasome. Neurobiol Dis. 2005;18:509–522. doi: 10.1016/j.nbd.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillee S, Rule M, McMahon AP, Doucette W, Siwek D, Ferrante RJ, Brown RH, Jr, Julien JP, Goldstein LS, Cleveland DW. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 2003;302:113–117. doi: 10.1126/science.1086071. [DOI] [PubMed] [Google Scholar]
- DeMartino GN, Slaughter CA. The proteasome, a novel protease regulated by multiple mechanisms. J Biol Chem. 1999;274:22123–22126. doi: 10.1074/jbc.274.32.22123. [DOI] [PubMed] [Google Scholar]
- Di Noto L, Whitson LJ, Cao X, Hart PJ, Levine RL. Proteasomal degradation of mutant superoxide dismutases linked to amyotrophic lateral sclerosis. J Biol Chem. 2005;280:39907–39913. doi: 10.1074/jbc.M506247200. [DOI] [PubMed] [Google Scholar]
- Diaz-Hernandez M, Hernandez F, Martin-Aparicio E, Gomez-Ramos P, Moran MA, Castano JG, Ferrer I, Avila J, Lucas JJ. Neuronal induction of the immunoproteasome in Huntington’s disease. J Neurosci. 2003;23:11653–11661. doi: 10.1523/JNEUROSCI.23-37-11653.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong YH, Parsadanian AS, Andreeva A, Snider WD, Elliott JL. Restricted expression of G86R Cu/Zn superoxide dismutase in astrocytes results in astrocytosis but does not cause motoneuron degeneration. J Neurosci. 2000;20:660–665. doi: 10.1523/JNEUROSCI.20-02-00660.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Dalton MJ, Gurney ME, Kopito RR. Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2000;97:12571–12576. doi: 10.1073/pnas.220417997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, Durham HD. Failure of protein quality control in amyotrophic lateral sclerosis. Biochim Biophys Acta. 2006;1762:1038–1050. doi: 10.1016/j.bbadis.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Kabashi E, Agar JN, Taylor DM, Minotti S, Durham HD. Focal dysfunction of the proteasome: a pathogenic factor in a mouse model of amyotrophic lateral sclerosis. J Neurochem. 2004;89:1325–1335. doi: 10.1111/j.1471-4159.2004.02453.x. [DOI] [PubMed] [Google Scholar]
- Kisselev AF, Garcia-Calvo M, Overkleeft HS, Peterson E, Pennington MW, Ploegh HL, Thornberry NA, Goldberg AL. The caspase-like sites of proteasomes, their substrate specificity, new inhibitors and substrates, and allosteric interactions with the trypsin-like sites. J Biol Chem. 2003;278:35869–35877. doi: 10.1074/jbc.M303725200. [DOI] [PubMed] [Google Scholar]
- Martin S, Gee JR, Bruce-Keller AJ, Keller JN. Loss of an individual proteasome subunit alters motor function but not cognitive function or ambulation in mice. Neurosci Lett. 2004;357:76–78. doi: 10.1016/j.neulet.2003.10.085. [DOI] [PubMed] [Google Scholar]
- Peters B, Janek K, Kuckelkorn U, Holzhutter HG. Assessment of proteasomal cleavage probabilities from kinetic analysis of time-dependent product formation. J Mol Biol. 2002;318:847–862. doi: 10.1016/S0022-2836(02)00167-5. [DOI] [PubMed] [Google Scholar]
- Puttaparthi K, Elliott JL. Non-neuronal induction of immunoproteasome subunits in an ALS model: possible mediation by cytokines. Exp Neurol. 2005;196:441–451. doi: 10.1016/j.expneurol.2005.08.027. [DOI] [PubMed] [Google Scholar]
- Puttaparthi K, Wojcik C, Rajendran B, DeMartino GN, Elliott JL. Aggregate formation in the spinal cord of mutant SOD1 transgenic mice is reversible and mediated by proteasomes. J Neurochem. 2003;87:851–860. doi: 10.1046/j.1471-4159.2003.02028.x. [DOI] [PubMed] [Google Scholar]
- Puttaparthi K, Gitomer WL, Krishnan U, Son M, Rajendran B, Elliott JL. Disease progression in a transgenic model of familial amyotrophic lateral sclerosis is dependent on both neuronal and non-neuronal zinc binding proteins. J Neurosci. 2002;22:8790–8796. doi: 10.1523/JNEUROSCI.22-20-08790.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, Beal MF, Brown RH, Jr, Scott RW, Snider WD. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet. 1996;13:43–47. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- Ripps ME, Huntley GW, Hof PR, Morrison JH, Gordon JW. Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 1995;92:689–693. doi: 10.1073/pnas.92.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Shibata N, Hirano A, Kobayashi M, Siddique T, Deng HX, Hung WY, Kato T, Asayama K. Intense superoxide dismutase-1 immunoreactivity in intracytoplasmic hyaline inclusions of familial amyotrophic lateral sclerosis with posterior column involvement. J Neuropathol Exp Neurol. 1996;55:481–490. doi: 10.1097/00005072-199604000-00011. [DOI] [PubMed] [Google Scholar]
- Stohwasser R, Kuckelkorn U, Kraft R, Kostka S, Kloetzel PM. 20S proteasome from LMP7 knock out mice reveals altered proteolytic activities and cleavage site preferences. FEBS Lett. 1996;383:109–113. doi: 10.1016/0014-5793(96)00110-x. [DOI] [PubMed] [Google Scholar]
- Urushitani M, Kurisu J, Tsukita K, Takahashi R. Proteasomal inhibition by misfolded mutant superoxide dismutase 1 induces selective motor neuron death in familial amyotrophic lateral sclerosis. J Neurochem. 2002;83:1030–1042. doi: 10.1046/j.1471-4159.2002.01211.x. [DOI] [PubMed] [Google Scholar]
- Van Kaer L, Ashton-Rickardt PG, Eichelberger M, Gaczynska M, Nagashima K, Rock KL, Goldberg AL, Doherty PC, Tonegawa S. Altered peptidase and viral-specific T cell response in LMP2 mutant mice. Immunity. 1994;1:533–541. doi: 10.1016/1074-7613(94)90043-4. [DOI] [PubMed] [Google Scholar]
- Wang J, Xu G, Borchelt DR. High molecular weight complexes of mutant superoxide dismutase 1: age-dependent and tissue-specific accumulation. Neurobiol Dis. 2002;9:139–148. doi: 10.1006/nbdi.2001.0471. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Dykes-Hoberg M, Culotta VC, Price DL, Wong PC, Rothstein JD. Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol Dis. 2001;8:933–941. doi: 10.1006/nbdi.2001.0443. [DOI] [PubMed] [Google Scholar]
- Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]