

Abstract
Endocannabinoids are lipid signaling molecules that regulate a wide range of mammalian behaviors, including pain, inflammation, and cognitive/emotional state. The endocannabinoid anandamide is principally degraded by the integral membrane enzyme fatty acid amide hydrolase (FAAH), and there is currently much interest in developing FAAH inhibitors to augment endocannabinoid signaling in vivo. Here we report the discovery and detailed characterization of a highly efficacious and selective FAAH inhibitor PF-3845. Mechanistic and structural studies confirm that PF-3845 is a covalent inhibitor that carbamylates FAAH's serine nucleophile. PF-3845 selectively inhibits FAAH in vivo as determined by activity-based protein profiling and raises brain anandamide levels for up to 24 hrs, resulting in profound cannabinoid receptor-dependent reductions in inflammatory pain. These data thus designate PF-3845 as a valuable pharmacological tool for in vivo characterization of the endocannabinoid system.
The endogenous cannabinoid (endocannabinoid) system is composed of two G-protein coupled receptors, CB1 and CB2, their cognate lipid ligands N-arachidonoyl ethanolamine (anandamide, AEA) and 2-arachidonoylglycerol (2-AG), and the metabolic enzymes responsible for AEA and 2-AG biosynthesis and degradation (Fowler, 2006; Pacher et al., 2006; Di Marzo et al., 2007; Ahn et al., 2008). CB1 and CB2 are also activated by Δ9-tetrahydrocannabinol (THC), the psychoactive substance in marijuana (Mechoulam, 1986). THC and other direct CB1 agonists have long been recognized to possess medicinally beneficial properties, such as pain relief; however, these agents also produce myriad side effects, including impairments in cognition and motor control, that limit their broad clinical utility. In contrast, inhibitors of the principal AEA-degrading enzyme fatty acid amide hydrolase (FAAH (Cravatt et al., 1996; McKinney and Cravatt, 2005)) and FAAH(-/-) mice have been found to display analgesia (Cravatt et al., 2001; Kathuria et al., 2003; Lichtman et al., 2004; Lichtman et al., 2004; Chang et al., 2006; Russo et al., 2007), anti-inflammation (Cravatt et al., 2004; Massa et al., 2004; Holt et al., 2005), anxiolysis (Kathuria et al., 2003; Naidu et al., 2007; Moreira et al., 2008), and anti-depression (Gobbi et al., 2005; Naidu et al., 2007) without disruptions in motility, cognition, or body temperature (Boger et al., 2000; Cravatt et al., 2001; Kathuria et al., 2003; Lichtman et al., 2004). These initial findings suggest that FAAH/AEA signaling pathways regulate a discrete subset of the behavioral processes affected by direct CB1 agonists. The selective pharmacological blockade of FAAH has, therefore, emerged as an exciting strategy to discern the endogenous functions of AEA-mediated endocannabinoid pathways and gauge their therapeutic potential.
Several classes of FAAH inhibitors have been reported, including reversibly (e.g., α-ketoheterocycles (Boger et al., 2000; Leung et al., 2003; Lichtman et al., 2004); trifluoromethyl ketones (Koutek et al., 1994; Boger et al., 1999; Leung et al., 2003)) and irreversibly (e.g., carbamates (Kathuria et al., 2003); fluorophosphonates (Deutsch et al., 1997)) acting agents. These inhibitors each possess distinct sets of attributes and deficiencies that reflect some of the most challenging aspects of FAAH inhibitor development. Reversible inhibitors, such as the α-ketoheterocycle OL-135, have been found to display excellent selectivity for FAAH relative to other serine hydrolases in mammalian proteomes, but produce only transient elevations in AEA in vivo (Lichtman et al., 2004). The submaximal efficacy of reversible FAAH inhibitors may be due to their rapid metabolism, as well as the fact that near-complete (>85%) blockade of FAAH activity is required to maintain elevated AEA levels in vivo (Fegley et al., 2005). Another challenge with developing reversible inhibitors is that inhibition of FAAH leads to elevated levels of several N-acyl ethanolamine (NAE) substrates, which can diminish the efficiency and potency of the inhibitor by mass-action competition with the substrate (Swinney, 2004). Irreversible inhibitors, such as the carbamate URB597 (Kathuria et al., 2003; Lichtman et al., 2004), produce a more complete blockade of FAAH in vivo, but also inhibit other serine hydrolases in peripheral tissues, including several carboxylesterases (Lichtman et al., 2004; Alexander and Cravatt, 2005; Zhang et al., 2007). Considering that FAAH is a serine hydrolase (Patricelli et al., 1999) and that there are > 200 members of this enzyme class in the human proteome, inhibitor selectivity represents a major challenge.
Toward the goal of developing FAAH inhibitors that display optimal efficacy and selectivity, we recently reported a new class of piperidine/piperazine ureas that inhibit FAAH with high specificity (Ahn et al., 2007). Here, we report a detailed mechanistic, structural, and pharmacological characterization of the FAAH piperidine urea inhibitor PF-3845. Our data indicate that PF-3845 displays an unprecedented combination of in vivo activity and selectivity, designating this agent as a valuable pharmacological tool for studying FAAH-regulated endocannabinoid pathways.
Results and Discussion
Optimization of potency of piperidine/piperazine urea FAAH inhibitors
Recently, we (Ahn et al., 2007) and others (Keith et al., 2008) have described a new class of FAAH inhibitors that contain a piperidine/piperazine urea moiety. These agents were found to inhibit FAAH by a covalent, irreversible mechanism involving carbamylation of FAAH's catalytic S241 nucleophile. Lead urea inhibitors, such as PF-750 (Table 1), however, showed only moderate potency for FAAH, as determined by measurements of kinact/Ki values (∼ 800 M-1s-1). In an effort to improve the potency of piperidine/piperazine urea inhibitors, we explored a series of analogs where the quinoline group of PF-750 was replaced by a biaryl ether group (Table 2). This medicinal chemistry effort culminated in the discovery of PF-3845 (compound 10, Table 2), a biaryl ether piperidine that displayed a 10-20-fold higher kinact/Ki value for FAAH compared to PF-750 or the well-studied carbamate FAAH inhibitor URB597 (Kathuria et al., 2003) (Tables 1 and 2). Key features of PF-3845 that contribute to enhanced potency include the p-trifluoromethyl substituent on the biaryl ether moiety and substitution of the aniline leaving group of PF-750 with 3-aminopyridine.
Table 1.
Potency (kinact/Ki values) comparison of FAAH inhibitors PF-3845, PF-750, and URB597. The kinact and Ki values were obtained as described in the Supplemental Methods. Values are averages ± SD of three independent experiments.
| Inhibitor |
kinact (s-1) |
Ki (μM) |
kinact/Ki (M-1s-1) |
|---|---|---|---|
|
PF-3845, 10 |
0.0033 ± 0.0002 | 0.23 ± 0.03 | 14,310 |
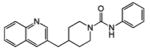 PF-750 |
- | - | 791* |
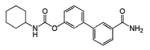 URB597 |
0.0033 ± 0.0003 | 2.0 ± 0.3 | 1,590 |
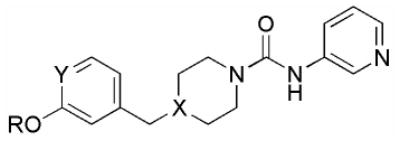
Table 2.
Structure-activity relationship of biarylether urea FAAH inhibitors.
 | ||||||
|---|---|---|---|---|---|---|
| kinact/Ki (M-1s-1) | ||||||
| Compound | R | Y | X | hFAAH | rFAAH | h/rFAAH |
| 1 | 4-MePh | C | N | 1440 ± 260 | 303 ± 41 | 974 ± 490 |
| 2 | 4-OMePh | C | N | 1370 ± 300 | 1190 ± 600 | 1600 ± 340 |
| 3 | 4-CF3Ph | C | N | 4070 ± 22 | 1340 ± 370 | - |
| 4 | 4-CF3-2-pyr | C | N | 4160 ± 570 | 1520 ± 220 | - |
| 5 | Ph | C | C | 1280 ± 390 | 314 ± 55 | - |
| 6 | 4-MePh | C | C | 4640 ± 170 | 531 ± 37 | - |
| 7 | 4-CF3Ph | C | C | 9230 ± 3600 | 2140 ± 660 | - |
| 8 | 4-FPh | C | C | 3460 ± 390 | 445 ± 110 | 2470 ± 130 |
| 9 | 2-pyr | C | C | 1090 ± 340 | 76 ± 1 | - |
| 10 (PF-3845) | 4-CF3-2-pyr | C | C | 12,600 ± 3000 | 3900 ± 780 | 13,300 ± 1300 |
| 11 | 3-CF3-2-pyr | C | C | 267 ± 51 | 86 ± 7 | - |
| 12 | 6-CF3-2-pyr | C | C | 438 ± 260 | 69 ± 25 | - |
| 13 | 4-OMe-2-pyr | C | C | 4780 ± 390 | 2850 ± 170 | - |
| 14 | 4-NH2-2-pyr | C | C | 368 ± 140 | 64 ± 3 | - |
| 15 | 4-MePh | N | N | 17 ± 1 | 11 ± 3 | - |
| 16 | 4-(HC ≡ C)-2-pyr | C | C | 5230 ± 890 | 11,800 ± 3100 | - |
We next examined the mechanism of FAAH inhibition by PF-3845 in more detail using an enzyme-coupled assay with oleamide as a substrate, where stoichiometric quantities of NAD+ are formed upon generation of ammonia from oleamide by FAAH hydrolysis (Ahn et al., 2007). Linear production of NAD+ was observed over a 40 min time period in the absence of PF-3845 (Figure S1A). In the presence of PF-3845, the progress curves for oleamide hydrolysis by FAAH exhibited curvature (Figure S1A), consistent with an irreversible mechanism of inhibition. The data were fit to a pseudo-first-order decay equation to determine kobs values at each inhibitor concentration. Plotting these kobs values as a function of PF-3845 concentration revealed saturation (Figure S1B), thus supporting a two-step mechanism for FAAH inactivation involving reversible binding of PF-3845 to FAAH followed by the second chemical step that results in covalent bond formation. Based on this model, kinact and Ki values for FAAH inhibition by PF-3845 were calculated to be 0.0033 ± 0.0002 s-1 and 0.23 ± 0.03 μM (Table 1). Similar progress and saturation curves were plotted for inhibition of FAAH by the carbamate inhibitor URB597 (Figure S1C and D), generating kinact and Ki values of 0.0033 ± 0.0003 and 2.0 ± 0.3 μM, respectively (Table 1). These data thus indicate that most, if not all of the increased potency observed for PF-3845 over URB597 is due to an improvement in binding affinity (Ki value).
Crystal Structure of a PF-3845-FAAH Complex
To gain more detailed insights into the basis for improved potency of PF-3845, we next determined the crystal structure of a PF-3845-FAAH complex. Here, we used a recently described ‘humanized’ rat-FAAH (h/rFAAH) protein, where the active site of rat FAAH (rFAAH) has been mutagenically converted to match the active site of human FAAH (hFAAH) (Mileni et al., 2008). Previous studies have confirmed that h/rFAAH maintains the high recombinant expression levels of rFAAH and inhibitor sensitivity profile of hFAAH (Mileni et al., 2008), thus providing a versatile model for structure-activity relationship (SAR) studies relevant to the human enzyme.
A crystal structure of the PF-3845-h/rFAAH complex was determined at 2.8 Å resolution (see Table S1 for data collection and refinement statistics), revealing a dimeric enzyme with an overall fold that matched those reported in previous FAAH structures (Bracey et al., 2002; Mileni et al., 2008). PF-3845 was found to be covalently attached to the catalytic S241 nucleophile (Patricelli et al., 1999) of h/rFAAH through a carbamate linkage (Figure 1), similar to the previously reported structure of h/rFAAH complexed with PF-750 (Mileni et al., 2008). The 3-aminopyridine leaving group was not observed in the h/rFAAH structure, but the remaining piperidine portion of the parent molecule occupied the acyl chain-binding pocket (Figure 1A). As was observed in the PF-750-h/rFAAH structure (Mileni et al., 2008), a strong aromatic C-H…π-interaction (Brandl et al., 2001) can be seen between F192 and the phenyl aromatic ring of PF-3845 (Figure 1A). The ∼20-fold improvement in potency for PF-3845 appears to derive from a more extended set of van der Waals interactions between this inhibitor's 4-trifluoromethyl-2-pyridyl group and the hydrophobic acyl chain-binding pocket of FAAH (Figure 1B). In this respect, the biaryl ether piperidine moiety of PF-3845 binds in a fashion that more closely resembles the arachidonyl chain of the original methyl arachidonyl phosphonate (MAP)-rFAAH crystal structure (Bracey et al., 2002) (Figure 1C).
Figure 1.
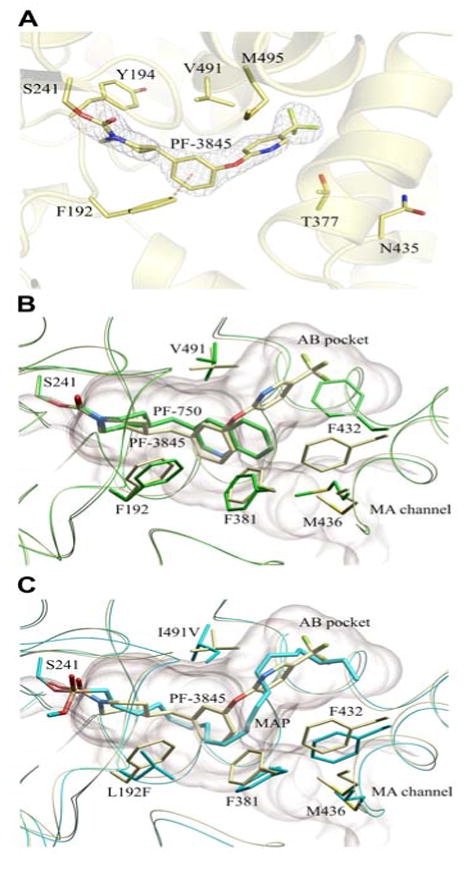
Crystal structure of a PF-3845-h/rFAAH complex. (A) Active site image of PF-3845-h/rFAAH complex, showing the S241-carbamylated adduct and six residues that have been mutated in h/rFAAH. (B) Overlap of the crystal structures of the PF-3845 (gray) and PF-750 (green) complexes with h/rFAAH, showing the different modes of binding that lead to distinct conformations for the F432 residue that toggles between the membrane access (MA) channel (F432 in gray) and AB pocket (F432 in green). (C) Overlap of crystal structures of PF-3845-h/rFAAH and MAP-rFAAH complexes, showing similar binding modes for PF-3845 (gray) and MAP (blue).
One marked difference that we previously observed between the crystal structures of FAAH bound to MAP (Bracey et al., 2002) and PF-750 (Mileni et al., 2008) was that the shorter PF-750 group allowed F432 to undergo a conformational shift that moved this residue out of the membrane access channel and into the acyl chain-binding pocket (Figure 1B). In the PF-3845-h/rFAAH structure, F432 resides in the membrane access channel at a site similar to its position in the MAP-rFAAH structure (Figure 1C). Adjacent to F432, residue M436 also undergoes a rotation of about 135° along the Cα-Cβ axis in both the PF-3845- and MAP-FAAH structures (Figure 1C). The structural rearrangements of F432 and M436, thus, appear to occur in a coordinated manner in these three structures and establish that inhibitors can promote conformational shifts that alter the relative size of FAAH's acyl chain-binding pocket and membrane access channel. The dynamic nature of this region of FAAH could facilitate entrance of lipid substrates from the cell membrane into the enzyme's active site.
The PF-3845-FAAH structure also aided our understanding of the SAR within the biaryl ether urea series by means of Monte Carlo simulations and energy calculations. For example, these calculations suggest that the higher potency of piperidine ureas (compounds 6, 7, 10; Table 2) over the corresponding piperazine ureas (compounds 1, 3, 4; Table 2) is likely due to unfavorable electrostatic interactions of the protonated piperazine within the hydrophobic acyl chain-binding pocket. Furthermore, replacement of the central phenyl group of PF-3845 with a pyridyl group drastically reduces inhibitor potency (compound 15; Table 2), which may reflect disruption of the aromatic-CH…π interaction with F192 caused by the higher electron withdrawing character of the pyridyl nitrogen, which lessens the electron density of the π ring. Finally, the crystal structure provides an attractive model to explain the improved potency gained by insertion of a trifluoromethyl group at the 4-position of the 2-pyridyl ring that resides in the acyl chain-binding pocket. Reducing the size of this substituent to a methyl or fluoro group decreased inhibitor potency (compounds 6 and 8, respectively; Table 2), which can be rationalized by simulations that predict suboptimal Van der Waals contacts for these smaller groups. Conversely, moving the trifluoromethyl group to the 3- or 6-position, which also impairs potency (compounds 11 and 12, respectively; Table 2), is predicted to introduce steric clashes with the FAAH protein.
PF-3845 Selectively Inhibits FAAH In Vivo for up to 24 hrs
We next administered PF-3845 to mice (10 mg/kg, i.p.) and monitored FAAH inactivation and substrate levels in the brain over a time course from 1 to 24 hrs. For comparison, we also analyzed mice treated with URB597 (10 mg/kg, i.p.). Both PF-3845- and URB597-treated mice showed rapid and complete inactivation of FAAH in the brain as judged by competitive activity-based protein profiling (ABPP (Liu et al., 1999; Leung et al., 2003)) with the serine hydrolase-directed probe fluorophosphonate (FP)-rhodamine (Patricelli et al., 2001) (Figure 2A). PF-3845 showed a longer duration of action than URB597, as judged by the rate of recovery of FP-labeling of FAAH. While URB597-treated animals showed nearly wild type levels of brain FAAH activity by 24 hr, FAAH was still mostly inhibited (> 75%) at this time point in PF-3845-treated animals (Figure 2A). These ABPP studies confirmed that PF-3845 and URB597 were both highly selective for FAAH in the brain, since none of the other FP-reactive serine hydrolases in this tissue were inhibited by these agents. In contrast, URB597, but not PF-3845, blocked FP-labeling of several additional liver serine hydrolases (Figure 2B), which have previously been identified as carboxylesterases (Alexander and Cravatt, 2005; Zhang et al., 2007). We furthermore tested PF-3845 for effects on FAAH-2, a recently identified FAAH homologue that is selectively expressed in higher mammals (Wei et al., 2006), but not rodents. In contrast to URB597, which has been found to inhibit human FAAH-2 with high potency (Wei et al., 2006), PF-3845 showed negligible activity against this FAAH variant (IC50 > 10 μM; Figure S2).
Figure 2.
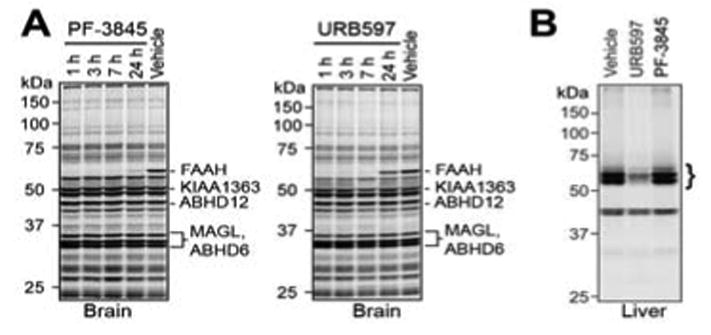
In vivo selectivity analysis for PF-3845 and URB597 as determined by competitive ABPP. (A) Gel profiles of FP-rhodamine labeled brain serine hydrolase activities from mice treated with PF-3845 or URB597 (10 mg/kg, i.p.) for the indicated times. Note that selective blockade of FP-rhodamine labeling of FAAH is observed by both inhibitors. Representative additional brain serine hydrolases are designated based on previous ABPP studies (Blankman et al., 2007; Nomura et al., 2008). (B) Gel profiles of FP-rhodamine-labeled liver serine hydrolase activities from mice treated with PF-3845 or URB597 (10 mg/kg, i.p., 2 hr). Note that URB597, but not PF-3845, blocks FP-rhodamine labeling of several liver serine hydrolase activities (bracket), which have previously been identified as carboxylesterases (Alexander and Cravatt, 2005; Zhang et al., 2007). For both (A) and (B), fluorescent gel images are shown in grayscale. For (B), a lower scanning intensity is shown due to the highly abundant activity signals for carboxylesterases present in the liver proteome.
To confirm the in vivo selectivity of PF-3845, we synthesized an alkyne analogue of this inhibitor, termed PF3845yne (Figure 3A), which maintained high potency for FAAH (kinact/Ki value of 5,230 M-1s-1). We then directly analyzed the protein targets of PF3845yne in vivo by click chemistry (CC)-ABPP (Speers et al., 2003; Speers and Cravatt, 2004; Alexander and Cravatt, 2005). For these studies, we compared the labeling profile of PF3845yne to those of JP104 (Figure 3B), a previously described alkyne analogue of URB597 (Alexander and Cravatt, 2005). PF3845yne and JP104 were administered to FAAH(++) and (-/-) mice (10 mg/kg, i.p.), and, after 2 hr, animals were sacrificed and their brain and liver proteomes analyzed by CC-ABPP using an azide-rhodamine tag. Consistent with the results of our competitive ABPP experiments (Figure 2), PF3845yne and JP104 selectively reacted with a single protein in mouse brain that was confirmed as FAAH based on its absence in FAAH(-/-) mice (Figure 3C). In liver, however, PF3845yne and JP104 showed strikingly different profiles, with the former agent once again showing selective reactivity with FAAH and the latter inhibitor labeling a number of proteins that were found in both FAAH(+/+) and (-/-) mice (Figure 3D). A single faint ∼60 kDa labeling event was observed in liver proteomes from PF3845-treated FAAH(-/-) mice, but a similar signal was also detected in vehicle-treated animals, suggesting that this protein may represent a non-specific target of the azide-rhodamine tag. Collectively, these data indicate that PF-3845 inhibits FAAH in vivo with exceptional efficacy and selectivity.
Figure 3.
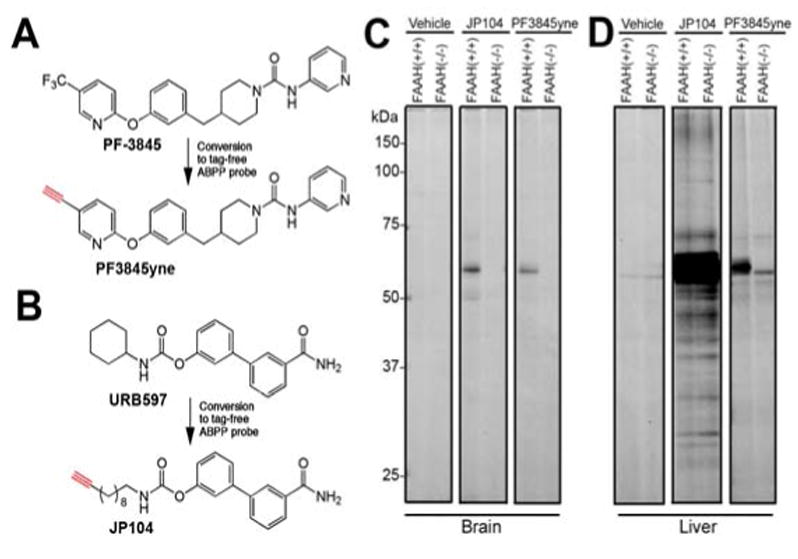
Direct analysis of in vivo protein targets of alkyne analogues of PF-3845 and URB597 by CC-ABPP. (A) Structure of PF3845yne, an alkyne analogue of PF-3845. (B) Structure of JP104, an alkyne analogue of URB597 (Alexander and Cravatt, 2005). (C & D) Gel profiles of CC-ABPP experiments where brain (C) or liver (D) proteomes from PF3845yne- and JP104-treated mice (10 mg/kg, i.p., 2 hr) were treated with a rhodamine-azide tag under CC conditions and analyzed by in-gel fluorescence scanning (shown in grayscale). PF3845yne selectively labels FAAH in both brain and liver tissue [60 kDa band absent in FAAH(-/-) mice], while JP104 labels several additional proteins in liver [protein bands present in both FAAH(+/+) and (-/-) mice]. Note that the 55 kDa protein band observed in liver proteome from PF-3845-treated FAAH(-/-) mice was also detected in liver proteomes from vehicle-treated FAAH(+/+) and (-/-) mice and therefore likely represents a background protein cross-reactive with the azide-rhodamine tag.
FAAH Inhibition by PF-3845 Causes a Dramatic and Sustained Elevation in AEA
Previous studies with FAAH(-/-) mice (Cravatt et al., 2001; Saghatelian et al., 2004) and rodents treated with FAAH inhibitors (Kathuria et al., 2003; Saghatelian et al., 2004) have confirmed a key role for FAAH in regulating tissue levels of AEA and other NAEs. We found that PF-3845-treated mice (10 mg/kg, i.p.) also showed dramatic (> 10-fold) elevations in brain levels of AEA (Figure 4A) and other NAEs [N-pamitoyl ethanolamine (PEA) (Figure 4B) and N-oleoyl ethanolamine (OEA) (Figure 4C)]. These animals also showed significant elevations in AEA, PEA, and OEA levels in liver tissue (Figures S3A). In contrast, PF-3845-treated animals did not show changes in the levels of 2-AG in brain or liver (Figure S3B), consistent with previous findings indicating that distinct enzymes regulate this endocannabinoid in vivo (Kathuria et al., 2003; Blankman et al., 2007; Long et al., 2009). We also confirmed that PF-3845 did not alter endocannabinoid (AEA or 2-AG) levels in FAAH(-/-) mice (Figure S3C).
Figure 4.
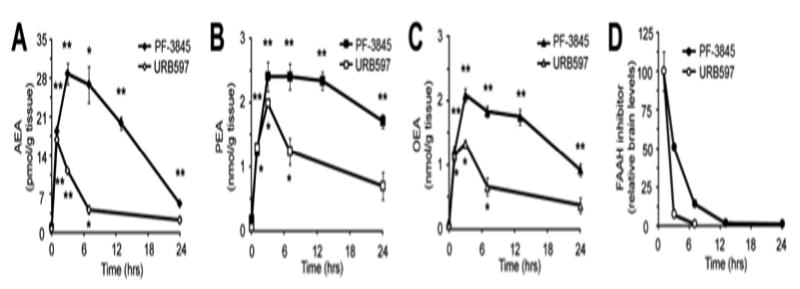
Brain levels of FAAH substrates and inhibitors. (A-C) Brain levels of AEA (A), PEA (B), and OEA (C) measured at the indicated time points following treatment with PF-3845 or URB597 (10 mg/kg, i.p.). *p < 0.05; **p < 0.01 for inhibitor- versus vehicle-treated groups. n = 4 mice/group. (D) Brain levels of PF-3845 and URB597 measured at the indicated time points following inhibitor treatment.
PF-3845-induced elevations in NAEs peaked at ∼3 hours and were maintained at maximal levels for up to 7-12 hrs, after which they slowly declined. This decline was faster for AEA than PEA or OEA, possibly reflecting that AEA is a much preferred substrate for FAAH (Cravatt et al., 1996; Wei et al., 2006) and therefore more sensitive to low levels of enzyme recovery. The slightly faster rate of recovery for OEA versus PEA is also consistent with the relative activity that FAAH displays for these substrates in vitro (Wei et al., 2006). Despite FAAH's preference for hydrolyzing AEA, significant elevations in this NAE (as well as PEA and OEA) were still observed at 24 hrs post-treatment with PF-3845, consistent with the substantial level of FAAH inhibition maintained at this time point (Figure 2A). URB597-treated mice also showed elevations in NAEs, but these increases were more transient, with AEA levels returning to near-baseline levels by 7 hrs post-treatment (Figure 4A-C). The different rates of recovery of both FAAH activity (Figure 2A) and NAE levels (Figure 4A-C) in mice treated with PF-3845 versus URB597 likely reflects the distinct half-lives of these inhibitors in vivo, since higher relative brain levels of PF-3845 were maintained over the time-course analysis (Figure 4D).
PF-3845 Produces Cannabinoid Receptor-Dependent Reductions in Inflammatory Pain
We next assessed whether PF-3845 was efficacious in a rat model of inflammatory pain. Subcutaneous injection of complete Freund's adjuvant (CFA) into the plantar surface of the hind paw produced a significant decrease in mechanical paw weight threshold (PWT) at 5 days post injection (Figure 5A). PF-3845 [1 – 30 mg/kg, oral administration (p.o),] caused a dose-dependent inhibition of mechanical allodynia with a minimum effective dose (MED) of 3 mg/kg (rats were analyzed at 4 hr post-dosing with PF-3845). At higher doses (10 and 30 mg/kg), PF-3845 inhibited pain responses to an equivalent, if not greater degree than the nonsteroidal anti-inflammatory drug naproxen (10 mg/kg, p.o.) (Figure 5A). We next determined the relationship between the in vivo efficacy and modulation of FAAH activity and substrates (i.e., NAEs) by PF-3845 in central (brain) and peripheral systems (peripheral blood leukocytes/plasma, liver). Robust, near-complete inhibition of FAAH activity (Figure 5B and Figure S4A) with concomitant elevations in AEA (Figure 5C) and other NAEs (Figures S4B and C and S5) were observed in brain, peripheral blood leukocytes/plasma, and liver from PF-3845-treated animals. Interestingly, we observed a slightly left-shifted dose-response profile for measurements of FAAH activity and NAEs compared to efficacy in the CFA model. For instance, at 1 mg/kg, PF-3845 was found to produce a near-complete blockade of FAAH activity (Figure 5B) and close to maximal elevations in AEA (Figure 5C), but efficacy was not observed at this dose of inhibitor (Figure 5A). These data may indicate the need to fully block FAAH (and fully elevate AEA levels) in order to observe anti-allodynic effects in the CFA model. Alternatively, the kinetics of FAAH blockade may have been slower at lower doses of PF-3845, which could reduce efficacy in pain models that require sustained elevations in AEA. It is also noteworthy that the maximal elevations observed for AEA in PF-3845-treated animals were similar in brain and plasma (∼10-fold), while OEA and PEA were increased to a lower extent in plasma (∼3-fold) compared to brain (∼10-fold). This might suggest the existence of alternative degradative (Tsuboi et al., 2005) [or biosynthetic (Leung et al., 2006)] pathways for AEA versus OEA/PEA in peripheral tissues.
Figure 5.
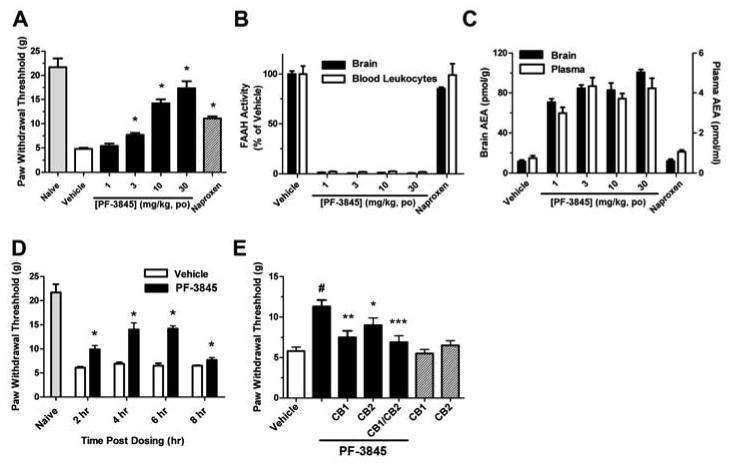
Anti-hyperalgesic effects of PF-3845 in the CFA model of inflammatory pain. (A) PF-3845 produces a dose-dependent reduction of mechanical allodynia (hyperalgesia) in rats (black bars). The effect of the non-steroidal anti-inflammatory drug naproxen (10 mg/kg, p.o., hatched bar) is shown for comparison. Paw withdrawal thresholds were measured 4 hours post-administration of drugs (B and C) PF-3845-treated rats show significant blockade of FAAH activity (B) and elevated AEA levels (C) in brain tissue and blood leukocytes/plasma. All FAAH activity and NAE measurements were determined at 4 hr following drug treatment and were significantly different between PF-3845- and vehicle-treated groups (p < 0.001, n = 3 rats/group). (D) Time-course for anti-hyperalgesic effects of PF-3845. (E) Blockade of anti-hyperalgesic effects of PF-3845 (10 mg/kg, p.o.) by CB1 and CB2 antagonists (SR141716 and SR144528, respectively; 10 and 30 mg/kg, i.p., respectively administered 1 hr prior to treatment with PF-3845). Note that neither the CB1 nor CB2 antagonist displayed significant effects on mechanical allodynia in rats not treated with PF-3845 (hatched bars). #p<0.001, for PF-3845- versus vehicle-treated groups. *p < 0.05; **p < 0.01; ; ***p<0.001, for vehicle-PF-3845 versus CB1/CB2 antagonist-PF-3845 treated groups. n = 8 rats/group.
Considering that mice treated with PF-3845 showed high (> 10-fold) elevations in AEA that lasted for at least 7 hrs, we next evaluated the duration of action of this inhibitor in the CFA model from 2-8 hrs post-administration. PF-3845 (10 mg/kg, p.o.) exhibited significant inhibition of mechanical allodynia at all time points tested (Figure 5D), although this effect appeared to diminish in magnitude by 8 hrs. This loss in activity could reflect gradual recovery of FAAH activity in PF-3845-treated rats, as may be reflected in the decline in AEA levels that was observed at a similar time point in mice treated with this inhibitor (see Figure 4A). However, further time-course studies in rats will be required to accurately estimate the duration of action of PF-3845 in these animals. We next examined the involvement of cannabinoid receptors in the PF-3845-induced anti-allodynia. Specific antagonists for central CB1 (SR141716) and peripheral CB2 (SR144528) receptors each partially, but significantly reduced the anti-allodynia activity of PF-3845 (Figure 5E). Notably, dual treatment with SR141716 and SR144528 produced a near-complete ablation of the efficacy of PF-3845 in the CFA model (Figure 5E). These data indicate that PF-3845 blocks inflammatory pain responses in the CFA model by a cannabinoid receptor-dependent mechanism that likely involves both CB1 and CB2 receptors, as has been shown for other FAAH inhibitors (Jayamanne et al., 2006).
Conclusion
Endocannabinoids, like other lipid signaling molecules, are thought to be produced on-demand by neurons, rather than being stored in synaptic vesicles like classical neurotransmitters (Ahn et al., 2008; Di Marzo, 2008). Although protein-mediated uptake pathways may exist for endocannabinoids (Hillard and Jarrahian, 2003; McFarland and Barker, 2004; Glaser et al., 2005; Fowler, 2006), the hydrophobic nature of these lipids also grants them the ability to freely diffuse across cell membranes. This feature assigns primary responsibility for signal termination to degradative enzymes, such as FAAH. The pharmacological blockade of FAAH has, thus, emerged as a strategy to elevate endocananbinoid signaling in vivo and study the behavioral consequences. Over the past decade, several classes of FAAH inhibitors have been described that vary in their mechanism of action, potency, selectivity, and in vivo efficacy. Arguably the most well-studied FAAH inhibitor to date is URB597 (Kathuria et al., 2003), a carbamate that irreversibly inhibits FAAH with high selectivity in the nervous system (Alexander and Cravatt, 2005). However, URB597 displays some notable shortcomings, including the inactivation of other serine hydrolases in peripheral tissues (Alexander and Cravatt, 2005; Zhang et al., 2007) and a relatively limited duration of action in vivo. Development of a FAAH inhibitor that displays optimal combination of efficacy and selectivity has, therefore, remained an intensely pursued objective.
We have described herein the detailed characterization of an advanced piperidine urea inhibitor of FAAH, PF-3845. Our data argue PF-3845 displays remarkable selectivity for FAAH and robust efficacy in vivo, as reflected in sustained elevations of brain AEA that correlate with cannabinoid receptor-dependent reductions in inflammatory pain responses. PF-3845 also induces elevations in other non-cannabinoid NAEs (e.g., OEA, PEA), which may have additional effects on inflammation and other physiological processes (Lambert et al., 2002). Structural and kinetic studies indicate that the improved potency of PF-3845 compared to previously described inhibitors (URB597, PF-750) likely originates from more extended Van der Waals contacts with the hydrophobic FAAH acyl-chain binding tunnel. This premise is also supported by SAR data generated with a set of biaryl ether analogues of PF-3845, where the introduction of polar groups within certain regions of the inhibitor scaffold severely impaired activity (e.g., compound 16, Table 2).
In summary, the properties of PF-3845 defined in this study indicate that this agent should be considered a frontline inhibitor for the pharmacological investigation of FAAH-regulated endocannabinoid pathways. Differentiating properties of PF-3845 include its high target selectivity, extended duration of action, and excellent oral bioavailability (> 80%, data not shown). These features should make PF-3845 particularly well suited for studying the role of the endocannabinoid system in both central and peripheral physiological processes, as well as chronic pathological syndromes (e.g., neuropathic pain, depression) that may require sustained elevations in endocannabinoid tone for rectification.
Significance
The endocannabinoid system regulates a number of important physiological processes in mammals and is the principal site of action for THC, the psychoactive ingredient in marijuana. THC and other direct agonists of the CB1 receptor have potentially useful medicinal properties, such as pain relief, but these activities are accompanied by a variety of untoward side effects, including impairments in motor control and cognitive function. In contrast, blockade of endocannabinoid-metabolizing enzymes such as FAAH has been shown to reduce pain sensation, inflammation, and anxiety/depression without substantial effects on motility or cognition. Despite considerable advances in the development of FAAH inhibitors, agents that combine high target selectivity with exceptional in vivo efficacy are still needed. Here, we report the discovery and detailed characterization of PF-3845, a piperidine urea FAAH inhibitor that satisfies these criteria. PF-3845 was shown by mechanistic and structural studies to covalently inactivate FAAH by carbamylation of the enzyme's serine nucleophile. PF-3845 completely inhibited FAAH activity, but did not react with other serine hydrolases in vivo, as determined by activity-based proteomics, raised brain levels of the endocannabinoid AEA for up to 24 hrs, and produced significant CB1/CB2-dependent anti-hyperalgesic effects in the CFA model of inflammatory pain. From these data, we conclude that PF-3845 is a highly selective and efficacious inhibitor of FAAH that should prove to be a valuable pharmacological tool for investigating the function of the endocannabinoid system.
Experimental Procedures
Materials
N-phenyl-4-(quinolin-3-ylmethyl)piperidine-1-carboxamide (PF-750) was synthesized as described previously (Ahn et al., 2007). URB597 and JP104 were purchased from Cayman Chemicals (Ann Arbor, MI). Inhibitors were stored as dry powders at RT and dissolved in DMSO to prepare concentrated stock solutions for the in vitro potency measurements. SR141716 and SR144528, selective CB1 and CB2 receptor antagonists, were synthesized at Pfizer. Polystyrene 96- and 384-microplates were purchased from Rainin and Evergreen Scientific (Los Angeles, CA), respectively. All reagents used were the highest quality commercially available.
Synthesis of PF-3845 and structural analogues
The synthesis of PF-3845 and other biaryl ether piperidine/piperazine ureas was performed as described in the Supplemental Methods.
FAAH expression and purification
All three forms of FAAH (hFAAH, rFAAH, and h/rFAAH) were expressed and purified following previously described procedures (Mileni et al., 2008) and as described in the Supplemental Methods.
Determination of inhibitor potency
Inhibitor potencies were determined following previously described procedures (Mileni et al., 2008) and as described in the Supplemental Methods.
h/rFAAH crystallography
Crystallization and structure determination were performed as described in the Supplemental Methods.
Monte Carlo simulations of FAAH-inhibitor adducts
The program ICM-Pro (Molsoft, L.L.C.) was used to simulate covalent docking of biaryl ether urea inhibitors into the active site of the h/rFAAH as described in the Supplemental Methods. As a positive control, we performed the docking of the PF-3845 ligand, which provided a conformation that closely matched the conformation observed in the PF-3845-FAAH crystal structure.
Competitive ABPP studies
PF-3845 or URB597 (neat) were dissolved at 1 mg/mL by sonication and vortexing directly into a solution of 18:1:1 v/v/v saline:emulphor:ethanol. Male C57Bl/6J mice (6–12 weeks old, 20–28 g) were intraperitoneally (i.p.) administered PF-3845, URB597, or an 18:1:1 v/v/v saline:emulphor:ethanol vehicle at a volume of 10 μL/g weight (10 mg/kg final dose). After the indicated amount of time, mice were anesthetized with isofluorane and sacrificed by decapitation. Brains were removed, hemisected along the midsagittal plane, and each half was then flash frozen in liquid N2. One half of the brain was homogenized in PBS (pH 7.4, 2 mL), diluted to 1 mg protein/mL, treated with FP-rhodamine (1 μM, 0.5 hr), and analyzed by gel-based ABPP following previously described methods (Alexander and Cravatt, 2005). The other half of brain was used for lipid and drug measurements as detailed in the Supplementary Methods.
CC-ABPP studies
CC-ABPP studies were performed following previously described procedures (Speers et al., 2003; Speers and Cravatt, 2004; Alexander and Cravatt, 2005) and as detailed in the Supplementary Methods.
Measurement of FAAH activity and lipid levels from rat tissues and blood leukocytes
Preparation of rat tissue (brain and liver) and blood leukocytes for measuring FAAH activity and lipid levels is described in Supplementary Methods.
Analysis of PF-3845 effects in the CFA model of inflammatory pain
Male Sprague-Dawley rats (200g- 250g) were used for all experiments. Rats were given free access to food and water and were maintained on a 12/12-hr light/dark cycle. For the inflammatory pain model, 0.15 ml of CFA (Sigma) was injected into the plantar surface of the left hind paw of the rat. The CFA injection immediately induces local inflammation, paw swelling, and pain that persist for at least two weeks post-injection. To assess mechanical allodynia, mechanical paw withdrawal thresholds (PWTs) were measured using a set of Von Frey Hairs on day 5 post injection as illustrated by the Dixon Up and Down Method (Dixon, 1980). Animals that exhibit the pain criteria of 9 g or less were then placed on study. Test compound was administered via either oral or intraperitoneal (i.p.) routes at the indicated concentrations (mg/kg) with the dosing vehicle 5% N,N-dimethylacetamide and 95% (40% 2-hydroxypropyl-β-cyclodextrin (Sigma) in water). PWTs were evaluated again at 4 hr post dosing. PWT measurements were averaged and statistical comparisons between groups were made using analysis of variance and unpaired T-tests. The Pfizer Institutional Animal Care and Use Committee reviewed and approved the animal use in these studies. The animal care and use program is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, International.
Supplementary Material
Acknowledgments
We thank Kimberly Masuda and Jessica P. Alexander for technical assistance with ABPP studies, Max Totrov from Molsoft for helpful suggestions and discussions on the docking and simulation work, Steve Kesten for the scale-up of PF-3845, Mike Walters, Michael Connolly, Yuntao Song, Mark Morris, and Sue Kesten for early chemistry contributions to the biaryl ether series, and Zhigang Wang and Daniel Everdeen for their assistance on the measurement of inhibitor potencies. This work was supported in part by the NIH (DA017259), the Helen L. Dorris Institute for the Study of Neurological and Psychiatric Disorders in Children and Adolescents, and The Skaggs Institute for Chemical Biology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn K, Johnson DS, Fitzgerald LR, Liimatta M, Arendse A, Stevenson T, Lund ET, Nugent RA, Nomanbhoy TK, Alexander JP, Cravatt BF. Novel mechanistic class of fatty acid amide hydrolase inhibitors with remarkable selectivity. Biochemistry. 2007;46:13019–30. doi: 10.1021/bi701378g. [DOI] [PubMed] [Google Scholar]
- Ahn K, McKinney MK, Cravatt BF. Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem Rev. 2008;108:1687–707. doi: 10.1021/cr0782067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander JP, Cravatt BF. Mechanism of carbamate inactivation of FAAH: implications for the design of covalent inhibitors and in vivo functional probes for enzymes. Chem Biol. 2005;12:1179–87. doi: 10.1016/j.chembiol.2005.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankman JL, Simon GM, Cravatt BF. A Comprehensive Profile of Brain Enzymes that Hydrolyze the Endocannabinoid 2-Arachidonoylglycerol. Chem Biol. 2007;14:1347–56. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boger DL, Sato H, Lerner AE, Austin BJ, Patterson JE, Patricelli MP, Cravatt BF. Trifluoromethyl ketone inhibitors of fatty acid amide hydrolase: a probe of structural and conformational features contributing to inhibition. Bioorg Med Chem Lett. 1999;9:265–270. doi: 10.1016/s0960-894x(98)00734-3. [DOI] [PubMed] [Google Scholar]
- Boger DL, Sato H, Lerner AE, Hedrick MP, Fecik RA, Miyauchi H, Wilkie GD, Austin BJ, Patricelli MP, Cravatt BF. Exceptionally potent inhibitors of fatty acid amide hydrolase: the enzyme responsible for degradation of endogenous oleamide and anandamide. Proc Natl Acad Sci USA. 2000;97:5044–5049. doi: 10.1073/pnas.97.10.5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracey MH, Hanson MA, Masuda KR, Stevens RC, Cravatt BF. Structural adaptations in a membrane enzyme that terminates endocannabinoid signaling. Science. 2002;298:1793–1796. doi: 10.1126/science.1076535. [DOI] [PubMed] [Google Scholar]
- Brandl M, Weiss MS, Jabs A, Suhnel J, Hilgenfeld R. C-H…pi-interactions in proteins. J Mol Biol. 2001;307:357–77. doi: 10.1006/jmbi.2000.4473. [DOI] [PubMed] [Google Scholar]
- Chang L, Luo L, Palmer JA, Sutton S, Wilson SJ, Barbier AJ, Breitenbucher JG, Chaplan SR, Webb M. Inhibition of fatty acid amide hydrolase produces analgesia by multiple mechanisms. Br J Pharmacol. 2006;148:102–13. doi: 10.1038/sj.bjp.0706699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, Lichtman AH. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci USA. 2001;98:9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Saghatelian A, Hawkins EG, Clement AB, Bracey MH, Lichtman AH. Functional disassociation of the central and peripheral fatty acid amide signaling systems. Proc Natl Acad Sci U S A. 2004;101:10821–6. doi: 10.1073/pnas.0401292101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch DG, Omir R, Arreaza G, Salehani D, Prestwich GD, Huang Z, Howlett A. Methyl arachidonyl fluorophosphonate: a potent irreversible inhibitor of anandamide amidase. Biochem Pharmacol. 1997;53:255–260. doi: 10.1016/s0006-2952(96)00830-1. [DOI] [PubMed] [Google Scholar]
- Di Marzo V. Endocannabinoids: synthesis and degradation. Rev Physiol Biochem Pharmacol. 2008;160:1–24. doi: 10.1007/112_0505. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Bisogno T, De Petrocellis L. Endocannabinoids and Related Compounds: Walking Back and Forth between Plant Natural Products and Animal Physiology. Chemistry & Biology. 2007;14:741–756. doi: 10.1016/j.chembiol.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–62. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- Fegley D, Gaetani S, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. Characterization of the fatty acid amide hydrolase inhibitor cyclohexyl carbamic acid 3′-carbamoyl-biphenyl-3-yl ester (URB597): effects on anandamide and oleoylethanolamide deactivation. J Pharmacol Exp Ther. 2005;313:352–8. doi: 10.1124/jpet.104.078980. [DOI] [PubMed] [Google Scholar]
- Fowler CJ. The cannabinoid system and its pharmacological manipulation--a review, with emphasis upon the uptake and hydrolysis of anandamide. Fundam Clin Pharmacol. 2006;20:549–62. doi: 10.1111/j.1472-8206.2006.00442.x. [DOI] [PubMed] [Google Scholar]
- Glaser ST, Kaczocha M, Deutsch DG. Anandamide transport: a critical review. Life Sci. 2005;77:1584–604. doi: 10.1016/j.lfs.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Gobbi G, Bambico FR, Mangieri R, Bortolato M, Campolongo P, Solinas M, Cassano T, Morgese MG, Debonnel G, Duranti A, Tontini A, Tarzia G, Mor M, Trezza V, Goldberg SR, Cuomo V, Piomelli D. Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc Natl Acad Sci U S A. 2005;102:18620–5. doi: 10.1073/pnas.0509591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillard CJ, Jarrahian A. Cellular accumulation of anandamide: consensus and controversy. Br J Pharmacol. 2003;140:802–8. doi: 10.1038/sj.bjp.0705468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt S, Comelli F, Costa B, Fowler CJ. Inhibitors of fatty acid amide hydrolase reduce carrageenan-induced hind paw inflammation in pentobarbital-treated mice: comparison with indomethacin and possible involvement of cannabinoid receptors. Br J Pharmacol. 2005;146:467–76. doi: 10.1038/sj.bjp.0706348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayamanne A, Greenwood R, Mitchell VA, Aslan S, Piomelli D, Vaughan CW. Actions of the FAAH inhibitor URB597 in neuropathic and inflammatory chronic pain models. Br J Pharmacol. 2006;147:281–8. doi: 10.1038/sj.bjp.0706510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- Keith JM, Apodaca R, Xiao W, Seierstad M, Pattabiraman K, Wu J, Webb M, Karbarz MJ, Brown S, Wilson S, Scott B, Tham CS, Luo L, Palmer J, Wennerholm M, Chaplan S, Breitenbucher JG. Thiadiazolopiperazinyl ureas as inhibitors of fatty acid amide hydrolase. Bioorg Med Chem Lett. 2008;18:4838–43. doi: 10.1016/j.bmcl.2008.07.081. [DOI] [PubMed] [Google Scholar]
- Koutek B, Prestwich GD, Howlett AC, Chin SA, Salehani D, Akhavan N, Deutsch DG. Inhibitors of arachidonyl ethanolamide hydrolysis. J Biol Chem. 1994;269:22937–22940. [PubMed] [Google Scholar]
- Lambert DM, Vandevoorde S, Jonsson KO, Fowler CJ. The palmitoylethanolamide family: a new class of anti-inflammatory agents? Curr Med Chem. 2002;9:663–674. doi: 10.2174/0929867023370707. [DOI] [PubMed] [Google Scholar]
- Leung D, Hardouin C, Boger DL, Cravatt BF. Discovering potent and selective inhibitors of enzymes in complex proteomes. Nat Biotechnol. 2003;21:687–691. doi: 10.1038/nbt826. [DOI] [PubMed] [Google Scholar]
- Leung D, Saghatelian A, Simon GM, Cravatt BF. Inactivation of N-acyl ethanolamine phospholipase D reveals multiple mechanisms for the biosynthesis of endocannabinoids. Biochemistry. 2006;45:4720–4726. doi: 10.1021/bi060163l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman AH, Leung D, Shelton C, Saghatelian A, Hardouin C, Boger D, Cravatt BF. Reversible inhibitors of fatty acid amide hydrolase that promote analgesia: evidence for an unprecedented combination of potency and selectivity. J Pharmacol Exp Ther. 2004;311:441–448. doi: 10.1124/jpet.104.069401. [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Shelton CC, Advani T, Cravatt BF. Mice lacking fatty acid amide hydrolase exhibit a cannabinoid receptor-mediated phenotypic hypoalgesia. Pain. 2004;109:319–327. doi: 10.1016/j.pain.2004.01.022. [DOI] [PubMed] [Google Scholar]
- Liu Y, Patricelli MP, Cravatt BF. Activity-based protein profiling: The serine hydrolases. Proc Natl Acad Sci USA. 1999;96:14694–14699. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavon FJ, Serrano AM, Selley DE, Parsons LH, Lichtman AH, Cravatt BF. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol. 2009;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massa F, Marsicano G, Hermann H, Cannich A, Monory K, Cravatt BF, Ferri GL, Sibaev A, Storr M, Lutz B. The endogenous cannabinoid system protects against colonic inflammation. J Clin Invest. 2004;113:1202–9. doi: 10.1172/JCI19465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland MJ, Barker EL. Anandamide transport. Pharmacol Ther. 2004;104:117–35. doi: 10.1016/j.pharmthera.2004.07.008. [DOI] [PubMed] [Google Scholar]
- McKinney MK, Cravatt BF. Structure and function of Fatty acid amide hydrolase. Annu Rev Biochem. 2005;74:411–32. doi: 10.1146/annurev.biochem.74.082803.133450. [DOI] [PubMed] [Google Scholar]
- Mechoulam R. The pharmacohistory of Cannabis sativa. Boca Raton, Florida: CRS Press; 1986. [Google Scholar]
- Mileni M, Johnson DS, Wang Z, Everdeen DS, Liimatta M, Pabst B, Bhattacharya K, Nugent RA, Kamtekar S, Cravatt BF, Ahn K, Stevens RC. Structure-guided inhibitor design for human FAAH by interspecies active site conversion. Proc Natl Acad Sci U S A. 2008;105:12820–4. doi: 10.1073/pnas.0806121105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira FA, Kaiser N, Monory K, Lutz B. Reduced anxiety-like behaviour induced by genetic and pharmacological inhibition of the endocannabinoid-degrading enzyme fatty acid amide hydrolase (FAAH) is mediated by CB1 receptors. Neuropharmacology. 2008;54:141–50. doi: 10.1016/j.neuropharm.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Naidu PS, Varvel SA, Ahn K, Cravatt BF, Martin BR, Lichtman AH. Evaluation of fatty acid amide hydrolase inhibition in murine models of emotionality. Psychopharmacology (Berl) 2007;192:61–70. doi: 10.1007/s00213-006-0689-4. [DOI] [PubMed] [Google Scholar]
- Nomura DK, Blankman JL, Simon GM, Fujioka K, Issa RS, Ward AM, Cravatt BF, Casida JE. Activation of the endocannabinoid system by organophosphorus nerve agents. Nat Chem Biol. 2008;4:373–8. doi: 10.1038/nchembio.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patricelli MP, Giang DK, Stamp LM, Burbaum JJ. Direct visualization of serine hydrolase activities in complex proteome using fluorescent active site-directed probes. Proteomics. 2001;1:1067–1071. doi: 10.1002/1615-9861(200109)1:9<1067::AID-PROT1067>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Patricelli MP, Lovato MA, Cravatt BF. Chemical and mutagenic investigations of fatty acid amide hydrolase: evidence for a family of serine hydrolase with distinct catalytic features. Biochemistry. 1999;38:9804–9812. doi: 10.1021/bi990637z. [DOI] [PubMed] [Google Scholar]
- Russo R, Loverme J, La Rana G, Compton TR, Parrott J, Duranti A, Tontini A, Mor M, Tarzia G, Calignano A, Piomelli D. The fatty acid amide hydrolase inhibitor URB597 (cyclohexylcarbamic acid 3′-carbamoylbiphenyl-3-yl ester) reduces neuropathic pain after oral administration in mice. J Pharmacol Exp Ther. 2007;322:236–42. doi: 10.1124/jpet.107.119941. [DOI] [PubMed] [Google Scholar]
- Saghatelian A, Trauger SA, Want EJ, Hawkins EG, Siuzdak G, Cravatt BF. Assignment of endogenous substrates to enzymes by global metabolite profiling. Biochemistry. 2004;43:14332–9. doi: 10.1021/bi0480335. [DOI] [PubMed] [Google Scholar]
- Speers AE, Adam GC, Cravatt BF. Activity-based protein profiling in vivo using a copper(I)-catalyzed azide-alkyne [3 + 2] cycloaddition. J Amer Chem Soc. 2003;125:4686–4687. doi: 10.1021/ja034490h. [DOI] [PubMed] [Google Scholar]
- Speers AE, Cravatt BF. Profiling enzyme activities in vivo using click chemistry methods. Chem Biol. 2004;11:535–546. doi: 10.1016/j.chembiol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- Swinney DC. Biochemical mechanisms of drug action: what does it take for success? Nat Rev Drug Discov. 2004;3:801–8. doi: 10.1038/nrd1500. [DOI] [PubMed] [Google Scholar]
- Tsuboi K, Sun YX, Okamoto Y, Araki N, Tonai T, Ueda N. Molecular characterization of N-acylethanolamine-hydrolyzing acid amidase, a novel member of the choloylglycine hydrolase family with structural and functional similarity to acid ceramidase. J Biol Chem. 2005;280:11082–92. doi: 10.1074/jbc.M413473200. [DOI] [PubMed] [Google Scholar]
- Wei BQ, Mikkelsen TS, McKinney MK, Lander ES, Cravatt BF. A second fatty acid amide hydrolase with variable distribution among placental mammals. J Biol Chem. 2006;281:36569–78. doi: 10.1074/jbc.M606646200. [DOI] [PubMed] [Google Scholar]
- Zhang D, Saraf A, Kolasa T, Bhatia P, Zheng GZ, Patel M, Lannoye GS, Richardson P, Stewart A, Rogers JC, Brioni JD, Surowy CS. Fatty acid amide hydrolase inhibitors display broad selectivity and inhibit multiple carboxylesterases as off-targets. Neuropharmacology. 2007;52:1095–105. doi: 10.1016/j.neuropharm.2006.11.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.