

Summary
We contrast the efforts to treat ovarian cancer and cervical cancer through vaccination because of their different pathobiology. A plethora of approaches have been developed for therapeutic vaccination against cancer, many of which target defined tumor-associated antigens (TAAs). Persistent infection with oncogenic human papillomavirus (HPV) types is necessary cause of cervical cancer. Furthermore, cervical cancer patients frequently mount both humoral and T cell immune responses to the HPV E6 and E7 oncoproteins, whose expression is required for the transformed phenotype. Numerous vaccine studies target these viral TAAs, including recent trials that may enhance clearance of pre-malignant disease. By contrast little is known about the etiology of epithelial ovarian cancer. Although it is clear that p53 mutation or loss is a critical early event in the development of epithelial ovarian cancer, no precursor lesion has been described for the most common serous histotype, and even the location of its origin is debated. These issues have complicated the selection of appropriate ovarian TAAs and the design of vaccines. Here we focus on mesothelin as a promising ovarian TAA because it is overexpressed and immunogenic at high frequency in patients, is displayed on the cell surface and potentially contributes to ovarian cancer biology.
Keywords: vaccine, cervical cancer, human papillomavirus, E6, E7, mesothelin, ovarian cancer
The distinct pathobiologies of cervical and ovarian cancer impact approaches to immunotherapy
Cervical cancer is the second leading cancer killer of women worldwide, with nearly half a million women diagnosed each year (1). However its impact is not even globally. Approximately 80% of diagnoses and 85% of deaths worldwide from cervical cancer occur in Developing countries. The primary reason for this difference has been the institution in developed countries of effective national programs for cytologic (Pap) screening for the precursor lesion, high-grade cervical intraepithelial neoplasia (CIN), and ablation of these pre-invasive lesions by conization or loop electrosurgical excision procedure (LEEP) etc. Unfortunately there are few such screening programs in Developing countries. It is estimated that cytologic screening, at a cost of $6 billion per annum, has reduced the incidence of cervical cancer by 70–80% in the US, such that there are now ~5000 cervical cancer deaths each year (2). Furthermore, in contrast to ovarian cancer, cervical cancer is more frequently diagnosed at a lower stage. The primary treatment for cervical cancer is radical hysterectomy and surgical debulking, followed by chemoradiation therapy. Unfortunately, patients with advanced cervical cancer still have a poor prognosis despite undergoing conventional therapy with significant side effects. It is for this reason that new, better targeted approaches to treatment, such as immunotherapy, remain a priority.
It is clear that persistent infection with oncogenic HPV is a necessary, but not sufficient cause of uterine cervical carcinoma, both squamous cell carcinoma and adenocarcinoma (3–5). Molecular testing for oncogenic HPV infection has now been licensed as an adjunct to cytologic screening e.g. HCII test (6). HPV testing provides screening with a greater sensitivity than cytology. However HPV testing can be overly sensitive as many patients with oncogenic HPV infections do not have high grade CIN. These patients are typically followed rather than treated, but a therapeutic vaccine would be valuable to ensure viral clearance.
Importantly, expression of the viral oncoproteins E6 and E7 is necessary to maintain the transformed phenotype of cervical cancer cells (7,8). As a result of a viral origin and oncogenicity respectively, E6 and E7 are neither subject to central immunologic tolerance nor can be lost during tumor evasion. These properties make E6 and E7 very attractive target TAAs for immunotherapy. Furthermore, a significant fraction of HPV infections and pre-invasive CIN are spontaneously cleared, although regression rates are substantially lower in immunosuppressed individuals. This provides a strong rationale for immune control of HPV and HPV-related disease and therefore therapeutic vaccination against cervical cancer. A plethora of vaccination approaches designed to generate regression of CIN and/or invasive carcinoma have been tested. However there have been no convincing reports of a completely effective vaccine therapy for either CIN or invasive cervical cancer, although most vaccines proved immunogenic. This suggests that simply generating an immune response to the relevant antigens is necessary, but not enough to trigger tumor rejection and that the local tumor microenvironment must be modified to permit effective immunotherapy. Nevertheless, there are hints of potential for success that may be realized with further development of E6/E7-specific vaccines and possibly their combination with other treatment modalities.
Given the difficulties in developing therapeutic vaccines that are effective against HPV-related disease, it is not surprising that this approach has proven even more challenging in ovarian cancer. However, important lessons may be learned from examining the successes and failures of antigen-specific immunotherapy in cervical cancer treatment and applied to ovarian cancer.
Ovarian cancer has the highest mortality rate among gynecologic cancers and ranks in the top five causes of cancer death among women in the United States (9). There are approximately 15000 deaths each year due to ovarian cancer and there has been little change in the mortality rates despite intensive efforts. Currently there are no simple preventive measures to significantly reduce risk for ovarian cancer (10), whereas cervical cancer prevention programs, and now vaccines, have proven very effective. Unfortunately the prognosis for women with advanced ovarian (or cervical) cancer remains poor, with five-year survival rates drastically falling as the cancer spreads into the peritoneal cavity (stage III) and beyond (stage IV). Conventional treatment with surgery and chemo or radiotherapy has a poor success rate for such disseminated cancers and produces harsh side effects, suggesting the importance of developing effective immunotherapies for advanced cervical and ovarian cancers.
Prospects of patients with early-stage ovarian cancer confined to the ovaries (Stage I) are more optimistic; with surgical removal of the ovaries ~90% survive five years after diagnosis. However, 70% of patients with ovarian cancer have already advanced disseminated disease at the time of initial diagnosis (9) because there is no diagnostic tool for reliable screening and detection of pre-malignant or localized ovarian cancer when treatment is most effective. Furthermore, these figures are deceptive as the distribution of histotypes, which vary in their aggressiveness, differs between early stage and advanced ovarian cancer. For example serous carcinoma is rarely detected at stage I and forms the bulk of advanced disease, whereas less aggressive histotypes are more common diagnosed as early stage disease. Therefore, it is more likely that immunotherapy of the aggressive histotypes would be initiated after surgical debulking and initial standard chemotherapy (taxol plus cisplatin) with the intention of targeting minimal residual disease.
Fortunately, conventional treatment of stage I ovarian cancer with surgery, and chemotherapy if necessary, has a high cure rate. There is however currently no test with sufficient predictive value for use in screening. CA125 is a serum marker approved only to monitor ovarian cancer. Detection of abnormally elevated CA125 in plasma is correlated to tumor diameter; only 21% of patients with microscopic disease, but >70% with a tumor diameter of 1–2 cm have elevated values (11). The CA125 test alone lacks the specificity necessary for use as a population screen for early stage ovarian cancer. Approximately 1% of normal healthy donors have plasma CA125 concentrations greater than the cut-point of 35U/ml, resulting in a relatively low predictive value since the incidence of ovarian cancer is ~40/100,000 women of >45 years of age per annum in the USA. A number of other physiologic and pathologic changes, including menstruation, first trimester pregnancy and endometriosis, are associated in some cases with elevated CA125. Sensitivity is also lacking; although elevated levels of CA125 are detected >90% of sera of disseminated ovarian cancer cases (stages II–IV), only 50% of patients with stage I disease are detected by this assay (11). Thus the identification of novel diagnostic serum biomarkers specific to, and also widely expressed by, early-stage ovarian cancers is crucial for the development of diagnostic tests for early detection, and these biomarkers may also prove useful targets for immunotherapy. Should a suitable assay be developed, regular screening could identify at-risk patients to be treated with prophylactic oophorectomy and possibly immunotherapy. However, this assumes a window for detection of incipient ovarian cancer. Current estimates suggest a mean duration of 1.9±0.4 years for the duration of pre-clinical ovarian cancer based upon fitting a longitudinal change point model to CA125 values in stage I patients to estimate the time from tumor inception to clinical detection (12). This is a dramatic difference to the 10–20 year lead time for the development of cervical cancer and the current availability of screening tests with a very high positive predictive value.
It must also be noted that ovarian cancer is not a single disease. Rather it encompasses many tumors of distinct histogenesis that arise from the ovarian surface epithelium, the gonadal stroma or germ cells (13). Approximately 90% of malignant ovarian tumors are of epithelial origin. These epithelial tumors are further classified by their predominant histologic differentiation pattern (Table 1). Serous carcinoma is the most common histologic type of epithelial ovarian cancer and is responsible for most deaths, and therefore is the focus of this review. However, much of what is discussed herein is likely to apply regardless of histotype. Genetic analysis of primary ovarian cancer and its metastases indicates that disease is clonal in >90% of epithelial ovarian cancer cases (14). There is no evidence of a causative infectious agent despite extensive efforts to identify one. Furthermore, while immunosuppressed patients are afflicted with cervical cancer at a greater rate than the immune competent, this effect is not apparent for ovarian cancer (15). This suggests that an infectious agent is probably not associated with ovarian cancer.
Table 1. Frequency and five-year survival rates according to histologic subtype of obviously malignant epithelial ovarian neoplasms.
Malignant tumors of the ovary derived from the celomic epithelim constitute 85% of all ‘ovarian cancers’.
| Histologic Subtype | Frequency (%)# | 5-year survival rate (%) from diagnosis* |
|---|---|---|
| Serous | 50 | 20–35 |
| Mucinous | 10–15 | 40–60 |
| Endometrioid | 10–15 | 40–60 |
| Clear cell | 2–5 | 35–50 |
| Undifferentiated | 15 | 15–20 |
Frequency information from Chapter 10 “Tumors of the Ovary: celomic epithelium” in Synopsis of Gynecologic Oncology 5th Edition edited by Morrow CP and JP Curtin. Information on very rare subtypes such as malignant Brenner cell, transitional cell and small cell carcinomas is omitted.
Survival data is taken from (13).
Molecular events of carcinogenesis
The molecular events underlying the genesis of the histologic subtypes of ovarian cancer are far from clear, and may differ between each. For high grade serous carcinoma, mutation of p53 is apparently a key early event in its development (16). Unfortunately, p53 mutation can occur by any of a large number of changes, or even loss of expression, suggesting that vaccinating against a particular p53 mutation could only potentially be effective against a very small subset of cases. However, mutant p53 is stabilized and therefore present at much greater levels than wild type. Furthermore, there are distinct variants of serous carcinoma (17). While conventional high grade (type I) serous carcinoma is associated with mutated p53, a more indolent form (type II) is associated with K-ras or B-raf mutation and wild type p53 expression. The type II form may represent as many as 30% of serous carcinoma cases, and unlike type I disease, is generally not sensitive to chemotherapy. The more indolent course and insensitivity to conventional chemotherapy suggest that type II serous carcinoma may be appropriate to target with immunotherapy. Thus even within a single histotype there is significant variation in key molecular events of carcinogenesis (17). This contrast to the molecular carcinogenesis in the uterine cervix, which is initiated by persistent oncogenic HPV infection in 99% of cases, has important implications (18). Since no infectious agent or consistent, required molecular change has been identified for ovarian cancer, it remains unclear which antigens can be successfully targeted by immunotherapy.
Targeting of pre-invasive or local tumors
There is no consensus on the nature or existence of precursor lesions of type I ovarian serous carcinoma (19). This is in stark contrast the well-defined histopathogenesis of cervical cancer; persistent oncogenic HPV infection of the epithelium typically at the transformation zone of the cervix progresses from a virion-producing low grade cervical intraepithelial neoplasia (CIN) to a less well differentiated high grade CIN which can then exhibit microinvasion and eventually frank metastasis (Figure 1). There has been much discussion as to where ovarian serous carcinoma originates (19). This may be the ovarian surface epithelium, the fimbriae of the fallopian tube or the mesothelium lining the peritoneum, or all of the above (17). The most common and deadly form of serous carcinoma (Type I) is aggressive and high grade even in the smallest lesions observed, and is likely directly shedding metastatic tumor cells that seed the peritoneal cavity at this early stage, resulting in rapid disease progression. There is some suggestion that apparently normal epithelial cells in the fallopian tube can exhibit mutant p53 expression, and that these may be precursors of class I ovarian serous carcinoma. Type II ovarian serous carcinoma appears to arise in stepwise fashion out of a benign serous cyst as an ‘atypical proliferative serous tumor’ and then acquires a more aggressive phenotype as a micropapillary serous carcinoma (17). The lack of well-defined precursor lesions for type I ovarian serous carcinoma prevents vaccination against the precursor and necessitates the development of therapies for advanced stage disease.
Figure 1. Morphological progression of squamous cell carcinoma of the cervix and high grade serous carcinoma of the ovary.
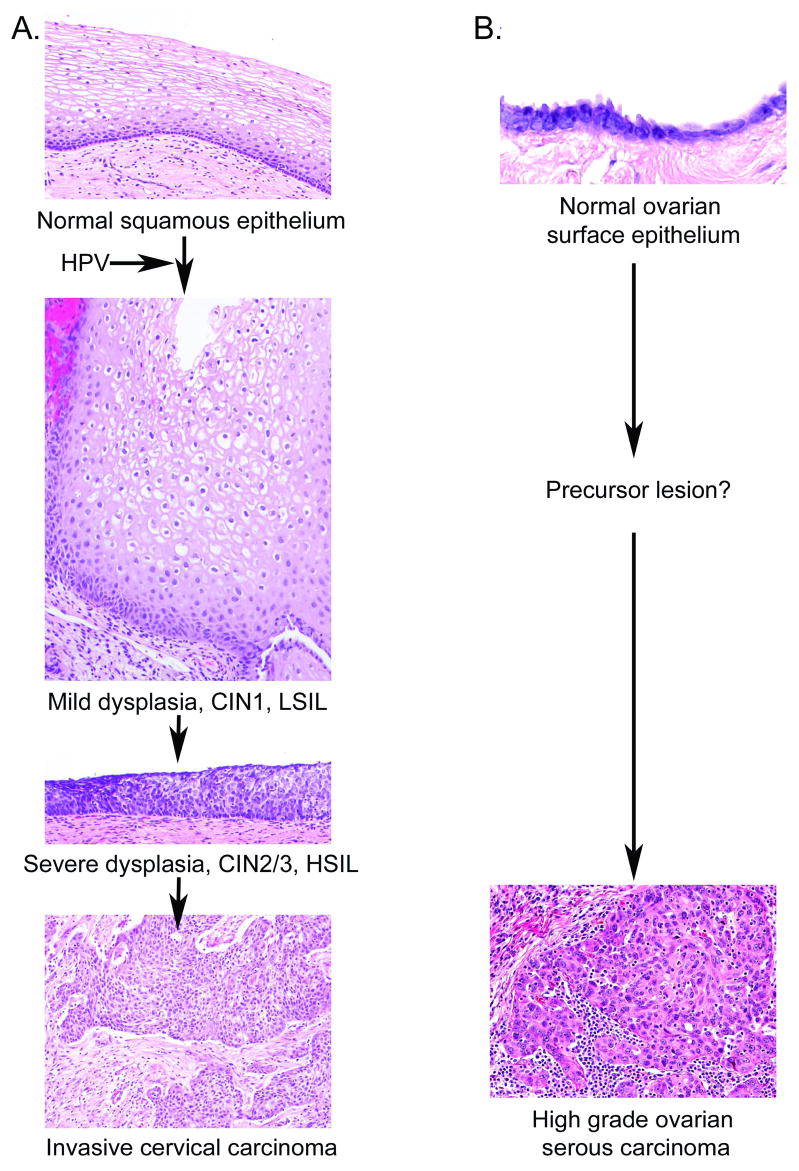
A. Cervical carcinogenesis is initiated by infection of the normal cervical squamous epithelium with an oncogenic type HPV and its persistance. Productive lesions produce mild dysplasia and are termed CIN1 or LSIL. These lesions become progressively less differentiated resulting in a severe displasia termed CIN2/3 or HSIL. Progression is associated with integration of the viral genome, loss of E2 expression and consequent upregulation of E6 and E7 expression, and genomic instability. Microinvasive carcinoma in situ metastasizes. B. Ovarian carcinogenesis. In contrast to cervical cancer, the timing of carcinogenesis, the cell types from which it arises, the critical molecular events and precursor lesions of high grade ovarian serous carcinoma are far less clear. It is believed to arise from the ovarian surface epithelium, or from the mesothelium or the fallopian tube. Certain hereditary genetic conditions, notably BRCA 1or 2 mutation, predispose women to ovarian carcinoma. Both the hereditary and sporadic forms exhibit p53 mutations and genomic instability (16,219).
This confused and potentially rapid pathogenesis of ovarian serous carcinoma is in contrast to cervical cancer, which may be addressed throughout its 10–20 year development since initial oncogenic type HPV infection (3). Oncogenic HPV infection can be prevented by vaccination with Gardasil or Cervarix, the multivalent HPV L1 virus-like particle (VLP) vaccines (2). Although these vaccines are highly effective for prevention, they do not alter the course of pre-existing infections. Pre-malignant HPV disease can be detected through cytological screening with a Papanicolou test for high grade CIN and more recently with a molecular HPV DNA test (Digene’s Hybrid Capture Assay, or Roche’s Line blot test). These high-grade CINs are treated by ablation, typically surgical conization or LEEP to remove the lesion and permit examination of the margins for microinvasion and completeness of resection. This treatment is generally curative, although conization is not an entirely benign procedure and may have implications for carrying pregnancies to full term. Invasive cervical cancer is initially treated with surgery including a hysterectomy and staging. For early stage disease this may be curative (approximately half of stage IB1 cervical cancers), but more advanced disease is currently treated with limited success using chemo- and radiotherapy.
Importance of innate immunity
Low grade CIN is not generally treated because the majority of infections spontaneously resolve over an average of 9 months. To speed their resolution, benign cutaneous warts and external genital warts are typically treated by creating local inflammation by freezing, local topical treatment with trichloroacetic acid or podophylotoxin (which is a potent irritant and induces complete clearance in 93% of cases with recurrence in 22%), and more recently with Imiquimod (1-{2-methylpropyl}-1H-imidazo{4,5-c}quinolin-4-amine) (20). Imiquimod is an immune response modifier that acts as a ligand for Toll-like receptor 7 and induces clearance of genital warts in 50% of cases with a 20% recurrence rate. Interferon has been used to treat for genital warts. Subcutaneous injection of interferon α2a or b (but not interferon beta 1) enhanced the spontaneous clearance of genital warts but did not enhance the success of treatment in combination with surgical removal of lesions, or in combination with podophyllotoxin. These clinical findings support the importance of modifying the local tumor microenvironment, and in particular targeting the innate immune system via TLR signaling and the production of type I interferon. No such approaches are available for the treatment of ovarian cancer.
Dendritic cell subtypes
HPV infection is not systemic, but localized in the discrete regions of the epithelium and is outside of the basement membrane. Ovarian carcinoma develops from the ovarian surface epithelium and/or mesothelium and possibly the epithelia of the fimbriae (19). The critical antigen presenting cell of the epithelium is the Langerhans cell, which has a distinct biology to other systemic dendritic cell subsets, such as myeloid and plasmacytoid dendritic cells with which ovarian cancer precursors might interact. However this difference is lost for metastatic cervical cancer with is directly available to the systemic immune system. HPV has apparently evolved to avoid detection by the innate immune system. HPV virions effectively activate immature myeloid dendritic cells (MDCs) and plasmacytoid dendritic cells (PDCs), but not Langerhans cells (21,22). Activation of murine MDCs is dependent upon MyD88, and TLR4 has also been suggested to contribute to recognition of HPV particles. Conversely, the failure of HPV virions to activate Langerhans cells is associated with PI3kinase activation. This difference is only relevant in CIN, because invasive cervical cancers do not express L1 or L2 capsid proteins, and they are exposed to MDCs and PDCs. Furthermore, HPV infection causes a reduction in the number of Langerhans cells at the site of infection. This is associated with reduced production of MIP3α by keratinocytes expressing E6/E7. Conversely, treatment of lesions with interferon is associated with a rapid influx of Langerhans cells. Furthermore, accumulation of a high density of Langerhans cells has been correlated with a favorable prognosis for cervical cancers. Taken together, the data point to the importance of Langerhans cells in mediating the clearance of pre-invasive HPV lesions. Furthermore, the success of destructive treatments in triggering regression of warts may reflect their exposure to MDC/PDC.
Immunosuppression demonstrates the importance of tumor immunity
As mentioned above, patients undergoing immunosuppressive therapy during to solid organ transplantation, and HIV+ patients present far more frequently with HPV-related lesions (15). Furthermore, these lesions are more severe and long lasting in the immunosuppressed and HIV+ patients as compared to otherwise healthy individuals. Regression of warts in immunosuppressed patients is frequently observed upon cessation of the immunosuppressive regimen. This effect is much more pronounced in pre-invasive disease suggesting that the influence of the immune system is less profound in more advanced disease (23). Indeed, regression of low grade CIN is a frequent occurrence, but regression rates are progressively lower for more advanced disease. In addition, the spectrum of HPV types that cause disease is also skewed in the immunocompromised. For example, HPV types that generally cause no disease in healthy individuals are associated with skin cancers in transplant and HIV+ patients at dramatically higher frequencies. The profound effect of immunosuppression clearly implicates the importance of antigen-specific T cell immunity in the control of HPV-related lesions, although the effect is less marked for invasive carcinoma (23). In contrast there is no evidence that higher rates of ovarian cancer are observed amongst the immunocompromised (15), there are no well-defined and identifiable precursor lesions of ovarian cancer in which to address this question. Nevertheless, some ovarian cancer patients fare better than others, and several studies relate different aspects of immunity to survival.
Immune cell subsets and their correlation with clinical outcome
Early clinical and histological studies by Tagami et al demonstrated that regression was associated with sudden and systemic onset of inflammation in every flat wart such that within 2–6 weeks they were completely involuted (24). Importantly, a mononuclear cell infiltration with epidermal invasion was observed in each biopsy further supporting the importance of cellular immunity in wart regression and probably provides a useful natural experimental model of rejection of CIN. Coleman et al described significantly more T lymphocytes and macrophages infilitrating regressing warts than non-regressing warts. CD4 T cells predominated in regression, both in the stroma below the wart and also within the epithelium, resulting in a significant increase in the ratio of CD4+ to CD8+ T cells (25). T cells in regressing lesions also exhibited a greater expression of activation markers, consistent with an “antigen-experienced” phenotype. However, the numbers of Langerhans cells in regressing versus persistent warts was unchanged. An induction of the immune accessory molecules HLA-DR and ICAM1 was observed on keratinocytes, as well as E-selectin and VCAM1 on endothelial cells in regressing as compared with persistent warts. These changes associated with wart regression are consistent with a delayed-type hypersensitivity response a foreign antigen, and this may contribute clearance of HPV lesions.
Grassegger et al investigated the cytokine expression patterns and immunohistochemical characteristics of persistent versus regressing anogenital warts (26). As described by Coleman et al, invasion of CD4 T cells into the warts and HLA-DR and ICAM-1 expression on keratinocytes was intensified in regressing lesions (25). The cytokine expression patterns were compatible with a predominant TH1 or balanced TH1/TH2 cytokine profile whereas these phenomena were not observed in recalcitrant warts. Indeed, recurrent warts were associated with IL4 and IL5 expression, suggestive of a TH2 response.
Trimble et al observed a high rate of spontaneous histologic regression of 28% in women with high-grade CIN with residual visible lesions after a colposcopically-directed biopsy within 15 weeks (27). Regression was associated with viral clearance, and this high rate may reflect biopsy-induced inflammation triggering anti-viral immunity. Women with HPV16+ only CIN2/3 were one third less likely to regress as compared to those with types other than HPV16 and HPV16+ high-grade CIN patients had similar outcomes regardless of HLA*A201 status. Conversely, for those women with CIN2/3 containing HPV types other than HPV16, those carrying HLA*A201 were the least likely to resolve. Such interactions between HPV type, HLA status, and disease regression are consistent with HLA-restricted HPV-specific CD8 T cell responses in effecting spontaneous clearance of disease. In the same cohort, Peng et al identified an HPV16 E7-specific CD4 T cell epitope (aa 71–85) restricted by HLA-DQB1*0201 (28). Analysis of systemic responses in HLA-DQB1*02 patients with HPV-16+ HSILs showed that the HPV16 E7 71-85 peptide-specific CD4 T cell immune response was significantly higher in women whose lesion had regressed as compared to those with persistent disease.
Cervical cancer is preceded by persistent HPV infection during which the host immune system fails to eliminate the virus, whereas most genital HPV infections and low-grade lesions are cleared. When de Jong et al. analyzed CD4+ T-helper responses to HPV16 E6, E7 and E2 antigens in healthy women they observed strong proliferative E2 and E6 (but typically not E7)-specific responses and secretion of both IFNγ and IL5 (29). Thus, the natural virus-specific immune response in patients who have cleared their HPV16 infections is consistent with a mixed Th1/Th2 response. In contrast, one half of the HPV16+ cervical cancer patients failed to mount a detectable Th response and the remainder demonstrated both weak HPV16-specific proliferative responses and typically an absence of IFNγ and IL5 secretion. Thus in contrast to the healthy patients who had presumably cleared or suppressed their prior HPV16 infections, the HPV16-specific CD4 T helper response in cervical cancer patients was either absent or severely impaired, despite a relatively intact responses against other recall antigens. This suggests that an appropriate CD4 T helper response is an important component of an effective cellular immune response against HPV disease. Thus, the failure of functional HPV-specific CD4 T helper responses in cervical cancer patients may contribute to the progression of incipient disease.
Importantly, CD4 T cells also play a critical role in self-tolerance. CD4 regulatory T cells suppress autoimmunity but also frequently suppress cancer-specific T cell immune responses. Van de Burg et al have demonstrated that CD4 regulatory T cells not only suppress responses to tumor-associated self-antigens, but also to HPV antigens expressed by cervical cancers (30). HPV-specific CD4 T cells isolated from lymph node biopsies of cervical cancer patients suppressed proliferation and IFNγ and IL2 production by responder T cells. This suppression was dependent upon their activation by cognate HPV antigen and on proximity to responder T cells. Furthermore, HPV-specific CD4 regulatory T cells were obtained from cervical cancer biopsies, suggesting that the possibility of CD4 regulatory T cell interference with the induction of anti-tumor immune response as well as the effector response. These observations provide a plausible explanation for the observed failure of the tumor-specific immune response in patients with cervical carcinoma.
Piersma et al examined both local and systemic tumor-specific immune responses in a prospective study of cervical cancer patients (31). They observed a significantly greater infiltration of the tumors by CD8 T cells, a higher CD8 to CD4 T cell ratio, and a higher CD8 T cell to regulatory T cell ratio in those patients with no evidence of tumor metastasis to the draining lymph node. Furthermore, the highest number of tumor-infiltrating CD8 T cells occurred in patients with no evidence of lymph node metastasis as well as a concomitant systemic tumor-specific immune response. CD8 T-cell infiltration of tumor was comparable in patients with no detectable systemic tumor-specific immune response regardless of lymph node status. These studies suggest that an anti-tumor cellular immune response is present in cervical cancer patients, especially in those with no evidence of lymph node metastasis and is consistent with their better prognosis.
Gambhira et al recently compared the immune responses of healthy volunteers and women with long term persistent HPV16+ vaginal and vulval intraepithelial neoplasia (VAIN and VIN) to vaccination with HPV16 L2-E6-E7 fusion protein (32). Although the vaccinations were not performed head-to-head, significantly weaker HPV-specific humoral and T cell responses were observed in the VAIN/VIN patients as compared with the healthy women. These findings suggested that the persistent VAIN/VIN patients exhibit compromised HPV-specific immunity, possibly due to immune suppression mechanisms similar to those discussed above.
In sum, clearance of premalignant HPV lesions and better prognosis for cervical cancer is associated with a robust antigen-specific CD4 T helper 1 or balanced Th1/Th2 type response and activated CD8 CTL cell infiltration but minimal or no immunosuppression by CD4 T regulatory cells. In contrast, cervical cancer is associated with a robust infiltrate of antigen-specific CD4 T regulatory cells and weak HPV-specific T cell responses.
Immune responses influence outcome of ovarian cancer
CD4 regulatory T cells maintain peripheral tolerance by suppressing self-reactive T cell responses. Tumor development and progression may be a consequence of the suppression of a patient’s anti-tumor immunity by tipping the balance towards tumor-associated antigen-reactive CD4 regulatory T cell responses. Ovarian cancers are able to prime tumor-specific T cell immune responses that can be detected in peripheral blood, as tumor infiltrates, and in the lymph nodes of patients. Purified TAA–specific T cells can effectively lyze autologous ovarian cancer cells ex vivo and such functional T cell lines have been established. In addition, increases in TAA–specific cytotoxic CD8 T cells have been described in ovarian cancer patients yet there is little evidence of disease regression, only differences in survival. This suggests that TAA–specific T cells might be functionally inhibited in cancer patients and that approaches to vaccination will have to address these suppressive mechanisms to effect tumor rejection (for review, see (33)).
Zhang et al correlated increased tumor infiltration by T cells with improved survival in epithelial ovarian cancer (34). Sato et al. (2005) observed that patients with higher frequencies of infiltrating CD8 T cells demonstrated longer survival as compared to those women with less significant infiltrates (35). No correlation was observed for other subtypes of intraepithelial or stromal tumor-infiltrating lymphocytes. Those patients with a high CD8 to CD4 tumor-infiltrating lymphocyte ratios were strongly associated with prolonged survival, suggesting that CD4 T cell subsets regulate CD8 T cell immunity. A shortened survival associated with CD4 T cells was also associated with high levels of CD25+ FOXP3+ CD4 regulatory T cells. Unfortunately, the beneficial effects of CD8 T cell infiltration on patient survival did not correlate with concurrent expression of several known serologically defined TAAs, including NY-ESO-1 and MAGE antigens.
Curiel et al found evidence that CD25+FOXP3+ CD4 regulatory T cells in patients with ovarian carcinoma suppress tumor-specific T cell immunity and contribute to growth of human tumors in vivo (36). These CD25+FOXP3+ CD4 regulatory T cells are associated with reduced survival and preferentially infiltrate tumors and ascites, but were rarely found in draining lymph nodes in late stage cancer. Both tumor cells and macrophages produce the chemokine CCL22 within the tumor microenvironment, which attracts infiltrates of regulatory T cells thereby potentially contributing to local immune suppression. Overcoming this suppression by regulatory T cell infiltrates is clearly a challenge for cancer vaccination strategies.
Wei et al studied the interaction between dendritic cells and T cells in the tumor environment of patients with ovarian carcinoma (37). Immature myeloid dendritic cells infiltrate in ovarian tumors and ascites and induce TAA-specific CD8 T cells with effector function. However, plasmacytoid dendritic cells (PDCs) also accumulated in ovarian tumors and induced, independently of CD25+ CD4 T cells, IL10-producing CD8 regulatory T cells. The IL10 produced by these CD8 regulatory T cells significantly suppresses MDC-induced TAA-specific T cell effector functions. These CD8 regulatory T cells also express functional CCR7, and efficiently migrate with lymphoid homing chemokine MIP-3β. Thus, tumor-associated PDCs can contribute to suppressive environment within the tumor by inducing these IL10-producing CD8 regulatory T cells.
In addition to suppression via release of cytokines such as IL10, direct interaction with B7 family co-suppressor molecules such as B7-H1 and B7-H4 may contribute to the failure of TAA-specific immunity. A subset of MDCs express B7-H1 on their surface and its expression could be further elevated by tumor-derived factors. Indeed most MDCs isolated from the tissues or draining lymph nodes of ovarian carcinoma patients express B7-H1. Blockade of B7-H1 enhances MDC-mediated T-cell activation and reduces IL10 production of IL10 by CD4 T cells and increased production of IL2 and IFNγ. Pretreatment of T cells with B7-H1-blocked MDCs potentiated their ability to control autologous human ovarian carcinoma growth upon reconstitution in SCID-NOD mice. These findings suggest that the tumor environment triggers elevated B7-H1 expression on MDCs which directly interacts with T cells to suppress TAA-specific immunity and that of B7-H1 in vivo may potentiate cancer immunotherapy.
In addition to the B7-H1-dependent suppression, both ovarian tumor cells and infiltrating macrophages express B7-H4; the latter acts with CD25+FOXP3+ CD4 regulatory T cells to suppress TAA-specific T cell immunity. Kryczek et al examined B7-H4 expression in ovarian carcinoma and tumor-associated macrophages and observed that the intensity of B7-H4 expression in macrophages was significantly correlated with the numbers of regulatory CD4 T cells within the tumor (38). Furthermore, elevated numbers of regulatory CD4 T cells and elevated macrophage B7-H4, but not tumor B7-H4 expression, were associated with shorter patient survival. The regulatory CD4 T cells triggered production of IL10 and IL6 by the tumor-infiltrating macrophages, which then elevated their B7-H4 expression via IL10 and IL6 autocrine signaling. Since tumor-associated macrophages secrete the chemokine CCL22 that attracts regulatory T cells to accumulate within the tumor, and then induced B7-H4 on adjacent antigen-presenting cells including macrophages, these observations suggest a mechanism for suppression of antigen presentation in human ovarian cancer.
In addition to secretion of immunosuppressive cytokines, and interactions with co-suppressor molecules, there are a number of other potentially immunosuppressive activities of MDCs in ovarian cancer patients. These include the production of arginase, and inducible nitric oxide synthetase (iNOS). However, Kryczek et al (2006) found that blockade B7-H4 signaling, but not arginase, iNOS or B7-H1 restored the T cell stimulating capacity of the ovarian tumor associated macrophages and contributes to tumor regression in vivo (39). Furthermore, overexpression of COX-2 and iNOS in ovarian carcinoma is not significantly associated with survival (40), and other groups find their expression is associated with improved outcome (41,42).
Given the extensive evidence that the immune system can control tumor growth and possibly even eliminate cancers described above, there has been considerable effort placed upon the development of cancer vaccines. Many approaches to vaccine development are possible, including whole tumor cell vaccines. Since here we focus upon the development of antigen-specific immunotherapies for gynecologic cancers, we examine firstly the selection of an appropriate TAA, secondly, selection of an appropriate preclinical model and thirdly, the construction of a vaccine for delivery of the TAA and preclinical testing.
1. SELECTION OF TAAs
The choice of target TAA is clearly central in therapeutic vaccine design. The ideal antigen for immunotherapy should be 1) non-self (i.e. a foreign antigen like HPV proteins) or not tolerized, 2) expressed only in the tumor, 3) common to all tumors of this type, 4) required for tumor viability, so that it cannot be lost (e.g. E6 and E7 since cervical cancer cells die in their absence), 5) immunogenic, and 6) cell surface (i.e. can be targeted by antibody). Below we describe several approaches to the identification of ovarian tumor antigens that might be useful in antigen-specific vaccine therapies.
TAAs in cervical cancer
The HPV early proteins (E1-E7) are obvious target antigens for cervical cancer since they are expressed throughout the viral life cycle, are present only in diseased cells, and help regulate progression of the disease. In particular, the HPV-encoded proteins E6 and E7 represent ideal targets for the development of therapeutic cervical cancer vaccines. Firstly, E6 and E7 proteins are constitutively expressed by HPV-associated tumors, and their expression is typically upregulated due to the loss of E2 repression during viral integration. Secondly, because E6 and E7 are critical for the induction and maintenance of cellular transformation in HPV-infected cells, it is unlikely that the tumor cells can escape immune attack through antigen loss. Thirdly, since E6 and E7 are foreign proteins, immunization against HPV-associated tumors circumvents some common cancer vaccine-associated problems such as immune tolerance. Thus, many therapeutic HPV vaccine strategies have focused primarily on stimulating the production and activation of T cells by targeting E6 and/or E7 proteins. One potential disadvantage of E6 and E7 as targets of immunotherapy is that they are not accessible on the cell surface. Thus cellular immune responses are likely to be critical for successful vaccines targeting E6 and E7.
Defining TAAs for ovarian cancer
A plethora of approaches have been used to identify potential TAAs by screening for genes commonly expressed by cancer cells and absent from normal cells. These include differential display, microarray and SAGE analyses that generate long lists of genes showing differential expression, but it is unclear how to select appropriate proteins that are immunogenic. Another approach has been to generate monoclonal antibodies from mice vaccinated with ovarian cancer cells or membranes. Monoclonal antibodies are then selected on their ability to react with a large fraction of cancer cases, and an absence of reactivity with normal tissues. One of the most important ovarian cancer antigens identified by generating a monoclonal antibody to tumor cells is CA125 (11). The CA125 test is approved to monitor disease recurrence for ovarian cancer patients after treatment, the value of CA125 as a vaccine antigen is less clear. MUC16 encoding the CA125 antigen is a very large gene, and produces a highly glycosylated protein that is shed into the milieu. Furthermore, there is little evidence that it is immunogenic and no description of CA125-specific autoantibodies suggesting that it may not be an ideal therapeutic vaccine antigen. Thus this approach may identify molecules that are over expressed on the cancer cell surface but like many of the gene profiling and proteomic approaches, it does not specifically target those that are immunogenic in the host.
Identification of the target antigens recognized by cancer-specific T cell lines
Another more direct approach to antigen identification has been the generation of cytotoxic or helper T cell lines that selectively kill tumor cells. The antigen recognized by these T cell lines can be identified using proteomic or genetic approaches. Specifically, the relevant HLA molecule is immunoprecipitated from detergent lysates of the cell line and the peptide epitope is eluted and sequenced by mass spectrometry. For example, the naturally occurring peptides associated with HLA-A2 in ovarian cell lines that are identified by mass spectrometry would be the targets of HLA-A2-restricted cytotoxic T cells (43).
Alternatively, the antigens may be identified by transduction of cell lines insensitive to killing by the CTL line using a cDNA expression library derived from the tumor cells killed by the CTL line. Reverse immunology, that is the procedure of predicting and identifying immunogenic peptides from the sequence of a gene product of interest, is a particularly efficient approach for the discovery of tumor antigens (for review see (44)).
Screening for TAAs with autologous antibody
Cancers arise through accumulation of a series of genetic and epigenetic changes that disrupt normal control of cell growth (45). These molecular changes cause aberrant gene expression and alteration in the structure, function and immunogenicity of mutated gene products. The immune system constantly surveys the body for such novel, ‘non-self’ antigens, and generates a response in the appropriate context. Thus many cancer patients produce a humoral response against tumor-associated antigens (46,47). Autologous antibodies have been documented in patients afflicted with a variety of different cancers, including melanoma, and cervical, breast, head and neck, colon, lung, and renal cancer patients (48–50). Ovarian tumor-reactive antibodies have been detected in patient serum and ascites (51). Tumor-associated antigens are expressed both on tumor cells and on certain normal cells, and in certain cases are lineage-specific antigens, such as Epcam and mesothelin in ovarian cancer. Despite the frequent expression of TAAs in normal tissues (often testis or embryonic), the antibody responses to many TAAs are generally confined to patients with cancer. These autoantibodies can be utilized to identify TAAs and may perhaps also be used for a successful detection test. It is unclear what triggers the immune recognition of such TAAs, although investigators have suggested expression over a critical threshold or in an inappropriate location, or in the context of apoptotic/necrotic cells break tolerance.
Autologous antibodies generated by cancer patients have been used to identify TAAs by several approaches. Many TAAs have been cloned by screening cancer cDNA expression libraries for serum antibody derived from a cancer patient. This technique, originally described by Sahin et al and termed SEREX (serological identification of antigens by recombinant expression cloning), has been used to identify numerous TAAs that may be categorized as shown in Table 2. SEREX has been applied to tumors of many organs (52) and antibody specific for antigens identified by SEREX in other cancer types have been demonstrated in ovarian cancer patients (53,54).
Table 2. A selection ofexample TAAs that generate autoantibody in patients with gynecologic and other cancers.
(adapted from (46)). Many TAAs have been documented in the cancer immunome database (http://www.licr.org/D_programs/d4_immunology.php).
| Antigen type | TAA | Tumor type | Reference |
|---|---|---|---|
| Mutational | p53 | Ovarian cancer | (50) |
|
| |||
| Differentiation | Epcam | Ovarian cancer | (220) |
| Mesothelin | Ovarian cancer | (71) | |
|
| |||
| Post-translational modification | MUC1 | Ovarian cancer | (221) |
| Cathepsin D | Ovarian cancer | (58) | |
|
| |||
| Amplified/over-expressed | Her-2 | Breast cancer | (222) |
| HSP90 | Ovarian cancer | (223) | |
| Folate receptor | Ovarian cancer | (224) | |
| HoxB7 | Ovarian cancer | (225) | |
|
| |||
| Viral | HPV E6 | Cervical cancer | (226) |
| HPV E7 | Cervical cancer | (227) | |
|
| |||
| Splice variant | Restin | Hodgkins disease | (48) |
| NY-CO-38 | Colon cancer | (50) | |
|
| |||
| Cancer/testis | MAGE-1 | Melanoma | (54) |
| NY-ESO-1 | Ovarian Cancer | (54) | |
Another approach is to identify TAAs is display of the cancer cDNA library on T7 phage. The recombinant phages are then subject to bio-panning with patient serum (55,56). However there are disadvantages of these prokaryotic screening systems. Firstly, the antibodies may recognize specific protein conformations that are not faithfully recapitulated in prokaryotes. Secondly, they would fail to detect antibodies to secondary modifications of proteins that are associated with cancer (57). Therefore, several investigators have used human serum antibodies to directly immunoprecipitate TAAs from tumor cells. The TAAs are separated on one (58) or 2D gels (57) followed by identification of the TAA using mass spectroscopy of tryptic peptides. The disadvantage of this approach is that the TAA gene must then be cloned once it has been identified by mass spectrometry. A recent study employed such an immunoproteomics approach to identify and categorize antigens that are involved in both humoral and cell-mediated immunity against ovarian cancer (59). They used mass spectrometry to identify novel autoantibody-based serum biomarkers for ovarian cancer by immunoprecipitating TAAs by autoantibodies from the sera of cancer patients. They also identified the endogenous MHC-presented peptides from ovarian tumor cell lines and correlated the results of the two approaches, they were able to identify common antigens that were involved in both humoral and cell-mediated immunity (59).
Autologous antibodies specific for TAAs are prevalent in cancer patients but absent from controls, and therefore have potential as serum biomarkers (54). The prevalence of autologous antibody responses to TAA is variable in cancer patients and dependent upon the antigen. A proportion of both patients and controls have antibodies to some SEREX-identified autoantigens e.g. restin (48). Often these autoantibodies occur in other contexts unrelated to cancer e.g. anti-NY-CO-40/hsp70 antibodies have been identified both in patients with colon cancer and in those with ulcerative colitis (50). Indeed, an initial survey of 20 antigens identified from colon cancer by SEREX revealed that autologous antibodies to 14 were present in patients and controls (50). These autologous antibodies may result from some process other than carcinogenesis.
In contrast, cancer patients but not healthy controls generate antibody to other SEREX-identified antigens. The relationship between carcinogenesis and most tumor-specific antigens identified by SEREX is unclear. However autologous antibodies have been demonstrated in cancer patients specific for such regulators of cell growth as mutant p53, ras, c-myc, c-myb, and Her-2/neu (60). This is clear in the case of cervical cancer; the majority of cervical cancer patients generate antibody specific to HPV E6 and E7. Viscidi et al tested sera of women with invasive cervical cancer associated with HPV16 or other HPV types and women with HPV16-associated CIN3 or controls. Serum antibodies to HPV16 E6 and E7 proteins were detected by radio-immunoprecipitation of in vitro translated proteins in 56% and 43%, respectively, of invasive cases and 1.7% and 4.1%, respectively, of controls. Antibodies to either E6 or E7 protein were present in 72% of sera from invasive cases and 5.8% of sera from controls and high titer antibody was found almost exclusively in invasive cancer cases. Importantly, the frequency of antibodies to the E6 protein and the E7 protein among CIN3 cases did not differ significantly from the controls, suggesting that the E6/E7-specific antibody response is associated with invasive disease (61).
Typically only a faction of cancer patients generates antibody to one particular TAA. For example only 10–30% of cancer patients generate antibody to the SEREX antigen NY-ESO-1 but this likely reflects the small proportion of tumor expressing this antigen, rather than infrequent immune response to cancer antigens. Although 10% of melanoma patients produce NY-ESO-1-specific antibody, only 20–40% of melanomas express NY-ESO-1. This suggests that patients with melanoma that express NY-ESO-1 frequently generate autoantibody to this antigen (54). NY-ESO-1-specific autologous antibody was only detected in those patients with melanoma that expressed NY-ESO-1. Neither controls nor patients with melanoma that did not express NY-ESO-1 had autologous antibody specific for NY-ESO-1, demonstrating the specificity of this assay (54). NY-ESO-1 expression is not limited to melanoma. We detected antibody to NY-ESO-1 in sera of 33/85 ovarian cancer patients and 0/23 controls, similar to previous findings (54). However, the proportion of responders likely differs between antigens. Other tumor autoantigens that are common among ovarian cancer patients have been identified; 48% of ovarian cancer patients generate antibody to cathepsin D and 40% to glucose-regulated protein 78 (GRP78) (58). Neither ovarian cancer antigen was recognized by antibody in sera of normal controls. Furthermore, it appears that both of these antigens contain tumor-specific epitopes since neither cathepsin D nor GRP78 derived from normal tissue were recognized by sera from ovarian cancer patients (58).
Perhaps the autoantigen best studied in ovarian cancer patients is p53 (62). Circulating antibodies against p53 were identified in patients with ovarian cancer and correlated to clinical and pathologic features and survival. Approximately 24% of sera from ovarian cancer patients had antibody to p53 (63,64). In stage I/II ovarian disease 22% of patients had antibody, 31% in stage III and 50% in stage IV (65). However, this difference may reflect changing ratios of serous carcinoma by stage rather than less frequent induction of p53-specific antibody by early versus late stage disease. Although there was no association of p53 antibody with clinical stage, tumor histologic type or overall patient survival (63,66), detection of autologous antibody to some ovarian cancer antigens appears to have prognostic significance (67). However, the clinical significance of detection of tumor-specific autologous antibodies requires further analysis and must be assessed antigen by antigen.
In sum, each approach to the identification of TAAs has advantages, and they are not necessarily mutually exclusive. Indeed, several antigens have been identified by multiple approaches and these might be considered the most promising. For example, mesothelin was identified first using monoclonal antibodies, but has also been identified serologically as an autologous TAA in ovarian cancer patients, and has been demonstrated as a target of cancer-specific T cell lines. Thus, while there is general consensus that targeting of HPV E6 and E7 is most appropriate for cervical cancer immunotherapy, it is far from clear which TAAs are appropriate for vaccines against ovarian cancer. While we have described several interesting candidates above as examples (and the list is not intended to be exhaustive), here we will focus on one TAA, mesothelin, which we believe shows promise as a target in ovarian cancer and is the focus of many of our vaccine studies.
Mesothelin
Mesothelin is a 40 kDa surface glycoprotein that is proteolytically cleaved from a 70 kDa precursor. This cleavage also releases a 32 kDa soluble fragment called the megakaryocyte potentiating factor (MPF) that may be useful in diagnosis. Mesothelin is anchored on the cell surface by a glycosylphosphatidyl inositol linkage. Its exact function is unclear since knockout of mesothelin does not produce any phenotype in mice. However, there is evidence that mesothelin plays a role in cell adhesion by interaction with MUC16/CA-125 via its N-linked oligosaccharides. Binding of mesothelin with MUC16 occurs with high affinity (kd ~5nM) and enhances cell adhesion between MUC16 and mesothelin expressing cells. It has been hypothesized that the MUC16-mesothelin interaction may play a role in the peritoneal spread of ovarian cancer.
Importantly, mesothelin is a differentiation antigen whose expression is limited to mesothelial cells covering the pleura, pericardium and peritoneum in the healthy patient, whereas mesothelin is widely expressed in ovarian cancer, especially serous carcinoma. In a large study by Yen et al., mesothelin expression was detected in 55% of ovarian serous carcinomas, although other studies have described up to 100% of cases expressing mesothelin (68,69).
Hassan et al. detected circulating mesothelin in 77% of sera from patients with ovarian cancer, but not in sera of healthy volunteers (70). Ho et al. identified autologous mesothelin-specific antibody in the sera of 10 of 24 ovarian cancer patients and 0 of 44 controls (71). Over a half of all patients whose tumor expressed mesothelin generated a detectable humoral response. Furthermore, mesothelin-specific CD8+ T cell responses in cancer patients with vaccine-induced DTH responses were found to correlate with prolonged disease-free survival, suggesting that mesothelin might represent an important target antigen for active immunotherapy (72). Thus, mesothelin is antigenic in cancer patients and has therefore received considerable attention as a target for immunotherapy.
In contrast to E6 and E7, mesothelin is accessible on the cell surface and therefore can be targeted using monoclonal antibodies for antibody-dependent cell-mediated cytotoxity (ADCC). Mesothelin, however, is not uniquely expressed by the cancer cells. It also represents a self-antigen and there is no evidence of mutation or aberrant modification in cancer suggesting that tolerance must be broken to generate an effective cellular immune response. There is also no demonstration that mesothelin is required for the viability of cancer cells, although the interaction between CA125 and mesothelin suggests a potential role in cancer cell adhesion or spread. Furthermore, because mesothelin deficient mice show no apparent phenotype, a tumor might be able to evade a mesothelin-specific cellular immune response through selection against mesothelin expression (73).
2. PRECLINICAL TESTING
Cervical cancer models for vaccine testing
Once the relevant TAA has been selected for vaccine development, it is critical to have representative animal models for pre-clinical testing and optimization. There are several important models for developing therapeutic HPV vaccines. The most commonly used are mouse-derived cancer lines expressing HPV oncoproteins. Examples include TC-1, which was derived by transforming lung cells with HPV16 E6 and E7 and activated H-ras, or the C3 line that was transformed using the full-length HPV16 genome, both in the C57BL/6 background. Because of the availability of reagents and genetically modified mice, these two models have become mainstays for therapeutic vaccine development. Similar cell lines are available in the guinea pig, but less frequently used. Unfortunately, several vaccines that have proven highly effective in the murine models for protection against tumor challenge have not proven efficacious in cancer patients. This suggests that these transplantable models should be used differently; for example vaccines should be tested for their ability to induce regression of sizable pre-existing tumors. Other models including prevention or treatment of E6/E7-expressing tumors in spontaneous models and the rejection of E6/E7-expressing transgenic skin grafts from wild type mice after vaccination are being developed. These may also be better able to predict the clinical outcome of therapeutic vaccine trials. E6/E7-transgenic mice are a challenging model because they are likely tolerized.
Another important model is vaccination to eliminate squamous skin cancer that develops in a significant fraction of domestic rabbits infected with cottontail rabbit papillomavirus (CRPV). This model recapitulates many more aspects of HPV-related cancer, but utilizes a different, though related, virus (CRPV) and there are many fewer reagents and genetically modified animals. There are a number of animal papillomaviruses that have been used to model cervical cancer, such as BPV4 that induces alimentary tract tumors synergistically with the bracken carcinogen quecertin, but they are not commonly used and are costly.
Ovarian cancer models for vaccine testing
One of the major limitations to the development of ovarian cancer immunotherapies is the difficulty of generating ovarian cancer models. Without suitable ovarian cancer models in immune intact mice, it is difficult to bring new vaccines for ovarian cancers into the clinic. Mouse ovarian surface epithelial cells (MOSEC), in immune intact mice were developed by Roby at al (74). MOSEC ovarian cancer cells were created by isolation of mouse ovarian surface epithelial cells and in vitro culture for more than 20 passages. Injection of these cells into the peritoneal cavity of immune intact mice resulted in the formation of ascitic fluid and multiple tumor implants. However, the genetic events underlying the spontaneous transformation of MOSEC cells are unclear, and the histopathology of these tumors does not resemble serous carcinoma. In addition, a more aggressive ovarian cancer line, Defb29Vegf was generated by stable transfection of VEGF and defensin in the MOSEC cell line (75) because of the slow development of tumor utilizing the parental line.
Another approach taken by Cheng et al. to generate a mouse model for immunotherapy studies has been to transform peritoneal cells of C57BL6 mice with HPV16 E6, E7 and v-Ha-ras (76). These cells were injected i.p. into immunocompetent mice, and subsequently passaged multiple times as ascites. While ovarian cancer does not contain HPV or generally exhibit Ha-ras mutations, this model does express mesothelin at high levels.
Another major limitation for the development of ovarian cancer therapies is the determination of ovarian tumor loads in the peritoneal cavity of mice. Without accurate methods to measure tumor loads in the peritoneal cavity, it is difficult to assess the effects of therapies at an early time point and investigators have to depend on measuring the body weight or the abdominal girdle of the tumor challenged mice or measuring the survival rate of the therapies. Recently, several groups (77,78) have developed non-invasive luminescence images to measure the amount of ovarian tumors in the peritoneal cavity of mice. They transduced the luciferase gene into mouse ovarian cancer lines including Mosec and Defb29Vegf and found that luciferase activities correlated with the tumor loads of ovarian cancer injected in the peritoneal cavity of mice. Furthermore, the luminescence activity was shown to correlate well with mouse survival rate.
A spontaneous ovarian cancer mouse model has been generated (79). The transgenic mice express the transforming region of SV40 under the control of the Mullerian inhibitory substance type II receptor gene promoter typically bilateral ovarian tumors. The MISIIR transgenic micehave been shown to develop ovarian carcinomas spontaneously within 6–13 weeks after birth (79). It is also seen that 100% of the MISIIR transgenic mice developed ovarian tumors within 4 months after birth. Unfortunately T antigen is not an antigen relevant to ovarian cancer and these tumors do not replicate the histopathology of serous carcinoma.
Another approach has been to introduce known mutations into the mouse ovarian surface epithelium using recombinant avian retroviruses. Orsulic et al. (2002) utilizedovarian surface epithelial cells from mice transgenic for the avian receptor TVA to deliver multiple vectors expressing human oncogenes (80). Target cells that were derived from TVA transgenic mice deficient for p53, were transformed by the introduction of any two of the oncogenes c-myc, K-ras, and Akt. These cell lines formed ovarian tumor formed upon injection subcutaneously, or within the peritoneum or ovaries. Xing and Orsulic (2006) utilized the same approach to generate primary mouse ovarian surface epithelial (OSE) cell lines lacking functional Brca1 and p53, as is thought to occur in the hereditary ovarian cancer patients (81). Importantly the introduction of Myc is sufficient to transform murine OSE cells that lack both Brca1 and p53, but interestingly is not sufficient to transform OSE deficient for either Brca1 or p53. Further, immunocompetent mice injected with the Myc transformed Brca1 and p53-deficient OSE cells develop tumors that are histologically similar to metastatic serous ovarian carcinoma in patients, suggesting that this is an important new model for the evaluation of vaccines therapies that target serous carcinoma. However, this is not a spontaneous model, but requires ex vivo culture.
Some progress has been made for ovarian endometriod adenocarcinoma (82). Recent studies show that mutation in the Wnt/Beta-catenin and PI3K/Pten pathways frequently occur in human endometriod carcinoma. Disruption of these two pathways in the murine ovarian surface epithelium by conditional inactivation of the Pten and Apc tumor suppressor genes results in the synergistic formation of adenocarcinomas that are morphologically consistent with human ovarian endometriod adenocarcinomas. The penetrance is complete and the tumors exhibit a short latency and rapidly progress to metastatic disease in ~ 75% of mice. The biological behavior and gene expression patterns of the murine cancers are consistent with human ovarian endometriod adenocarcinoma with defects in the Wnt/beta-catenin and PI3K/Pten pathways. A similar mouse model has been developed by targeting K-ras and Pten (83).
Unfortunately, these murine models do not necessarily model the human immune system, and it is unclear if these models express similar TAAs as the human disease. These problems could be partially addressed in mice carrying components of the human immune system, such as HLA molecules, and by generating mice that are transgenic for known human ovarian tumor-associated antigens. For example, the latter approach has been used in rats transgenic for human Her-2/neu as a model of breast cancer.
Finally, another possible approach partially reconstructs the human immune system in female immunodeficient mice. Primary ovarian (or cervical) tumor cells are injected and tumor size is monitored after challenge. However, this approach is limited by the availability of human tissues and the success of vaccination has not been tested.
3. APPROACHES TO IMMUNOTHERAPY
Immunotherapeutic strategies aim to eliminate preexisting lesions and even malignant tumors by generating cell-mediated immunity and in some cases humoral immunity against TAAs. Current approaches include live vector-based, peptide- and protein-based, nucleic acid-based, and cell-based vaccines. Table 3 summarizes the strengths and weaknesses of each strategy. This review discusses the future directions of therapeutic vaccine approaches for the treatment of established gynecologic malignancies, with emphasis on current progress of TAA-specific vaccine clinical trials.
Table 3.
Summary of the pros and cons of the numerous approaches to therapeutic cancer vaccines
| Strategy | Pros | Cons |
|---|---|---|
| Passive Antibody | Well tolerated Simple production |
Surface exposure required Repeated infusion Neutralizing antibody Cost |
| Peptide vaccine | Well tolerated Direct synthesis Stable |
Weak immunogenicity HLA restriction |
| Protein vaccine | No HLA restriction Easy to produce |
Strong adjuvant needed Better induction of antibody than CTL response Cold chain and injection |
| Live Vector | High immunogenicity Wide variety of vectors available Simple delivery |
Potential pre-existing immunity Neutralizing Ab inhibit boosting Risk of toxicity Potential to spread Dominance of viral epitopes |
| Naked DNA vaccine | Simple to produce, store and transport Multiple boosts possible Extended Ag production |
Low immunogenicity Integration into host genome Best delivery method unclear |
| RNA-based vaccines | Non-infectious Multiple immunizations possible RNA replicons replicate in the cell and enhance Ag expression |
Unstable Difficult to produce Delivery method unclear |
| Dendritic cell vaccines | High immunogenicity Generation of large quantities of DCs |
Cost Labor-intensive Massive tissue culture |
| Whole Tumor cell vaccines | Express TAAs in relevant form Valuable when TAA is unknown |
Safety concerns Difficult to produce Weak Ag presentation by tumor cells |
Antibody-based Therapy
Conceptually, the simplest approach to antigen-specific immunotherapy is passive infusion of TAA-specific antibodies. In this case, however, the antibody must recognize a TAA that is accessible on the surface of the tumor cell. This approach may lead to the killing of tumor cells by Antibody-Dependent Cell-mediated Cytotoxicity (ADCC) mediated by Natural Killer (NK) cells, via antibody-dependent fixation of complement factors and their induction of cell lysis (CDC), and possibly by blockade of a critical signal. One prominent example of this approach is the use of Herceptin to target Her-2 on the surface of breast cancers. If antibody alone is not sufficient for a therapeutic effect, then toxins, radioisotopes or other cytolytic agents may be directly coupled to the antibody and thereby concentrated on the surface of tumor cells. Potential downsides of this approach are the constant requirement for infusions, the development of neutralizing antibody responses against the immunotherapeutic agent, inhibition by soluble TAA shed from the tumor surface and low tumor uptake. The development of antibodies to the immunotherapeutic monoclonal antibody is minimized by its humanization (e.g. Herceptin), or development of fully human monoclonal antibodies to the TAA. NK cells express the Fcγ RIII receptor (CD16) on their surface which binds with high affinity to the immunoglobulin isotypes IgG1 and IgG3. This mechanism does not kill all cells expressing the TAA on their surface. NK cell killing is activated by the recognition of an ‘altered-self’ in the target cell. An ‘altered-self’ is determined by the balance of interactions between activating and inhibitory NK cell receptors that recognize MHCI molecules and carbohydrate on the target cell.
In the case of cervical cancer, the only HPV antigen that has the potential to be accessible on the cell surface is the oncogene E5. E5 is predominantly associated with the golgi apparatus, but binds to the PDGF and EGF receptors inhibiting their recycling, thus elevating surface levels. Interestingly, E5 also reduces surface levels of MHC class I, suggesting that cervical cancer cells would activate NK cells if targeted with a specific antibody. However, E5 is a very small protein, and is predominantly buried within the lipid bilayer. As a consequence it is poorly immunogenic and little if any is available for antibody binding on the cell surface, and thus it has proven a poor target to date. Therefore targeting of another TAA might be required to target cervical cancer by this approach. One possibility is targeting with the EGF-R1 specific monoclonal antibody Cetuximab, since E5 upregulates surface expression of this receptor and promotes cell growth through prolonged activation of this receptor (84).
Unfortunately, neither Her-2 nor EGFR are frequently over-expressed in ovarian cancer. The folate receptor α shows promise and a humanized monoclonal antibody has recently entered clinical trial in ovarian cancer patients (85). Mesothelin has also been targeted with monoclonal antibody therapy. Initial efforts utilized the monoclonal antibody K1 with which mesothelin was initially identified. Unfortunately this antibody is of low affinity and produced no antitumor effects in early studies. K1 coupled to the toxin PE38, derived from Pseudomonas endotoxin A, exhibited some antitumor activity, but its low affinity and large size reduced tumor penetration. Therefore, a single chain Fv of higher affinity for mesothelin was developed by phage display and coupled to PE38. This product, termed SS1P, was tested in 34 patients with tumors expressing mesothelin (including 12 with ovarian cancer). The treatment was generally well tolerated, but self-limited pleuritis was the dose limiting toxicity (presumably caused by SS1P binding to mesothelin expressed on normal pleural mesothelial cells and an inflammatory response). Of the 33 evaluable patients treated, 4 had minor responses, 19 had stable disease (including 2 with resolution of ascites), and 10 had progressive disease. It is noteworthy that most of patients received only one cycle of therapy because of the rapid development of neutralization of SS1P activity (86). Thus it may be appropriate to test this approach using unmodified, high affinity and humanized mesothelin-specific antibody as reliance on ADCC may avoid the toxicity of the PE38 and limit the development of antibodies that neutralize the antitumor effect of the monoclonal antibody.
These findings also suggest that the use of active vaccination to trigger such humoral responses may be more beneficial. However, this may also be associated with autoimmune disease in the case of self-antigens like mesothelin, and unlike passive antibody therapy, cannot ready be terminated once an immune response has been generated. As one approach to produce ADCC for ovarian cancer, the generation of CA125-specific antibodies has been triggered by the administration of anti-ideotypic CA125 antibody (87). The development of CA125-reactive antibodies and ADCC of CA125-positive tumor cells was observed in 50% and 27% of treated patients, respectively. Anti-ideotype reactive patients showed a significantly longer survival even when controlling for other prognostic factors and the immunization was well tolerated.
Another promising approach to break tolerance and achieve therapeutic (or protective) levels of antibody is through display of the TAA on the surface of virus-like particles. Indeed, a recent study was conducted using chimeric virus-like particle (VLP)-human mesothelin as a vaccine candidate for immunotherapy in pancreatic cancer (88). It was observed that VLP-human mesothelin immunization in mice significantly regressed the pre-existing pancreatic tumor and prolonged the survival.
Live vector-based vaccines
Live vector-based vaccines typically employ bacterial vectors, such as Salmonella typhimurium and Listeria monocytogenes, or viral vectors, typically adenovirus and vaccinia virus. This approach has been used extensively to express HPV E6 and/or E7 as a potential treatment of HPV-associated malignancies, and more recently has been tested for mesothelin to treat ovarian and other mesothelin-expressing cancer. Many live vector vaccines are highly immunogenic because they can replicate within host cells and facilitate intercellular spread of antigen. Furthermore, some vectors replicate preferentially in tumor cells providing a potential oncolytic effect and thereby trigger cross-priming of released TAAs. For example, the tropism of vaccinia virus for the ovary has been well described, and recently demonstrated for ovarian cancer. Cells infected with the vector typically die and release the TAAs for uptake by dendritic cells and provide danger signals.
Though live vector-based vaccines have strong immunogenicity and continue to produce antigen for a significant period, they are not without limitations. The production of neutralizing antibodies in the host during vaccination could reduce the potency of repeat booster immunizations. Their inherent immunogenicity can dominate poorly immunogenic vaccine antigens, and also can potentially trigger autoimmune reactions. However, their major potential issue is the risk of toxicity associated with poorly controlled vector replication in patients, especially in those with immune suppression. Vaccination with live vectors may also elicit immunosuppressive factors in the host; eliminating these factors may improve both the efficacy of these vaccines and the safety profile.
Bacterial vectors
Attenuated bacterial vector-based vaccines have been shown to elicit potent TAA-specific T cell mediated immune responses. Specifically, Listeria monocytogenes has been used to generate both CD8+ and CD4+ immune responses and induce regression of established tumors expressing TAAs. Vaccination with live recombinant Listeria-based vaccines expressing E7 overcomes central tolerance by expanding low avidity CD8+ T cells specific for E7 thereby induce the regression of spontaneous tumors in mice transgenic for thyroid-specific expression of HPV 16 E6 and E7 oncoproteins (89). Numerous other preclinical studies have been reported employing Listeria-based vaccines targeting HPV E6 and E7 antigens (90,91). A phase I/II clinical trial is currently ongoing using recombinant attenuated Listeria expressing the HPV E7 antigen as a fusion with LLO (Lovaxin C) in stage IV cervical cancer patients, and the treatments have been well tolerated to date (http://www.advaxis.com/2007oct9.htm).
Listeria-based vaccines have also been explored for the treatment of mesothelin-expressing for the treatment of pancreatic and ovarian cancers. Several preclinical studies have employed a live attenuated strain of Listeria that encodes human mesothelin called CRS-207 (LmΔ actA/Δ inlB/hMeso) (92). Furthermore, recent preclinical studies have shown that CRS-207 elicits human mesothelin-specific CD4+/CD8+ immunity in mice and in cynomolgus monkeys and exhibits therapeutic efficacy in tumor bearing mice (93). Phase I clinical trials using CRS-207 in patients with mesothelin-expressing cancers are currently being planned (http://www.advaxis.com).
Viral vectors
Numerous viral vector-based vaccines have been tested in mice (for a review, see (94)). Vaccinia virus expressing HPV E7 antigen linked to proteins that enhance antigen presentation in DCs generate potent E7-specific immune responses that regress E7-expressing tumors in mice (95–97). A replication-deficient adenovirus vector was employed to deliver a fusion protein encoding calreticulin linked to HPV-16 E7 antigen. Vaccination with this construct was shown to protect mice against E7-expressing tumor challenge and induce a therapeutic effect against established tumors (98). Furthermore, adeno-associated virus vectors expressing E7 linked to M. tuberculosis hsp70 (99) or the cytokine interleukin (IL)-12 (100) also trigger E7-specific immune responses in vivo.
In the clinic, a recombinant vaccinia vector encoding an HPV-16/18 E6/E7 fusion protein, termed TA-HPV, has been evaluated in several phase I/II trials. It was well tolerated and induced T cell responses in patients with CIN (101–103) and also vulval intraepithelial neoplasia (VIN) (104,105) and vaginal intraepithelial neoplasia (VAIN) (106). In an uncontrolled study in VAIN patients vaccinated with TA-HPV, five out of twelve patients showed at least a 50% reduction in lesion diameter over a 24-week period, and one patient showed complete regression of the lesion (106).
Several preclinical studies have employed viral vector-based vectors for the control of ovarian cancer. For example, Sindbis viral vectors have been used to control human ovarian tumors in C.B-17-SCID (SCID) mice suggesting an oncolytic mechanism rather than cross-priming (107). Other potentially oncolytic viruses, such as measles virus, herpes simplex virus and Yaba-like disease virus have been tested against ovarian cancer and may also trigger anti-tumor immunity (108–110). A recent study has employed vaccinia virus-based vaccine to effectively infect and kill human and murine ovarian tumors (111). Vaccinia virus administered to mice intraperitoneally was specifically targeted to the murine or human ovarian tumors and led to antitumor responses. Thus, intraperitoneal injection with vaccinia virus may provide a potentially effective strategy for treating mesothelin-expressing ovarian cancers. Adenoviruses have also been employed in preclinical studies targeting mesothelin-expressing ovarian cancers. Studies have focused on the use of mesothelin for transcriptional as well as transductional targeting strategies for ovarian cancer gene therapy. Transductional targeting of adenovirus via anti-mesothelin antibody was shown to increase transgene expression in ovarian cancer cells (112).
Viral-vector based vaccines targeting mesothelin have not yet been explored in clinical studies. However there have been several clinical studies performed with other serologically defined TAAs. One study utilized MVA expressing a string of CTL epitopes including one from NY-ESO-1. However, the responses in patients were dominated by poxviral (i.e. vector) epitopes (113). To minimize such issues, another trial utilized vaccination with MVA expressing NY-ESO-1 to prime patients followed by a boost with recombinant fowlpox also expressing NY-ESO-1. This approach generated TAA-specific immunity in the majority of patients and favorable clinical outcomes were observed in a few cancer patients in this non-controlled study (114). A similar approach is being utilized in PANVAC that targets CEA and MUC1. In addition to the antigen, some viral vector vaccines also encode cytokines and other factors that might enhance TAA-specific immunity. For example, TG-4010 is a second-generation MVA encoding MUC1 and IL-2 for the potential treatment of a variety of cancer types (115,116).
Peptide/Protein-based vaccines
Peptide-based vaccines
Peptide-based vaccines are typically safe, easy to produce, and stable. However, peptide-based vaccines are typically specific to particular MHC specific, requiring that the relevant immunogenic epitopes of TAAs are first identified. The highly polymorphic nature of HLA generally results in a vaccines that is potentially only effective in a subset of patients. Another drawback is that peptide vaccines tend to be poorly immunogenic and thus need to be administered in combination with potent adjuvants (for a review, see (117)). The peptide vaccine potency has been enhanced by linking peptides with DC-activating agents such as CpG oligodeoxynucleotide (CpG ODN) or MPL (118,119), or to immunostimulatory molecules (120) and using GM-CSF to promote CTL responses (121). The potency of peptide-based vaccines can also be enhanced by oil-based adjuvants or modifying the epitopes to prevent peptide degradation. This has been demonstrated in a study using E6 and E7 long peptides in a rabbit papillomavuirus model wherein vaccination was shown to control both the established virus-induced lesions and latently infected sites (122).
Several trials have demonstrated the safety and tolerability of peptide-based vaccines (123,124). Vaccination with lipidated HPV-16 E7 peptides was safe in patients with high grade cervical or vulvar intraepithelial neoplasia. Six of 18 patients had partial colposcopically measured regression of their cervical intraepithelial neoplastic lesions in addition to the three complete responders in this uncontrolled study (125). A recent study demonstrated that vaccination with 30–35 mer long overlapping peptides of HPV-16 E6 and E7 sequences in Montamide ISA 51 adjuvant generated HPV-16-specific Th1/Th2 cells infiltration in both the vaccination site and the lesions in VIN3 patients (126).
There have been no preclinical studies of peptide-based vaccines targeting mesothelin. However, a study identified novel mesothelin HLA-A2 CTL epitopes that could efficiently activate human T cells to lyse human tumors by mesothelin-specific T cells (127). Another study by Hung et al. created a mesothelin-positive luciferase-expressing ovarian cancer model, MOSEC/luc and identified an H-2D(b)-restricted mesothelin peptide-specific cytotoxic T-lymphocyte (CTL) epitope (amino acid (aa) 406–414) that was endogenously processed and presented by MOSEC/luc tumor cells (128). They also showed that adoptive transfer of mesothelin peptide (aa406-414)-specific CD8(+) T cells led to the control of MOSEC/luc tumor cells.
Protein-based vaccines
Protein vaccines are less subject to the issues of MHC restriction because they contain all of the epitopes but are more complex to produce than peptide vaccines (129–131). Typically protein-based vaccines are weak immunogens and adjuvant and fusion protein strategies are thus frequently used to enhance their potency.
Several preclinical studies have used the saponin-based adjuvant ISCOMATRIX (132) and the liposome-polycation-DNA (LPD) adjuvant (133). Linkage of a TAA to Bordetella pertussis adenylate cyclase (CyaA), that targets APCs through specific interaction with αMβ2 integrin (134), the translocation domain of bacterial exotoxin Pseudomonas aeruginosa exotoxin A (EXA) (135) or heat-shock proteins, which target the antigen to APCs (136,137), have been used.
Several clinical studies have explored fusion proteins containing HPV early proteins. TA-GW, which is a fusion of HPV-6 L2 and E7 absorbed onto Alhydrogel was well tolerated by patients but not effective in clearing HPV genital warts (138,139). Another vaccine containing an HPV-16 E6/E7 fusion protein mixed with the ISCOMATRIX adjuvant has been shown to be safe and immunogenic and resulted in significantly enhanced CD8+ T cell responses to both E6 and E7 in vaccinated patients in phase I trials (140). A third vaccine, comprising a fusion of HPV-16 L2, E6 and E7 (termed TA-CIN), safely induced antibodies and T cell immunity in a subset of vaccinated women (29). However there is no clear evidence of efficacy to date, but this product was used without adjuvant. PD-E7, comprised of mutated HPV16 E7 fused with a fragment of Haemophilus influenzae protein D and formulated in the adjuvant AS02B has been examined in Phase I/II trials. This vaccine induced detectable E7-specific CTL responses in patients with CIN1 and CIN3 lesions (141). Vaccination with HPV16 E7 fused to M. bovis hsp65 was well tolerated in patients with high-grade anal intraepithelial neoplasia (AIN) (142,143). However, it produced no significant difference in regression rates in HPV 16+ CIN III infected women compared to unvaccinated patients (144).
Nucleic acid-based vaccines
DNA-based vaccines
Naked DNA is generally considered to be safe, stable, and relatively easy to manufacture vaccine. It can potentially sustain expression of the TAA for longer periods than RNA or protein vaccines. Furthermore, unlike live vector vaccines, DNA vaccines do not elicit neutralizing antibody production in the patient, and thus can be repeatedly administered. However one significant caveat is that the administered DNA may potentially integrate into the host genome. DNA vaccines expressing oncogenes are of particular concern. For the HPVE6 and E7 oncoproteins this has been addressed through inactivating mutations or shuffling peptides to abolish their ability to transform cells. This is less of a concern for mesothelin, but the strong promoter utilized in the expression vector still might integrate near a growth regulatory gene. Further, DNA vaccines have proven relatively poorly immunogenic in patients, and this may reflect issues with their delivery.
DNA vaccines targeting both HPV antigen as well as mesothelin have been extensively studied in preclinical models. For example, a recent study utilized a DNA vaccine encoding human mesothelin (pcDNA3-Hmeso) to treat C57BL/6 mice challenged with luciferase-expressing, human mesothelin-expressing ovarian cancer cell line, Defb29 Vegf-luc/Hmeso (145). It was observed that vaccination with pcDNA3-Hmeso DNA vaccine generated significant antitumor effects and promotes survival in tumor-challenged mice via both T cell-mediated immunity as well as antibody-mediated immunity. Another study in a rabbit model demonstrated that intradermal injection of plasmid DNA encoding full length human mesothelin into rabbits resulted in high titers of anti-mesothelin antibodies in the sera (146).
Although DNA vaccines have been very promising in many preclinical studies, they have demonstrated limited immunogenicity in human studies to date. Several strategies have been developed in preclinical models to enhance DNA vaccine potency, and are summarized below and in Table 4 (for reviews, see (147,148)).
Table 4. Strategies to enhance the immunogenicity of TAA delivered via naked DNA vaccines.
| Strategies | Methods |
|---|---|
| Increase the number of TAA-presenting APCs |
|
| Greater TAA expression, and enhanced peptide processing, and presentation by APCs |
|
| Facilitate and extend productive APC and T cell interaction |
|
Enhancing vaccine delivery to APCs
Delivery of DNA vaccines via the most effective routes is essential to enhance the number of antigen-expressing APCs. Intramuscular injection of DNA has not proven very effective. New approaches such as in vivo electroporation (149) and vaccination via gene gun is highly effective for DNA vaccination in animals (150). The gene gun is used to fire DNA-coated gold particles into the epidermis and efficiently transfect intradermal DCs that can mature and migrate to the lymphoid organs for T cell priming. Methods for vaccination of patients with naked DNA still require optimization.
The efficacy of DNA vaccines might be enhanced by allowing the release and spread of TAA between DCs by linkage with proteins that facilitate intercellular transport. For example fusion of E7 with VP22, a viral protein with intercellular trafficking properties, in a DNA vaccine dramatically enhanced E7-specific CD8+ T cell responses and generated greater antitumor effects than DNA vaccines encoding E7 alone in vaccinated mice (151) (152).
Enhancing TAA presentation by APCs
Some antigens express poorly, including the HPV oncoproteins, and codon modification can greatly improve their expression by DNA vaccines. Replacing rarely used codons with more commonly utilized codons can also eliminate problematic secondary RNA structures and enhance TAA expression in transduced cells, thereby boosting the CTL response induced by DNA vaccination when expression levels are limiting (153–155).
Linkage of TAAs to various MHC class I proteins or domains that facilitate class I processing and presentation, including M. tuberculosis hsp70 (156), γ-tubulin (157), the extracellular domain of Flt3 (Fms-like tyrosine kinase 3)-ligand (158), and the translocation domain of Pseudomonas aeruginosa exotoxin A (159) has greatly enhanced immunity against E7-expressing tumor challenge in vaccinated mice. Among the various MHC class I proteins, DNA vaccines expressing E7 fused with calreticulin (CRT) has proven one of the most effective in generating E7-specific CD8+ T cell responses among all the strategies that we have tested (160).
Fusing of a TAA to targeting molecules that redirect the TAA in to the class II pathway to enhance CD4+ T cell responses can thereby augment the development of CTL responses (for a review, see (161)). This has been demonstrated for HPV E7 by its linkage of to a signal peptide for entry to the endoplasmic reticulum (Sig) and a C terminal fusion with the transmembrane and cytoplasmic domains of LAMP-1 for targeting to the lysosomes (Sig/E7/LAMP-1) enhances its MHC class II antigen presentation (162). Expression of Sig/E7/LAMP-1 via a DNA vaccine and produced greater numbers of E7-specific CD4+ T cells and also higher E7-specific CTL activity in mice than vaccines composed of Sig/E7 or wild-type E7 DNA alone (163).
Another approach to enhance MHCII presentation of TAAs expressed via DNA vaccination is fusion to the MHC class II invariant chain (Ii). Ii binds with MHC class II molecules in the endoplasmic reticulum such that the CLIP region of the Ii blocks the peptide-binding groove. Within the lysosomes the CLIP is displaced by antigenic peptide allowing the MHC class II/peptide complex to be presented on the APC’s surface. If the CLIP peptide of Ii is replaced by a T helper (Th) epitope of the TAA or an epitope like PADRE (Ii-PADRE), the TAA’s epitope can then be presented by MHCII very efficiently and with less peptide competition, e.g. vaccination of mice with a DNA encoding Ii-PADRE dramatically enhances PADRE-specific CD4+ T cell immune responses. Interestingly, coadministration of the DNA encoding Ii-PADRE with a DNA vaccine expressing another TAA enhances the TAA-specific CD8+ T cell responses (164).
A similar approach has been developed to circumvent MHCI antigen processing (165) by linking of a TAA peptide to β2-microglobulin and co-expressing the MHC class I heavy chain, produces a ‘single-chain trimer (SCT)’. Huang et al created a DNA vaccine employing an SCT targeting human mesothelin and characterized the ensuing antigen-specific CD8+ T cell-mediated immune responses and anti-tumor effects against human mesothelin-expressing tumors in HLA-A2 transgenic mice. Vaccination with DNA employing an SCT of HLA-A2 linked to human mesothelin epitope aa540-549 generated strong mesothelin peptide-specific CD8+ T cell responses and prevent the growth of mesothelin-expressing tumors in mice (77). Likewise, mice vaccinated with a DNA vaccine encoding an SCT displaying an HPV16 E6 immunodominant class I epitope (pIRES-E6-β2m-Kb) generated a greatly enhanced E6 peptide-specific CD8+ T cell response (165).
Improved interaction with APC and T cell activation
An extended and productive synapse between APCs and T cells is critical for T cell activation. Thus, by enhancing the duration of this interaction, T cells activation could potentially be enhanced after DNA vaccination. Co-expression of inhibitors of apoptosis (e.g. BCL-xL, BCL-2, XIAP, and dominant-negative caspase) with the TAA has been utilized to prolong the life of APCs transduced with the DNA vaccine (160,166). Mice vaccinated in the same site with DNA expressing E7 and anti-apoptotic proteins on separate constructs exhibited more potent E7-specific CD8+ T cell responses (167). However, constituative expression of anti-apoptotic factors is potentially oncogenic. A shorter term approach utilizing RNA interference (RNAi) was has been employed to knockdown key pro-apoptotic protein expression. Indeed, co-administration via gene gun of E7 DNA vaccines and siRNA for Bak and Bax both helped DC to resist apoptotic cell death and enhanced E7-specific CD8+ T cell responses in immunized mice (168).
Since activation of naïve antigen-specific T cells is modulated by co-stimulatory signals and cytokines produced by DCs, several studies have explored coadministration of DNA vectors expressing the TAA with vector expressing cytokines or costimulatory molecules. Indeed, co-administration of vectors encoding granulocyte-macrophage colony stimulating factor (GMCSF) (169), IL-2 (170) or IL-12 (171) with DNA vaccines has enhanced antigen-specific immune responses in preclinical models.
Clinical Trials of DNA Vaccines
Several DNA vaccines have been translated into clinical trials. For example, administration of a microencapsulated DNA vaccine termed ZYC-101, that encodes multiple HLA-A2-restricted epitopes derived from E7, was well tolerated in patients with high grade CIN lesions (172) or high-grade anal intra-epithelial neoplasia (AIN) (173). A new version of the vaccine, termed ZYC-101a, encodes epitopes derived from E6 and E7 of HPV16 and HPV18 and the high grade CIN of a subset of vaccinated women resolved (174). A phase I trial of a DNA vaccine encoding modified HPV-16 E7 DNA (which abolished the Rb binding site) linked to M. tuberculosis hsp70 (pNGVL4a-Sig/E7(detox)/hsp70) in HPV16+ high grade CIN patients has recently completed enrollment (http://pathology2.jhu.edu/ccspore/index.html). A similar phase I trial using the same Sig/E7detox/Hsp70 DNA vaccine in patients with HPV16+ advanced head and neck squamous cell carcinoma has also completed enrollment (Maura Gillison, personal communication). Another DNA vaccine encoding calreticulin fused to a mutated HPV16 E7 (E7detox) will be tested shortly in a phase I trial in cervical cancer patients (http://pathology2.jhu.edu/ccspore/index.html).
RNA replicon vectors
The use of self-replicating RNA replicons as vectors to deliver TAA to APCs has several potential advantages. Typically they replicate in many cell types resulting in high level but short term TAA expression. For example subgenomic alphavirus RNA vectors replicate rapidly but are self-limiting because they typically kill the target cell and do not contain the viral structural genes. Alphavirus replicons can be administered in several ways; as naked RNA or DNA that produces the RNA replicons on transcription, or as a replication defective particle capable of delivering the RNA replicon but not production of new infectious particles. Delivery of RNA replicon vaccines as naked RNA or DNA permits repeated immunization of patients, although this is not the case for the replication-defective particles. RNA replicons generally have no DNA intermediate, and therefore do not integrate into the host genome, and typically kill transduced cells. This is advantageous for expression of oncoproteins like HPV E6 and E7. Conversely, RNA replicons are likely to produce shorter term TAA expression than DNA vaccines. Flavivirus replicons based on Kunjin (KUN) have been used to deliver TAA, including E7 (175). A new DNA-based KUN replicon that does not trigger apoptosis, potentially prolonging antigen presentation has been developed (176). RNA replicons launched by all three approaches have been used successfully for expression of E7 to induce anti-tumor immunity in preclinical models (177–182). This approach has yet to be tested for mesothelin, but VEE replicon-based vaccines expressing HIV Gag are now being tested in the clinic (http://www.alphavax.com/products/hiv.aspx).
Cell-based Vaccines
Dendritic cell-based vaccines
DC-based vaccines represent a potentially effective strategy for therapeutic vaccines against gynecologic malignancies (for a review, see (183)). A possible advantage of DC-based vaccines is their potential to circumventing tumor-mediated immune suppression (184,185). However, the preparation of individualized DC-based vaccines is costly and cumbersome to generate because it requires large-scale culture. Furthermore, the route of administration is likely to be important for DC-based vaccination because the DCs must home to the lymphoid organs to interact with the majority of naïve T cells.
We have already described several approaches to enhance the potency of DNA-based TAA vaccines by prolonging the survival of APCs to promote T cell priming (186) and these could also be combined with improved MHC class I/II presentation of antigen by direct loading of peptides to enhance vaccine potency (187). Subcutaneous injection of HPV-18 E7-pulsed DCs has been tested in clinical trials in patients with cervical cancer (188,189). Furthermore, in a clinical pilot study, autologous DCs pulsed with E7 protein were shown to induce T cell responses in some late stage cervical cancer patients but no objective clinical responses (190).
Tumor cell-based vaccines
One advantage of tumor cell-based vaccines is the convenience that tumor antigens need not be clearly identified, although this is less relevant in the case of cervical cancer. The immunogenicity of tumor cells can also be improved by ex vivo transduction to express cytokines or costimulatory molecules. For example the immunogenicity of HPV-transformed tumor vaccines has been boosted by ectopic expression of IL-2 (191), IL-12 (192,193), or GM-CSF (193,194). A potential safety concern of tumor cell-based vaccines involves the risk of introducing new cancers. In addition, individualized large-scale autologous vaccine production is difficult, although other tumor cells of the same type might be used.
HPV transformed cell-based vaccines have been tested in preclinical models. Vaccination of mice with E7-positive tumor cells that ectopically express GM-CSF triggered an E7-specific CTL response and potent antitumor immunity to E7-expressing tumors (194). A recent study demonstrated that mice challenged with heat shock protein 70 (Hsp70)-secreting murine ovarian cancer cells expressing luciferase (MOSEC/luc) generate significant antigen-specific CD8(+) T-cell immune responses (195). Hsp70 has been shown to target and concentrate antigenic peptides in dendritic cells and is also able to activate dendritic cells. Furthermore, mice vaccinated with irradiated MOSEC/luc cells expressing Hsp70 also showed significant therapeutic effects against MOSEC/luc cells. In addition, it was observed that CD8 T cells, NK cells and CD4 T cells were important for protective antitumor effects generated by irradiated tumor cell-based vaccines expressing Hsp70. CD40 receptor and Toll-like receptor 4 were also important for inhibiting in vivo tumor growth (195). Although tumor cell-based vaccines, such as GM-CSF-transduced autologous or allogeneic tumor cells have been used in clinical trials for other cancers such as pancreatic cancer, renal cell carcinoma, melanoma, and prostate carcinoma (http://www.cellgenesys.com/products.shtml), tumor cell-based vaccines have not been tested against HPV-associated malignancies in the clinical arena.
Tumor cell-based vaccines have been employed in the treatment of mesothelin expressing cancer. For instance, a phase I trial tested a novel GM-CSF-secreting pancreatic tumor-based vaccine in patients with surgically resected adenocarcinoma of the pancreas (196). Mesothelin-specific cellular immunity was shown to correlate with positive clinical outcomes observed in patients with pancreatic carcinoma after vaccination with tumor-cell based vaccine. Another clinical trial of vaccination with granulocyte macrophage-colony stimulating factor-transduced pancreatic cancer lines was designed to test whether cross-presentation by locally recruited APCs can activate pancreatic tumor-specific CD8(+) T cells (72). Mesothelin-specific CD8+ T cell responses in pancreatic cancer patients with vaccine-induced DTH responses were found to correlate with prolonged disease-free survival, suggesting that mesothelin might represent an important target antigen for active immunotherapy for cancers, including ovarian cancer, that express mesothelin (72).
Combination Approaches
Many vaccination strategies have been developed for the treatment of gynecological malignancies but to date no single approach has proven effective. However, appropriate combinations of these vaccination strategies as prime-boost regimens might be more effective than either alone, and indeed some combinations are highly synergistic in animal models. For example, priming with a DNA or RNA vaccine and then boosting with a viral vector vaccine is synergistic for immune responses relative to the individual approach in many cases (197,198) (97). The efficacy of other treatment strategies, such as protein-based vaccination, can also be enhanced by prime-boost approaches (199). Prime-boost heterologous HPV vaccination (HPH16 L2E7E6 (TA-CIN) protein followed by vaccinia virus expressing HPV16 and HPV18 E6/E7 (TA-HPV)) enhanced HPV-16 specific T cell responses without serious adverse effects in AIN patients (200,201). Vaccination in the reverse order was not as effective. Patients primed with TA-HPV and then boosted with TA-CIN developed HPV 16-specific immune and/or serological responses, and three showed a reduction in lesion size or experienced symptomatic relief (202). A phase I trial in which women with high grade HPV16+ CIN are primed with pNGVL4a/Sig/E7(detox)/Hsp70 DNA and then boosted with MVA expressing E6, E7 and IL-2 has recently been proposed (R21CA123876).
Vaccination strategies could also potentially be combined with other forms of treatment such as chemotherapy, radiation or other biotherapeutic agents (203,204). For example, synergy against established E7-expressing tumors was found for vaccination with E7 protein in CpG oligonucleotide adjuvant and cisplatin chemotherapy (203). Also the chemotherapeutic agent epigallocatechin-3-Gallate (EGCG) triggers apoptosis of tumor cells and enhances TAA-specific T cell immune responses produced by DNA vaccination (105).
Vaccines targeting gynecological malignancies might be improved by co-administration of antibody that blocks immune suppressive signaling, such as that by CTLA-4 or PD-1, to promote anti-tumoral T cell responses (for review, see (205,206)). Several factors in the tumor microenvironment have been shown to contribute to immunosuppression, e.g. B7-H1 (207), STAT3 (208), MIC-A and MIC-B (209), indoleamine 2,3-dioxygenase (IDO) enzymic activity and reactive oxygen species (210), and galectin-1 (211) on the tumor cell surface, immune suppressive cytokines like IL-10 (212) and TGF-β (213), T regulatory cells (36), myeloid-derived suppressor cells (214). Thus, vaccination might be combined with drugs or antibodies that combat local immune suppression in the tumor microenvironment by modulating the activity of these immunosuppressive factors e.g. local application of imiquimod after vaccination. Some combination of these strategies may be applied to generate robust anti-tumor cellular immune responses that home to and are effective in the tumor microenvironment, and thereby produce meaningful therapeutic activity against gynecological malignancies.
Possibility of Prevention
Is there a possibility of using these therapeutic vaccines in prevention of gynecologic malignancy? In the case of HPV-specific therapeutic vaccines, this is very appropriate to consider. There is currently a molecular test for HPV in cytologic specimens, but for patients infected with high risk HPV without evidence of high grade CIN, there is no virus-specific therapy. These patients are followed carefully and many, but not all infections are spontaneously cleared. Furthermore, with the advent of preventive HPV vaccines, this is likely to become a less frequent occurrence. Nevertheless, there may be an opportunity to vaccinate patients with high risk HPV infections and/or low grade CIN to control the infection and this may speed the impact of preventive HPV L1 VLP vaccination upon cervical cancer rates.
However, the case for preventive vaccination against ovarian cancer is uncertain. There is evidence suggesting that humoral immunity to MUC1 correlates inversely with risk factors for ovarian cancer leading to suggestions that triggering of MUC1-specific immunity might be protective against ovarian cancer (215,216). We have also observed humoral responses negatively correlated with ovarian cancer (217). Furthermore there is considerable evidence that mesothelin-specific antibodies and T cell responses are able to kill ovarian cancer cells and thus the notion of a vaccine induced TAA-specific immune response eliminating ovarian cancer at its earliest stages if it develops is an attractive notion. However, ovarian cancer is a relatively infrequent disease, and therefore the number of side effects from a preventive vaccine are likely to far out way the number of cases prevented. Nevertheless, there are groups of women with substantially higher likelihood of contracting ovarian cancer, and they might be candidates for a preventive vaccination. It is clear that patients carrying BRCA mutations and some other hereditary cancer syndromes have a significantly increased risk of ovarian cancer. Some patients choose to undergo prophylactic oophorectomy to lessen the chances of developing ovarian cancer, and yet peritoneal carcinoma still occurs in a fraction of these patients. Furthermore, women with a strong family history of ovarian cancer are also at an elevated risk, albeit not as high as known BRCA mutation carriers. Patients with stage I ovarian cancer who are treated with surgery alone might also be considered. Perhaps the strongest case for vaccines is in women who have undergone standard chemotherapy and surgery and have no detectable disease based on imaging and tumor marker studies i.e. minimal residual disease.
Summary and Future Directions
We have attempted to underline the importance of the pathobiology in determining the relevant targets for, and approaches to, antigen-specific immunotherapy of gynecologic cancer. It is likely that the biomarkers sought for early detection of cancers and the molecular targets for therapy overlap the antigens relevant for immunotherapy. This is clear in the case of cervical cancer in which E6 and E7 expression is critical to driving and maintaining the transformed phenotype and these antigens frequently trigger a humoral response in cancer patients. These viral oncogenes are the primary targets for diagnosis or directed molecular therapies and the logical targets of antigen-specific immunotherapy.
However, this is less clear in the case of ovarian serous carcinoma. The only consistently detected and causative genetic change that has been described for high-grade serous carcinoma is mutation of p53. Unfortunately, this is difficult to target because p53 expression may be lost, or even if mutated, there are a plethora of possible mutations, although mutant p53 is typically present at high levels. In contrast, low-grade serous carcinoma expresses wild type p53 and is driven by mutation in the B-Raf or K-ras. Thus one approach to immunotherapy may be the identification of these driver mutations for each patient and vaccinating against their particular mutation (218). Another approach may be to screen for TAAs using autologous anti-tumor antibody responses, and through the use of vaccines invigorate and redirect the patient’s immune response to these TAAs. In this regard, mesothelin is an attractive TAA because its expression is limited to and low in the epithelia of the pleura, pericardium and peritoneum, and knockout of mesothelin produces no discernable phenotype in mice. Importantly, mesothelin is highly over expressed in the majority of serous carcinoma (such that it shows promise as a serum biomarker), it is immunogenic in these patients and these responses have been correlated with better clinical outcome. Furthermore, mesothelin may contribute to the pathology of serous carcinoma and is accessible on the cell surface to antibodies. Other such TAAs, notably NY-ESO-1 and MUC1 also show promise as targets of antigen-specific immunotherapy in ovarian cancer, and it is not clear which may be optimal.
Another critical part of the pathobiology of cancer is the tumor microenvironment and its suppression of antigen-specific immunity. Although much can be gleaned from the study of patient materials, the development of spontaneous tumor models in the mouse is critical to better understand these events. This is a significant issue for ovarian serous carcinoma because so little is known about the causative events. It is possible to augment in cervical cancer patients the antigen-specific cellular immune responses targeting the relevant viral antigens through vaccination, but so far there has been little impact on clinical outcome. While there is progress in treatment of pre-malignant lesions with such antigen-specific immunotherapy, an effective immunotherapy for invasive cervical cancer remains elusive. Consistent with this issue, the detection of antigen-specific cellular immune responses in the peripheral blood of cancer patients has weakly correlated with clinical regression. Therefore, continued efforts to support homing of antigen-specific T cells to tumor sites, and preventing their suppression in the tumor microenvironment remain of vital importance. Cervical cancer represents an important disease in which to develop cancer immunotherapic strategies because its pathobiology and target antigens are relatively well understood in comparison to most other epithelial cancers like ovarian cancer.
Acknowledgments
Given its scope, this review is not intended to be encyclopedic, but the authors apologize to those whose work is relevant but not cited. The work is supported by the NCI SPORE in Cervical Cancer P50 CA098252, CA122581, CA118790, CA114978, CA114425 and CA118790, The Alliance for Cancer Gene Therapy, American Cancer Society, U19 CA113341. We thank Ie-Ming Shih for photomicrographs.
References
- 1.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 2.Roden R, Wu TC. How will HPV vaccines affect cervical cancer? Nat Rev Cancer. 2006;6:753–763. doi: 10.1038/nrc1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nature Rev Cancer. 2002;2:342–350. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 4.Schiffman MH, et al. Epidemiologic evidence showing that human papillomavirus infection causes most cervical intraepithelial neoplasia [see comments] J Natl Cancer Inst. 1993;85:958–964. doi: 10.1093/jnci/85.12.958. [DOI] [PubMed] [Google Scholar]
- 5.Walboomers JM, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189:12–19. doi: 10.1002/(SICI)1096-9896(199909)189:1<12::AID-PATH431>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 6.Schiffman M, Castle PE, Jeronimo J, Rodriguez AC, Wacholder S. Human papillomavirus and cervical cancer. Lancet. 2007;370:890–907. doi: 10.1016/S0140-6736(07)61416-0. [DOI] [PubMed] [Google Scholar]
- 7.Goodwin EC, DiMaio D. Repression of human papillomavirus oncogenes in HeLa cervical carcinoma cells causes the orderly reactivation of dormant tumor suppressor pathways. Proc Natl Acad Sci U S A. 2000;97:12513–12518. doi: 10.1073/pnas.97.23.12513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goodwin EC, DiMaio D. Induced senescence in HeLa cervical carcinoma cells containing elevated telomerase activity and extended telomeres. Cell Growth Differ. 2001;12:525–534. [PubMed] [Google Scholar]
- 9.Rosenthal A, Jacobs I. Ovarian cancer screening. Semin Oncol. 1998;25:315–325. [PubMed] [Google Scholar]
- 10.Daly M, Obrams GI. Epidemiology and risk assessment for ovarian cancer. Semin Oncol. 1998;25:255–264. [PubMed] [Google Scholar]
- 11.Jacobs I, Bast RC., Jr The CA 125 tumour-associated antigen: a review of the literature. Hum Reprod. 1989;4:1–12. doi: 10.1093/oxfordjournals.humrep.a136832. [DOI] [PubMed] [Google Scholar]
- 12.Bast RC., Jr Status of tumor markers in ovarian cancer screening. J Clin Oncol. 2003;21:200–205. doi: 10.1200/JCO.2003.01.068. [DOI] [PubMed] [Google Scholar]
- 13.Friedlander ML. Prognostic factors in ovarian cancer. Semin Oncol. 1998;25:305–314. [PubMed] [Google Scholar]
- 14.Jacobs IJ, et al. Clonal origin of epithelial ovarian carcinoma: analysis by loss of heterozygosity, p53 mutation, and X-chromosome inactivation. J Natl Cancer Inst. 1992;84:1793–1798. doi: 10.1093/jnci/84.23.1793. [DOI] [PubMed] [Google Scholar]
- 15.Grulich AE, van Leeuwen MT, Falster MO, Vajdic CM. Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: a meta-analysis. Lancet. 2007;370:59–67. doi: 10.1016/S0140-6736(07)61050-2. [DOI] [PubMed] [Google Scholar]
- 16.Salani R, et al. Assessment of TP53 mutation using purified tissue samples of ovarian serous carcinomas reveals a higher mutation rate than previously reported and does not correlate with drug resistance. Int J Gynecol Cancer. 2007 doi: 10.1111/j.1525-1438.2007.01039.x. [DOI] [PubMed] [Google Scholar]
- 17.Shih Ie M, Kurman RJ. Ovarian tumorigenesis: a proposed model based on morphological and molecular genetic analysis. Am J Pathol. 2004;164:1511–1518. doi: 10.1016/s0002-9440(10)63708-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walboomers JM, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189:12–19. doi: 10.1002/(SICI)1096-9896(199909)189:1<12::AID-PATH431>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 19.Dubeau L. The cell of origin of ovarian epithelial tumors and the ovarian surface epithelium dogma: does the emperor have no clothes? Gynecol Oncol. 1999;72:437–442. doi: 10.1006/gyno.1998.5275. [DOI] [PubMed] [Google Scholar]
- 20.Brodell LA, Mercurio MG, Brodell RT. The diagnosis and treatment of human papillomavirus-mediated genital lesions. Cutis. 2007;79:5–10. [PubMed] [Google Scholar]
- 21.Fausch SC, Da Silva DM, Kast WM. Heterologous papillomavirus virus-like particles and human papillomavirus virus-like particle immune complexes activate human Langerhans cells. Vaccine. 2005;23:1720–1729. doi: 10.1016/j.vaccine.2004.09.035. [DOI] [PubMed] [Google Scholar]
- 22.Fausch SC, Da Silva DM, Kast WM. Differential uptake and cross-presentation of human papillomavirus virus-like particles by dendritic cells and Langerhans cells. Cancer Res. 2003;63:3478–3482. [PubMed] [Google Scholar]
- 23.Palefsky JM, Gillison ML, Strickler HD. Chapter 16: HPV vaccines in immunocompromised women and men. Vaccine. 2006;24 (Suppl 3):S140–146. doi: 10.1016/j.vaccine.2006.05.120. [DOI] [PubMed] [Google Scholar]
- 24.Tagami H, Takigawa M, Ogino A, Imamura S, Ofugi S. Spontaneous regression of plane warts after inflammation: clinical and histologic studies in 25 cases. Arch Dermatol. 1977;113:1209–1213. [PubMed] [Google Scholar]
- 25.Coleman N, et al. Immunological events in regressing genital warts. Am J Clin Pathol. 1994;102:768–774. doi: 10.1093/ajcp/102.6.768. [DOI] [PubMed] [Google Scholar]
- 26.Grassegger A, et al. Spontaneous or interferon-gamma-induced T-cell infiltration, HLA-DR and ICAM-1 expression in genitoanal warts are associated with TH1 or mixed TH1/TH2 cytokine mRNA expression profiles. Arch Dermatol Res. 1997;289:243–250. doi: 10.1007/s004030050187. [DOI] [PubMed] [Google Scholar]
- 27.Trimble CL, et al. Spontaneous regression of high-grade cervical dysplasia: effects of human papillomavirus type and HLA phenotype. Clin Cancer Res. 2005;11:4717–4723. doi: 10.1158/1078-0432.CCR-04-2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peng S, et al. HLA-DQB1*02-restricted HPV-16 E7 peptide-specific CD4+ T-cell immune responses correlate with regression of HPV-16-associated high-grade squamous intraepithelial lesions. Clin Cancer Res. 2007;13:2479–2487. doi: 10.1158/1078-0432.CCR-06-2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Jong A, et al. Enhancement of human papillomavirus (HPV) type 16 E6 and E7-specific T-cell immunity in healthy volunteers through vaccination with TA-CIN, an HPV16 L2E7E6 fusion protein vaccine. Vaccine. 2002;20:3456–3464. doi: 10.1016/s0264-410x(02)00350-x. [DOI] [PubMed] [Google Scholar]
- 30.van der Burg SH, et al. Association of cervical cancer with the presence of CD4+ regulatory T cells specific for human papillomavirus antigens. Proc Natl Acad Sci U S A. 2007;104:12087–12092. doi: 10.1073/pnas.0704672104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Piersma SJ, et al. High number of intraepithelial CD8+ tumor-infiltrating lymphocytes is associated with the absence of lymph node metastases in patients with large early-stage cervical cancer. Cancer Res. 2007;67:354–361. doi: 10.1158/0008-5472.CAN-06-3388. [DOI] [PubMed] [Google Scholar]
- 32.Gambhira R, Gravitt PE, Bossis I, Stern PL, Viscidi RP, Roden RB. Vaccination of healthy volunteers with human papillomavirus type 16 L2E7E6 fusion protein induces serum antibody that neutralizes across papillomavirus species. Cancer Res. 2006;66:11120–11124. doi: 10.1158/0008-5472.CAN-06-2560. [DOI] [PubMed] [Google Scholar]
- 33.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5:263–274. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 34.Zhang L, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348:203–213. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 35.Sato E, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A. 2005;102:18538–18543. doi: 10.1073/pnas.0509182102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Curiel TJ, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 37.Wei S, et al. Plasmacytoid dendritic cells induce CD8+ regulatory T cells in human ovarian carcinoma. Cancer Res. 2005;65:5020–5026. doi: 10.1158/0008-5472.CAN-04-4043. [DOI] [PubMed] [Google Scholar]
- 38.Kryczek I, et al. Relationship between B7-H4, regulatory T cells, and patient outcome in human ovarian carcinoma. Cancer Res. 2007;67:8900–8905. doi: 10.1158/0008-5472.CAN-07-1866. [DOI] [PubMed] [Google Scholar]
- 39.Kryczek I, et al. B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J Exp Med. 2006;203:871–881. doi: 10.1084/jem.20050930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ozols RF. Challenges for chemotherapy in ovarian cancer. Ann Oncol. 2006;17 (Suppl 5):v181–v187. doi: 10.1093/annonc/mdj978. [DOI] [PubMed] [Google Scholar]
- 41.Raspollini MR, et al. COX-2 status in relation to tumor microvessel density and VEGF expression: analysis in ovarian carcinoma patients with low versus high survival rates. Oncol Rep. 2004;11:309–313. [PubMed] [Google Scholar]
- 42.Anttila MA, Voutilainen K, Merivalo S, Saarikoski S, Kosma VM. Prognostic significance of iNOS in epithelial ovarian cancer. Gynecol Oncol. 2007;105:97–103. doi: 10.1016/j.ygyno.2006.10.049. [DOI] [PubMed] [Google Scholar]
- 43.Ramakrishna V, et al. Naturally occurring peptides associated with HLA-A2 in ovarian cancer cell lines identified by mass spectrometry are targets of HLA-A2-restricted cytotoxic T cells. Int Immunol. 2003;15:751–763. doi: 10.1093/intimm/dxg074. [DOI] [PubMed] [Google Scholar]
- 44.Viatte S, Alves PM, Romero P. Reverse immunology approach for the identification of CD8 T-cell-defined antigens: advantages and hurdles. Immunol Cell Biol. 2006;84:318–330. doi: 10.1111/j.1440-1711.2006.01447.x. [DOI] [PubMed] [Google Scholar]
- 45.Auersperg N, Edelson MI, Mok SC, Johnson SW, Hamilton TC. The biology of ovarian cancer. Semin Oncol. 1998;25:281–304. [PubMed] [Google Scholar]
- 46.Old LJ, Chen YT. New paths in human cancer serology. J Exp Med. 1998;187:1163–1167. doi: 10.1084/jem.187.8.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Finn OJ. Immune response as a biomarker for cancer detection and a lot more. New England Journal of Medicine. 2005;353:1288–1290. doi: 10.1056/NEJMe058157. [DOI] [PubMed] [Google Scholar]
- 48.Sahin U, et al. Human neoplasms elicit multiple specific immune responses in the autologous host. Proc Natl Acad Sci U S A. 1995;92:11810–11813. doi: 10.1073/pnas.92.25.11810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brass N, Heckel D, Sahin U, Pfreundschuh M, Sybrecht GW, Meese E. Translation initiation factor eIF-4gamma is encoded by an amplified gene and induces an immune response in squamous cell lung carcinoma. Hum Mol Genet. 1997;6:33–39. doi: 10.1093/hmg/6.1.33. [DOI] [PubMed] [Google Scholar]
- 50.Scanlan MJ, et al. Characterization of human colon cancer antigens recognized by autologous antibodies. Int J Cancer. 1998;76:652–658. doi: 10.1002/(sici)1097-0215(19980529)76:5<652::aid-ijc7>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 51.Dawson JR, Lutz PM, Shau H. The humoral response to gynecologic malignancies and its role in the regulation of tumor growth: a review. Am J Reprod Immunol. 1983;3:12–17. doi: 10.1111/j.1600-0897.1983.tb00205.x. [DOI] [PubMed] [Google Scholar]
- 52.Sahin U, Tureci O, Pfreundschuh M. Serological identification of human tumor antigens. Curr Opin Immunol. 1997;9:709–716. doi: 10.1016/s0952-7915(97)80053-2. [DOI] [PubMed] [Google Scholar]
- 53.Tureci O, et al. The SSX-2 gene, which is involved in the t(X;18) translocation of synovial sarcomas, codes for the human tumor antigen HOM-MEL-40. Cancer Res. 1996;56:4766–4772. [PubMed] [Google Scholar]
- 54.Stockert E, et al. A survey of the humoral immune response of cancer patients to a panel of human tumor antigens [see comments] J Exp Med. 1998;187:1349–1354. doi: 10.1084/jem.187.8.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fernandez-Madrid F, et al. Autoantibodies to Annexin XI-A and Other Autoantigens in the Diagnosis of Breast Cancer. Cancer Res. 2004;64:5089–5096. doi: 10.1158/0008-5472.CAN-03-0932. [DOI] [PubMed] [Google Scholar]
- 56.Popkov M, Rader C, Barbas CF., 3rd Isolation of human prostate cancer cell reactive antibodies using phage display technology. J Immunol Methods. 2004;291:137–151. doi: 10.1016/j.jim.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 57.Hong SH, et al. An autoantibody-mediated immune response to calreticulin isoforms in pancreatic cancer. Cancer Res. 2004;64:5504–5510. doi: 10.1158/0008-5472.CAN-04-0077. [DOI] [PubMed] [Google Scholar]
- 58.Chinni SR, Falchetto R, Gercel-Taylor C, Shabanowitz J, Hunt DF, Taylor DD. Humoral immune responses to cathepsin D and glucose-regulated protein 78 in ovarian cancer patients. Clin Cancer Res. 1997;3:1557–1564. [PubMed] [Google Scholar]
- 59.Philip R, et al. Shared immunoproteome for ovarian cancer diagnostics and immunotherapy: potential theranostic approach to cancer. J Proteome Res. 2007;6:2509–2517. doi: 10.1021/pr0606777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Canevari S, Pupa SM, Menard S. 1975–1995 revised anti-cancer serological response: biological significance and clinical implications. Ann Oncol. 1996;7:227–232. doi: 10.1093/oxfordjournals.annonc.a010564. [DOI] [PubMed] [Google Scholar]
- 61.Viscidi RP, Sun Y, Tsuzaki B, Bosch FX, Munoz N, Shah KV. Serologic response in human papillomavirus-associated invasive cervical cancer. Int J Cancer. 1993;55:780–784. doi: 10.1002/ijc.2910550515. [DOI] [PubMed] [Google Scholar]
- 62.Angelopoulou K, Diamandis EP, Sutherland DJ, Kellen JA, Bunting PS. Prevalence of serum antibodies against the p53 tumor suppressor gene protein in various cancers. Int J Cancer. 1994;58:480–487. doi: 10.1002/ijc.2910580404. [DOI] [PubMed] [Google Scholar]
- 63.Angelopoulou K, Rosen B, Stratis M, Yu H, Solomou M, Diamandis EP. Circulating antibodies against p53 protein in patients with ovarian carcinoma. Correlation with clinicopathologic features and survival Cancer. 1996;78:2146–2152. [PubMed] [Google Scholar]
- 64.Angelopoulou K, Yu H, Bharaj B, Giai M, Diamandis EP. p53 gene mutation, tumor p53 protein overexpression, and serum p53 autoantibody generation in patients with breast cancer. Clin Biochem. 2000;33:53–62. doi: 10.1016/s0009-9120(99)00084-3. [DOI] [PubMed] [Google Scholar]
- 65.Gadducci A, et al. Preoperative serum antibodies against the p53 protein in patients with ovarian and endometrial cancer. Anticancer Res. 1996;16:3519–3523. [PubMed] [Google Scholar]
- 66.Gadducci A, et al. Assessment of the prognostic relevance of serum anti-p53 antibodies in epithelial ovarian cancer. Gynecol Oncol. 1999;72:76–81. doi: 10.1006/gyno.1998.5101. [DOI] [PubMed] [Google Scholar]
- 67.Chinni SR, Gercel-Taylor C, Conner GE, Taylor DD. Cathepsin D antigenic epitopes identified by the humoral responses of ovarian cancer patients. Cancer Immunol Immunother. 1998;46:48–54. doi: 10.1007/s002620050459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yen MJ, et al. Diffuse mesothelin expression correlates with prolonged patient survival in ovarian serous carcinoma. Clin Cancer Res. 2006;12:827–831. doi: 10.1158/1078-0432.CCR-05-1397. [DOI] [PubMed] [Google Scholar]
- 69.Frierson HF, Jr, et al. Large-scale molecular and tissue microarray analysis of mesothelin expression in common human carcinomas. Hum Pathol. 2003;34:605–609. doi: 10.1016/s0046-8177(03)00177-1. [DOI] [PubMed] [Google Scholar]
- 70.Hassan R, Bera T, Pastan I. Mesothelin: a new target for immunotherapy. Clin Cancer Res. 2004;10:3937–3942. doi: 10.1158/1078-0432.CCR-03-0801. [DOI] [PubMed] [Google Scholar]
- 71.Ho M, et al. Humoral immune response to mesothelin in mesothelioma and ovarian cancer patients. Clin Cancer Res. 2005;11:3814–3820. doi: 10.1158/1078-0432.CCR-04-2304. [DOI] [PubMed] [Google Scholar]
- 72.Thomas AM, et al. Mesothelin-specific CD8(+) T cell responses provide evidence of in vivo cross-priming by antigen-presenting cells in vaccinated pancreatic cancer patients. J Exp Med. 2004;200:297–306. doi: 10.1084/jem.20031435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bera TK, Pastan I. Mesothelin is not required for normal mouse development or reproduction. Mol Cell Biol. 2000;20:2902–2906. doi: 10.1128/mcb.20.8.2902-2906.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Roby KF, et al. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis. 2000;21:585–591. doi: 10.1093/carcin/21.4.585. [DOI] [PubMed] [Google Scholar]
- 75.Conejo-Garcia JR, et al. Ovarian carcinoma expresses the NKG2D ligand Letal and promotes the survival and expansion of CD28- antitumor T cells. Cancer Res. 2004;64:2175–2182. doi: 10.1158/0008-5472.can-03-2194. [DOI] [PubMed] [Google Scholar]
- 76.Cheng WF, et al. Generation and characterization of an ascitogenic mesothelin-expressing tumor model. Cancer. 2007 doi: 10.1002/cncr.22781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hung CF, Calizo R, Tsai YC, He L, Wu TC. A DNA vaccine encoding a single-chain trimer of HLA-A2 linked to human mesothelin peptide generates anti-tumor effects against human mesothelin-expressing tumors. Vaccine. 2007;25:127–135. doi: 10.1016/j.vaccine.2006.06.087. [DOI] [PubMed] [Google Scholar]
- 78.Tseng JC, et al. Using sindbis viral vectors for specific detection and suppression of advanced ovarian cancer in animal models. Cancer Res. 2004;64:6684–6692. doi: 10.1158/0008-5472.CAN-04-1924. [DOI] [PubMed] [Google Scholar]
- 79.Connolly DC, et al. Female mice chimeric for expression of the simian virus 40 TAg under control of the MISIIR promoter develop epithelial ovarian cancer. Cancer Res. 2003;63:1389–1397. [PubMed] [Google Scholar]
- 80.Orsulic S. An RCAS-TVA-based approach to designer mouse models. Mamm Genome. 2002;13:543–547. doi: 10.1007/s00335-002-4003-4. [DOI] [PubMed] [Google Scholar]
- 81.Xing D, Orsulic S. A mouse model for the molecular characterization of brca1-associated ovarian carcinoma. Cancer Res. 2006;66:8949–8953. doi: 10.1158/0008-5472.CAN-06-1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wu R, et al. Mouse model of human ovarian endometrioid adenocarcinoma based on somatic defects in the Wnt/beta-catenin and PI3K/Pten signaling pathways. Cancer Cell. 2007;11:321–333. doi: 10.1016/j.ccr.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 83.Dinulescu DM, Ince TA, Quade BJ, Shafer SA, Crowley D, Jacks T. Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat Med. 2005;11:63–70. doi: 10.1038/nm1173. [DOI] [PubMed] [Google Scholar]
- 84.Bellone S, et al. Overexpression of epidermal growth factor type-1 receptor (EGF-R1) in cervical cancer: implications for Cetuximab-mediated therapy in recurrent/metastatic disease. Gynecol Oncol. 2007;106:513–520. doi: 10.1016/j.ygyno.2007.04.028. [DOI] [PubMed] [Google Scholar]
- 85.Ebel W, et al. Preclinical evaluation of MORAb-003, a humanized monoclonal antibody antagonizing folate receptor-alpha. Cancer Immun. 2007;7:6. [PMC free article] [PubMed] [Google Scholar]
- 86.Hassan R, et al. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin Cancer Res. 2007;13:5144–5149. doi: 10.1158/1078-0432.CCR-07-0869. [DOI] [PubMed] [Google Scholar]
- 87.Reinartz S, et al. Vaccination of patients with advanced ovarian carcinoma with the anti-idiotype ACA125: immunological response and survival (phase Ib/II) Clin Cancer Res. 2004;10:1580–1587. doi: 10.1158/1078-0432.ccr-03-0056. [DOI] [PubMed] [Google Scholar]
- 88.Li M, et al. Mesothelin: A malignant factor and a novel therapeutic vaccine target for pancreatic cance. Journal of Surgical Research. 2007;137:194. [Google Scholar]
- 89.Souders NC, et al. Listeria-based vaccines can overcome tolerance by expanding low avidity CD8+ T cells capable of eradicating a solid tumor in a transgenic mouse model of cancer. Cancer Immun. 2007;7:2. [PMC free article] [PubMed] [Google Scholar]
- 90.Sewell DA, Douven D, Pan ZK, Rodriguez A, Paterson Y. Regression of HPV-positive tumors treated with a new Listeria monocytogenes vaccine. Arch Otolaryngol Head Neck Surg. 2004;130:92–97. doi: 10.1001/archotol.130.1.92. [DOI] [PubMed] [Google Scholar]
- 91.Sewell DA, Shahabi V, Gunn GR, 3rd, Pan ZK, Dominiecki ME, Paterson Y. Recombinant Listeria vaccines containing PEST sequences are potent immune adjuvants for the tumor-associated antigen human papillomavirus-16 E7. Cancer Res. 2004;64:8821–8825. doi: 10.1158/0008-5472.CAN-04-1958. [DOI] [PubMed] [Google Scholar]
- 92.Brockstedt DG, et al. Listeria-based cancer vaccines that segregate immunogenicity from toxicity. Proc Natl Acad Sci U S A. 2004;101:13832–13837. doi: 10.1073/pnas.0406035101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Brockstedt DG, et al. Live-attenuated L. monocytogenes encoding mesothelin for immunotherapy of patients with pancreas and ovarian cancers. AACR Annual Meeting; Los Angeles, CA. 2007. [Google Scholar]
- 94.Monahan SJ, Salgaller ML. Viral vectors for gene transfer into antigen presenting cells. Curr Opin Mol Ther. 1999;1:558–564. [PubMed] [Google Scholar]
- 95.Hsieh CJ, et al. Enhancement of vaccinia vaccine potency by linkage of tumor antigen gene to gene encoding calreticulin. Vaccine. 2004;22:3993–4001. doi: 10.1016/j.vaccine.2004.03.057. [DOI] [PubMed] [Google Scholar]
- 96.Lamikanra A, Pan ZK, Isaacs SN, Wu TC, Paterson Y. Regression of established human papillomavirus type 16 (HPV-16) immortalized tumors in vivo by vaccinia viruses expressing different forms of HPV-16 E7 correlates with enhanced CD8(+) T-cell responses that home to the tumor site. J Virol. 2001;75:9654–9664. doi: 10.1128/JVI.75.20.9654-9664.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lin CT, et al. Boosting with recombinant vaccinia increases HPV-16 E7-Specific T cell precursor frequencies and antitumor effects of HPV-16 E7-expressing Sindbis virus replicon particles. Mol Ther. 2003;8:559–566. doi: 10.1016/s1525-0016(03)00238-7. [DOI] [PubMed] [Google Scholar]
- 98.Gomez-Gutierrez JG, Elpek KG, Montes de Oca-Luna R, Shirwan H, Sam Zhou H, McMasters KM. Vaccination with an adenoviral vector expressing calreticulin-human papillomavirus 16 E7 fusion protein eradicates E7 expressing established tumors in mice. Cancer Immunol Immunother. 2007;56:997–1007. doi: 10.1007/s00262-006-0247-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu DW, et al. Recombinant adeno-associated virus expressing human papillomavirus type 16 E7 peptide DNA fused with heat shock protein DNA as a potential vaccine for cervical cancer. J Virol. 2000;74:2888–2894. doi: 10.1128/jvi.74.6.2888-2894.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jin HS, et al. Immunization with adenoviral vectors carrying recombinant IL-12 and E7 enhanced the antitumor immunity to human papillomavirus 16-associated tumor. Gynecol Oncol. 2005;97:559–567. doi: 10.1016/j.ygyno.2005.01.046. [DOI] [PubMed] [Google Scholar]
- 101.Borysiewicz LK, et al. A recombinant vaccinia virus encoding human papillomavirus types 16 and 18, E6 and E7 proteins as immunotherapy for cervical cancer. Lancet. 1996;347:1523–1527. doi: 10.1016/s0140-6736(96)90674-1. [DOI] [PubMed] [Google Scholar]
- 102.Adams M, et al. In vivo induction of HPV 16 specific CTL and T helper responses in patients with advanced cervical cancer using autologous dendritic cells pulsed with tumour lysate. 19th International Papillomavirus Conference; Florianopolis, Brazil. 2001. p. P-99. [Google Scholar]
- 103.Kaufmann AM, et al. Safety and immunogenicity of TA-HPV, a recombinant vaccinia virus expressing modified human papillomavirus (HPV)-16 and HPV-18 E6 and E7 genes, in women with progressive cervical cancer. Clin Cancer Res. 2002;8:3676–3685. [PubMed] [Google Scholar]
- 104.Davidson EJ, et al. Immunological and clinical responses in women with vulval intraepithelial neoplasia vaccinated with a vaccinia virus encoding human papillomavirus 16/18 oncoproteins. Cancer Res. 2003;63:6032–6041. [PubMed] [Google Scholar]
- 105.Kang TH, et al. Epigallocatechin-3-gallate enhances CD8+ T cell-mediated antitumor immunity induced by DNA vaccination. Cancer Res. 2007;67:802–811. doi: 10.1158/0008-5472.CAN-06-2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Baldwin PJ, et al. Vaccinia-expressed human papillomavirus 16 and 18 e6 and e7 as a therapeutic vaccination for vulval and vaginal intraepithelial neoplasia. Clin Cancer Res. 2003;9:5205–5213. [PubMed] [Google Scholar]
- 107.Tseng JC, et al. Systemic tumor targeting and killing by Sindbis viral vectors. Nat Biotechnol. 2004;22:70–77. doi: 10.1038/nbt917. [DOI] [PubMed] [Google Scholar]
- 108.Peng KW, TenEyck CJ, Galanis E, Kalli KR, Hartmann LC, Russell SJ. Intraperitoneal therapy of ovarian cancer using an engineered measles virus. Cancer Res. 2002;62:4656–4662. [PubMed] [Google Scholar]
- 109.Nakamori M, et al. Effective therapy of metastatic ovarian cancer with an oncolytic herpes simplex virus incorporating two membrane fusion mechanisms. Clin Cancer Res. 2003;9:2727–2733. [PubMed] [Google Scholar]
- 110.Hu Y, et al. Yaba-like disease virus: an alternative replicating poxvirus vector for cancer gene therapy. J Virol. 2001;75:10300–10308. doi: 10.1128/JVI.75.21.10300-10308.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hung CF, et al. Vaccinia virus preferentially infects and controls human and murine ovarian tumors in mice. Gene Ther. 2007;14:20–29. doi: 10.1038/sj.gt.3302840. [DOI] [PubMed] [Google Scholar]
- 112.Breidenbach M, et al. Mesothelin-mediated targeting of adenoviral vectors for ovarian cancer gene therapy. Gene Ther. 2005;12:187–193. doi: 10.1038/sj.gt.3302404. [DOI] [PubMed] [Google Scholar]
- 113.Smith CL, et al. Immunodominance of poxviral-specific CTL in a human trial of recombinant-modified vaccinia Ankara. J Immunol. 2005;175:8431–8437. doi: 10.4049/jimmunol.175.12.8431. [DOI] [PubMed] [Google Scholar]
- 114.Jager E, et al. Recombinant vaccinia/fowlpox NY-ESO-1 vaccines induce both humoral and cellular NY-ESO-1-specific immune responses in cancer patients. Proc Natl Acad Sci U S A. 2006;103:14453–14458. doi: 10.1073/pnas.0606512103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Liu M, et al. Gene-based vaccines and immunotherapeutics. Proc Natl Acad Sci U S A. 2004;101 (Suppl 2):14567–14571. doi: 10.1073/pnas.0404845101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Scholl S, et al. Metastatic Breast Tumour Regression Following Treatment by a Gene-Modified Vaccinia Virus Expressing MUC1 and IL-2. J Biomed Biotechnol. 2003;2003:194–201. doi: 10.1155/S111072430320704X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Roden RB, Monie A, Wu TC. Opportunities to improve the prevention and treatment of cervical cancer. Curr Mol Med. 2007;7:490–503. doi: 10.2174/156652407781387127. [DOI] [PubMed] [Google Scholar]
- 118.Chen YF, Lin CW, Tsao YP, Chen SL. Cytotoxic-T-lymphocyte human papillomavirus type 16 E5 peptide with CpG-oligodeoxynucleotide can eliminate tumor growth in C57BL/6 mice. J Virol. 2004;78:1333–1343. doi: 10.1128/JVI.78.3.1333-1343.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Daftarian P, et al. Eradication of established HPV 16-expressing tumors by a single administration of a vaccine composed of a liposome-encapsulated CTL-T helper fusion peptide in a water-in-oil emulsion. Vaccine. 2006;24:5235–5244. doi: 10.1016/j.vaccine.2006.03.079. [DOI] [PubMed] [Google Scholar]
- 120.Qin Y, et al. Human papillomavirus type 16 E7 peptide(38–61) linked with an immunoglobulin G fragment provides protective immunity in mice. Gynecol Oncol. 2005;96:475–483. doi: 10.1016/j.ygyno.2004.10.028. [DOI] [PubMed] [Google Scholar]
- 121.Zwaveling S, et al. Established human papillomavirus type 16-expressing tumors are effectively eradicated following vaccination with long peptides. J Immunol. 2002;169:350–358. doi: 10.4049/jimmunol.169.1.350. [DOI] [PubMed] [Google Scholar]
- 122.Vambutas A, et al. Therapeutic vaccination with papillomavirus E6 and E7 long peptides results in the control of both established virus-induced lesions and latently infected sites in a pre-clinical cottontail rabbit papillomavirus model. Vaccine. 2005;23:5271–5280. doi: 10.1016/j.vaccine.2005.04.049. [DOI] [PubMed] [Google Scholar]
- 123.Ressing ME, et al. Detection of T helper responses, but not of human papillomavirus-specific cytotoxic T lymphocyte responses, after peptide vaccination of patients with cervical carcinoma. J Immunother. 2000;23:255–266. doi: 10.1097/00002371-200003000-00010. [DOI] [PubMed] [Google Scholar]
- 124.van Driel WJ, et al. Vaccination with HPV16 peptides of patients with advanced cervical carcinoma: clinical evaluation of a phase I-II trial. Eur J Cancer. 1999;35:946–952. doi: 10.1016/s0959-8049(99)00048-9. [DOI] [PubMed] [Google Scholar]
- 125.Muderspach L, et al. A phase I trial of a human papillomavirus (HPV) peptide vaccine for women with high-grade cervical and vulvar intraepithelial neoplasia who are HPV 16 positive. Clin Cancer Res. 2000;6:3406–3416. [PubMed] [Google Scholar]
- 126.Welters MJ, et al. Long peptide vaccine-induced migration of HPV16 specific type 1 and 2T-cells into the lesions of VIN 3 patients . 23rd International Papillomavirus Conference & Clinical Workshop 2006; 2006. [Google Scholar]
- 127.Yokokawa J, et al. Identification of novel human CTL epitopes and their agonist epitopes of mesothelin. Clin Cancer Res. 2005;11:6342–6351. doi: 10.1158/1078-0432.CCR-05-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hung CF, Tsai YC, He L, Wu TC. Control of mesothelin-expressing ovarian cancer using adoptive transfer of mesothelin peptide-specific CD8(+) T cells. Gene Ther. 2007;14:921–929. doi: 10.1038/sj.gt.3302913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Greenstone HL, et al. Chimeric papillomavirus virus-like particles elicit antitumor immunity against the E7 oncoprotein in an HPV16 tumor model. Proc Natl Acad Sci U S A. 1998;95:1800–1805. doi: 10.1073/pnas.95.4.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Peng S, Frazer IH, Fernando GJ, Zhou J. Papillomavirus virus-like particles can deliver defined CTL epitopes to the MHC class I pathway. Virology. 1998;240:147–157. doi: 10.1006/viro.1997.8912. [DOI] [PubMed] [Google Scholar]
- 131.Schafer K, et al. Immune response to human papillomavirus 16 L1E7 chimeric virus-like particles: induction of cytotoxic T cells and specific tumor protection. Int J Cancer. 1999;81:881–888. doi: 10.1002/(sici)1097-0215(19990611)81:6<881::aid-ijc8>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 132.Stewart TJ, et al. ISCOMATRIX adjuvant: an adjuvant suitable for use in anticancer vaccines. Vaccine. 2004;22:3738–3743. doi: 10.1016/j.vaccine.2004.03.026. [DOI] [PubMed] [Google Scholar]
- 133.Cui Z, Huang L. Liposome-polycation-DNA (LPD) particle as a carrier and adjuvant for protein-based vaccines: Therapeutic effect against cervical cancer. Cancer Immunol Immunother. 2005 doi: 10.1007/s00262-005-0685-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Preville X, Ladant D, Timmerman B, Leclerc C. Eradication of established tumors by vaccination with recombinant Bordetella pertussis adenylate cyclase carrying the human papillomavirus 16 E7 oncoprotein. Cancer Res. 2005;65:641–649. [PubMed] [Google Scholar]
- 135.Liao CW, et al. Fusion protein vaccine by domains of bacterial exotoxin linked with a tumor antigen generates potent immunologic responses and antitumor effects. Cancer Res. 2005;65:9089–9098. doi: 10.1158/0008-5472.CAN-05-0958. [DOI] [PubMed] [Google Scholar]
- 136.Qian X, Lu Y, Liu Q, Chen K, Zhao Q, Song J. Prophylactic, therapeutic and anti-metastatic effects of an HPV-16mE6Delta/mE7/TBhsp70Delta fusion protein vaccine in an animal model. Immunol Lett. 2006;102:191–201. doi: 10.1016/j.imlet.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 137.Chu NR, Wu HB, Wu T, Boux LJ, Siegel MI, Mizzen LA. Immunotherapy of a human papillomavirus (HPV) type 16 E7-expressing tumour by administration of fusion protein comprising Mycobacterium bovis bacille Calmette-Guerin (BCG) hsp65 and HPV16 E7. Clin Exp Immunol. 2000;121:216–225. doi: 10.1046/j.1365-2249.2000.01293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lacey CJ, et al. Phase IIa safety and immunogenicity of a therapeutic vaccine, TA-GW, in persons with genital warts. J Infect Dis. 1999;179:612–618. doi: 10.1086/314616. [DOI] [PubMed] [Google Scholar]
- 139.Thompson HS, et al. Phase I safety and antigenicity of TA-GW: a recombinant HPV6 L2E7 vaccine for the treatment of genital warts. Vaccine. 1999;17:40–49. doi: 10.1016/s0264-410x(98)00146-7. [DOI] [PubMed] [Google Scholar]
- 140.Frazer IH, et al. Phase 1 study of HPV16-specific immunotherapy with E6E7 fusion protein and ISCOMATRIX adjuvant in women with cervical intraepithelial neoplasia. Vaccine. 2004;23:172–181. doi: 10.1016/j.vaccine.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 141.Hallez S, et al. Phase I/II trial of immunogenicity of a human papillomavirus (HPV) type 16 E7 protein-based vaccine in women with oncogenic HPV-positive cervical intraepithelial neoplasia. Cancer Immunol Immunother. 2004;53:642–650. doi: 10.1007/s00262-004-0501-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chu NR. Therapeutic vaccination for the treatment of mucosotropic human papillomavirus-associated disease. Expert Opin Biol Ther. 2003;3:477–486. doi: 10.1517/14712598.3.3.477. [DOI] [PubMed] [Google Scholar]
- 143.Palefsky JM, et al. A trial of SGN-00101 (HspE7) to treat high-grade anal intraepithelial neoplasia in HIV-positive individuals. Aids. 2006;20:1151–1155. doi: 10.1097/01.aids.0000226955.02719.26. [DOI] [PubMed] [Google Scholar]
- 144.Einstein MH, et al. Heat shock fusion protein-based immunotherapy for treatment of cervical intraepithelial neoplasia III. Gynecol Oncol. 2007 doi: 10.1016/j.ygyno.2007.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chang CL, Wu TC, Hung CF. Control of human mesothelin-expressing tumors by DNA vaccines. Gene Ther. 2007;14:1189–1198. doi: 10.1038/sj.gt.3302974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chowdhury PS, Gallo M, Pastan I. Generation of high titer antisera in rabbits by DNA immunization. J Immunol Methods. 2001;249:147–154. doi: 10.1016/s0022-1759(00)00353-7. [DOI] [PubMed] [Google Scholar]
- 147.Hung CF, Wu TC. Improving DNA vaccine potency via modification of professional antigen presenting cells. Curr Opin Mol Ther. 2003;5:20–24. [PubMed] [Google Scholar]
- 148.Tsen SW, Paik AH, Hung CF, Wu TC. Enhancing DNA vaccine potency by modifying the properties of antigen-presenting cells. Expert Rev Vaccines. 2007;6:227–239. doi: 10.1586/14760584.6.2.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Luckay A, et al. Effect of plasmid DNA vaccine design and in vivo electroporation on the resulting vaccine-specific immune responses in rhesus macaques. J Virol. 2007;81:5257–5269. doi: 10.1128/JVI.00055-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Trimble C, et al. Comparison of the CD8+ T cell responses and antitumor effects generated by DNA vaccine administered through gene gun, biojector, and syringe. Vaccine. 2003;21:4036–4042. doi: 10.1016/s0264-410x(03)00275-5. [DOI] [PubMed] [Google Scholar]
- 151.Hung C-F, et al. Improving vaccine potency through intercellular spreading and enhanced MHC class I presentation of antigen. J Immunol. 2001;166:5733–5740. doi: 10.4049/jimmunol.166.9.5733. [DOI] [PubMed] [Google Scholar]
- 152.Hung CF, He L, Juang J, Lin TJ, Ling M, Wu TC. Improving DNA Vaccine Potency by Linking Marek’s Disease Virus Type 1 VP22 to an Antigen. J Virol. 2002;76:2676–2682. doi: 10.1128/JVI.76.6.2676-2682.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Cheung YK, Cheng SC, Sin FW, Xie Y. Plasmid encoding papillomavirus Type 16 (HPV16) DNA constructed with codon optimization improved the immunogenicity against HPV infection. Vaccine. 2004;23:629–638. doi: 10.1016/j.vaccine.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 154.Liu WJ, et al. Codon modified human papillomavirus type 16 E7 DNA vaccine enhances cytotoxic T-lymphocyte induction and anti-tumour activity. Virology. 2002;301:43–52. doi: 10.1006/viro.2002.1584. [DOI] [PubMed] [Google Scholar]
- 155.Lin CT, et al. A DNA Vaccine Encoding a Codon-Optimized Human Papillomavirus Type 16 E6 Gene Enhances CTL Response and Anti-tumor Activity. J Biomed Sci. 2006 doi: 10.1007/s11373-006-9086-6. [DOI] [PubMed] [Google Scholar]
- 156.Chen CH, et al. Enhancement of DNA vaccine potency by linkage of antigen gene to an HSP70 gene. Cancer Res. 2000;60:1035–1042. [PubMed] [Google Scholar]
- 157.Hung CF, et al. Enhancing major histocompatibility complex class I antigen presentation by targeting antigen to centrosomes. Cancer Res. 2003;63:2393–2398. [PubMed] [Google Scholar]
- 158.Hung C-F, et al. Enhancement of DNA vaccine potency by linkage of antigen gene to a gene encoding the extracellular domain of Flt3-ligand. Cancer Research. 2001;61:1080–1088. [PubMed] [Google Scholar]
- 159.Hung C-F, et al. Cancer immunotherapy using a DNA vaccine encoding the translocation domain of a bacterial toxin linked to a tumor antigen. Cancer Research. 2001;61:3698–3703. [PubMed] [Google Scholar]
- 160.Kim JW, et al. Comparison of HPV DNA vaccines employing intracellular targeting strategies. Gene Ther. 2004;11:1011–1018. doi: 10.1038/sj.gt.3302252. [DOI] [PubMed] [Google Scholar]
- 161.Moniz M, Ling M, Hung CF, Wu TC. HPV DNA vaccines. Front Biosci. 2003;8:d55–68. doi: 10.2741/936. [DOI] [PubMed] [Google Scholar]
- 162.Wu TC, et al. Engineering an intracellular pathway for major histocompatibility complex class II presentation of antigens. Proc Natl Acad Sci U S A. 1995;92:11671–11675. doi: 10.1073/pnas.92.25.11671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ji H, et al. Targeting HPV-16 E7 to the endosomal/lysosomal compartment enhances the antitumor immunity of DNA vaccines against murine HPV-16 E7-expressing tumors. Hum Gene Ther. 1999;10:2727–2740. doi: 10.1089/10430349950016474. [DOI] [PubMed] [Google Scholar]
- 164.Hung CF, Tsai YC, He L, Wu TC. DNA Vaccines Encoding Ii-PADRE Generates Potent PADRE-specific CD4(+) T-Cell Immune Responses and Enhances Vaccine Potency. Mol Ther. 2007;15:1211–1219. doi: 10.1038/sj.mt.6300121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Huang CH, et al. Cancer immunotherapy using a DNA vaccine encoding a single-chain trimer of MHC class I linked to an HPV-16 E6 immunodominant CTL epitope. Gene Ther. 2005;12:1180–1186. doi: 10.1038/sj.gt.3302519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kim TW, et al. Enhancing DNA vaccine potency by combining a strategy to prolong dendritic cell life with intracellular targeting strategies. J Immunol. 2003;171:2970–2976. doi: 10.4049/jimmunol.171.6.2970. [DOI] [PubMed] [Google Scholar]
- 167.Kim TW, et al. Enhancing DNA vaccine potency by coadministration of DNA encoding antiapoptotic proteins. J Clin Invest. 2003;112:109–117. doi: 10.1172/JCI17293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kim TW, Lee JH, He L, Boyd DA, Hung CF, Wu TC. DNA vaccines employing intracellular targeting strategies and a strategy to prolong dendritic cell life generate a higher number of CD8+ memory T cells and better long-term antitumor effects compared with a DNA prime-vaccinia boost regimen. Hum Gene Ther. 2005;16:26–34. doi: 10.1089/hum.2005.16.26. [DOI] [PubMed] [Google Scholar]
- 169.Leachman SA, et al. Granulocyte-macrophage colony-stimulating factor priming plus papillomavirus E6 DNA vaccination: effects on papilloma formation and regression in the cottontail rabbit papillomavirus--rabbit model. J Virol. 2000;74:8700–8708. doi: 10.1128/jvi.74.18.8700-8708.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Chen Y, Hu D, Eling DJ, Robbins J, Kipps TJ. DNA vaccines encoding full-length or truncated Neu induce protective immunity against Neu-expressing mammary tumors. Cancer Res. 1998;58:1965–1971. [PubMed] [Google Scholar]
- 171.Kim MS, Sin JI. Both antigen optimization and lysosomal targeting are required for enhanced anti-tumour protective immunity in a human papillomavirus E7-expressing animal tumour model. Immunology. 2005;116:255–266. doi: 10.1111/j.1365-2567.2005.02219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Sheets EE, et al. Immunotherapy of human cervical high-grade cervical intraepithelial neoplasia with microparticle-delivered human papillomavirus 16 E7 plasmid DNA. Am J Obstet Gynecol. 2003;188:916–926. doi: 10.1067/mob.2003.256. [DOI] [PubMed] [Google Scholar]
- 173.Klencke B, et al. Encapsulated plasmid DNA treatment for human papillomavirus 16-associated anal dysplasia: a Phase I study of ZYC101. Clin Cancer Res. 2002;8:1028–1037. [PubMed] [Google Scholar]
- 174.Garcia F, et al. ZYC101a for treatment of high-grade cervical intraepithelial neoplasia: a randomized controlled trial. Obstet Gynecol. 2004;103:317–326. doi: 10.1097/01.AOG.0000110246.93627.17. [DOI] [PubMed] [Google Scholar]
- 175.Herd KA, Harvey T, Khromykh AA, Tindle RW. Recombinant Kunjin virus replicon vaccines induce protective T-cell immunity against human papillomavirus 16 E7-expressing tumour. Virology. 2004;319:237–248. doi: 10.1016/j.virol.2003.10.032. [DOI] [PubMed] [Google Scholar]
- 176.Varnavski AN, Young PR, Khromykh AA. Stable high-level expression of heterologous genes in vitro and in vivo by noncytopathic DNA-based Kunjin virus replicon vectors. J Virol. 2000;74:4394–4403. doi: 10.1128/jvi.74.9.4394-4403.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Cheng WF, et al. Enhancement of sindbis virus self-replicating RNA vaccine potency by targeting antigen to endosomal/lysosomal compartments. Hum Gene Ther. 2001;12:235–252. doi: 10.1089/10430340150218387. [DOI] [PubMed] [Google Scholar]
- 178.Cheng WF, et al. Sindbis virus replicon particles encoding calreticulin linked to a tumor antigen generate long-term tumor-specific immunity. Cancer Gene Ther. 2006;13:873–885. doi: 10.1038/sj.cgt.7700956. [DOI] [PubMed] [Google Scholar]
- 179.Cheng W-F, et al. Enhancement of Sindbis virus self-replicating RNA vaccine potency by linkage of herpes simplex virus type 1 VP22 protein to antigen. J Virol. 2001;75:2368–2376. doi: 10.1128/JVI.75.5.2368-2376.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Cheng W-F, et al. Enhancement of Sindbis virus self-replicating RNA vaccine potency by linkage of Mycobacterium tuberculosis heat shock protein 70 gene to an antigen gene. J Immunol. 2001;166:6218–6226. doi: 10.4049/jimmunol.166.10.6218. [DOI] [PubMed] [Google Scholar]
- 181.Cassetti MC, et al. Antitumor efficacy of Venezuelan equine encephalitis virus replicon particles encoding mutated HPV16 E6 and E7 genes. Vaccine. 2004;22:520–527. doi: 10.1016/j.vaccine.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 182.Velders MP, et al. Eradication of established tumors by vaccination with Venezuelan equine encephalitis virus replicon particles delivering human papillomavirus 16 E7 RNA. Cancer Res. 2001;61:7861–7867. [PubMed] [Google Scholar]
- 183.Santin AD, Bellone S, Roman JJ, Burnett A, Cannon MJ, Pecorelli S. Therapeutic vaccines for cervical cancer: dendritic cell-based immunotherapy. Curr Pharm Des. 2005;11:3485–3500. doi: 10.2174/138161205774414565. [DOI] [PubMed] [Google Scholar]
- 184.Doan T, Herd KA, Lambert PF, Fernando GJ, Street MD, Tindle RW. Peripheral tolerance to human papillomavirus E7 oncoprotein occurs by cross-tolerization, is largely Th-2-independent, and is broken by dendritic cell immunization. Cancer Res. 2000;60:2810–2815. [PubMed] [Google Scholar]
- 185.Murakami M, Gurski KJ, Steller MA. Human papillomavirus vaccines for cervical cancer. J Immunother. 1999;22:212–218. doi: 10.1097/00002371-199905000-00003. [DOI] [PubMed] [Google Scholar]
- 186.Peng S, et al. Vaccination with dendritic cells transfected with BAK and BAX siRNA enhances antigen-specific immune responses by prolonging dendritic cell life. Hum Gene Ther. 2005;16:584–593. doi: 10.1089/hum.2005.16.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kim TW, et al. A DNA vaccine co-expressing antigen and an anti-apoptotic molecule further enhances the antigen-specific CD8+ T-cell immune response. J Biomed Sci. 2004;11:493–499. doi: 10.1007/BF02256098. [DOI] [PubMed] [Google Scholar]
- 188.Santin AD, Bellone S, Gokden M, Cannon MJ, Parham GP. Vaccination with HPV-18 E7-pulsed dendritic cells in a patient with metastatic cervical cancer. N Engl J Med. 2002;346:1752–1753. doi: 10.1056/NEJM200205303462219. [DOI] [PubMed] [Google Scholar]
- 189.Santin AD, et al. HPV16/18 E7-pulsed dendritic cell vaccination in cervical cancer patients with recurrent disease refractory to standard treatment modalities. Gynecol Oncol. 2006;100:469–478. doi: 10.1016/j.ygyno.2005.09.040. [DOI] [PubMed] [Google Scholar]
- 190.Ferrara A, et al. Dendritic cell-based tumor vaccine for cervical cancer II: results of a clinical pilot study in 15 individual patients. J Cancer Res Clin Oncol. 2003;129:521–530. doi: 10.1007/s00432-003-0463-5. [DOI] [PubMed] [Google Scholar]
- 191.Bubenik J, et al. Interleukin 2 gene therapy of residual disease in mice carrying tumours induced by HPV 16. Int J Oncol. 1999;14:593–597. doi: 10.3892/ijo.14.3.593. [DOI] [PubMed] [Google Scholar]
- 192.Hallez S, et al. Interleukin-12-secreting human papillomavirus type 16-transformed cells provide a potent cancer vaccine that generates E7-directed immunity. Int J Cancer. 1999;81:428–437. doi: 10.1002/(sici)1097-0215(19990505)81:3<428::aid-ijc17>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 193.Mikyskova R, et al. Treatment of minimal residual disease after surgery or chemotherapy in mice carrying HPV16-associated tumours: Cytokine and gene therapy with IL-2 and GM-CSF. Int J Oncol. 2004;24:161–167. [PubMed] [Google Scholar]
- 194.Chang EY, et al. Antigen-specific cancer immunotherapy using a GM-CSF secreting allogeneic tumor cell-based vaccine. Int J Cancer. 2000;86:725–730. doi: 10.1002/(sici)1097-0215(20000601)86:5<725::aid-ijc19>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 195.Chang CL, Tsai YC, He L, Wu TC, Hung CF. Cancer immunotherapy using irradiated tumor cells secreting heat shock protein 70. Cancer Res. 2007;67:10047–10057. doi: 10.1158/0008-5472.CAN-07-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Jaffee EM, et al. Novel allogeneic granulocyte-macrophage colony-stimulating factor-secreting tumor vaccine for pancreatic cancer: a phase I trial of safety and immune activation. J Clin Oncol. 2001;19:145–156. doi: 10.1200/JCO.2001.19.1.145. [DOI] [PubMed] [Google Scholar]
- 197.Wlazlo AP, Deng H, Giles-Davis W, Ertl HC. DNA vaccines against the human papillomavirus type 16 E6 or E7 oncoproteins. Cancer Gene Ther. 2004 doi: 10.1038/sj.cgt.7700723. [DOI] [PubMed] [Google Scholar]
- 198.Chen CH, Wang TL, Hung CF, Pardoll DM, Wu TC. Boosting with recombinant vaccinia increases HPV-16 E7-specific T cell precursor frequencies of HPV-16 E7-expressing DNA vaccines. Vaccine. 2000;18:2015–2022. doi: 10.1016/s0264-410x(99)00528-9. [DOI] [PubMed] [Google Scholar]
- 199.Mackova J, et al. Prime/boost immunotherapy of HPV16-induced tumors with E7 protein delivered by Bordetella adenylate cyclase and modified vaccinia virus Ankara. Cancer Immunol Immunother. 2006;55:39–46. doi: 10.1007/s00262-005-0700-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Fiander AN, et al. Prime-boost vaccination strategy in women with high-grade, noncervical anogenital intraepithelial neoplasia: clinical results from a multicenter phase II trial. Int J Gynecol Cancer. 2006;16:1075–1081. doi: 10.1111/j.1525-1438.2006.00598.x. [DOI] [PubMed] [Google Scholar]
- 201.Smyth LJ, et al. Immunological responses in women with human papillomavirus type 16 (HPV-16)-associated anogenital intraepithelial neoplasia induced by heterologous prime-boost HPV-16 oncogene vaccination. Clin Cancer Res. 2004;10:2954–2961. doi: 10.1158/1078-0432.ccr-03-0703. [DOI] [PubMed] [Google Scholar]
- 202.Davidson EJ, et al. Effect of TA-CIN (HPV 16 L2E6E7) booster immunisation in vulval intraepithelial neoplasia patients previously vaccinated with TA-HPV (vaccinia virus encoding HPV 16/18 E6E7) Vaccine. 2004;22:2722–2729. doi: 10.1016/j.vaccine.2004.01.049. [DOI] [PubMed] [Google Scholar]
- 203.Bae SH, Park YJ, Park JB, Choi YS, Kim MS, Sin JI. Therapeutic synergy of human papillomavirus E7 subunit vaccines plus cisplatin in an animal tumor model: causal involvement of increased sensitivity of cisplatin-treated tumors to CTL-mediated killing in therapeutic synergy. Clin Cancer Res. 2007;13:341–349. doi: 10.1158/1078-0432.CCR-06-1838. [DOI] [PubMed] [Google Scholar]
- 204.Ye GW, Park JB, Park YJ, Choi YS, Sin JI. Increased Sensitivity of Radiated Murine Cervical Cancer Tumors to E7 Subunit Vaccine-driven CTL-mediated Killing Induces Synergistic Anti-tumor Activity. Mol Ther. 2007 doi: 10.1038/sj.mt.6300149. [DOI] [PubMed] [Google Scholar]
- 205.Peggs KS, Quezada SA, Korman AJ, Allison JP. Principles and use of anti-CTLA4 antibody in human cancer immunotherapy. Curr Opin Immunol. 2006;18:206–213. doi: 10.1016/j.coi.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 206.Blank C, Mackensen A. Contribution of the PD-L1/PD-1 pathway to T-cell exhaustion: an update on implications for chronic infections and tumor evasion. Cancer Immunol Immunother. 2007;56:739–745. doi: 10.1007/s00262-006-0272-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Goldberg MV, et al. Role of PD-1 and its ligand, B7-H1, in early fate decisions of CD8 T cells. Blood. 2007;110:186–192. doi: 10.1182/blood-2006-12-062422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7:41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 209.Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature. 2002;419:734–738. doi: 10.1038/nature01112. [DOI] [PubMed] [Google Scholar]
- 210.Munn DH, Mellor AL. IDO and tolerance to tumors. Trends Mol Med. 2004;10:15–18. doi: 10.1016/j.molmed.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 211.Rubinstein N, et al. Targeted inhibition of galectin-1 gene expression in tumor cells results in heightened T cell-mediated rejection; A potential mechanism of tumor-immune privilege. Cancer Cell. 2004;5:241–251. doi: 10.1016/s1535-6108(04)00024-8. [DOI] [PubMed] [Google Scholar]
- 212.Yue FY, et al. Interleukin-10 is a growth factor for human melanoma cells and down-regulates HLA class-I, HLA class-II and ICAM-1 molecules. Int J Cancer. 1997;71:630–637. doi: 10.1002/(sici)1097-0215(19970516)71:4<630::aid-ijc20>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 213.Gorelik L, Flavell RA. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat Med. 2001;7:1118–1122. doi: 10.1038/nm1001-1118. [DOI] [PubMed] [Google Scholar]
- 214.Nagaraj S, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med. 2007;13:828–835. doi: 10.1038/nm1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Terry KL, Titus-Ernstoff L, McKolanis JR, Welch WR, Finn OJ, Cramer DW. Incessant ovulation, mucin 1 immunity, and risk for ovarian cancer. Cancer Epidemiol Biomarkers Prev. 2007;16:30–35. doi: 10.1158/1055-9965.EPI-06-0688. [DOI] [PubMed] [Google Scholar]
- 216.Cramer DW, et al. Conditions associated with antibodies against the tumor-associated antigen MUC1 and their relationship to risk for ovarian cancer. Cancer Epidemiol Biomarkers Prev. 2005;14:1125–1131. doi: 10.1158/1055-9965.EPI-05-0035. [DOI] [PubMed] [Google Scholar]
- 217.Erkanli A, et al. Application of Bayesian modeling of autologous antibody responses against ovarian tumor-associated antigens to cancer detection. Cancer Res. 2006;66:1792–1798. doi: 10.1158/0008-5472.CAN-05-0669. [DOI] [PubMed] [Google Scholar]
- 218.Carbone DP, et al. Immunization with mutant p53- and K-ras-derived peptides in cancer patients: immune response and clinical outcome. J Clin Oncol. 2005;23:5099–5107. doi: 10.1200/JCO.2005.03.158. [DOI] [PubMed] [Google Scholar]
- 219.Nakayama K, et al. Amplicon profiles in ovarian serous carcinomas. Int J Cancer. 2007;120:2613–2617. doi: 10.1002/ijc.22609. [DOI] [PubMed] [Google Scholar]
- 220.Kim JH, et al. Identification of epithelial cell adhesion molecule autoantibody in patients with ovarian cancer. Clin Cancer Res. 2003;9:4782–4791. [PubMed] [Google Scholar]
- 221.Richards ER, Devine PL, Quin RJ, Fontenot JD, Ward BG, McGuckin MA. Antibodies reactive with the protein core of MUC1 mucin are present in ovarian cancer patients and healthy women. Cancer Immunol Immunother. 1998;46:245–252. doi: 10.1007/s002620050484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Chapman C, et al. Autoantibodies in breast cancer: their use as an aid to early diagnosis. Ann Oncol. 2007;18:868–873. doi: 10.1093/annonc/mdm007. [DOI] [PubMed] [Google Scholar]
- 223.Luo LY, Herrera I, Soosaipillai A, Diamandis EP. Identification of heat shock protein 90 and other proteins as tumour antigens by serological screening of an ovarian carcinoma expression library. Br J Cancer. 2002;87:339–343. doi: 10.1038/sj.bjc.6600439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Knutson KL, et al. T-cell immunity to the folate receptor alpha is prevalent in women with breast or ovarian cancer. J Clin Oncol. 2006;24:4254–4261. doi: 10.1200/JCO.2006.05.9311. [DOI] [PubMed] [Google Scholar]
- 225.Naora H, Yang YQ, Montz FJ, Seidman JD, Kurman RJ, Roden RB. A serologically identified tumor antigen encoded by a homeobox gene promotes growth of ovarian epithelial cells. Proc Natl Acad Sci U S A. 2001;98:4060–4065. doi: 10.1073/pnas.071594398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Bleul C, et al. Human papillomavirus type 18 E6 and E7 antibodies in human sera: increased anti-E7 prevalence in cervical cancer patients. J Clin Microbiol. 1991;29:1579–1588. doi: 10.1128/jcm.29.8.1579-1588.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Muller M, et al. Antibodies to HPV-16 E6 and E7 proteins as markers for HPV-16-associated invasive cervical cancer. Virology. 1992;187:508–514. doi: 10.1016/0042-6822(92)90453-v. [DOI] [PubMed] [Google Scholar]