

Summary
The arduous task of rationally designing small molecule enzyme inhibitors is complicated by the inherent flexibility of the protein scaffold. To gain insight into the changes in dynamics associated with small molecule based inhibition, we have characterized, using NMR spectroscopy, E. coli dihydrofolate reductase in complex with two drugs: methotrexate and trimethoprim. The complexes allowed the intrinsic dynamic effects of drug binding to be revealed within the context of the “closed” structural ensemble. Binding of both drugs results in an identical decoupling of global motion on the micro- to millisecond timescale. Consistent with a change in overall dynamic character, the drugs’ perturbations to pico- to nanosecond backbone and side-chain methyl dynamics are also highly similar. These data show that the inhibitors simultaneously modulate slow concerted switching and fast motions at distal regions of DHFR, providing a dynamic link between the substrate binding site and distal loop residues known to affect catalysis.
Introduction
Proteins adopt ensembles of conformations in solution. Although this is often demonstrated with enzymes, the dynamic equilibria between structural substates in either catalytic or non-catalytic proteins have a direct impact on substrate or ligand binding, catalysis, and product release (Boehr et al., 2006; Careri et al., 1979; Eisenmesser et al., 2002; Frederick et al., 2007; Karplus and McCammon, 2002; Lee et al., 2000; Watt et al., 2007; Zidek et al., 1999). Understanding how protein dynamics influences small molecule binding and release should therefore greatly enhance efficient rational drug design (Teague, 2003). Surprisingly, few experimental studies have focused on the dynamical consequences of drug inhibition. Herein, we detail a structural and multi-timescale dynamical study of Escherichia coli dihydrofolate reductase bound to its cofactor NADPH and to either of the small molecule drugs methotrexate (MTX) or trimethoprim (TMP). NMR spectroscopy is uniquely suited to study protein structure and dynamics in solution with atomic resolution and was used extensively in this report.
Dihydrofolate reductase (DHFR) is a classic drug target and one of the most comprehensively studied enzymes (Schnell et al., 2004b). It catalyzes the NADPH-dependent reduction of dihydrofolate to tetrahydrofolate. Tetrahydrofolate is required for the biosynthesis of DNA bases and key amino acids. DHFR thus plays a vital role in cell proliferation, and is a popular target for chemotherapeutic agents. MTX and TMP are established antifolates against DHFR, sharing a common 2,4-diamino pyrimidine moiety (Figure 1A). MTX, the first successful anti-cancer drug (2006; Huennekens, 1994), not only binds to human DHFR, but it also binds with low nanomolar affinity to the E. coli enzyme (Sasso et al., 1994). TMP is an antibiotic that specifically targets the bacterial enzyme, also binding with high affinity (Kd = 6 pM) (Sasso et al., 1994). We sought to determine how binding of these different drugs impacts the motions of this functionally dynamic enzyme.
Figure 1.
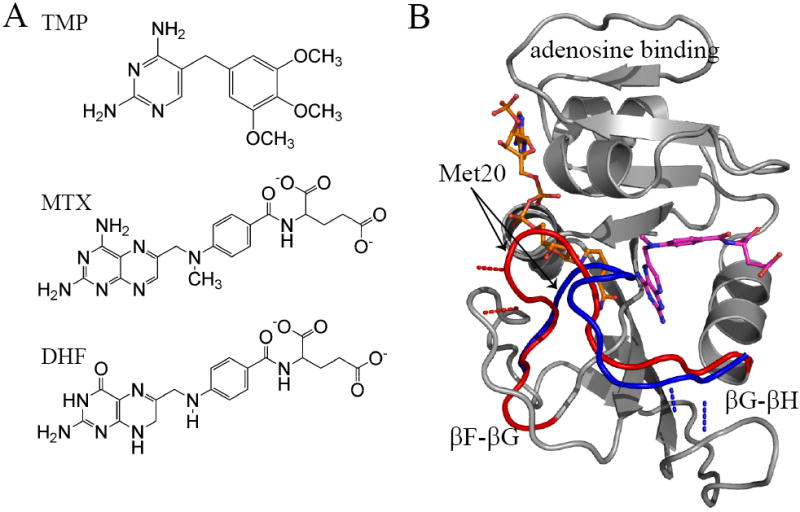
The structure of DHFR, its inhibitors, and substrate. A) The chemical structures of trimethoprim, methotrexate, and dihydrofolate. B) DHFR transitions between the closed (red; PDB 1rx3) and occluded (blue; PDB 1rx5) Met20 loop conformations. This transition involves breaking hydrogen bonds with the F-G loop and creating interactions with the G-H loop illustrated by the red (closed) and blue (occluded) dashed lines. Regions of the DHFR structure that do not undergo significant conformational change are rendered in grey. Methotrexate is shown in magenta and NADPH in orange. NADPH is illustrated occupying the active site, which can only occur in the “closed” conformation.
During its catalytic cycle, E. coli DHFR switches between “closed” and “occluded” conformations (Sawaya and Kraut, 1997). This global exchange is largely defined by the conformation of the Met20 loop flanking the active site (Figure 1B). In the closed conformation, the nicotinamide moiety of NADPH is buried within the active site. The Met20 loop packs against the cofactor, closing the active site to solvent. The closed conformation is partially stabilized by hydrogen bond interactions between the Met20 loop and the F-G loop (Figure 1B)(Sawaya and Kraut, 1997). Upon hydride transfer, DHFR undergoes a conformational switch to the occluded conformation. The Met20 loop forms a short 310 helix, projecting the side chain of M16 into the active site, thus occluding the cofactor binding pocket. To form the occluded conformation, hydrogen bond interactions between the Met20 loop and the F-G loop are broken, and new interactions with the residues on the G-H loop are formed (Sawaya and Kraut, 1997). While the work of Wright and colleagues have focused on the catalytic cycle and how dynamics are modulated by the conformation of the Met20 loop (Boehr et al., 2006; McElheny et al., 2005; Osborne et al., 2001; Schnell et al., 2004a, b), less has been reported on how inhibitors alter E. coli DHFR dynamics. In fact, even though the same (closed) conformation is present before and after dihydrofolate or drug binding, there have been few studies of the effect of ligand binding in the context of the closed state. Most studies have either involved binding at the cofactor site or the study of major conformational change in the loops flanking the active site of DHFR (for a review see (Schnell et al., 2004b)). The study of drug binding within the closed state allows intrinsic drug binding effects to be separated from subsequent conformational changes.
In this paper, we present the results of a comprehensive dynamical investigation of DHFR inhibition brought about by small molecule drugs. Use of MTX and TMP ligands in this system allows for NMR-monitored dynamics to report on both the sampling of minor semi-stable conformations within the closed ensemble, as well as smaller, faster motions that may lead to those conformational excursions. Experiments sensitive to micro- to millisecond (μs-ms) conformational dynamics indicate localized regions of drug-bound DHFR experience slow motion analogous to the reactive complex (vide infra). Moreover, MTX and TMP alter the collective enzyme motions in exactly the same manner: residues lining the drugs retain their μs-ms switching, whereas distal loops stop switching altogether. Thus, as a whole, the inhibited protein is dynamically dysfunctional. Drug-bound DHFR appears to be on the brink of a global transition, but its restricted loops prevent the transition from occurring, leaving a “half-switching” enzyme. Changes in pico- to nanosecond (ps-ns) backbone amide and side-chain methyl dynamics indicate drug binding is “felt” throughout the protein. These changes in fast dynamics show structural connectivity to distal loops implicated in function and provide insight into the possible linkage between fast fluctuations that may precede or contribute to slower switching events. Our results are consistent with experimental and theoretical studies that indicate long-range coupling between the active site and distal regions of DHFR (Hammes-Schiffer and Benkovic, 2006; Ohmae et al., 1996; Pan et al., 2000).
Results and Discussion
Predominance of the closed conformation in solution upon drug binding
Crystal structures exist for the holoenzyme (E:NADPH) and the MTX complex (E:NADPH:MTX) (Sawaya and Kraut, 1997), but not for the TMP complex (E:NADPH:TMP). Although the holoenzyme has been crystallized in both closed and occluded states as shown in Figure 1B, recent solution NMR work shows it to be predominantly closed in solution (Osborne et al., 2003). The MTX complex, considered a model for DHFR’s transition state, is also found in the closed conformation (Matthews et al., 1977; Sawaya and Kraut, 1997). While the TMP complex has been solved for L. casei DHFR (Polshakov et al., 2002), the structural features of the corresponding E. coli complex were unclear. Thus, we sought to determine the structural state of the TMP complex and detect any differences in structure between the three complexes using NMR chemical shifts and residual dipolar couplings (RDCs).
DHFR amide chemical shift changes are exquisitely sensitive to ligand binding and to the active site loop conformation (Osborne et al., 2003). Chemical shift changes in response to MTX and TMP binding were found to be primarily proximal to drug, thus reporting on ligand binding, with similar patterns for the two drugs (Figure 2A). If either ternary protein complex were to undergo a closed-occluded conformational change, one would expect clusters of sizable chemical shift changes in the Met20 (residues 9-23), F-G (residues 117-131), and G-H (residues 142-149) loops (Osborne et al., 2003). In particular, the chemical shift changes in key marker residues 121 and 149 are 10% and 20%, respectively, of the expected amount for the occluded conformation. Although the conformational change is not observed, we note that significant drug-dependent chemical shift change is observed at one position in the F-G loop (119) in the case of MTX (Figure 2A). While the analogous change is not recognized as statistically significant in the TMP complex, an obvious deviation from baseline is observed. These data indicate the ensemble conformations of the drug bound complexes are nearly identical to the holoenzyme.
Figure 2.
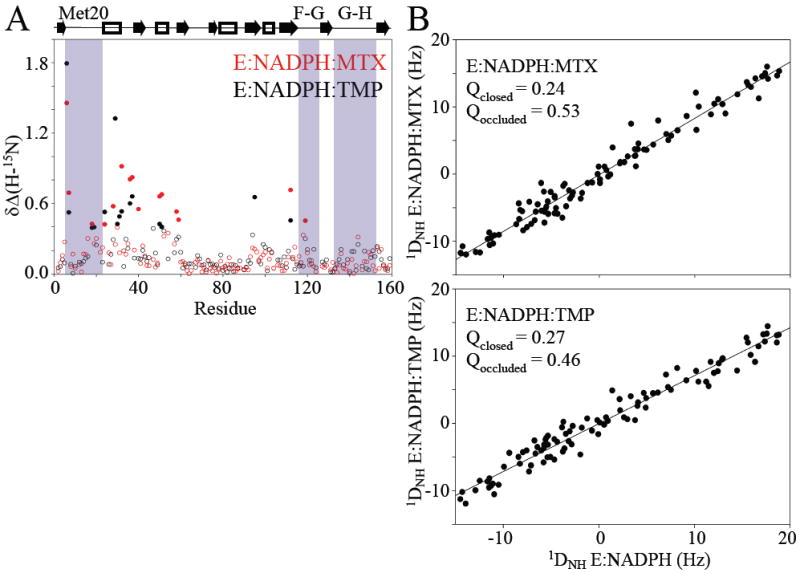
Conformational effects of DHFR due to drug binding. A) The combined change in 1H and 15N chemical shifts, relative to holoenzyme, are plotted for the MTX (red) and TMP (black) complexes. Significant changes are indicated by filled circles. B) Linear correlation between 1H-15N RDC values for the drug complexes and the holoenzyme suggest they are in the same conformation. Q-factors indicate that RDCs agree closely with the closed conformation. PDB IDs 1rx3 and 1rx5 were used for the closed and occluded conformations, respectively.
RDC data confirm structural homogeneity between the three complexes. The correlation of RDCs for either drug complex is strikingly linear with respect to the holoenzyme, indicative of no significant conformational change (Figure 2B). This was corroborated upon calculation of “quality factors” (Cornilescu et al., 1998), Q. RDCs of either drug complex show good agreement with the closed form, Q = 0.24-0.27, and poor agreement with the occluded form, Q = 0.46-0.53 (Figure 2B). Overall, the chemical shift and RDC data indicate the three DHFR complexes are in the closed conformation, and that any large-scale structural differences are imperceptible. Minor changes in chemical shift are observed in the F-G loop, suggestive of propagation of subtle changes in local conformation and/or dynamics.
Drug-induced decoupling of a μs-ms functional global switch
As DHFR moves through its catalytic cycle, it undergoes slow, μs-ms collective motions that are dependent on substrate, product, and cofactor ligands (Boehr et al., 2006). To see how these functional motions are modulated by drug binding, 15N CPMG relaxation dispersion experiments (Loria et al., 1999b) were carried out on the three closed complexes. The dispersion data for all complexes were fitted to the general equation for the two-site exchange model to yield the exchange rate (kex = k1+k-1), the change in chemical shift (Δω), and populations pa and pb (Palmer et al., 2001). The residues that show dispersion in addition to representative dispersion curves are shown in Figure 3.
Figure 3.

R2 relaxation dispersion of DHFR, holo and bound to drugs. A) Relaxation dispersion of the holoenzyme was recorded at two temperatures, 284 K and 298 K. Due to the fast exchange rates, only 7 residues show significant dispersions at the higher temperature (orange spheres). At 284 K, the same residues were observed (orange spheres) in addition to the residues indicated by red spheres. The R2 dispersion profiles for G121 at 298 K (orange) and 284 K (red) are plotted. B) Residues that exhibit significant R2 dispersion in MTX and TMP complexes at 298 K only (blue and green spheres, respectively). In the case of the MTX complex, A8 (orange sphere) also exhibited dispersion at 284 K. Conformational exchange is localized to the active site in both complexes. The fitted R2 dispersion profiles for active site residue F31 are shown (blue and green). In stark contrast to the holoenzyme, drug binding quenches exchange in the functional loops of DHFR, as indicated by the flat dispersion profiles (black lines) of G121 (black spheres).
The holoenzyme was previously characterized at 281 K, with supportive data at 284 K (Boehr et al., 2006). Our holoenzyme data at 284 K are in agreement, showing 23 sites that yield significant R2 dispersion (data not shown). At 298 K, a total of 7 sites yielded dispersions indicative of slow switching, all within the substrate binding region (Figure 3A). Initial fits indicate the rate of motion to be fast (2000-4000 s-1) relative to the lower temperature (Boehr et al., 2006). While R2 dispersion can be extremely sensitive for detecting the sampling of minor states, the accessible time window is relatively narrow; thus, the reduction of sites showing significant dispersions at 298 K may be due to a modest increase in exchange rates (Palmer et al., 2001). Upon inspection, at 298 K, residues such as G121, a marker for global conformational exchange (McElheny et al., 2005; Osborne et al., 2001; Osborne et al., 2003), indeed show slight R2 dispersion curvature as a result of μs motion (Figure 3). Model-free analysis of R1, R2, and 15N-{1H} NOE data at 298 K provide an additional, independent evaluation of μs-ms timescale motion within DHFR. As shown in Figure 4A, models that account for motion on the μs-ms timescale (i.e., models 3 and 4 that use an Rex term) are required to accurately fit these data for many residues in the holoenzyme, indicating widespread presence of μs-ms conformational exchange. Both Met20 and F-G loop residues require usage of Rex terms. Thus, at 298 K, the holoenzyme active site and loops appear to be involved in functional conformational switching (Figures 3A, 4A; Tables S1-S2), as observed previously (Boehr et al., 2006).
Figure 4.

The “model-free” dynamic response of DHFR to drug binding. A) Model selection results for E:NADPH, E:NADPH:MTX, and E:NADPH:TMP. Structures are colored as follows (models 1-5): S2 only in blue; S2 and τe in cyan; S2 and Rex in yellow; S2, Rex, and τe in green; and S2 f, S2 s, and τe in red. Residues that did not fit to a model are orange, and residues that could not be fit due to spectral overlap are gray. B) The change in backbone order parameter due to MTX (red) and TMP (black) binding to the holoenzyme. Residues that experience consensus significant change are indicated by filled circles. G121 (blue fill) shows a dynamic response in the MTX complex. Residues 67-69 (green stippling) exhibit a consistent, slight increase in rigidity. The inset shows the correlation in the dynamic responses of DHFR (relative to holoenzyme) to binding both drugs.
Binding of MTX and TMP to the holoenzyme at 298 K results in ~10 residues exhibiting R2 dispersion in these complexes (Figure 3B, Table S1). These sets of residues are nearly identical (8, 9, 28, 29, 30, 31, 111, 112 in both), cluster around the substrate/drug binding site, and exhibit similar switching. The global kex rates are indistinguishable at 430 ± 150 s-1 and 460 ± 170 s-1 for MTX and TMP, respectively, with both excited state populations at 2%. Thus, MTX and TMP binding result in the same collective slow motion in DHFR, albeit slower than in the holoenzyme. To test whether fast microsecond motions are present but difficult to detect at 298 K, R2 dispersion curves were collected for the MTX ternary complex at 284 K. In stark contrast to the holoenzyme, only one residue (A8) exhibited significant R2 dispersion, indicating that much of the dynamics of the ternary complex is indeed quenched by drug (Figure 3B). It is striking that binding either drug leads to complete loss of R2 dispersion in the F-G and G-H loops, while retaining slow motions around the substrate binding site (Figure 3). At 298 K, considering all indicators of slow motion (i.e. from R2 dispersion and model-free analysis), the holoenzyme shows 47 sites experiencing microsecond motion, and the MTX and TMP complexes show 18 and 23 sites experiencing μs-ms motion, respectively (Tables S1-S4).
It should be mentioned that the E:NADP+:folate complex has been used as a model for the closed, reactive Michaelis complex (McElheny et al., 2005). In this complex, the substrate binding and loop regions undergo conformational change, apparently in a concerted, global, closed-occluded conformational exchange, characterized by kex = 477 s-1 (reported at 303.4 K), although the observed structural change is manifested primarily in the loops and not the substrate binding pocket (Sawaya and Kraut, 1997). The calculated forward closed-to-occluded rate of the E:NADP+:folate complex at 298 K is 11 s-1 (McElheny et al., 2005), which, coincidentally, matches the product release rate (Boehr et al., 2006). From our drug complex data, the forward ground-to-excited rates of the substrate binding residues (see above) in the closed ternary drug complexes (at 298 K) are similarly calculated to be 7.4 ± 2.7 s-1 and 10.6 ± 3.8 s-1 for MTX and TMP complexes, respectively. Because these sites show the same forward rate as for the corresponding region in the also-closed E:NADP+:folate complex, this suggests that drug binding initiates similar “micro-switching” in the substrate binding pocket to that which occurs in the global closed-to-occluded transition. It has recently been proposed that the closed-occluded conformational change is an intrinsic feature of the E. coli DHFR structure (Schnell et al., 2004b). While it is not clear whether a true closed-to-occluded transition is occurring in the holoenzyme (Boehr et al., 2006) or at least partially in the substrate binding pockets of the closed drug complexes, it is evident that drug binding slows down motion in the binding pocket and removes functional loop conformational changes.
Taking these data together, we conclude that in these two drug complexes, a dynamic decoupling occurs. The active site (substrate binding pocket) behaves as if the enzyme is poised to undergo the global transition that it has evolved to make, but MTX and TMP manage to dampen and slow the larger concerted motion, probably via formation of stable contacts with the nicotinamide moiety or protein (Sawaya and Kraut, 1997). In effect, the enzyme is “in a dynamic straightjacket” and the loops cannot perform conformational switching, even though the active site is initiating μs-ms timescale switching events. Thus, the coupled and temporally correlated active site and loop motions, which are present in every physiological complex (Boehr et al., 2006), have been decoupled by binding either MTX or TMP. We propose that filling the active site sends a signal through the enzyme to initiate conformational change, but these complexes are locked in the closed conformation and therefore the signal becomes ineffective, resulting in a dysfunctional, “half-switching” enzyme (Figures 3, 4A).
Consensus drug-binding effects on ps-ns motions: pre-transitional connectivity to distal loops that regulate DHFR activity
To expand the timescale range of the effects that MTX and TMP binding have on DHFR dynamics, 15N dipolar and methyl 2H quadrupolar relaxation experiments were carried out on all three complexes to characterize backbone and side-chain ps-ns dynamics at 298 K. The relaxation data were interpreted in terms of the Lipari-Szabo “model-free” formalism, in which the squared generalized order parameter indicates the motional restriction of the backbone N-H bond (S2) or, in the case of side-chain methyl groups, the methyl symmetry axis (S2 axis) (Lipari and Szabo, 1982). The order parameter can range between 0 and 1, corresponding to complete disorder and a fixed orientation in the molecular frame, respectively. Because the observed μs-ms dynamics are between a highly populated ground state and a lowly populated excited state, the relaxation rates have negligible contribution from the excited state and therefore report on conformational fluctuations (i.e., dynamics) within the closed, ground state of each complex. In principle, isolating motion within the ground state ensemble may allow identification of key, pre-transitional, motions that facilitate larger, slower conformational changes, thus potentially enabling study of both cause and effect (of a conformational change). To the extent that MTX (or even TMP) mimics the closed transition state along the reaction coordinate (Sawaya and Kraut, 1997), the fluctuations will correspond to motions promoting conformational changes associated with reactivity (Agarwal et al., 2002).
Backbone Dynamics
Backbone amide order parameters (S2) were extracted from 15N R1, R2, and 15N-{1H} NOE data collected at two fields. Model selection was carried out using global anisotropic tumbling (see Experimental Procedures) and yielded quite different models for the holoenzyme vs. ternary drug complexes (Figure 4A). Specifically, model 3 (S2, Rex) was selected for a large number of residues in the holoenzyme, indicating widespread μs-ms motion, in contrast with significantly fewer residues in the drug complexes (see discussion above). In addition, model 5 (S2 f, S2 s, τs), as well as complex motions that did not fit well to any model, was frequently required in the F-G loop of the holoenzyme, with few complex motions detected in the ternary complexes. Thus, drug binding simplifies the motional landscape of the holoenzyme over a wide timescale range.
The model-selected backbone order parameters were found to reflect consistent dynamic changes upon binding MTX and TMP. Although overall backbone flexibility is similar for the three complexes, clear changes in order parameters (ΔS2) emerge for specific residues in response to binding (Figure 4B). These changes follow similar trends for the two drugs, with r = 0.74 (see insert). Because of the high quality of these data, we set a criterion for consensus changes in dynamics: residues that showed significant change in S2 (≥ 2x error), in the same direction, upon binding either drug. These consensus residues are 33, 51, 73, 134, and 159 (Figures 4B, 6). In addition, the adenosine binding loop residues 67-69 are individually noisy but collectively show average rigidification of about 0.05 in response to drug binding. These consensus changes in backbone dynamics impart high confidence in altered flexibility observed at sites that extend far beyond the substrate binding site. Finally, though it does not respond to TMP, the catalytically important and distal residue G121 (in the F-G loop) undergoes an increase in flexibility (by 0.06) in response to MTX binding (Figure 4B), 15 Å away.
Figure 6.
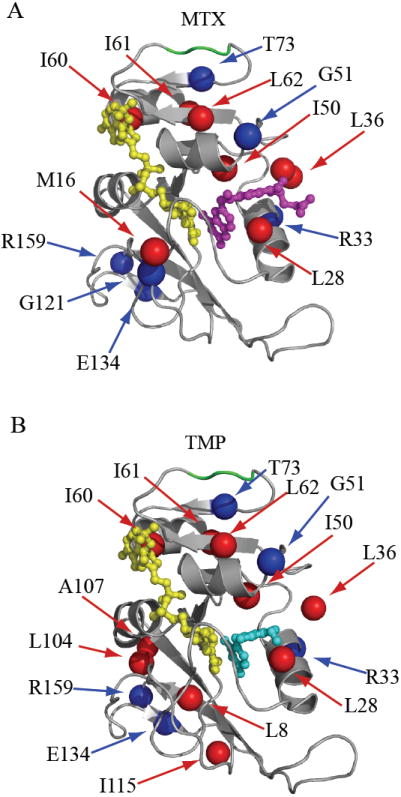
Summary of ps-ns dynamic changes. The dynamic response as a result of binding MTX or TMP are illustrated in A) and B), respectively. Residues with significant change in backbone amide (blue) and side-chain methyl (red) order parameter (complex – E:NADPH) are mapped onto the DHFR structure. Residues 67-69 are highlighted in green. The ligands MTX, TMP, and NADPH are shown in magenta, cyan, and yellow, respectively.
Side-chain dynamics
Side-chain methyl order parameters (S2 axis) are exquisitely sensitive probes to dynamic changes as a result of ligand binding (Fuentes et al., 2004; Lee et al., 2000) and complement the backbone dynamics by increasing coverage. As shown in Figure 5, methyl side chains are evenly distributed in both the catalytic and adenosine binding subdomains of DHFR providing a uniform probe of dynamics. The changes in S2 axis indicate DHFR methyl groups generally become more rigid on the ps-ns timescale (Figures 5, S4), and the MTX- and TMP-induced changes are correlated (r = 0.65). As expected, residues making contact with drug show the largest changes. Interestingly, residues 60-62 in the adenosine binding domain display significant changes in S2 axis in both ternary drug complexes. Drug binding rigidifies I60δ1 and L62δ2, whereas I61δ1 becomes more flexible (Figure S4). Although these residues are outside the substrate binding pocket, they are conserved in DHFR, are prominent in a statistical coupling analysis (SCA) based network, and may be involved in promoting catalysis (Chen et al., 2007). Spatially, they also lie in-between the substrate binding site and loop residues 67-69. The changes in S2 axis provide additional evidence that the bulk of the adenosine binding subdomain is dynamically linked to the substrate binding pocket. Of special note, the dynamics of M20ε is very different between MTX and TMP complexes (0.20 vs. 0.50, Figure S4), although the methyl resonance was overlapped in the holoenzyme. Thus, the dynamics of M20 is clearly perturbed upon binding one or both drugs.
Figure 5.
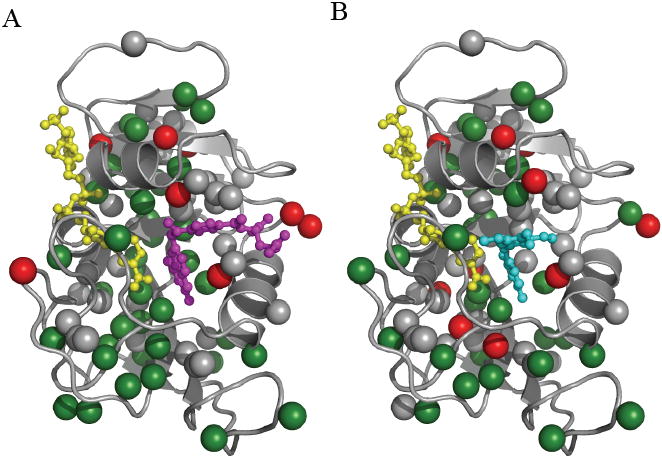
Distribution of methyl bearing side chains in DHFR. Methyl sites within DHFR are represented by the colored spheres. Residues that show significant change in order parameter (> 2x error) as a result of binding A) MTX or B) TMP to the holoenzyme are shown in red. Residues that do not change are represented by green spheres. Methyl sites that were not comparable due to spectral overlap in the drug bound and/or holo enzyme are colored gray. The ligands MTX, TMP, and NADPH are shown in magenta, cyan, and yellow, respectively.
The significant changes in side-chain and backbone ps-ns order parameters are summarized in Figure 6. It is clear that local perturbation by small molecules has far reaching dynamic consequences in this enzyme on the ps-ns timescale. Drug binding induces ps-ns dynamics changes in residues 50, 51, 60, 61, 62 and 73, which essentially form a pathway to the base of the loop containing the perturbed G67 (Figure 6). Similarly, dynamics and subtle chemical shift changes are observed in the F-G loop, in particular, upon MTX binding. These findings complement reports that demonstrate distal regions of DHFR to be linked to catalysis: mutation of either G121 (F-G loop) or G67 (adenosine binding loop) have significant effects on kcat/KM (Cameron and Benkovic, 1997; Gekko et al., 1994; Ohmae et al., 1996). Furthermore, G121 and G67 have been shown to be thermodynamically coupled with each other with respect to kcat (Ohmae et al., 1998), despite their physical separation by 28 Å; and theoretical studies suggest these residues are correlated to the active site (Agarwal et al., 2002; Pan et al., 2000; Radkiewicz and Brooks, 2000). Our experimental data demonstrate a direct dynamic connectivity, within the confines of the closed ensemble, between the active site and functional distal regions in DHFR. This supports the idea that dynamics provides a mechanism for propagating functional change observed upon mutation or ligand binding. Indeed, previous NMR studies have shown that changes in ps-ns side-chain dynamics can reveal propagation pathways linking distal sites (Clarkson and Lee, 2004; Fuentes et al., 2004), and in some cases, linking distal functional sites (Igumenova et al., 2005; Namanja et al., 2007).
It is important to emphasize a key difference between the ps-ns dynamics comparison made here and in previous studies (Osborne et al., 2001; Schnell et al., 2004a). In those studies, dynamics changed in response to a change in the Met20 loop conformation. By contrast, here, alterations in ps-ns dynamics is a direct result of ligand binding in the absence of conformational change. Because the binding of antifolates such as MTX and TMP appears to be the only way to study substrate binding effects (by NMR) without inducing a net conformational change, such small molecules can be important tools for isolating pure dynamic changes within the closed state, and potentially for capturing motions that trigger – or quench, depending on how one perceives the reference state – larger, slower dynamic events. From our data, we can speculate that because the ps-ns motions of G121 are sensitive to ligand occupancy in the context of the closed state, these results are consistent with G121, and perhaps its nearest neighbors, participating in rapid, pre-transitional, initiating movements to the occluded conformation (Chen et al., 2007; Radkiewicz and Brooks, 2000).
Sensitivity of closed conformational dynamics to ligand structure
Depending on ligand occupancy in its two binding sites, DHFR undergoes different μs-ms switching dynamics (Boehr et al., 2006). The data here show that the high-affinity drugs MTX and TMP cause novel “half-switching” behavior on the μs-ms timescale, with additional perturbations to ps-ns fluctuations. While the Met20 loop conformation is clearly an important factor in regulating DHFR dynamics (Osborne et al., 2001; Schnell et al., 2004a), we propose that, even with a fixed Met20 loop conformation, motions across a wide range of timescales can be regulated by the specific nature of ligands bound. Occupation of the active site by small ligands of different shapes and physical characteristics places differential stresses on the enzyme, resulting in differential thermal fluctuations that propagate through the structure. In this view, enzymes, through evolution, develop sensitivities to ligand properties from which mechanisms for organizing and building such fluctuations into useful work can arise. The collective results on DHFR dynamics support the notion of a so-called “allosteric wiring network” that connects distal regions of the protein (Chen et al., 2007; Hammes-Schiffer and Benkovic, 2006). It has been suggested that coordinated motions within the network are responsible for controlling the large conformational transition (Chen et al., 2007) and the chemical step of catalysis (Agarwal et al., 2002) in DHFR. Binding MTX or TMP may disrupt these motions and reduce connectivity within the network, preventing the transition from occurring.
Concluding remarks
In this report, we have presented a dynamic survey of DHFR with reduced cofactor in the presence and absence of two potent inhibitory drugs, methotrexate (MTX) and trimethoprim (TMP). Binding either drug results in essentially identical behavior: quenched μs-ms conformational exchange in the Met20, F-G, and G-H loops, and slowed substrate binding site μs-ms dynamics. MTX and TMP partially suppress the global conformational switching that is found in every physiological complex (Boehr et al., 2006); however, this suppression takes place within the closed conformational ensemble. On the ps-ns timescale, both backbone amides and methyl side chains show sparse, yet largely consistent changes in their dynamics as a result of drug binding. The changes in these fast motions show that, despite a lack of overall conformational change, changes propagate to the distal F-G and adenosine binding loops that contain residues implicated in DHFR function. The emergent picture is that upon binding high-affinity drug, the enzyme is on the brink of a structural transition, yet trapped in the closed conformation. It is left undergoing slow, correlated switching in the substrate binding site. Because the affected loop structures are primarily not in contact with drug, it is reasonable to envision inhibitory small molecule drugs that act by allosterically modulating dynamic motions.
Experimental Procedures
Protein Purification
DHFR was expressed in M9 minimal media supplemented with combinations of 15NH4Cl, D-glucose (U-13C6-99%), and 2H2O. The cells were harvested by centrifugation and suspended in lysis buffer (20 mM sodium phosphate, 50 mM sodium chloride, 1 mM EDTA, and 1 mM DTT pH 6.8). DHFR was purified using methods similar to those previously described (Rajagopalan et al., 2002). Cell lysate was loaded onto a MTX affinity column (Sigma). The column was washed with lysis buffer containing 750 mM NaCl, and protein was eluted with buffer containing 20 mM sodium borate, 750 mM NaCl, 1 mM EDTA, 1 mM DTT, 5 mM folate pH 9.0. The eluent was passed over a G-50 gel filtration column (GE Biosciences) equilibrated with KPE (50 mM potassium phosphate, 150 mM KCl, 1 mM EDTA, and 1mM DTT pH 6.8). Any remaining folate was removed by dialysis against KPE containing 5 M deionized urea. Finally, DHFR was passed over a G-50 column equilibrated with NMR Buffer (70 mM HEPES, 20 mM KCl, 1 mM EDTA, and 1 mM DTT pH 7.6). Pure DHFR was concentrated, lyophilized, and stored at 4 °C. Sample purity was estimated by SDS-PAGE and verified by ESI-MS to be greater than 97%.
NMR Samples
NMR experiments were performed on 1 mM DHFR in NMR Buffer containing 15 mM NADPH, 10 mM glucose-6-phosphate and 10 U glucose-6-phosphate dehydrogenase and, when applicable, 3-5 mM MTX or 2-3 mM TMP. The protein was placed in amber NMR tubes and flame sealed under argon.
NMR Spectroscopy
All NMR experiments were conducted at 298 or 284 K on Varian spectrometers equipped with room-temperature (500 and 600 MHz) or cryogenic probes (700 MHz). Backbone and side-chain resonances were assigned using standard triple resonance methods. NMR data were processed with NMRPipe (Delaglio et al., 1995) and visualized with NMRDraw and NMRView (Johnson and Blevins, 1994). Combined chemical shifts for the purposes of chemical shift mapping were calculated using the Euclidean distance (Schumann et al., 2007). Significant changes in combined chemical shift were identified using the boxplot function in Matlab R2006b (Mathworks).
Relaxation dispersion measurements were performed at 500, 600, and 700 MHz using relaxation-compensated CPMG experiments (Loria et al., 1999a). The total CPMG period was 40 ms in all experiments. The effective field strength was modulated by changing the delay time, τ. Sixteen HSQC-type spectra composed of 13 τ values between 0.556 and 10 ms, two duplicates, and a reference experiment were collected interleaved. Peak intensities were extracted using the NMRDraw module nlinLS. Effective R2 rates were calculated as described (Mulder et al., 2001). Highly deuterated DHFR was used for measuring E:NADPH and E:NADPH:MTX relaxation dispersion while protonated DHFR was used for E:NADPH:TMP.
Standard backbone relaxation experiments were used to collect the R1, R2, and {1H}-15N NOE data at 500 and 600 MHz (Farrow et al., 1994). Side-chain 2H Dz and Dy relaxation experiments (Millet et al., 2002) were collected using multi-coherence relaxation pulse sequences, similar to previously described (Fuentes et al., 2004).
Relaxation Analysis
Motions occurring on the μs-ms timescale were characterized assuming a two-state model. Only residues for which R2,eff changed >2 s-1 over the series of time points were considered for further analysis. The data were best-fitted to a model assuming no exchange (R2=R2 o) and a simple two state model. An F-test (alpha critical = 0.01) was used to identify residues with statistically significant exchange. To determine exchange kinetics, effective R2 rates as a function of τ-1 at two fields were fit simultaneously to the Carver-Richards equation (Palmer et al., 2001) using the in-house program exrate2.0 (by R.V.M). The errors in fitted parameters were estimated using Monte Carlo simulation. Relaxation dispersion data often report on a global exchange process and can be fitted using shared kex and pa values. To identify these residues, we followed the procedure outlined by Kay and coworkers (Mulder et al., 2001). As previously described, residues 129-134 and 155-159 in E. coli DHFR report on ligand independent exchange (Boehr et al., 2006). These residues were not included in any global fit and were not considered in this study.
Motions on the ps-ns timescale were characterized using the model-free formalism (Lipari and Szabo, 1982). Using the strategy outlined by Dellwo and Wand (Dellwo and Wand, 1989) the isotropic rotational correlation times for the binary and ternary complexes were determined to be 11.2 and 10.5 ns/rad, respectively. While the ternary correlation times are shorter than the binary, the effect is small and could be explained by deviation from isotropic tumbling, a change in hydration patterns around the active site, or a small degree of non-specific aggregation in the binary complex. Rotational diffusion anisotropy was calculated via the local Di method (Lee et al., 1997) using the in-house program qfit and the PDB file 1rx3. The values of D∥/D┴ for the E:NADPH, E:NADPH:MTX, and E:NADPH:TMP complexes are 1.15, 1.18, and 1.14, respectively. Rotational anisotropy has been shown to have a large impact on backbone model selection and was used in the subsequent analyses (Osborne and Wright, 2001). Backbone relaxation rates were fitted to the five “model-free” models using the in-house program relxn2.2 (Clarkson et al., 2006; Lee et al., 1999). The values used for the effective N-H bond distance and the 15N chemical shift anisotropy were 1.02 Å and -170 ppm, respectively. Akaike’s information criterion was used to select the model that best describes the motion (d’Auvergne and Gooley, 2003). Side-chain methyl dynamics parameters were carried out as described previously (Fuentes et al., 2004). The quadrupolar coupling constant was set to 165 kHz (Muhandiram et al., 1995).
Residual Dipolar Coupling
DHFR was partially aligned using a 6 mm stretched gel alignment kit (6% acrylamide) (Chou et al., 2001). Cross-peak position of isotropic and anisotropic IPAP-HSQC spectra were adjusted and fitted using the ipap.tcl and nlinLS modules provided in NMRPipe. The 1DHN value was calculated by subtracting the measured coupling in the isotropic experiment from the anisotropic experiment. The PDB file 1RX3 (closed) and 1RX5 (occluded) were used to calculate Q-factors (Cornilescu et al., 1998). The data were analyzed using the program REDCAT (Valafar and Prestegard, 2004).
Acknowledgments
The authors would like to thank Karl Koshlap (UNC Eshelman School of Pharmacy NMR Facility) and Greg Young (UNC Biomolecular NMR Facility) for their technical assistance; Scott Singleton (UNC Eshelman School of Pharmacy) for invaluable discussion and providing the DHFR expression construct; and Patrick Loria (Yale University) for providing the relaxation dispersion pulse sequence. This work was funded by NIH grant GM08359 (to A.L.L.).
Abbreviations
- CPMG
Carr–Purcell–Meiboom–Gill
- DHFR
dihydrofolate reductase
- MTX
methotrexate
- μs-ms
microsecond-to-millisecond
- NADPH
nicotinamide adenine dinucleotide phosphate
- NMR
nuclear magnetic resonance
- NOE
nuclear Overhauser enhancement
- ps-ns
picosecond-to-nanosecond
- RDCs
residual dipolar couplings
- S2
generalized order parameter
- S2axis
order parameter of methyl symmetry axis
- τe
effective correlation time
- τm
global tumbling correlation time
- TMP
trimethoprim
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agarwal PK, Billeter SR, Rajagopalan PT, Benkovic SJ, Hammes-Schiffer S. Network of coupled promoting motions in enzyme catalysis. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:2794–2799. doi: 10.1073/pnas.052005999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehr DD, McElheny D, Dyson HJ, Wright PE. Science. Vol. 313. (New York, N.Y): 2006. The dynamic energy landscape of dihydrofolate reductase catalysis; pp. 1638–1642. [DOI] [PubMed] [Google Scholar]
- Burton LL, Lazo JS, Parker KL. Goodman & Gilman’s the Pharmacological Basis of Therapeutics. 11. New York: McGraw-Hill; 2006. [Google Scholar]
- Cameron CE, Benkovic SJ. Evidence for a functional role of the dynamics of glycine-121 of Escherichia coli dihydrofolate reductase obtained from kinetic analysis of a site-directed mutant. Biochemistry. 1997;36:15792–15800. doi: 10.1021/bi9716231. [DOI] [PubMed] [Google Scholar]
- Careri G, Fasella P, Gratton E. Enzyme dynamics: the statistical physics approach. Annual review of biophysics and bioengineering. 1979;8:69–97. doi: 10.1146/annurev.bb.08.060179.000441. [DOI] [PubMed] [Google Scholar]
- Chen J, Dima RI, Thirumalai D. Allosteric communication in dihydrofolate reductase: signaling network and pathways for closed to occluded transition and back. Journal of molecular biology. 2007;374:250–266. doi: 10.1016/j.jmb.2007.08.047. [DOI] [PubMed] [Google Scholar]
- Chou JJ, Gaemers S, Howder B, Louis JM, Bax A. A simple apparatus for generating stretched polyacrylamide gels, yielding uniform alignment of proteins and detergent micelles. J Biomol NMR. 2001;21:377–382. doi: 10.1023/a:1013336502594. [DOI] [PubMed] [Google Scholar]
- Clarkson MW, Gilmore SA, Edgell MH, Lee AL. Dynamic coupling and allosteric behavior in a nonallosteric protein. Biochemistry. 2006;45:7693–7699. doi: 10.1021/bi060652l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson MW, Lee AL. Long-range dynamic effects of point mutations propagate through side chains in the serine protease inhibitor eglin c. Biochemistry. 2004;43:12448–12458. doi: 10.1021/bi0494424. [DOI] [PubMed] [Google Scholar]
- Cornilescu G, Marquardt JL, Ottiger M, Bax A. Validation of Protein Structure from Anisotropic Carbonyl Chemical Shifts in a Dilute Liquid Crystalline Phase. J Am Chem Soc. 1998;120:6836–6837. [Google Scholar]
- d’Auvergne EJ, Gooley PR. The use of model selection in the model-free analysis of protein dynamics. J Biomol NMR. 2003;25:25–39. doi: 10.1023/a:1021902006114. [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. Nmrpipe - a Multidimensional Spectral Processing System Based on Unix Pipes. Journal of Biomolecular Nmr. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Dellwo MJ, Wand AJ. Model-independent and model-dependent analysis of the global and internal dynamics of cyclosporin A. Journal of the American Chemical Society. 1989;111:4571–4578. [Google Scholar]
- Eisenmesser EZ, Bosco DA, Akke M, Kern D. Science. Vol. 295. (New York, N.Y): 2002. Enzyme dynamics during catalysis; pp. 1520–1523. [DOI] [PubMed] [Google Scholar]
- Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Forman-Kay JD, Kay LE. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- Frederick KK, Marlow MS, Valentine KG, Wand AJ. Conformational entropy in molecular recognition by proteins. Nature. 2007;448:325–329. doi: 10.1038/nature05959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes EJ, Der CJ, Lee AL. Ligand-dependent dynamics and intramolecular signaling in a PDZ domain. Journal of molecular biology. 2004;335:1105–1115. doi: 10.1016/j.jmb.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Gekko K, Kunori Y, Takeuchi H, Ichihara S, Kodama M. Point mutations at glycine-121 of Escherichia coli dihydrofolate reductase: important roles of a flexible loop in the stability and function. J Biochem. 1994;116:34–41. doi: 10.1093/oxfordjournals.jbchem.a124499. [DOI] [PubMed] [Google Scholar]
- Hammes-Schiffer S, Benkovic SJ. Relating protein motion to catalysis. Annu Rev Biochem. 2006;75:519–541. doi: 10.1146/annurev.biochem.75.103004.142800. [DOI] [PubMed] [Google Scholar]
- Huennekens FM. The methotrexate story: a paradigm for development of cancer chemotherapeutic agents. Advances in enzyme regulation. 1994;34:397–419. doi: 10.1016/0065-2571(94)90025-6. [DOI] [PubMed] [Google Scholar]
- Igumenova TI, Lee AL, Wand AJ. Backbone and side chain dynamics of mutant calmodulin-peptide complexes. Biochemistry. 2005;44:12627–12639. doi: 10.1021/bi050832f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BA, Blevins RA. Nmr View - a Computer-Program for the Visualization and Analysis of Nmr Data. Journal of Biomolecular Nmr. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- Karplus M, McCammon JA. Molecular dynamics simulations of biomolecules. Nature structural biology. 2002;9:646–652. doi: 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]
- Lee AL, Flynn PF, Wand AJ. Comparison of 2H and 13C NMR Relaxation Techniques for the Study of Protein Methyl Group Dynamics in Solution. Journal of the American Chemical Society. 1999;121:2891–2902. [Google Scholar]
- Lee AL, Kinnear SA, Wand AJ. Redistribution and loss of side chain entropy upon formation of a calmodulin-peptide complex. Nature structural biology. 2000;7:72–77. doi: 10.1038/71280. [DOI] [PubMed] [Google Scholar]
- Lee LK, Rance M, Chazin WJ, Palmer AG., 3rd Rotational diffusion anisotropy of proteins from simultaneous analysis of 15N and 13C alpha nuclear spin relaxation. J Biomol NMR. 1997;9:287–298. doi: 10.1023/a:1018631009583. [DOI] [PubMed] [Google Scholar]
- Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1 Theory and range of validity. Journal of the American Chemical Society. 1982;104:4546–4559. [Google Scholar]
- Loria JP, Rance M, Palmer AG. A Relaxation-Compensated Carr-Purcell-Meiboom-Gill Sequence for Characterizing Chemical Exchange by NMR Spectroscopy. J Am Chem Soc. 1999a;121:2331–2332. [Google Scholar]
- Loria JP, Rance M, Palmer AG., 3rd Transverse-relaxation-optimized (TROSY) gradient-enhanced triple-resonance NMR spectroscopy. J Magn Reson. 1999b;141:180–184. doi: 10.1006/jmre.1999.1891. [DOI] [PubMed] [Google Scholar]
- Matthews DA, Alden RA, Bolin JT, Freer ST, Hamlin R, Xuong N, Kraut J, Poe M, Williams M, Hoogsteen K. Science. Vol. 197. (New York, N.Y): 1977. Dihydrofolate reductase: x-ray structure of the binary complex with methotrexate; pp. 452–455. [DOI] [PubMed] [Google Scholar]
- McElheny D, Schnell JR, Lansing JC, Dyson HJ, Wright PE. Defining the role of active-site loop fluctuations in dihydrofolate reductase catalysis. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:5032–5037. doi: 10.1073/pnas.0500699102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millet O, Muhandiram DR, Skrynnikov NR, Kay LE. Deuterium spin probes of side-chain dynamics in proteins. 1 Measurement of five relaxation rates per deuteron in (13)C-labeled and fractionally (2)H-enriched proteins in solution. Journal of the American Chemical Society. 2002;124:6439–6448. doi: 10.1021/ja012497y. [DOI] [PubMed] [Google Scholar]
- Muhandiram DR, Yamazaki T, Sykes BD, Kay LE. Measurement of 2H T1 and T1.rho. Relaxation Times in Uniformly 13C-Labeled and Fractionally 2H-Labeled Proteins in Solution. Journal of the American Chemical Society. 1995;117:11536–11544. [Google Scholar]
- Mulder FA, Mittermaier A, Hon B, Dahlquist FW, Kay LE. Studying excited states of proteins by NMR spectroscopy. Nature structural biology. 2001;8:932–935. doi: 10.1038/nsb1101-932. [DOI] [PubMed] [Google Scholar]
- Namanja AT, Peng T, Zintsmaster JS, Elson AC, Shakour MG, Peng JW. Substrate recognition reduces side-chain flexibility for conserved hydrophobic residues in human Pin1. Structure. 2007;15:313–327. doi: 10.1016/j.str.2007.01.014. [DOI] [PubMed] [Google Scholar]
- Ohmae E, Iriyama K, Ichihara S, Gekko K. Effects of point mutations at the flexible loop glycine-67 of Escherichia coli dihydrofolate reductase on its stability and function. J Biochem. 1996;119:703–710. doi: 10.1093/oxfordjournals.jbchem.a021299. [DOI] [PubMed] [Google Scholar]
- Ohmae E, Iriyama K, Ichihara S, Gekko K. Nonadditive effects of double mutations at the flexible loops, glycine-67 and glycine-121, of Escherichia coli dihydrofolate reductase on its stability and function. J Biochem. 1998;123:33–41. doi: 10.1093/oxfordjournals.jbchem.a021913. [DOI] [PubMed] [Google Scholar]
- Osborne MJ, Schnell J, Benkovic SJ, Dyson HJ, Wright PE. Backbone dynamics in dihydrofolate reductase complexes: role of loop flexibility in the catalytic mechanism. Biochemistry. 2001;40:9846–9859. doi: 10.1021/bi010621k. [DOI] [PubMed] [Google Scholar]
- Osborne MJ, Venkitakrishnan RP, Dyson HJ, Wright PE. Diagnostic chemical shift markers for loop conformation and substrate and cofactor binding in dihydrofolate reductase complexes. Protein Sci. 2003;12:2230–2238. doi: 10.1110/ps.03219603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne MJ, Wright PE. Anisotropic rotational diffusion in model-free analysis for a ternary DHFR complex. J Biomol NMR. 2001;19:209–230. doi: 10.1023/a:1011283809984. [DOI] [PubMed] [Google Scholar]
- Palmer AG, 3rd, Kroenke CD, Loria JP. Nuclear magnetic resonance methods for quantifying microsecond-to-millisecond motions in biological macromolecules. Methods Enzymol. 2001;339:204–238. doi: 10.1016/s0076-6879(01)39315-1. [DOI] [PubMed] [Google Scholar]
- Pan H, Lee JC, Hilser VJ. Binding sites in Escherichia coli dihydrofolate reductase communicate by modulating the conformational ensemble. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:12020–12025. doi: 10.1073/pnas.220240297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polshakov VI, Smirnov EG, Birdsall B, Kelly G, Feeney J. NMR-based solution structure of the complex of Lactobacillus casei dihydrofolate reductase with trimethoprim and NADPH. J Biomol NMR. 2002;24:67–70. doi: 10.1023/a:1020659713373. [DOI] [PubMed] [Google Scholar]
- Radkiewicz JL, Brooks CL., 3rd Protein dynamics in enzymatic catalysis: Exploration of dihydrofolate reductase. Journal of the American Chemical Society. 2000;122:225–231. [Google Scholar]
- Rajagopalan PT, Lutz S, Benkovic SJ. Coupling interactions of distal residues enhance dihydrofolate reductase catalysis: mutational effects on hydride transfer rates. Biochemistry. 2002;41:12618–12628. doi: 10.1021/bi026369d. [DOI] [PubMed] [Google Scholar]
- Sasso SP, Gilli RM, Sari JC, Rimet OS, Briand CM. Thermodynamic study of dihydrofolate reductase inhibitor selectivity. Biochim Biophys Acta. 1994;1207:74–79. doi: 10.1016/0167-4838(94)90053-1. [DOI] [PubMed] [Google Scholar]
- Sawaya MR, Kraut J. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: crystallographic evidence. Biochemistry. 1997;36:586–603. doi: 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- Schnell JR, Dyson HJ, Wright PE. Effect of cofactor binding and loop conformation on side chain methyl dynamics in dihydrofolate reductase. Biochemistry. 2004a;43:374–383. doi: 10.1021/bi035464z. [DOI] [PubMed] [Google Scholar]
- Schnell JR, Dyson HJ, Wright PE. Structure, dynamics, and catalytic function of dihydrofolate reductase. Annu Rev Biophys Biomol Struct. 2004b;33:119–140. doi: 10.1146/annurev.biophys.33.110502.133613. [DOI] [PubMed] [Google Scholar]
- Schumann FH, Riepl H, Maurer T, Gronwald W, Neidig KP, Kalbitzer HR. Combined chemical shift changes and amino acid specific chemical shift mapping of protein-protein interactions. J Biomol NMR. 2007;39:275–289. doi: 10.1007/s10858-007-9197-z. [DOI] [PubMed] [Google Scholar]
- Teague SJ. Implications of protein flexibility for drug discovery. Nature reviews. 2003;2:527–541. doi: 10.1038/nrd1129. [DOI] [PubMed] [Google Scholar]
- Valafar H, Prestegard JH. REDCAT: a residual dipolar coupling analysis tool. J Magn Reson. 2004;167:228–241. doi: 10.1016/j.jmr.2003.12.012. [DOI] [PubMed] [Google Scholar]
- Watt ED, Shimada H, Kovrigin EL, Loria JP. The mechanism of rate-limiting motions in enzyme function. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:11981–11986. doi: 10.1073/pnas.0702551104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zidek L, Novotny MV, Stone MJ. Increased protein backbone conformational entropy upon hydrophobic ligand binding. Nature structural biology. 1999;6:1118–1121. doi: 10.1038/70057. [DOI] [PubMed] [Google Scholar]