

Abstract
The problem of increasing bacterial resistance to the current generation of antibiotics is well documented. This includes such pathogens as methicillin–resistant Staphylococcus aureus and the potential for developing drug–resistant pathogens for use as bioweapons, such as Bacillus anthracis. The biphenyl ether, antibacterial triclosan exhibits broad–spectrum activity and provides a potential scaffold for the development of new, broad–spectrum antibiotics targeting the fatty acid biosynthetic pathway, via inhibition of enoyl–acyl carrier protein reductase (ENR). We have utilized a structure–based approach to develop novel aryl ether analogs of triclosan that target ENR, the product of the FabI gene, from Bacillus anthracis (BaENR). Structure–based design methods were used for the expansion of the compound series including X-ray crystal structure determination, molecular docking, and QSAR methods. Structural modifications were made to both phenyl rings of the 2-phenoxyphenyl core. A number of compounds were derived that exhibited improved potency against BaENR and increased efficacy against both the Sterne strain of B. anthracis and the methicillin–resistant strain of S. aureus. X-ray crystal structures of BaENR in complex with triclosan and two other compounds help explain the improved efficacy of the new compounds and suggest future rounds of optimisation that might be used to improve their potency.
Keywords: Triclosan, Enoyl-Reductase, Inhibitors, B. anthracis, Structure-activity relationships
Introduction
The increasing prevalence of antibiotic resistance of certain bacteria is well–documented. The 2004 monograph by the Infectious Diseases Society of America noted that the incidence of methicillin–resistant Staphylococcus aureus (MRSA) in particular, has increased quite rapidly over the last two decades.[1] Recent summaries have documented that staphylococci are among the most common causes of nosocomial infections and that resistance to beta–lactams and glycopeptides is complicating treatment of those infections.[2] Particularly alarming is a recent JAMA article and accompanying editorial, which noted that deaths from invasive MRSA in 2005 were comparable to, or exceeded those from HIV/AIDS.[3,4] This rapid increase in bacterial resistance to current antibiotics is a strong motivation for the development of new antibacterials with alternate modes of action.
Drug resistance is also an issue for pathogens that might be used as bioweapons. For instance, the literature indicates that natural isolates of B. anthracis exist that are resistant to the following antibiotics: penicillin G, amoxicillin, erythromycin, cefuroxime, sulfamethoxazole, trimethoprim, cefotaxime–sodium, aztreonam and deftazidime[5–8] and ofloxacin,[9] as well as tetracycline and penicillin.[10] Further, two reports have shown that it is feasible to select for strains of B. anthracis that are resistant to all of the common antibiotics through standard microbial selection procedures.[11,12] Thus, whereas the 2001 bioterrorism attacks used a B. anthracis strain that was susceptible to conventional antibiotics, it must be considered possible that future repeats of such attacks could involve B. anthracis strains selected for resistance to one or more conventional antibiotics. Since anthrax has been rated first or second in potential bioterrorism impact, comparable to smallpox,[13,14] and since anthrax can be readily adapted to biowarfare applications,[14] there is incentive for the development of antibiotics with novel modes of action that could be used to combat such potential drug–resistant biothreats.[15]
To develop new antibiotics we have considered targeting of the fatty acid biosynthesis pathways, as these pathways are essential for bacterial growth and they represent validated targets for antibiotic development for several reasons.[16,17] Fatty acids are synthesized by mammals (FAS I) and bacteria (FAS II) via substantially different biosynthetic mechanisms, thus providing the possibility of bacteria–specific drug targeting. FAS I involves a single multifunctional enzyme–acyl carrier protein (ACP) complex, whereas FAS II utilizes several small monofunctional enzymes that operate in conjunction with ACP–associated substrates.[18] Recent studies have revealed that the genes responsible for FAS II are essential in Bacillus subtilis, a close relative of B. anthracis.[19]
Enoyl–ACP reductase (ENR), the product of the FabI gene in Bacillus anthracis (BaENR), is an NADH–dependent, key enzyme in FAS II that catalyzes the final and rate–determining step of chain elongation.[20] Considerable research over the past few years has shown that enoyl–ACP reductase in a number of pathogens is efficiently inhibited by antibacterial agents including isoniazid,[21] diazaboranes,[22–24] triclosan,[25–28] and several other small molecule inhibitors.[29–35] API-1252, a recently developed FabI inhibitor shows excellent in vitro activity against clinical isolates of Staphylococcus epidermidis and Staphylococcus aureus.[36a] Another novel FabI inhibitor, CG400462, was recently reported to show efficacy against Staphylococcus aureus infected mice.[36b] These studies clearly indicate that inhibition of enoyl–ACP reductase is a viable approach to develop new antibacterials with novel modes of action.
Triclosan, a 2-phenoxyphenol, is a well–known, broad–spectrum antibacterial that is used in a number of consumer products, such as toothpastes, soaps and plastics. It has been shown to inhibit the growth of Escherichia coli,[26,37] Pseudomonas aeruginosa,[38] and Staphylococcus aureus.[32] Originally, it was believed that triclosan was a non-specific antibacterial that attacks bacterial cell membranes. Later it was shown to have a specific mode of action that involves inhibition of bacterial fatty acid synthesis at the enoyl–acyl carrier protein reductase step.[39,40] Triclosan has since been shown to inhibit ENR from the genes of FabI, FabL and InhA in a number of microorganisms, which has lead to several attempts to develop new triclosan derived antibacterials.[26,41–45] Improvements in the broad–spectrum activity of triclosan–like compounds have been deterred by the variability of effectiveness of triclosan against different species. For example, the IC50 values range from 70 nM and 73 nM respectively in S. aurues[32] and malaria (PfENR)[42] to only 7.25 μM in E. coli.[26,37] Due to this large range in activity, we believed that it would be valuable to explore the inhibitory action of additional triclosan–like aryl ether analogs against individual organisms, to maximize the specificity for each organism. Initially focusing on B. anthracis, we have determined the IC50 of triclosan against BaENR to be 0.6 μM with a MIC of 3.1 μg/mL.[46] In the current report, we describe our approaches to improve its efficacy through structural modifications to the 2-phenoxyphenol core utilizing a structure–based design approach that relies on our crystal structure of BaENR with triclosan bound in the active site.[46] Additional BaENR crystal structures involving newly designed diphenyl ethers are also presented and discussed. We find that at least two compounds exhibit improved activity against both the ΔANR and Sterne strain of B. anthracis as well as methicillin–resistant Staphylococcus aureus. The ΔANR strain of B. anthracis is used for the enzymatic assays as it has both the pXO1 (toxin) and pXO2 (capsule) removed. The Sterne strain of B. anthracis is used in subsequent antibacterial testing as it contains the pXO1 toxin but not the pXO2.
Results and Discussion
Synthesis of the inhibitors
The general synthesis of 2-phenoxyphenol core involved preparation of the corresponding methoxy substituted aryl ethers, made from commercially available materials via nucleophilic aromatic substitution reaction (Method A) or through Cu catalyzed coupling reactions (Methods B and C)[47] followed by demethylation of the methoxy group. Method A (Scheme 1) involves the reaction of an appropriate phenol with a fluoro–aromatic compound in the presence of K2CO3 and was used to prepare a variety of 2-phenoxyphenol derivatives bearing an electron withdrawing group on the ring B, namely NO2, or CN groups (4–6, 8–12, and 14) via the intermediates 4a–6a, 8a–12a, 14a. Compound 14 was synthesized by alkaline hydrolysis of the intermediate benzonitrile 14a in refluxing ethanol. [48] The benzylic alcohol 16 was prepared by sodium borohydride reduction of the acid 14 in the presence of BF3·Et2O.[49] An attempt to demethylate the methoxy derivative 13b using excess boron tribromide resulted in the formation of brominated analog 13 as a major product. Carboxamides 15 and 17 were prepared by the hydrolysis of the corresponding benzonitriles in alkaline medium containing hydrogen peroxide. [48]
Scheme 1.

Synthesis of compounds 1–17: When X = F and R4 = R5 = H, NO2, CN, Cl, Method A: K2CO3, DMSO, 100 °C, 8–12 h. When X = Br or I and R4 = R5 = H, OMe, Ph, Method B: KOtBu, DMF, (CuOTf)2·PhH, 140 °C, 16–20 h. a) BH3·THF, 3M NaOH, H2O2, rt, 4–6 h; b) Excess BBr3, CH2Cl2, −78 °C to rt, 2–6 h; c) 35% H2O2, 3N NaOH, EtOH, 30 °C, 18 h; d) 14a, 25% NaOH, EtOH, reflux, 20 h; e) NaBH4, BF3·Et2O, THF, rt to reflux, 1 h.
Anilines 20 and 21 were synthesized via catalytic hydrogenation followed by demethylation of nitro intermediates 4a and 8a respectively (Scheme 2). The aniline intermediates 19a and 20a were converted to the corresponding acetamides (19 and 18, respectively) by reacting with acetic anhydride followed by demethylation of the methoxy derivatives. Similarly, sulfonamide 22 was prepared by treating 20a with 4-toluenesulfonyl chloride followed by demethylation using excess boron tribromide.
Scheme 2.

Synthesis of compounds 18–22: a) Pd/C, H2, EtOH, rt, 2–6 h; b) Ac2O, DMAP, Et3N, CH2Cl2, rt, 3–6 h; c) Excess BBr3, CH2Cl2, −78 °C to rt, 2–6 h; d) 4-toluenesulfonyl chloride, Et3N, CH2Cl2, 0 °C to rt, 3 h.
Method B involves Cu catalyzed coupling reaction of an appropriate phenol with a variety of aromatic halides under thermal conditions. This method is versatile, and was used to synthesize electron rich diphenyl ethers 1, 3, and 7 via corresponding intermediates 1a, 3a, and 7a respectively (Scheme 1). Heteroaromatic ring B analogs (23–25) were prepared by following similar protocol starting from commercially available materials (Scheme 3).
Scheme 3.

Synthesis of compounds 23–25: a) Method B: KOtBu, DMF, (CuOTf)2·PhH, 140 °C, 14–18 h; b) Excess BBr3, CH2Cl2, −78 °C to rt, 2–6 h.
Method C is a mild copper promoted C–O coupling reaction of arylboronic acids and phenols (Schemes 4–7) that was employed to synthesize a variety of aryl ethers. This coupling reaction complements nucleophilic aromatic substitutions (Method A) and Ullman type couplings (Method B) described above for the synthesis of aryl ethers. Although this reaction worked well with meta- and para-substituted phenylboronic acids, coupling of 2-methoxyphenols with ortho substituted phenylboronic acids, as well as with heteroaromatic boronic acids failed, presumably due to steric hindrance.
Scheme 4.

Synthesis of compounds 26–31: a) Phenylboronic acid, Cu(OAc)2, Et3N, CH2Cl2, air, rt, 16 h; b) OsO4, NMO, THF, rt, overnight; c) BBr3, CH2Cl2, −78 °C, 2h; d) H2, Pd/C, EtOAc, rt, 2h; e) NaBH4, MeOH, 0 °C, 1h; f) amine, NaBH(OAc)3, AcOH, CH2Cl2, rt, overnight.
Scheme 7.

Synthesis of compounds 47–51: a) Cu(OAc)2, 3-formylphenylboronic acid, Et3N, CH2Cl2, air, rt, 16 h; b) BBr3, CH2Cl2, −78 °C to rt, 2h; c) Ph3P=CHCO2Me, THF, reflux, overnight; d) H2, Pd/C, EtOAc, rt, 2h; e) LiOH·H2O, MeOH, H2O, rt, 2h.
In Scheme 4, coupling of phenylboronic acid with eugenol and vanillin yields respective diphenyl ether intermediates 27a and 28a in high yields. Allyl and aldehyde functional groups were tolerated under these mild reaction conditions. The allyl group was further elaborated to the vicinal diol 26 via dihydroxylation and demethylation. Subjecting the allyl group to catalytic hydrogenation followed by demethylation of the resulting methoxy substituted diaryl ether resulted in the compound 27. The aldehyde group from vanillin was either reduced to an alcohol, or transformed to a series of alkyl amines by reductive amination reactions. Thus, compounds 28 and 29–31 were obtained after subsequent demethylation of the corresponding methoxy substituted diaryl ethers using boron tribromide.
Scheme 5 illustrates the coupling of 2-methoxy-4-chlorophenol or 2-methoxy-4-propylphenol with commercially available meta- and para-substituted phenyldiboronic acids. While the coupling of meta-substituted phenyldiboronic acid and phenols resulted in high yields, para-substituted phenyldiboronic acid did not react very well (less than 10% yield). The intermediate triphenyl ethers 32a–35a were demethylated to give compounds 32–35.
Scheme 5.

Synthesis of compounds 32–35: a) Cu(OAc)2, meta- or para-phenyldiboronic acid, Et3N, CH2Cl2, air, rt, 16 h; b) BBr3, CH2Cl2, −78 °C, 2h.
Schemes 6 and 7 illustrate the synthetic routes to meta- and para-substituted ring B diphenyl ether analogs from commercially available boronic acids. The reaction works equally well for arylboronic acids bearing electron donating as well as electron withdrawing functional groups. The functional groups on the resulting diphenyl ethers were further elaborated to obtain diverse substitutions on the ring B.
Scheme 6.

Synthesis of compounds 36–46: a) Cu(OAc)2, substituted phenylboronic acids, Et3N, CH2Cl2, air, rt, 16 h; b) BBr3, CH2Cl2, −78 °C to rt, 2h; c) m-CPBA, CH2Cl2, 0 °C, 10 min; d) NaBH4, MeOH, 0 °C, 1h; e) LiOH·H2O, MeOH, H2O, rt, 2h; f) H2, Pd/C, EtOAc, rt, 2h.
Schemes 6 illustrates the synthesis of compounds 36–38, 40, and 41 via coupling of 2-methoxy-4-propylphenol with appropriate arylboronic acids, and subsequent demethylation of the corresponding methoxy diphenyl ethers using boron tribromide. Benzoic acid 39 was obtained by hydrolysis of the corresponding methyl benzoate 37 and compound 42 was obtained by the reduction of the ketone 40 using sodium borohydride. The methylthioether group in 41a was oxidized by m-CPBA to afford methyl sulfoxide (43a) and methyl sulfone (44a), these were demthylated to afford compounds 43 and 44, respectively. The allylic intermediates 45a and 46a were prepared by coupling eugenol with corresponding arylboronic acids. Reduction of the allylic side chain via catalytic hydrogenation, and subsequent demethylation led to compounds 45 and 46.
In Scheme 7, 2-methoxy-4-propylphenol was coupled with 3-formylphenylboronic acid to give the aldehyde 47a. The intermediate 47a was converted to compound 47 via demethylation reaction using boron tribromide. The meta-aldehyde group in 47a was subjected to Wittig reaction, and two–carbon elongated side chain analogs 48–51 were obtained by following similar hydrogenation and hydrolysis protocols.
Initial assay results indicate that triclosan is very effective in the inhibition of BaENR and has good antibacterial activity against B. anthracis (Table 1). We conducted structure–activity relationship studies on a number of aryl ether derivatives to explore the possibility of further improvement of potency of this lead compound, with the goal of optimising the activity against BaENR. The first structural modifications were directed at ring A of triclosan. As expected, the hydrogen bonding interaction of the phenolic hydroxyl group on ring A is critical for BaENR inhibitory activity. Conversion of the phenolic hydroxyl group to ethers resulted in complete loss of activity (52 and 53). Attempts to replace the hydroxyl group by an amino functionality (compound 54), carboxylic acid (compound 14), or a carboxamide (compound 17) resulted in no inhibition at 1μM concentrations. Replacing the phenolic hydroxyl group with an elongated hydroxy-methyl group (compound 16) gave no improvement in activity.
Table 1.
Ba ENR inhibition and antibacterial activity for modifications of ring A.
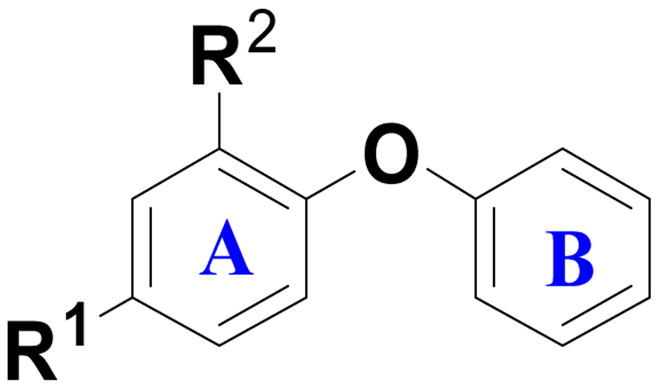
| ||||
|---|---|---|---|---|
| Compound | R1 | R2 | IC50 (μM) or % inhibition at 1μM | MIC[b] (μg/mL) |
| Triclosan | 0.6 ± 0.0 | 3.1 | ||
| 52 | Cl |
|
0.5% | NT |
| 53 | Cl | OMe | 0% | NT |
| 54 | Cl | NH2 | 4.4% | NT |
| 14 | Cl | CO2H | 0% | NT |
| 16 | Cl | CH2OH | 1.8% | NT |
| 17 | Cl | CONH2 | 0% | NT |
| 55 | Cl | OH | 0.5 ± 0.1 | 32 |
| 1 | OH | OH | 6.3 ± 0.4 | 64 |
| 2 | NO2 | OH | >50 | 5.8 |
| 26 | CH2CH(OH)CH2OH | OH | 9.8% | >104 |
| 28 | CH2OH | OH | 5.0% | 43 |
| 29 | CH2NHCH(CH3)CH2CH2CH3 | OH | 0% | >109 |
| 30 | CH2–(1–piperidine) | OH | 0% | >113 |
| 31 | CH2NHCH2Ph | OH | 7.9% | >122 |
| 27 | n-propyl | OH | >0.8[a] | 22.8 |
Saturation with inhibitor was not obtained over the concentration range tested. The percent inhibition of BaENR showed a linear response to increasing inhibitor concentrations.
MIC values are against ΔANR B. anthracis. NT = Not tested.
Evaluation of the active site
The X-ray structure of triclosan bound to BaENR that was recently reported by us,[46] helps explain the lack of activity for this set of compounds. The binding geometry of the triclosan, shown in Figure 1A, is similar to that seen in the structures from other organisms. The phenolic ‘ring A’ of triclosan binds face-to-face with the nicotinamide ring of NAD+ involving in a π – π stacking interaction. The hydroxyl group on ring A is involved in two hydrogen bonds, one with Tyr 157 (OH) and the other with the 2′-hydroxyl group of nicotinamide ribose. Thus, removal of these hydrogen bonds via alternate substitutions at the 2–position would be expected to have a substantial impact on the binding energy of these compounds with the active site and is borne out by the experimental results (Table 1). The ether linkage is also within hydrogen bonding distance to the 2′-hydroxyl group of nicotinamide ribose, thereby adding increased binding energy between BaENR and the NAD+–triclosan complex. Based on these findings, our design efforts focused on 2-phenoxyphenol as the key starting scaffold, and on finding optimal substituents for rings A and B that could maximize the van der Waals, electrostatic, and hydrogen bonding interactions in the active site of BaENR.
Figure 1.
Comparisons of ligand–bound crystal structures of BaENR: side chains are shaded gray, NAD+ is shaded blue–gray (behind the ligand). (A) The triclosan crystal structure is shaded in coral. The GOLD docking conformation for triclosan is in cyan. (B) Compound 11 crystal structure is shaded coral and GOLD conformation is cyan. (C) Compound 43 crystal structure is shown in coral and the GOLD conformation is cyan. Hydrogen bonding is shown in green, between the ligands and Tyr 157, as well as the multiple hydrogen bonding of the 2′-hydroxyl of NAD+ with both the 2-hydroxy and the ether linkage of the ligands. Also shown is a hydrogen bond between 2′-chloro of triclosan and Ser 197.
Connolly surface maps with various properties were generated for the active site of BaENR, see Figure 2A–C. Connolly surfaces allow the van der Waals space to be shown. Figure 2A maps the charge distribution to the surface, Figure 2B maps the lipophilicity and Figure 2C maps the hydrogen bonding opportunities found on the active site surface. Full evaluation and consideration of these active site surface maps contribute significantly to the structure–based design approach. For this system, the shape of the active site indicates that the para position on ring A and the meta and/or para positions on ring B, are suitable sites for introducing bulky substitutions. Further analysis of the ring A pocket indicates that it is an essentially neutral region in terms of charge (Figure 2A), and that there is also a lack of hydrogen bonding opportunities (Figure 2C). The lipophilicity map in Figure 2B suggests the ring A pocket to be dominated by a high amount of hydrophobicity. Thus, we focused our attention on substitutions that could add hydrophobic interactions within the ring A pocket.
Figure 2.
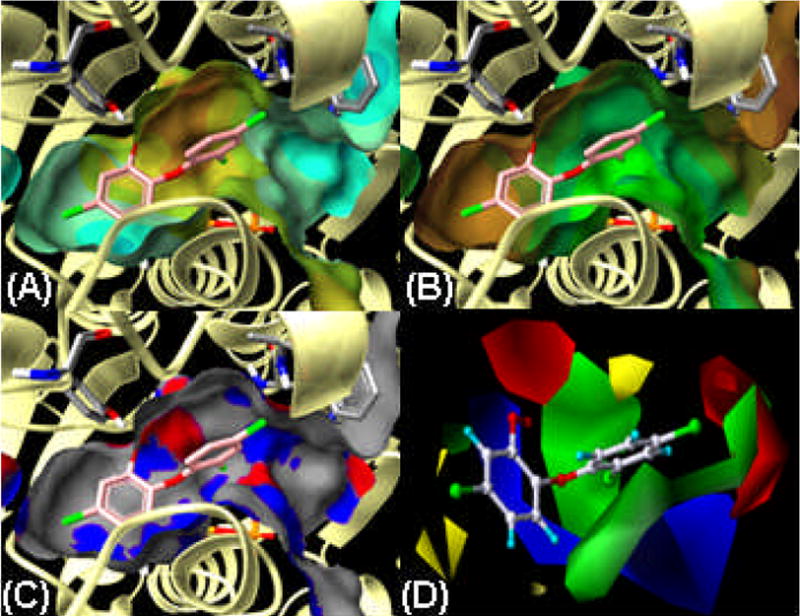
Surface maps of the BaENR active site. (A) Electrostatic Potential surface map, areas shaded orange/yellow indicates regions of moderate positive charge on BaENR, whereas cyan shading indicates areas of neutral charge. (B) Lipophilic map, with a brown to green scale, where brown depicts the more hydrophobic areas. (C) Hydrogen bonding map, where the blue and red areas indicate places for either hydrogen bonding acceptors or donors, respectively. (D) Steric and electrostatic fields from CoMFA based on IC50 values. Steric fields are green and yellow, indicating regions of favorable and unfavorable steric expansion. Electrostatic fields are red and blue, indicating preferred regions of negative and positive charge. Surface maps were generated using the Benchware software and CoMFA fields are generated within Sybyl 7.2, both softwares from Tripos, Inc., St. Louis, MO.
Substitutions on Ring A
We limited modifications of ring A to the para-substituent, R1, due to steric constraints observed in the crystal structure of triclosan bound to BaENR. Compound 55,[37] bereft of substituents on ring B, was earlier shown to be a potent inhibitor of ENR from E. Coli. Compound 55 shows activity similar to that of triclosan against BaENR (Table 1), although its MIC value against ΔANR B. anthracis is an order of magnitude lower. To reveal the true effect on binding affinities of the different substitutions, compound 55 was used as a reference for comparison of activities among this set of compounds. Replacement of the chlorine atom located at the R1 position on ring A with polar hydrophilic functionalities such as OH and NO2 decreased the inhibitory activity against BaENR (compounds 1 and 2, Table 1). Although compound 2 has a reasonable antibacterial activity (MIC = 5.8 μg/mL), its poor enzyme inhibitory activity suggests that it may be working through some other mechanism. Because of poor solubilities of many of these diphenyl ether derivatives, we were forced to introduce polar hydrophilic groups at the R1 position. Unfortunately, this resulted in compounds with lower inhibitory activity (compounds 26, 28, and 29–31, Table 1). The design of compounds in this investigation was also based on comparision of calculated molecular properties (CLogP, ALOGpS and TPSA) with those of triclosan.[50]
The weaker inhibitions of compounds with polar side chains at the R1 position is in agreement with the surface–map property analysis of the active site from the BaENR–triclosan X-ray structure (Figures 2A–2C). The surface maps show the importance of the hydrophobic pocket, which is approximately 6 Å in diameter near the para position of ring A. Moreover, the maps explain the experimental results whereby there are no opportunities for additional hydrogen bonds to occur within the ring A pocket. From the crystal structure shown in Figure 1, it is clear that hydrophobic residues dominate the region surrounding ring A. Sullivan et al. recently reported potent inhibition of ENR (InhA) from Mycobacterium tuberculosis by triclosan analogs bearing long aliphatic chains at the R1 position.[43,51] Since the ring A pocket in BaENR is much smaller than in the InhA, numerous aliphatic chain lengths (2–8 carbons) were evaluated via GOLD docking prior to synthesis, to optimise the chain length for our active site. We are confident that the GOLD docking can accurately predict the binding conformation of these diphenyl ether analogs. An overlay of the GOLD docking conformation of triclosan with the crystal structure is seen in Figure 1A, and shows it to be in very good agreement (RMSD = 0.32 Å). Compound 27, possessing an n-propyl group was suggested by the docking result (conformation and scoring) to be the best prospect, thus it was synthesized and tested. Compound 27 turned out to have an IC50 value higher than compound 55 as judged by the fact that it could not saturate the BaENR active site under the experimental concentrations attempted. These compounds had sparing solubility so it was difficult to accurately quantitate IC50 values. However, compound 27 had an MIC value similar to that of compound 55, so we chose to expand upon two series of compounds. The first was based on a chloro group at R1 of ring A (compound 55), and the second was based on the n-propyl group at R1 (compound 27).
Substitutions on Ring B
In our preliminary search to identify the optimum substitutions on ring B, the first series of modifications to this ring were made while retaining ring A as the starting 2-hydroxy-4-chlorophenyl moiety. The goal was to improve the affinity of these compounds for BaENR by increasing the hydrogen bonding to the protein residues in the ring B pocket, specifically to the backbone residues in the region of Ala 95–Ala 97. These residues were judged to be potential hydrogen bond donors/acceptors based on the modeling and crystal structure analysis presented in Figures 1 and 2. Thus, we introduced several functional groups at the 4′-position of ring B that have the potential to form hydrogen bonds. Compound 55, with an IC50 of 0.5 μM and an MIC of 32 μg/mL was our reference material for comparing the activities of this set of structures. Introduction of a hydroxyl group at the para position on ring B did not improve the activity (compound 3, Table 2). Compound 20, bearing an amino group, showed improved antibacterial activity, but its IC50 against BaENR was only 7 μM. Further acetylation or sulfonylation of the amino group was not effective in improving the BaENR activity (compounds 18, 19, and 22).
Table 2.
Ba ENR inhibition and antibacterial activity for modification of ring B.
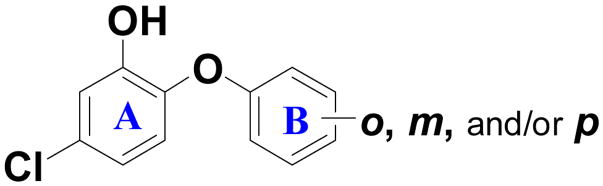
| |||||
|---|---|---|---|---|---|
| Compound | ortho | meta | para | IC50 (μM) or %inhibition at 1μM | MIC[b] (μg/mL) |
| Triclosan | Cl | --- | Cl | 0.6 ± 0.0 | 3.1 |
| 55 | --- | --- | --- | 0.5 ± 0.1 | 32 |
| 3 | --- | --- | OH | 2.6% | >64 |
| 20 | --- | --- | NH2 | 7.1 ± 1.2 | 12 |
| 18 | --- | --- |
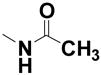
|
1.6% | 111 |
| 19 |
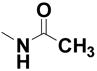
|
--- | --- | 0% | 111 |
| 22 | --- | --- |
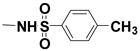
|
>12[a] | 4.9 |
| 4 | --- | --- | NO2 | 7.2% | 3.3 |
| 5 | NO2 | --- | --- | 7.7 ± 0.7 | 13.3 |
| 6 | Cl | --- | NO2 | 0.3 ± 0.0 | <0.1 – 3.1 |
| 7 | --- | --- | Ph | >6.25 | 1.9 |
| 32 | --- | --- |
|
15.5% | >145 |
| 33 | --- |
|
--- | 9.9% | 2.3 |
Saturation with inhibitor was not obtained over the concentration range tested. The percent inhibition of BaENR showed a linear response to increasing inhibitor concentrations.
MIC values are against ΔANR B. anthracis.
Compound 6, possessing ortho-chloro and para-nitro groups on ring B, proved to be the best inhibitor in this series (IC50 = 290 nM, and MIC = 3.1 μg/mL). This compound has earlier been shown to be active against the malarial ENR from P. falciparum.[41] The presence of a nitro group not only adds a strong electron withdrawing group to ring B, but perhaps more importantly, it adds two potential hydrogen bond acceptor atoms close to the aromatic ring. Compound 7, with a 4′-phenyl group, is more active against the bacterium (MIC = 1.9 μg/mL) than triclosan, but it is not as effective against BaENR, indicating that its action may be non-specific or that the biphenyl ring improves bacterial membrane penetration. Similarly, the symmetrical triaryl ethers 32 and 33 were ineffective against BaENR, even though 33 showed improved antibacterial activity.
Considerable improvement in the IC50 values of compound 6 over 4 (less than 20% inhibition at 1 μM) suggests that the 2′-chloro group contributes considerably to the affinity of these compounds. From the crystal structure of triclosan bound to BaENR (see Figure 1A), the 2′-chloro is 2.6 Å from the side chain hydroxyl of Ser 197 which is in favourable position for a halogen–oxygen interaction. [52] The Ser 197 (OH) is also 2.7 Å from one of the oxygen atoms of the bridging phosphate group of NAD+. It is possible that the improved activity of the compounds possessing a 2′-chloro group is due to this hydrogen–bonding network, rendering ring B in a ‘locked’ orientation thereby positioning para substituents optimally to form hydrogen bonds with the backbone of Ala 97. Hydrogen–bonding to Ser 197 (OH) may also stabilize positioning of the substrate binding or “flipping” loop, which is documented as being flexible in a number of organisms. [16,17,46]
To potentially improve the solubility of these diphenyl ethers, we synthesized and studied the activity of derivatives bearing a heteroaromatic ring as ring B, such as the pyridine and pyrazine analogs of 55 (data shown in supplemental material). These compounds failed to show promising activity. Compound 24, containing an acetamidopyridine ring, gave a moderate IC50 value (IC50 = 4.1 μM), but proved to be inactive against the bacterium (MIC > 111.5 μg/mL). Therefore, introduction of a heteroatom into ring B of these aryl ether analogs was ineffective, and further pursuit of such modifications was terminated.
Having established a preliminary SAR at this stage, we set out to combine our knowledge obtained from our study of the ring A and B modifications, in order to produce compounds of optimal activity. We therefore synthesized compounds whereby ring A is fixed with the more hydrophobic 2-hydroxy-4-n-propylphenyl moiety (Compound 27, Table 1) and ring B is modified with various substituents that provide additional hydrogen bond donors and/or acceptor atoms, both at the meta and the para positions of ring B. The goal once again was to design compounds to interact with the residues in the region of Ala 97, positioning the ring B substituents toward the entrance of the active site, closer to the protein surface.
Table 3 shows the results from the n-propyl series of compounds. Overall, an improvement in both the IC50 and MIC values were found over the values listed in Tables 1 and 2. Introduction of small, hydrophilic hydrogen–bond acceptor groups that have the ability to withdraw electron density from the aromatic ring B produces improved activity. The best results were obtained when ring B contained a nitro or a cyano substituent (compounds 8, 9, 10–12, Table 3). In line with the results obtained in Table 2 discussed above, the presence of a 2′-chloro group contributed to the binding affinity of these inhibitors (compound 8 vs 11). Compounds with either an amide or methyl ketone group also had inhibitory activity similar to that of triclosan (compounds 15 and 40). The compounds with an electron rich amino group at the para position were found to be weak inhibitors of BaENR (e.g., compare compound 8 vs 21), consistent with the results of compound 55 vs 20 (Table 2).
Table 3.
Ba ENR inhibition and antibacterial activity for modification of ring B, where ring A is a 2-hydroxy-4-propylphenyl group.
| |||||
|---|---|---|---|---|---|
| Compound | ortho | meta | para | IC50 (μM) or % inhibition at 1μM | MIC[c] (μg/mL) |
| Triclosan | 0.6 ± 0.0 | 3.1 | |||
| 27 | --- | --- | --- | >0.8[a] | 22.8 |
| 8 | --- | --- | NO2 | 1.1 ± 0.1 | 3.4 |
| 9 | --- | NO2 | --- | >0.8[a] | 1.7 |
| 10 | NO2 | --- | NO2 | 3.6 ± 0.8 | 4 |
| 11 | Cl | --- | NO2 | 0.5 ± 0.0 | 1.9–3.1 |
| 12 | Cl | --- | CN | >0.8[a] | 1.8 |
| 15 | Cl | --- | –C(=O)NH2 | 1.1 ± 0.1 | 30.6 |
| 40 | --- | --- | –C(=O)Me | 0.8 ± 0.1 | 13.5 |
| 43 | --- | --- | –S(=O)Me | 3.6 ± 0.3 | 116 |
| 44 | --- | --- | –SO2Me | 2.2 ± 0.3 | 61.3 |
| 42 | --- | --- | CH(OH)Me | 17.1% | 54.5 |
| 21 | --- | --- | NH2 | 8.8 ± 1.0 | >97 |
| 41 | --- | --- | SMe | 0.6 ± 0.0 | 13.5 |
| 45 | --- | N(Me)2 | --- | 12.6% | 13.6 |
| 46 | --- | CF3 | --- | 1.24 | 7.4 |
| 37 | --- | CO2Me | --- | 2.0 ± 0.3 | 3.6 |
| 39 | --- | CO2H | --- | 9.2% | 6.8 |
| 38 | --- | CH2OH | --- | 20.3 ± 1.3 | 12.9 |
| 36 | --- | Ph | --- | 0.5 ± 0.1[b] | 1.9 |
| 34 | --- |
|
--- | 20.6% | 1.2 |
| 35 | --- | --- |
|
10.0% | >151 |
| 48 | --- |
|
--- | >6.25 | 2 |
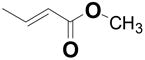
| 49 | --- | CH2CH2CO2Me | --- | 7.5 ± 1.9 | 7.9 |
| 50 | --- | CH=CHCO2H | --- | 17.9% | 14.9 |
| 51 | --- | CH2CH2CO2H | --- | 17.6% | 60.1 |
| 13 | --- | CH2CH2Br | --- | 6.0 ± 1.5 | 1.7 |
Saturation with inhibitor was not obtained over the concentration range tested. The percent inhibition of BaENR showed a linear response to increasing inhibitor concentrations.
100% inhibition was not observed. The response of the enzyme to inhibitor showed maximum saturation at ~50% inhibition.
MIC values are against ΔANR B. anthracis.
The correlation of inhibitory activities of compounds according to the electron donating vs withdrawing ability of functional groups such as NH2 vs NO2 at the para position of ring B, does not hold for the sulfur containing substituent groups. Improved IC50 values were seen with the more electron donating methylthioether group (41, IC50 = 0.6 μM), while compounds bearing relatively stronger electron withdrawing groups (a methylsulfoxide in 43, IC50 = 3.6 μM or the methylsulfone in 44, IC50 = 2.2 μM) were only moderately active. It appears that, along with an electrostatic effect, a steric contribution and/or orientation of the hydrogen bonding acceptor group in the active site is contributing to the inhibitory activity of these compounds. Since the geometry around the sulfur atom of a sulfone is not the same as the geometry around the nitrogen atom of a nitro group, the two oxygens attached to the sulfur or nitrogen would be positioned very differently, and could result in varied hydrogen bonding capabilities. It is notable here that methyl ketone 40, which is a close structural analog of methylsulfoxide 43, exhibits improved inhibitory activity against BaENR and ΔANR B. anthracis (IC50 = 0.8 μM, MIC = 13.5 μg/mL).
To better understand the binding interactions of these inhibitors at the active site, we determined the X-ray crystal structures of inhibitors 11 and 43 bound to BaENR in the presence of NAD+, both at resolutions of 2.3 Å (see Figures 1B and 1C), and compared them to the structure of BaENR in complex with triclosan. As expected from modeling, the binding geometries of these inhibitors were nearly identical to that of triclosan. The most significant difference between the structures was the interaction of the ring B para substituent and the residues in the enzyme. Improved activity of compound 11 could be attributed to the ‘additional’ hydrogen bonding occurring between the para nitro group of 11 and Ala 97(NH) (~2.2 Å). GOLD docking accurately predicted the orientation of this nitro group, as well as the proposed orientation of the 2′-Cl group within hydrogen bonding distance to the sidechain of Ser 197. Figure 1B is an overlay of X-ray crystal structure of compound 11 and its GOLD docking conformation in the active site of BaENR (RMSD = 0.45 Å). Figure 1C is an overlay of X-ray crystal structure of compound 43 and its GOLD docking conformation in the active site of BaENR (RMSD = 0.69 Å). As in compound 11, (Figure 1B), there is very good agreement between the two, as well as the correct prediction of the sulfoxide’s oxygen hydrogen bonding to Ala 97(NH) (~1.8 Å).
Ring B modifications at the meta position involved the addition of heteroatoms very close to the aromatic ring. This included compounds 45 (NMe2), 46 (CF3), and 38 (CH2OH). Of these, only compound 46 showed improved activities against BaENR and the bacterium. Compounds 36 and 34, with a meta-phenyl group substitution on ring B, were more active toward the bacterium than triclosan itself (MIC = 1.9 and 1.2 μg/mL respectively), but exhibited lower activity against the BaENR enzyme. It is well documented that the ENR from multiple organisms has a very flexible substrate binding loop.[46, 53] It is possible that the above compounds with larger groups induce a shift in this flexible substrate–binding loop, opening up the active site. This would likely lead to a decrease in the binding of the ligand and possibly result in lower affinity of the compound for BaENR.
Compounds 37–39, and 48–51 are closely related ester/acid analogs chemically extended from the meta-position of ring B. As previously noted, modeling and structural analysis suggests that opportunities for hydrogen bonding between substituents on ring B and the active site residues exists in the region of the Ala 97 on BaENR. The best opportunities for this hydrogen bonding to occur are with the hydrogen bonding group being located approximately 1–2 atoms away from ring B, as seen in compounds 37–39. Similar hydrogen bonding opportunities are not as likely for the longer chain substituents found in 48–51, and results in weaker inhibition. Although this extension proved disappointing, these results do indicate that the acids were less inhibitory than esters toward the BaENR enzyme (37 vs 39, 49 vs 51).
Overall, the assay results indicated that a few of the meta substituted analogs in Table 3 show IC50 values near 1μM, with the majority having little activity against BaENR (less than 20% inhibition at 1 μM). This is in contrast to the para substituted analogs, which in general had IC50 values less than 3.6 μM. Antibacterial activity for this series in Table 3 suggested that the meta compounds have better MICs than similar para derivatives (compare compounds 34 and 35). Figure 3 summarizes the SAR of the diphenyl ethers studied, showing the key pharmacophoric elements required for effective BaENR inhibition.
Figure 3.

SAR of diaryl ether BaENR inhibitors.
Regardless of antibacterial activity, the n-propyl series of compounds show better inhibitory activity against BaENR than the chloro series shown in Table 2. More importantly, this series has resulted in nine compounds (8, 9, 11, 12, 15, 27, 36, 40, and 41) that are equipotent or nearly equipotent with triclosan. Of these, 15, 40, and 41 have potential for future structural modification and expansion. Pressence of a chloro group at the 2′-position on ring B has shown to increase activity against BaENR. Interestingly, we observed a similar improvement in the binding affinity of 2-pyridone derivatives to the BaENR active site in a recent study.[54] Thus, introduction of a chloro group to 40 and 41 is likely to be beneficial in improving the activity of these inhibitors. Additionally, compounds 15, 40, and 41 ideally possess functional groups at the para position of ring B, a carboxamide in 15, a methyl ketone in 40, and a thioether in 41 that are useful in the design of the next generation of compounds.
CoMFA Maps and future design implications
Even though GOLD did a good job at predicting the binding conformations of simple substitutions, it is unable to account for the flexibility in the BaENR’s substrate binding loop. To aid in future design, we turned to a ligand–based approach, a 3D–QSAR, or CoMFA. We generated a CoMFA for our results against BaENR presented in Tables 1–3 and Supplemental Material. The CoMFA model has a q2 = 0.831 and an r2 = 0.929. Figure 2D shows the steric and electrostatic fields generated from the CoMFA. One can see the favorability of a negative charge around the 2-hydroxy position of ring A (red regions), as well as at the para position of ring B. Sterically, the CoMFA map suggests expansion to be concentrated at the meta/para position of the ring B. Expansions on ring A should be limited, as shown by the yellow regions here. One potential misleading region is the large green region behind the molecule. From the crystal structure, we know this to be the region where NAD+ binds, and modifications to the structure in these regions are not likely to be beneficial since the binding of triclosan and analogs relies on the co–binding of the product, NAD+, for maximal binding energy.[37,46,55] The CoMFA model visually illustrates sites for future structural expansion of these aryl ether compounds, and will be used for further predictions of activity for the next generation of compounds.
Antibacterial testing of lead compounds
Table 4 shows the MICs and MBCs of two of our BaENR inhibitors, 6 and 11, against several bacterial pathogens. The broth microdilution method of the National Committee for Laboratory Standards was validated using a panel of ATCC strains of bacterial pathogens with known MICs to ciprofloxacin. This method was employed for the MIC and MBC determinations for these two inhibitors. From the results, these two compounds are equally active and bactericidal against Sterne and ΔANR strains of B. anthracis. These compounds also have noticeable activity against both Gram (+) and Gram (−) bacterial pathogens, suggesting that these types of compounds could be expanded for a more broad–spectrum application. The notable exception is the high–level resistance of Pseudomonas aeruginosa, which has been reported for triclosan,[56] and may be explained by bacterial efflux pump activity.[29] Of particular note is the impressive activity against MRSA, with an MIC = 0.3 μg/mL for both compounds and an MBC/MIC ratio of 1.6–1.9, compared with a ratio of approximately 20 for the tested strain of methicillin–sensitive S. aureus (MSSA). The observation of comparable MICs for MRSA and MSSA is consistent with previous comparisons of large numbers of both types of S. aureus isolates.[56] If the 10–fold increase in bactericidal activity against MRSA vs. MSSA is reproducible upon testing multiple strains of each, it might be interesting to pursue mechanistically. Previous studies have indicated that the bactericidal activity of triclosan against S. aureus isolates (both MRSA and MSSA) may be unrelated to MIC and might involve multiple bacterial targets of such compounds.[57] It is likely that compounds 6 and 11 target FabI in S. aureus too. In the absence of a reported X-ray crystal structure of ENR from S. aureus, explanation of increased activity should rely upon secondary structures. Sequence alignment of the ENR from S. aureus with B. anthracis indicates a 62% identity, with all residues in the substrate–binding loop (residues 190–210) completely conserved. In addition, all the residues within 5 Å radius of the ENR active site are conserved in both S. aureus and B. anthracis. The only exception being a Met 99 present in S. aureus ENR, this position is occupied by Arg 99 in BaENR. The location of the Arg 99 in B. anthracis is near the surface of ENR, where the arginine side chain partially covers the entrance to the active site. Presence of a methionine in its place in S. aureus ENR is likely to leave the active site more accessible for the ligands, and could partially explain the increased activity found against S. aureus for these types of compounds, as compared with B. anthracis. Therefore, further study will be needed to assess the possible strain specificity and ENR pathway relatedness of these MBC/MIC findings. A preliminary cytotoxicity testing for compounds 6 and 11 (see footnote in Table 4) showed an EC50/MIC ratio of approximately 15–20 for both compounds against HeLa cells. Further testing is needed to validate these values, and whether structural modifications are necessary to minimize toxicity.
Table 4.
MICs and MBCs of ENR inhibitors against B. anthracis and other bacterial pathogens.
| Bacteria | Cipro[a] | Compound 6[e] | Compound 11[e] | ||||
|---|---|---|---|---|---|---|---|
| MIC[b] | MIC | MBC | MBC/MIC | MIC | MBC | MBC/MIC | |
| ΔANR[c] | 0.1 ± 0.0 | 2.2 ± 0.4 | 3.1 ± 1.1 | 1.4 | 2.2 ± 0.0 | 3.9 ± 0.8 | 1.8 |
| Sterne[d] | 0.1 ± 0.0 | 1.9 ± 0.3 | 3.9 ± 0.8 | 2.1 | 1.9 ± 0.0 | 3.9 ± 0.8 | 2.1 |
| S. aureus | 0.4 ± 0.0 | 0.1 ± 0.1 | 2.9 ± 1.2 | 20.9 | 0.1 ± 0.1 | 2.9 ± 1.2 | 20.9 |
| MRSA | 0.5 ± 0.2 | 0.3 ± 0.3 | 0.4 ± 0.1 | 1.6 | 0.3 ± 0.3 | 0.5 ± 0.2 | 1.9 |
| E. fecalis | 0.7 ± 0.1 | 4.4 ± 0.8 | 31.3 ± 6.3 | 7.1 | 4.4 ± 0.5 | 12.5 ± 0.0 | 2.9 |
| VRE | 0.6 ± 0.2 | 5.7 ± 0.6 | 37.5 ± 12.5 | 6.6 | 5.7 ± 0.3 | 15.6 ± 3.1 | 2.7 |
| Listeria mono. | 0.9 ± 0.3 | 2.8 ± 0.3 | 14.1 ± 3.9 | 5.0 | 2.8 ± 0.9 | 14.1 ± 3.9 | 5.0 |
| Pseud. aerug. | 0.3 ± 0.1 | >25 | NT | >25 | NT | ||
| Klebs. pneum. | 0.5 ± 0.1 | 1.8 ± 0.4 | 12.5 | 7.1 | 1.8 ± 0.9 | 8.3 ± 2.1 | 4.7 |
| E. coli | 0.1 ± 0.0 | 0.3 ± 0.1 | 12.50 | 48.0 | 0.3 ± 0.2 | 1.2 ± 0.4 | 4.5 |
Drug concentrations = μg/mL. Results = mean ± SEM, n = 3.
MIC = minimum inhibitory concentration; MBC = minimum bactericidal concentration; MBC/MIC: bactericidal drug ≤ 4.
Plasmid–negative strain of anthrax, lacking both pXO1 and pXO2 plasmids.
Sterne strain of anthrax that contains pXO1 (toxin production) but lacks pXO2 (capsule).
The EC50 against HeLa cells is 29.3 μg/mL for 6 and 41.4 μg/mL for 11. EC50 against MHS is 9.9 μg/mL for 6 and 22.6 μg/mL for 11.
Conclusion
A number of novel aryl ethers, including triphenyl ethers and heteroaromatic analogs have been prepared and tested for inhibition of purified BaENR and cultured ΔANR Bacillus anthracis. These efforts have led to an improved understanding of the enzyme active site and provided a clear SAR for these inhibitors. The X-ray crystal structures, coupled with molecular modeling studies, have demonstrated the importance of hydrophobic interactions of substituents located at the para position on ring A with the enzyme active site and underscore the H–bonding contribution of the ortho and para substituents on ring B with Ser 197 (OH) and Ala 97. The importance of the ortho-chloro group on ring B for the improvement of activity warrants further investigation. These results highlight the subtleness of an appropriate electronic distribution in either of the aromatic rings of the 2-phenoxyphenol core to allow for optimal interaction with the active site. For example, although incorporation of an n-propyl group in place of the chlorine atom at the para-position of ring A was advantageous in improving the effectiveness of the inhibitors, this should also be simultaneously complemented with an appropriate substitution pattern on ring B. We are continuing our structure–based design efforts, with newer efforts focused on modifications at the para position of ring B, with linking groups such as thioethers, amides, and esters, as well as modification of the nitro group.
Several compounds have been synthesized that exhibit varied structural diversity, show improved antibacterial activity over triclosan, and offer an opportunity to identify new inhibitory pathways and drug candidates. The encouraging inhibitory activities of these compounds against a number of pathogens suggest their potential for broad–spectrum application. The intriguing activity shown by compounds 6 and 11 against methicillin–resistant S. aureus provides potential leads against a serious and increasingly common pathogen. To advance these compounds further, pharmacokinetic and drug metabolism studies are planned.
Experimental Section
General
1H NMR and 13C NMR spectra were recorded on Bruker spectrometer with TMS as an internal standard. Standard abbreviation indicating multiplicity was used as follows: s = singlet, d = doublet, t = triplet, q = quadruplet, quin = quintuplet, m = multiplet and br = broad. HRMS experiment was performed on Q–TOF–2TM (Micromass). The progress of all reactions was monitored by TLC on precoated silica gel plates (Merck Silica Gel 60 F254). Preparative TLC was performed with Analtech 1000–mm silica gel GF plates. Column chromatography was performed using Merck silica gel (40 –60 mesh). HPLC was carried out on an ACE AQ columns (100 × 4.6 mm and 250 × 10 mm), with detection at 254 nm on a Shimadzu SPD–10A VP detector; flow rate = 2.0–3.5 mL·min−1; from 10% acetonitrile in water to 100% acetonitrile with 0.05% TFA. Compound 54 is commercially available. Compounds 53, 55,[37] and 28[26,37] are known in the literature.
Synthesis of inhibitors
General Methods
Method A. The aryl halide (1 mmol), phenol (1 mmol), and K2CO3 (2–4 mmol) in DMSO (1.5 mL) were heated to 100 °C under nitrogen until the reaction was shown to be complete by TLC (8–12 h). After cooling to rt, the reaction mixture was diluted with ethyl acetate, and washed with 5% aqueous NaOH solution. The aqueous layer was further extracted with ethyl acetate, and the combined organic layers were washed with brine. The organic layer was dried over Na2SO4, and concentrated under vacuum to give the crude product, which was subsequently purified by flash chromatography on silica gel. Method B. To the phenol (1 mmol) dissolved in DMF (1.75 mL) was added KOtBu (1.1 mmol) in one portion and the mixture was heated at 45 °C under mild vacuum. After 2 h, the reaction mixture was cooled to rt. Aryl halide (1 mmol) and (CuOTf)2·PhH (0.05 mmol) were added and the mixture was heated to reflux at 145 °C for 16–20 h. After cooling to rt, the reaction mixture was diluted with ethyl acetate, filtered over Celite, and extracted with ethyl acetate. The extract was washed with 5% aqueous NaOH solution. The aqueous layer was then extracted with ethyl acetate, and the combined organic layers were washed with brine. The organic layer was dried over Na2SO4, and concentrated under vacuum to give the crude product, which was subsequently purified by flash chromatography on silica gel. Method C. The arylboronic acid (1–2 mmol), phenol (1 mmol), Cu(OAc)2 (2–5 mmol), triethyl amine (5–10 mmol), and powdered 5 Å molecular sieves were mixed in dichloromethane (10 mL), and were stirred in the presence of atmospheric air until the reaction was shown to be complete by TLC (3–16 h). The reaction mixture was diluted with ethyl acetate, filtered over Celite, and extracted. The extract was washed with 5% aqueous NaOH solution. The aqueous layer was further extracted with ethyl acetate, and the combined organic layers were washed with brine. The organic layer was dried over Na2SO4, and concentrated under vacuum to give the crude product, which was subsequently purified by flash chromatography on silica gel. Method D. In each case, the methoxy diarylether derivatives obtained by the above methods were converted to the corresponding phenols using boron tribromide according to the following procedure: A solution of boron tribromide (2–8 mmol of 1.0 M solution in dichloromethane) was added to a solution of the corresponding methoxy diphenylether analog (1 mmol) in dry dichloromethane (4 mL) maintained at −78 °C under nitrogen. The reaction mixture was stirred at the same temperature (1 h), and then at rt (3–8 h) while monitoring by TLC. Following completion, the reaction was quenched with methanol at −78 °C and concentrated under vacuum. The concentrate was further dissolved in ethyl acetate, washed with 10% aqueous sodium bicarbonate solution, and the organic layer was separated and washed with water and then with brine. The aqueous layer was extracted twice with ethyl acetate. The combined organic layers were then dried over Na2SO4, concentrated under vacuum, and the crude product was purified by flash chromatography on silica gel.
4-Phenoxybenzene-1,3-diol (1)
Method B was used to prepare the intermediate 1a from 2,4-dimethoxyphenol (1.00 g, 6.5 mmol), KOtBu (0.87 g, 7.8 mmol), (CuOTf)2·PhH (0.17 g, 0.3 mmol) and iodobenzene (1.59 g, 7.8 mmol) in 50% yield, and method D was used to convert it to the title compound. Purification by flash chromatography (SiO2, 10% EtOAc/Hexanes) gave analytically pure 1 as viscous brown oil (80%). 1H NMR (400 MHz, CDCl3): δ = 4.86 (s, 1H), 5.56 (s, 1H), 6.36 (dd, J = 9.0, J = 3.0 Hz, 1H), 6.58 (d, J = 2.8 Hz, 1H), 6.83 (d, J = 8.7 Hz, 1H), 7.08 (d, J = 7.6 Hz, 2H), 7.09 (t, J = 7.4 Hz, 1H), 7.33 ppm (t, J = 7.4 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ = 103.4, 107.3, 116.4, 120.6, 122.7, 129.5, 136.3, 148.0, 152.5, 157.3 ppm; HRMS (ESI-positive): calcd for C12H10O3 ([M−H]+): 201.0557, found: 201.0556.
5-Nitro-2-phenoxyphenol (2)
Method A was used to prepare the methoxy intermediate 2a from phenol (0.66 g, 7.0 mmol), commercially available 1-fluoro-2-methoxy-4-nitrobenzene (1.12 g, 7.0 mmol) and K2CO3 (1.80 g, 12.8 mmol) in 48% yield, and method D was used to convert it to the title compound. Purification by flash chromatography (SiO2, 15% EtOAc/Hexanes) gave the analytically pure product 2 as a greenish yellow oil (75%). 1H NMR (400 MHz, CDCl3): δ = 6.53 (br s, 1H), 7.08–7.14 (m, 3H), 7.26 (t, J = 7.6 Hz, 1H), 7.43 (t, J = 7.6 Hz, 2H), 7.70 (d, J = 2.8 Hz, 1H), 7.97 ppm (dd, J = 4.0, J = 2.0 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ = 112.5, 115.3, 118.8, 120.1, 124.9, 130.0, 140.7, 143.9, 154.5, 152.4 ppm; HRMS (ESI-positive): calcd for C12H9NO4 ([M−H]+): 230.0459, found: 230.0458.
5-Chloro-2-(4-hydroxyphenoxy)phenol (3)
Method B was used to prepare the dimethoxy intermediate 3a from 4-chloro-2-methoxy-phenol (1.00 g, 6.3 mmol), KOtBu (0.85 g, 7.6 mmol), (CuOTf)2·PhH (0.17 g, 0.3 mmol), and 4-iodoanisole (1.80 g, 7.6 mmol) in 49% yield, and method D was used to convert it to the phenol 3. Purification by flash chromatography (10% EtOAc/Hexanes) gave the analytically pure product as a colourless oil (82%). 1H NMR (400 MHz, CDCl3): δ = 4.68 (s, 1H), 5.72 (s, 1H), 6.70 (d, J = 8.8 Hz, 1H), 6.79 (dd, J = 8.0, J = 2.0 Hz, 1H), 6.84 (dd, J = 7.0, J = 2.0 Hz, 2H), 6.94 (dd, J = 7.0, J = 2.0 Hz, 2H), 7.04 ppm (d, J = 2.4 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ = 115.8, 116.1, 117.4, 119.8, 119.9, 128.2, 143.3, 147.1, 149.1, 151.7 ppm; HRMS (ESI-positive): calcd for C12H9ClO3 ([M−H]+): 235.0168, found: 235.0166.
5-Chloro-2-(4-nitrophenoxy)phenol (4)
Method A was used to prepare the methoxy intermediate 4a in 96% yield, as described in the synthesis of 20. Method D was used to convert it to the required phenol 4. Purification by flash chromatography (3% MeOH/CHCl3) gave the analytically pure product as a muddy brown solid (70%). 1H NMR (400 MHz, CDCl3): δ = 5.42 (s, 1H), 6.95 (s, 2H), 7.14–7.02 (m, 3H), 8.26 ppm (d, J = 9.2 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ = 116.5, 117.3, 120.9, 121.1, 125.7, 131.2, 139.7, 142.9, 147.9, 161.7 ppm; HRMS (ESI-positive): calcd for C12H8ClNO4 ([M−H]+): 264.0069, found: 264.0069.
5-Chloro-2-(2-nitrophenoxy)phenol (5)
Method A was used to prepare the methoxy intermediate 5a in 96% yield, as described in the synthesis of 19, and method D was used to convert it to the phenol 5. Purification by flash chromatography (1% MeOH/CHCl3) gave the analytically pure product as an off white solid (76%). 1H NMR (400 MHz, CDCl3): δ = 6.89 (dd, J = 4.0, J = 2.0 Hz, 1H), 6.98 (d, J = 4.0 Hz, 1H), 7.12–7.08 (m, 2H), 7.28–7.23 (m, 1H), 7.57–7.53 (m, 1H), 7.94 ppm (dd, J = 4.0, J = 1.0 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ = 117.8, 118.9, 120.6, 121.3, 123.9, 125.9, 131.5, 134.6, 140.9, 148.7, 150.0 ppm; HRMS (ESI-positive): calcd for C12H8ClNO4 ([M−H]+): 264.0069, found: 264.0068.
5-Chloro-2-(2-chloro-4-nitrophenoxy)phenol (6)
Method A was used to prepare the corresponding methoxy intermediate 6a in 91% yield from 4-chloro-2-methoxyphenol (1.00 g, 6.3 mmol), 2-chloro-1-fluoro-4-nitrobenzene (1.10 g, 6.3 mmol), and K2CO3 (1.80 g, 12.6 mmol) by heating the mixture in DMSO (10 mL) at 100 °C (12 h). Method D was used to convert it to the target compound. Purification by flash chromatography (3% MeOH/CHCl3) gave the analytically pure product 6 as a light yellow solid (79%). 1H NMR (400 MHz, CDCl3): δ = 5.73 (s, 1H), 6.97–6.91(m, 3H), 7.14 (d, J = 1.2 Hz, 1H), 8.11 (dd, J = 4.0, J = 1.0 Hz, 1H), 8.41 ppm (d, J = 2.0 Hz, 1H); 13C NMR (400 MHz, CDCl3): δ = 116.1, 117.5, 120.8, 121.0, 123.4, 124.2, 126.3, 131.5, 139.7, 142.9, 147.7, 157.4 ppm; HRMS (ESI-positive): calcd for C12H7Cl2NO4 ([M−H]+): 297.9679, found: 297.9679.
2-(Biphenyl-4-yloxy)-5-chlorophenol (7)
Method B was used to prepare the methoxy intermediate 7a from 4-chloro-2-methoxyphenol (1.00 g, 6.3 mmol), KOtBu (0.78 g, 6.9 mmol), (CuOTf)2·PhH (0.16 g, 0.3 mmol), and 4-bromobiphenyl (1.47 g, 6.3 mmol) in 69% yield, and method D was used to convert it to the phenol 7. Purification by flash chromatography (2% EtOAc/Hexanes) gave the analytically pure product as a white solid (65%). 1H NMR (400 MHz, CDCl3): δ = 5.83 (br s, 1H), 6.90 (s, 2H), 7.12 (d, J = 8.8 Hz, 3H), 7.40 (t, J = 7.2 Hz, 1H), 7.49 (t, J = 7.2 Hz, 2H), 7.61 ppm (d, J = 8.0 Hz, 4H); 13C NMR (100 MHz, CDCl3): δ = 116.3, 117.9, 119.1, 120.3, 126.6, 126.9, 128.3, 128.5, 129.39, 129.2, 136.8, 139.8, 142.9, 147.7, 155.5 ppm; HRMS (ESI-positive): calcd for C18H13ClO2 ([M+H]+): 297.0621, found: 297.0652.
2-(4-Nitrophenoxy)-5-propylphenol (8)
Method A was used to prepare the methoxy intermediate 8a in 93% yield from 2-methoxy-4-propylphenol (1.00 g, 6.3 mmol), 1-fluoro-4-nitrobenzene (0.89 g, 6.3 mmol) and K2CO3 (1.80 g, 12.6 mmol) by heating the mixture in DMSO (10 mL) at 100 °C (12 h), and method D was used to convert it to the title compound. Purification by flash chromatography (10% EtOAc/Hexanes) gave the analytically pure product 8 as an yellow solid (96%). 1H NMR (400 MHz, CDCl3): δ = 0.99 (t, J = 3.2 Hz, 3H), 1.65 (q, J = 3.6 Hz, 2H), 2.59 (t, J = 3.6 Hz, 2H), 6.77 (d, J = 8.0 Hz, 1H), 6.90–6.91 (m, 2H), 7.07 (d, J = 9.2 Hz, 2H), 8.21 ppm (d, J = 1.2 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ = 13.4, 23.9, 37.2, 116.1, 116.8, 120.3, 120.9, 125.6, 138.6, 141.5, 146.9, 162.5 ppm; HRMS (ESI-positive): calcd for C15H15NO4 ([M−H]+): 272.0928, found: 272.0925.
2-(3-Nitrophenoxy)-5-propylphenol (9)
Method A was used to prepare the methoxy precursor 9a in 65% yield from 2-methoxy-4-propylphenol (1.00 g, 6.3 mmol), 1-fluoro-3-nitrobenzene (0.89 g, 6.3 mmol) and K2CO3 (1.8 g, 12.6 mmol) by heating the mixture in DMSO (10 mL) at 100 °C (10 h), and method D was used to convert it to the title compound. Purification by flash chromatography (15% EtOAc/Hexanes) gave the analytically pure product 9 as an yellow solid (83%). 1H NMR (400 MHz, CDCl3): δ = 0.99 (t, J = 7.2 Hz, 3H), 1.67 (quin, J = 7.6 Hz, 2H), 2.59 (t, J = 7.6 Hz, 2H), 5.37 (s, 1H), 6.76 (dd, J = 4.0, J = 2.0 Hz, 1H), 6.89 (d, J = 8.0 Hz, 1H), 6.93 (d, J = 1.6 Hz, 1H), 7.36 (dd, J = 5.0, J = 2.0 Hz, 1H), 7.50 (t, J = 8.2 Hz, 1H), 7.83 (t, J = 2.0 Hz, 1H), 7.96 ppm (dd, J = 4.0, J = 1.0 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ = 13.4, 23.9, 37.1, 111.5, 116.7, 117.3, 119.6, 120.8, 122.7, 129.9, 139.2, 141.0, 146.9, 148.8, 157.9 ppm; HRMS (ESI-positive): calcd for C15H15NO4 ([M−H]+): 272.0928, found: 272.0926.
2-(2,4-Dinitrophenoxy)-5-propylphenol (10)
Method A was used to prepare the methoxy precursor 10a in 93% yield from 2-methoxy-4-propylphenol (0.50 g, 3.0 mmol), 1-fluoro-2,4-dinitrobenzene (0.56 g, 3.0 mmol), and K2CO3 (0.84 g, 6.0 mmol) by heating the mixture in DMSO (10 mL) at 100 °C (8 h), and method D was used to convert it to the title compound. Purification by flash chromatography (10% EtOAc/Hexanes) gave the analytically pure product 10 (81%). 1H NMR (400 MHz, CDCl3): δ = 0.95 (t, J = 7.2 Hz, 3H), 1.65 (quin, J = 7.2 Hz, 2H), 2.59 (t, J = 7.2 Hz, 2H), 5.72 (s, 1H), 6.83 (d, J = 8.2 Hz, 1H), 7.02 (s, 1H), 7.06–7.04 (m, 1H), 7.18–7.13 (m, 1H), 8.36 (dd, J = 4.0, J = 3.0 Hz, 1H), 8.84 ppm (t, J = 2.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ = 13.3, 24.0, 36.9, 117.4, 117.5, 120.8, 121.2, 121.6, 127.6, 128.7, 136.2, 137.8, 141.2, 142.9, 144.8, 146.8, 155.4 ppm; HRMS (ESI-positive): calcd for C15H14N2O6 ([M−H]+): 317.0779, found: 317.0776.
2-(2-Chloro-4-nitrophenoxy)-5-propylphenol (11)
Method A was used to prepare the methoxy intermediate 11a in 93% yield from 2-methoxy-4-propylphenol (0.50 g, 3.0 mmol), 2-chloro-1-fluoro-4-nitrobenzene (0.53 g, 3.0 mmol), and K2CO3 (0.84 g, 6.1 mmol) by heating the mixture in DMSO (10 mL) at 100 °C (12 h), and method D was used to convert it to the title compound. Purification by flash chromatography (10% EtOAc/Hexanes) gave the analytically pure product 11 (82%). 1H NMR (400 MHz, CDCl3): δ = 0.97 (t, J = 7.2 Hz, 3H), 1.66 (quin, J = 7.6 Hz, 2H), 2.57 (t, J = 7.6 Hz, 2H), 5.55 (s, 1H), 6.77 (dd, J = 4.0, J = 2.0 Hz, 1H), 6.91–6.89 (m, 3H), 8.02 (dd, J = 5.0, J = 2.0 Hz, 1H), 8.33 ppm (d, J = 2.0 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ = 13.4, 23.9, 37.1, 115.3, 117.1, 120.2, 121.1, 123.3, 123.5, 125.9, 138.5, 141.9, 142.1, 146.7, 158.3 ppm; HRMS (ESI-positive): calcd for C15H14ClNO4 ([M−H]+): 306.0539, found: 306.0541.
3-Chloro-4-(2-hydroxy-4-propylphenoxy)benzonitrile (12)
Method A was used to prepare the methoxy intermediate 12a in 80% yield from 2-methoxy-4-propylphenol (1.00 g, 6.3 mmol), 3-chloro-4-fluorobenzonitrile (0.94 g, 6.0 mmol), and K2CO3 (1.7 g, 12.0 mmol) by heating the mixture in DMSO (20 mL) at 100 °C (14 h), and method D was used to convert it to the target compound. Purification by flash chromatography (10% EtOAc/Hexanes) gave the analytically pure product 12 (79%). 1H NMR (400 MHz, CDCl3): δ = 7.76 (d, J = 2.0 Hz, 1H), 7.47 (dd, J = 4.0, J = 2.0 Hz, 1H), 6.93–6.87 (m, 3H), 6.78–6.75 (m, 1H), 5.42 (s, 1H), 2.58 (t, J = 7.6 Hz, 2H), 1.67 (quin, J = 7.6 Hz, 2H), 0.98 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ = 13.4, 23.9, 37.1, 106.3, 116.4, 117.0, 120.1, 120.9, 124.1, 131.8, 133.8, 138.5, 141.7, 146.9, 156.9 ppm; HRMS (ESI-positive): calcd for C16H14ClNO2 ([M−H]+): 286.0640, found: 286.0637.
2-[3-(2-Hydroxy-ethyl)phenoxy]-5-propylphenol (13)
Method B was used to prepare the methoxy-vinyl compound 13a from the appropriate phenol, and 3-bromostyrene (62%). This methoxy-vinyl compound 13a was subjected to hydroboration/alkaline hydrolysis, wherein it was treated with 1 M borane tetrahydrofuran complex in anhydrous tetrahydrofuran (0.5 equivalents), and the mixture was stirred at rt (3 h). Aqueous 3M NaOH solution (7 mL) followed by 30% hydrogen peroxide (4 mL) were added slowly drop wise to the above reaction mixture at 0 °C, and stirring continued (12 h) at rt. Usual workup followed by purification using flash column chromatography (2% MeOH/CHCl3) resulted in the methoxy compound 13b (68%). Method D was used to convert this methoxy precursor to 13. Purification by flash chrom atography (5% EtOAc/Hexanes) gave the analytically pure product as a colorless oil (75%). 1H NMR (400 MHz, CDCl3): δ = 0.98 (t, J = 7.2 Hz, 3H), 1.67 (t, J = 7.6 Hz, 2H), 2.57 (t, J = 7.6 Hz, 2H), 3.16 (t, J = 7.6 Hz, 2H), 3.57 (t, J = 7.2 Hz, 2H), 5.47 (s, 1H), 6.69 (dd, J = 4.0 Hz, J = 2.0 Hz, 1H), 6.84 (d, J = 8.4 Hz, 1H), 6.93–6.89 (m, 3H), 6.97 (d, J = 7.2 Hz, 1H), 7.30 ppm (t, J = 7.2 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ = 13.9, 24.5, 32.6, 37.6, 39.1, 116.0, 116.3, 117.8, 119.1, 120.7, 123.6, 129.9, 140.0, 140.9, 141.0, 147.3, 157.5 ppm; HRMS (ESI-positive): calcd for C17H19BrO2 ([M−H]+): 333.0496, found: 333.0492.
5-Chloro-2-phenoxybenzoic acid (14)
Method A was used to prepare the intermediate nitrile 14a. 5-chloro-2-fluorobenzonitrile (1.00 g, 6.4 mmol), phenol (0.66 g, 7.0 mmol) and K2CO3 (1.8 g, 12.8 mmol) were mixed in anhydrous DMSO (10 mL), and heated at 100 °C (8 h). A mixture of this nitrile 14a (0.20 g, 0.9 mmol) and 25% aqueous NaOH (0.4 mL) in ethanol (2 mL) was refluxed with stirring (20 h). After cooling, the reaction mixture was acidified with dilute hydrochloric acid. The precipitate was filtered, dried, and purified by column chromatography (SiO2, 6% MeOH/CHCl3) to yield the title compound (143 mg, 64%) as a white solid. 1H NMR (400 MHz, CD3OD): δ = 6.95 (m, 3H), 7.1 (t, J = 8.0 Hz, 1H), 7.33 (t, J = 8.0 Hz, 2H), 7.46 (dd, J = 9.0, J = 3.0 Hz, 1H), 7.84 ppm (d, J = 2.0 Hz, 1H); 13C NMR (100 MHz, CD3OD): δ = 119.6, 123.1, 124.8, 129.6, 131.1, 132.4, 134.4, 156.5, 158.6 ppm; MS (ESI) m/z: 249 [M + H]+; HRMS (ESI-positive): calcd for C13H9ClO3Na ([M + Na]+): 271.0133, found: 271.0129.
3-Chloro-4-(2-hydroxy-4-propylphenoxy)benzamide (15)
A mixture of nitrile 12 (0.38 g, 1.3 mmol), 30% hydrogen peroxide (1 mL), and aqueous 3N NaOH (0.18 mL) in ethanol was stirred at 35 °C (20 h). The resulting mixture was acidified with 1N sulfuric acid. Ethanol was evaporated in vacuum, and the white precipitate was dissolved in ethyl acetate, and washed with water followed by brine. Evaporation of the solvent resulted in the crude product which was purified by flash chromatography (3% MeOH/CHCl3) to give analytically pure product 15 as a white solid (65%). 1H NMR (400 MHz, CD3OD): δ = 0.91 (t, J = 7.6 Hz, 3H), 1.58 (quin, J = 7.6 Hz, 2H), 2.50 (t, J = 7.6 Hz, 2H), 6.62 (d, J = 8.0 Hz, 1H), 6.68 (dd, J = 4.0, J = 2.0 Hz, 1H), 6.82 (d, J = 2.0 Hz, 1H), 6.95 (d, J = 8.0 Hz, 1H), 7.39 (br s, 1H), 7.73 (dd, J = 4.0, J = 2.0 Hz, 1H), 7.97 (br s, 1H), 8.03 ppm (d, J = 2.0 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ = 115.5, 117.7, 120.2, 121.5, 122.3, 128.3, 129.1, 129.9, 139.7, 141.1, 149.1, 156.3, 166.6 ppm; HRMS (ESI-positive): calcd for C16H16ClNO3 ([M−H]+): 304.0746, found: 304.0743.
5-Chloro-2-phenoxyphenylmethanol (16)
A solution of BF3·Et2O (17 μL, 0.13 mmol) in THF (2 mL) was slowly added to a mixture of NaBH4 (0.012 g, 0.31 mmol) and the acid 14 (0.051 g, 0.21 mmol) in THF (1 mL) at rt under an inert atmosphere. The mixture was heated to reflux (1 h). The reaction mixture was cooled to 0 °C, and quenched with water. After 10 min, THF was removed under reduced pressure, CH2Cl2 was added, and the stirring was continued (1 h). The organic layer was separated, washed with brine, dried over anhydrous Na2SO4, and solvent was removed under reduced pressure. Purification of the crude residue by column chromatography (18% EtOAc/Hexanes) afforded the title compound 16 (39 mg, 80%) as a white solid. 1H NMR (400 MHz, CDCl3): δ = 2.11 (br s, 1H), 4.72 (s, 2H), 6.79 (d, J = 9.0 Hz, 1H), 6.96 (d, J = 8.0 Hz, 2H), 7.12 (t, J = 8.0 Hz, 1 H), 7.20 (dd, J = 8.0, J = 2.0 Hz, 1H), 7.34 (t, J = 8.0 Hz, 2H), 7.47 ppm (d, J = 3.0 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ = 60.7, 118.6, 119.9, 123.4, 129.0, 129.1, 130.2, 133.9, 153.3, 157.0 ppm; MS (ESI) m/z: 217 [M − H2O + H]+.
5-Chloro-2-phenoxybenzamide (17)
A mixture of the intermediate nitrile 14a (0.20 g, 0.9 mmol), 35% hydrogen peroxide (0.37 mL) and 3N aqueous NaOH (0.36 mL) in ethanol (5 mL) was stirred at 30 °C (18 h). The resulting mixture was acidified with 1N sulphuric acid, the precipitate was filtered, dried and purified by column chromatography (60% EtOAc/Hexanes) to afford the title compound 17 (171 mg, 70%) as a white solid. 1H NMR (400 MHz, CDCl3): δ = 6.55 (br s, 1H), 6.76 (d, J = 8.0 Hz, 1H), 7.07 (d, J = 6.0 Hz, 2H), 7.26 (t, J = 8.0 Hz, 1H), 7.34 (dd, J = 9.0, J = 3.0 Hz, 1H), 7.42 (t, J = 8.0 Hz, 2H), 7.55 (br s, 1H), 8.24 ppm (d, J = 2.0 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ = 119.6, 120.0, 125.4, 129.2, 130.6, 132.4, 133.2, 154.9, 133.2, 165.0 ppm; MS (ESI) m/z: 248 [M+H]+; HRMS (ESI-positive): calcd for C13H10ClNO2Na ([M + Na]+): 270.0293, found 270.0286.
N-[4-(4-Chloro-2-hydroxy-phenoxy)phenyl]acetamide (18)
The corresponding methoxy intermediate 20a (0.14 g, 0.6 mmol), prepared as described in the synthesis of 20, was acetylated by treating with acetic anhydride (0.07 g, 0.7 mmol), catalytic DMAP and Et3N (0.07 g, 0.7 mmol) in methylene chloride (2 mL) at 0 °C (3h). Method D was used to convert it to the target compound. Purification by flash chromatography (3% MeOH/CHCl3) gave the analytically pure product 18 as a white solid (92%). 1H NMR (400 MHz, CD3OD): δ = 2.11 (s, 3H), 7.00–6.78 (m, 5H), 7.52–7.45 ppm (m, 2H); 13C NMR (400 MHz, CD3OD): δ = 21.9, 116.4, 116.9, 119.0, 120.6, 121.2, 121.3, 124.2, 128.6, 133.1, 142.6, 149.2, 153.6, 169.8 ppm; HRMS (ESI-positive): calcd for C14H12ClNO3 ([M−H]+): 276.0433, found: 276.0432.
N-[2-(4-Chloro-2-hydroxy-phenoxy)phenyl]acetamide (19)
Method A was used to prepare the methoxy intermediate 5a, from 4-chloro-2-methoxyphenol (1.00 g, 6.3 mmol), 1-fluoro-2-nitrobenzene (0.89 g, 6.3 mmol), and K2CO3 (1.80 g, 12.6 mmol) in 96% yield. A mixture of 5a (1.70 g, 6.1 mmol), 10% Pd/C (0.30 mg), and EtOH (24 mL) was stirred at rt under H2 atmosphere. After 6 h, the reaction mixture was filtered through Celite, and the filter pad was washed with EtOH. The filtrate was concentrated in vacuo. The residue was purified by flash chromatography on silica gel (10% EtOAc/Hexane) to give the amino intermediate 19a (0.82 g, 92%) as a brown liquid. This intermediate 19a was acetylated by treating with acetic anhydride, catalytic DMAP and Et3N in methylene chloride at 0 °C in 3h. Method D was used to convert it to the target compound. Purification by flash chromatography (3% MeOH/CHCl3) gave the analytically pure product 19 as a white solid (92%). 1H NMR (400 MHz, CD3OD): δ = 2.17 (s, 3H), 6.73 (dd, J = 4.0, J = 1 Hz, 1H), 6.84 (dd, J = 4.0, J = 2.0 Hz, 1H), 6.98–6.93 (m, 2H), 7.09–7.03 (m, 2H), 7.76 ppm (dd, J = 3.0, J = 1 Hz, 1H); 13C NMR (100 MHz, CD3OD): δ = 21.7, 115.1, 116.7, 119.1, 121.7, 122.2, 123.8, 125.1, 127.1, 129.5, 141.5, 149.0, 149.3, 170.3 ppm; HRMS (ESI-positive): calcd for C14H12ClNO3 ([M−H]+): 276.0433, found: 276.0429.
2-(4-Aminophenoxy)-5-chlorophenol (20)
Method A was used to prepare the methoxy intermediate 4a, from 4-chloro-2-methoxyphenol (1.00 g, 6.3 mmol), 1-fluoro-4-nitrobenzene (0.89 g, 6.3 mmol), and K2CO3 (1.8 g, 12.6 mmol) in 96% yield. A mixture of 4a (1.00 g, 3.6 mmol), 10% Pd/C (0.36 g), and EtOH (20 mL) was stirred at rt under H2 atmosphere. After 4 h, the reaction mixture was filtered through Celite, and the filter pad was washed with MeOH. The filtrate was concentrated in vacuo. The residue was purified by flash chromatography on silica gel (20% EtOAc/Hexanes) to give the amino intermediate 20a (0.82 g, 92%) as a brown liquid. Method D was used to convert this amino intermediate 20a to the required compound 20. Purification by flash chromatography (20% EtOAc/Hexanes) gave the analytically pure product as brown solid (82%). 1H NMR (400 MHz, (CD3)2SO): δ = 4.89 (br s, 2H), 6.54 (d, J = 8.8 Hz, 2H), 6.67 (d, J = 8.8 Hz, 2H), 6.74 (dd, J = 4.0, J = 2.0 Hz, 2H), 6.90 ppm (d, J = 2.4 Hz, 2H); 13C NMR (100 MHz, CD3OD): δ = 115.3, 116.9, 119.3, 119.5, 119.7, 119.9, 126.8, 145.2, 145.5, 147.3, 149.5 ppm; HRMS (ESI-positive): calcd for C12H10ClNO2 ([M+H]+): 236.0473, found: 236.0471.
2-(4-Aminophenoxy)-5-propylphenol (21)
Method A was used to prepare the methoxy intermediate 8a in 93% yield from 2-methoxy-4-propylphenol (1.00 g, 6.3 mmol), 1-fluoro-4-nitrobenzene (0.89 g, 6.3 mmol), and K2CO3 (1.80 g, 12.6 mmol) by heating the mixture in DMSO (10 mL) at 100 °C (12 h). A mixture of 8a (1.00 g, 3.6 mmol), 10% Pd/C (0.36 g), and MeOH (20 mL) was stirred at rt under H2 atmosphere. After 4 h, the reaction mixture was filtered through Celite, and the filter pad was washed with MeOH. The filtrate was concentrated in vacuo. The residue was purified by flash chromatography on silica gel (20% EtOAc/Hexanes) to give the amino intermediate 21a (0.82 g, 92%) as a brown liquid. Method D was used to convert the intermediate 21a to the title compound. Purification by flash chromatography (3% MeOH/CHCl3) gave the analytically pure product 21 (84%). 1H NMR (400 MHz, CDCl3): δ = 0.96 (t, J = 7.6 Hz, 3H), 1.65 (quin, J = 7.6 Hz, 2H), 2.53 (t, J = 7.6 Hz, 2H), 6.62 (d, J = 8.0 Hz, 1H), 6.71–6.67 (m, 3H), 6.89–6.87 ppm (m, 3H); 13C NMR (100 MHz, CDCl3): δ = 13.4, 24.1, 37.1, 115.3, 115.9, 116.6, 119.4, 119.8, 138.1, 142.1, 142.5, 146.2, 148.6 ppm; HRMS (ESI-positive): calcd for C15H17NO2 ([M−H]+): 242.1236, found: 242.1238.
4-(4-Chloro-2-hydroxyphenoxy)-1-(4 methylphenylsulphonamido) benzene (22)
Amino intermediate 20a (0.16 g, 0.63 mmol) was dissolved in anhydrous methylene chloride (3 mL), and cooled to 0 °C and then triethylamine (0.2 mL, 1.25 mmol) was added, after 10 minutes, tosyl chloride (0.18 g, 0.94 mmol) was added, and the reaction mixture was stirred at the rt (3 h). The reaction was quenched by adding 10 mL of 1M HCl, and diluted with ethyl acetate (100 mL), the organic layer was separated, and the aqueous layer was extracted twice with ethyl acetate. All the organic layers were combined and washed with water, brine, and dried over anhydrous Na2SO4, and concentrated in vacuo. The crude reaction mixture was subjected to subsequent reaction without further purification. General method D was applied for demethylation. Purification of the crude reaction mixture by column chromatography (22% EtOAc/Hexanes) afforded the title compound 22 (122 mg, 50%) as a white solid. 1H NMR (300 MHz, CDCl3): δ = 2.41 (s, 3H), 5.66 (s, 1H), 6.68 (s, 1H), 6.72 (d, J = 9.0 Hz, 1H), 6.81 (dd, J = 9.0, J = 2.0 Hz, 1H), 6.90 (m, 2H), 7.04 (m, 3H), 7.26 (d, J = 8.0 Hz, 2H), 7.64 ppm (d, J = 9.0 Hz, 2H); 13C NMR (75 MHz, CDCl3): δ = 21.8, 116.9, 118.9, 119.6, 120.8, 124.7, 127.5, 129.9, 136.2, 142.3, 144.2, 154.5 ppm; MS (ESI) m/z: 412 [M + Na]+. HRMS (ESI-positive): calcd for C19H15ClNO4S ([M−H]+): 388.0416, found 388.0415.
5-Chloro-2-(pyridin-3-yloxy)phenol (23)
Method B was used to prepare the methoxy precursor 3-(4-chloro-2-methoxyphenoxy)pyridine in 48% from 4-chloro-2-methoxyphenol (1 g, 6.3 mmol), KOtBu (0.85 g, 7.6 mmol), (CuOTf)2·PhH (0.33 g, 0.63 mmol), and 3-iodopyridine (1.60 g, 7.6 mmol), and method D was used to convert it to the phenol 23. Purification by flash chromatography (3% MeOH/CHCl3) gave the analytically pure product as an off white solid (52%). 1H NMR (400 MHz, CD3OD): δ = 4.83 (s, 1H), 5.56 (s, 1H), 6.36 (dd, J = 8.0, J = 2.0 Hz, 1H), 6.58 (d, J = 2.8 Hz, 1H), 6.88 (dd, J = 4.0, J = 2.0 Hz, 1H), 7.03–6.97 (m, 2H), 7.39–7.29 (m, 2H), 8.23 ppm (br s, 2H); 13C NMR (100 MHz, CD3OD): δ = 116.7, 119.3, 122.2, 123.3, 124.1, 130.2, 137.8, 140.6, 141.9, 149.8 ppm; HRMS (ESI-positive): calcd for C11H8ClNO2 ([M−H]+): 220.0171, found: 220.0170.
N-[5-(4-Chloro-2-hydroxyphenoxy)-pyridin-2-yl]acetamide (24)
The methoxy precursor N-[5-(4-chloro-2-methoxyphenoxy)-pyridin-2-yl]acetamide was prepared from commercially available 4-chloro-2-methoxyphenol (1.00 g, 6.3 mmol), N-(5-iodo-pyridin-2-yl)acetamide (1.4 g, 5.3 mmol), KOtBu (0.78 g, 7.0 mmol), and (CuOTf)2·PhH (0.49 g, 0.95 mmol) using method B (15%), and method D was used to convert it to the phenol 24. Purification by flash chrom atography (8% MeOH/CHCl3) gave the analytically pure product as a white solid (60%). 1H NMR (400 MHz, CDCl3): δ = 2.16 (s, 3H), 6.84 (dd, J = 4.0, J = 2.0 Hz, 1H), 6.93–6.98 (m, 2H), 7.31(dd, J = 8.0, J = 3.0 Hz, 1H), 7.98 (d, J = 2.8 Hz, 1H), 8.02 ppm (d, J = 8.9 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ = 114.9, 117.0, 119.6, 119.9, 120.7, 121.6, 125.4, 125.9, 126.0, 129.9, 136.6, 136.7, 142.2, 146.6, 148.9, 149.8, 151.1, 170.3 ppm; HRMS (ESI-positive): calcd for C13H11ClN2O3 ([M−H]+): 277.0385, found: 277.0384.
5-Chloro-2-(pyrazin-2-yloxy)phenol (25)
Method B was used to prepare the methoxy precursor 2-(4-chloro-2-methoxyphenoxy)pyrazine in 67% yield from commercially available 4-chloro-2-methoxyphenol (1.00 g, 6.3 mmol), 2-iodopyrazine (1.10 g, 5.3 mmol), KOtBu (0.78 g, 7.0 mmol) and (CuOTf)2·PhH (0.49 g, 0.95 mmol), and method D was used to convert it to the phenol 25. Purification by flash chromatography (3% MeOH/CHCl3) gave the analytically pure product as a white solid (73%). 1H NMR (400 MHz, CD3OD): δ = 6.88 (dd, J = 4.0, J = 3.0 Hz, 1H), 6.96 (d, J = 2.4 Hz, 1H), 7.09 (d, J = 8.8 Hz, 1H), 8.1 (dd, J = 2.0, J = 1.0 Hz, 1H), 8.25 (d, J = 2.8 Hz, 1H), 8.41 ppm (d, J = 1.6 Hz, 1H); 13C NMR (100 MHz, CD3OD): δ = 116.4, 117.2, 118.9, 122.5, 123.3, 125.7, 130.5, 134.4, 137.4, 137.5, 140.6 ppm; HRMS (ESI-positive): calcd for C10H7ClN2O2 ([M−H]+): 221.0123, found: 221.0123.
3-(3-Hydroxy-4-phenoxyphenyl)propane-1,2-diol (26)
To a stirred solution of intermediate 27a (0.10 g, 0.41 mmol) in THF (20 mL) were added N-methyl-morpholine N-oxide (0.20 g, 1.66 mmol) and a solution of 4 wt.% OsO4/H2O (0.24 mL, 0.038 mmol, 4 mol%) and the reaction was allowed to stir (12 h), at which time all staring material had been consumed as monitored by TLC. The reaction mixture was poured into a solution of 15% Na2SO3 (50 mL), extracted with CH2Cl2. The organic extract was washed with brine, dried with MgSO4, and evaporated in vacuo. The crude material was subjected to subsequent demethylation according to the general procedure described in method D, to yield 31 mg (28%) of 26. 1H NMR (400 MHz, CDCl3): δ = 2.75 (t, J = 7.7 Hz, 2H), 3.58–3.54 (m, 1H), 3.75–3.72 (m, 1H), 3.97 (br s, 1H), 5.47 (s, 1H), 6.68 (dd, J = 8.0, J = 1.0 Hz, 1H), 6.82 (d, J = 8.1 Hz, 1H), 5.66 (s, 1H), 6.72 (dd, J = 8.0, J = 1.0 Hz, 1H), 6.85 (d, J = 8.2 Hz, 1H), 6.95 (s, 1H), 7.04 (d, J = 8.0 Hz, 2H), 7.14 (t, J = 7.3 Hz, 1H), 7.36 ppm (t, J = 7.8 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ = 39.2, 66.0, 72.8, 116.8, 117.8, 118.9, 121.3, 123.6, 129.8, 134.4, 142.1, 147.4 ppm; HRMS (ESI-positive): calcd for C15H16O4 ([M−H]+): 259.0975, found: 259.0978.
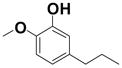
2-Phenoxy-5-propylphenol (27)
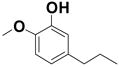
A suspension of intermediate 27a (0.10 g, 0.41 mmol) and 10% Pd/C (0.02 g) in EtOAc was stirred under a hydrogen atmosphere at rt (4 h). The catalyst was removed by filtration through a pad of Celite, and the residue was thoroughly washed with EtOAc. The solvent was evaporated to give a crude residue, which was directly subjected to demethylation using the standard procedure described in method D above. The crude product was purified by preparative TLC (30% EtOAc/Hexanes) to afford 54 mg (71%) of 27. 1H NMR (400 MHz, CDCl3): δ = 0.97 (t, J = 7.3 Hz, 3H), 1.69–1.62 (m, 2H), 2.56 (t, J = 7.5 Hz, 2H), 5.49 (s, 1H), 6.68 (dd, J = 8.0, J = 1.0 Hz, 1H), 6.83 (d, J = 8.2 Hz, 1H), 6.90 (d, J = 1.6 Hz, 1H), 7.03 (d, J = 7.8 Hz, 2H), 7.12 (t, J = 7.3 Hz, 1H), 7.35 ppm (t, J = 7.7 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ = 13.7, 24.4, 37.5, 116.0, 117.5, 118.9, 120.5, 123.2, 129.7, 139.8, 147.2, 141.0, 157.1 ppm.
5-Hydroxymethyl-2-phenoxyphenol (28)
A mixture of intermediate 28a (0.08 g, 0.34 mmol), and BBr3 (1M, 6.9 mL, 6.9 mmol) was subjected to the general demethylation procedure described in method D above. The crude product was purified by preparative TLC (40% EtOAc/Hexanes) to afford 64 mg (85%) of 28. 1H NMR (400 MHz, CDCl3): δ = 4.48 (s, 2H), 5.66 (s, 1H), 6.82 (d, J = 7.3 Hz, 1H), 6.89 (dd, J = 8.0, J = 2.0 Hz, 1H), 7.06 (d, J = 8.3 Hz, 2H), 7.12 (d, J = 2.0 Hz, 1H), 7.17 (t, J = 7.3 Hz, 1H), 7.38 (t, J = 7.8 Hz, 2H) ppm; 13C NMR (100 MHz, CDCl3): δ = 33.2, 116.7, 118.2, 118.4, 121.3, 124.0, 129.9, 134.0, 143.9, 147.2, 156.1 ppm.
5-[(1-Methyl-butylamino)methyl]-2-phenoxyphenol (29)
A mixture of intermediate 29a (0.10 g, 0.33 mmol), and BBr3 (1M, 6.6 mL, 6.6 mmol) were subjected to the general demethylation procedure outlined in method D above. The crude product was purified by preparative TLC (10% MeOH/CH2Cl2) to afford 65 mg (68%) of 29. 1H NMR (400 MHz, CDCl3): δ = 0.91 (t, J = 7.4 Hz, 3H), 1.09 (d, J = 11.0 Hz, 3H), 1.41–1.34 (m, 1H), 1.58–1.51 (m, 1H), 2.67–2.62 (m, 1H), 3.78–3.67 (m, 2H), 6.79–6.77 (m, 1H), 6.84 (d, J = 8.1 Hz, 1H), 6.99 (d, J = 7.9 Hz, 2H), 7.01 (d, J = 1.4 Hz, 1H), 7.10 (d, J = 7.3 Hz, 1H), 7.33 ppm (t, J = 7.8 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ = 10.2, 19.5, 29.6, 50.7, 53.9, 116.6, 117.5, 119.2, 120.1, 123.1, 129.7, 137.4, 142.3, 147.7, 157.1, 165.0 ppm.
2-Phenoxy-5-piperidin-1-ylmethylphenol (30)
The intermediate 30a (0.07 g, 0.23 mmol) was subjected to the general demethylation procedure outlined in method D above using 4.7 mL of 1M BBr3 in dichloromethane solution. The residue was purified by preparative TLC using 1:10 MeOH/CH2Cl2 to afford 30 (34 mg, 50%). 1H NMR (400 MHz, CDCl3): δ = 1.47 (br s, 2H), 1.70–1.60 (m, 4H), 2.47 (br s, 4H), 3.49 (s, 2H), 6.83 (s, 2H), 7.03 (d, J = 7.8 Hz, 2H), 7.12 (t, J = 7.3 Hz, 1H), 7.07 (s, 1H), 7.35 ppm (t, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ = 24.1, 25.5, 54.2, 63.0, 117.2, 117.8, 118.6, 121.4, 123.4, 129.8, 142.5, 147.3, 156.9 ppm.
5-(Benzylaminomethyl)-2-phenoxyphenol (31)
A mixture of intermediate 31a (0.08 g, 0.25 mmol) and BBr3 (1M, 5 mL, 5.0 mmol) were subjected to the general demethylation procedure outlined in method D above. The crude product was purified by preparative TLC (10% MeOH/CH2Cl2) to afford 42 mg (54%) of 31. 1H NMR (400 MHz, CDCl3): δ = 3.78 (s, 2H), 3.86 (s, 2H), 6.84 (s, 2H), 7.00 (d, J = 8.0 Hz, 2H), 7.14–7.10 (m, 2H), 7.42–7.28 ppm (m, 7H); 13C NMR (100 MHz, CDCl3): δ = 51.5, 52.0, 116.6, 117.8, 119.0, 120.6, 123.4, 127.5, 128.5, 128.6, 128.7, 129.8, 134.4, 137.6, 142.9, 147.7, 156.8 ppm. HRMS (ESI-positive): calcd for C20H19NO2 ([M−H]+): 304.1343, found: 304.1352.
2-[4-(2-Hydroxy-4-Chlorophenoxy)phenoxy]-5-chlorophenol (32)
A mixture of 2-methoxy-4-propylphenol (0.20 g, 1.2 mmol), and benzene-1, 4-diboronic acid (0.20 g, 1.2 mmol) were subjected to the general biaryl ether coupling procedure described in method C. The crude product was purified by preparative TLC (30% EtOAc/Hexanes) to afford 35 mg (7%) of intermediate 32a. A mixture of 32a (0.03 g, 0.076 mmol) and BBr3 (1M, 1.5 mL, 1.5 mmol) was subjected to the general demethylation procedure outlined above in method D. The crude product was purified by preparative TLC (40% EtOAc/Hexanes) to afford 12 mg (43%) of 32. 1H NMR (400 MHz, (CD3)2SO): δ = 6.86–6.83 (m, 6H), 6.93 (d, J = 7.5 Hz, 2H), 6.98 (d, J = 2.3 Hz, 2H), 10.06 ppm (s, 2H); 13C NMR (100 MHz, (CD3)2SO): δ = 117.3, 118.3, 119.7, 122.6, 128.5, 143.1, 150.4, 152.9 ppm. HRMS (ESI-positive): calcd for C18H12Cl2O4 ([M−H]+): 361.0039, found: 361.0046.
2-[3-(2-Hydroxy-4-chlorophenoxy)phenoxy]-5-chlorophenol (33)
A mixture of 2-methoxy-4-propylphenol (0.20 g, 1.20 mmol), and benzene-1, 3-diboronic acid (0.20 g, 1.20 mmol) were subjected to the general biaryl ether coupling procedure described in method C. The crude product was purified by preparative TLC (30% EtOAc/Hexanes) to afford 207 mg (41%) of intermediate 33a. A mixture of 33a (0.1 g, 0.25 mmol) and BBr3 (1M, 5.1 mL, 5.1 mmol) was subjected to the general demethylation procedure outlined above in method D. The crude product was purified by preparative TLC (40% EtOAc/Hexanes) to afford 65 mg (70%) of 33. 1H NMR (400 MHz, CDCl3): δ = 0.96 (t, J = 7.3 Hz, 6H), 1.70–1.60 (m, 4H), 2.55 (t, J = 7.4 Hz, 4H), 5.68 (s, 2H), 6.76–6.71 (m, 3H), 6.86 (s, 4H), 7.08 (t, J = 1.2 Hz, 2H), 7.30 ppm (t, J = 8.2 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ = 107.2, 111.9, 112.2, 116.4, 116.5, 119.1, 119.7, 120.4, 129.7, 130.5, 141.1, 147.7, 157.6 ppm. HRMS (ESI-positive): calcd for C18H12Cl2O4 ([M−H]+): 361.0039, found: 361.0050.
2-[3-(2-Hydroxy-4-propylphenoxy)phenoxy]-5-Propylphenol (34)
A mixture of 2-methoxy-4-propylphenol (0.20 g, 1.2 mmol), and benzene-1,3-diboronic acid (0.20 g, 1.2 mmol) was subjected to the general biaryl ether coupling procedure described in method C. The crude product was purified by preparative TLC (30% EtOAc/Hexanes) to afford 27 mg (5%) of intermediate 34a. A mixture of 34a (0.10 g, 0.24 mmol), and BBr3 (1M, 4.9 mL, 4.9 mmol) was subjected to the general demethylation procedure outlined in method D above. The crude product was purified by preparative TLC (40% EtOAc/Hexanes) to afford 67 mg (71%) of 34. 1H NMR (400 MHz, CDCl3): δ = 0.97 (t, J = 7.3 Hz, 6H), 1.70–1.60 (m, 4H), 2.56 (t, J = 7.4 Hz, 4H), 5.40 (s, 2H), 6.75–6.65 (m, 5H), 6.86 (s, 1H), 6.88 (t, J = 1.8 Hz, 3H), 7.27–7.22 ppm (m, 1H); 13C NMR (100 MHz, CDCl3): δ = 13.3, 24.0, 37.1, 106.4, 111.3, 115.9, 119.0, 120.3, 130.1, 139.9, 146.8, 158.2 ppm; HRMS (ESI-positive): calcd for C24H26O4 ([M−H]+): 377.1758, found: 377.1763.
2-[4-(2-Hydroxy-4-propylphenoxy)phenoxy]-5-propylphenol (35)
A mixture of 2-methoxy-4-propylphenol (0.20 g, 1.2 mmol), and benzene-1,4-diboronic acid (0.20 g, 1.2 mmol) was subjected to the general biaryl ether coupling procedure outlined in method C above. The crude product was purified by preparative TLC (30% EtOAc/Hexanes) to afford 27 mg (5%) of 35a. A mixture of 35a (0.02 g, 0.049 mmol), and BBr3 (1M, 0.98 mL, 0.98 mmol) was subjected to the general demethylation procedure outlined above. The crude product was purified by preparative TLC (40% EtOAc/Hexanes) to afford 13 mg (69%) of 35. 1H NMR (400 MHz, CDCl3): δ = 0.96 (t, J = 7.3 Hz, 6H), 1.70–1.60 (m, 4H), 2.55 (t, J = 7.4 Hz, 4H), 5.51 (s, 2H), 6.67 (dd, J = 8.0, J = 2.0 Hz, 2H), 6.79 (d, J = 8.2 Hz, 2H), 6.89 (d, J = 1.8 Hz, 2H), 7.00 ppm (s, 4H); 13C NMR (100 MHz, CDCl3): δ = 13.3, 24.0, 37.1, 115.6, 117.8, 118.7, 120.1, 139.2, 141.1, 146.5, 152.3 ppm; HRMS (ESI-positive): calcd for C26H26O5 ([M−H]+): 377.1758, found: 377.1747.
2-(Biphenyl-3-yloxy)-5-propylphenol (36)
A mixture of 4-propyl-2-methoxyphenol (0.10 g, 0.6 mmol), and 3-biphenylboronic acid (0.36 g, 1.8 mmol) was subjected to the general biaryl ether coupling procedure outlined in the method C above to obtain intermediate 36a in 46% yield. A mixture of 36a (0.08 g, 0.25 mmol), and BBr3 (1M, 5.0 mL, 5.0 mmol) was subjected to the general demethylation procedure described in method D above. The crude product was purified by preparative TLC (40% EtOAc/Hexanes) to afford 34 mg (44%) of 36. 1H NMR (400 MHz, CDCl3): δ = 0.99 (t, J = 7.3 Hz, 3H), 1.75–1.65 (m, 2H), 2.58 (t, J = 7.4 Hz, 2H), 5.56 (s, 1H), 6.71 (d, J = 8.2 Hz, 1H), 6.90 (d, J = 8.2 Hz, 1H), 6.93 (s, 1H), 7.01 (d, J = 6.7 Hz, 1H), 7.42–7.35 (m, 3H), 7.46 (t, J = 7.3 Hz, 2H), 7.59 ppm (d, J = 7.4 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ = 13.4, 24.0, 37.1, 115.7, 115.9, 116.0, 118.6, 120.2, 121.7, 126.7, 127.2, 128.4, 129.7, 139.5, 139.9, 140.6, 142.8, 146.8, 157.2 ppm; HRMS (ESI-positive): calcd for C29H34N4O5 ([M−H]+): 519.2602, found: 519.2595.
3-(2-Hydroxy-4-propylphenoxy)benzoic acid methyl ester (37)
A mixture of 4-propyl-2-methoxyphenol (0.10 g, 0.6 mmol), and 3-(ethoxycarbonyl) phenylboronic acid (0.32 g, 1.8 mmol) was subjected to the general biaryl ether coupling procedure outlined in method C above to obtain intermediate 37a in 41% yield. A mixture of 37a (0.07 g, 0.23 mmol) and BBr3 (1M, 4.6 mL, 4.6 mmol) was subjected to the general demethylation procedure described in method D. The crude product was purified by preparative TLC (50% EtOAc/Hexanes) to afford 44 mg (65%) of 37. 1H NMR (400 MHz, CDCl3): δ = 0.97 (t, J = 7.3 Hz, 3H), 1.69–1.62 (m, 2H), 2.57 (t, J = 7.5 Hz, 2H), 3.91 (s, 3H), 5.56 (s, 1H), 6.72 (dd, J = 8.0, J = 2.0 Hz, 1H), 6.82 (d, J = 8.2 Hz, 1H), 6.91 (d, J = 1.8 Hz, 1H), 7.22–7.19 (m, 1H), 7.40 (t, J = 8.0 Hz, 1H), 7.68–7.67 (m, 1H), 7.77 ppm (d, J = 7.7 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ = 13.7, 24.4, 37.5, 52.2, 116.4, 118.4, 119.0, 120.7, 121.9, 124.3, 129.8, 131.9, 140.3, 140.6, 147.2, 157.3, 166.4 ppm; HRMS (ESI-positive): calcd for C17H18O4 ([M−H]+): 285.1132, found: 285.1139.
2-(3-Hydroxymethyl-phenoxy)-5-propylphenol (38)
A mixture of 4-propyl-2-methoxyphenol (0.10 g, 0.6 mmol) and 3-(hydroxymethyl) phenylboronic acid (0.27 g, 1.8 mmol) was subjected to the general biaryl ether coupling procedure in method C above to obtain intermediate 38a in 18% yield. A mixture of 38a (0.03 g, 0.11 mmol), and BBr3 (1M, 2.2 mL, 2.2 mmol) was subjected to the general demethylation procedure outlined in method D. The crude product was purified by preparative TLC (60% EtOAc/Hexanes) to afford 12 mg (42%) of 38. 1H NMR (300 MHz, CDCl3): δ = 0.98 (t, J = 7.3 Hz, 3H), 1.78–1.62 (m, 2H), 2.57 (t, J = 7.4 Hz, 2H), 5.45 (s, 1H), 6.70 (dd, J = 8.0, J = 2.0 Hz, 1H), 6.84 (d, J = 8.2 Hz, 1H), 6.91 (d, J = 1.8 Hz, 1H), 6.94 (dd, J = 8.0, J = 2.0 Hz, 1H), 7.06 (d, J = 1.9 Hz, 1H), 7.14 (d, J = 7.6 Hz, 1H), 7.29 ppm (d, J = 8.8 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ = 13.8, 24.4, 32.7, 37.5, 116.2, 117.3, 117.9, 119.1, 120.7, 123.8, 125.8, 130.1, 139.7, 140.2, 140.5, 147.2, 157.4 ppm.
3-(2-Hydroxy-4-propylphenoxy)benzoic acid (39)
A mixture of 37 (0.02 g, 0.073 mmol), and LiOH·H2O (0.03 g, 0.73 mmol) in methanol and water (5 mL + 5 mL) was stirred at rt for 2 h. The reaction mixture was acidified with saturated KHSO4 solution, and extracted with EtOAc. The organic layer was washed with brine, dried over Na2SO4, filtered, and evaporated under reduced pressure. The residue was purified by preparative TLC using 1:10 MeOH/CH2Cl2 to afford 11 mg (55%) of 39. 1H NMR (300 MHz, CDCl3): δ = 0.98 (t, J = 7.3 Hz, 3H), 1.70–1.60 (m, 2H), 2.57 (t, J = 7.5 Hz, 2H), 6.71 (dd, J = 8, J = 1 Hz, 1H), 6.84 (d, J = 8.2 Hz, 1H), 6.92 (d, J = 1.5 Hz, 1H), 7.27 (dd, J = 8, J = 2 Hz, 1H), 7.45 (t, J = 8.0 Hz, 1H), 7.74 (s, 1H), 7.86 ppm (d, J = 7.6 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ = 13.7, 24.4, 37.5, 116.4, 118.8, 119.1, 120.8, 122.8, 124.9, 129.9, 130.2, 131.0, 140.4, 147.2, 157.4, 171.2; HRMS (ESI-positive): calcd for C16H16O4 ([M−H]+): 271.0975, found: 271.0982.
1-[4-(2-Hydroxy-4-propylphenoxy)phenyl]ethanone (40)
General method D was employed to prepare the methoxy intermediate 40a in 39% yield from 2-methoxy-4-propylphenol (0.10 g, 0.6 mmol), and 4-acetylphenylboronic acid (0.30 g, 1.8 mmol). A mixture of 40a (0.06 g, 0.21 mmol), and BBr3 (1M, 4.2 mL, 4.2 mmol) were subjected to the general demethylation procedure outlined in method D above. The crude product was purified by preparative TLC (40% EtOAc/Hexanes) to afford 42 mg (73%) of 40. 1H NMR (400 MHz, CDCl3): δ = 0.98 (t, J = 7.2 Hz, 3H), 1.75–1.62 (m, 2H), 2.60–2.50 (m, 5H), 5.37 (s, 1H), 6.74 (d, J = 8.0 Hz, 1H), 6.90 (d, J = 8.5 Hz, 1H), 6.92 (s, 1H), 7.05 (d, J = 8.5 Hz, 2H), 7.96 ppm (d, J = 8.5 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ = 13.7, 24.4, 26.4, 37.5, 116.4, 116.6, 120.1, 120.9, 130.6, 132.2, 139.5, 141.1, 147.4, 161.5, 196.6 ppm; HRMS (ESI-positive): calcd for C17H18O3 ([M−H]+): 269.1183, found: 269.1188.
2-(4-Methylsulfanylphenoxy)-5-propylphenol (41)
A mixture of 41a (0.05 g, 0.17 mmol) and BBr3 (1M, 3.4 mL, 3.4 mmol) was subjected to the general demethylation procedure outlined in method D above. The crude product was purified by preparative TLC (40% EtOAc/Hexanes) to afford 32 mg of 41 (67%). 1H NMR (400 MHz, CDCl3): δ = 0.97 (t, J = 7.2 Hz, 3H), 1.72–1.62 (m, 2H), 2.49 (s, 3H), 2.56 (t, J = 6.4 Hz, 2H), 5.47 (s, 1H), 6.67 (d, J = 8.2 Hz, 1H), 6.80 (d, J = 8.2 Hz, 1H), 6.89 (s, 1H), 6.97 (d, J = 8.7 Hz, 2H), 7.29–7.27 ppm (m, 2H); 13C NMR (100 MHz, CDCl3): δ = 13.7, 17.1, 24.4, 37.5, 116.1, 118.3, 118.6, 120.5, 125.8, 129.2, 132.3, 139.8, 141.1, 147.0, 155.2 ppm; HRMS (ESI-positive): calcd for C16H18O2S ([M−H]+): 273.0954, found: 273.0958.
2-[4-(1-Hydroxyethyl)phenoxy]-5-propylphenol (42)
A mixture of 40 (0.02 g, 0.073 mmol) and NaBH4 (.008 g, 0.22 mmol) was subjected to the general reduction procedure outlined above. The crude product was purified by preparative TLC (50% EtOAc/Hexanes) to afford 14 mg (69%) of 42. 1H NMR (300 MHz, CDCl3): δ = 0.96 (t, J = 7.3 Hz, 3H), 1.70–1.55 (m, 2H), 2.56 (t, J = 7.4 Hz, 2H), 4.91 (q, J = 6.4 Hz, 1H), 5.50 (s, 1H), 6.67 (dd, J = 8, J = 2 Hz, 1H), 6.82 (d, J = 8.2 Hz, 1H), 6.89 (d, J = 1.7 Hz, 1H), 7.00 (d, J = 8.2 Hz, 2H), 7.35 ppm (d, J = 8.5 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ = 13.7, 24.4, 25.1, 37.5, 69.8, 116.1, 117.5, 118.9, 120.5, 126.9, 139.8, 140.7, 141.0, 147.1, 156.5 ppm; HRMS (ESI-positive): calcd for C17H20O3 ([M−H]+): 271.1339, found: 271.1342.
2-(4-Methanesulfinylphenoxy)-5-propylphenol (43)
A mixture of 2-methoxy-4-propylphenol (0.20 g, 1.2 mmol), and 4-(methylsulfanyl) phenylboronic acid (0.61 g, 3.61 mmol) were subjected to the general biaryl ether coupling procedure outlined in method C above to obtain intermediate 41a in 59% yield. 41a (0.15 g, 0.52 mmol) was subjected to oxidation using m-CPBA (80%, 0.17 g, 0.78 mmol, in 3 mL dichloromethane) in dichloromethane (20 mL) at 0 °C to afford 35 mg (22%) of 43a, and 54 mg (32%) of 44a. A mixture of 43a (0.03 g, 0.098 mmol), and BBr3 (1M, 1.9 mL, 1.9 mmol) was subjected to the general demethylation protocol described in method D above. The crude product was purified by preparative TLC (60% EtOAc/Hexanes) to afford 19 mg (66%) of 43. 1H NMR (400 MHz, CDCl3): δ = 0.97 (t, J = 7.1 Hz, 3H), 1.70–1.58 (m, 2H), 2.57 (t, J = 6.7 Hz, 2H), 2.73 (s, 3H), 5.86 (s, 1H), 6.72 (d, J = 8.0 Hz, 1H), 6.88 (d, J = 8.1 Hz, 1H), 6.92 (s, 1H), 7.12 (d, J = 6.9 Hz, 2H), 7.60 ppm (d, J = 6.9 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ = 13.7, 24.4, 37.5, 43.9, 116.8, 117.6, 120.1, 120.8, 125.5, 138.9, 139.8, 141.0, 147.5, 160.1 ppm; HRMS (ESI-positive): calcd for C16H18O3S ([M−H]+): 289.0903, found: 289.0910.
2-(4-Methanesulfonylphenoxy)-5-propylphenol (44)
A mixture of 44a (0.05 g, 0.15 mmol) and BBr3 (1M, 3.1 mL, 3.1 mmol) was subjected to the general demethylation procedure outlined above in method D. The crude product was purified by preparative TLC (60% EtOAc in hexane) to afford 24 mg (50%) of 44. 1H NMR (400 MHz, CDCl3): δ = 0.98 (t, J = 7.2 Hz, 3H), 1.75–1.65 (m, 2H), 2.59 (t, J = 6.6 Hz, 2H), 3.06 (s, 3H), 5.41 (s, 1H), 6.76 (d, J = 8 Hz, 1H), 6.91 (d, J = 8 Hz, 1H), 6.93 (s, 1H), 7.12 (d, J = 6.7 Hz, 2H), 7.89 (d, J = 6.6 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ = 13.7, 24.3, 37.5, 44.7, 116.9, 117.0, 120.4, 121.1, 129.7, 134.4, 139.1, 141.6, 147.4, 162.0 ppm; HRMS (ESI-positive): calcd for C16H18O4S ([M−H]+): 305.0853, found: 305.0858.
2-(3-Dimethylaminophenoxy)-5-propylphenol (45)
A mixture of 45b (0.09 g, 0.31 mmol), and BBr3 (1M, 6.3 mL, 6.3 mmol) was subjected to the general demethylation procedure outlined above. The crude product was purified by flash chromatography over silica gel (40% EtOAc/Hexanes) to afford 70 mg (81%) of 45. 1H NMR (400 MHz, CDCl3): δ = 0.97 (t, J = 7.3 Hz, 3H), 1.68–1.63 (m, 2H), 2.56 (t, J = 7.5 Hz, 2H), 2.95 (s, 6H), 5.52 (s, 1H), 6.32 (d, J = 7.6 Hz, 1H), 6.44 (s, 1H), 6.49 (d, J = 8.3 Hz, 1H), 6.66 (d, J = 7.1 Hz, 1H), 6.85 (d, J = 8.2 Hz, 1H), 6.86 (s, 1H), 7.18 ppm (t, J = 8.1 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ = 13.4, 24.0, 37.1, 40.0, 101.7, 104.8, 107.2, 115.4, 118.4, 120.2, 129.6, 139.0, 140.9, 146.7, 151.6, 157.7 ppm; HRMS (ESI-positive): calcd for C17H21NO2 ([M−H]+): 270.1499, found: 270.1501.
5-Propyl-2-(3-trifluoromethylphenoxy)phenol (46)
A mixture of intermediate 46b (0.08 g, 0.25 mmol) and BBr3 (1M, 5.1 mL, 5.1 mmol) was subjected to the general demethylation procedure outlined above. The crude product was purified by preparative TLC (40% EtOAc/Hexanes) to afford 45 mg (58%) of 46. 1H NMR (400 MHz, CDCl3): δ = 0.98 (t, J = 7.3 Hz, 3H), 1.72–1.62 (m, 2H), 2.58 (t, J = 6.8 Hz, 2H), 5.40 (s, 1H), 6.72 (d, J = 8.1 Hz, 1H), 6.85 (d, J = 8.1 Hz, 1H), 6.92 (s, 1H), 7.17 (d, J = 8.1 Hz, 1H), 7.37 (d, J = 7.3 Hz, 1H), 7.45 ppm (t, J = 7.9 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ = 13.3, 24.0, 37.1, 113.9, 113.9, 116.1, 118.9, 119.4, 119.4, 119.9, 120.5, 121.8, 124.5, 130.0, 131.4, 131.7, 132.0, 139.7, 140.4, 146.8, 157.2 ppm; HRMS (ESI-positive): calcd for C16H19F3O2 ([M−H]+): 295.0951, found: 295.0973.
3-(2-Hydroxy-4-propylphenoxy)benzaldehyde (47)
A mixture of 47a (0.25 g, 0.92 mmol), and BBr3 (1M, 18.4 mL, 18.4 mmol) was subjected to the general demethylation procedure outlined above. The crude product was purified by flash chromatography over silica gel (60% EtOAc/Hexanes) to afford 187 mg (78%) of 47. 1H NMR (300 MHz, CDCl3): δ = 0.98 (t, J = 7.2 Hz, 3H), 1.70–1.63 (m, 2H), 2.57 (t, J = 6.7 Hz, 2H), 6.71 (dd, J = 8, J = 2 Hz, 1H), 6.85 (d, J = 8.3 Hz, 1H), 6.97 (d, J = 1.7 Hz, 1H), 7.32–7.29 (m, 1H), 7.49 (s, 1H), 7.53 (d, J = 7.9 Hz, 1H), 7.63 (d, J = 7.3 Hz, 1H), 9.98 (s, 1H) ppm.
3-[3-(2-Hydroxy-4-propylphenoxy)phenyl]acrylic acid methyl ester (48)
A mixture of 47 (0.18 g, 0.70 mmol), and methyl (triphenylphosphoranylidene) acetate (0.47 g, 1.40 mmol) in 20 mL of THF was refluxed overnight. The solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (1:3 EtOAc/Hexanes) to afford 177 mg (80%) of 48. 1H NMR (400 MHz, CDCl3): δ = 0.98 (t, J = 7.3 Hz, 3H), 1.72–1.60 (m, 2H), 2.57 (t, J = 7.0 Hz, 2H), 3.82 (s, 3H), 5.43 (s, 1H), 6.40 (d, J = 16.0 Hz, 1H), 6.70 (d, J = 8.1 Hz, 1H), 6.84 (d, J = 8.1 Hz, 1H), 6.91 (s, 1H), 7.05 (d, J = 8.3 Hz, 1H), 7.16 (s, 1H), 7.36 (t, J = 7.8 Hz, 1H), 7.64 ppm (d, J = 16.0 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ = 13.7, 24.4, 37.5, 51.7, 116.3, 116.4, 118.7, 119.2, 120.7, 123.0, 130.2, 136.2, 140.3, 140.4, 144.0, 147.2, 157.7, 167.1 ppm; HRMS (ESI-positive): calcd for C19H20O4 ([M−H]+): 311.1288, found: 311.1299.
3-[3-(2-Hydroxy-4-propylphenoxy)phenyl]propionic acid methyl ester (49)
A mixture of 48 (0.10 g, 0.32 mmol) and 10% Pd/C (0.02 g) was subjected to the general hydrogenation to afford 49 (88 mg, 87%). 1H NMR (400 MHz, CDCl3): δ = 0.97 (t, J = 7.3 Hz, 3H), 1.69–1.62 (m, 2H), 1.64 (t, J = 7.8 Hz, 1H), 2.56 (t, J = 7.4 Hz, 2H), 2.63 (t, J = 7.9 Hz, 2H), 2.94 (t, J = 7.6 Hz, 2H), 5.47 (s, 1H), 3.68 (s, 3H), 6.68 (dd, J = 8, J = 2 Hz, 1H), 6.82 (d, J = 8.1 Hz, 1H), 6.87–6.85 (m, 2H), 6.89 (d, J = 1.5 Hz, 1H), 6.95 (d, J = 7.4 Hz, 1H), 7.25 ppm (t, J = 7.8 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ = 13.7, 24.4, 30.7, 35.4, 37.5, 51.6, 115.3, 116.0, 117.3, 118.9, 120.5, 123.1, 129.8, 139.8, 140.9, 142.6, 147.2, 157.3, 173.1 ppm; HRMS (ESI-positive): calcd for C19H22O4 ([M−H]+): 313.1445, found: 313.1451.
3-[3-(2-Hydroxy-4-propylphenoxy)phenyl]propionic acid (50)
A mixture of 48 (0.05 g, 0.16 mmol), and LiOH·H2O (0.07 g, 1.62 mmol) was subjected to the hydrolysis reaction to afford acid 50 (29 mg, 60%). 1H NMR (400 MHz, CDCl3): δ = 0.98 (t, J = 7.3 Hz, 6H), 1.70–1.60 (m, 4H), 2.58 (t, J = 7.4 Hz, 4H), 6.41 (d, J = 16.0 Hz, 1H), 6.71 (d, J = 8.0 Hz, 1H), 6.85 (d, J = 8.0 Hz, 1H), 6.92 (s, 1H), 7.07 (d, J = 8.0 Hz, 1H), 7.18 (s, 1H), 7.29 (d, J = 8.0 Hz, 1H), 7.38 (t, J = 7.6 Hz, 1H), 7.62 ppm (d, J = 16.0 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ = 13.8, 24.4, 37.5, 116.4, 116.6, 118.1, 119.2, 119.6, 120.8, 123.2, 130.3, 135.8, 140.3, 140.4, 146.2, 147.2, 157.8, 171.7 ppm; HRMS (ESI-positive): calcd for C18H18O4 ([M−H]+): 297.1132, found: 297.1137.
3-[3-(2-Hydroxy-4-propylphenoxy)phenyl]propionic acid (51)
A mixture of 49 (0.05 g, 0.16 mmol), and LiOH·H2O (0.07 g, 1.62 mmol) was subjected to the hydrolysis reaction to afford acid 51 (37 mg, 75%). 1H NMR (300 MHz, CDCl3): δ = 0.97 (t, J = 7.3 Hz, 3H), 1.72–1.60 (m, 2H), 2.56 (t, J = 7.4 Hz, 2H), 2.68 (t, J = 7.8 Hz, 2H), 2.95 (t, J = 7.5 Hz, 2H), 6.67 (dd, J = 8, J = 2 Hz, 1H), 6.82 (d, J = 8.2 Hz, 1H), 6.92–6.85 (m, 3H), 6.93 (d, J = 7.4 Hz, 1H), 7.24 ppm (d, J = 7.8 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ = 13.7, 24.4, 30.4, 35.1, 37.5, 115.4, 116.1, 117.4, 119.0, 120.5, 123.1, 129.8, 139.9, 140.8, 142.2, 147.2, 157.3, 177.7 ppm; HRMS (ESI-positive): calcd for C18H20O4 ([M−H]+): 299.1288, found: 299.1298.
3-[5-Chloro-2-(2,4-dichlorophenoxy)phenoxy]pyridine (52)
Method C was used to prepare 52. A suspension of triclosan (0.10 g, 0.34 mmol), 3-pyridineboronic acid (0.13 g, 1.03 mmol), Cu(OAc)2 (0.31 g, 1.72 mmol), Et3N (0.35 g, 3.45 mmol) and 5 Å molecular sieves (0.6 g) in dichloromethane (20 mL) was stirred for 16 h at rt. Purification by preparative TLC using 1:3 EtOAc/Hexanes gave 45 mg (35%) of the title compound 52. 1H NMR (400 MHz, CDCl3): δ = 6.83 (d, J = 8.7 Hz, 1H), 6.94 (d, J = 8.6 Hz, 1H), 7.20–7.13 (m, 3H), 7.27–7.26 (m, 2H), 7.42 (d, J = 2.4 Hz, 1H), 8.33 (s, 1H), 8.39 ppm (d, J = 2.4 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ = 119.5, 121.1, 122.0, 124.0, 124.4, 125.5, 125.6, 127.9, 129.3, 129.9, 130.5, 140.2, 144.7, 145.8, 146.5, 150.8, 153.2 ppm; HRMS (ESI-positive): calcd for C17H11O2N1Cl3 ([M + H]+): 365.9849, found: 365.9843.
X-ray Crystallography
Details on the crystal structure of the triclosan–bound complex have been reported elsewhere.[46] The atomic coordinates and structure factors have been deposited in the Protein Data Bank (http://www.rcsb.org/pdb) under the PDB code 2QIO. Details on the crystal structure of BaENRI bound to inhibitors 11 and 43 will be reported elsewhere. A summary of crystallographic data collection and refinement information for these inhibitors is provided in the supporting information.
Computational Methods
All of the compounds synthesized in Tables 2–6 were docked using the GOLD docking program[58] into our BaENR crystal structure that was co–crystallized with triclosan. Within GOLD, the triclosan crystal structure was used as a reference ligand, and default parameters were chosen. The scoring function within GOLD chosen was ChemScore, after testing out numerous algorithms, and finding that ChemScore performed the best. A CoMFA was performed using up to 10 docking conformations of each of our ligands. Initial alignment of the compounds began by choosing the best scoring conformation. For each compound, multiple alternative conformations were systematically replaced to determine whether a better alignment could be achieved over the initial alignment, by way of a cross–validated PLS analysis. After all the conformations for one compound are evaluated, the conformation associated with the highest q2 was kept in the alignment. Using a training set of 22 compounds, an acceptable model was found, shown by the PLS results of q2 = 0.831, an r2 = 0.929, and an SEE = 0.36. The steric and electrostatic fields were graphed and were used in the design of new compounds.
Purification of BaENR and Activity Assays
Enoyl reducase (ENR) from Bacillus anthracis was expressed and purified from E. coli according to the procedures of Klein et al.[46] The ENR reaction converts one molecule of NADH and crotonyl–CoA into NAD+ and butyryl–CoA. The assay was conducted at 30 °C in a 96–well plate with 200 μL of buffer containing 20 mM Tris–HCl, 150 mM NaCl, 175 μM NADH, 200 μM crotonyl–CoA and 1 μM of purified BaENR. Crotonyl–CoA was added last to initiate the reaction. BaENR activity was recorded using a Molecular Devices SpectraMax384 Plus plate reader by measuring the decrease in NADH absorbance at 340 nm (ε = 6220 M−1 cm−1). The path length of the well in the 96–well microtiter plate cell was determined by measuring the rate (ΔAU/min) in a 1 mL reaction volume with a 1.00 cm cuvette, and then calculating the rate of reaction (NADH/min) using the Beer–Lambert equation (AU = ε·L·[NADH]) where L = path length. The reaction was then split into 200 μL fractions, added to 5 wells of the 96–well plate, and the reaction rates were measured (ΔAU/min). The ΔAU/min values, in units of [NADH]/min, and the value of ε (6220 M−1 cm−1) allowed us to determine a path length for the reactions of 0.45 cm.
Inhibition of BaENR by triclosan and other compounds
For the synthetic compounds, the maximum solubility of these compounds were determined first by adding 2 μL of a 10 mM stock solution of the compound that was dissolved in DMSO to the reaction well. The final volume was 200 μL, and if precipitation was visible in the reaction well, then sequential two–fold dilutions of the stock were created in DMSO and tested in the same manner until precipitation was no longer visible. Creating the stock solutions in this manner ensured a constant 1% (v/v) of DMSO to total solution. The percent inhibition (%I) was tested at the maximum solubility concentration using the equation %I = ((AC − AI)/AC) * 100 where AC = activity of the control (uninhibited) and AI = activity with the inhibitor. If a percent inhibition value greater than 50% was attainable then the IC50 values were determined. IC50 values were determined by fixing the non–varied substrates at subsaturating concentrations as described above, and then varying the inhibitor concentrations. The reactions were allowed to preincubate for 30 minutes before adding crotonyl–CoA. An equal concentration of DMSO was added to the control experiments to negate the effect of solvent on enzyme activity. The data were fit to the equation %I = %Imax/(IC50/[I]+1) using non–linear regression and the Enzyme Kinetics Module 1.0 of the program Sigma Plot (SYSTAT Inc).
Bacterial Growth and MIC Determination
The minimum inhibitory concentration (MIC) of triclosan and other compounds was tested against the B. anthracis ΔANR strain (plasmid–cured Ames Strain), Sterne strain B. anthracis and reference strains of the following bacteria: Staphylococcus aureus (ATCC strain 29213), methicillin–resistant Staphylococcus aureus (ATCC strain 43300), Enterococcus fecalis (ATCC strain 29212), vancomycin–resistant Enterococcus (VRE) (ATCC strain 51299), Listeria monocytogenes (10403S, provided by Nancy E. Freitag, University of Illinois, Chicago),[59] Pseudomonas aeruginosa (ATCC strain 27853), Klebsiella pneumoniae (ATCC strain 700603) and Escherchia coli (ATCC strain 25922). Luria–Bertani (LB) medium was added to each well in a row on a sterile, Falcon MICROTEST™ 96–well U–bottom tissue culture plate; 96 μL was added to each well of the first column, and 50 μL was added to all subsequent wells. The compounds or control antibiotic to be tested were added to the first column for a final well volume of 100 μL. The inhibitors were then serially diluted (2–fold) across the columns of wells by pipetting and mixing 50 μL of solution; the extra 50 μL at the end of the row was discarded. Prior to setting up the MIC plates, the appropriate bacterial culture was grown to mid log–phase (OD600 = 0.4–0.6) and subsequently diluted to OD600 = 0.004 with fresh LB medium. 50 μL of the bacterial culture was added to each well of the plate and the plate was then incubated at 37 °C overnight (~14 h) without shaking. For each compound or antibiotic control, the first clear well with no signs of visible growth was reported as the MIC value. Medium from all clear wells was then inoculated onto plates of LB agar medium that were incubated overnight. The first clear–medium dilution from which no bacterial colonies could be grown was reported as the MBC value.
Acknowledgments
This work was funded by a grant from NIH/NIAID (U19 AI056575) and a contract from DOD (W81XWH-07-1-0445). X-ray diffraction intensities were collected at Southeast Regional Collaborative Access Team (SER–CAT) 22–ID and 22–BM beamlines at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at www.ser-cat.org/members.html. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38. Work in the Cook Laboratory was supported in part by the James A. and Marion C. Grant Fund.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemmedchem.org or from the author.
References
- 1.Infectious Disease Society of America. Bad Bugs, No Drugs. http://www.idsociety.org/WorkArea/showcontent.aspx?id=5554.
- 2.Casey AL, Lambert PA, Elliott TS. Int J Antimicrob Agents. 2007;29:S23–S32. doi: 10.1016/S0924-8579(07)72175-1. [DOI] [PubMed] [Google Scholar]
- 3.Bancroft EA. J Am Med Assoc. 2007;298:1803–1804. [Google Scholar]
- 4.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK. J Am Med Assoc. 2007;298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 5.Weisblum B. Antimicrob Agents Chemother. 1995;39:577–585. doi: 10.1128/AAC.39.3.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bartlett JG, Inglesby TV, Jr, Borio L. Clin Infect Dis. 2002;35:851–858. doi: 10.1086/341902. [DOI] [PubMed] [Google Scholar]
- 7.Cavallo JD, Ramisse F, Girardet M, Vaissaire J, Mock M, Hernandez E. Antimicrob Agents Chemother. 2002;46:2307–2309. doi: 10.1128/AAC.46.7.2307-2309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coker PR, Smith KL, Hugh–Jones ME. Antimicrob Agents Chemother. 2002;46:3843–3845. doi: 10.1128/AAC.46.12.3843-3845.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vester B, Douthwaite S. Antimicrob Agents Chemother. 2001;45:1–12. doi: 10.1128/AAC.45.1.1-12.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stepanov AV, Marinin LI, Pomerantsev AP, Staritsin NA. J Biotechnol. 1996;44:155–160. doi: 10.1016/0168-1656(95)00092-5. [DOI] [PubMed] [Google Scholar]
- 11.Price LB, Vogler A, Pearson T, Busch JD, Schupp JM, Keim P. Antimicrob Agents Chemother. 2003;47:2362–2365. doi: 10.1128/AAC.47.7.2362-2365.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Athamna A, Athamna M, bu-Rashed N, Medlej B, Bast DJ, Rubinstein E. J Antimicrob Chemother. 2004;54:424–428. doi: 10.1093/jac/dkh258. [DOI] [PubMed] [Google Scholar]
- 13.MacIntyre CR, Seccull A, Lane JM, Plant A. Mil Med. 2006;171:589–594. doi: 10.7205/milmed.171.7.589. [DOI] [PubMed] [Google Scholar]
- 14.Rotz LD, Khan AS, Lillibridge SR, Ostroff SM, Hughes JM. Emerg Infect Dis. 2002;8:225–230. doi: 10.3201/eid0802.010164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burnett JC, Henchal EA, Schmaljohn AL, Bavari S. Nat Rev Drug Discov. 2005;4:281–297. doi: 10.1038/nrd1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.White SW, Zheng J, Zhang YM, Rock CO. Annu Rev Biochem. 2005;74:791–831. doi: 10.1146/annurev.biochem.74.082803.133524. [DOI] [PubMed] [Google Scholar]
- 17.Zhang YM, Lu YJ, Rock CO. Lipids. 2004;39:1055–1060. doi: 10.1007/s11745-004-1330-3. [DOI] [PubMed] [Google Scholar]
- 18.Rock CO, Jackowski S. Biochem Biophys Res Commun. 2002;292:1155–1166. doi: 10.1006/bbrc.2001.2022. [DOI] [PubMed] [Google Scholar]
- 19.Kobayashi K, Ehrlich SD, Albertini A, Amati G, Andersen KK, Arnaud M, Asai K, Ashikaga S, Aymerich S, Bessieres P, Boland F, Brignell SC, Bron S, Bunai K, Chapuis J, Christiansen LC, Danchin A, Dâebarbouille M, Dervyn E, Deuerling E, Devine K, Devine SK, Dreesen O, Errington J, Fillinger S, Foster SJ, Fujita Y, Galizzi A, Gardan R, Eschevins C, Fukushima T, Haga K, Harwood CR, Hecker M, Hosoya D, Hullo MF, Kakeshita H, Karamata D, Kasahara Y, Kawamura F, Koga K, Koski P, Kuwana R, Imamura D, Ishimaru M, Ishikawa S, Ishio I, Le Coq D, Masson A, Mauèel C, Meima R, Mellado RP, Moir A, Moriya S, Nagakawa E, Nanamiya H, Nakai S, Nygaard P, Ogura M, Ohanan T, O’Reilly M, O’Rourke M, Pragai Z, Pooley HM, Rapoport G, Rawlins JP, Rivas LA, Rivolta C, Sadaie A, Sadaie Y, Sarvas M, Sato T, Saxild HH, Scanlan E, Schumann W, Seegers JF, Sekiguchi J, Sekowska A, Sâeror SJ, Simon M, Stragier P, Studer R, Takamatsu H, Tanaka T, Takeuchi M, Thomaides HB, Vagner V, van Dijl JM, Watabe K, Wipat A, Yamamoto H, Yamamoto M, Yamamoto Y, Yamane K, Yata K, Yoshida K, Yoshikawa H, Zuber U, Ogasawara N. Proc Natl Acad Sci U S A. 2003;100:4678–4683. doi: 10.1073/pnas.0730515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heath RJ, Rock CO. J Biol Chem. 1995;270:26538–26542. doi: 10.1074/jbc.270.44.26538. [DOI] [PubMed] [Google Scholar]
- 21.Rozwarski DA, Grant GA, Barton DH, Jacobs WR, Jr, Sacchettini JC. Science. 1998;279:98–102. doi: 10.1126/science.279.5347.98. [DOI] [PubMed] [Google Scholar]
- 22.Baldock C, de Boer GJ, Rafferty JB, Stuitje AR, Rice DW. Biochem Pharm. 1998;55:1541–1549. doi: 10.1016/s0006-2952(97)00684-9. [DOI] [PubMed] [Google Scholar]
- 23.Baldock C, Rafferty JB, Sedelnikova SE, Baker PJ, Stuitje AR, Slabas AR, Hawkes TR, Rice DW. Science. 1996;274:2107–2110. doi: 10.1126/science.274.5295.2107. [DOI] [PubMed] [Google Scholar]
- 24.Levy CW, Baldock C, Wallace AJ, Sedelnikova S, Viner RC, Clough JM, Stuitje AR, Slabas AR, Rice DW, Rafferty JB. J Mol Biol. 2001;309:171–180. doi: 10.1006/jmbi.2001.4643. [DOI] [PubMed] [Google Scholar]
- 25.McMurry LM, McDermott PF, Levy SB. Antimicrob Agents Chemother. 1999;43:711–713. doi: 10.1128/aac.43.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heath RJ, Yu YT, Shapiro MA, Olson E, Rock CO. J Biol Chem. 1998;273:30316–30320. doi: 10.1074/jbc.273.46.30316. [DOI] [PubMed] [Google Scholar]
- 27.Levy CW, Roujeinikova A, Sedelnikova S, Baker PJ, Stuitje AR, Slabas AR, Rice DW, Rafferty JB. Nature. 1999;398:383–384. doi: 10.1038/18803. [DOI] [PubMed] [Google Scholar]
- 28.McLeod R, Muench SP, Rafferty JB, Kyle DE, Mui EJ, Kirisits MJ, Mack DG, Roberts CW, Samuel BU, Lyons RE, Dorris M, Milhous WK, Rice DW. Int J Parasitol. 2001;31:109–113. doi: 10.1016/s0020-7519(01)00111-4. [DOI] [PubMed] [Google Scholar]
- 29.Moir DT. Curr Drug Targets Infect Disord. 2005;5:297–305. doi: 10.2174/1568005054880154. [DOI] [PubMed] [Google Scholar]
- 30.He X, Alian A, Stroud R, Ortiz de Montellano PR. J Med Chem. 2006;49:6308–6323. doi: 10.1021/jm060715y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Broussy S, Bernardes-Gâenisson V, Quâemard A, Meunier B, Bernadou J. J Org Chem. 2005;70:10502–10510. doi: 10.1021/jo051901z. [DOI] [PubMed] [Google Scholar]
- 32.Miller WH, Seefeld MA, Newlander KA, Uzinskas IN, Burgess WJ, Heerding DA, Yuan CC, Head MS, Payne DJ, Rittenhouse SF, Moore TD, Pearson SC, Berry V, DeWolf WE, Jr, Keller PM, Polizzi BJ, Qiu X, Janson CA, Huffman WF. J Med Chem. 2002;45:3246–3256. doi: 10.1021/jm020050+. [DOI] [PubMed] [Google Scholar]
- 33.Seefeld MA, Miller WH, Newlander KA, Burgess WJ, DeWolf WE, Jr, Elkins PA, Head MS, Jakas DR, Janson CA, Keller PM, Manley PJ, Moore TD, Payne DJ, Pearson S, Polizzi BJ, Qiu X, Rittenhouse SF, Uzinskas IN, Wallis NG, Huffman WF. J Med Chem. 2003;46:1627–1635. doi: 10.1021/jm0204035. [DOI] [PubMed] [Google Scholar]
- 34.Protasevich II, Brouillette CG, Snow ME, Dunham S, Rubin JR, Gogliotti R, Siegel K. Biochemistry. 2004;43:13380–13389. doi: 10.1021/bi0492509. [DOI] [PubMed] [Google Scholar]
- 35.Seefeld MA, Miller WH, Newlander KA, Burgess WJ, Payne DJ, Rittenhouse SF, Moore TD, DeWolf WE, Jr, Keller PM, Qiu X, Janson CA, Vaidya K, Fosberry AP, Smyth MG, Jaworski DD, Slater-Radosti C, Huffman WF. Bioorg Med Chem Lett. 2001;11:2241–2244. doi: 10.1016/s0960-894x(01)00405-x. [DOI] [PubMed] [Google Scholar]
- 36.a) Karlowsky JA, Laing NM, Baudry T, Kaplan N, Vaughan D, Hoban DJ, Zhanel GG. Antimicrob Agents Chemother. 2007;51:1580–1581. doi: 10.1128/AAC.01254-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Park HS, Yoon YM, Jung SJ, Yun INR, Kim CM, Kim JM, Kwak J-H. Int J Antimicrob Ag. 2007;30:446–451. doi: 10.1016/j.ijantimicag.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 37.Sivaraman, Sullivan TJ, Johnson F, Novichenok P, Cui G, Simmerling C, Tonge PJ. J Med Chem. 2004;47:509–518. doi: 10.1021/jm030182i. [DOI] [PubMed] [Google Scholar]
- 38.Escalada MG, Harwood JL, Maillard JY, Ochs D. J Antimicrob Chemother. 2005;55:879–882. doi: 10.1093/jac/dki123. [DOI] [PubMed] [Google Scholar]
- 39.McMurry LM, Oethinger M, Levy SB. Nature. 1998;394:531–532. doi: 10.1038/28970. [DOI] [PubMed] [Google Scholar]
- 40.Regèos J, Zak O, Solf R, Vischer WA, Weirich EG. Dermatol. 1979;158:72–79. doi: 10.1159/000250746. [DOI] [PubMed] [Google Scholar]
- 41.a) Freundlich JS, Anderson JW, Sarantakis D, Shieh HM, Yu M, Valderramos JC, Lucumi E, Kuo M, Jacobs WR, Jr, Fidock DA, Schiehser GA, Jacobus DP, Sacchettini JC. Bioorg Med Chem Lett. 2005;15:5247–5252. doi: 10.1016/j.bmcl.2005.08.044. [DOI] [PubMed] [Google Scholar]; b) Freundlich JS, Wang F, Han-Chun Tsai, Kuo M, Hong-Ming Shieh, Anderson JW, Nkrumah LJ, Valderramos Juan-Carlos, Yu M, Kumar TRS, Valderramos SG, Jacobs WR, Jr, Schiehser GA, Jacobus DP, David P, Fidock DA, Sacchettini JC. J Biol Chem. 2007;282:25436–25444. doi: 10.1074/jbc.M701813200. [DOI] [PubMed] [Google Scholar]
- 42.Freundlich JS, Yu M, Lucumi E, Kuo M, Tsai HC, Valderramos JC, Karagyozov L, Jacobs WR, Jr, Schiehser GA, Fidock DA, Jacobus DP, Sacchettini JC. Bioorg Med Chem Lett. 2006;16:2163–2169. doi: 10.1016/j.bmcl.2006.01.051. [DOI] [PubMed] [Google Scholar]
- 43.a) Lu H, Tonge PJ. Acc Chem Res. 2008;41:11–20. doi: 10.1021/ar700156e. [DOI] [PubMed] [Google Scholar]; b) Sullivan TJ, Truglio JJ, Boyne ME, Novichenok P, Zhang X, Stratton CF, Li HJ, Kaur T, Amin A, Johnson F, Slayden RA, Kisker C, Tonge PJ. ACS Chem Biol. 2006;1:43–53. doi: 10.1021/cb0500042. [DOI] [PubMed] [Google Scholar]
- 44.Perozzo R, Kuo M, Sidhu AS, Valiyaveettil JT, Bittman R, Jacobs WR, Jr, Fidock DA, Sacchettini JC. J Biol Chem. 2002;277:13106–13114. doi: 10.1074/jbc.M112000200. [DOI] [PubMed] [Google Scholar]
- 45.Chhibber M, Kumar G, Parasuraman P, Ramya TN, Surolia N, Surolia A. Bioorg Med Chem. 2006;14:8086–8098. doi: 10.1016/j.bmc.2006.07.034. [DOI] [PubMed] [Google Scholar]
- 46.Klein GM, Bernard DS, Tipparaju SK, Pegan S, Bishop MH, Kozikowski AP, Mesecar AD. Biochemistry. 2007 Accepted with minor revisions. [Google Scholar]
- 47.Evans DA, Katz JL, West TR. Tetrahedron Lett. 1998;39:2937–2940. [Google Scholar]
- 48.Papamicael C, Queguiner G, Bourguignon J, Dupas G. Tetrahedron. 2001;57:5385–5391. [Google Scholar]
- 49.Su-Dong Cho, Yong-Dae Park, Jeum-Jong Kim, Falck JR, Yoon Y-J. Bull Korean Chem Soc. 2004;25:407–409. [Google Scholar]
- 50.Molecular properties were calculated using a free web-based program (http://146.107.217.178/lab/alogps/index.html). A table showing comparision of values of synthesized compounds with those of triclosan is included in supplementary material.
- 51.Boyne ME, Sullivan TJ, amEnde CW, Lu H, Gruppo V, Heaslip D, Amin AG, Chatterjee D, Lenaerts A, Tonge PJ, Slayden RA. Antimicrob Agents Chemother. 2007;51:3562–3567. doi: 10.1128/AAC.00383-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Auffinger P, Hays FA, Westhof E, Ho PS. Proc Natl Acad Sci U S A. 2004;101:16789–16794. doi: 10.1073/pnas.0407607101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pidugu LS, Kapoor M, Surolia N, Surolia A, Suguna K. J Mol Biol. 2004;343:147–155. doi: 10.1016/j.jmb.2004.08.033. [DOI] [PubMed] [Google Scholar]
- 54.Tipparaju SK, Joyasawal S, Forrester S, Mulhearn DC, Pegan S, Johnson ME, Mesecar AD, Kozikowski AP. Bioorg Med Chem Lett. 2008 doi: 10.1016/j.bmcl.2008.05.004. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kapoor M, Mukhi PL, Surolia N, Suguna K, Surolia A. Biochem J. 2004;381:725–733. doi: 10.1042/BJ20040228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lambert RJ. J Appl Microbiol. 2004;97:699–6711. doi: 10.1111/j.1365-2672.2004.02345.x. [DOI] [PubMed] [Google Scholar]
- 57.Suller MT, Russell AD. J Antimicrob Chemother. 2000;46:11–8. doi: 10.1093/jac/46.1.11. [DOI] [PubMed] [Google Scholar]
- 58.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Proteins. 2003;52:609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 59.Mueller KJ, Freitag NE. Infect Immun. 2005;73:1917–1926. doi: 10.1128/IAI.73.4.1917-1926.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]