

Abstract
Recessive mutations in Pink1 lead to a selective degeneration of dopaminergic neurons in the substantia nigra that is characteristic of Parkinson disease. Pink1 is a kinase that is targeted in part to mitochondria, and loss of Pink1 function can alter mitochondrial morphology and dynamics, thus supporting a link between mitochondrial dysfunction and Parkinson disease etiology. Here, we report the unbiased identification and confirmation of a mitochondrial multi-protein complex that contains Pink1, the atypical GTPase Miro, and the adaptor protein Milton. Our screen also identified an interaction between Pink1 and Mitofilin. Based on previously established functions for Miro and Milton in the trafficking of mitochondria along microtubules, we postulate here a role for Pink1 in mitochondrial trafficking. Using subcellular fractionation, we show that the overexpression of Miro and Milton, both of which are known to reside at the outer mitochondrial membrane, increases the mitochondrial Pink1 pool, suggesting a function of Pink1 at the outer membrane. Further, we document that Pink1 expressed without a mitochondrial targeting sequence can still be targeted to a mitochondria-enriched subcellular fraction via Miro and Milton. The latter finding is important for the interpretation of a previously reported protective effect of Pink1 expressed without a mitochondrial targeting sequence. Finally, we find that Miro and Milton expression suppresses altered mitochondrial morphology induced by loss of Pink1 function in cell culture. Our findings suggest that Pink1 functions in the trafficking of mitochondria in cells.
Parkinson disease (PD) is the second most common neurodegenerative disorder and results largely from the progressive loss of dopaminergic neurons in the pars compacta of the substantia nigra. Mitochondrial dysfunction, oxidative damage, and abnormal protein accumulation likely play key roles in the etiology of sporadic PD and also rare familial forms of PD (1). Originally, mitochondrial dysfunction was implicated in PD because Complex I inhibitors induce PD-like phenotypes in animals and humans. Later, demonstration of the involvement in normal mitochondrial function of the three autosomal recessive PD (ARPD) gene products, Parkin, DJ-1 and Pink1, strengthened the link between PD and mitochondrial biology (2). But exactly how these three proteins influence mitochondrial function is unclear.
Pink1 contains an N-terminal mitochondrial targeting sequence (MTS) and a large serine/threonine kinase domain (3). Reduced Pink1 kinase function can presumably precipitate PD in humans (4). The mitochondrial localization of Pink1 (5, 6), alterations in mitochondrial morphology, dynamics and function caused by Pink1 deficiency (7-11), and the discovery of a mitochondrial Pink1 substrate, TRAP1 (12), all point to a central role of Pink1 in normal mitochondrial function. However, recent data suggest that Pink1 also acts outside of the mitochondria. In this regard, we and others have shown that both the full-length precursor (Pink1 66 kDa) and the mature, processed isoform (Pink1 55 kDa) not only exist in mitochondria but are also detectable in cytosol and microsomes (5, 6, 13). In addition, a recent study showed that the kinase domain of mitochondrial Pink1 faces the cytosol (14). Moreover, an artificial cytosolic form of Pink1, i.e., expressed without its MTS (ΔMTS-Pink1), protects against the PD-causing Complex I inhibitor, MPTP, in vivo and in vitro (13). Thus, it is important to elucidate whether and how a cytosolic form of Pink1 can affect mitochondria.
In this study, we sought to identify in an unbiased fashion mitochondrial proteins that interact with Pink1. Through a mass spectrometry screen, we have identified a novel multi-protein complex that points strongly to an apparent function of Pink1 in mitochondrial trafficking. Further, we report experiments that establish the relevance of this new protein complex for the subcellular distribution of Pink1, for the recently described protective cytosolic form, ΔMTS-Pink1, and for alterations in mitochondrial morphology upon Pink1 silencing.
EXPERIMENTAL PROCEDURES
Plasmids
Pink1, Pink1-FLAG and Pink1-FLAG KD expression vectors have been described before (6). To produce an ΔMTS-Pink1 expression vector, the sequence corresponding to Pink1 amino acids 112−581 was PCR amplified and cloned into pcDNA3.1+ (Invitrogen). Primers were designed with a BamH1 site and start codon at the 5’ end and an EcoRI site after the stop codon at the 3’ end. ΔMTS-Pink1-FLAG was cloned by adding a FLAG epitope tag into the 3’ primer before a stop codon. pRK5Myc Miro-2 and Miro-1 constructs were generous gifts of P. Aspenström (15), and Xpress-tagged Milton-1 (OIP106) (16) has been described before. GST-mouse Pink1 was a generous gift of Huaibin Cai.
Antibodies
Polyclonal rabbit anti-Pink1 antibody (BC100−494) was purchased from Novus Biologicals (Littleton, CO, USA) and used at dilutions of 1:1,000 for infrared and 1:10,000 for chemiluminescent immunoblotting. The following antibodies were used at dilutions recommended by their manufacturers: Monoclonal mouse M2 anti-FLAG and polyclonal rabbit (A14) anti-Myc from Sigma Aldrich (St Louis, MO, USA), monoclonal mouse anti-Actin (8226) from Abcam (Cambridge, MA, USA), polyclonal anti-VDAC from Affinity Bioreagents (Golden, CO, USA), polyclonal rabbit anti-calnexin from Stressgene (San Diego, CA, USA), mouse monoclonal anti-Myc (9E10) from Santa Cruz Biotechnology (Santa Cruz, CA, USA), polyclonal rabbit anti-Miro-2 (RHOT2) and anti-Mitofilin (IMMT) from Proteintech Group Inc (Chicago, IL, USA), and anti-Xpress from Invitrogen, (Carlsbad, CA, USA).
Cell culture and Pink1 shRNA silencing
COS-7 and HEK-293FT cells were cultured in DMEM containing 10% fetal bovine serum, penicillin (100 U/ml), Streptomycin (100 μg/ml) and L-Glutamine. Transient transfections were performed using Fugene6 (Roche) according to the manufacturer's protocol. In order to knockdown endogenous human PINK1, we made two separate shRNA constructs (target sequences 5’-GCTGGAGGAGTATCTGATAGG, starting at nucleotide 550 of human PINK1 and 5’-GGGAGCCATCGCCTATGAAAT, starting at nucleotide 1411) and a control shRNA (5’-CCTAGACGCGATAGTATGGAC). These sequences were cloned into pLenti6, packaged and used to transduce M17 cells (5). Knockdown was confirmed using qRT-PCR methods described previously (5). Transduced M17 cells were maintained in phenol-red free Opti-MEM (Invitrogen) with 10% FBS and 5ug/ml Blasticidin with no additional supplements.
Cell fractionation and its quantitative analysis, immunoprecipitation and immunoblotting
Cell fractionations, immunoprecipitations and immunoblotting were done as described in detail previously (6), with the following minor modifications. For the double co-ip approach, transfected cells were lysed in 1% TX-100, PBS, 1mM EDTA, plus protease and phosphatase inhibitors, and anti-FLAG immunoprecipitates were eluted by incubation for 1 hr with 0.3 μ/ml FLAG peptide in 1% TX-100, PBS and 1mM EDTA at room temperature. Quantitative analysis of subcellular fractionation was done as described previously (6). In short, protein levels of Pink1 66 kDa, Pink1 55 kDa or ΔMTS-Pink1 were determined in S100k (cytosolic), P10k (mitochondrial) and P100k (microsomal) fractions by quantitative infrared immunoblotting (Licor). The sum of the three fractions represents the total protein. The relative distribution of the protein of interest among the three fractions was then calculated as a percentage of total protein. Statistical analysis was carried out using Prism 4.0 (GraphPad)
Immunoisolation and MS/MS analysis of mitochondrial Pink1 complexes
Production of mitochondrial fractions and MS/MS analysis of Pink1 complexes were performed as described before, with minor modifications (6). Briefly, crude mitochondrial fractions were isolated by homogenizing HEK293-FT cells transduced with lentivirus expressing Pink1-FLAG IRES GFP, followed by differential centrifugation and washing. Subsequently, Pink1-FLAG complexes were immunoisolated from solubilized mitochondria using anti-FLAG Agarose beads (Sigma). After intensive washing, specific Pink1-FLAG complexes were eluted from the beads using a FLAG peptide and analyzed using MS/MS sequencing. As a negative control, the same procedure was done simultaneously with an equal number of cells expressing only IRES GFP. Only proteins, which were covered by 6 or more peptides in the Pink1-FLAG sample but were completely absent from the mock control, were designated potential Pink1 interacting proteins.
Confocal Microscopy
Cells were seeded in LabTekII-Borosilicate chambered coverglass (Nunc) and were transiently transfected with 0.5 μg of mitochondrial matrix-localized YFP in serum-free, phenol red-free OptiMEM. Live cells were imaged using a 100× Plan-Apochromat 1.4/Oil DIC objective lens (Carl Zeiss) with an excitation wavelength set to 514nm.
RESULTS
Isolation and MS/MS analysis of Pink1 complexes derived from isolated mitochondria reveals new potential Pink1 binding partners
The mechanism by which loss of Pink1 function in flies and HeLa cells causes alterations in mitochondrial morphology is unknown (7, 8, 10). In order to obtain new information about Pink1 function in mitochondria per se, we immunoisolated Pink1 complexes from a mitochondria-enriched fraction of HEK293-FT cells transduced with lentivirus expressing human Pink1 with a C-terminal FLAG epitope. These complexes were then analyzed by MS/MS sequencing. As a negative control, a mock isolation was performed through an identical procedure on mitochondria derived from HEK293-FT cells transduced with mock lentivirus. The MS/MS data revealed, aside from Pink1, two other predicted mitochondrial proteins that were both covered by more than 5 tryptic peptides but completely absent in the mock eluate (Fig. 1a). These proteins are the atypical GTPase, Miro-2, that is implicated in mitochondrial homeostasis and trafficking (15, 17, 18), and Mitofilin, which controls crista morphology (19). To confirm that endogenous Miro-2 and Mitofilin interact with Pink1, we transiently expressed Pink1-FLAG or mock DNA into HEK-293FT cells and probed FLAG immunoprecipitates (ip's) from theses lysates for endogenous Miro-2 and Mitofilin. As shown in Fig. 1b, endogenous Miro-2 and Mitofilin were only found in the ip when Pink1-FLAG was co-expressed, confirming our MS/MS data. It is important to emphasize that there are no known antibodies which can detectably immunoprecipitate endogenous Pink1, as its cellular levels are very low in cell culture. In the following work, we focus on the newly revealed Pink1/Miro-2 interaction and its functional relevance.
Figure 1.

Identification of Miro-2 and Mitofilin as mitochondrial Pink1 binding proteins. (A) MS/MS sequencing results of Pink1-FLAG pull down from mitochondria. Relative peak area comparison between Pink1-FLAG (PF) and negative control (−) reveals Miro-2 and Mitofilin as specific Pink1 binding partners. (B) Confirmation of the novel Pink1 binding partners. Pink1-FLAG or mock vector was transiently expressed in HEK-293FT cells. Cells extracts were probed for Miro-2, Mitofilin and Pink1 (Input) and then subjected to immunoprecipitation (IP) with an anti-FLAG antibody. Western blotting for endogenous Miro-2 and Mitofilin confirmed the detection by MS/MS sequencing.
Pink1 forms a protein complex with Miro and Milton
The atypical GTPase, Miro, and the adapter protein, Milton, are known to be part of a protein complex that is attached at the mitochondrial outer membrane facing the cytosol. This complex links the heavy chain of conventional kinesin to mitochondria for anterograde axonal transport of mitochondria along microtubules in Drosophila (20, 21). Our newly identified Pink1/Miro interaction raised the question of whether Pink1 is part of the Miro-Milton complex and thus potentially involved in the regulation of mitochondrial trafficking. To address this question, we first sought to demonstrate the Pink1-Miro interaction in a heterologous expression system. We transiently co-transfected Pink1-FLAG and Myc-Miro-2 into COS7 cells and then probed FLAG ip's for Myc-Miro-2. Whereas no Myc-Miro-2 was detected in FLAG ip's in the absence of Pink1-FLAG expression (Fig. 2a, lane 1), Miro-2 was detected in FLAG ip's upon Pink1-FLAG co-expression (Fig. 2a, lane 2). A kinase dead (KD) mutant version of Pink1 had similar Miro-2 binding (Fig. 2a, lane 3). The reverse co-ip, i.e., Myc-Miro ip's probed for Pink1, revealed a specific interaction between Miro-2 and both Pink1 isoforms (66 and 55 kDa) (Fig. 2b). Pink1 also co-immunoprecipitated with Miro-1, which shares 54% identity and 68% similarity with Miro-2 (data not shown). As pointed out above, available Pink1 antibodies have been shown not to be sensitive enough to detect all endogenous protein interactions of Pink1 (14), and we carried out extensive attempts to detect endogenous Pink1 by ip, co-ip and/or Western blotting and confirmed this problem. To determine whether exogenous Pink1 is part of the Miro-2/Milton complex, we used a double co-ip protocol. COS7 cells co-expressing combinations of Xpress-Milton-1 with Pink1-FLAG and/or Myc-Miro-2 were subjected to an anti-FLAG (Pink1) ip followed by elution with a FLAG peptide (i.e., non-denaturing). The eluate was then subjected to a 2nd ip with an anti-Myc resin (for Miro-2). As shown in Figure 2c, Xpress-Milton was only present in the 2nd co-ip when Pink1-FLAG and Myc-Miro-2 were co-expressed, whereas the presence of Xpress-Milton-1 in the 1st co-ip depended only on Pink1-FLAG expression. Interestingly, Xpress-Milton-1 co-ip'd more efficiently with Pink1-FLAG when Myc-Miro-2 was co-expressed (Fig. 2c, compare lanes 2 and 3). Moreover, Miro-2 co-expression increased the Pink1 66 kDa/55 kDa ratio (6) by decreasing the Pink1 55 kDa level (Fig. 2b and c, top panels). Taken together, these findings demonstrate a protein complex that contains exogenous Pink1-FLAG, Myc-Miro-2 and Xpress-Milton-1.
Figure 2.

Pink1 forms a protein complex with Miro-2 and Milton-1. (A) Pink1-FLAG, Pink1 kinase-dead (KD)-FLAG, or mock vector were transiently co-transfected with Myc-Miro-2 into COS7 cells. Cell extracts were probed for transfected proteins (Input) and then subjected to IP with an anti-FLAG antibody (M2). Immunoprecipitates were then probed for Myc-Miro-2 and Pink1-FLAG. (B) Myc-Miro-2 or mock vector was co-transfected with Pink1 into COS7 cells. Cell extracts (Input) were analyzed for expression of transfected proteins using Western blotting and then subjected to IP with an anti-Myc antibody (A14). Immunoprecipitates were probed for Pink1-FLAG and Myc-Miro-2. (C) Sequential co-IPs reveal a Pink1/Miro-2/Milton-1 multi-protein complex. Xpress-Milton-1 was transiently co-expressed with Miro-2 and/or Pink1-FLAG in COS7 cells. Cell extracts were subjected to an anti-FLAG IP. Immunoprecipitates were then eluted using a FLAG peptide and subjected to an anti-Myc IP. Cell extracts (Input) and both the immunoprecipitates were probed for Pink1-FLAG, Myc-Miro-2 and Xpress-Milton-1, revealing a multi-protein comprised of Pink1, Miro-2 and Milton-1 (middle lane). Names of the antibodies applied are shown in parentheses; * indicates a cross-reactive band.
Overexpression of either Miro-2 or Milton-1 markedly increases the percentage of Pink1 in the mitochondrial pool
The Milton/Miro protein complex is known to localize to mitochondria despite the lack of an obvious mitochondrial targeting sequence on either protein (15, 21). Miro is anchored facing the cytosol in the outer mitochondrial membrane by its extreme C-terminal transmembrane domain and thereby couples the Miro/Milton complex to the mitochondrion (22). Having shown above that Pink1 is part of the Miro/Milton protein complex, we asked next whether increasing either Milton or Miro protein levels enhances the mitochondrial localization of Pink1. To this end, we used subcellular fractionation by differential centrifugation to ascertain the distribution of transiently expressed Pink1 among cytosolic (S100k), mitochondria-rich (P10k) and microsome-rich (P100k) fractions in COS-7 cells transiently expressing Miro-2 or Milton-1 (Figs. 3 a-c). The subcellular fractionation was verified by immunoblotting for marker proteins such as actin (cytosol), VDAC (mitochondria) and calnexin (endoplasmatic reticulum). Immunoblotting for Miro-2 (Fig. 3b) and Milton-1 (Fig. 3c) confirmed their predominantly mitochondrial (P10k) localization. Pink1 co-expression did not alter their subcellular distribution (data not shown). We then calculated the percentage distribution of the Pink1 66 kDa and 55 kDa isoforms by applying quantitative infrared immunoblotting, as described previously (6). First, we compared the distribution among the three subcellular fractions of Pink1 66 kDa in mock (Fig. 3a), Miro-2 (Fig. 3b) or Milton-1 (Fig. 3c) transfected cells (results quantified in Fig. 3d). The mitochondria-enriched (P10K) pool of Pink1 66 kDa dramatically increased from 29.1 ± 3.4% (mock) to 74.2 ± 2.1% of the total pool after Miro-2 co-expression or to 57.7 ± 4.0% after Milton-1 co-expression (means ± SEM, n=3). Conversely, Miro-2 and Milton-1 expression led to a significant decrease of Pink1 66 kDa in the cytosolic pool (11.7 ± 2% (mock) to 2.9 ± 0.9% with Miro-2 and to 5.1 ± 0.3% with Milton-1, means ± SEM, n=3) and also a decrease in the microsome-rich fraction (64.3 ± 4.3% (mock) to 23.1 ± 1% for Mito-2 and to 33.6 ± 2.1% for Milton-1, means ± SEM, n=3). Similarly, Pink1 55 kDa levels (quantified in Fig. 3e) in the mitochondria-rich fraction increased from 38.4 ± 2.8% (mock) to 82.8 ± 9% with Miro-2 and 81.7± 1.8% with Milton-1 (means ± SEM, n=3), whereas they decreased in the cytosolic pool (20.6 ± 2.2% (mock) to 6.5 ± 4.7% with Miro-2 and 6.9± 0.9% with Milton-1 co-expression; means ± SEM, n=3) and likewise in the microsome-rich fraction (38.99 ± 4.2% (mock) to 13.4 ± 7.1% with Miro-2 and 11.2 ± 1.2% with Milton-1 co-expression; means ± SEM, n=3). These findings reveal that Miro-2 or Milton-1 expression can robustly recruit Pink1 to the mitochondria-rich fraction.
Figure 3.

Overexpression of Miro-2 or Milton-1 increases the percentage of Pink1 in the mitochondrial pool. Subcellular fractionation using differential centrifugation of COS7 cells that transiently co-express (A) mock, (B) Myc-Miro-2 or (C) Xpress-Milton-1 vectors together with Pink1. Fractions were probed for Pink1, Myc-Miro-2, Xpress-Milton-1 and compartment marker proteins by Western blotting. (D) Quantitative analysis of relative subcellular distributions among cytosolic (S100k), mitochondria-rich (P10k) and microsome-rich (P100k) fractions of Pink1 66 kDa (D) and Pink1 55 kDa (E) after mock, Myc-Miro-2 or Xpress-Milton-1 expression (n=3, means ± SEM). Significant changes vs. mock are indicated (* p<0.05, ** p<0.01 and *** p<0.001; one-way ANOVA with Newman-Keuls Multiple Comparison Test.).
Pink1 lacking its MTS localizes to the outer mitochondrial membrane
Overexpression of Pink1 lacking its mitochondrial targeting sequence (ΔMTS-Pink1) has been reported to protect against MPTP toxicity in cultured cells and in vivo (13). However, reports regarding the subcellular localization of ΔMTS-Pink1 are conflicting (13, 23). Intuitively, it is plausible that lack of its MTS abolishes the mitochondrial localization of Pink1 and that this N-terminally truncated Pink1 form then resides in the cytosol, as suggested by data from Haque et al (13). On the other hand, a confocal microscopy study clearly showed that a ΔMTS-Pink1 was at least partially localized to mitochondria, suggesting that mitochondrial localization can occur independently of the MTS targeting mechanism (23). In light of our new findings (above), we hypothesized that this apparently persistent mitochondrial localization of ΔMTS-Pink1 could be due at least in part to the formation of a Miro/Milton/ΔMTS-Pink1 complex at the outer mitochondrial membrane. Here, we report experiments supporting this hypothesis.
First, we asked whether ΔMTS-Pink1 is still capable of binding to Miro-2. We generated Δ1−111 mutant Pink1 that migrates very close to the size of mature, processed Pink1 55 kDa on SDS-PAGE (Fig. 4a and data not shown) and is thus reasonable to use; it is the same length as a ΔMTS-Pink1 construct reported previously (13). We transiently co-transfected Myc-Miro-2 with Pink1-FLAG or else with ΔMTS-Pink1-FLAG into COS7 cells and probed FLAG immunoprecipitates for Myc-Miro-2. Both the wt and N-terminally truncated Pink1 specifically co-ip'd with Myc-Miro-2 (Fig. 4a), showing that ΔMTS-Pink1 can be part of the Miro/Milton protein complex. Next, we wanted to address the controversial subcellular localization of ΔMTS-Pink1, using our established subcellular fractionation protocol (6). Supporting the confocal microscopy data of Plun-Favreau et al. (23), we found that there was a readily detectable amount of ΔMTS-Pink1 in the mitochondria-rich (P10k) and microsome-rich (P100k) subcellular fractions (Fig. 4b, open bars). As expected, the cytosolic pool of ΔMTS-Pink1 is higher and the mitochondrial pool is lower than is the case with the wt Pink1 66 kDa and 55 kDa species (compare open bars in Fig. 4b and Figs. 3d, e). Based on our data and hypothesis stated above, we predicted that the percentage of mitochondrial localization of ΔMTS-Pink1 would increase upon Miro-2 or Milton-1 expression. Indeed, as shown in Figure 4b, we found that both Miro-2 and Milton-1 expression dramatically increased the relative levels of ΔMTS-Pink1 in the mitochondria-rich pool (19.3 ± 3.4% (mock) to 70.8 ± 1% for Miro-2 co-expression and to 42.2 ± 5.9% for Milton-1 co-expression), while they significantly decreased the cytosolic ΔMTS-Pink1 pool (39.3 ± 1.6% (mock) to 10.1 ± 2.4% for Miro-2 and to 28.5 ± 4.1% for Milton-1) and the microsome-rich ΔMTS-Pink1 pool (41.2 ± 4.7% (mock) to 18.9 ± 2.7% for Miro-2 and to 29.3 ± 2.6% for Milton-1). These findings suggest that Pink1 expressed without its MTS can nevertheless be partially localized to mitochondria, and this localization is markedly enhanced by expressing Miro or Milton.
Figure 4.
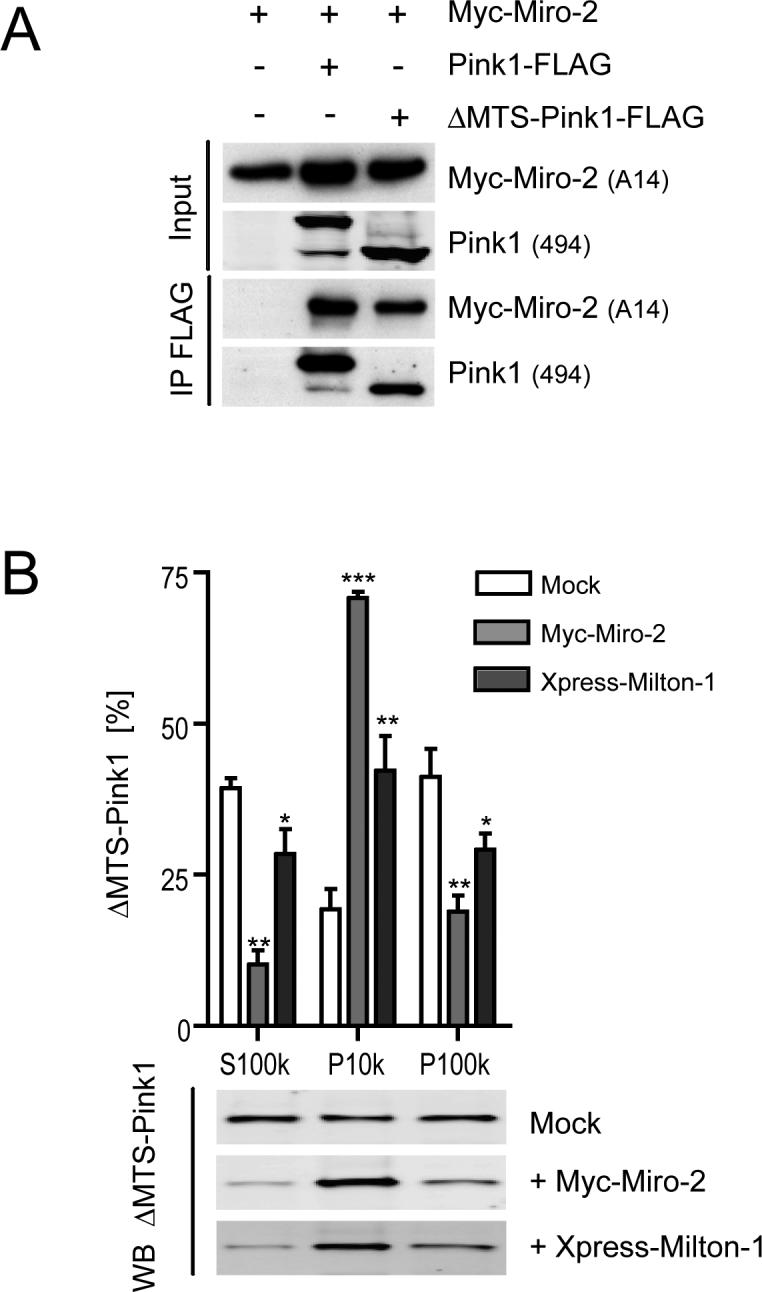
Miro-2 and Milton-1 recruit ΔMTS-Pink1 to mitochondria. (A) ΔMTS-Pink1 interacts with Miro-2. Transient co-transfections of Myc-Miro-2 into COS7 cells with mock, Pink1-FLAG, or ΔMTS-Pink1-FLAG (Pink1 aa 112−581) vectors. Cell extracts were probed (Input) for Pink1 and Myc-Miro-2 and then subjected to co-ip with an anti-FLAG antibody (M2). Western blotting of precipitates probed for Myc-Miro-2 and Pink1 demonstrated specific binding of both Pink1-FLAG and ΔMTS-Pink1-FLAG to Myc-Miro-2. (B) Quantitative analysis of the relative subcellular distributions across cytosolic (S100k), mitochondria-rich (P10k) and microsome-rich (P100k) fractions of ΔMTS-Pink1 transfectants after mock, Myc-Miro-2 or Xpress-Milton-1 co-expression (n=3, means ± SEM). Significant changes are indicated (* p<0.05, ** p<0.01 and *** p<0.001; one-way ANOVA with Newman-Keuls Multiple Comparison Test). A representative infrared ΔMTS-Pink1 immunoblot of the S100k, P10k and P100k fractions upon mock, Myc-Miro-2 or Xpress-Milton-1 transfection is shown below the graph.
Miro and Milton-1 expression rescues the altered mitochondrial morphology induced by Pink1 loss-of-function
RNAi-mediated silencing of Pink1 results in altered mitochondrial morphology in HeLa cells, and this alteration can be rescued by Parkin overexpression (8). We used confocal microscopy to probe for a potential effect of Miro and Milton expression on the altered mitochondrial morphology induced by loss of Pink1 function. First, we imaged mitochondrial morphology in M17 neuroblastoma cells stably expressing either scrambled control or Pink1 shRNA. Mitochondrial morphology was visualized by transient expression of mito-YFP and fluorescence microscopy. Efficient Pink1 silencing by shRNA was confirmed using RT-PCR (Supporting information Fig. S1), again because no available antibodies can detect endogenous Pink-1 protein in M17 cell lines. For quantitative analysis of morphologically altered mitochondria, we distinguished between cells with elongated, aggregated or fragmented mitochondria, as exemplified in Figure 5a. We found that Pink1 silencing significantly altered mitochondrial morphology: M17 cells with elongated mitochondrial morphology decreased from 67.5 ± 2.1% to 32.5 ± 1.6%, while cells with a fragmented mitochondrial morphology increased from 32.5 ± 2.1% to 67.5 ±1.6% upon Pink1 shRNA-mediated silencing (Fig. 5c). Expression of mouse Pink1 (which cannot be targeted by our Pink-1 shRNA) completely rescued this altered mitochondrial morphology, as expected (Supporting information: Fig. S2 a and b). This rescue experiment shows that the observed morphology alterations are not due to non-specific shRNA effects. Next, we asked if Miro-2 or Milton-1 expression had an effect on these alterations. Expression of each protein was confirmed by Western blotting (Fig. 5b). In the presence of Miro-2 or Milton-1, there were no longer any significant differences in mitochondrial morphology between cells expressing control or Pink1 shRNA (Fig. 5c). That is, co-expression of Miro-2 or Milton-1 returned the morphological alterations of mitochondria induced by Pink1 silencing to normal. Notably, the Miro-2 or Milton-1 co-expressing cells display a small proportion of aggregated mitochondria, in addition to elongated and fragmented ones (Fig. 5a, c). Similar results were obtained with Miro-1 expression (data now shown). These results show a functional interaction between Pink1, Miro and Milton-1 as regards the maintenance of proper mitochondrial morphology.
Figure 5.

Both Miro-2 and Milton-1 overexpression suppress alterations of mitochondrial morphology induced by Pink1 silencing in M17 cells. (A) Examples of mitochondrial morphology from control shRNA lines showing mito-YFP labeled cells with elongated, aggregated or fragmented mitochondria (Scale bar, 2 μm). (B) Transient transfections of Myc-Miro-2 or Xpress-Milton-1 into M17 cells stably expressing either scrambled control or Pink1 shRNA. Cell extracts were then probed for transfected proteins by Western blot analysis. (C) Quantitative analysis of mitochondrial morphology after Pink1 silencing without vs. with Miro-2 or Milton-1 overexpression (n=4, means ± SEM, 30 cells counted per experiment). Significant changes (vs. the control shRNA condition) are indicated (* p<0.05, ** p<0.01 and *** p<0.001; one-way ANOVA with Newman-Keuls Multiple Comparison Test). Absence of the first two bars (Control shRNA and Pink1 shRNA) in the Aggregated category indicates that essentially no aggregated mitochondria were observed in these cells.
DISCUSSION
PD is one of several neurodegenerative diseases linked to mitochondrial dysfunction. The functions of specific mitochondrial proteins implicated in these diverse diseases include modifiers of respiratory chain complexes, metabolic enzymes, chaperones, and regulators of mitochondrial dynamics (24). Here, we provide evidence that Pink1, which is genetically linked to PD, may participate in mitochondrial trafficking.
Milton and Miro are part of an essential protein complex that is known to link kinesin-1 to mitochondria for anterograde transport of mitochondria (18, 20, 25). Milton (i.e., Milton-1 (OIP106) and Milton-2 (GRIF-1) in humans (26)) serves as the adapter molecule that connects microtubules via kinesin to the mitochondrial-anchored Miro protein (20). Miro (i.e., Miro-1 and Miro-2 in humans) is an atypical GTPase that contains tandem GTP-binding domains separated by a linker region with putative calcium-binding EF hand domains (15). Genetic studies in yeast show that all domains of Gem1p (i.e., yeast Miro) are required for proper mitochondrial morphology (22). Calcium stops mitochondrial movement, and it has been speculated that Miro's calcium-binding EF hand domains could be important for this effect (20).
Taken together, these various published findings indicate that a function of the Miro/Milton complex in mitochondrial trafficking is well established. Thus, our identification of Pink1 as a member of this multi-protein complex makes it very likely that Pink1 is involved in the function of the complex, namely mitochondrial trafficking. To strengthen this finding, it is now critically important for the field to develop better Pink1 antibodies (14) that will allow the detection and characterization of endogenous Pink1 protein complexes.
Pink1 expressed without its mitochondrial targeting sequence protects neurons against the dopaminergic neurotoxin MPTP (13). These data strongly suggest that Pink1 can play a role relevant to PD mechanisms outside of mitochondria. Confocal immunofluorescence studies have suggested that ΔMTS-Pink1 partially overlaps with a mitochondrial marker stain (23), although this result has been controversial (13). Our study now provides data that help resolve this debate. First, we show by subcellular fractionation using differential centrifugation that about 20% of ΔMTS-Pink1 expressed in COS7 cells is found in the mitochondria-rich subcellular fraction. Our data is in agreement with the very recently published study showing that around 26% of exogenous Pink1 Δ1−91 in SH-SY5Y cells is mitochondrial (14). The failure of Haque et al. (13) to detect ΔMTS-Pink1 in the mitochondrial fraction using differential centrifugation could be explained by their use of NIH 3T3 cells. As they report, even wt Pink1 is only detectable at very low levels in the mitochondrial fraction of NIH 3T3 cells. Zhou et al. (14) recently provided evidence that the kinase domain of mitochondrial Pink1 faces the cytosol. Their experiments suggest that Pink1 is anchored at the mitochondria by a transmembrane domain. Small amounts of full-length Pink1 have been reported in cytosolic fractions of overexpressing cells, suggesting that the full-length protein is not solely membrane-anchored (5, 6, 13). Our new findings offer an alternative mechanism: that Pink1 66 and 55 kDa and ΔMTS all bind to the Miro-2/Milton-1 complex, which is well-known to reside at the outer mitochondrial membrane facing the cytosol. Indeed, we show that increasing either Miro or Milton retains more Pink1 in the mitochondria-rich fraction. Thus, we propose that Pink1 can also act in an MTS-independent fashion at the mitochondrion and can be retained on the surface of the mitochondrion by the Miro/Milton complex. Future experiments will determine whether Pink1 being part of the Miro/Milton complex is sufficient for its localization at the outer mitochondrial membrane and also whether the ΔMTS-Pink1/Miro/Milton complex is involved in the reported protective property of ΔMTS-Pink1 against toxicity by the mitochondrial toxin, MPTP.
Our mitochondrial morphology imaging data show increased mitochondrial fragmentation after Pink1 silencing that is fully suppressed by Miro overexpression. These data provide a functional link for mitochondria between Miro and Pink1. Although this rescue experiment apparently places the GTPase Miro downstream of Pink1, overexpression of Miro leads to an increased Pink1 66/55 kDa ratio, suggesting also potential upstream effects of Miro on Pink1. A similar phenomenon regarding Pink1 has been observed previously for Parkin (6). The striking increase in mitochondrial fragmentation when Pink1 is silenced points to a defect in mitochondrial fusion. But one important concept in mitochondrial dynamics is that mitochondrial trafficking and fusion are not completely independent of one another (27). Because proper mitochondrial fusion requires mitochondrial trafficking, it is possible that the observed fragmentation phenotype is mostly due to an impaired function of the Pink1/Miro/Milton protein complex. Future experiments will be necessary to confirm this hypothesis.
Alterations in the morphology of mitochondrial cristae have been found in Pink1 loss-of-function models (7, 8). In this regard, our identification of Mitofilin (also referred to as HMG or inner membrane mitochondrial protein) as a novel Pink1 binding protein may have special importance. Indeed, Mitofilin is known to be a critical organizer of mitochondrial cristae morphology and is indispensable for normal mitochondrial function (19). The identification of Pink1 interacting proteins that are localized at different sites within mitochondria suggests multiple functional roles for Pink1. In order to better understand Pink1 in the context of pathogenic mechanisms of PD, it will therefore be important to analyze in more detail the Pink1/Miro/Milton protein complex in the context of the Pink1/Mitofilin interaction.
Supplementary Material
ACKNOWLEDGMENTS
We thank Thomas Schwarz (Children's Hospital, Boston) for providing an OIP106 expression plasmid and Pontus Aspenstrom (Uppsala University, Sweden) for providing Miro expression constructs and Huaibin Cai (NIH, Bethesda) for the GST-mouse Pink1 construct. We are grateful to Bryan Krastins (Harvard Partners Center for Genetics and Genomics, Cambridge, MA) for MS/MS sequencing.
Abbreviations
- PD
Parkinson Disease
- ARPD
Autosomal recessive Parkinson Disease
- MTS
mitochondrial targeting sequence
- ΔMTS-Pink1
Pink1 lacking its mitochondrial targeting sequence
- ip
immunoprecipitation
- KD
kinase-dead
Footnotes
This work is supported by NINDS Udall Center grant (NS038375) and the NINDS grant NS051318 to D.J.S, and by the Intramural Research Program of the NIH, National Institute on Aging (Project Z01-AG000953) to M.R.C and K.J.T.
SUPPORTING INFORMATION AVAILABLE
Figures: S1, Measuring of shRNA induced Pink1 silencing by quantitative RT-PCR; S2, Overexpression of mouse Pink1 rescues altered mitochondrial morphology upon human Pink1 silencing in M17 cells. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Thomas B, Beal MF. Parkinson's disease. Hum Mol Genet. 2007;16(2):R183–194. doi: 10.1093/hmg/ddm159. [DOI] [PubMed] [Google Scholar]
- 2.Dodson MW, Guo M. Pink1, Parkin, DJ-1 and mitochondrial dysfunction in Parkinson's disease. Curr Opin Neurobiol. 2007;17:331–337. doi: 10.1016/j.conb.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 3.Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 4.Cookson MR, Dauer W, Dawson T, Fon EA, Guo M, Shen J. The roles of kinases in familial Parkinson's disease. J Neurosci. 2007;27:11865–11868. doi: 10.1523/JNEUROSCI.3695-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beilina A, Van Der Brug M, Ahmad R, Kesavapany S, Miller DW, Petsko GA, Cookson MR. Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc. Natl. Acad. Sci. U S A. 2005;102:5703–5708. doi: 10.1073/pnas.0500617102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weihofen A, Ostaszewski B, Minami Y, Selkoe DJ. Pink1 Parkinson mutations, the Cdc37/Hsp90 chaperones and Parkin all influence the maturation or subcellular distribution of Pink1. Hum Mol Genet. 2008;17:602–616. doi: 10.1093/hmg/ddm334. [DOI] [PubMed] [Google Scholar]
- 7.Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- 8.Exner N, Treske B, Paquet D, Holmstrom K, Schiesling C, Gispert S, Carballo-Carbajal I, Berg D, Hoepken HH, Gasser T, Kruger R, Winklhofer KF, Vogel F, Reichert AS, Auburger G, Kahle PJ, Schmid B, Haass C. Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J Neurosci. 2007;27:12413–12418. doi: 10.1523/JNEUROSCI.0719-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gautier CA, Kitada T, Shen J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci U S A. 2008;105:11364–11369. doi: 10.1073/pnas.0802076105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- 11.Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pridgeon JW, Olzmann JA, Chin LS, Li L. PINK1 Protects against Oxidative Stress by Phosphorylating Mitochondrial Chaperone TRAP1. PLoS Biol. 2007;5:e172. doi: 10.1371/journal.pbio.0050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haque ME, Thomas KJ, D'Souza C, Callaghan S, Kitada T, Slack RS, Fraser P, Cookson MR, Tandon A, Park DS. Cytoplasmic Pink1 activity protects neurons from dopaminergic neurotoxin MPTP. Proc Natl Acad Sci U S A. 2008;105:1716–1721. doi: 10.1073/pnas.0705363105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou C, Huang Y, Shao Y, May J, Prou D, Perier C, Dauer W, Schon EA, Przedborski S. The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proc Natl Acad Sci U S A. 2008;105:12022–12027. doi: 10.1073/pnas.0802814105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fransson A, Ruusala A, Aspenstrom P. Atypical Rho GTPases have roles in mitochondrial homeostasis and apoptosis. J Biol Chem. 2003;278:6495–6502. doi: 10.1074/jbc.M208609200. [DOI] [PubMed] [Google Scholar]
- 16.Iyer SP, Akimoto Y, Hart GW. Identification and cloning of a novel family of coiled-coil domain proteins that interact with O-GlcNAc transferase. J Biol Chem. 2003;278:5399–5409. doi: 10.1074/jbc.M209384200. [DOI] [PubMed] [Google Scholar]
- 17.Fransson S, Ruusala A, Aspenstrom P. The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem Biophys Res Commun. 2006;344:500–510. doi: 10.1016/j.bbrc.2006.03.163. [DOI] [PubMed] [Google Scholar]
- 18.Guo X, Macleod GT, Wellington A, Hu F, Panchumarthi S, Schoenfield M, Marin L, Charlton MP, Atwood HL, Zinsmaier KE. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron. 2005;47:379–393. doi: 10.1016/j.neuron.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 19.John GB, Shang Y, Li L, Renken C, Mannella CA, Selker JM, Rangell L, Bennett MJ, Zha J. The mitochondrial inner membrane protein mitofilin controls cristae morphology. Mol Biol Cell. 2005;16:1543–1554. doi: 10.1091/mbc.E04-08-0697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glater EE, Megeath LJ, Stowers RS, Schwarz TL. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol. 2006;173:545–557. doi: 10.1083/jcb.200601067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stowers RS, Megeath LJ, Gorska-Andrzejak J, Meinertzhagen IA, Schwarz TL. Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron. 2002;36:1063–1077. doi: 10.1016/s0896-6273(02)01094-2. [DOI] [PubMed] [Google Scholar]
- 22.Frederick RL, McCaffery JM, Cunningham KW, Okamoto K, Shaw JM. Yeast Miro GTPase, Gem1p, regulates mitochondrial morphology via a novel pathway. J Cell Biol. 2004;167:87–98. doi: 10.1083/jcb.200405100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Plun-Favreau H, Klupsch K, Moisoi N, Gandhi S, Kjaer S, Frith D, Harvey K, Deas E, Harvey RJ, McDonald N, Wood NW, Martins LM, Downward J. The mitochondrial protease HtrA2 is regulated by Parkinson's disease-associated kinase PINK1. Nat Cell Biol. 2007;9:1243–1252. doi: 10.1038/ncb1644. [DOI] [PubMed] [Google Scholar]
- 24.Mandemakers W, Morais VA, De Strooper B. A cell biological perspective on mitochondrial dysfunction in Parkinson disease and other neurodegenerative diseases. J Cell Sci. 2007;120:1707–1716. doi: 10.1242/jcs.03443. [DOI] [PubMed] [Google Scholar]
- 25.Rice SE, Gelfand VI. Paradigm lost: milton connects kinesin heavy chain to miro on mitochondria. J Cell Biol. 2006;173:459–461. doi: 10.1083/jcb.200604071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brickley K, Smith MJ, Beck M, Stephenson FA. GRIF-1 and OIP106, members of a novel gene family of coiled-coil domain proteins: association in vivo and in vitro with kinesin. J Biol Chem. 2005;280:14723–14732. doi: 10.1074/jbc.M409095200. [DOI] [PubMed] [Google Scholar]
- 27.Baloh RH. Mitochondrial dynamics and peripheral neuropathy. Neuroscientist. 2008;14:12–18. doi: 10.1177/1073858407307354. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.