

Abstract
Many new vaccines under development consist of rationally designed recombinant proteins that are relatively poor immunogens unless combined with potent adjuvants. There is only one adjuvant in common use in the U.S., aluminum phosphate or hydroxide (e.g. alum). This adjuvant, however, has significant limitations, particularly regarding the generation of strong cell-mediated (T cell) immune responses. A novel adjuvant, JVRS-100, composed of cationic liposome-DNA complexes (CLDC) has been evaluated for immune enhancing activity. The JVRS-100 adjuvant has been shown to elicit robust immune responses compared to CpG oligonucleotides, alum, and MPL adjuvants, and efficiently enhances both humoral and cellular immune responses. Safety has been evaluated in preclinical studies, and the adjuvant is now in early-stage clinical development. One application of this novel adjuvant is to augment the immune responses to recombinant subunit antigens, which are often poorly immunogenic. The JVRS-100 adjuvant, when combined with a recombinant influenza hemagglutinin (H1), elicited increased specific antibody and T-cell responses in mice. Single-dose vaccination and prime/boost vaccinations with JVRS-100-H1 were both shown to be protective (i.e., survival, reduced weight loss) following H1N1 (PR/8/34) virus challenge. Enhanced immunological responses could be critically important for improved efficacy and dose-sparing of a recombinant influenza vaccine.
Keywords: Adjuvant, vaccine, influenza
Introduction
In the early 1990’s, cationic liposomes complexed to plasmid DNA were investigated for gene delivery with the goal of eliciting high-level expression of the delivered gene product in target tissues [1]. In clinical trials, this material induced interferon-like responses when delivered via intravenous administration at doses far below those required for effective gene expression. In experimental models, innate immune activation occurred whether or not the plasmid DNA contained a gene of interest. The responses were dependent upon formation of the cationic liposome-DNA complexes (CLDC) since neither liposome nor plasmid DNA alone had significant immunostimulatory properties. Stimulation of innate immunity, triggered by CLDC, is due in part to a liposome-mediated potentiation of the inherent responsiveness of the mammalian immune system to non-methylated CpG motifs within the bacterial DNA of the plasmids. CpG motifs function via interaction with the toll-like receptor 9 (TLR9), an interaction that requires internalization into the endosomal compartment of cells facilitated by the lipid component [2]. Recently there have been reports in the literature of CpG- independent pathways of innate immune activation, in which DNA activates the innate immune system [3] and that this effect is enhanced when the DNA is complexed with cationic liposomes [4]. Microarray analysis has also shown that there are significant qualitative differences in immune system gene expression in CLDC- versus CpG oligonucleotide (ODN)-treated mice, with a number of important molecules necessary for effective antigen presentation. In particular, IL-12 and IL-15, and type I and II interferon (IFN)-related genes were up-regulated in CLDC-treated, but not CpG ODN-treated animals. Overall, in vitro and animal studies suggest that CLDC activate the innate immune system more broadly and at a higher level than CpG ODN [5]. Activation of innate immunity by CLDC is modulated by dose and route of administration, but is not species specific. In multiple species (mouse, rabbit, dog, monkey), CLDC treatment results in an immediate up-regulation of a broad-array of soluble and cellular host defenses such as release of IL-12 and interferons with the accumulation of macrophages and NK cells [6–8]. In addition to the utility of CLDC in promoting an antiviral response [5,6,9], administration of CLDC is efficacious against tumor progression in mouse models of cancer and in dogs with naturally-occurring tumors [10].
The addition of CLDC to antigens has been shown to produce a potent adjuvant effect following subcutaneous or intramuscular vaccination, with induction of enhanced T-cell and antibody responses [11]. The enhanced adaptive immune responses are linked to the profound activation of innate immunity, as well as to the co-delivery of antigens (bound by charge interactions to the positively-charged liposomes) and adjuvant to antigen-presenting cells. The CLDC are virus-sized particulates, with a mean diameter of ~120 nm, which facilitates trafficking to antigen presenting cells in lymph nodes draining the site of inoculation [12]. The nature of the enhanced immune responses suggests that vaccines and specifically, recombinant vaccines, would be greatly improved by administration with CLDC as an adjuvant. CLDC-adjuvanted vaccines have previously been shown to produce a greater CD8+ T-cell specific response than Freund’s complete adjuvant, peptide-pulsed dendritic cells, vaccinia vectored, and DNA vaccines [11]. Additionally CLDC-based vaccines demonstrated a greater CD4+ T cell-specific response than lymphocytic choriomeningitis virus (LCMV) infection and greater protection in a model of aerosol challenge with Mycobacterium tuberculosis (Erdman strain) [11].
As novel recombinant and subunit vaccines are developed against infectious diseases and cancer, the acute need for new adjuvant technologies has become more apparent. New adjuvants must be safe and well-tolerated, but also are most useful when they can stimulate both humoral and cellular immune responses, orient the qualitative response to be specifically directed against the invading pathogen, and can provide a dose-sparing capability.
A high priority in influenza research is the development of vaccines that do not rely upon the use of embryonated eggs as the substrate for vaccine production. The challenge of novel influenza antigen production methods such as mammalian cells or baculovirus-based systems, is that the proteins frequently do not fold into the native conformation to allow for optimal antigen presentation. Thus, these proteins are generally less immunogenic, as evidenced by high-dose requirements to achieve an antibody response equivalent to native antigen. Therefore, there is a need for improved adjuvants to enhance new vaccines consisting of rationally-designed recombinant proteins. The current studies demonstrate the effectiveness of a lipid-DNA complex, termed the JVRS-100 adjuvant, combined with a recombinant influenza antigen.
Materials and Methods
Preparation of JVRS-100 adjuvant-antigen mixture
The JVRS-100 adjuvant was prepared by mixing DOTIM/cholesterol liposomes with plasmid DNA (pMB75.6) in the presence of lactose followed by lyophilization and storage at 2–8°C. JVRS-100 was reconstituted prior to use by the addition of 500μl of sterile water for injection. Dilutions of JVRS-100 were prepared in dextrose-5% in water (D5W), to which recombinant vaccine antigens were added and diluted appropriately to administer the indicated dosage.
Mouse immunization schedule
Groups (5 animals) of female Balb/c mice were immunized via the intramuscular (IM, quadriceps muscle) route of administration with JVRS-100 adjuvanted or unadjuvanted antigens at day 0 or days 0 and 14. Mice were sacrificed 14 days following the second vaccination to assess humoral and cellular immunity as described below. In some cases, immunized mice were challenged 14 days following the last vaccination and followed for survival and weight loss to assess response to the influenza viral challenge.
Antibody titer ELISA
Fluzone® vaccine (2006–2007 formulation, Sanofi-Pasteur) containing A/New Caledonia/20/99, A/Wisconsin/67/2005, and B/Malaysia/2506/2007 at 90ug/ml hemagglutinin (HA) was diluted to 0.5μg/ml HA and plated at 4°C overnight on Maxisorp plates (NUNC, Rochester, NY). Plates were washed three times with PBS containing 0.05% Tween-20 and blocked with phosphate buffered saline pH 7.0 (PBS) containing 1% bovine serum albumin (BSA) for a minimum of 1 hour. Plates were washed as described above and 100μl of serial 1:2 dilutions of serum from individual vaccinated mice were added and incubated for 2 hours at room temperature. Plates were subsequently washed and incubated with a 1:6000 dilution of the appropriate isotype-specific antibody conjugated to horseradish peroxidase (Southern Biotech, Birmingham, AL). Plates were visualized with the addition of 100μl of TMB substrate (Pierce, Rockford, IL) and incubation for 20–30 minutes. Fifty microliters of stop solution (1M H2SO4) was then added and the absorbance measured at 450nm and 570nm. The reading at 570nm was subtracted from the reading at 450nm to correct for plate abnormalities and bubbles in the analyte solution. The resulting data for each sample was plotted to obtain a curve of the reciprocal dilution versus the A450–A570 measurement. The antibody titer was determined as the midpoint of the dilution curve as defined by EC50 calculations using Prism statistical software (Graphpad Software, San Diego, CA). The average of the EC50 for each cohort was determined to be the final antibody titer.
Hemagglutinin titer determination
Chicken red blood cells (cRBC) in Alsever’s Solution (Colorado Serum Company, Denver, CO) were washed 3x with PBS and resuspended to a 10% stock solution. Serial 1:2 dilutions of antigens were added to an equal volume of 0.5% cRBC. Following 1 hour incubation at room temperature, the plates were read for hemagglutination. HA titer was determined to be the reciprocal of the last well exhibiting hemagglutination.
Hemagglutination Inhibition Assay
Sera from mice were pre-treated by incubation overnight at 37°C with three parts receptor destroying enzyme (RDEII, Denka Seiken, Tokyo, Japan). After inactivation at 56°C for 30–60 minutes, serial 1:2 dilutions in PBS were mixed with 4 HA units of antigen and incubated at room temperature for 15 minutes, followed by the addition of an equal volume of 0.5% cRBC. Samples were performed in duplicate and a control (no antigen) was included to rule out non-specific activity. Plates were read at 1 hour. Hemagglutination inhibiting (HAI) antibody titer was determined to be the reciprocal dilution of the last well that showed complete inhibition of hemagglutination.
Splenocyte restimulation assays
Splenocytes isolated from vaccinated and control mice were restimulated with H1 protein or a representative H1N1 virus (PR/8/34). Supernatants were collected after 48 hours of stimulation and assayed by ELISA for mouse IFN-γ (eBioscience). Controls included spleen cells cultured 48 hours without the addition of specific antigens and unvaccinated splenocytes incubated with antigen.
Mouse serum adoptive transfer
Groups (10 animals) of Balb/c mice were immunized as described above. Mice were sacrificed 14 days following the last vaccination, serum was isolated, pooled and 300μl transferred to naïve mice via intraperitoneal administration. Mice were challenged the day following adoptive transfer and monitored for survival and weight change to assess response to influenza viral challenge. Controls included no treatment and mice that received an equal amount of serum from unvaccinated mice.
Mouse influenza lethal challenge
Mice were lightly anesthetized with isofluorane and challenged via intranasal administration with 2xLD50 (100HA) or 6xLD50 (300HA) of PR/8/34. The mice were monitored for weight loss and mortality for up to 20 days following infection.
Results
The established standard measurement of efficacy for vaccination against influenza is demonstrated increases in HAI antibody titer. To test antibody-mediated protection from homologous and drifted influenza strains, sera from vaccinated mice were tested in the HAI assay using A/New Caledonia, A/Solomon Islands/3/2006, and PR/8/34 (all H1N1 viruses). Administration of the JVRS-100 adjuvant with recombinant H1 protein (JVRS-100-H1) resulted in a significant increase in H1-specific antibody and TH1 bias of the immune response to the candidate recombinant antigen. Administration of the JVRS-100-H1vaccine resulted in increases of IgG (~18 fold, P<0.01), IgG1 (~20 fold, P<0.01), and IgG2a (~100 fold, P<0.01) based on EC50 antibody titer (Figure 1). Vaccination with JVRS-100-H1 resulted in a higher IgG2a:IgG1 ratio (4.01) compared to the unadjuvanted H1 group (0.82), indicating that the adjuvant is effective at promoting TH1 immunity. Furthermore, HAI titers with both matched and unmatched H1 viruses showed a considerable increase in antibody that was sufficient to inhibit hemagglutination (Figure 2). T cell responses to both recombinant H1 and a related H1N1 virus (PR/8/34) were also enhanced with the addition of the JVRS-100 adjuvant as evidenced by increased interferon-gamma (IFN-γ) production from restimulated splenocytes (Figure 3).
Figure 1.

Vaccination with JVRS100-H1 results in an increase in TH1-biased antibody responses. Groups of Balb/c mice (n=5) were vaccinated via IM administration at days 0 and 14 with 5μg recombinant H1 hemagglutinin combined with 20μg of JVRS-100. At day 28, sera were collected and antibody titer determined as described in materials and methods.
Figure 2.

Vaccination with JVRS-100-H1 results in increased HAI antibody response versus matched and unmatched H1 virus. Groups of Balb/c mice (n=5) were vaccinated via IM administration at days 0 and 14 with 5μg recombinant H1 hemagglutinin combined with 20μg of JVRS-100. At day 28 serum was collected and HAI antibody titer was determined as described in materials and methods for matched virus (New Caledonia/20/99) and unmatched virus (PR/8/34).
Figure 3.
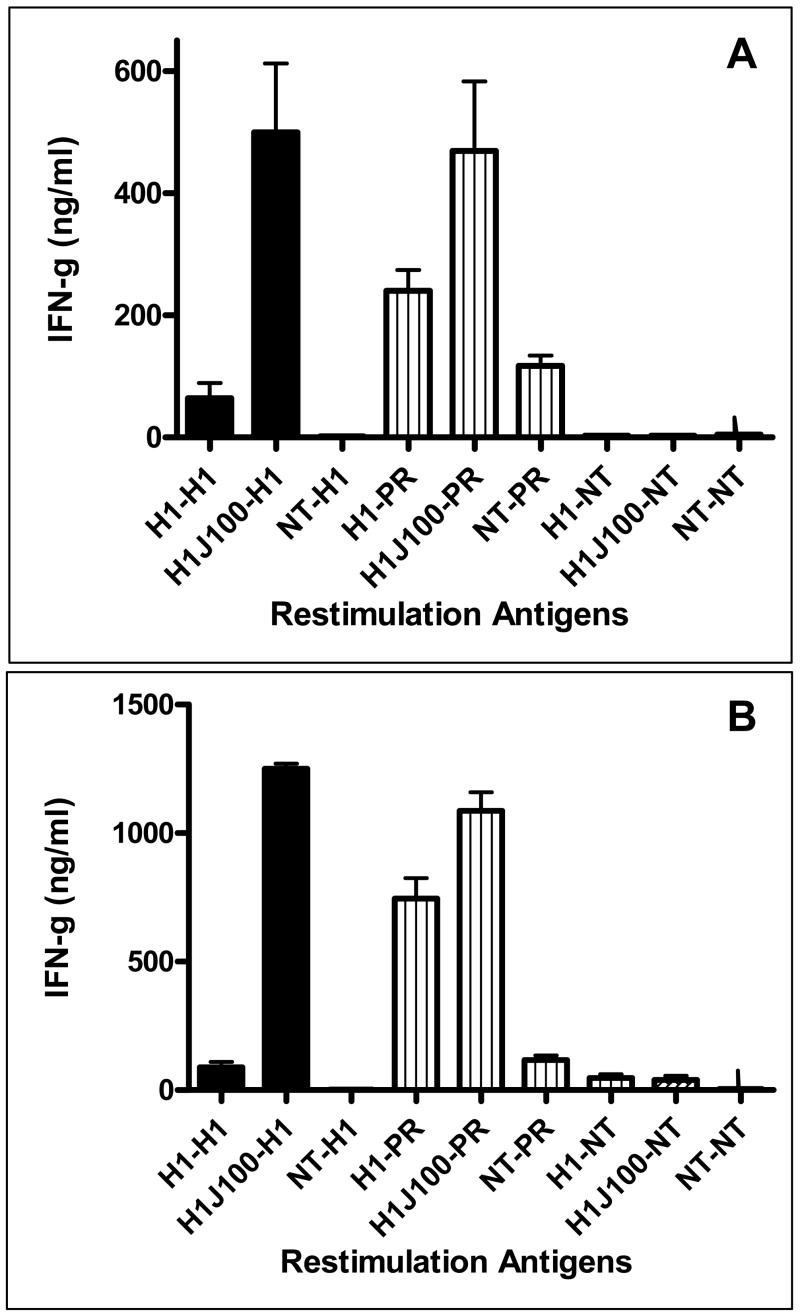
Vaccination with JVRS-100-adjuvanted H1 results in an increased splenocyte response following a single vaccination (Panel A) and prime/boost vaccination (Panel B). Groups of Balb/c mice (n=5) were vaccinated via IM administration at day 0 (single) or days 0 and 14 (prime/boost) with 5μg recombinant hemagglutinin combined with 20μg of JVRS-100. At day 28 splenocytes were collected and restimulated with matched virus (New Caledonia/20/99) and unmatched virus (PR/8/34) as described in materials and methods. The in vivo vaccination-in vitro stimulation combinations are listed (NT = no treatment).
A single vaccination with JVRS-100-H1 (A/New Caledonia/20/99) followed by challenge of mice with 2xLD50 of a related H1N1 virus (PR/8/34) resulted in decreased morbidity and mortality. As seen in Figure 4, the JVRS-100 adjuvanted group was completely protected from mortality and showed less weight loss due to influenza infection, which resolved in approximately 7 days. In contrast, the recombinant H1 vaccine alone was considerably less protective (i.e., 60% survival) and showed significant weight loss in the surviving mice, even at the 20-day post-challenge conclusion of the experiment. In subsequent studies, mice were given a prime-boost vaccination of recombinant H1 (A/New Caledonia/20/99) with or without the JVRS-100 adjuvant and were challenged with 6xLD50 of a related H1N1 virus (PR/8/34). The JVRS-100 adjuvanted group was 100% protected from mortality and weight loss following infection, which resolved in approximately 9 days (Figure 5). In comparison, the recombinant H1 vaccine alone was less protective (80% survival) and showed significant weight loss in the surviving mice even at the 20-day conclusion of the experiment.
Figure 4.

Protection following single-dose administration. A single vaccination with H1/JVRS-100 protects versus 2xLD50 lethal viral challenge. Balb/c mice (n=5) were vaccinated once IM with 5μg H1 or 5μg H1 combined with 20μg of JVRS-100. Fourteen days following vaccination, mice were challenged intranasally with 2xLD50 of PR/8/34 (H1N1) and monitored for body weight changes (A) and survival (B) over a 20-day period.
Figure 5.

Protection following prime/boost vaccination. A prime and boost vaccination with H1/JVRS-100 shows protection following a 6xLD50 lethal viral challenge. Balb/c mice (n=5) were vaccinated IM on day 0 and 14 with 5μg H1 or 5μg H1 combined 20μg JVRS-100. Fourteen days following the last vaccination, mice were challenged intranasally with 6xLD50 of PR/8/34 (H1N1) virus and monitored over 20 days for body weight (A) and survival (B).
To determine the potential mechanism of protection, sera were collected from mice following a prime/boost vaccination with H1 or JVRS-100-H1 and transferred to naïve mice that were subsequently challenged with 2xLD50 of H1N1 virus (PR/8/34). Mice receiving serum from JVRS-100-H1 vaccinated mice showed improved survival (100% vs. 40%) and reduced weight loss as compared with the unadjuvanted H1 group (Figure 6). Given that the amount of serum was more than sufficient for complete protection from weight loss and challenge based on HAI, the extent of morbidity and mortality in both the adjuvanted and unadjuvanted groups of mice following adoptive serum transfer suggested that both cellular and antibody-mediated immunity plays a role in protection from drifted strains following drifted influenza challenge. Subsequent experiments will examine adoptive transfer of CD4+ and CD8+ T-cells from vaccinated mice in addition to serum transfer to better elucidate the mechanism of protection from drifted influenza strain challenge.
Figure 6.

Adoptive transfer of immune serum and subsequent challenge with 2xLD50 of PR/8/34 (H1N1). Balb/c mice (n=5) were vaccinated IM on day 0 and 14 with 5μg H1 or 5μg H1 combined with 20μg JVRS-100. Fourteen days following the last vaccination, 300μl of serum from vaccinated or non-vaccinated (NT) mice was transferred intraperitoneally into naïve mice. One day following adoptive transfer, mice were challenged intranasally with 2xLD50 of PR/8/34 (H1N1) virus and monitored over 20 days for body weight (A) and survival (B).
Discussion
Addition of JVRS-100 as a vaccine adjuvant significantly increased the overall amount of antibody and HAI activity against matched and unmatched H1 influenza strains. These data suggest that not only is there an increase in homotypic protection, but also the protection against drifted strains, which has long been a shortcoming of trivalent influenza vaccines. Furthermore the addition of the JVRS-100 adjuvant to a candidate vaccine antigen has been shown to increase the TH1 bias of the antibody response. While it has not been shown by direct evidence in these experiments, it is plausible that IgG2a provides greater protection in vivo due to its superior ability to bind Fc receptors, which may play a role in defense against influenza [13]. Moreover, IgG2a is more effective at activating complement than IgG1, and such activation may enhance viral neutralization [14]. This suggests that a TH1-biased antibody response may be more protective than a TH2 biased antibody response, especially for influenza challenge.
In cases involving failure of protection due to insufficient or mismatched neutralizing antibodies, the resolution of influenza infection primarily depends on T-cells and in particular, CD8+ T-cells [15]. Enhanced T-cell responses to H1 were observed with the JVRS-100-adjuvanted recombinant antigens compared to the unadjuvanted vaccines. Previous reports of the adjuvant activity of JVRS-100 have shown the enhancement of both CD4+ and CD8+ T-cell responses of vaccine antigens [11]. The focus of future studies will be to demonstrate the relative importance of the CD4+ and CD8+ T-cell response in protection against heterosubtypic (i.e., drifted and shifted strains) and homosubtypic virus strains, compared to natural infection.
The protection of mice vaccinated with adjuvanted H1 antigen and challenged with a non-matched H1 virus (drifted) resulted in a substantial decrease in morbidity and mortality when compared with unadjuvanted H1 antigen. This ability to confer mismatched protection could be critical in the situation of an influenza pandemic or where fully or partially HA-matched vaccines may not be available due to incorrect viral strain choices, production issues, or vaccine shortages.
The role of Tcells and antibodies in heterosubtypic immunity has been a focus of some debate over the past few years in the literature. The adoptive transfer studies detailed herein, suggest that the protection observed from JVRS-100-adjuvanted recombinant H1 vaccine may due to a combination of antibody and T-cell immunity, however much work remains to be done to identify the exact protective mechanisms.
Conclusions
Addition of the JVRS-100 adjuvant to a recombinant influenza protein vaccine has been shown to increase immunogenicity, which resulted in robust protection following lethal challenge in a mouse model of influenza infection. These results indicate that the JVRS-100 adjuvant may increase the effectiveness of recombinant antigen-based vaccines, which have been shown to have poor immunogenicity and limited ability to induce cross-protective immune responses.
Acknowledgments
Work presented in this publication was supported by grant # 5R41AI068260-02 entitled “Lipid/DNA/Antigen Complexes for Influenza A Viral Vaccination”.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zhu N, Liggitt D, Liu Y, Debs R. Systemic gene expression after intravenous DNA delivery into adult mice. Science. 1993;261:209–211. doi: 10.1126/science.7687073. [DOI] [PubMed] [Google Scholar]
- 2.Gursel I, Gursel M, Ishii KJ, Klinman DM. Sterically stabilized cationic liposomes improve the uptake and immunostimulatory activity of CpG oligonucleotides. J Immunol. 2001;167:3324–3328. doi: 10.4049/jimmunol.167.6.3324. [DOI] [PubMed] [Google Scholar]
- 3.Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, Ohba Y, Taniguchi T. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- 4.Yasuda K, Ogawa Y, Kishimoto M, Takagi T, Hashida M, Takakura Y. Plasmid DNA activates murine macrophages to induce inflammatory cytokines in a CpG motif-independent manner by complex formation with cationic liposomes. Biochem Biophys Res Commun. 2002;293:344–348. doi: 10.1016/S0006-291X(02)00210-3. [DOI] [PubMed] [Google Scholar]
- 5.Gowen BB, Fairman J, Dow S, Troyer R, Wonga MH, Junga KH, Melby PC, Morrey JD. Prophylaxis with cationic liposome-DNA complexes protects hamsters from 1 phleboviral disease: Importance of Liposomal Delivery and CpG motifs. Antiviral Res. doi: 10.1016/j.antiviral.2008.09.001. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gowen BB, Fairman J, Smee DF, Wong MH, Jung JH, Pace AM, Heiner ML, Bailey KW, Dow SW, Sidwell RW. Protective immunity against acute phleboviral infection elicited through immunostimulatory cationic liposome-DNA complexes. Antiviral Res. 2006;69:165–172. doi: 10.1016/j.antiviral.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 7.Dow SW, Schwarze J, Heath TD, Potter TA, Gelfand EW. Systemic and local interferon gamma gene delivery to the lungs for treatment of allergen-induced airway hyperresponsiveness in mice. Hum Gene Ther. 1999;10:1905–1914. doi: 10.1089/10430349950017266. [DOI] [PubMed] [Google Scholar]
- 8.Freimark BD, Blezinger HP, Florack VJ, Nordstrom JL, Long SD, Deshpande DS, Nochumson S, Petrak KL. Cationic lipids enhance cytokine and cell influx levels in the lung following administration of plasmid: cationic lipid complexes. J Immunol. 1998;160:4580–4586. [PubMed] [Google Scholar]
- 9.Morrey JD, Motter NE, Taro B, Lay M, Fairman J. Efficacy of cationic lipid-DNA complexes (CLDC) on hepatitis B virus in transgenic mice. Antiviral Res. 2008;79(1):71–79. doi: 10.1016/j.antiviral.2008.01.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Higgins RJ, McKisic M, Dickinson PJ, Jimenez DF, Dow SW, Tripp LD, LeCouteur RA. Growth inhibition of an orthotopic glioblastoma in immunocompetent mice by cationic lipid-DNA complexes. Cancer Immunol Immunother. 2004;53:338–344. doi: 10.1007/s00262-003-0447-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zaks K, Jordan M, Guth A, Sellins K, Kedl R, Izzo A, Bosio C, Dow W. Efficient immunization and cross-priming by vaccine adjuvants containing TLR3 or TLR9 agonists complexed to cationic liposomes. J Immunol. 2006;176:7335–7345. doi: 10.4049/jimmunol.176.12.7335. [DOI] [PubMed] [Google Scholar]
- 12.Manolova V, Flace A, Bauer M, Schwarz K, Saudan P, Bachmann MF. Nanoparticles target distinct dendritic cell populations according to their size. Eur J Immunol. 2008;38(5):1404–13. doi: 10.1002/eji.200737984. [DOI] [PubMed] [Google Scholar]
- 13.Huber VC, Lynch JM, Bucher DJ, Le J, Metzger DW. Fc receptor-mediated phagocytosis makes a significant contribution to clearance of influenza virus infections. J Immunol. 2001;166:7381–7388. doi: 10.4049/jimmunol.166.12.7381. [DOI] [PubMed] [Google Scholar]
- 14.Beebe DP, Schreiber RD, Cooper NR. Neutralization of influenza virus by normal human sera: mechanisms involving antibody and complement. J Immunol. 1983;130:1317–1322. [PubMed] [Google Scholar]
- 15.Doherty PC, Topham DJ, Tripp RA, Cardin RD, Brooks JW, Stevenson PG. Effector CD4+ and CD8+ T-cell mechanisms in the control of respiratory virus infections. Immunol Rev. 1997;159:105–117. doi: 10.1111/j.1600-065x.1997.tb01010.x. [DOI] [PubMed] [Google Scholar]