

Abstract
Mitochondrial diseases have been shown to result from mutations in mitochondrial genes located in either the nuclear DNA (nDNA) or mitochondrial DNA (mtDNA). Mitochondrial OXPHOS complex I has 45 subunits encoded by 38 nuclear and 7 mitochondrial genes. Two male patients in a putative X-linked pedigree exhibiting a progressive neurodegenerative disorder and a severe muscle complex I enzyme defect were analyzed for mutations in the 38 nDNA and seven mtDNA encoded complex I subunits. The nDNA X-linked NDUFA1 gene (MWFE polypeptide) was discovered to harbor a novel missense mutation which changed a highly conserved glycine at position 32 to an arginine, shown to segregate with the disease. When this mutation was introduced into a NDUFA1 null hamster cell line, a substantial decrease in the complex I assembly and activity was observed. When the mtDNA of the patient was analyzed, potentially relevant missense mutations were observed in the complex I genes. Transmitochondrial cybrids containing the patient's mtDNA resulted in a mild complex I deficiency. Interestingly enough, the nDNA-encoded MWFE polypeptide has been shown to interact with various mtDNA-encoded complex I subunits. Therefore, we hypothesize that the novel G32R mutation in NDUFA1 is causing complex I deficiency either by itself or in synergy with additional mtDNA variants.
Keywords: Mitochondria, Mitochondrial disorders, complex I, mtDNA, NDUFA1
Introduction
Mitochondrial complex I or NADH-ubiquinone oxidoreductase (complex I, EC 1.6.5.3) is the largest complex of the oxidative phosphorylation (OXPHOS) system, containing at least 45 subunits [1]. It is the most common clinical mitochondrial defect, linked to a large variety of clinical phenotypes [2], [3]. Complex I is L-shaped with the base embedded in the inner membrane and a peripheral arm extending into the mitochondrial matrix. This complex can be fractionated by treatment with chaotropic agents into simpler subcomplexes, subcomplex Iα and Iλ contain most of the hydrophilic subunits and represent the peripheral part of complex I, subcomplexes Iβ and Iγ contain most of the hydrophobic subunits and correspond to the membrane arm [4, 5]. All of the seven mtDNA encoded subunits are confined to the hydrophobic membrane component of the complex [4].
The nuclear gene NDUFA1 located on chromosome Xq24 encompasses 3 exons [6] and 2 missense mutations (G8R and R37S) have been recently identified in male patients presenting with neurological syndromes [7]. The NDUFA1 subunit, essential for complex I assembly and function [8], has been shown to interact with the mtDNA encoded subunits during the complex I assembly process [9]. It may also be phosphorylated and thus involved in complex I regulation [10, 11].
When the nuclei of one species are combined with the mtDNA of another by the creation of transmitochondrial cybrids [12, 13], incompatibility is commonly observed [14-16]. Combining a human nucleus with chimp or gorilla mtDNA results in a specific 40% reduction of complex I activity [17]. Since the mtDNA mutation rate is at least one order of magnitude higher than that of comparable nDNA encoded OXPHOS subunits [18], incompatibility might be expected for certain combinations of nDNA and mtDNA polymorphisms within the same species, a phenomenon that has already been implicated in the primate speciation process [19]. This implies that nDNA and mtDNA incompatibility might be a common cause of OXPHOS defects in mammals. In this publication we are reporting a disease arising out of such a potential incompatibility among nuclear and mitochondrial variations.
Methods
Patient report
A 35 year old man (III-11), the first child of healthy non-consanguineous parents appeared to have normal developmental milestones during the first several years after birth (Figure 1). At 4 years of age, he lost the ability to form words and his motor skills began to deteriorate. Generalized tonic-clonic seizures began at age 5. An EEG was found abnormal showing bitemporal spike wave foci. At 7 years of age an examination revealed an unsteady gait with ataxia and retinitis pigmentosa and a MRI revealed mild cerebellar atrophy and increased signal intensity in the anterior medulla in relation with a neurodegenerative process. His unbalanced gait worsened with time with frequent falls and he developed a progressive behavior disorder. Karyotype did not show any chromosomal abnormalities, and lactate and pyruvate were normal in blood, cerebrospinal fluid (CSF) and urine. Hence, since 4 years of age he suffered from a progressive neurodegeneration associated with retinitis pigmentosa, generalized seizures, ataxia aphasia and dementia.
Figure 1.
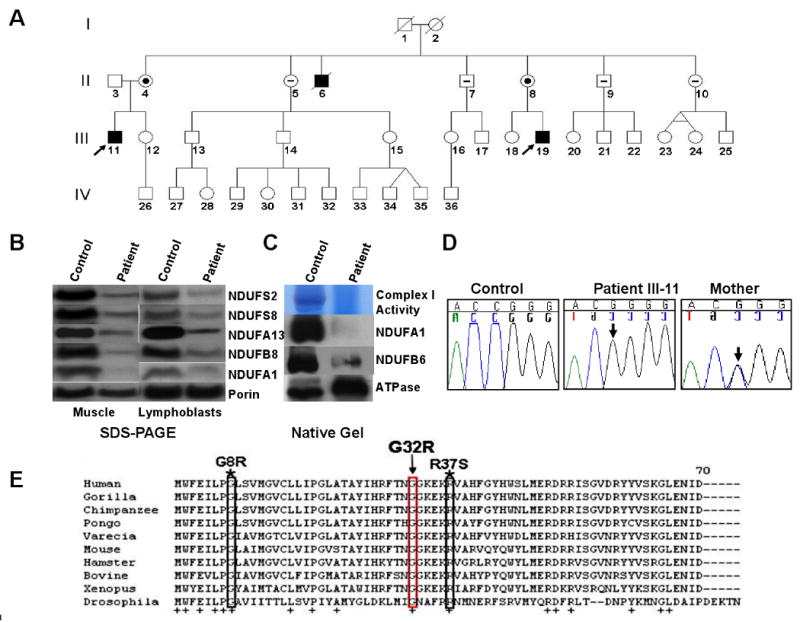
A Pedigree of the family. Affected individuals are represented by solid black symbols and open symbols represent unaffected individuals. Negative signs designate the absence of NDUFA1 mutation; carriers are represented by a symbol with a dot. Both probands are indicated by the arrows. B: Immunodetection of nuclear-encoded subunits of complex I from the proband III-11 and an age-matched control. Porin was used as a loading control. C: Blue Native Gel, Western analysis and in-gel activity assays of the complex-I and complex V from patient and control lymphoblast cells. D: Sequence analysis of NDUFA1 cDNA in the control, patient and patient's mother. The arrow shows the position of the c.111 G>C change in the NDUFA1 cDNA. E: Interspecies comparison of NDUFA1 subunit. Asterisks indicate the position of the 2 pathogenic mutations in NDUFA1 gene recently reported [7]. Plus signs designate conserved amino acid among all species.
The second proband of this family patient III-19 a maternal cousin of proband III-11, is a 30 year old man born from non-consanguineous parents after a normal pregnancy. Early motor milestones were within normal limits but he did not walk independently until 18 months. At age 5, a moderate hypotonia with mild proximal muscle weakness was noticed and he progressively became ataxic. Since then his movement control and cognitive function progressively deteriorated. At age 10 he had seizures with irregular myoclonic jerks more pronounced in the lower extremities and was diagnosed with a progressive bilateral sensorineural hearing loss. A mildly elevated plasma lactate was noted (3.0 mmol/l, normal <2.8) and lactate in the CSF was at the upper limit of normal (1.98 mmol/l, normal <2.2mmol/l).
Pedigree analysis (Figure 1A) revealed a maternal uncle II-6 who was thought to have a normal development until 4 years of age when he presented a severe gait imbalance with ataxia associated with mental retardation and committed suicide at age 35 (II-6). The proband's maternal grand mother I-2 developed dementia at age 65 and died at age 80 with a diagnosis of Alzheimer's disease. A muscle biopsy was collected from both probands and found to be negative for mtDNA rearrangements and the most common pathogenic mtDNA mutations. Histochemistry and electron microscopy performed on the skeletal muscle were unremarkable.
Cell lines and cybrid cell cultures
Lymphoblastoid cell lines were cultured in RPMI 1640 (Invitrogen, Carlsbad, CA) with high glucose (4.5 mg/ml) supplemented with 10% fetal calf serum (FCS) and 50 μg/ml uridine and 1mM sodium pyruvate [20]. The mtDNAs of the patient III-11 and control lymphoblasts were transferred into 143B rho° cells by fusion following nuclear inactivation with actinomycin D [21]. The patient origin of the cybrid mtDNA was confirmed by PCR amplification and restriction site polymorphism analysis. The resulting cybrid cells were subsequently expanded and mtDNA level was restored to normal level. Biochemical assays were performed on isolated mitochondria and/or permeabilized cells.
Mouse cell lines 3A-20-4 and 4A harbored null mutations in the ND5 and ND6 subunits, respectively, a gift from Drs. Attardi and Chomyn [22]. Chinese hamster cells harboring a null mutation for the NDUFA1 gene were cultured as described [9, 23].
Complex I cDNA analysis
RNA extraction from patient and control lymphoblast cells was performed using Trizol reagent followed by first strand cDNA synthesis using the SuperScript kit according to the manufacturer's recommendations (Invitrogen). Complex I nuclear genes were subjected to PCR amplification and sequencing (supplemental table 1). A list of oligonucleotide primers used for PCR amplification and sequencing is available (supplemental table 1). PCR products were purified by ExoSAP-IT (Amersham) and directly sequenced using the PRISM Ready Reaction Sequencing Kit (Applied Biosystems) on an automated sequencer (ABI 3130, Applied Biosystems). Sequence data were analyzed using Sequencher (version 4.8, Genecode Corp.) software.
Mitochondrial DNA analysis
Genomic DNA was extracted using the Puregene DNA Isolation Kit (Gentra System, Minneapolis, MN). The mtDNA was PCR amplified in 8 fragments using the protocol 96°C for 5 min, followed by 30 cycles of 96°C for 45 sec, 58°C for 30 sec and 72°C for 3 min, and a last extension at 72°C for 10 min (primers for mtDNA analysis available upon request). PCR products were purified and sequenced as described above.
Plasmids, Genes and Complementation Tests
The hamster NDUFA1 cDNA [8] was tagged with the HA epitope and cloned into a mammalian expression vector pTRIDENT-14 neo [9]. Site-directed mutagenesis (Quickchange XL site-directed mutagenesis kit, Stratagene La Jolla, CA) of the pTRIDENT-NDUFA1-HA template plasmid used primers for the G32R mutation: forward 5′-CATCCACAAGTACACCAACCGGGGCAAGGAAAAAAGAG-3′ and reverse 5′-CTCTTTTTTCCTTGCCCCGGTTGGTGTACTTGTGGATG-3′; for the G8R mutation: forward 5′-TTCGAGATTCTCCCTCGCCTCGCCGTCATGGGTGTGTGC 3′ and reverse 5′-ACACACCCATGACGGCGAGGCGAGGGAGAATCTCGAACC-3′; for the R37S mutation: forward 5′-GGGGGCAAGGAAAAAAGCGTTGGTCGCCTTCGTTACCAG-3′ and reverse 5′-CTGGTAACGAAGGCGACCAACGCTTTTTTCCTTGCCCCC-3′ and generated plasmid pIS-2057, which harbors a Neo selectable marker. Mutations were confirmed by sequencing. Transfection and complementation of the chinese hamster mutant cell lines were performed as reported elsewhere [9].
Measurement of Respiratory Activities
Mitochondria from muscle was isolated and used for OXPHOS enzyme analyses as previously described [24]. Respiration in Chinese hamster cells and transmitochondrial cybrids lines were analyzed by polarography [9, 25]. The cells were collected (350 × g), resuspended in 1× HSM buffer (20 mM Hepes, pH 7.1, 250 mM sucrose, 10 mM MgCl2) at density 2 × 107/ml, and permeabilized with 100 μg/ml digitonin until more than 90% of the cells are stained by trypan blue, about 1-3 min at 4 °C. The cell suspension was then diluted 10-fold with HSM buffer, and the cells were harvested by centrifugation, washed once with HSM buffer and resuspended at 3 × 107 cells/ml. Oxygen consumption of roughly 5 × 106 cells was measured using a Clark-type oxygen electrode (Hansatech, Norfolk, UK) at 37 °C.
Western blotting assays, Blue Native gel electrophoresis and in-gel activity assays
Mitochondrial samples (10 ug) were separated by SDS-PAGE and transferred to Immobilon-P (0.2 μm) membranes. Antibodies for NDUFA13, NDUFB6, NDUFB8 for complex I; 30 kDa Ip subunit for complex II; F1-α subunit for complex V and porin antibody were from Mitosciences (Eugene, OR); NDUFS2 and NDUFS8 antibodies provided by Prof. Lunardi and NDUFA1 antibody by Prof. Scheffler. Blue native gel (BNG) electrophoresis of mitochondrial respiratory complexes and in-gel histochemical assays were performed as reported previously [26].
Statistical Analyses
All data values are given as mean value of n experiments ± SEM with n=3. Statistical differences were determined by Student's t test with GraphPad Prism 4.0 software (San Diego, CA). In all cases, statistical significance was set at P ≤ 0.05.
Results
Biochemical studies on isolated mitochondria from muscle
The probands, III-11 and III-19, with a progressive neurodegenerative disease, were selected (Figure 1A). A muscle biopsy was performed on both probands III-11 and III-19 and the respiratory chain enzymes and citrate synthase were assayed from isolated muscle mitochondria. Complex I was specifically reduced to 5% to 10% residual activity of control values on probands III-11 and III-19, whether normalized either to protein concentration or to citrate synthase activity. By contrast, complex IV activity was identical to controls (Table 1).
Table 1.
Respiratory-chain enzyme activities in skeletal muscle of patients III-11 and III-19. CI: Complex I; CIV: Complex IV; CS: Citrate Synthase). Complex I and IV enzyme activities were normalized by citrate synthase. Controls values are Mean and ± 1 Standard Deviation (SD). Abnormal values are in bold.
| Mean specific activity | |||
|---|---|---|---|
| Complex | Patient 1 | Patient 2 | Control (mean ± SD) |
| III-11 | III-19 | (n = 25) | |
| Complex I | 5 | 8 | 106 ± 46 |
| Complex I+III | 32 | 77 | 262 ± 93 |
| Complex II+III | 622 | 1184 | 526 ± 140 |
| Complex IV | 1205 | 1201 | 1583 ± 370 |
| Citrate Synthase | 557 | 531 | 685 ± 157 |
| Ratios | |||
| Complex I/CS | 0.008 | 0.015 | 0.15 ± 0.03 |
| Complex IV/CS | 2.1 | 2.2 | 2.3 ± 0.01 |
Specific enzymes activities as nmol of substrate/min/mg/of mitochondrial protein
Complex I assembly in the proband muscle and cell line
To determine if the reduced complex I activity was due to of inhibition of activity or failure to assemble the enzyme, Western blots and BN-gel analyses were performed on muscle homogenate and on lymphoblastoid cell mitochondria from the proband III-11. Immunoblots were analyzed for the presence of complex I subunits known to be part of the peripheral arm (NDUFS2, NDUFA13, NDUFS8) or to the membrane arm (NDUFA1, NDUFB8) (Figure 1B). A significant reduction was found in the proband's muscle and lymphoblastoid cells of all nDNA encoded complex I subunits including NDUFA1. This alteration in subunit levels was correlated with a reduction in complex I activity within the BNG, and an absence of immunodetectable complex I subunits NDUFA1 and NDUFB6 in the expected region of the BN gel (Figure 1C). By contrast, the level of ATP synthase was significantly increased in the patient's sample, suggesting a compensatory response to the complex I defect.
Sequence analysis of nuclear complex I subunits
The clear X-linked recessive nature of the proband's pedigree (Figure 1A), led us to investigate the nDNA genes of complex I, with special attention being directed at the two X-linked subunits NDUFA1 and NDUFB10. All 38 structural subunits plus two additional genes known to be involved in complex I assembly (NDUFAF1 and NDUFA12L) were RT-PCR amplified from lymphoblast RNA of the proband III-11 and sequenced. The analysis revealed only a single nuclear variant in both probands, a c.111 G>C change in the NDUFA1 cDNA, which converted the highly conserved glycine at codon 32 to an arginine (G32R) (Figure 1D). Both proband's mothers were found heterozygous carriers for this mutation showing that the mutation segregates with the phenotype (Figure 1A). The G32R variant was not seen in more than 150 DNA controls from the same ethnic origin, though the change was reported in the SNP database (http://www.ncbi.nlm.nih.gov/SNP/; rs1801316) in a heterozygous female (1 of 41 female DNA samples) giving a heterozygosity level (0.026). The glycine 32 is located within a highly hydrophilic domain of NDUFA1 peptide close to the outer surface of the inner membrane and adjacent to the transmembrane anchor. The mutation replaces the hydrogen R group of glycine with the charged and bulky R group of arginine. Insertion of a charged group into the small hydrophilic domain (∼45 amino acids) of NDUFA1 peptide would be expected to affect its interaction with the other complex I integral membrane proteins [9]. The substitution G32R from our study, the G8R, and R37G mutations recently described [7] (Figure 1E) were further analysed using polyphen software (http://genetics.bwh.harvard.edu/pph/) and predicted as probably damaging substitutions.
Site-specific mutagenesis of the NDUFA1 mutations in hamster cell line
To determine if the NDUFA1 G32R substitution and the newly described NDUFA1 G8R and R37S mutations affect complex I activity [7], NDUFA1 null CCL16-B2 hamster cells were transfected with an expression plasmid in which the mutations had been introduced into the Chinese hamster NDUFA1 cDNA. Three independent stable transfectants for each selected NDUFA1 mutation (G8R, G32R and R37S) were obtained by selecting in G418 and further analyzed. The complex I activity was found to be reduced by 41% in the G32R mutant cell line, by 51% in the G8R and was undetectable in the R37S mutant (Figure 2A). Other electron transport chain enzyme activities were normal. Furthermore, measurements of growth rates of the NDUFA1 G32R and G8R mutant cells in DME-Gal medium showed a distinct reduction and no growth at all of the NDUFA1 R37S mutant compared to B2 null cell line transfected with NDUFA1 wild type used as a control (Figure 2B). A significant decrease in the complex I band was also seen by assays within BNG (Figure 2C).
Figure 2.
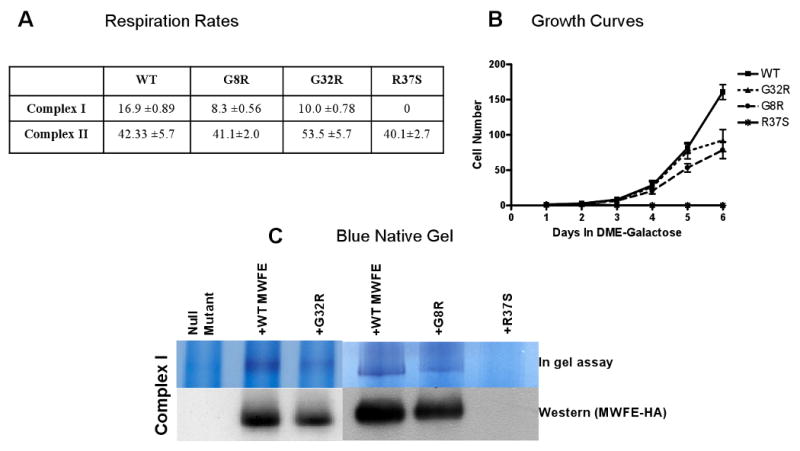
A: Complex-I and II activities measured by O2 consumption of the NDUFA1 G32R, G8R and R37S mutants and wild type transfectants. B: Mean growth rates in DME-Gal; P ≤ 0.0001; n=3. C: Blue Native Gel and Western analysis.
Mitochondrial DNA analysis
NDUFA1 is known to be dependent on mitochondrially encoded complex I subunits. To study the possibility of additional mtDNA variant or variants contributing to the complex I defect and the disease phenotype we sequenced the proband's muscle mtDNA that revealed an H haplogroup containing 24 nucleotide differences relative to the Cambridge reference sequence [27]. Of these differences, 17 were known synonymous polymorphisms [27] and the non-synonymous changes are listed in Table 2. Five of the mtDNA changes are haplogroup H hallmarks, including three substitutions in ND genes (ND1 m.3992C>T (T229M), ND1 m.4024A>G (T240A) and ND6 m.14582A>G (V31A)) plus the ATP6 m.8860A>G (T112A) and the tRNAPro m.16000G>A variant. Two additional mtDNA, ND1 m.3368T>C (M21T) and ND5 m.12599T>C (M88T) have rarely if ever been observed in mtDNA databases [27-29]. However, the conservation index (CI) of these was only 18% (7/39) for the ND1 M21T variant and 38% (15/39) for the ND5 M88T variant (Table 2).
Table 2.
Non-synonymous mtDNA changes in the proband. rCRS: Revised Cambridge Reference Sequence [38]; Conservation Index: conservation index between 39 different species as described [39]; mtDB database: Numbers in brackets indicate the total number of sequences found in the mitochondrial database.
| Nucleotide change CRS position | Amino acid change | Gene | Present in Mitomaster | Conservation Index | MtDB database (2704) | Comment |
|---|---|---|---|---|---|---|
| 3368T>C | M21T | ND1 | Not present | 7/39 | 1 | Rare variant |
| 3992C>T | T229M | ND1 | H | 10/39 | 24 | Common polymorphism |
| 4024A>G | T240A | ND1 | H | 12/39 | 22 | Common polymorphism |
| 8860A>G | T112A | ATP6 | H | 28/39 | 2698 | Common polymorphism |
| 12599T>C | M88T | ND5 | Not present | 15/39 | 0 | Not previously described |
| 14582A>G | V31A | ND6 | H | 17/39 | 23 | Common polymorphism |
| 16000G>A | - | tRNApro | H | 5/30 | 3 | Common polymorphism |
Transmitochondrial cybrid analysis
To determine if the proband's mtDNA with its haplogroup H variants plus the novel ND1 M21T and ND5 M88T variants adversely affected mitochondrial complex I activity, lymphoblastoid cells from the proband were chemically enucleated and fused to mtDNA-deficient 143B(TK-) ρ° cells. The resulting cybrid cells were subsequently expanded during several passages and the mtDNA nuclear ratio in the cybrid cells compared to controls was measured showing that the patient cybrid mtDNA had been restored to a normal level (data not shown). Three independent transmitochondrial cybrid clones were analyzed by polarography and revealed a mild complex I defect, resulting in a 29% reduction in respiration using site I substrates relative to the 143B(TK-) parental control cells (Figure 3A).
Figure 3.
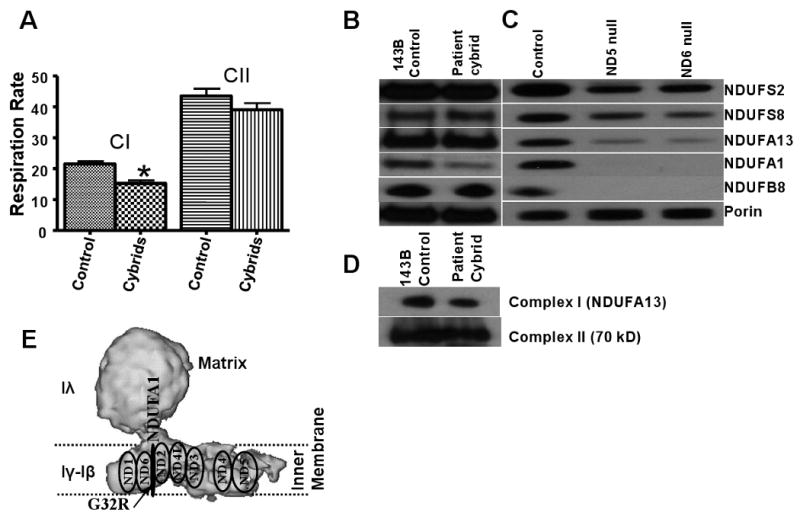
A: Respiratory rate on transmitochondrial cybrids. CI, CII: activities of complexes I and II. *Significantly different from control; P ≤ 0.0035; n=3. B: Immunodetection of nuclear complex I subunits of patient's cybrid cells and 143B parental cells. C: Immunodetection of complex I subunits of ND5 and ND6 mouse null mutants compared to LA9 control cell line. D: Blue Native Gel of the native complex-I and complex II from patient's cybrid and control cells. E: Predicted location of NADH mitochondrially-encoded and NDUFA1 nuclear-encoded subunits in the mitochondrial membrane. ND1; ND2; ND3; ND4; ND4L; ND5; ND6: NADH dehydrogenase subunits 1; 2; 3; 4; 4L; 5; 6. The arrow indicates the probable location of the observed G32R mutation.
Immunochemical analysis revealed a marked reduction of NDUFA1 in the patient cybrids compared to controls (Figure 3B), while the relative levels of the other complex I subunits tested were not significantly different. On BNG, the level of complex I in cybrid cells was reduced too (Figure 3D). Since NDUFA1 has been claimed to interact with ND subunits within the mitochondrial inner membrane, we looked at the relative levels of complex I polypeptides in cell lines harboring null mutations in ND5 and ND6 [22] by Western blot. The NDUFA1 and also the NDUFB8 subunits were absent from both null cell lines and the relative intensities of the other nDNA encoded subunits were markedly reduced (Figure 3C). Hence, the stability of the NDUFA1 subunit seems to be strongly dependent on the presence of normal mtDNA encoded subunits.
Discussion
Primary mitochondrial diseases due to an OXPHOS deficiency display a wide spectrum of clinical phenotypes and multiple complex I genes have been reported to be mutated in various cases [1]. In this study we describe two probands following an X-linked pedigree, both of whom experienced a relatively mild progressive encephalopathy, and exhibited a nearly complete complex I enzyme activity in muscle (Table 1). This deficiency was confirmed by BNG and by the loss of complex I subunits seen in Western blotting (Figure 1B and C). Detailed analysis of all nuclear complex I genes in the proband III-11 revealed a single novel missense variant (G32R) in the X-linked NDUFA1 gene. This was consistent with the X-linked nature of the pedigree, the biochemical defect generated by the NDUFA1 G32R in a hamster model cell culture system reduced the complex I activity by 41% (Figure 2A). Additional knock-in mutations in the hamster cell NDUFA1 gene at previously reported pathogenic residues suggests that mutations in highly conserved residues are often deleterious, though the severity of phenotype in hamster cell lines varied significantly from the reported patient phenotype. This also implies that the severity of G32R mutation in humans may be significantly different from the hamster model system.
Cell lines lacking the mtDNA-encoded ND5 or ND6 proteins also showed a complete loss of NDUFA1. There is published evidence that NDUFA1 is unstable in rho0 cells and in the absence of other membrane domain subunits like NDUFB11 [30]. This observation fits into the recently suggested model of complex I assembly by Lazarou et al [31] where mtDNA encoded ND subunits form a sub-assembly complex in the membrane domain along with various nuclear encoded subunits.
To determine if the mtDNA might also contribute to the observed complex I defect, we created transmitochondrial cybrids. Significantly, the cybrids were also found to have a 29% reduction in complex I activity in association with a marked reduction in the complex I level. Since all of the nucleotide variants in the mtDNA are linked and thus are inherited together, it is not clear which mtDNA variants or combination of variants accounts for the 29% reduction in complex I activity observed in the cybrid lines. The ND6 m.14582A>G (V31A) variant previously reported in association with the mild LHON ND6 missense mutation at m.14484T>C (M64V) was proposed to contribute to the manifestation of the disease [32]. A survey of a large cohort of patients with complex I deficiency revealed that the sequence variants in the mtDNA ND5 gene were common, [33] suggesting that the ND5 m.12599T>C M88T variant might also be a factor in causing the disease.
The NDUFA1 subunit is localized in the membrane at or near the junction of the membrane and the peripheral subcomplexes, thus placing it in the vicinity of the ND6 subunit (Figure 3E) (30). Therefore, the ND6 V31A polymorphism might be in a position to interact with the NDUFA1 G32R to generate the complex I defect. A direct interaction between ND5 and NDUFA1 is unlikely. Since ND5 subunit functions as a proton channel/pump [34] and it has been hypothesized that the membrane domain undergoes large conformational movements during proton pumping, it is likely that an interaction between NDUFA1 and ND5 does not necessarily require direct contact between the two proteins.
Recently, it was suggested that patient mitochondria with a specific ND5 mutation are increasingly dependent on glycolysis for maintaining the mitochondrial membrane potential in response to the OXPHOS deficiency [35]. Accordingly, the proband's mitochondria from lymphoblasts have shown a significant increase in ATP synthase expression which may represent a compensatory mechanism to offset the loss of complex I function.
Many patients with the clinical and biochemical features associated with mitochondrial disease arise “spontaneously” with no clear mendelian or maternal inheritance patterns. This suggests that aberrant interactions between the mtDNA and the nDNA encoded subunits of the OXPHOS complexes might be a common cause of mitochondrial disease. For diseases caused by complex I defects, interactions have already been documented between mtDNA haplogroup J and the milder primary Leber Hereditary Optic Neuropathy (LHON) mutations [36] and the variability in expression of the ND6 m.14459G>A (L60S) has been hypothesized to be modulated by nDNA variation [37].
Our results suggest that the NDUFA1 G32R mutation is deleterious and in combination with mtDNA variations may result in a synergistic defect. Analysing further cybrid experiments using different mtDNA backgrounds in our study is warranted in the future. The demonstration that mitochondrial diseases can arise from the random interaction of nuclear and mitochondrial DNA variants indicates that mitochondrial disease may be much more common than previously thought.
Supplementary Material
Supplemental Table 1: Primer Sequences for amplification and sequencing of the nuclear complex I cDNAs.
Acknowledgments
This research was supported in part by NIH grants RO1 NS21328, NS41850, AG24373 and FIS-PI08/0264.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Carroll J, Fearnley IM, Skehel JM, Shannon RJ, Hirst J, Walker JE. Bovine complex I is a complex of 45 different subunits. J Biol Chem. 2006;281:32724–32727. doi: 10.1074/jbc.M607135200. [DOI] [PubMed] [Google Scholar]
- 2.Janssen RJ, Nijtmans LG, van den Heuvel LP, Smeitink JA. Mitochondrial complex I: structure, function and pathology. J Inherit Metab Dis. 2006;29:499–515. doi: 10.1007/s10545-006-0362-4. [DOI] [PubMed] [Google Scholar]
- 3.Wallace DC, Lott MT, Procaccio V. Mitochondrial Genes in Degenerative Diseases, Cancer and Aging. In: Rimoin DL, Connor JM, Pyeritz RE, Korf BR, editors. Emery and Rimoin's Principles and Practice of Medical Genetics. 5th. Churchill Livingstone Elsevier; Philadelphia, PA: 2007. pp. 194–298. [Google Scholar]
- 4.Sazanov LA, Peak-Chew SY, Fearnley IM, Walker JE. Resolution of the membrane domain of bovine complex I into subcomplexes:implications for the structural organization of the enzyme. Biochemistry. 2000;39:7229–7235. doi: 10.1021/bi000335t. [DOI] [PubMed] [Google Scholar]
- 5.Hirst J, Carroll J, Fearnley IM, Shannon RJ, Walker JE. The nuclear encoded subunits of complex I from bovine heart mitochondria. Biochimica et Biophysica Acta. 2003;1604:135–150. doi: 10.1016/s0005-2728(03)00059-8. [DOI] [PubMed] [Google Scholar]
- 6.Zhuchenko O, Wehnert M, Bailey J, Sun ZS, Lee CC. Isolation, mapping, and genomic structure of an X-linked gene for a subunit of human mitochondrial complex I. Genomics. 1996;37:281–288. doi: 10.1006/geno.1996.0561. [DOI] [PubMed] [Google Scholar]
- 7.Fernandez-Moreira D, Ugalde C, Smeets R, Rodenburg RJ, Lopez-Laso E, Ruiz-Falco ML, Briones P, Martin MA, Smeitink JA, Arenas J. X-linked NDUFA1 gene mutations associated with mitochondrial encephalomyopathy. Ann Neurol. 2007;61:73–83. doi: 10.1002/ana.21036. [DOI] [PubMed] [Google Scholar]
- 8.Au HC, Seo BB, Matsuno-Yagi A, Yagi T, Scheffler IE. The NDUFA1 gene product (MWFE protein) is essential for activity of complex I in mammalian mitochondria. Proc Natl Acad Sci U S A. 1999;96:4354–4359. doi: 10.1073/pnas.96.8.4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yadava N, Potluri P, Smith EN, Bisevac A, Scheffler IE. Species-specific and mutant MWFE proteins. Their effect on the assembly of a functional mammalian mitochondrial complex I. Journal of Biological Chemistry. 2002;277:21221–21230. doi: 10.1074/jbc.M202016200. [DOI] [PubMed] [Google Scholar]
- 10.Chen R, Fearnley IM, Peak-Chew SY, Walker JE. The phosphorylation of subunits of complex I from bovine heart mitochondria. Journal of Biological Chemistry. 2004;279:26036–26045. doi: 10.1074/jbc.M402710200. [DOI] [PubMed] [Google Scholar]
- 11.Yadava N, Potluri P, Scheffler IE. Investigations of the potential effects of phosphorylation of the MWFE and ESSS subunits on complex I activity and assembly. Int J Biochem Cell Biol. 2007 doi: 10.1016/j.biocel.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 12.Bunn CL, Wallace DC, Eisenstadt JM. Cytoplasmic inheritance of chloramphenicol resistance in mouse tissue culture cells. Proceedings of the National Academy of Sciences of the United States of America. 1974;71:1681–1685. doi: 10.1073/pnas.71.5.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wallace DC, Bunn CL, Eisenstadt JM. Cytoplasmic transfer of chloramphenicol resistance in human tissue culture cells. Journal of Cell Biology. 1975;67:174–188. doi: 10.1083/jcb.67.1.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wallace DC, Eisenstadt JM. The expression of cytoplasmically inherited genes for chloramphenicol resistance in interspecific somatic cell hybrids and cybrids. Somatic Cell Genetics. 1979;5:573–396. [Google Scholar]
- 15.Giles RE, Blanc H, Cann HM, Wallace DC. Maternal inheritance of human mitochondrial DNA. Proceedings of the National Academy of Sciences of the United States of America. 1980;77:6715–6719. doi: 10.1073/pnas.77.11.6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McKenzie M, Trounce I. Expression of rattus norvegicus mtDNA in mus musculus cells results in multiple respiratory chain defects. Journal of Biological Chemistry. 2000;275:31514–31519. doi: 10.1074/jbc.M004070200. [DOI] [PubMed] [Google Scholar]
- 17.Barrientos A, Moraes CT. Titrating the effects of mitochondrial complex I impairment in the cell physiology. Journal of Biological Chemistry. 1999;274:16188–16197. doi: 10.1074/jbc.274.23.16188. [DOI] [PubMed] [Google Scholar]
- 18.Wallace DC, Ye JH, Neckelmann SN, Singh G, Webster KA, Greenberg BD. Sequence analysis of cDNAs for the human and bovine ATP synthase β-subunit: mitochondrial DNA genes sustain seventeen times more mutations. Current Genetics. 1987;12:81–90. doi: 10.1007/BF00434661. [DOI] [PubMed] [Google Scholar]
- 19.Mishmar D, Ruiz-Pesini E, Mondragon-Palomino M, Procaccio V, Gaut B, Wallace DC. Adaptive selection of mitochondrial complex I subunits during primate radiation. Gene. 2006;378:11–18. doi: 10.1016/j.gene.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 20.Trounce IA, Kim YL, Jun AS, Wallace DC. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods in Enzymology. 1996;264:484–509. doi: 10.1016/s0076-6879(96)64044-0. [DOI] [PubMed] [Google Scholar]
- 21.Bayona-Bafaluy MP, Manfredi G, Moraes CT. A chemical enucleation method for the transfer of mitochondrial DNA to rho(0) cells. Nucleic Acids Research. 2003;31:e98. doi: 10.1093/nar/gng100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chomyn A. Mitochondrial genetic control of assembly and function of complex I in mammalian cells. J Bioenerg Biomembr. 2001;33:251–257. doi: 10.1023/a:1010791204961. [DOI] [PubMed] [Google Scholar]
- 23.Scheffler IE, Yadava N. Molecular genetics of the mammalian NADH-ubiquinone oxidoreductase. J Bioenerg Biomembr. 2001;33:243–250. doi: 10.1023/a:1010739120891. [DOI] [PubMed] [Google Scholar]
- 24.Procaccio V, Wallace DC. Late-onset Leigh syndrome in a patient with mitochondrial complex I NDUFS8 mutations. Neurology. 2004;62:1899–1901. doi: 10.1212/01.wnl.0000125251.56131.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seo BB, Kitajima-Ihara T, Chan EK, Scheffler IE, Matsuno-Yagi A, Yagi T. Molecular remedy of complex I defects: rotenone-insensitive internal NADH-quinone oxidoreductase of Saccharomyces cerevisiae mitochondria restores the NADH oxidase activity of complex I-deficient mammalian cells. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:9167–9171. doi: 10.1073/pnas.95.16.9167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wittig I, Karas M, Schagger H. High resolution clear native electrophoresis for in-gel functional assays and fluorescence studies of membrane protein complexes. Mol Cell Proteomics. 2007;6:1215–1225. doi: 10.1074/mcp.M700076-MCP200. [DOI] [PubMed] [Google Scholar]
- 27.Ruiz-Pesini E, Lott MT, Procaccio V, Poole J, Brandon MC, Mishmar D, Yi C, Kreuziger J, Baldi P, Wallace DC. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Research. 2007;35:D823–D828. doi: 10.1093/nar/gkl927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ingman M, Gyllensten U. mtDB: Human Mitochondrial Genome Database, a resource for population genetics and medical sciences. Nucleic Acids Res. 2006;34:D749–751. doi: 10.1093/nar/gkj010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Munakata K, Tanaka M, Mori K, Washizuka S, Yoneda M, Tajima O, Akiyama T, Nanko S, Kunugi H, Tadokoro K, Ozaki N, Inada T, Sakamoto K, Fukunaga T, Iijima Y, Iwata N, Tatsumi M, Yamada K, Yoshikawa T, Kato T. Mitochondrial DNA 3644T-->C mutation associated with bipolar disorder. Genomics. 2004;84:1041–1050. doi: 10.1016/j.ygeno.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 30.Potluri P, Yadava N, Scheffler IE. The role of the ESSS protein in the assembly of a functional and stable mammalian mitochondrial complex I (NADH-ubiquinone oxidoreductase) Eur J Biochem. 2004;271:3265–3273. doi: 10.1111/j.1432-1033.2004.04260.x. [DOI] [PubMed] [Google Scholar]
- 31.Lazarou M, Thorburn DR, Ryan MT, McKenzie M. Assembly of mitochondrial complex I and defects in disease. Biochim Biophys Acta. 2008 doi: 10.1016/j.bbamcr.2008.04.015. [DOI] [PubMed] [Google Scholar]
- 32.Wissinger B, Besch D, Baumann B, Fauser S, Christ-Adler M, Jurklies B, Zrenner E, Leo-Kottler B. Mutation analysis of the ND6 gene in patients with Lebers hereditary optic neuropathy. Biochemical and Biophysical Research Communications. 1997;234:511–515. doi: 10.1006/bbrc.1997.6660. [DOI] [PubMed] [Google Scholar]
- 33.Blok MJ, Spruijt L, de Coo IF, Schoonderwoerd K, Hendrickx A, Smeets HJ. Mutations in the ND5 subunit of complex I of the mitochondrial DNA are a frequent cause of oxidative phosphorylation disease. J Med Genet. 2007;44:e74. doi: 10.1136/jmg.2006.045716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baranova EA, Holt PJ, Sazanov LA. Projection structure of the membrane domain of Escherichia coli respiratory complex I at 8 A resolution. J Mol Biol. 2007;366:140–154. doi: 10.1016/j.jmb.2006.11.026. [DOI] [PubMed] [Google Scholar]
- 35.McKenzie M, Liolitsa D, Akinshina N, Campanella M, Sisodiya S, Hargreaves I, Nirmalananthan N, Sweeney MG, Abou-Sleiman PM, Wood NW, Hanna MG, Duchen MR. Mitochondrial ND5 Gene Variation Associated with Encephalomyopathy and Mitochondrial ATP Consumption. J Biol Chem. 2007;282:36845–36852. doi: 10.1074/jbc.M704158200. [DOI] [PubMed] [Google Scholar]
- 36.Brown MD, Sun F, Wallace DC. Clustering of Caucasian Leber hereditary optic neuropathy patients containing the 11778 or 14484 mutations on an mtDNA lineage. American Journal of Human Genetics. 1997;60:381–387. [PMC free article] [PubMed] [Google Scholar]
- 37.Gropman A, Chen TJ, Perng CL, Krasnewich D, Chernoff E, Tifft C, Wong LJ. Variable clinical manifestation of homoplasmic G14459A mitochondrial DNA mutation. American Journal of Medical Genetics. 2004;124A:377–382. doi: 10.1002/ajmg.a.20456. [DOI] [PubMed] [Google Scholar]
- 38.Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nature Genetics. 1999;23:147. doi: 10.1038/13779. [DOI] [PubMed] [Google Scholar]
- 39.Ruiz-Pesini E, Mishmar D, Brandon M, Procaccio V, Wallace DC. Effects of purifying and adaptive selection on regional variation in human mtDNA. Science. 2004;303:223–226. doi: 10.1126/science.1088434. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1: Primer Sequences for amplification and sequencing of the nuclear complex I cDNAs.